
Synthesis and Cytotoxic Activity of Biphenylurea Derivatives Containing Indolin-2-one Moieties

 ,
,  ,
,  , and
, and
Abstract
:
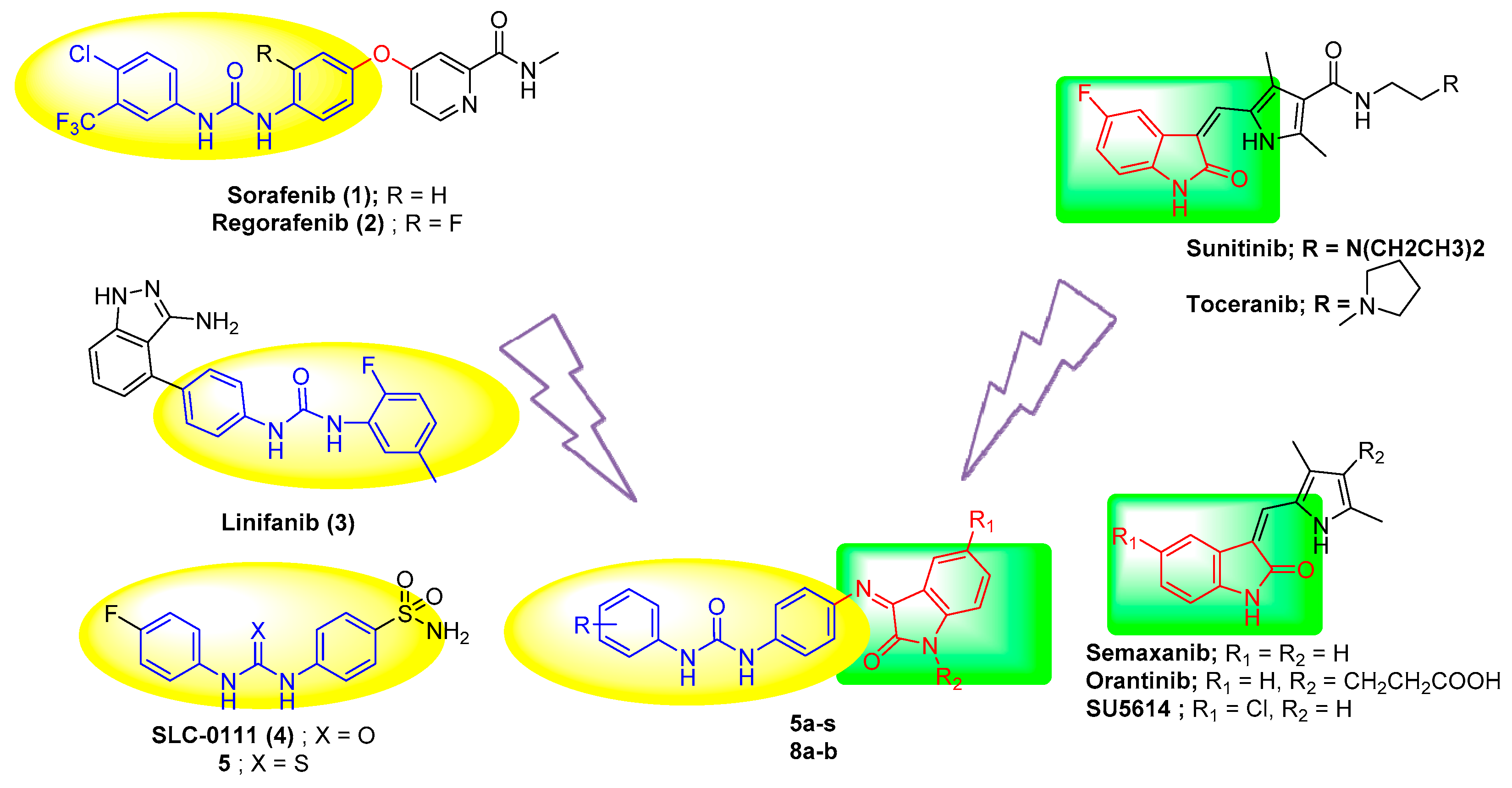
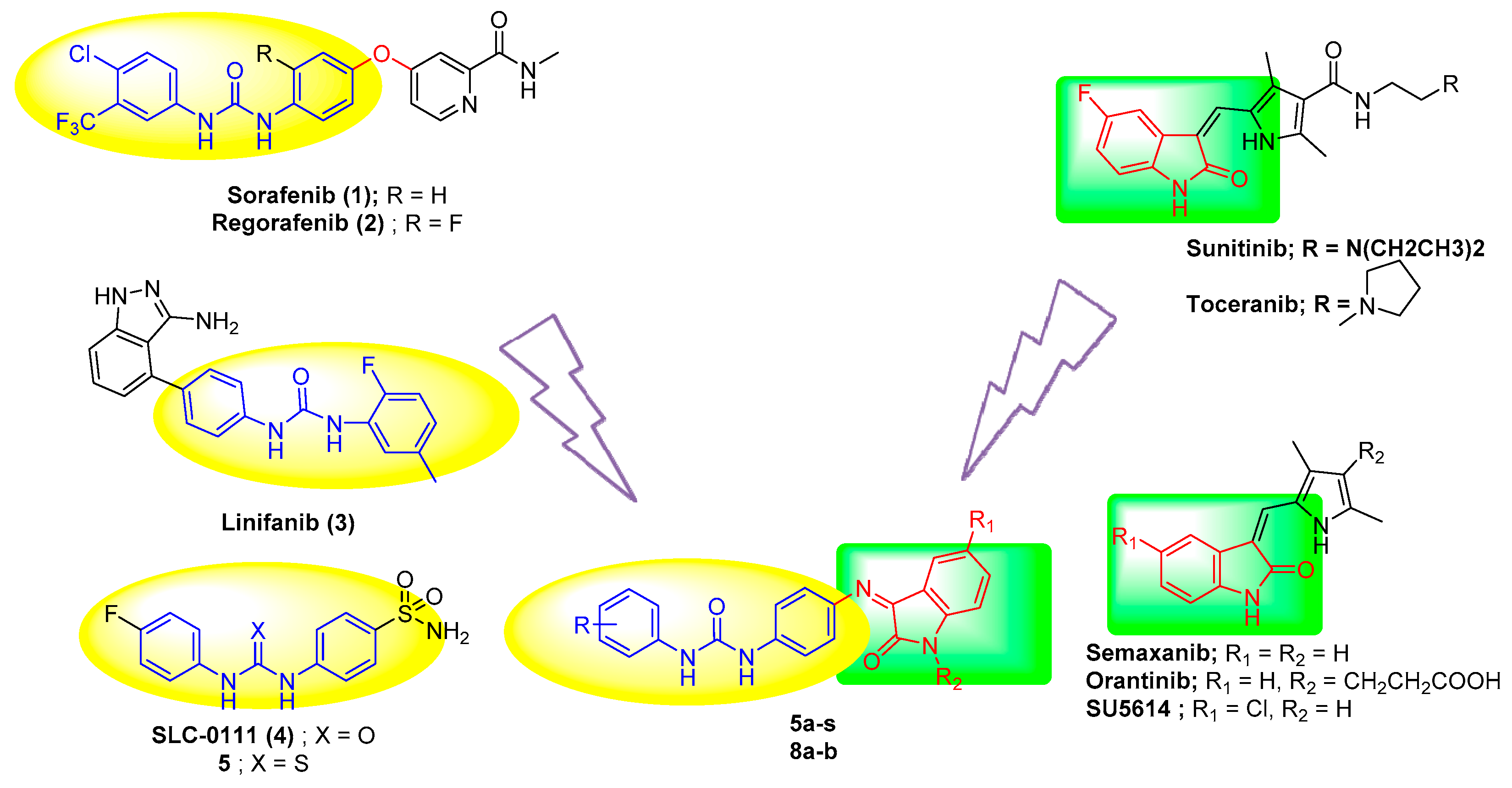
1. Introduction
2. Results and Discussion
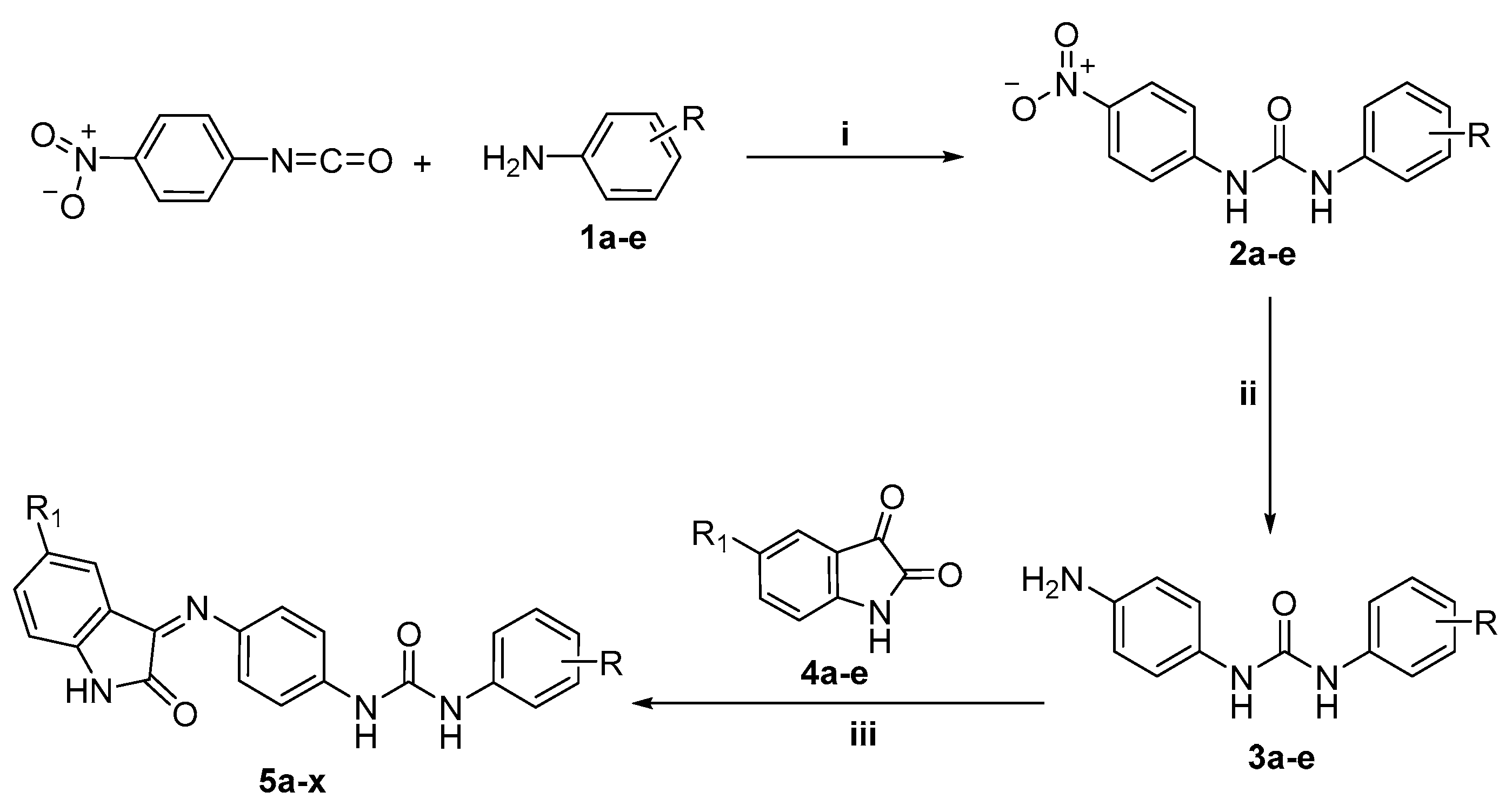
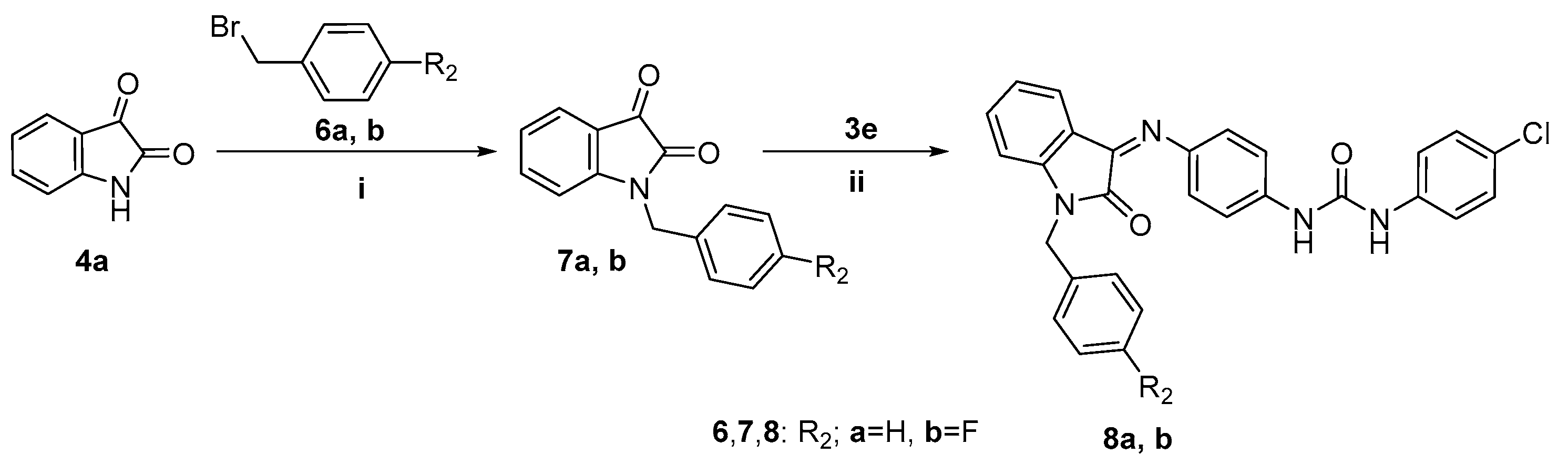
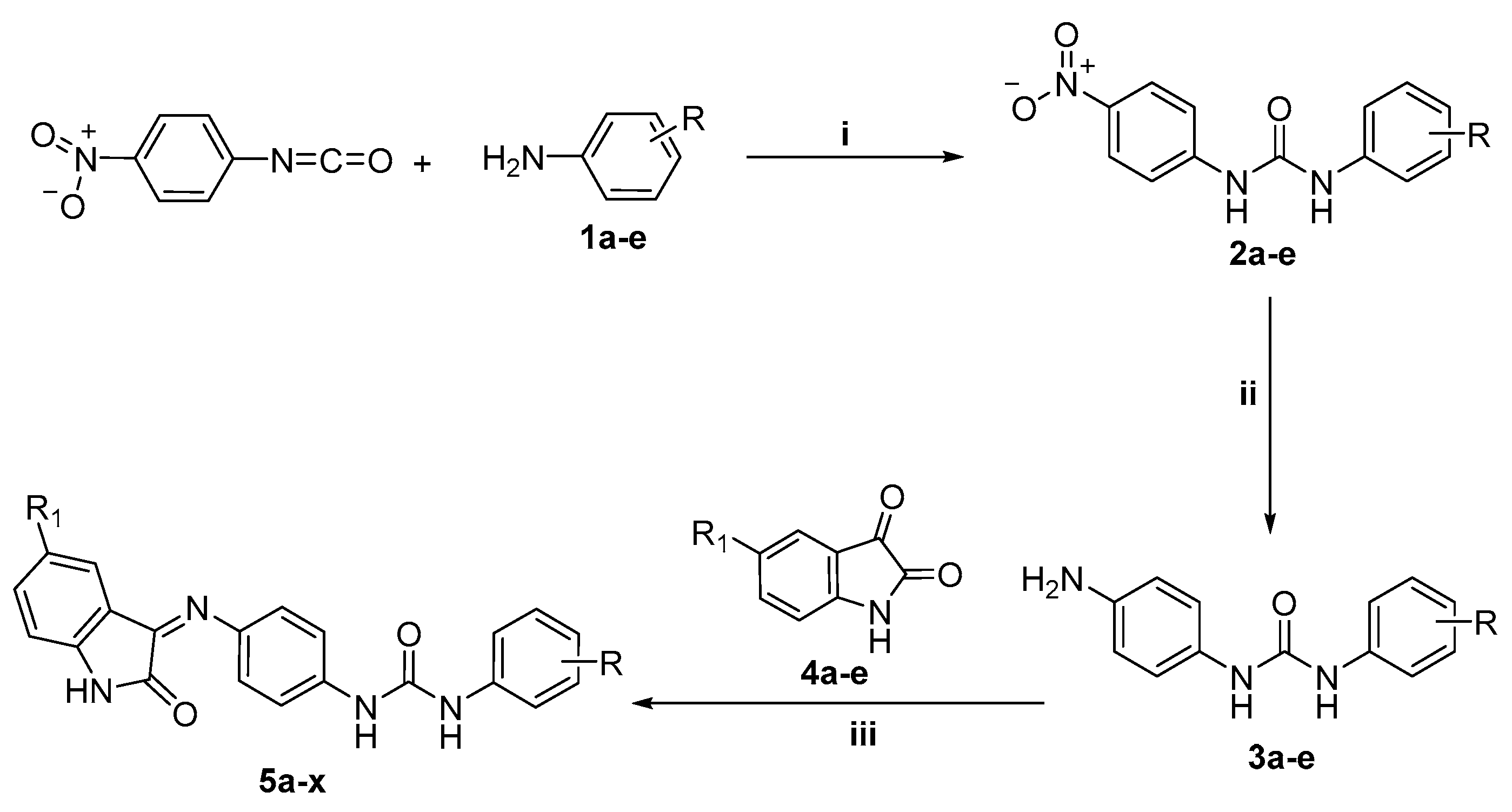
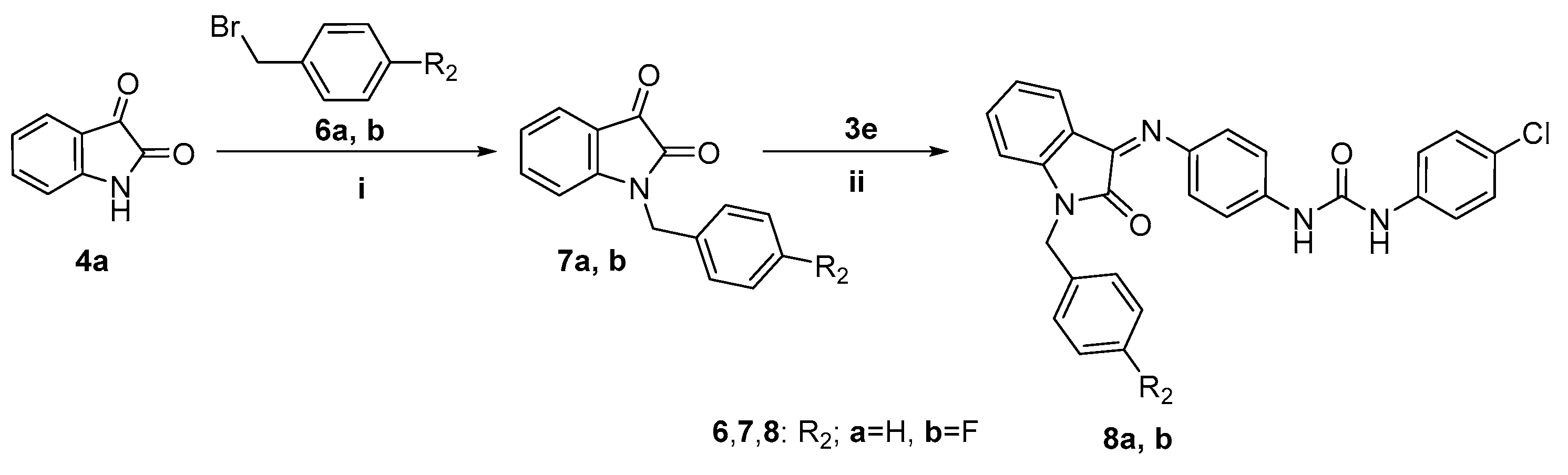
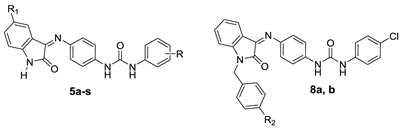
2.1. Chemistry
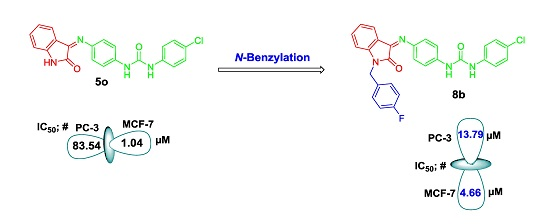
2.2. Biological Evaluation
2.3. Physicochemical Properties and ADME Profiling
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. 1-(4-Nitrophenyl)-3-phenyl/substituted phenylurea 2a–e
3.2.2. 1-(4-Aminophenyl)-3-phenyl/substituted phenylurea 3a–e
3.2.3. General Procedure for Preparation of Target Compounds 5a–x
3.2.4. N-Benzylindoline-2,3-diones 7a,b
3.2.5. General Procedure for Preparation of Target Compounds 8a,b
3.3. In Vitro Cytotoxic Activity
3.3.1. Cell Culture
3.3.2. In vitro Cytotoxic Activity
3.3.3. Physicochemical Properties and ADME Profiling
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Cancer Report 2014. Available online: http://www.iarc.fr/en/publications/books/wcr/wcr-order.php (accessed on 6 May 2016).
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA-Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Havrylyuk, D.; Kovach, N.; Zimenkovsky, B.; Vasylenko, O.; Lesyk, R. Synthesis and Anticancer Activity of Isatin-Based Pyrazolines and Thiazolidines Conjugates. Arch. Pharm. 2011, 344, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Aly, M.H.; Zada, S.; Ali, M.M.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; El Ella, D.A.A. Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: Synthesis, in vitro biological evaluation and molecular docking. Eur. J. Med. Chem. 2015, 100, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Ibrahim, H.S.; Abdel-Aziz, H.A.; Farrag, N.N.; Youssef, M.M. Design, synthesis and in vitro antitumor activity of novel N-substituted-4-phenyl/benzylphthalazin-1-ones. Eur. J. Med. Chem. 2015, 89, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.S.; Abou-seri, S.M.; Ismail, N.S.; Elaasser, M.M.; Aly, M.H.; Abdel-Aziz, H.A. Bis-isatin hydrazones with novel linkers: Synthesis and biological evaluation as cytotoxic agents. Eur. J. Med. Chem. 2016, 108, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Sławiński, J.; Grzonek, A.; Żołnowska, B.; Kawiak, A. Synthesis of Novel Pyrido[4,3-e][1,2,4] triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide Derivatives with Potential Anticancer Activity. Molecules 2015, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Fares, M.; Eldehna, W.M.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; Aly, M.H.; Tolba, M.F. Design, Synthesis and in Vitro Antiproliferative Activity of Novel Isatin-Quinazoline Hybrids. Arch. Pharm. 2015, 348, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.Y.; Heo, J. Sorafenib in liver cancer. Expert. Opin. Pharmacother. 2012, 13, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Dumas, J.; Ladouceur, G.; Lynch, M.; Scott, W. Diaryl Ureas with Kinase Inhibiting Activity. U.S. Patent 20070020704, 25 January 2007. [Google Scholar]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73–4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- DiGiulio, S. FDA Approves Stivarga for Advanced GIST. Oncol. Times 2013, 35, 12. [Google Scholar]
- Tan, E.-H.; Goss, G.D.; Salgia, R.; Besse, B.; Gandara, D.R.; Hanna, N.H.; Yang, J.C.-H.; Thertulien, R.; Wertheim, M.; Mazieres, J. Phase 2 trial of Linifanib (ABT-869) in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Winum, J.-Y. Designing carbonic anhydrase inhibitors for the treatment of breast cancer. Expert Opin. Drug Discov. 2015, 10, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.L.; Mahon, B.P.; McKenna, R.; Carta, F.; Supuran, C.T. Kinetic and X-ray crystallographic investigations on carbonic anhydrase isoforms I, II, IX and XII of a thioureido analog of SLC-0111. Bioorg. Med. Chem. 2016, 24, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Vine, K.; Matesic, L.; Locke, J.; Ranson, M.; Skropeta, D. Cytotoxic and anticancer activities of isatin and its derivatives: a comprehensive review from 2000–2008. Anti-Cancer Agents Med. Chem. 2009, 9, 397–414. [Google Scholar] [CrossRef]
- Vine, K.L.; Matesic, L.; Locke, J.M.; Skropeta, D. Recent highlights in the development of isatin-based anticancer agents. Adv. Anticancer Agents Med. Chem. 2013, 2, 254–312. [Google Scholar]
- Motzer, R.J.; Michaelson, M.D.; Redman, B.G.; Hudes, G.R.; Wilding, G.; Figlin, R.A.; Ginsberg, M.S.; Kim, S.T.; Baum, C.M.; DePrimo, S.E. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Cools, J.; Mentens, N.; Folens, C.; Sciot, R.; Schöffski, P.; Van Oosterom, A.; Marynen, P.; Debiec-Rychter, M. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin. Cancer Res. 2006, 12, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- London, C.A.; Malpas, P.B.; Wood-Follis, S.L.; Boucher, J.F.; Rusk, A.W.; Rosenberg, M.P.; Henry, C.J.; Mitchener, K.L.; Klein, M.K.; Hintermeister, J.G. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin. Cancer Res. 2009, 15, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Sun, Y.; Chen, P.; Sackett, D.L.; Hu, Z.; Rich, W.; Li, C.; Lewis, A.; Schaefer, K.; Li, P.-K. Structure–activity-relationship studies of conformationally restricted analogs of combretastatin A-4 derived from SU5416. Bioorg. Med. Chem. 2006, 14, 6492–6501. [Google Scholar] [CrossRef] [PubMed]
- Griffith, R.; Brown, M.; McCluskey, A.; Ashman, L. Small molecule inhibitors of protein kinases in cancer-how to overcome resistance. Mini Rev. Med. Chem. 2006, 6, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Sessa, C.; Viganò, L.; Grasselli, G.; Trigo, J.; Marimon, I.; Lladò, A.; Locatelli, A.; Ielmini, N.; Marsoni, S.; Gianni, L. Phase I clinical and pharmacological evaluation of the multi-tyrosine kinase inhibitor SU006668 by chronic oral dosing. Eur. J. Cancer 2006, 42, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.D.; Vajkoczy, P.; Shawver, L.K.; Thurnher, A.; Liang, C.; Mohammadi, M.; Schlessinger, J.; Ullrich, A.; Hubbard, S.R.; Blake, R.A. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000, 60, 4152–4160. [Google Scholar] [PubMed]
- Spiekermann, K.; Faber, F.; Voswinckel, R.; Hiddemann, W. The protein tyrosine kinase inhibitor SU5614 inhibits VEGF-induced endothelial cell sprouting and induces growth arrest and apoptosis by inhibition of c-kit in AML cells. Exp. Hematol. 2002, 30, 767–773. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Fares, M.; Ceruso, M.; Ghabbour, H.A.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; El Ella, D.A.A.; Supuran, C.T. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur. J. Med. Chem. 2016, 110, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer. Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Abou-Seri, S.M.; El Kerdawy, A.M.; Ayyad, R.R.; Hamdy, A.M.; Ghabbour, H.A.; Ali, M.M.; El Ella, D.A.A. Increasing the binding affinity of VEGFR-2 inhibitors by extending their hydrophobic interaction with the active site: Design, synthesis and biological evaluation of 1-substituted-4-(4-methoxybenzyl) phthalazine derivatives. Eur. J. Med. Chem. 2016, 113, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Presset, M.; Mohanan, K.; Hamann, M.; Coquerel, Y.; Rodriguez, J. 1, 3-Dipolar cycloaddition of hydrazones with α-oxo-ketenes: A three-component stereoselective entry to pyrazolidinones and an original class of spirooxindoles. Org. Lett. 2011, 13, 4124–4127. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of all compounds are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R | R1 | R2 | IC50 (μM) a | |
|---|---|---|---|---|---|
| MCF-7 | PC-3 | ||||
| 5a | H | H | - | 19.53 ± 1.05 | NA b |
| 5b | H | F | - | 17.14 ± 0.66 | NA b |
| 5c | H | Cl | - | 19.17 ± 1.94 | 20.43 ± 1.51 |
| 5d | 4-F | H | - | NA b | NA b |
| 5e | 4-F | F | - | NA b | NA b |
| 5f | 4-F | Cl | - | 48.99 ± 3.74 | NA b |
| 5g | 3-CF3 | H | - | 39.53 ± 4.02 | NA b |
| 5h | 3-CF3 | F | - | NA b | 51.76 ± 4.01 |
| 5i | 3-CF3 | Cl | - | 97.65 ± 7.39 | NA b |
| 5j | 3-Cl | H | - | NA b | NA b |
| 5k | 3-Cl | F | - | 75.92 ± 7.21 | NA b |
| 5l | 3-Cl | Cl | - | 1.93 ± 0.17 | NA b |
| 5m | 3-Cl | Me | - | 35.59 ± 3.27 | 73.22 ± 5.07 |
| 5n | 3-Cl | OMe | - | 9.60 ± 1.08 | 96.00 ± 6.85 |
| 5o | 4-Cl | H | - | 1.04 ± 0.10 | 83.54 ± 6.32 |
| 5p | 4-Cl | F | - | 26.02 ± 2.51 | NA b |
| 5q | 4-Cl | Cl | - | 3.87 ± 0.31 | NA b |
| 5r | 4-Cl | Me | - | 12.16 ± 1.17 | NA b |
| 5s | 4-Cl | OMe | - | 43.08 ± 4.22 | 97.00 ± 3.25 |
| 8a | - | - | H | 28.00 ± 2.93 | 18.40 ± 1.33 |
| 8b | - | - | F | 4.66 ± 0.42 | 13.79 ± 0.88 |
| Dox. | - | - | - | 7.30 ± 0.84 | 0.84 ± 0.079 |
| Compd. | H-bond Donor a,# | H-bond Acceptor b,# | Molecular Weight | AlogP d | No. of Rotatable Bonds e | Polar Surface Area f (Å) |
|---|---|---|---|---|---|---|
| 5a | 3 | 6 | 356.4 | 3.200 | 3 | 82.59 |
| 5b | 3 | 6 | 374.4 | 3.405 | 3 | 82.59 |
| 5c | 3 | 6 | 390.8 | 3.864 | 3 | 82.59 |
| 5d | 3 | 6 | 374.4 | 3.405 | 3 | 82.59 |
| 5e | 3 | 6 | 392.4 | 3.611 | 3 | 82.59 |
| 5f | 3 | 6 | 408.8 | 4.07 | 3 | 82.59 |
| 5g | 3 | 6 | 424.4 | 4.142 | 4 | 82.59 |
| 5h | 3 | 6 | 442.4 | 4.348 | 4 | 82.59 |
| 5i | 3 | 6 | 458.8 | 4.807 | 4 | 82.59 |
| 5j | 3 | 6 | 390.8 | 3.864 | 3 | 82.59 |
| 5k | 3 | 6 | 408.8 | 4.07 | 3 | 82.59 |
| 5l | 3 | 6 | 425.3 | 4.529 | 3 | 82.59 |
| 5m | 3 | 6 | 404.8 | 4.351 | 3 | 82.59 |
| 5n | 3 | 7 | 420.8 | 3.848 | 4 | 91.82 |
| 5o | 3 | 6 | 390.8 | 3.864 | 3 | 82.59 |
| 5p | 3 | 6 | 408.8 | 4.07 | 3 | 82.59 |
| 5q | 3 | 6 | 425.3 | 4.529 | 3 | 82.59 |
| 5r | 3 | 6 | 404.8 | 4.351 | 3 | 82.59 |
| 5s | 3 | 7 | 420.8 | 3.848 | 4 | 91.82 |
| 8a | 2 | 6 | 480.9 | 5.654 * | 5 | 73.8 |
| 8b | 2 | 6 | 498.9 | 5.859 * | 5 | 73.8 |
| Compd. | ADMET Absorption Level a | ADMET Solubility b | ADMET Solubility Level c | ADMET BBB d | ADMET BBB Level d | CYP2D6 Probability | CYP2D6 e |
|---|---|---|---|---|---|---|---|
| 5a | 0 | −4.438 | 2 | −0.500 | 2 | 0.722 | 1 |
| 5b | 0 | −4.899 | 2 | −0.436 | 2 | 0.594 | 1 |
| 5c | 0 | −5.299 | 2 | −0.294 | 2 | 0.584 | 1 |
| 5d | 0 | −4.878 | 2 | −0.436 | 2 | 0.633 | 1 |
| 5e | 0 | −5.331 | 2 | −0.373 | 2 | 0.584 | 1 |
| 5f | 0 | −5.732 | 2 | −0.231 | 2 | 0.485 | 0 |
| 5g | 0 | −6.048 | 1 | −0.208 | 2 | 0.495 | 0 |
| 5h | 0 | −6.477 | 1 | −0.145 | 2 | 0.465 | 0 |
| 5i | 0 | −6.877 | 1 | NA | 4 | 0.544 | 1 |
| 5j | 0 | −5.289 | 2 | −0.294 | 2 | 0.683 | 1 |
| 5k | 0 | −5.742 | 2 | −0.231 | 2 | 0.504 | 1 |
| 5l | 0 | −6.143 | 1 | −0.089 | 2 | 0.514 | 1 |
| 5m | 0 | −5.734 | 2 | −0.144 | 2 | 0.653 | 1 |
| 5n | 0 | −5.415 | 2 | −0.441 | 2 | 0.683 | 1 |
| 5o | 0 | −5.279 | 2 | −0.294 | 2 | 0.623 | 1 |
| 5p | 0 | −5.732 | 2 | −0.231 | 2 | 0.485 | 0 |
| 5q | 0 | −6.132 | 1 | −0.089 | 2 | 0.504 | 1 |
| 5r | 0 | −5.723 | 2 | −0.144 | 2 | 0.643 | 1 |
| 5s | 0 | −5.404 | 2 | −0.441 | 2 | 0.524 | 1 |
| 8a | 1 | -6.436 | 1 | NA | 4 | 0.712 | 1 |
| 8b | 1 | -6.737 | 1 | NA | 4 | 0.762 | 1 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Alsherbiny, M.A.; Aly, M.H.; Ghabbour, H.A.; Abdel-Aziz, H.A. Synthesis and Cytotoxic Activity of Biphenylurea Derivatives Containing Indolin-2-one Moieties. Molecules 2016, 21, 762. https://doi.org/10.3390/molecules21060762
Eldehna WM, Fares M, Ibrahim HS, Alsherbiny MA, Aly MH, Ghabbour HA, Abdel-Aziz HA. Synthesis and Cytotoxic Activity of Biphenylurea Derivatives Containing Indolin-2-one Moieties. Molecules. 2016; 21(6):762. https://doi.org/10.3390/molecules21060762
Chicago/Turabian StyleEldehna, Wagdy M., Mohamed Fares, Hany S. Ibrahim, Muhammad A. Alsherbiny, Mohamed H. Aly, Hazem A. Ghabbour, and Hatem A. Abdel-Aziz. 2016. "Synthesis and Cytotoxic Activity of Biphenylurea Derivatives Containing Indolin-2-one Moieties" Molecules 21, no. 6: 762. https://doi.org/10.3390/molecules21060762
APA StyleEldehna, W. M., Fares, M., Ibrahim, H. S., Alsherbiny, M. A., Aly, M. H., Ghabbour, H. A., & Abdel-Aziz, H. A. (2016). Synthesis and Cytotoxic Activity of Biphenylurea Derivatives Containing Indolin-2-one Moieties. Molecules, 21(6), 762. https://doi.org/10.3390/molecules21060762