
Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts

 , , and
, , and 
Abstract
:
1. Introduction
2. Results and Discussion
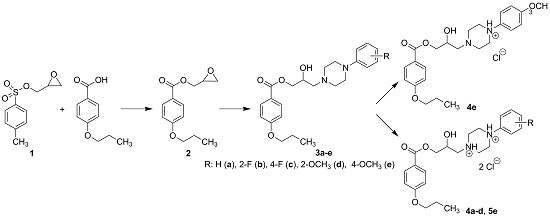
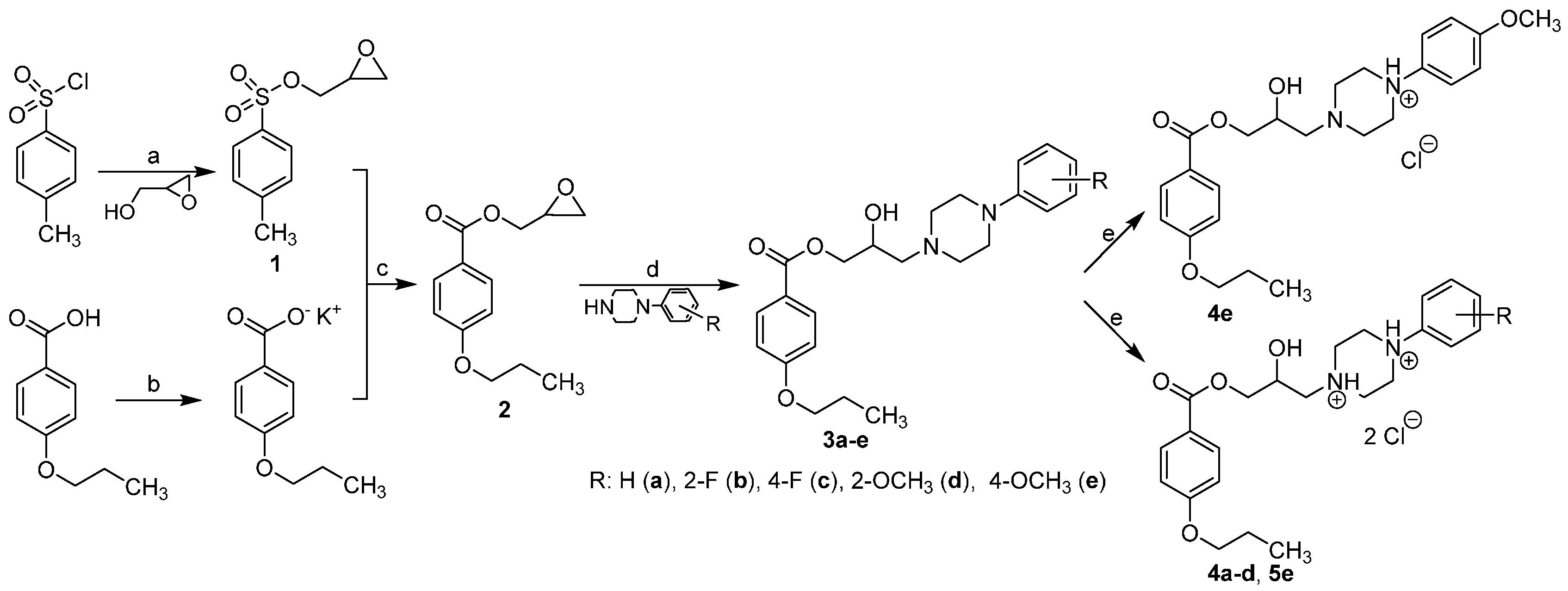
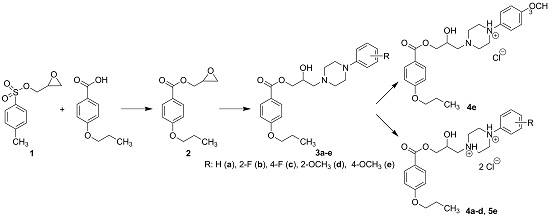
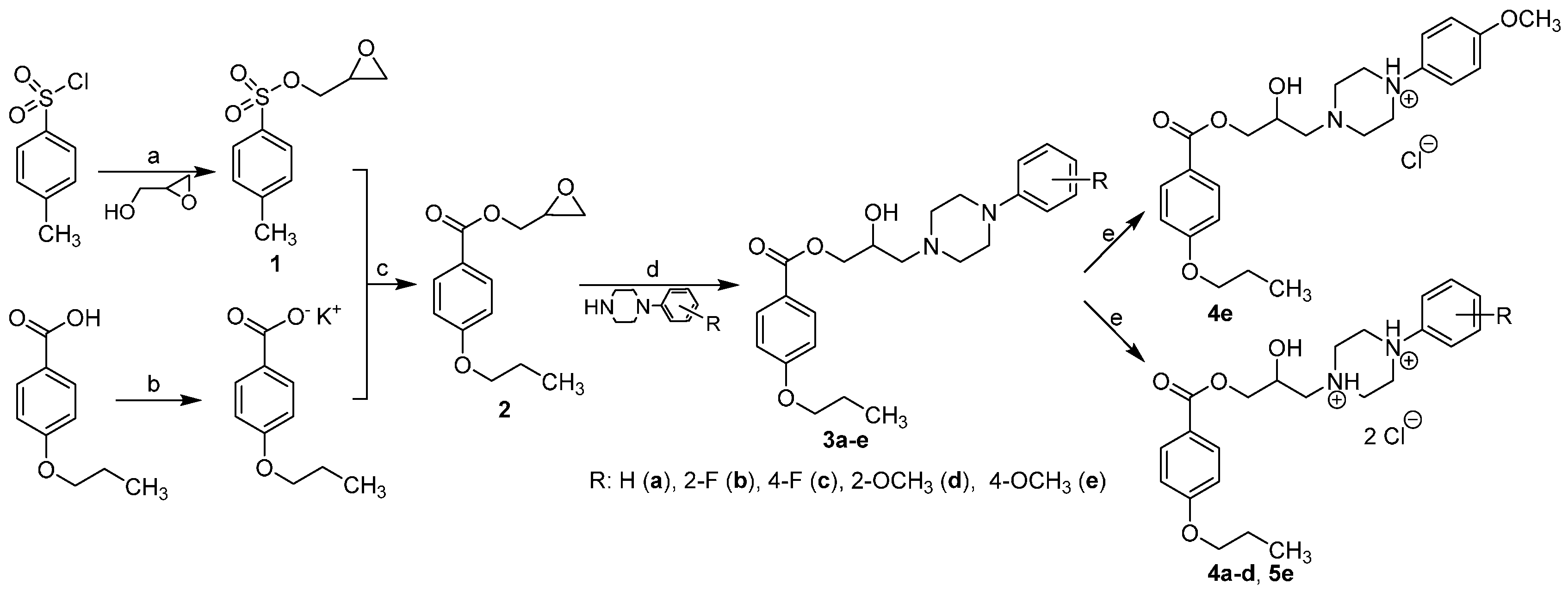
2.1. Chemistry
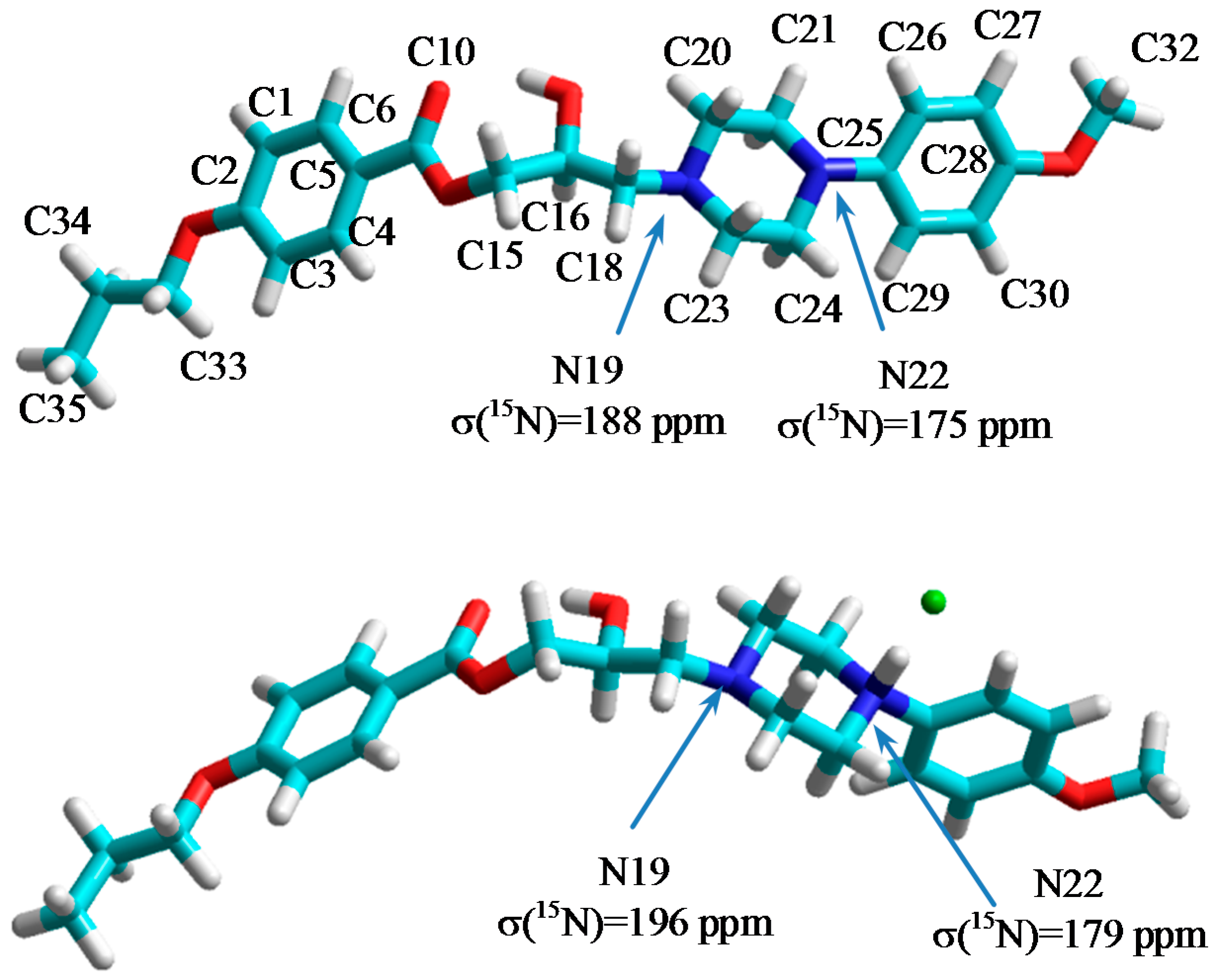
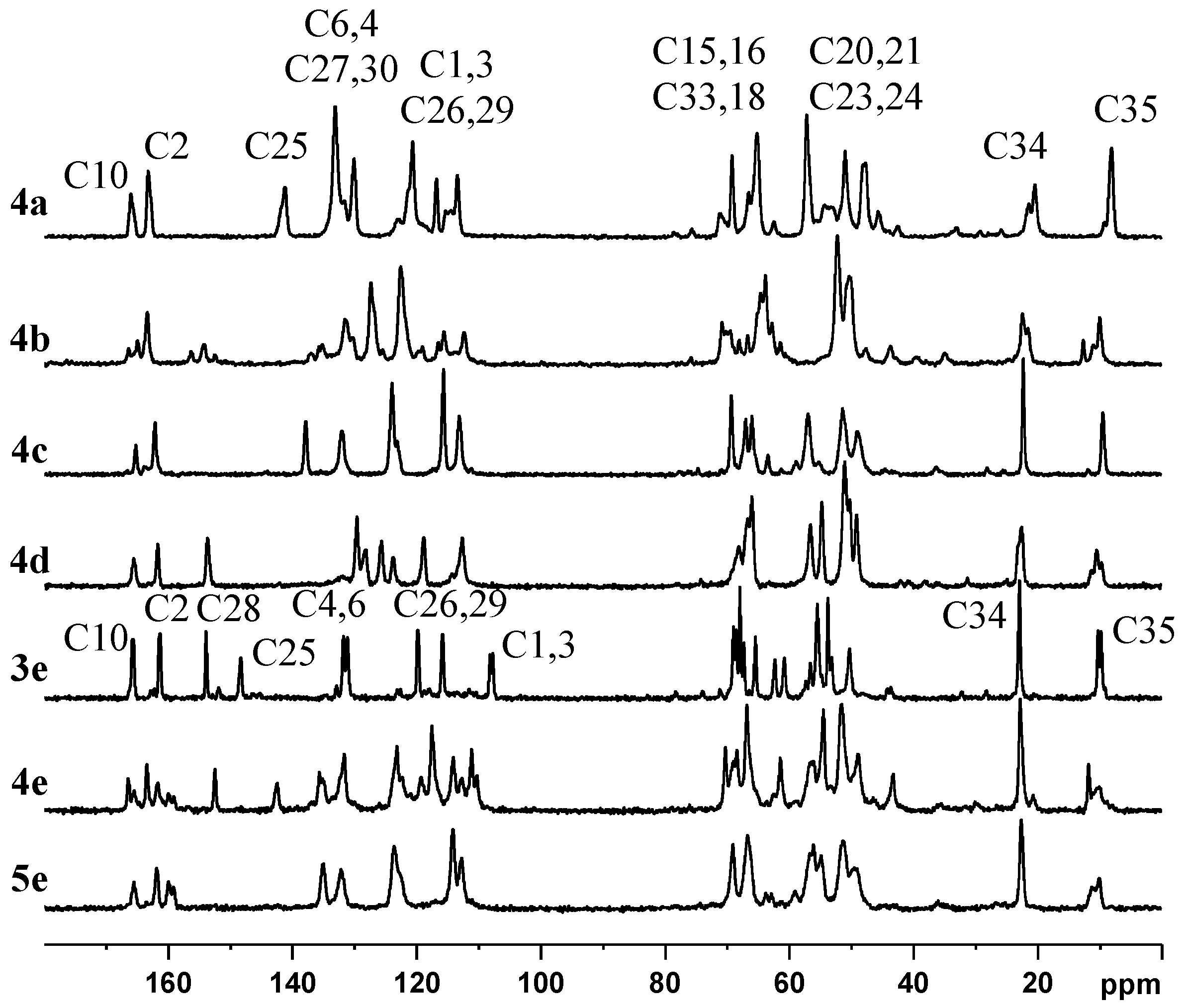
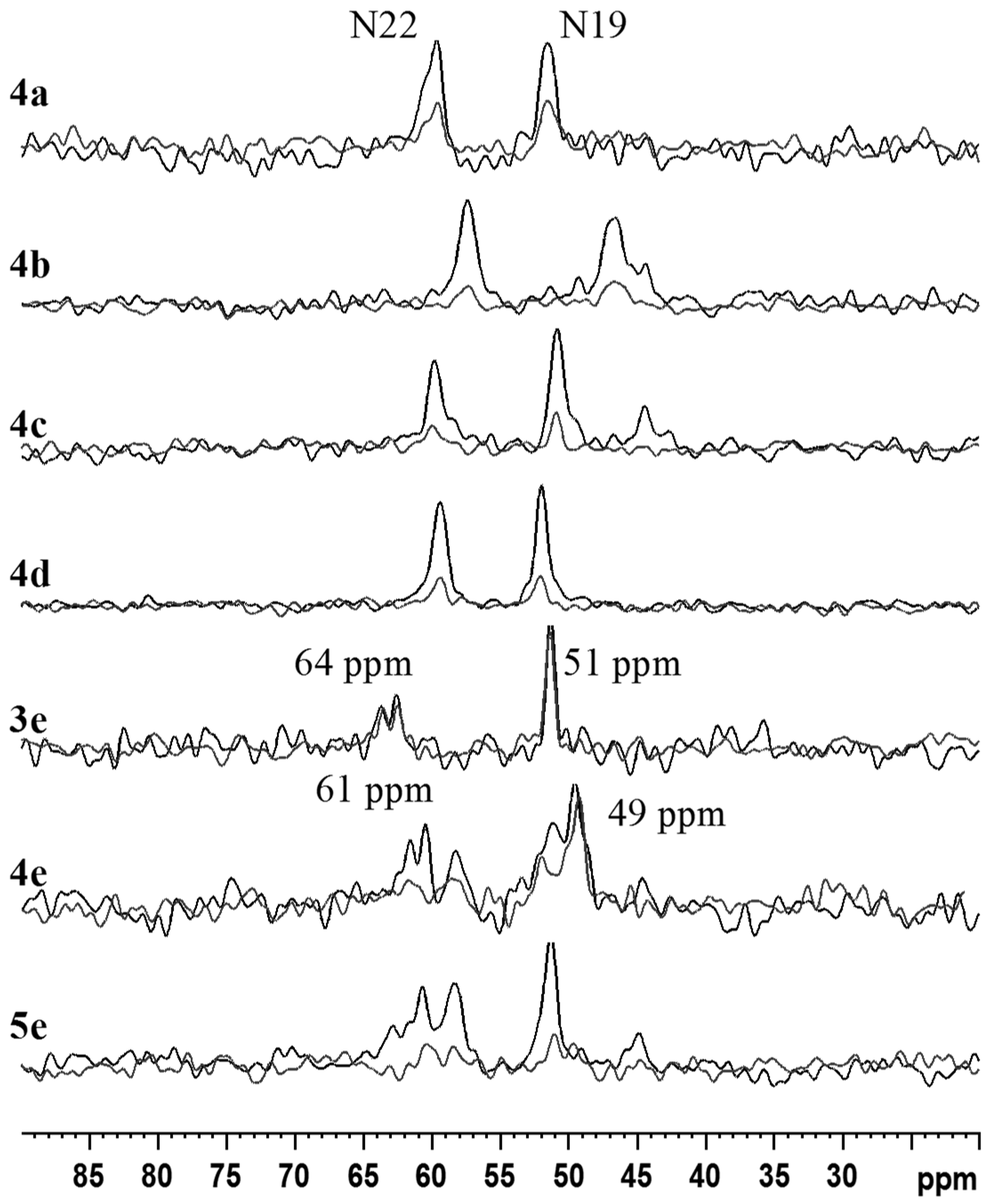
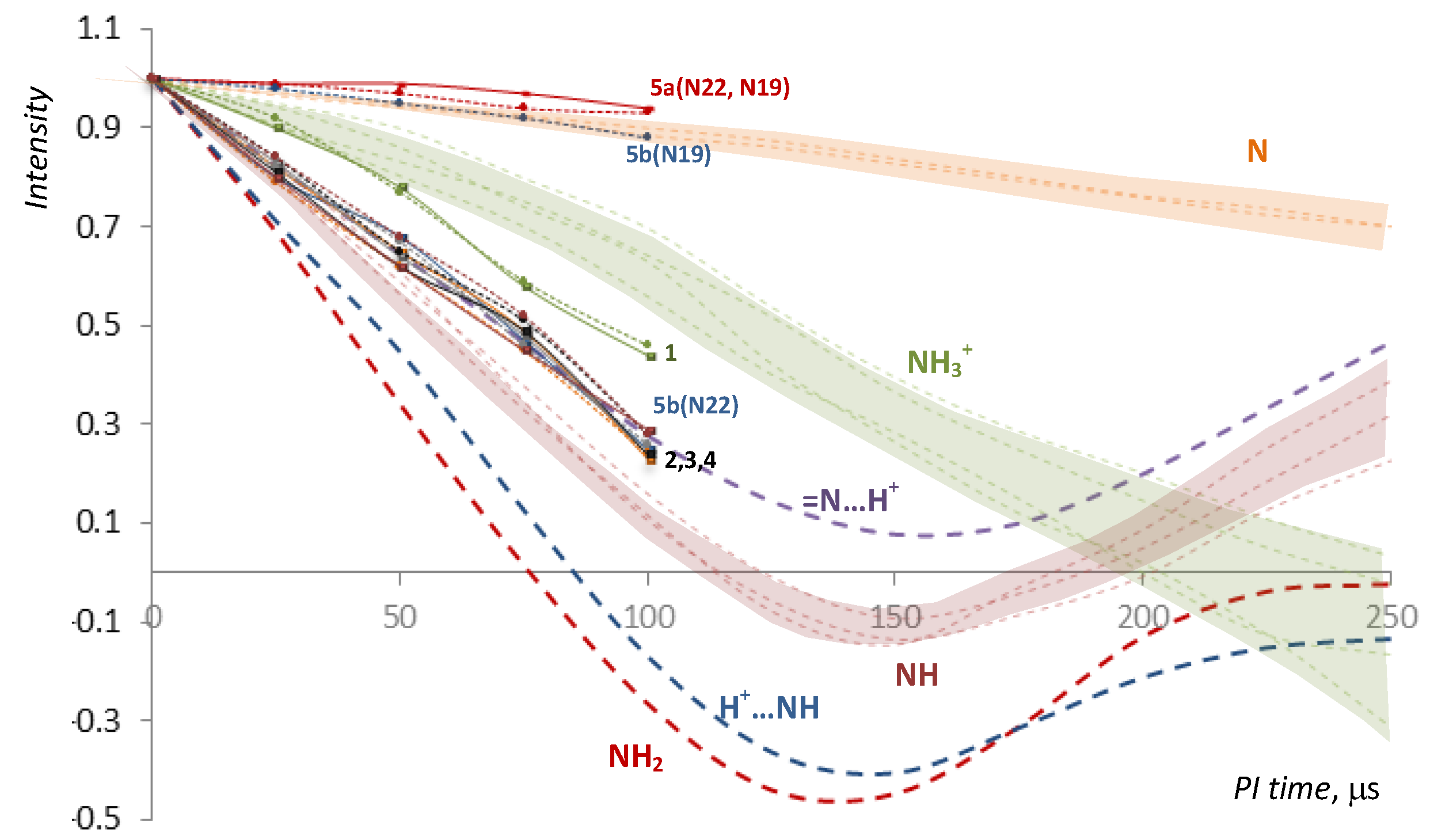
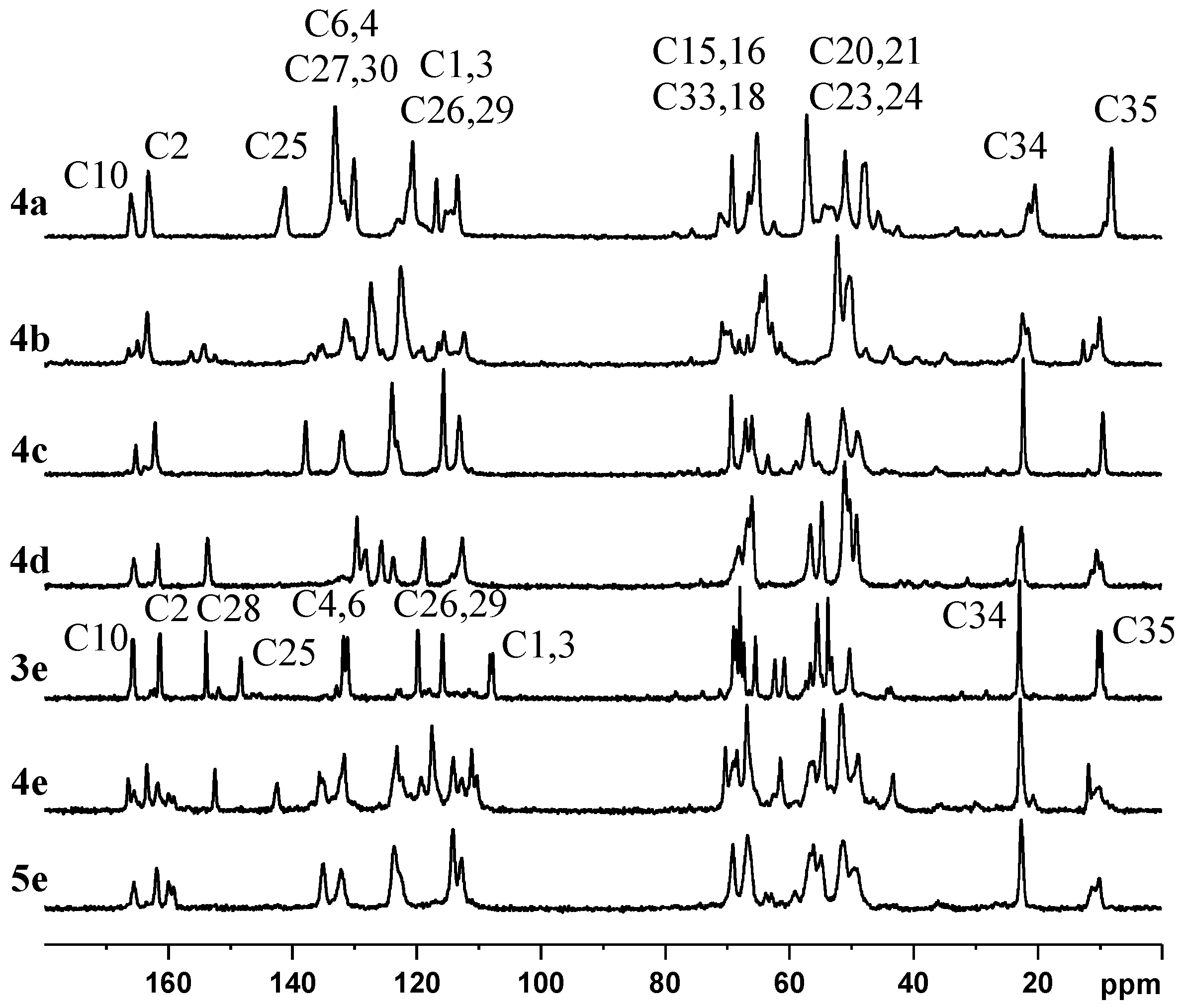
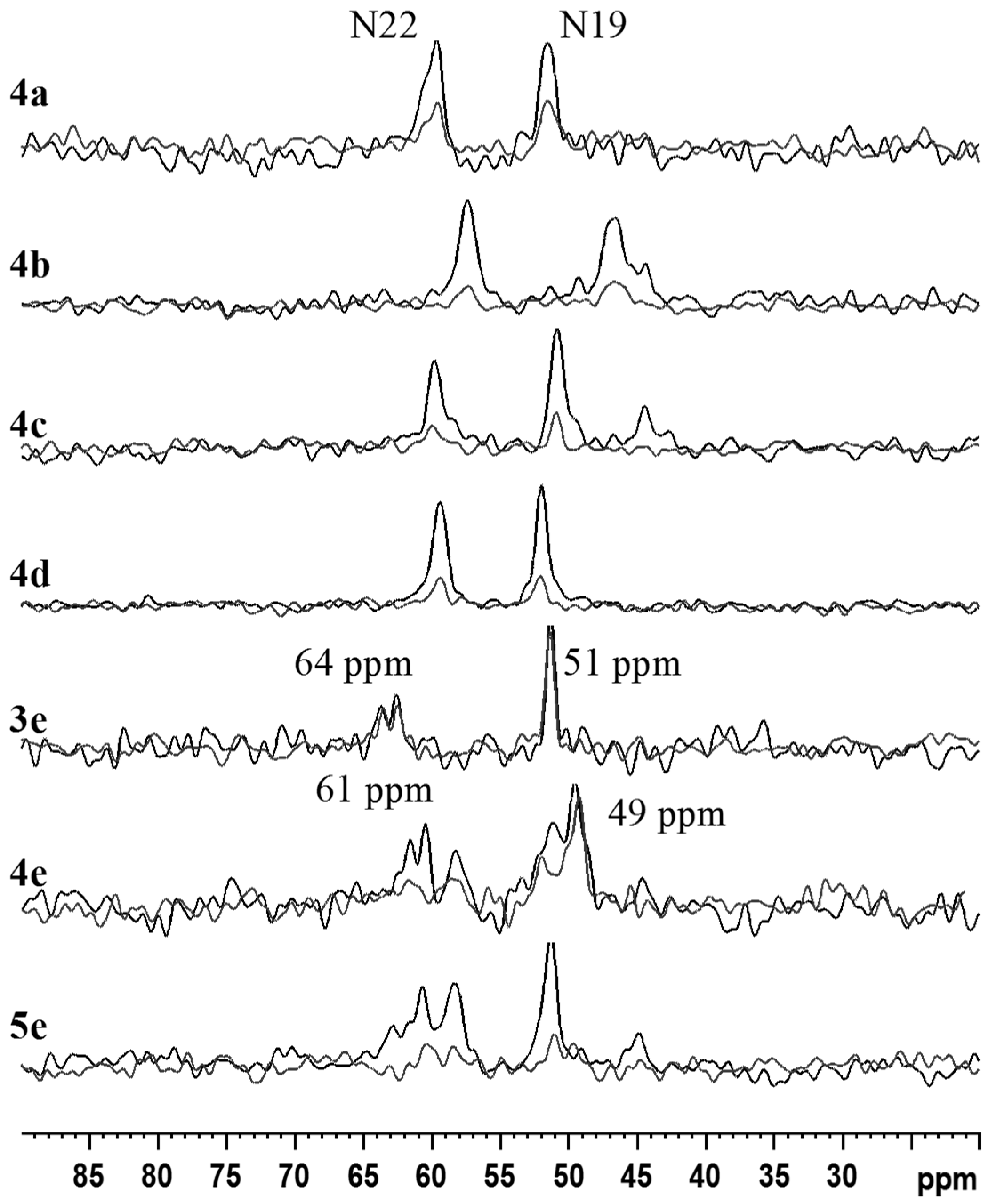
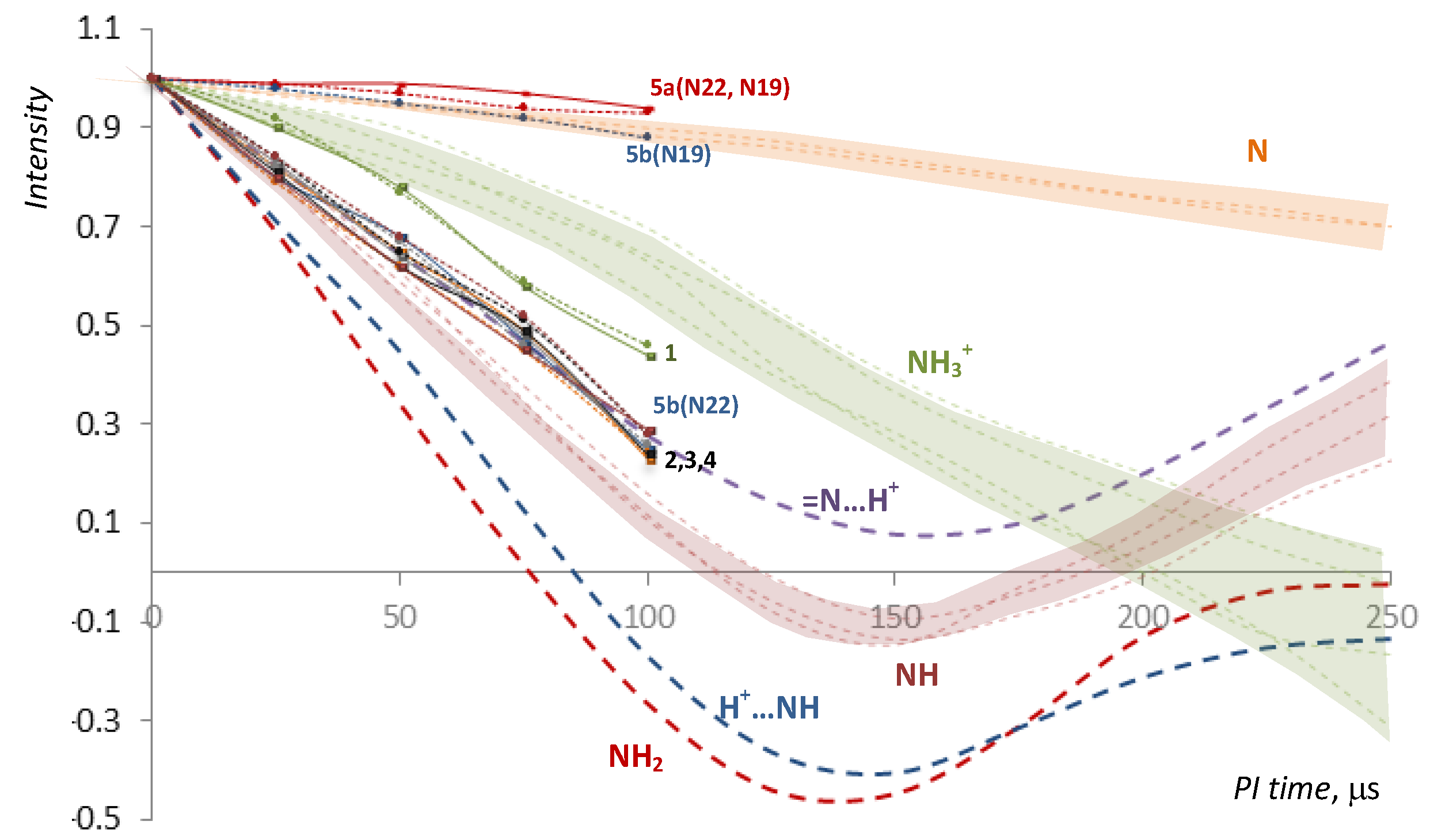
2.2. NMR, CP/MAS Spectra
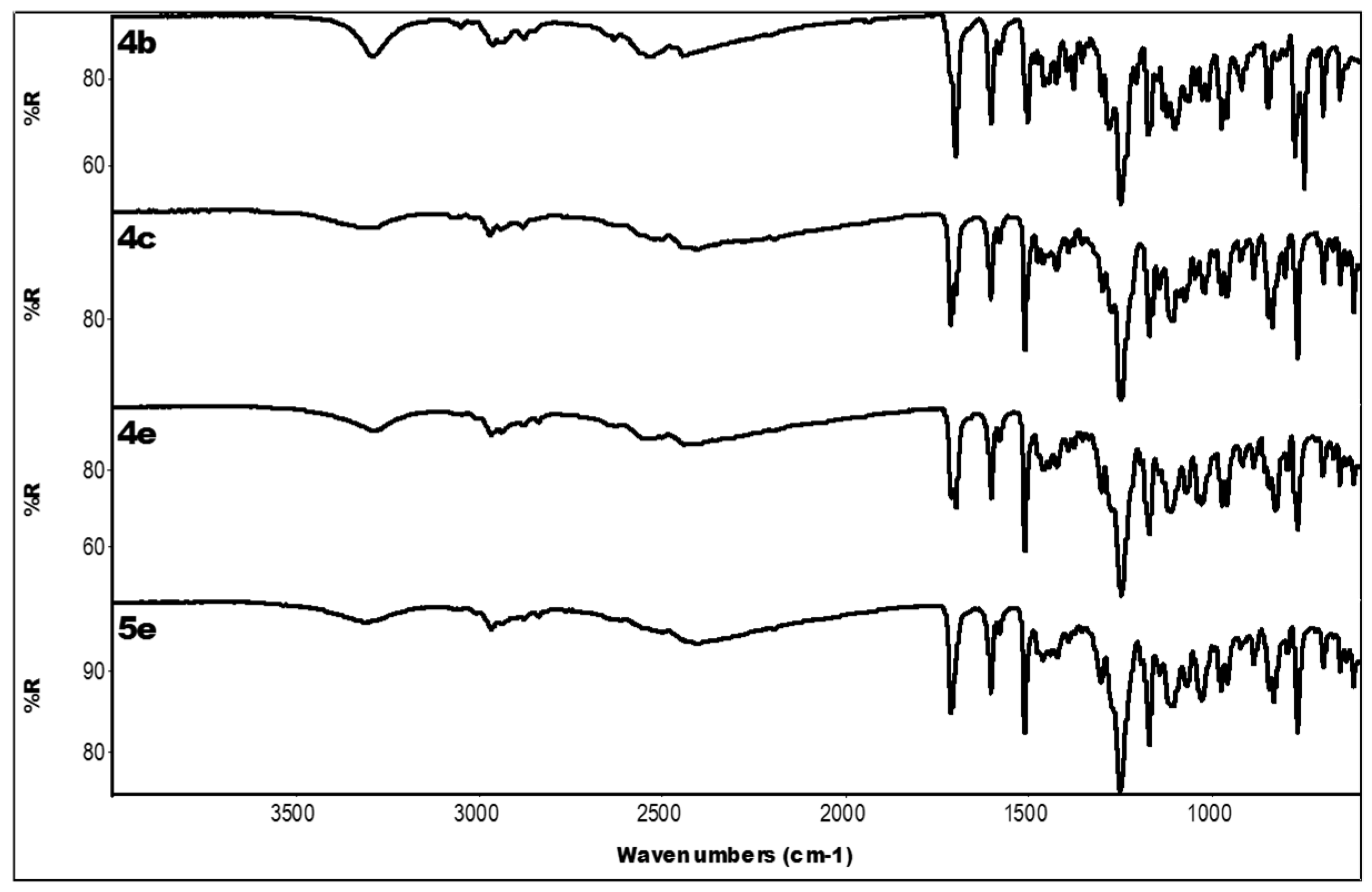
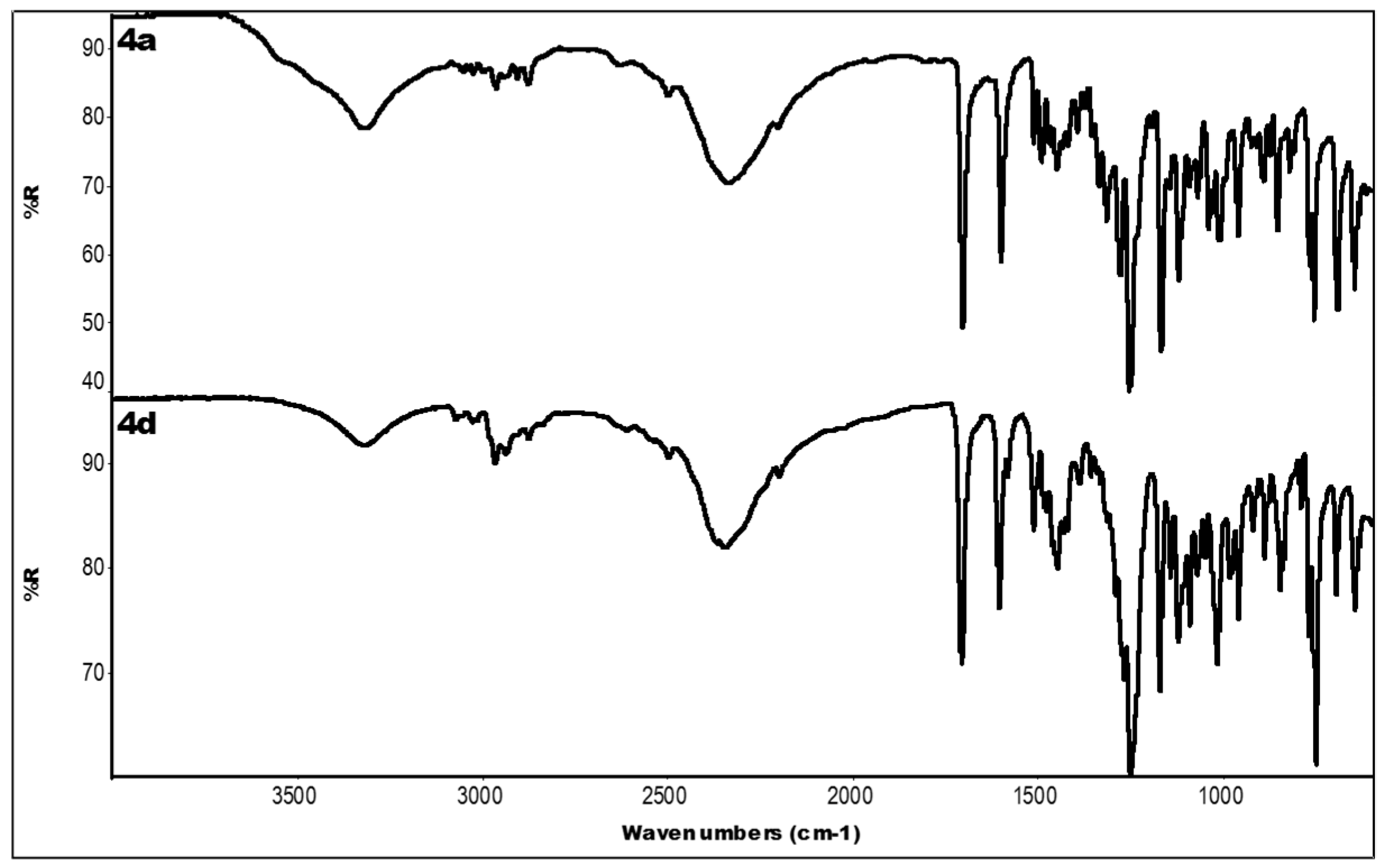
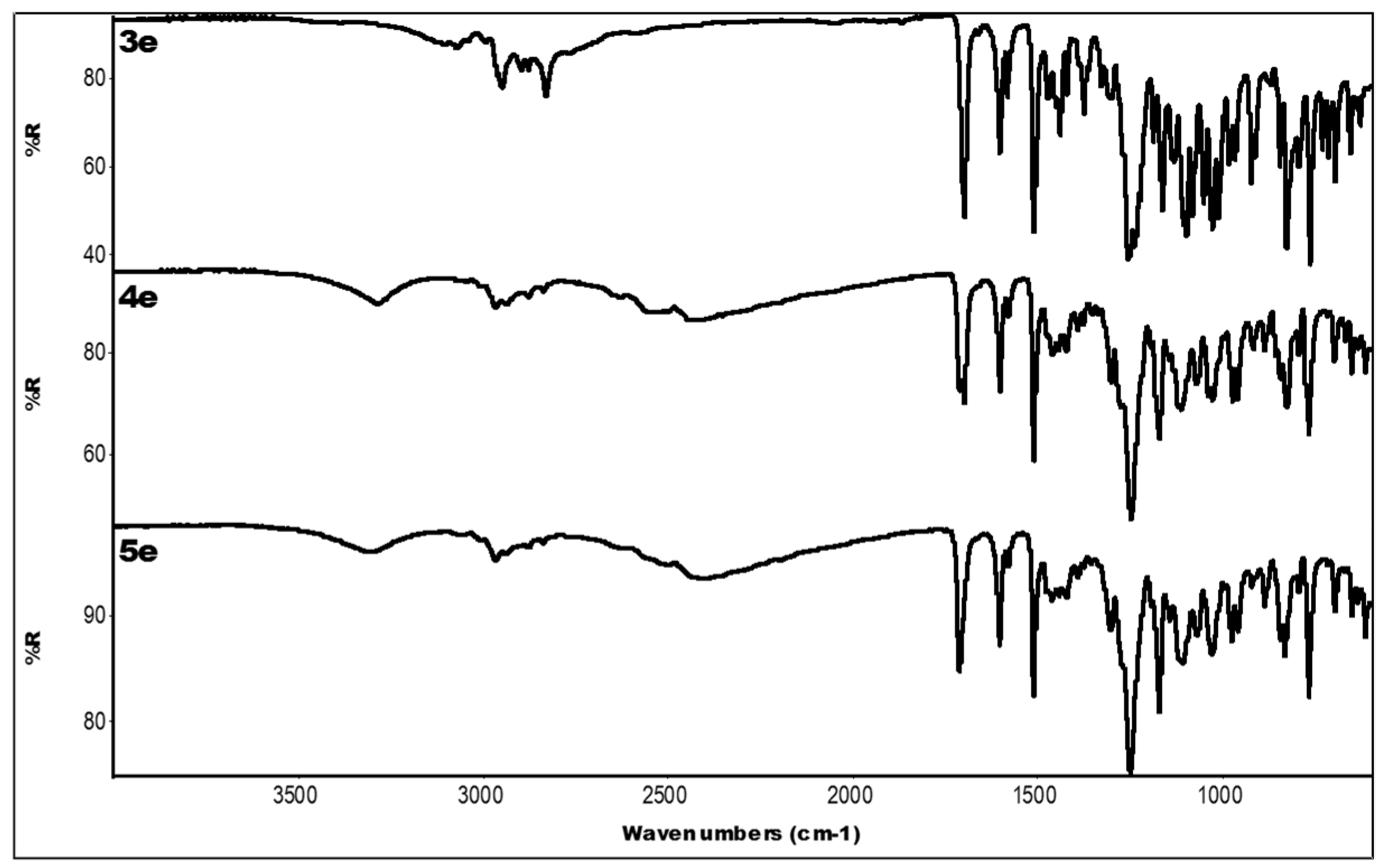
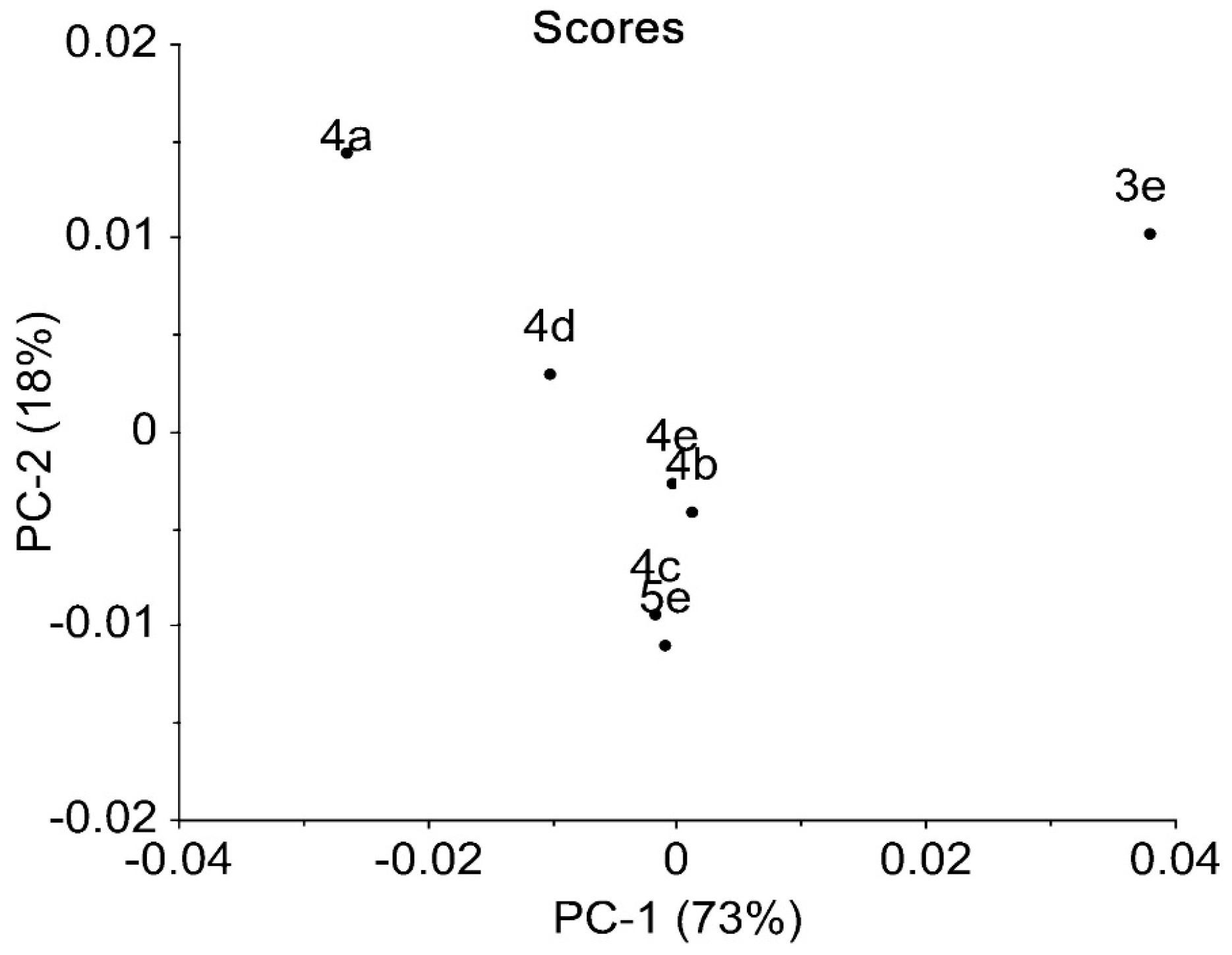
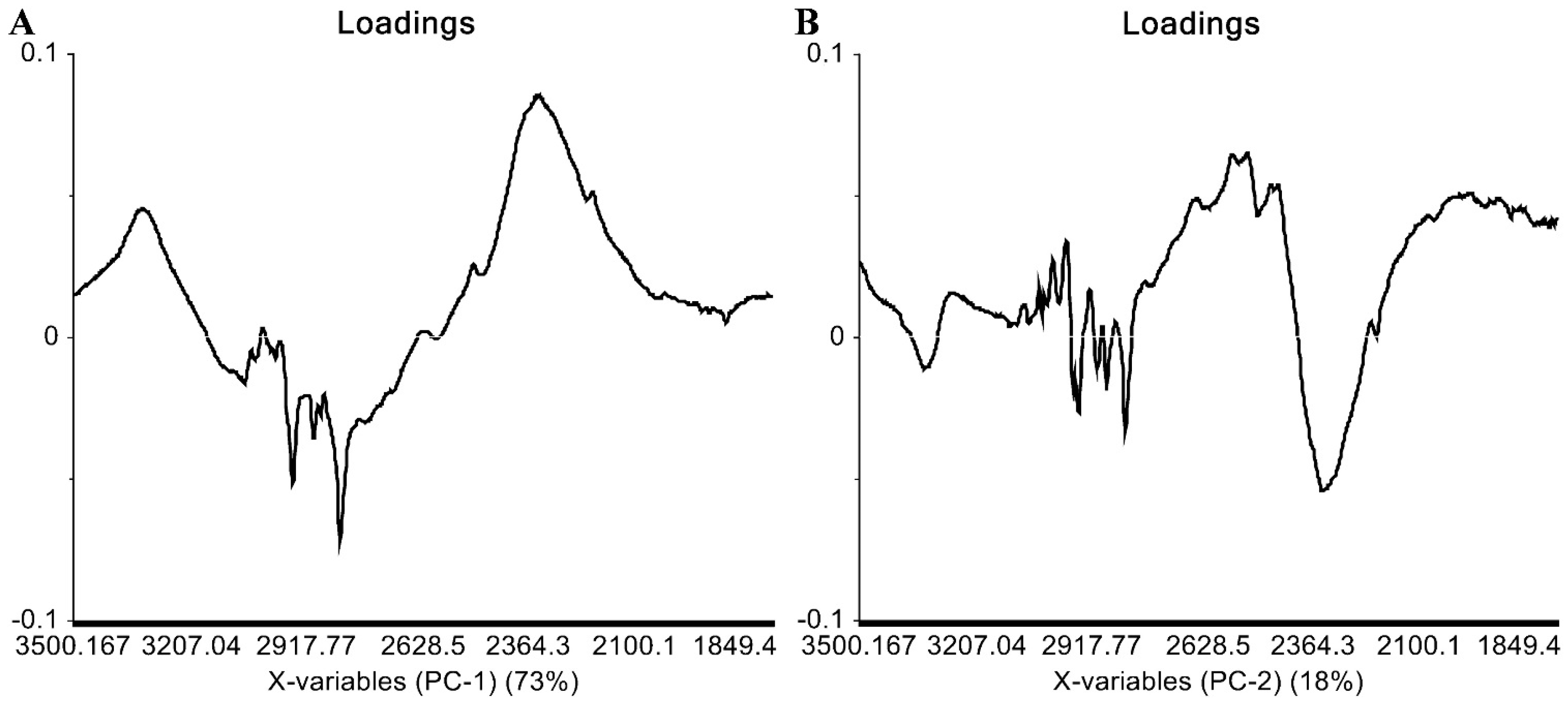
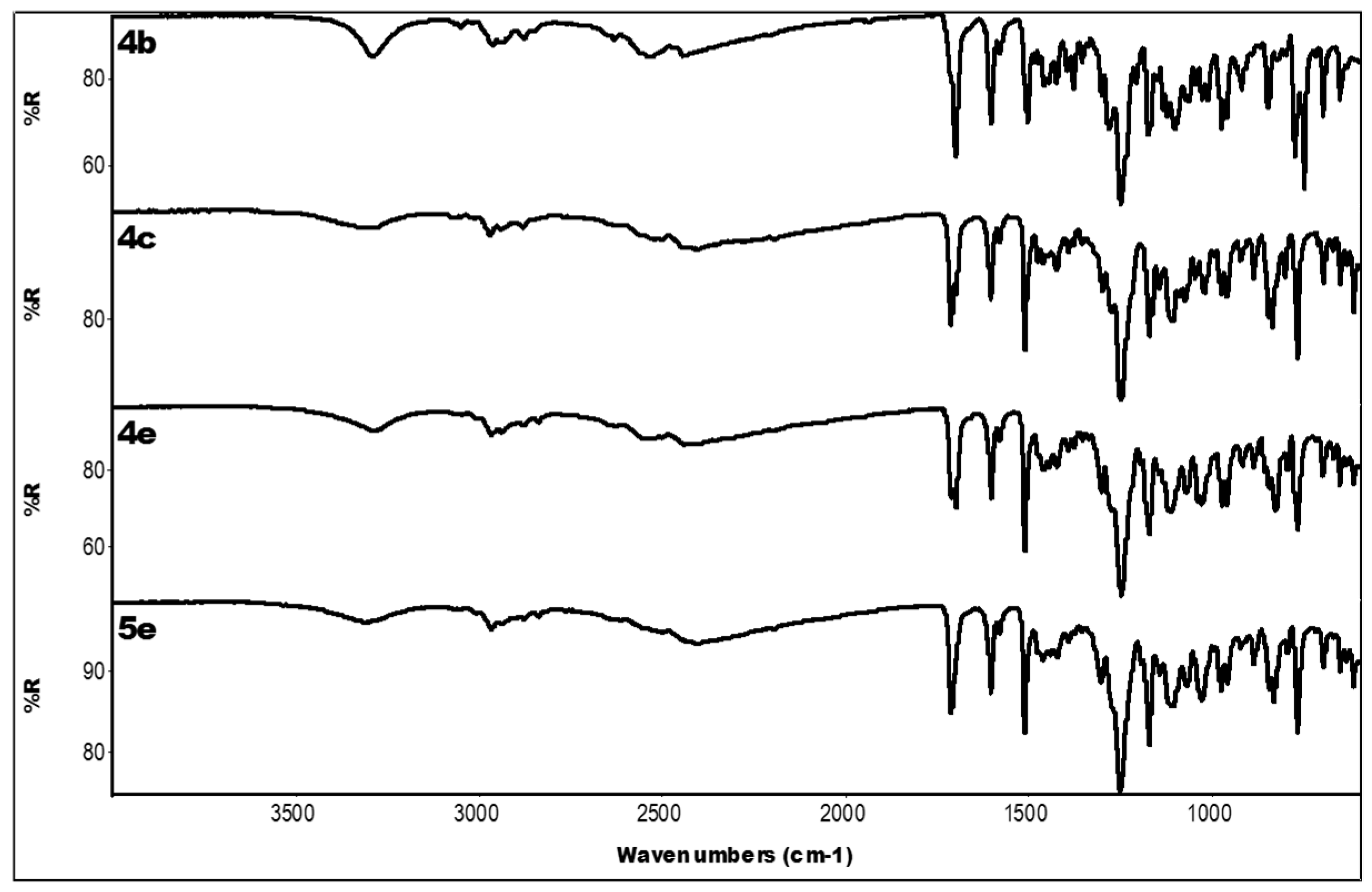
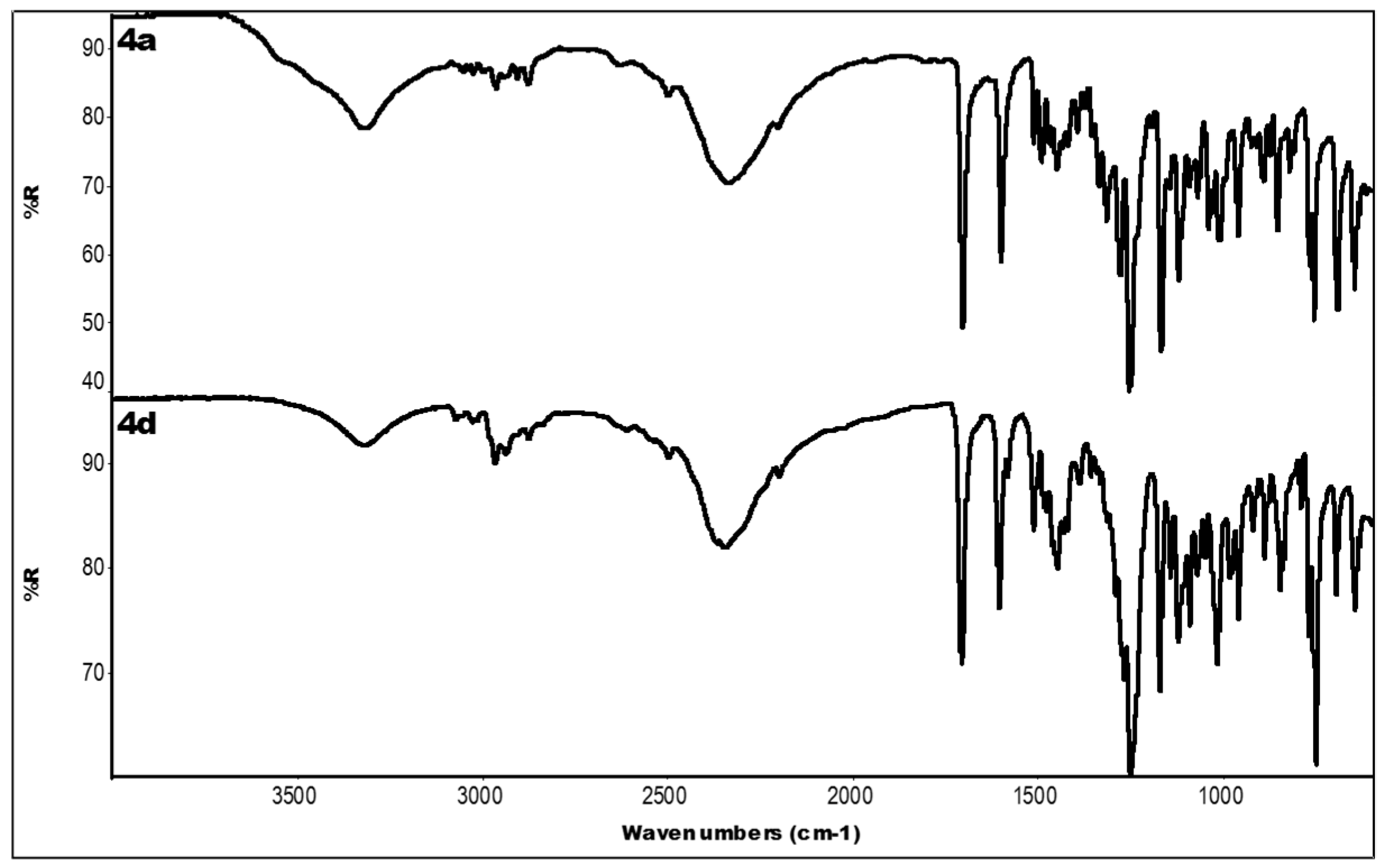
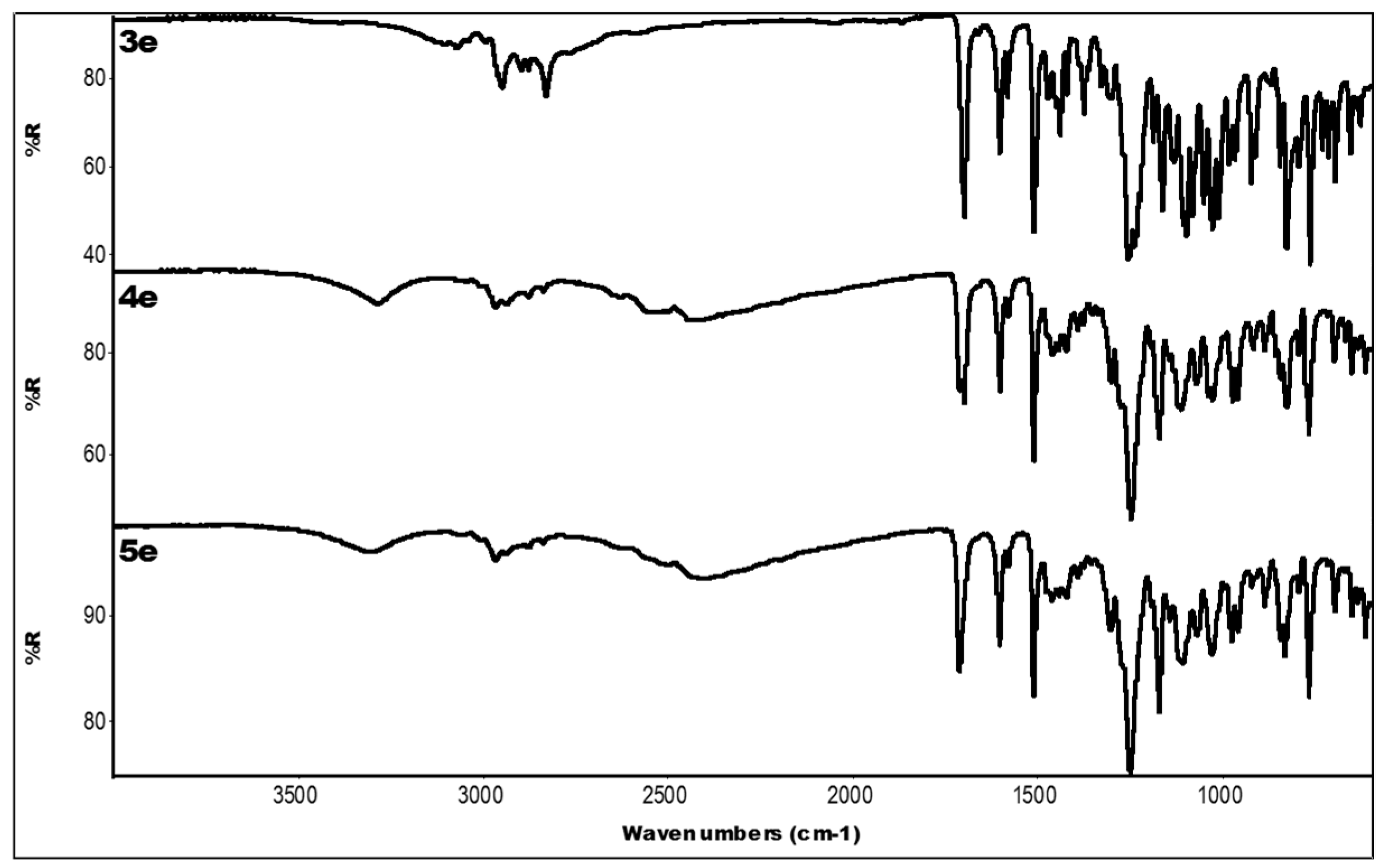
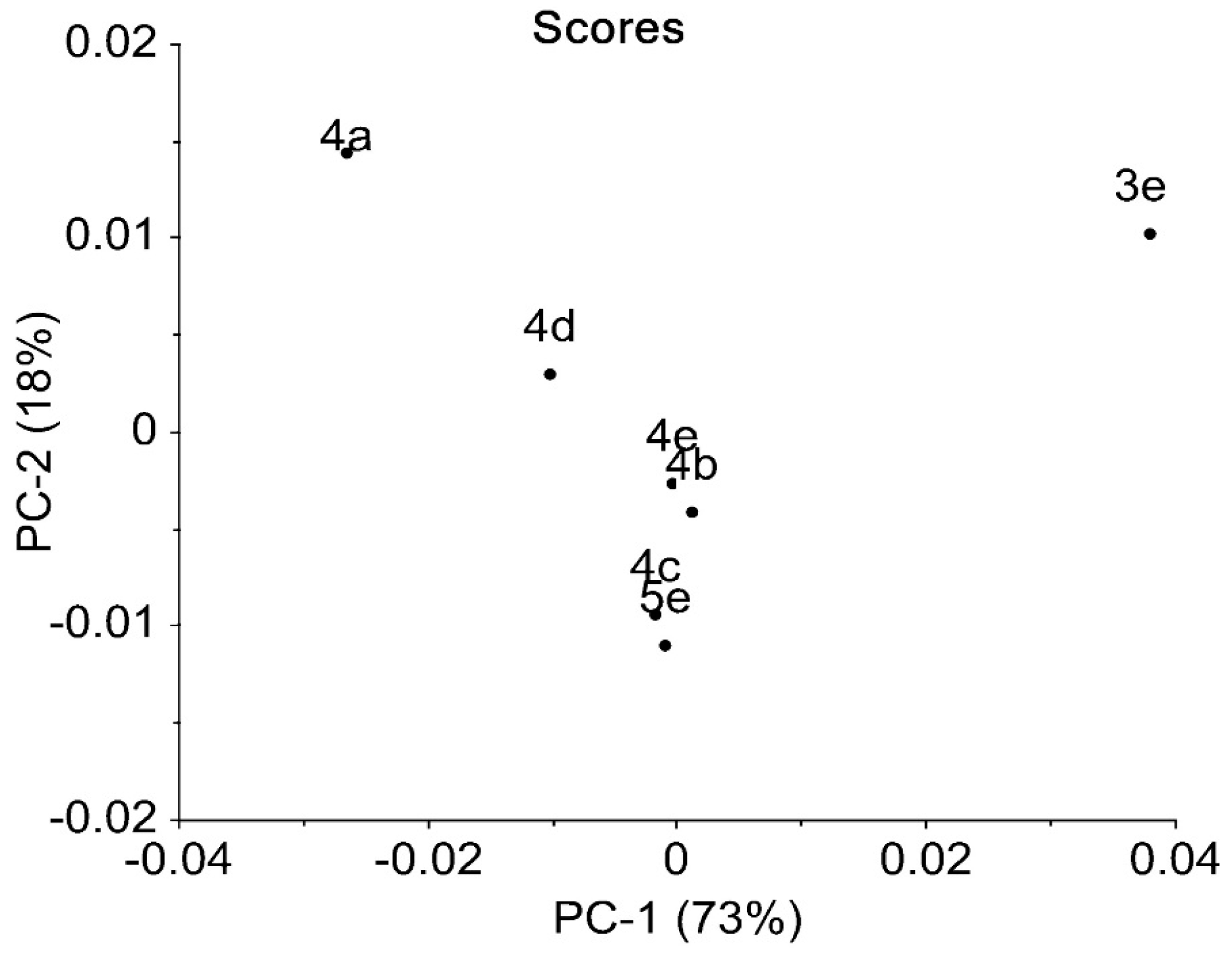
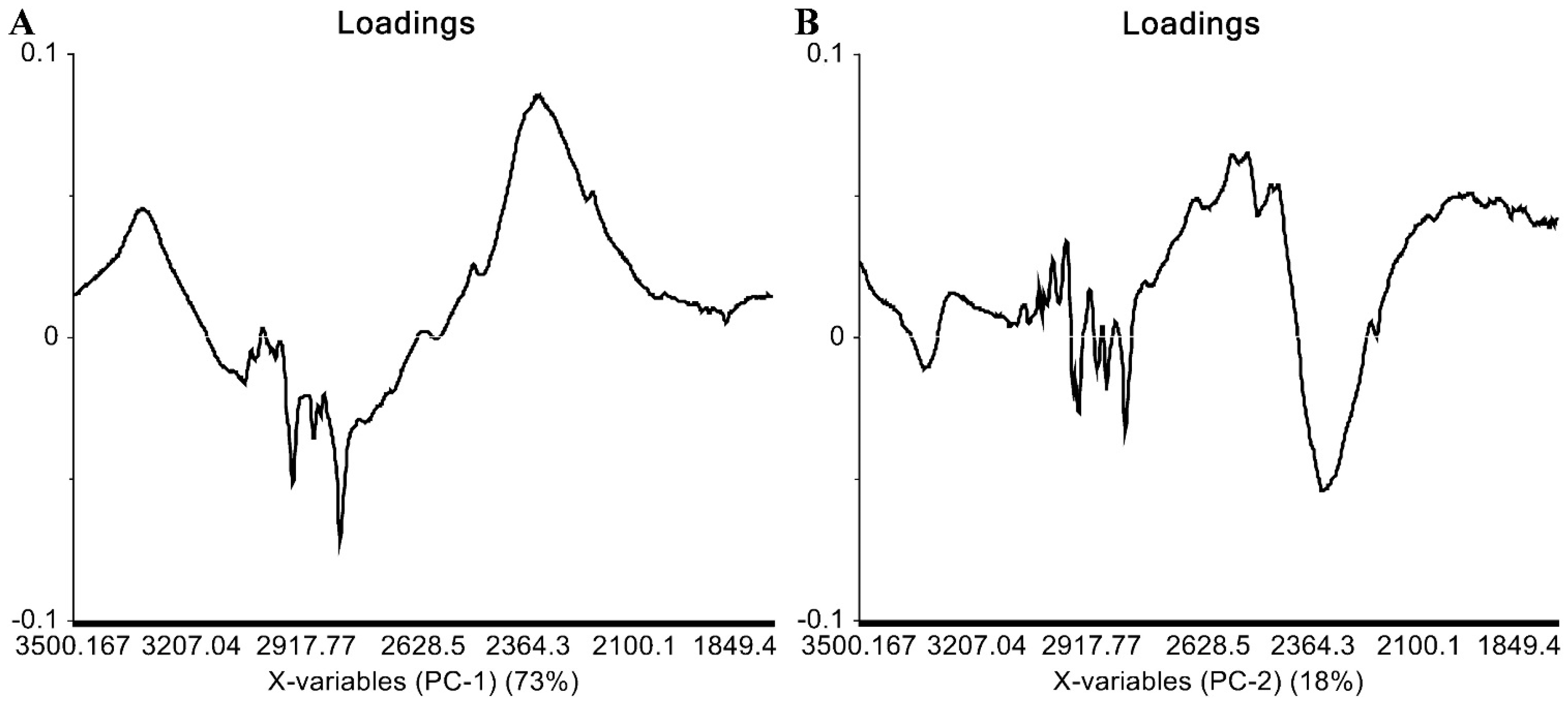
2.3. IR Data
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. Synthesis of Intermediates
3.2.2. Synthesis of Final Compounds
3.3. Quantum Chemical Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Prichard, B.N.C.; Graham, B.; Cruickshank, J.M. β-Blockers in the third millennium—When are they really indicated? J. Clin. Basic Cardiol. 2001, 4, 3–9. [Google Scholar]
- Roth, H.J.; Fenner, H. Adrenozeptoren-Blocker (Sympatholytika). In Arzneistoffe, 3rd ed.; Deutscher Apotheker Verlag: Stuttgart, Germany, 2000; pp. 389–402. [Google Scholar]
- Feuerstein, G.Z.; Ruffolo, R.R. Carvedilol, a novel multiple action antihypertensive agent with antioxidant activity and the potential for myocardial and vascular protection. Eur. Heart J. 1995, 16, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Wright, H.M. Nebivolol: A highly selective β1-adrenergic receptor blocker that causes vasodilatation by increasing nitric oxide. Cardiovasc. Ther. 2008, 26, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Wiest, D. Esmolol. A review of its therapeutic efficiacy and pharmacokinetic characteristics. Clin. Pharmacokinet. 1995, 28, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, H.; Kuruma, A.; Yashima, M.; Saitoh, H.; Ino, T.; Endoh, Y.; Hayakawa, H. Pharmacokinetics of landiolol hydrochloride, a new ultra-short-acting β-blocker, in patients with cardiac arrhythmias. Clin. Pharmacol. Ther. 2000, 68, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Tengler, J.; Stropnicky, O. Soft drugs and retrometabolic drug design. Chem. Listy 2014, 108, 25–31. [Google Scholar]
- Bodor, N.; Buchwald, P. Soft drug design: General principles and recent applications. Med. Res. Rev. 2000, 20, 58–101. [Google Scholar] [CrossRef]
- Mokry, P.; Zemanova, M.; Csollei, J.; Racanska, E.; Tumova, I. Synthesis and pharmacological evaluation of novel potential ultrashortacting β-blockers. Pharmazie 2003, 58, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Bartosova, L.; Frydrych, M.; Hulakova, G.; Berankova, K.; Strnadova, V.; Mokry, P.; Brunclik, V.; Kolevska, J.; Bebarova, M. Efficacy of newly synthesized 44Bu ultrashort-acting β-adrenergic antagonist to isoprenaline-induced tachycardia—Comparison with esmolol. Acta. Vet. Brno 2004, 73, 171–179. [Google Scholar] [CrossRef]
- Racanska, E.; Kurfurst, P.; Csollei, J.; Svec, P. Cardiovascular effects of newly sythesized hybrid heteroarylaminoethanols. Acta Fac. Pharm. Univ. Comen. 2004, 51, 182–191. [Google Scholar]
- Ammazzalorso, A.; Amoroso, R.; Bettoni, G.; Fantacuzzi, M.; De Filippis, B.; Giampietro, L.; Maccallini, C.; Paludi, D.; Tricca, M.L. Synthesis and antibacterial evaluation of oxazolidin-2-ones structurally related to linezolid. Farmaco 2004, 59, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Tengler, J.; Kapustikova, I.; Stropnicky, O.; Mokry, P.; Oravec, M.; Csollei, J.; Jampilek, J. Synthesis of new (arylcarbonyloxy)aminopropanol derivatives and the determination of their physico-chemical properties. Cent. Eur. J. Chem. 2013, 11, 1757–1767. [Google Scholar] [CrossRef]
- Pawlak, T.; Trzeciak-Karlikowska, K.; Czernek, J.; Ciesielski, W.; Potrzebowski, M.J. Computed and experimental chemical shift parameters for rigid and flexible YAF peptides in the solid state. J. Phys. Chem. B 2012, 116, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Apperley, D.C.; Basford, P.A.; Dallman, C.I.; Harris, R.K.; Kinns, M.; Marshall, P.V.; Swanson, A.G. Nuclear magnetic resonance investigation of the interaction of water vapor with sildenafil citrate in the solid state. J. Pharm. Sci. 2005, 94, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wilson, D.; Harbison, G.S. Solid-State NMR and the crystallization of aspartic and glutamic acids. Cryst. Growth Des. 2016, 16, 625–631. [Google Scholar] [CrossRef]
- Policianova, O.; Brus, J.; Hruby, M.; Urbanova, M.; Zhigunov, A.; Kredatusova, J.; Kobera, L. Structural diversity of solid dispersions of acetylsalicylic acid as seen by solid-state NMR. Mol. Pharm. 2014, 11, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Burns, S.T.; Zilm, K.W. Spectral editing in CPMAS NMR. Generating subspectra based on proton multiplicities. J. Magn. Reson. A 1994, 111, 29–36. [Google Scholar] [CrossRef]
- Brus, J. Heating of samples induced by fast magic-angle spinning. Solid State Nucl. Magn. Reson. 2000, 16, 151–160. [Google Scholar] [CrossRef]
- Proks, V.; Brus, J.; Pop-Georgievski, O.; Vecernikova, E.; Wisniewski, W.; Kotek, J.; Urbanova, M.; Rypacek, F. Thermal-induced transformation of polydopamine structures: An efficient route for the stabilization of the polydopamine surfaces. Macromol. Chem. Phys. 2013, 214, 499–507. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Kobera, L.; Czernek, J.; Streckova, M.; Urbanova, M.; Abbrent, S.; Brus, J. Structure and distribution of cross-links in boron-modified phenol–formaldehyde resins designed for soft magnetic composites: A multiple-quantum 11B–11B MAS NMR correlation spectroscopy study. Macromolecules 2015, 48, 4874–4881. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Sample Availability: Samples of compounds are available from authors P. Marvanova and P. Mokry.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IR ν (cm−1) |
|---|---|
| 4a | 3319 (OH); 2336 (N-H+); 1704 (C=O); 1601, 1450 (aromatics); 1253, 1168 (ester) |
| 4b | 3290 (OH); 2528, 2445 (N-H+); 1701 (C=O); 1604, 1458 (aromatics); 1249, 1175 (ester) |
| 4c | 3285 (OH); 2503, 2401 (N-H+); 1712 (C=O); 1605, 1458 (aromatics); 1249, 1171 (ester) |
| 4d | 3318 (OH); 2349 (N-H+); 1706 (C=O); 1606, 1448 (aromatics); 1250, 1171 (ester) |
| 3e | 3071 (OH + CH); 1699 (C=O); 1604, 1439 (aromatics); 1255, 1163 (ester) |
| 4e | 3287 (OH); 2527, 2442 (N-H+); 1699 (C=O); 1603, 1462 (aromatics); 1248, 1171 (ester) |
| 5e | 3312 (OH); 2401 (N-H+); 1711 (C=O); 1604, 1462 (aromatics); 1250, 1171 (ester) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marvanova, P.; Padrtova, T.; Pekarek, T.; Brus, J.; Czernek, J.; Mokry, P.; Humpa, O.; Oravec, M.; Jampilek, J. Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts. Molecules 2016, 21, 707. https://doi.org/10.3390/molecules21060707
Marvanova P, Padrtova T, Pekarek T, Brus J, Czernek J, Mokry P, Humpa O, Oravec M, Jampilek J. Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts. Molecules. 2016; 21(6):707. https://doi.org/10.3390/molecules21060707
Chicago/Turabian StyleMarvanova, Pavlina, Tereza Padrtova, Tomas Pekarek, Jiri Brus, Jiri Czernek, Petr Mokry, Otakar Humpa, Michal Oravec, and Josef Jampilek. 2016. "Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts" Molecules 21, no. 6: 707. https://doi.org/10.3390/molecules21060707
APA StyleMarvanova, P., Padrtova, T., Pekarek, T., Brus, J., Czernek, J., Mokry, P., Humpa, O., Oravec, M., & Jampilek, J. (2016). Synthesis and Characterization of New 3-(4-Arylpiperazin-1-yl)-2-hydroxypropyl 4-Propoxybenzoates and Their Hydrochloride Salts. Molecules, 21(6), 707. https://doi.org/10.3390/molecules21060707