
Neuroprotective Effects of Biochanin A against β-Amyloid-Induced Neurotoxicity in PC12 Cells via a Mitochondrial-Dependent Apoptosis Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
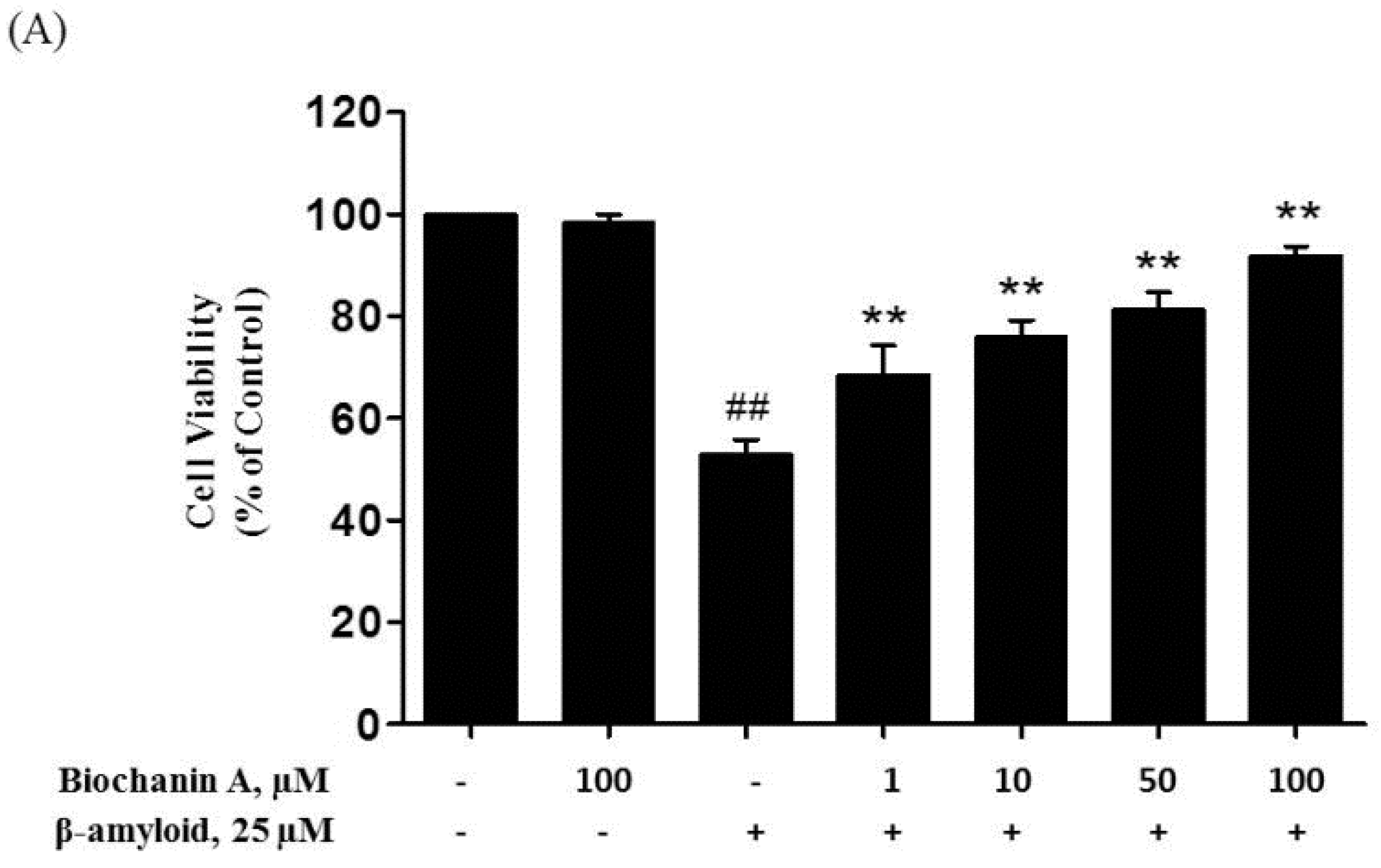
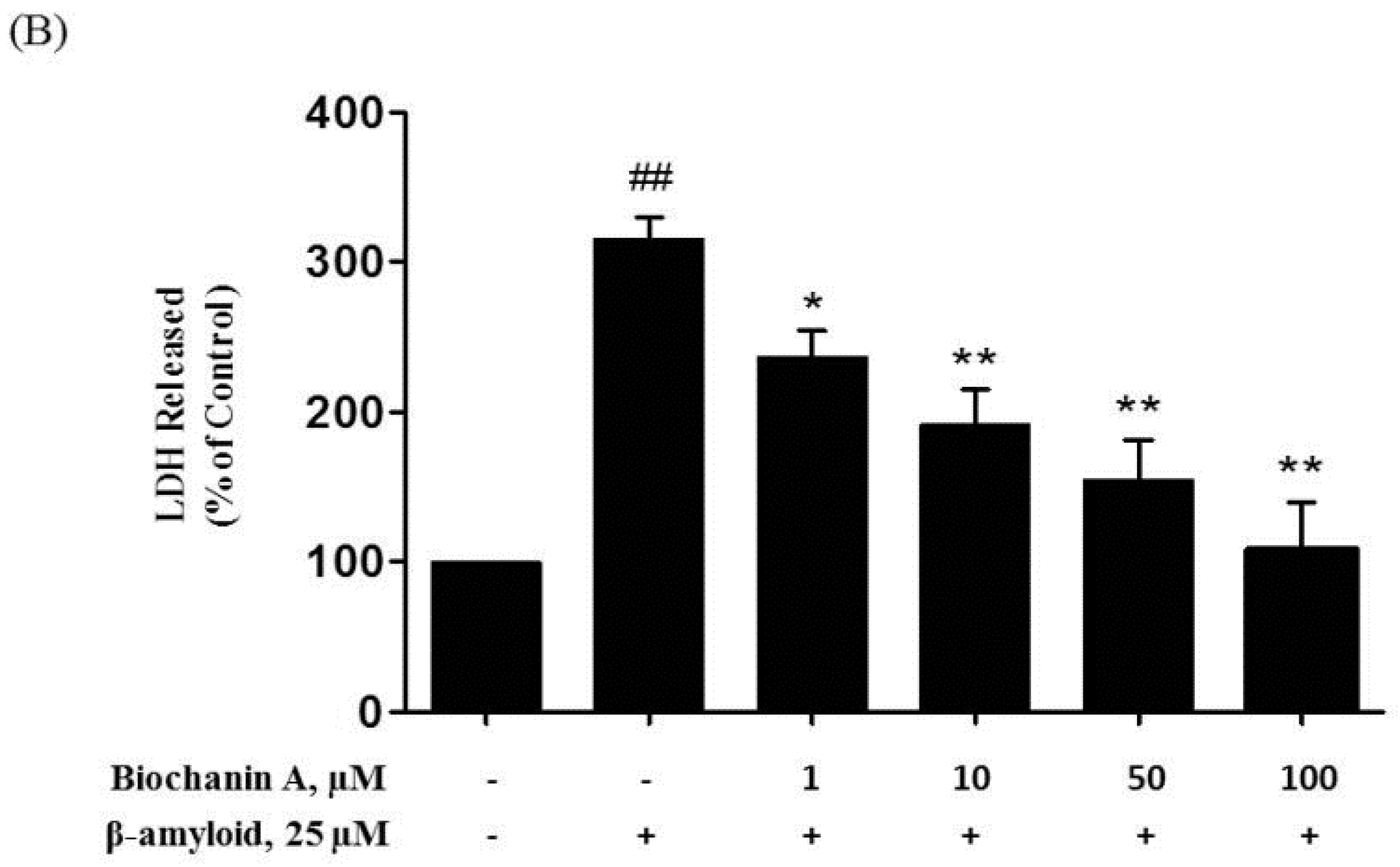
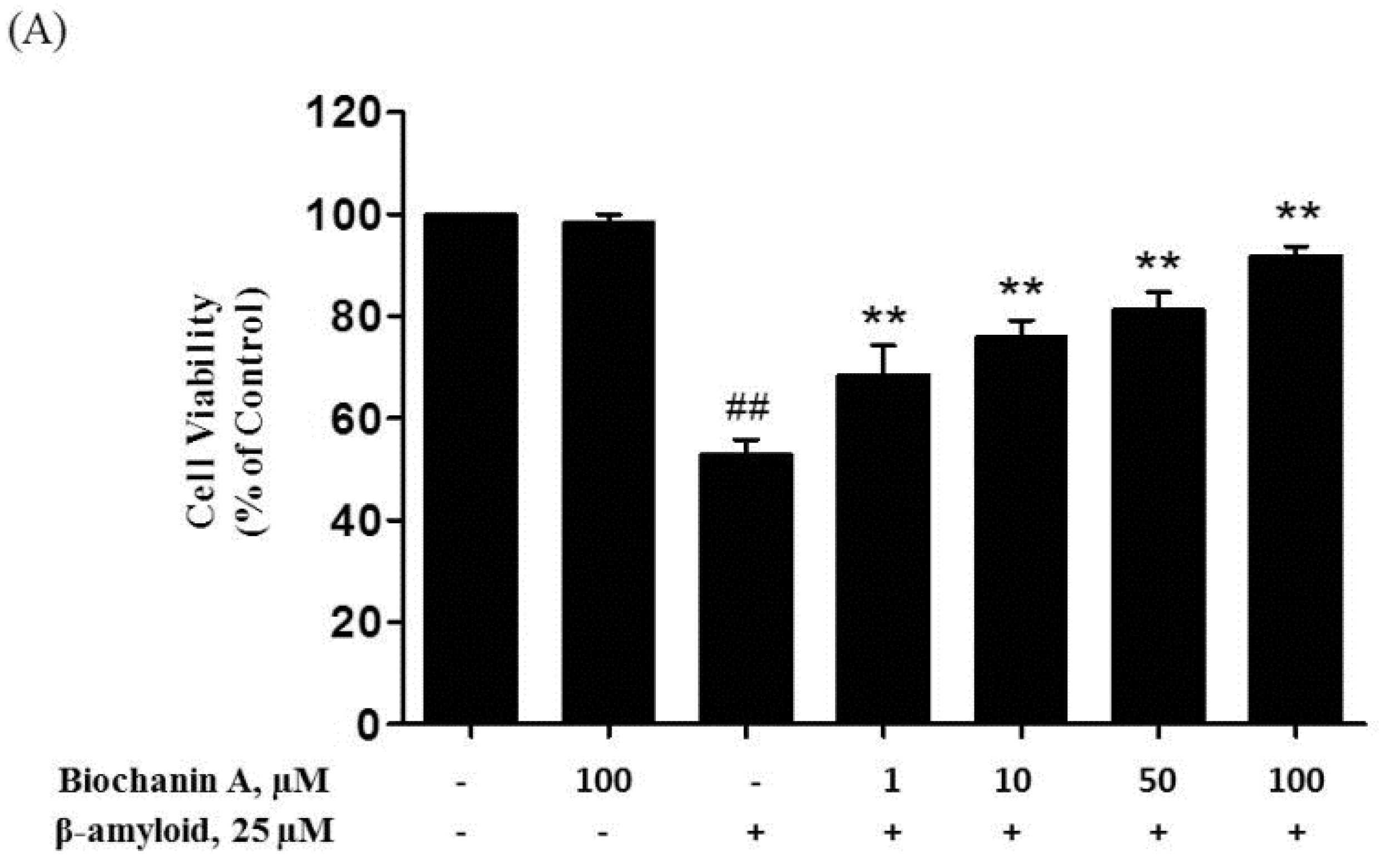
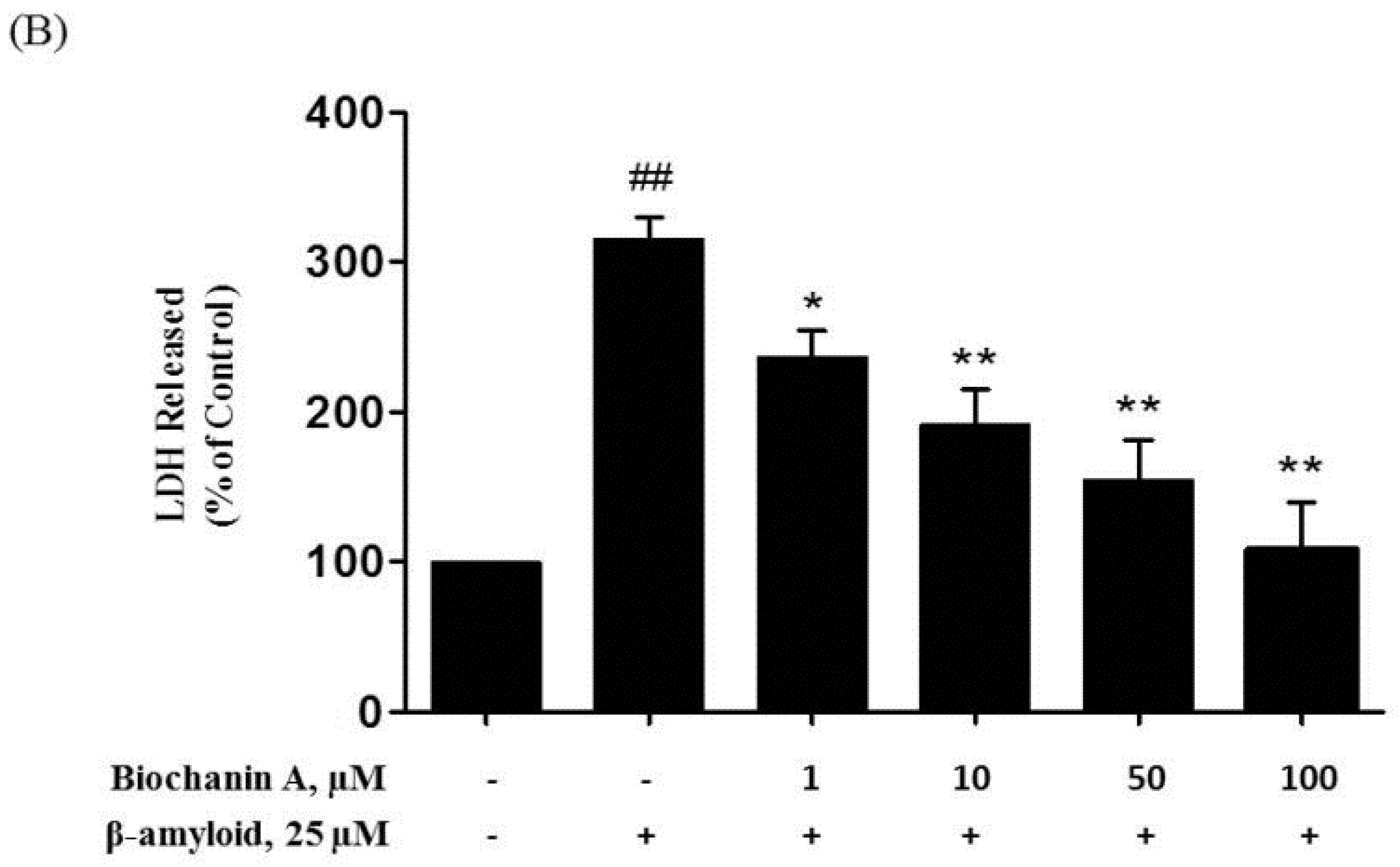
2.1. Protective Effect of Biochanin A against Aβ25–35-Induced Cytotoxicity
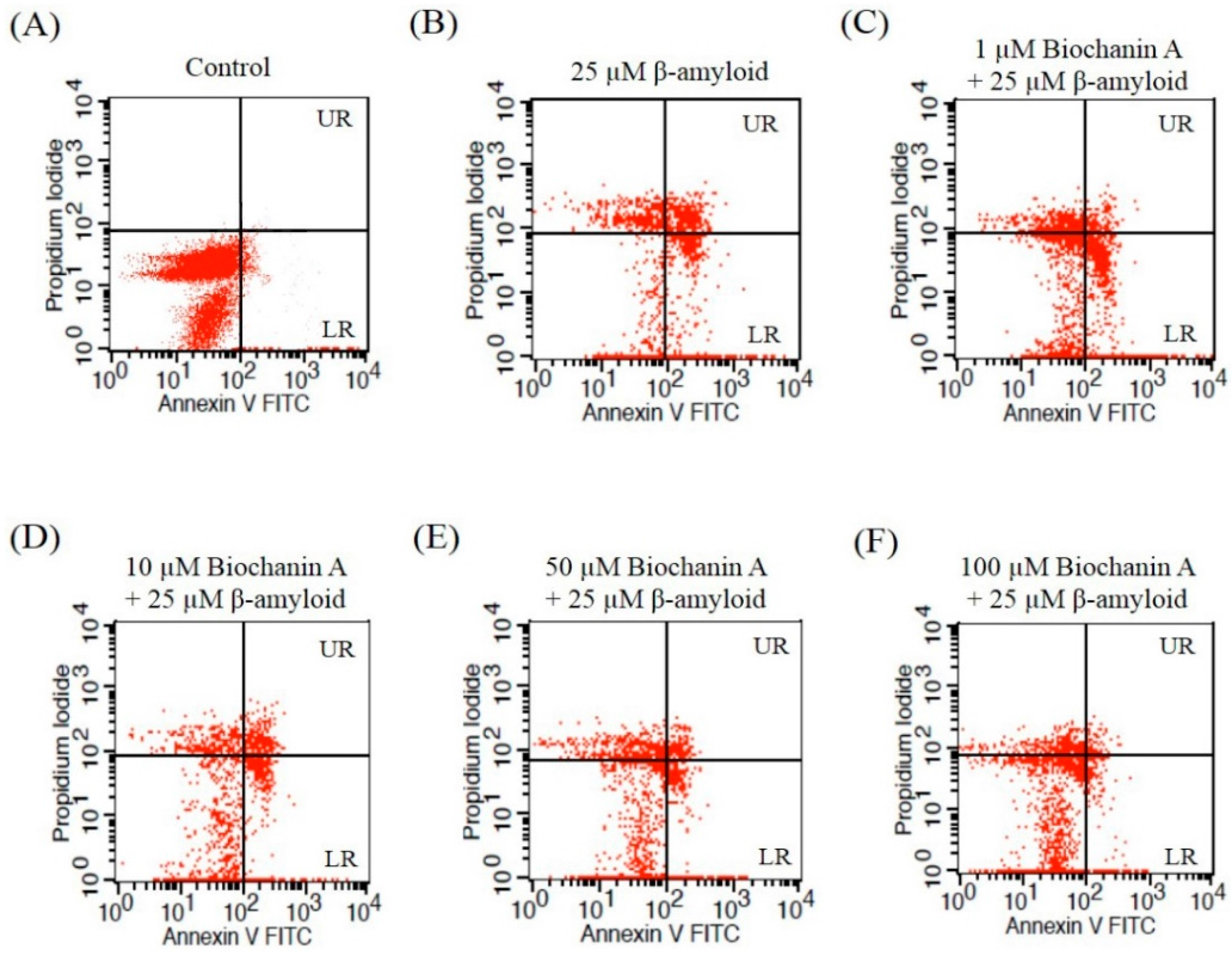
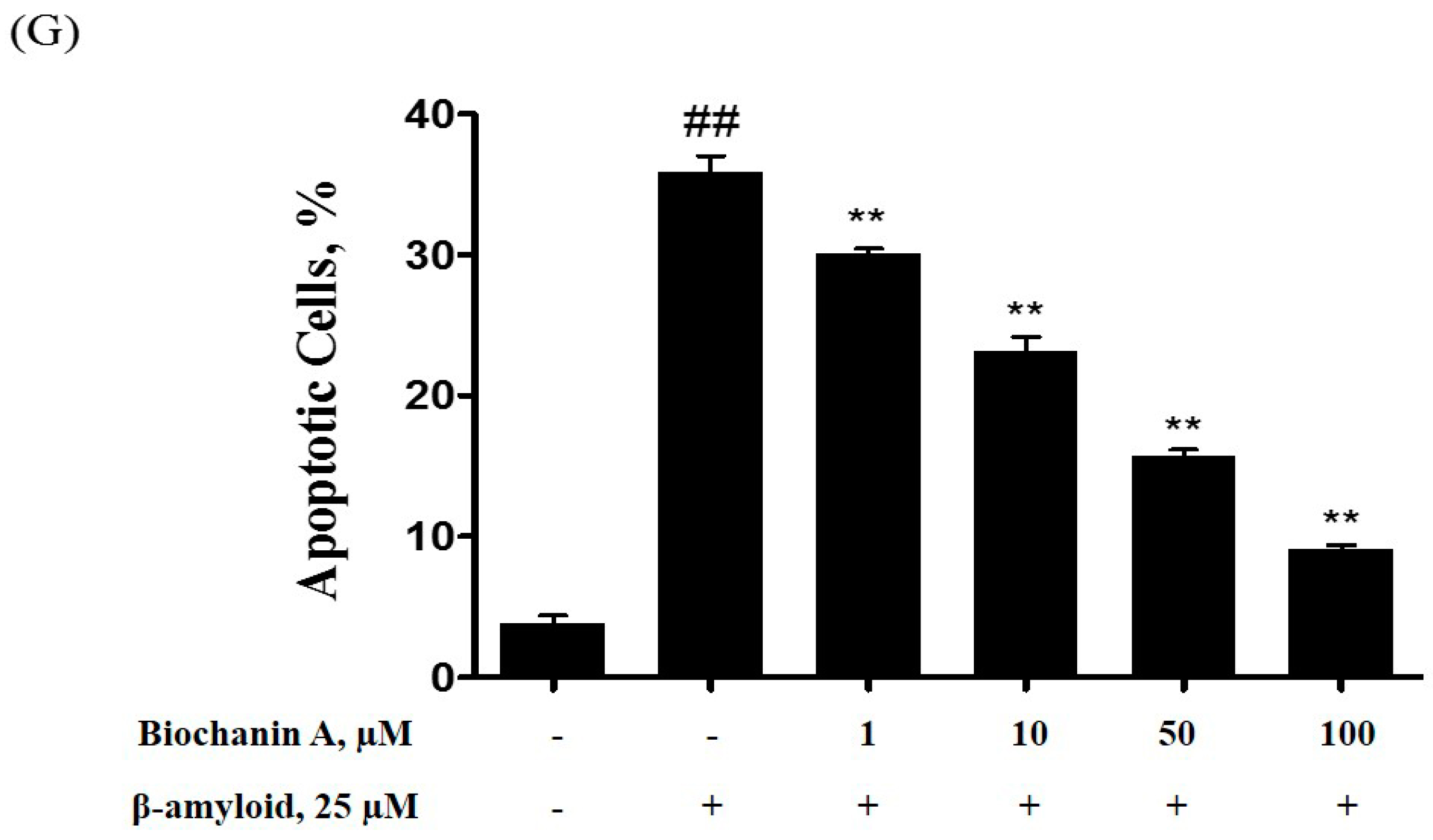
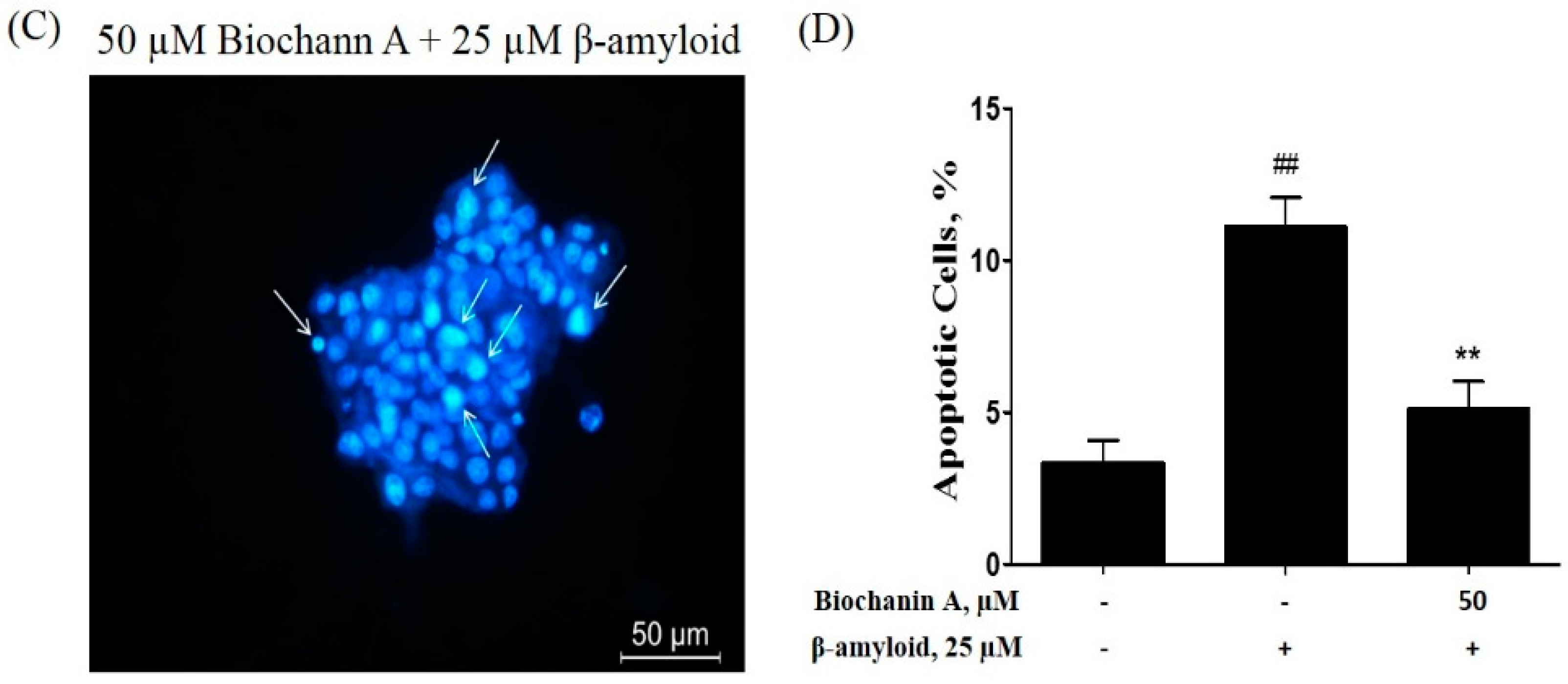
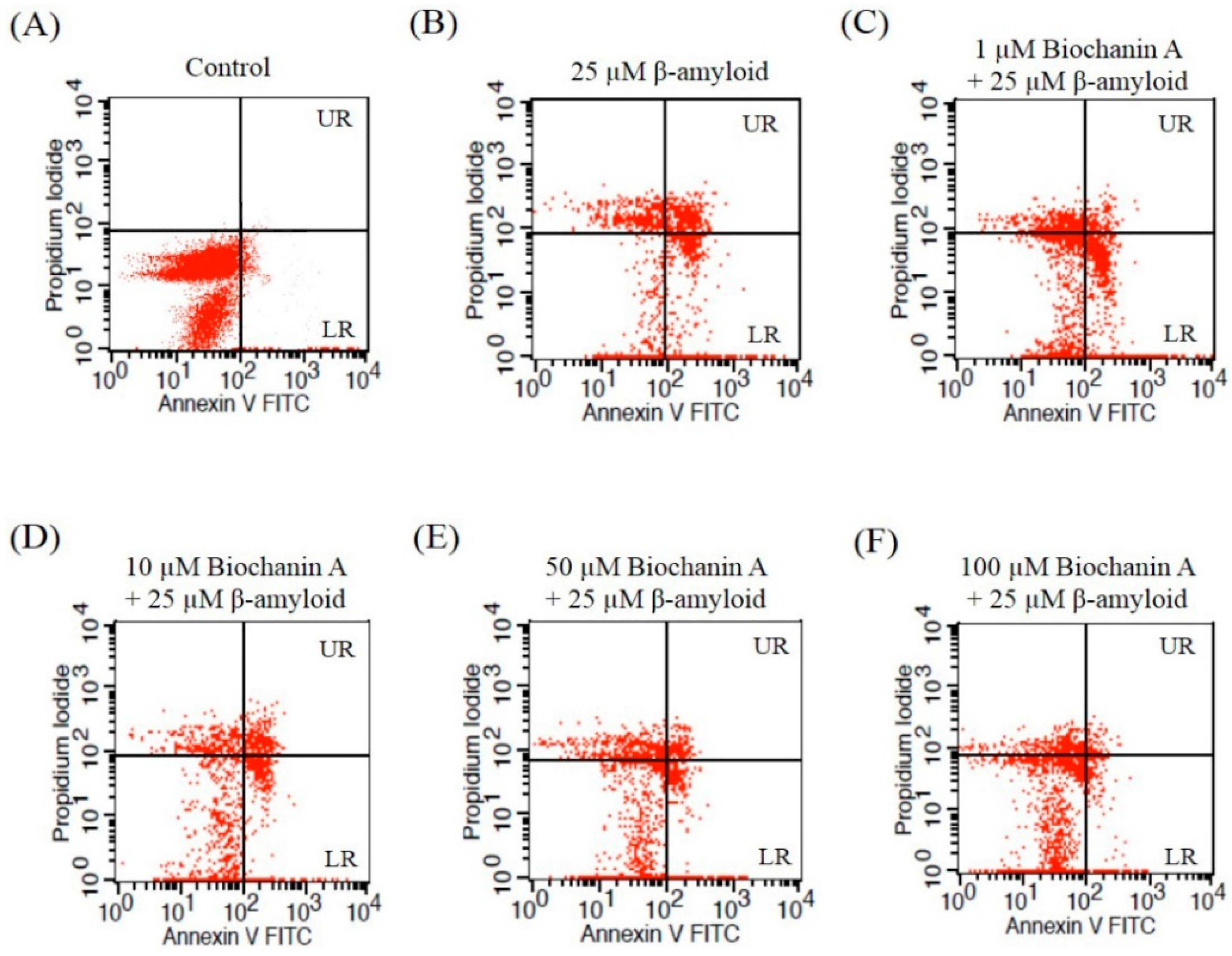
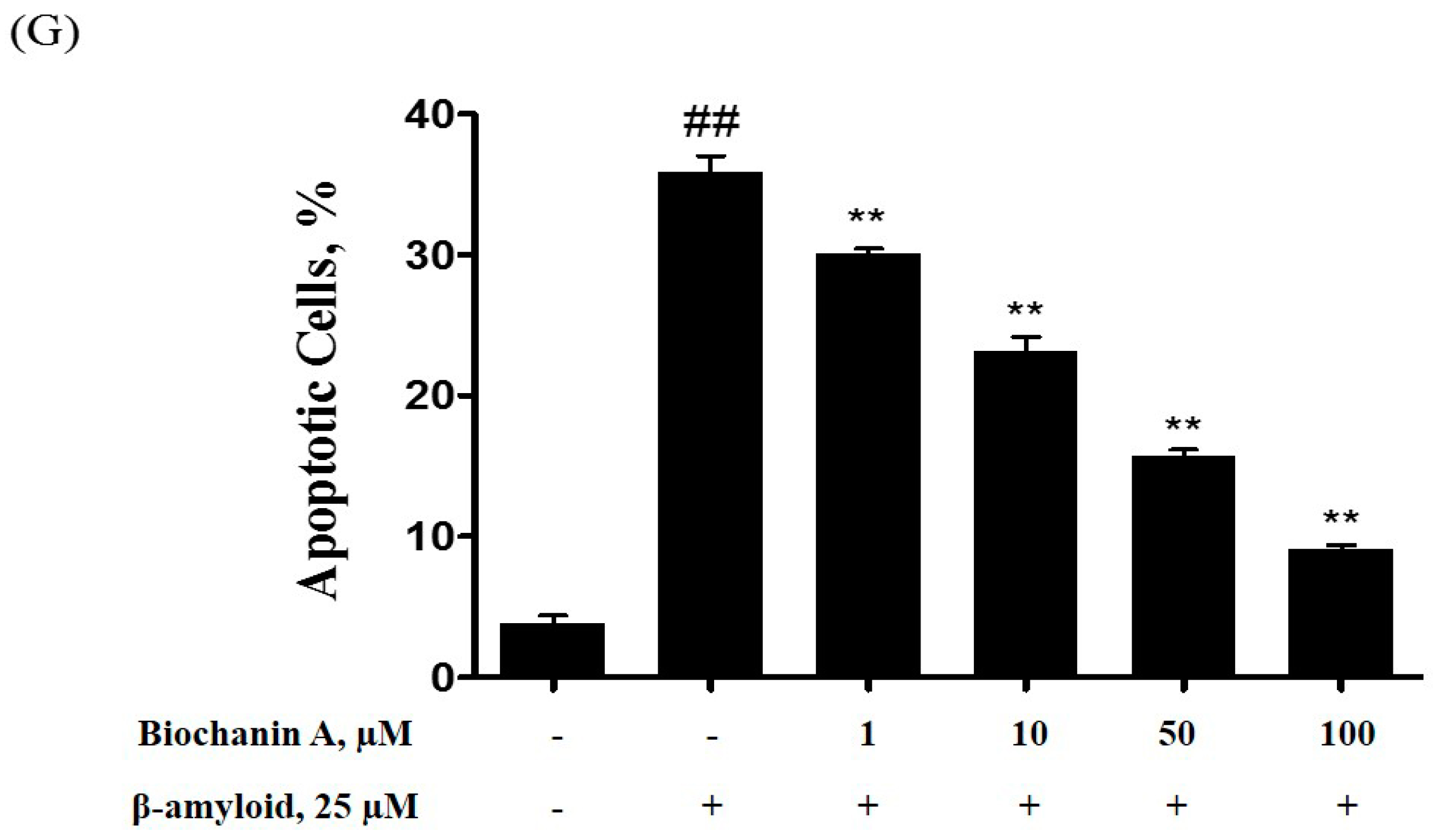
2.2. Effect of Biochanin A on Suppressing Cell Apoptosis Induced by Aβ25–35
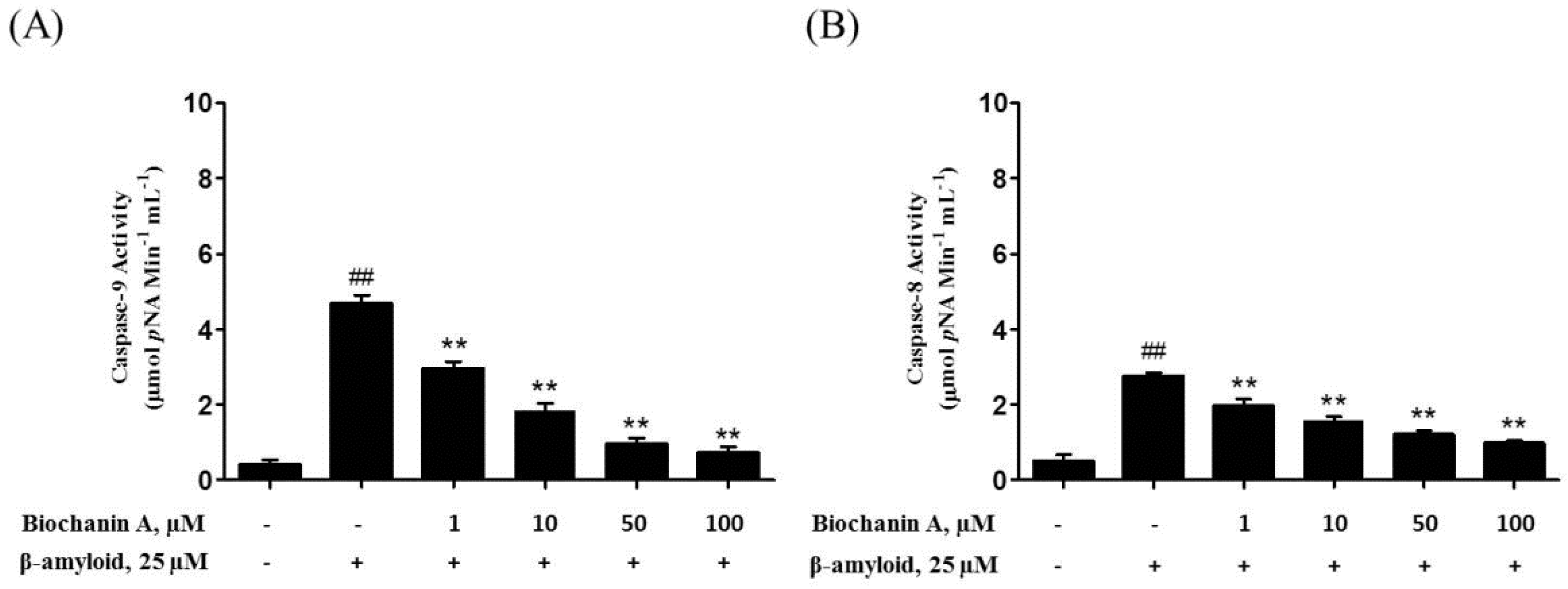
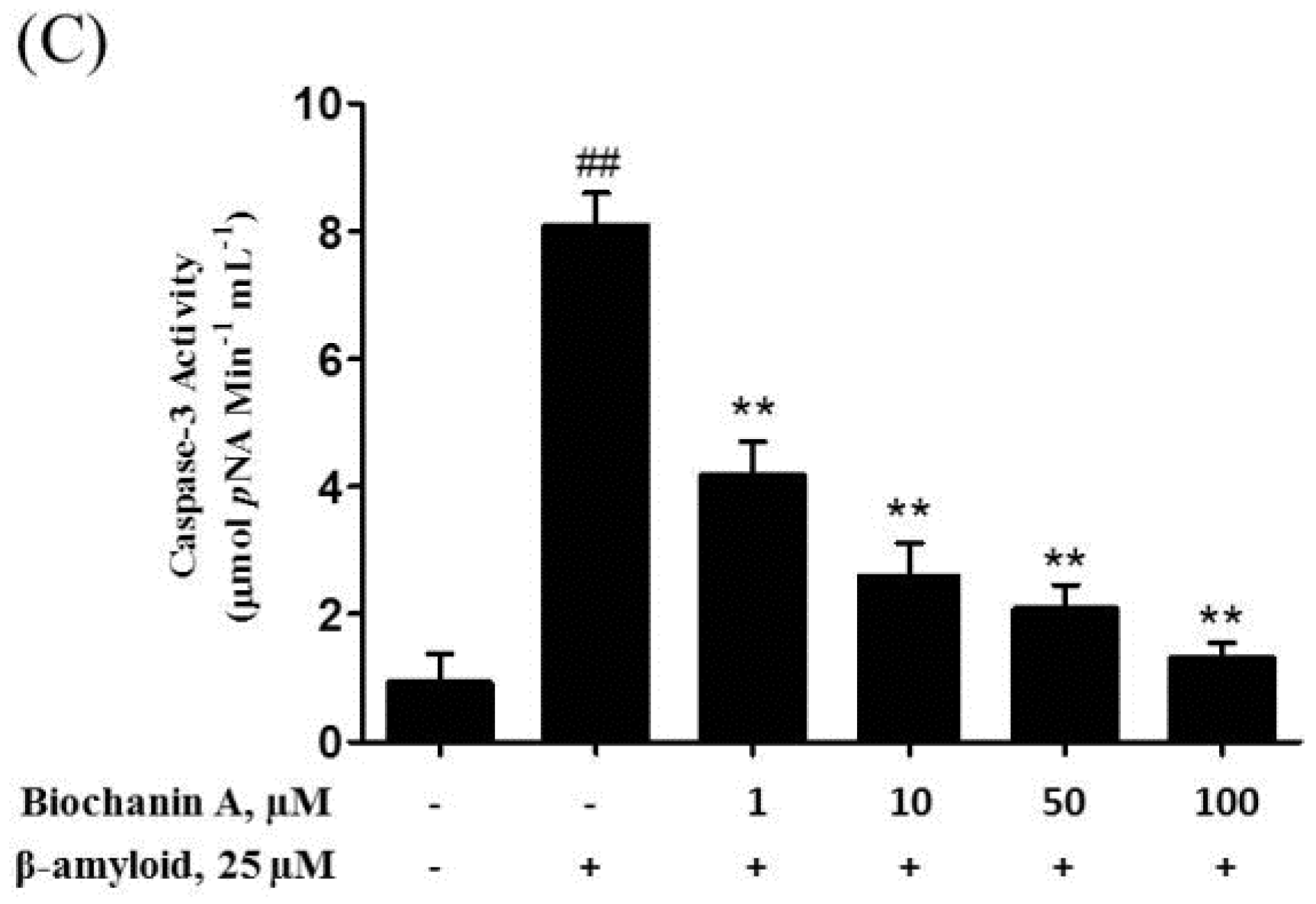
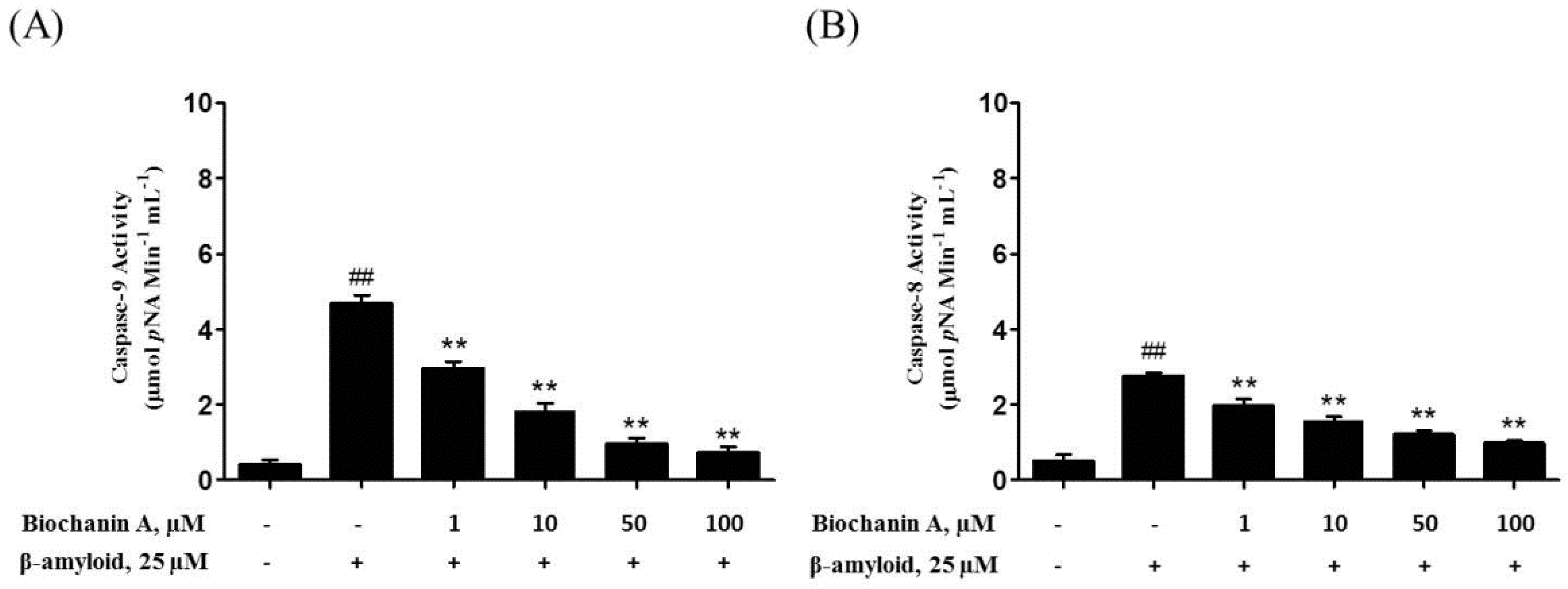
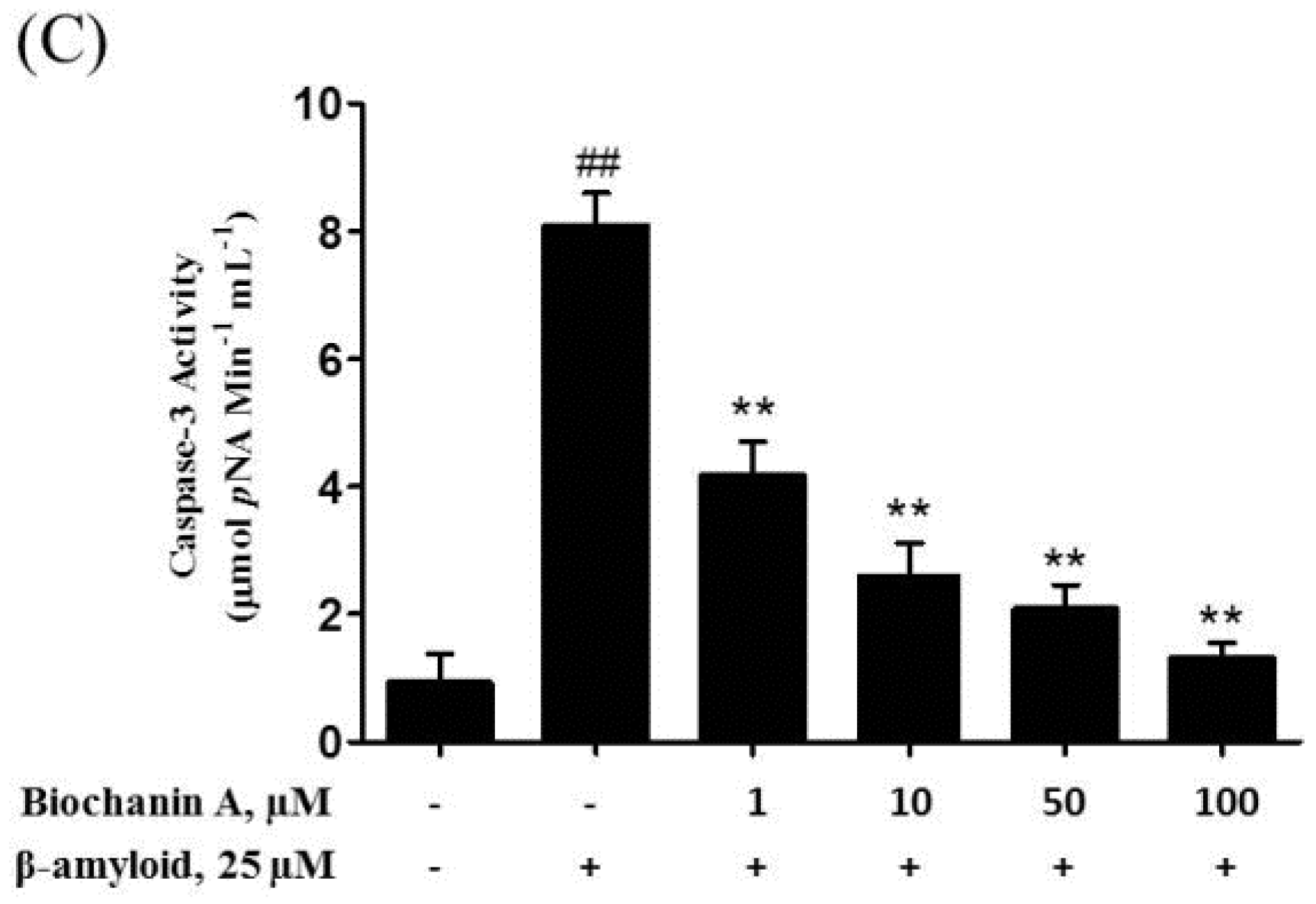
2.3. Biochanin A Suppressed Aβ25–35-Induced Caspase Activity
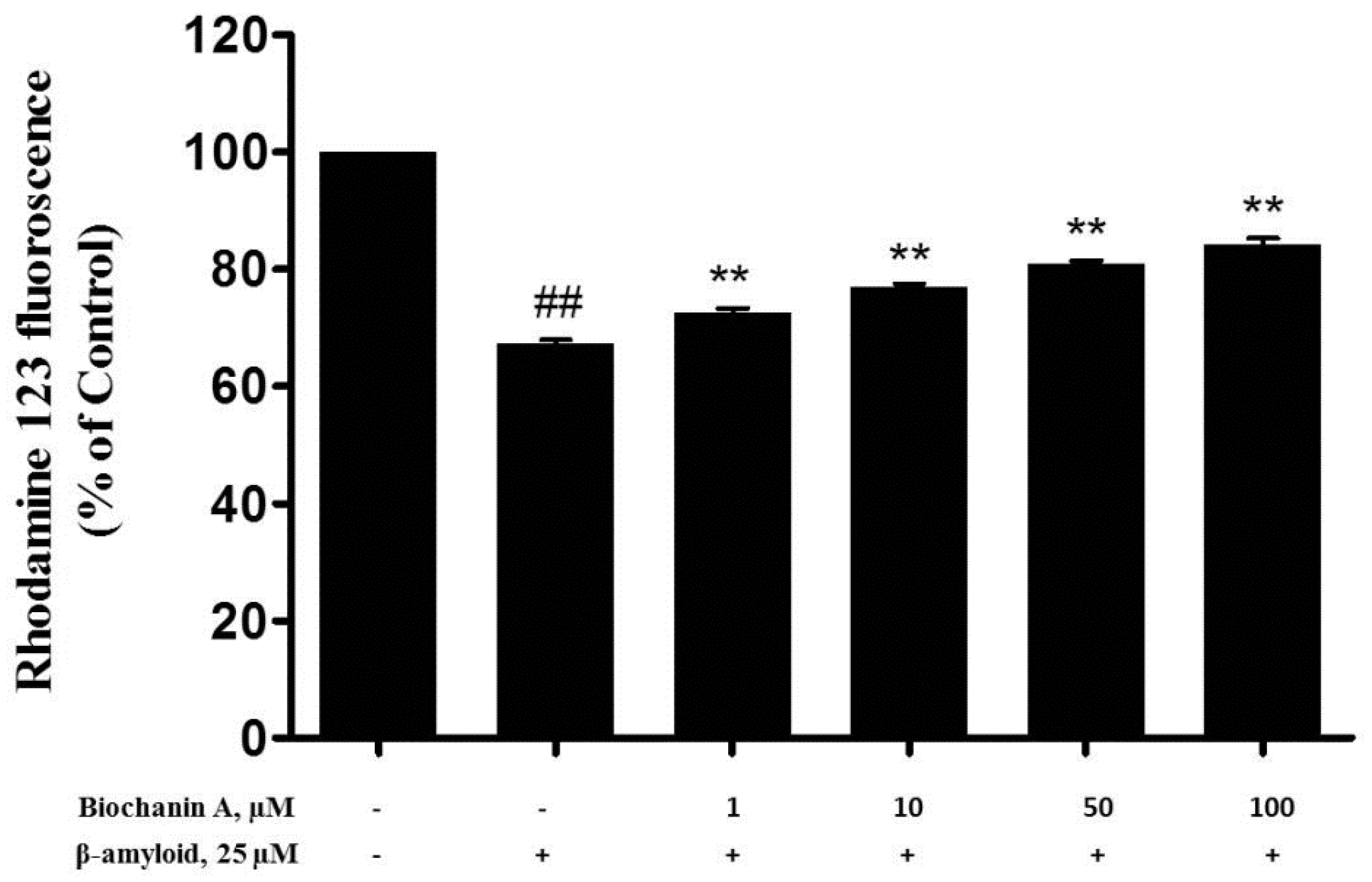
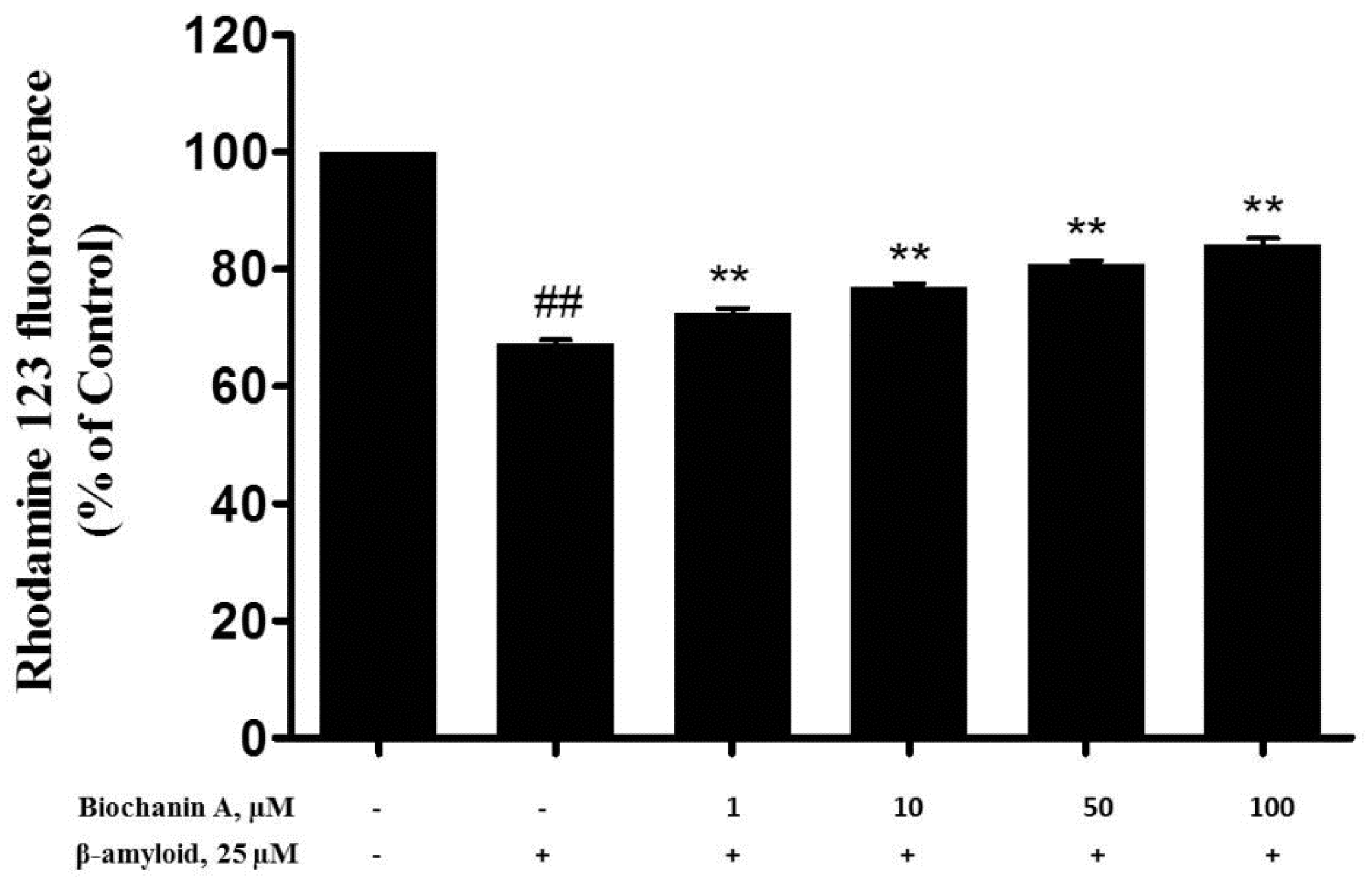
2.4. Effect of Biochanin A on Aβ25–35-Induced MMP Collapse
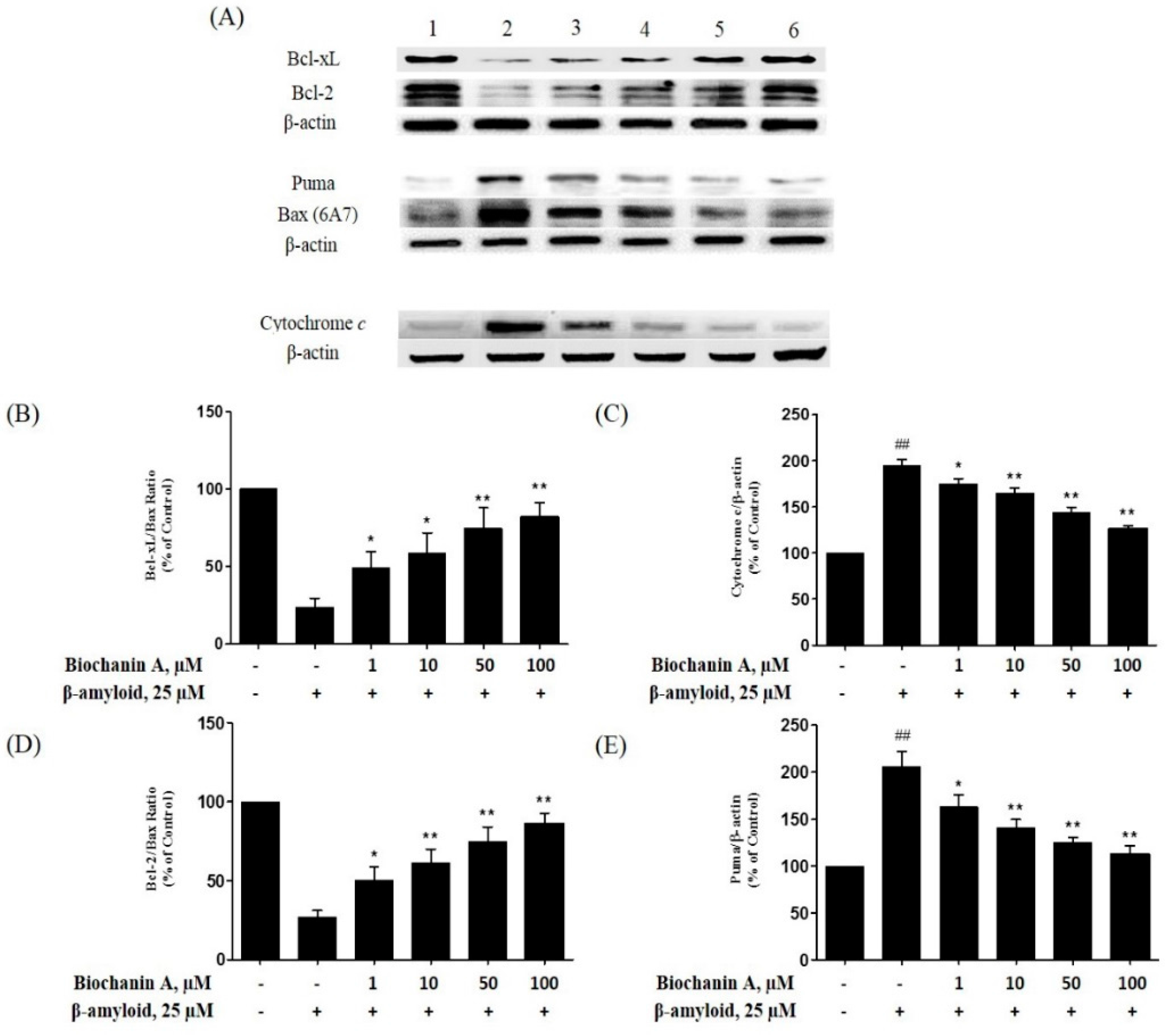
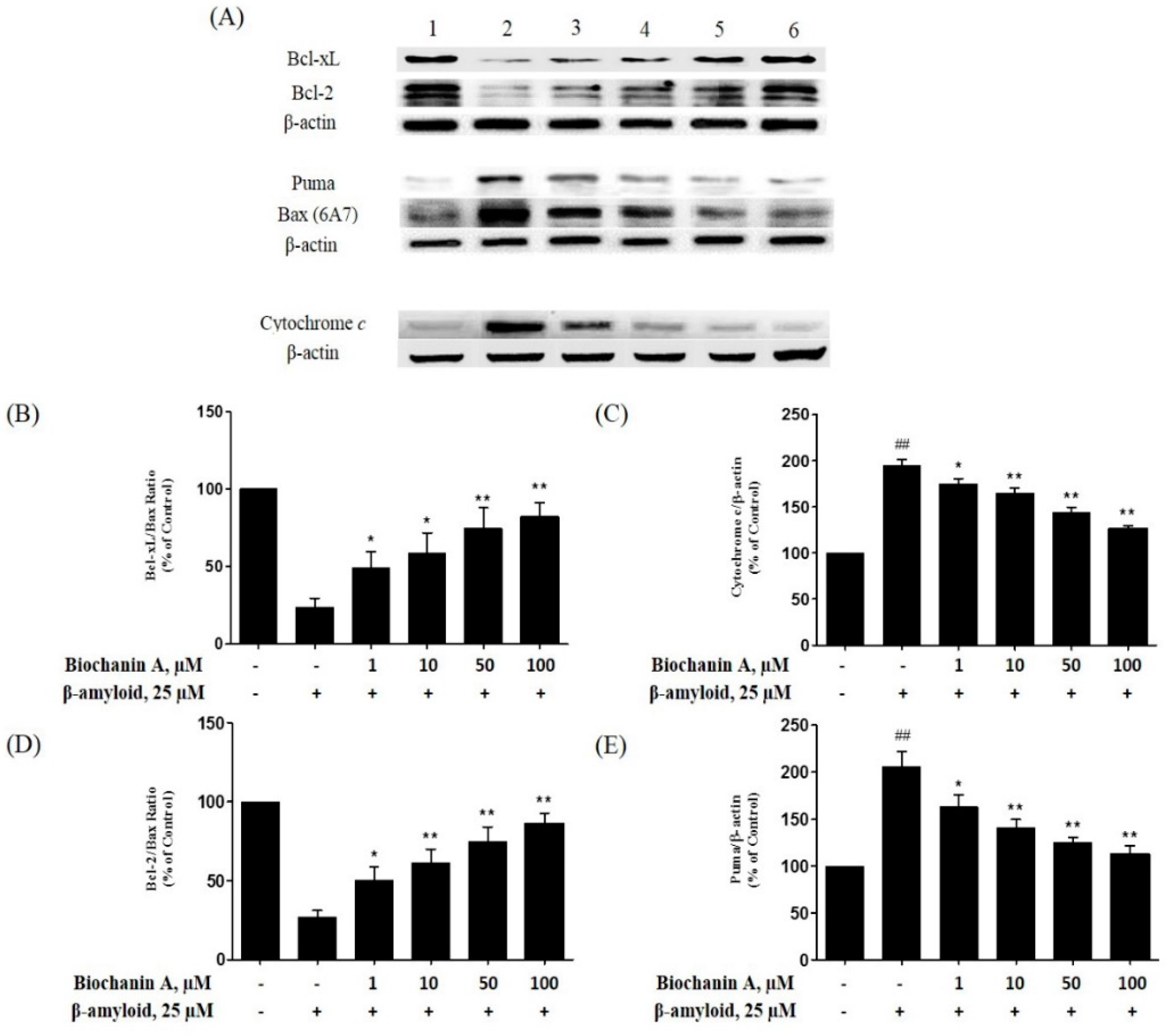
2.5. Effect of Biochanin A on the Expression of Pro- and Antiapoptotic Proteins in PC12 Cells Treated with Aβ25–35
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Peptides
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Lactate Dehydrogenase Release Assay
4.6. Apoptosis Analysis
4.7. Hoechst 33342 Staining Assay
4.8. Caspase Activity Measurement
4.9. Mitochondrial Membrane Potential (MMP) Measurement
4.10. Western Blot Analysis
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yamin, G.; Ono, K.; Inayathullah, M.; Teplow, D.B. Amyloid β-protein assembly as a therapeutic target of Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 3231–3246. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, W.M.; Scheltens, P. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76, v2–v7. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann. N. Y. Acad. Sci. 2000, 924, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid β peptide 25–35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [PubMed]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic exposure to sub-lethal β-amyloid (Aβ) inhibits the import of nuclear encoded proteins to mitochondria in differentiated PC12 cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A β accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Keil, U.; Marques, C.A.; Bonert, A.; Frey, C.; Schussel, K.; Muller, W.E. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem. Pharmacol. 2003, 66, 1627–1634. [Google Scholar] [CrossRef]
- Hauptmann, S.; Keil, U.; Scherping, I.; Bonert, A.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol. 2006, 41, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Hauptmann, S.; Scherping, I.; Drose, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer's disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Hemmelgarn, B.T.; Chuang, C.C.; Best, T.M. The role of oxidative stress-induced epigenetic alterations in amyloid-β production in Alzheimer's disease. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, P.; Fernandez, A.P.; Castro-Blanco, S.; Serrano, J.; Bentura, M.L.; Martinez-Murillo, R.; Martinez, A.; Rodrigo, J. Intra and extracellular A β and PHF in clinically evaluated cases of Alzheimer’s disease. Histol. Histopathol. 2004, 19, 823–844. [Google Scholar] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links A β to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [PubMed]
- Kim, H.S.; Lee, J.H.; Lee, J.P.; Kim, E.M.; Chang, K.A.; Park, C.H.; Jeong, S.J.; Wittendorp, M.C.; Seo, J.H.; Choi, S.H.; et al. Amyloid β peptide induces cytochrome C release from isolated mitochondria. Neuroreport 2002, 13, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Usui, T. Pharmaceutical prospects of phytoestrogens. Endocr. J. 2006, 53, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Horn-Ross, P.L.; John, E.M.; Canchola, A.J.; Stewart, S.L.; Lee, M.M. Phytoestrogen intake and endometrial cancer risk. J. Natl. Cancer Inst. 2003, 95, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Van de Weijer, P.H.; Barentsen, R. Isoflavones from red clover (Promensil®) significantly reduce menopausal hot flush symptoms compared with placebo. Maturitas 2002, 42, 187–193. [Google Scholar] [CrossRef]
- Chen, H.Q.; Jin, Z.Y.; Li, G.H. Biochanin A protects dopaminergic neurons against lipopolysaccharide-induced damage through inhibition of microglia activation and proinflammatory factors generation. Neurosci. Lett. 2007, 417, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Yamada, K.; Yin, H.; Ito, A.; Kataoka, T.; Dohi, K. Chemoprevention of N-nitroso-N-methylurea-induced rat mammary carcinogenesis by soy foods or biochanin A. Jpn. J. Cancer. Res. 1998, 89, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.J.; Shin, B.S.; An, G.; Morris, M.E. Biochanin A inhibits breast cancer tumor growth in a murine xenograft model. Pharm. Res. 2008, 25, 2158–2163. [Google Scholar] [CrossRef] [PubMed]
- Touny, L.H.E.; Henderson, F.; Djakiew, D. Biochanin A reduces drug-induced p75NTR expression and enhances cell survival: A new in vitro assay for screening inhibitors of p75NTR expression. Rejuv. Res. 2010, 13, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.W.; Tham, C.L.; Israf, D.A.; Lee, S.H.; Kim, M.K. Neuroprotective effects of biochanin A against glutamate-induced cytotoxicity in PC12 cells via apoptosis inhibition. Neurochem. Res. 2013, 38, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M.F. The release of calcium from the endoplasmic reticulum induced by amyloid-β and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B.; Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocrinol. 2010, 31, 400–419. [Google Scholar] [CrossRef] [PubMed]
- Dijsselbloem, N.; Vanden Berghe, W.; De Naeyer, A.; Haegeman, G. Soy isoflavone phyto-pharmaceuticals in interleukin-6 affections. Multi-purpose nutraceuticals at the crossroad of hormone replacement, anti-cancer and anti-inflammatory therapy. Biochem. Pharmacol. 2004, 68, 1171–1185. [Google Scholar] [CrossRef] [PubMed]
- Jefremov, V.; Rakitin, A.; Mahlapuu, R.; Zilmer, K.; Bogdanovic, N.; Zilmer, M.; Karelson, E. 17β-Oestradiol stimulation of G-proteins in aged and Alzheimer’s human brain: Comparison with phytoestrogens. J. Neuroendocrinol. 2008, 20, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.J.; Li, Z.; Zhang, L.; Chen, D.H.; Du, G.H.; Sun, L. Protective effects of trans-2,4-dimethoxystibene on cognitive, impairments induced by Aβ(25–35) in hypercholesterolemic rats. Brain Res. Bull. 2010, 82, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.H.; Dong, M.X.; Li, X.M.; Zou, Y.; Fan, L.; Wang, X.L.; Cai, D.F.; Li, C.C.; Zhou, L.; Liu, J.H.; et al. Neuroprotective effects of puerarin against beta-amyloid-induced neurotoxicity in PC12 cells via a PI3K-dependent signaling pathway. Brain Res. Bull. 2011, 85, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, Q.; Diaz Brinton, R. Neuroprotective and neurotrophic efficacy of phytoestrogens in cultured hippocampal neurons. Exp. Biol. Med. 2002, 227, 509–519. [Google Scholar]
- Ma, W.; Yuan, L.; Yu, H.; Ding, B.; Xi, Y.; Feng, J.; Xiao, R. Genistein as a neuroprotective antioxidant attenuates redox imbalance induced by β-amyloid peptides 25–35 in PC12 cells. Int. J. Dev. Neurosci. 2010, 28, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Lotocki, G.; Keane, R.W. Inhibitors of apoptosis proteins in injury and disease. IUBMB Life 2002, 54, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, T.E.; Wyatt, S.; Paterson, H.F.; Davies, A.M. The proto-oncogene Bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell 1993, 73, 295–307. [Google Scholar] [CrossRef]
- Boise, L.H.; González-García, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Mao, X.; Nuñez, G.; Thompson, C.B. Bcl-x, a Bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Sassone, J.; Maraschi, A.; Sassone, F.; Silani, V.; Ciammola, A. Defining the role of the Bcl-2 family proteins in Huntington’s disease. Cell Death Dis. 2013, 4, e772. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Ma, R.; Huang, C.F.; Tang, Q.; Fu, Q.; Liu, H.; Hu, B.R.; Xiang, J.Z. Protective effect of erythropoietin on β-amyloid-induced PC12 cell death through antioxidant mechanisms. Neurosci. Lett. 2008, 442, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Liu, T.; Wang, W.X.; Xu, J.H.; Yang, P.B.; Lu, H.X.; Sun, Q.R.; Hu, H.T. Protective effects of [Gly14]-humanin on β-amyloid-induced PC12 cell death by preventing mitochondrial dysfunction. Neurochem. Int. 2010, 56, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid induces cytochrome c release and apoptosis in a Bax/Bak-independent, permeability transition pore dependent fashion. Apoptosis 2009, 14, 191–202. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, T.J.; Beham, A.; Sarkiss, M.; Andersen, M.M.; Lo, P. Importance of the Bcl-2 family in cell death regulation. Experientia 1996, 52, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Kudo, W.; Lee, H.P.; Smith, M.A.; Zhu, X.; Matsuyama, S.; Lee, H.G. Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. 2012, 3, e309. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.W.; Kim, M.K. Neuroprotective Effects of Biochanin A against β-Amyloid-Induced Neurotoxicity in PC12 Cells via a Mitochondrial-Dependent Apoptosis Pathway. Molecules 2016, 21, 548. https://doi.org/10.3390/molecules21050548
Tan JW, Kim MK. Neuroprotective Effects of Biochanin A against β-Amyloid-Induced Neurotoxicity in PC12 Cells via a Mitochondrial-Dependent Apoptosis Pathway. Molecules. 2016; 21(5):548. https://doi.org/10.3390/molecules21050548
Chicago/Turabian StyleTan, Ji Wei, and Min Kyu Kim. 2016. "Neuroprotective Effects of Biochanin A against β-Amyloid-Induced Neurotoxicity in PC12 Cells via a Mitochondrial-Dependent Apoptosis Pathway" Molecules 21, no. 5: 548. https://doi.org/10.3390/molecules21050548
APA StyleTan, J. W., & Kim, M. K. (2016). Neuroprotective Effects of Biochanin A against β-Amyloid-Induced Neurotoxicity in PC12 Cells via a Mitochondrial-Dependent Apoptosis Pathway. Molecules, 21(5), 548. https://doi.org/10.3390/molecules21050548