
A Chemo-Enzymatic Road Map to the Synthesis of CoA Esters

Abstract
:
1. Introduction
2. Results
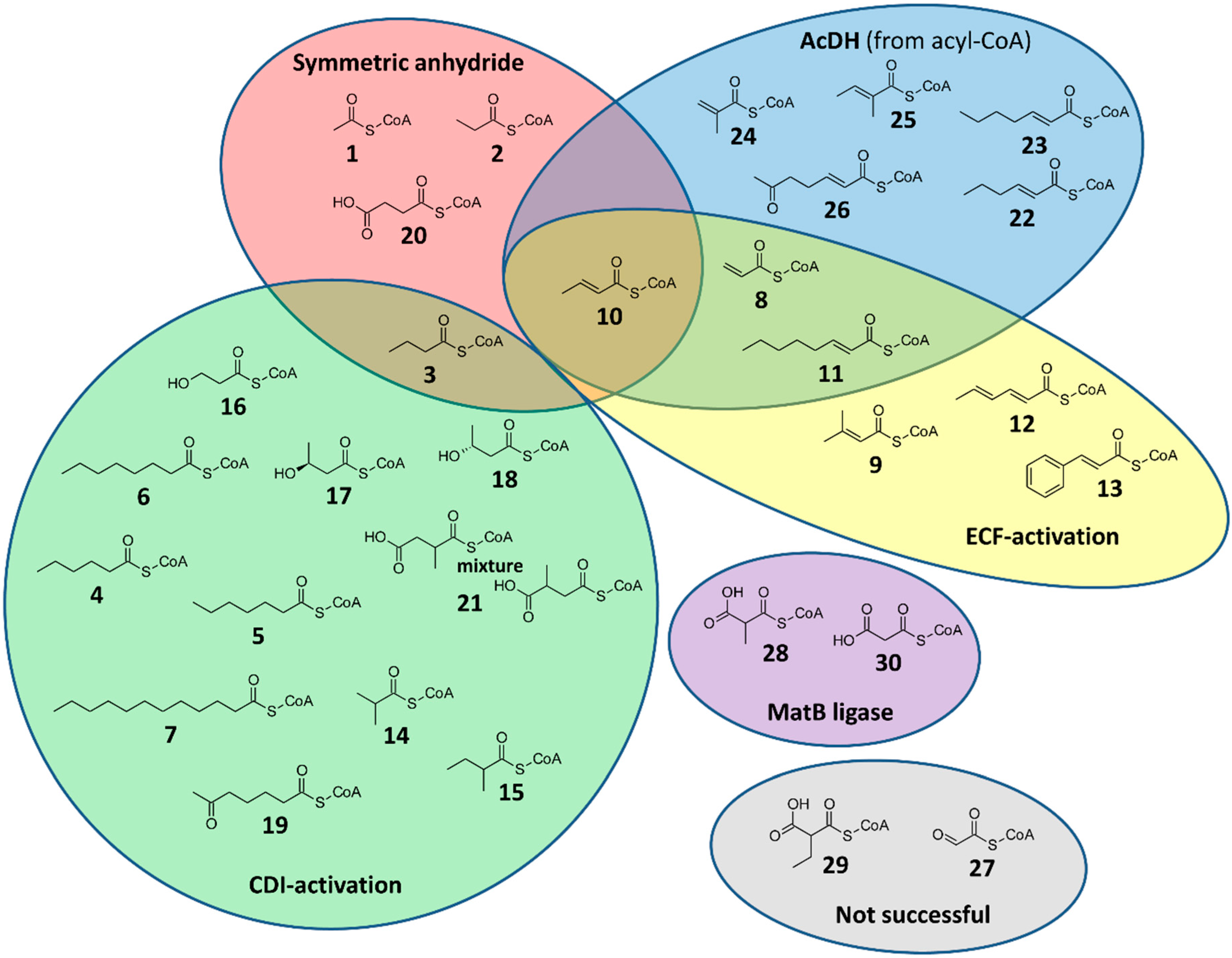
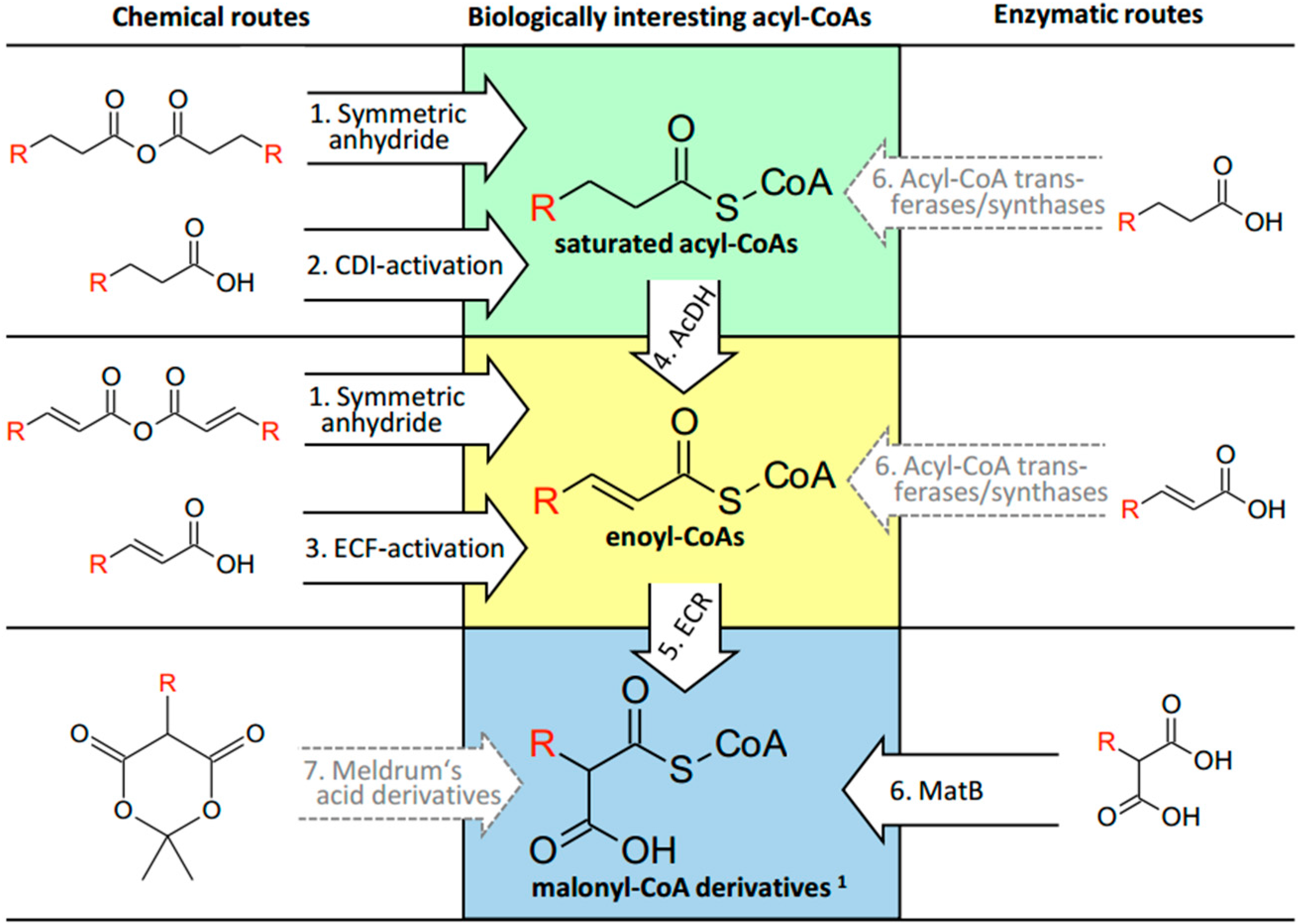
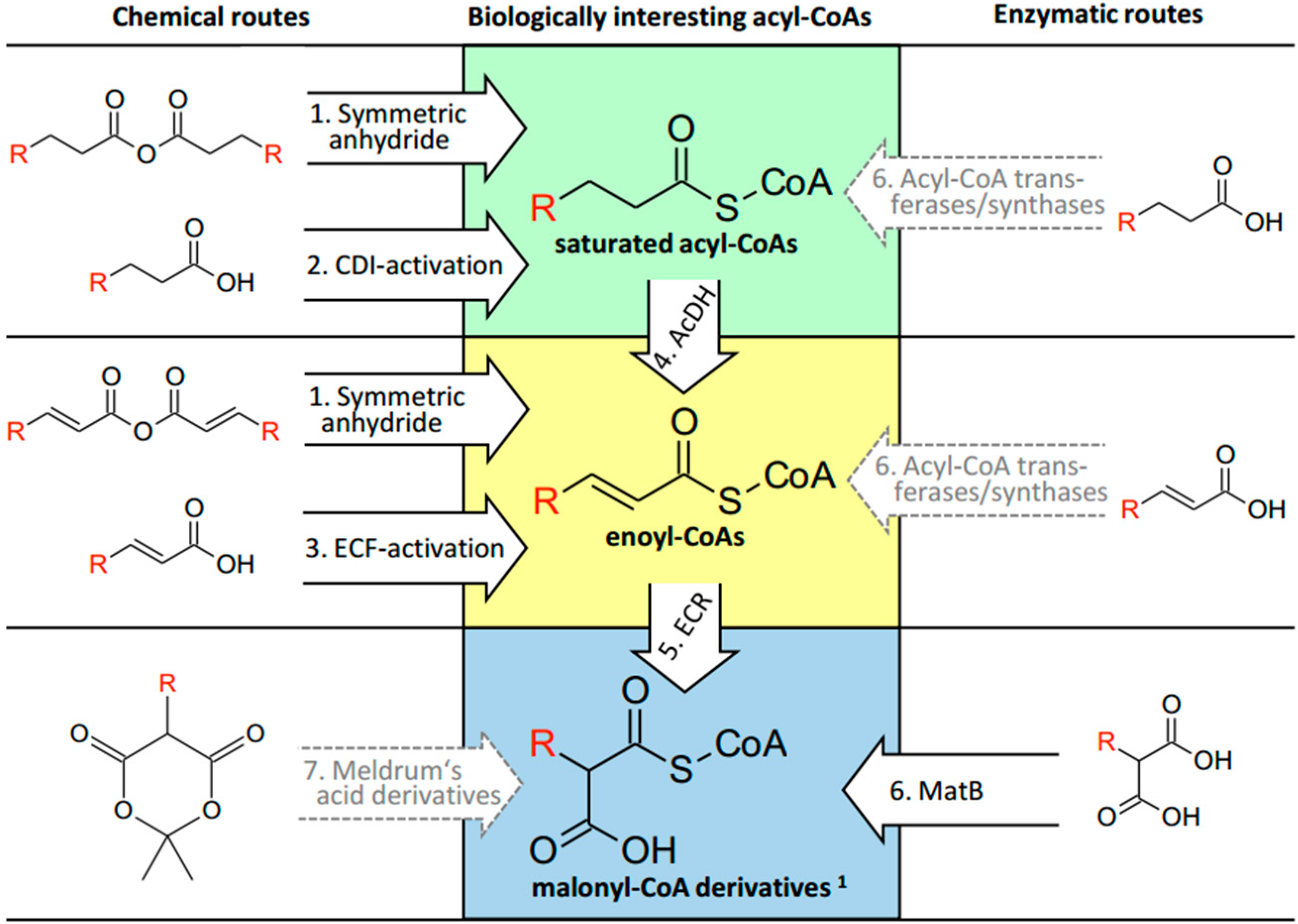
2.1. CoA Acylations by Symmetric Anhydride
2.2. CoA Acylations by Carbonyldiimidazole (CDI)
2.3. CoA Acylations by Ethylchloroformate (ECF)
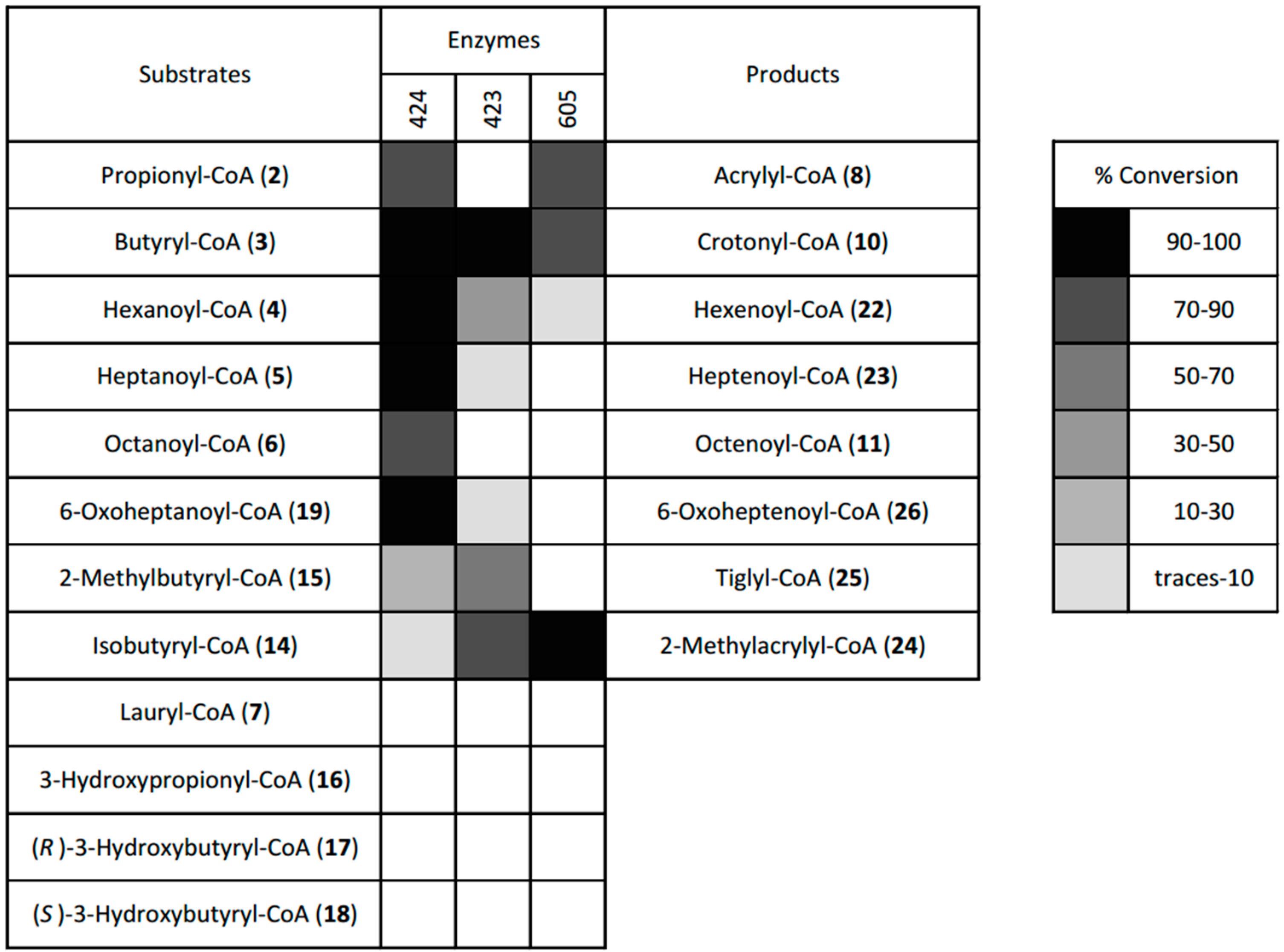
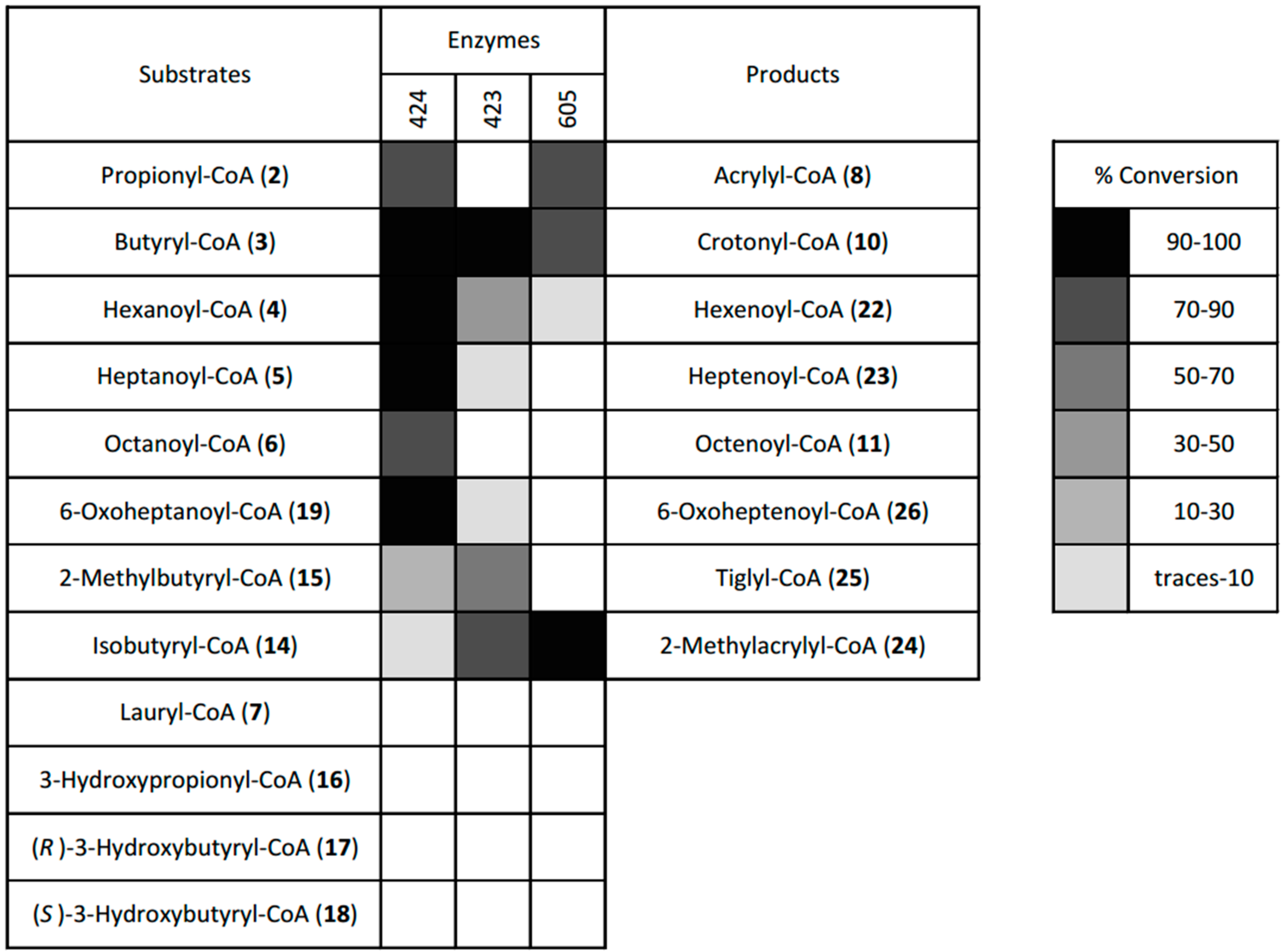
2.4. Desaturation of Acyl-CoAs
2.5. Enzymatic CoA Acylation by MatB
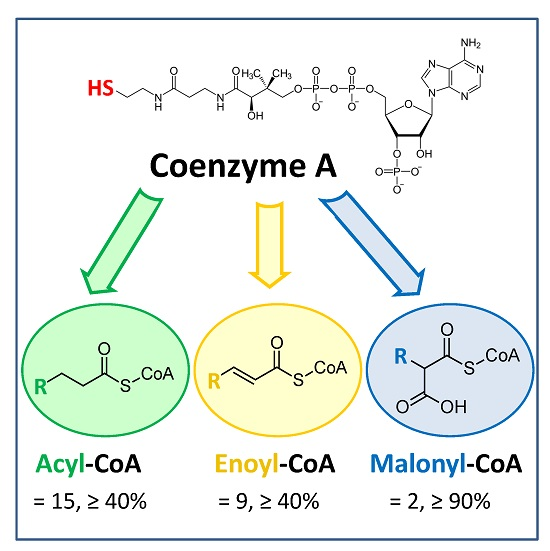
3. Discussion
3.1. Saturated Acyl-CoA Esters
3.2. α,β-Unsaturated Enoyl-CoA Esters
3.3. α-Carboxylated Malonyl-CoA Derivatives
4. Materials and Methods
4.1. Symmetric Anhydride Synthesis
4.2. Carbonyldiimidazole Synthesis
4.3. Ethylchloroformate Synthesis
4.4. Dehydrogenase Screen
4.4.1. Cloning
4.4.2. Expression and Purification
4.4.3. Screen of the Three Dehydrogenases
4.5. CoA Ligation by MatB and Determination of the Stereochemistry of Methylmalonyl-CoA
4.6. Yield Determination
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CoA | Coenzyme A |
| CDI | Carbonyldiimidazole |
| ECF | Ethylchloroformate |
| HPLC | High performance liquid chromatography |
| UV-Vis | Ultraviolet-visible light |
References
- Brenda—The Comprehensive Enzyme Information System. Available online: http://brenda-enzymes.de/index.php (accessed on 6 April 2016).
- Mishra, P.K.; Drueckhammer, D.G. Coenzyme a analogues and derivatives: Synthesis and applications as mechanistic probes of coenzyme a ester-utilizing enzymes. Chem. Rev. 2000, 100, 3283–3309. [Google Scholar] [CrossRef] [PubMed]
- Nazi, I.; Koteva, K.P.; Wright, G.D. One-pot chemoenzymatic preparation of coenzyme a analogues. Anal. Biochem. 2004, 324, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Diethelm, S.; Ray, L.; Garg, N.; Awakawa, T.; Dorrestein, P.C.; Moore, B.S. Chemoenzymatic synthesis of acyl coenzyme a substrates enables in situ labeling of small molecules and proteins. Org. Lett. 2015, 17, 4452–4455. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, A.; Yoshimura, T.; Okuda, S. A new method for the preparation of acyl-CoA thioesters. J. Biochem. 1981, 89, 337–339. [Google Scholar] [PubMed]
- Valenzano, C.R.; You, Y.O.; Garg, A.; Keatinge-Clay, A.; Khosla, C.; Cane, D.E. Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase. J. Am. Chem. Soc. 2010, 132, 14697–14699. [Google Scholar] [CrossRef] [PubMed]
- Pohl, N.L.; Hans, M.; Lee, H.Y.; Kim, Y.S.; Cane, D.E.; Khosla, C. Remarkably broad substrate tolerance of malonyl-CoA synthetase, an enzyme capable of intracellular synthesis of polyketide precursors. J. Am. Chem. Soc. 2001, 123, 5822–5823. [Google Scholar] [CrossRef] [PubMed]
- Peter, D.M.; Schada von Borzyskowski, L.; Kiefer, P.; Christen, P.; Vorholt, J.A.; Erb, T.J. Screening and engineering the synthetic potential of carboxylating reductases from central metabolism and polyketide biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 13457–13461. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Fuchs, G.; Alber, B.E. (2S)-methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-coa pathway for acetyl-CoA assimilation. Mol. Microbiol. 2009, 73, 992–1008. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Retey, J.; Fuchs, G.; Alber, B.E. Ethylmalonyl-CoA mutase from rhodobacter sphaeroides defines a new subclade of coenzyme b12-dependent acyl-CoA mutases. J. Biol. Chem. 2008, 283, 32283–32293. [Google Scholar] [CrossRef] [PubMed]
- Sohling, B.; Gottschalk, G. Purification and characterization of a coenzyme-a-dependent succinate-semialdehyde dehydrogenase from clostridium kluyveri. Eur. J. Biochem. 1993, 212, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, I.; McArthur, J.; Randall, S.; Draelos, M.M.; Musiol, E.M.; Muddiman, D.C.; Weber, T.; Williams, G.J. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem. Biol. 2013, 8, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Go, M.K.; Chow, J.Y.; Cheung, V.W.; Lim, Y.P.; Yew, W.S. Establishing a toolkit for precursor-directed polyketide biosynthesis: Exploring substrate promiscuities of acid-coa ligases. Biochemistry 2012, 51, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Vats, A.; Saxena, P.; Mohanty, D.; Gokhale, R.S. Promiscuous fatty acyl coa ligases produce acyl-CoA and acyl-snac precursors for polyketide biosynthesis. J. Am. Chem. Soc. 2005, 127, 9388–9389. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Berg, I.A.; Brecht, V.; Muller, M.; Fuchs, G.; Alber, B.E. Synthesis of c5-dicarboxylic acids from c2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-coa pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 10631–10636. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, I.; Williams, G.J. Mutant malonyl-CoA synthetases with altered specificity for polyketide synthase extender unit generation. ChemBioChem 2011, 12, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Erb, T.J.; Brecht, V.; Fuchs, G.; Muller, M.; Alber, B.E. Carboxylation mechanism and stereochemistry of crotonyl-CoA carboxylase/reductase, a carboxylating enoyl-thioester reductase. Proc. Natl. Acad. Sci. USA 2009, 106, 8871–8876. [Google Scholar] [CrossRef] [PubMed]
- Pohl, N.L.; Gokhale, R.S.; Cane, D.E.; Khosla, C. Synthesis and incorporation of an n-acetylcysteamine analogue of methylmalonyl-CoA by a modular polyketide synthase. J. Am. Chem. Soc. 1998, 120, 11206–11207. [Google Scholar] [CrossRef]
- Dawson, R.M.C.; Elliott, D.C.; Elliott, W.H.; Jones, K.M. Data for Biochemical Research, 3rd ed.; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Sample Availability: Expression plasmids of the three dehydrogenases are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acyl-CoA | Yield (%) | Used Method 1 |
|---|---|---|
| Acetyl-CoA 1 | 81 | anhydride |
| Propionyl-CoA 2 | 86 | anhydride |
| Butyryl-CoA 3 | 87 | anhydride |
| Hexanoyl-CoA 4 | 76 | CDI |
| Heptanoyl-CoA 5 | 74 | CDI |
| Octanoyl-CoA 6 | 68 | CDI |
| Lauryl-CoA 7 | 67 | CDI |
| Acrylyl-CoA 8 | 17 | ECF |
| 3,3-Dimethylacrylyl-CoA 9 | 39 | ECF |
| Crotonyl-CoA 10 | 80 | anhydride |
| Octenoyl-CoA 11 | 57 | ECF |
| Sorbityl-CoA 12 | 61 | ECF |
| Cinnamoyl-CoA 13 | 75 | ECF |
| Isobutyryl-CoA 14 | 68 | CDI |
| 2-Methylbutyryl-CoA 15 | 78 | CDI |
| 3-Hydroxypropionyl-CoA 16 | 66 | CDI |
| 3-(R)-Hydroxybutyryl-CoA 17 | 54 | CDI |
| 3-(S)-Hydroxybutyryl-CoA 18 | 57 | CDI |
| 6-Oxoheptanoyl-CoA 19 | 56 | CDI |
| Succinyl-CoA 20 | 86 | anhydride |
| Methylsuccinyl-CoA 21 | 40 | CDI |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, D.M.; Vögeli, B.; Cortina, N.S.; Erb, T.J. A Chemo-Enzymatic Road Map to the Synthesis of CoA Esters. Molecules 2016, 21, 517. https://doi.org/10.3390/molecules21040517
Peter DM, Vögeli B, Cortina NS, Erb TJ. A Chemo-Enzymatic Road Map to the Synthesis of CoA Esters. Molecules. 2016; 21(4):517. https://doi.org/10.3390/molecules21040517
Chicago/Turabian StylePeter, Dominik M., Bastian Vögeli, Niña Socorro Cortina, and Tobias J. Erb. 2016. "A Chemo-Enzymatic Road Map to the Synthesis of CoA Esters" Molecules 21, no. 4: 517. https://doi.org/10.3390/molecules21040517
APA StylePeter, D. M., Vögeli, B., Cortina, N. S., & Erb, T. J. (2016). A Chemo-Enzymatic Road Map to the Synthesis of CoA Esters. Molecules, 21(4), 517. https://doi.org/10.3390/molecules21040517