
Synthesis and Evaluation of Ester Derivatives of 10-Hydroxycanthin-6-one as Potential Antimicrobial Agents


Abstract
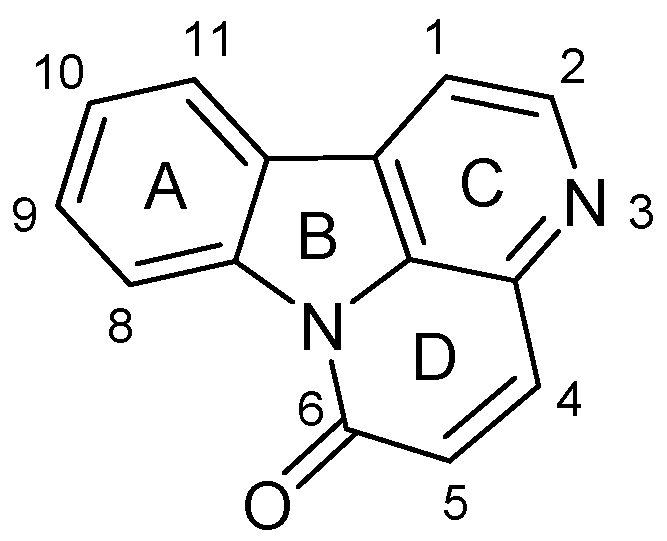
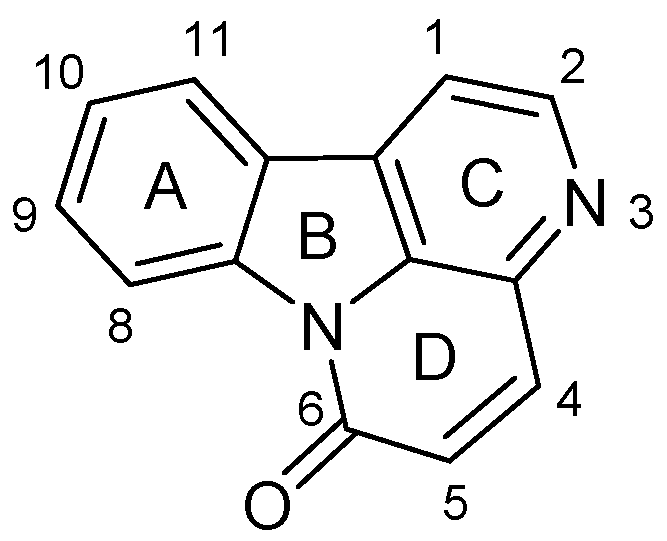
:1. Introduction
2. Results and Discussion
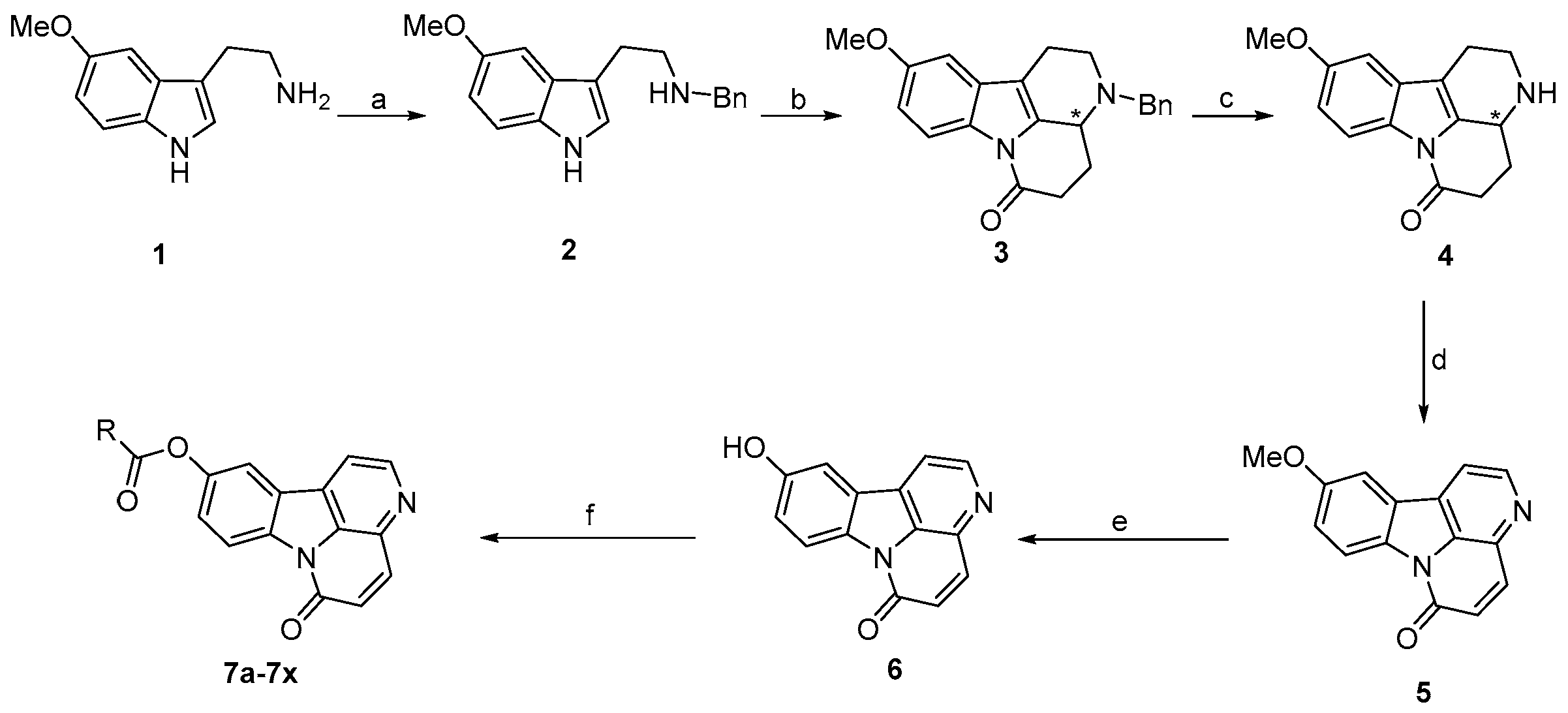
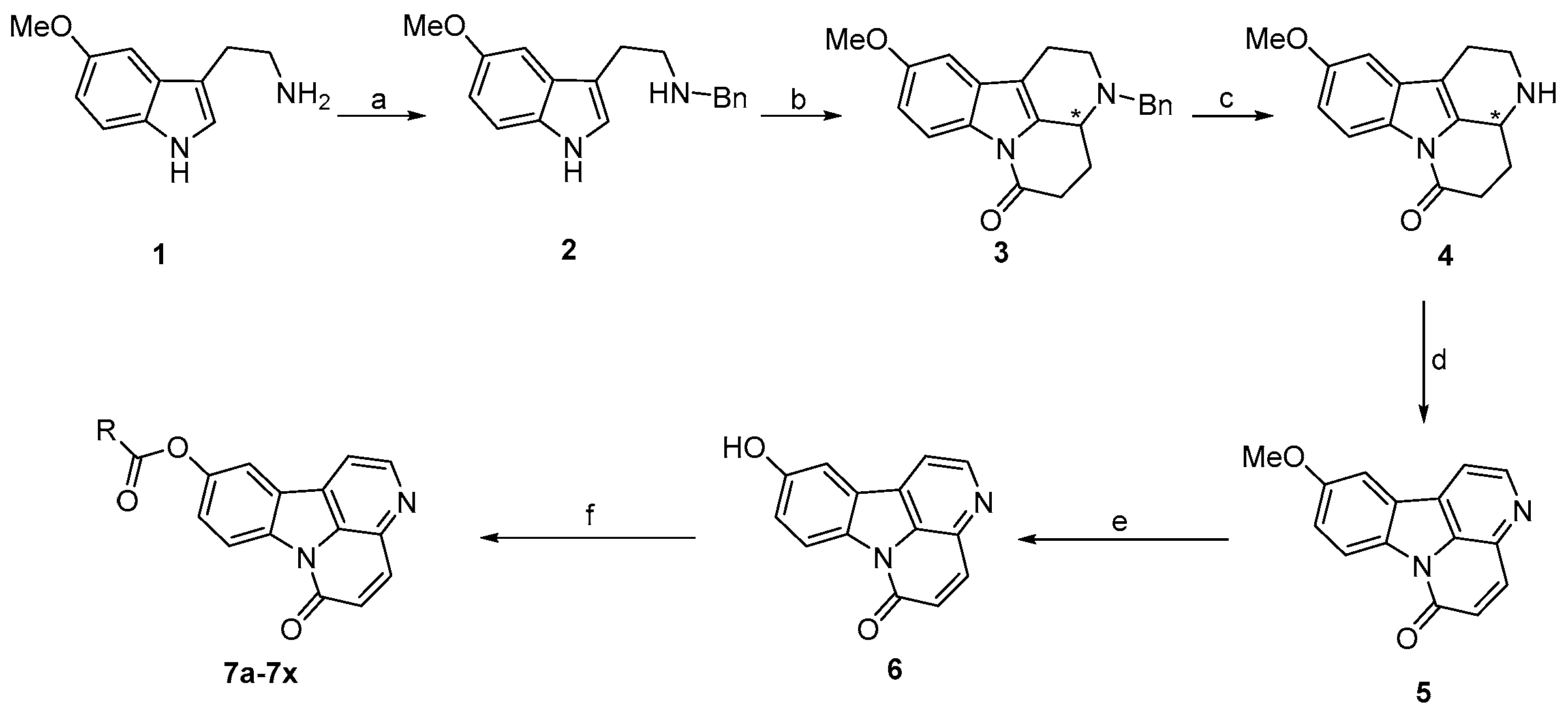
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Antifungal Activity
2.2.2. Antibacterial Activity
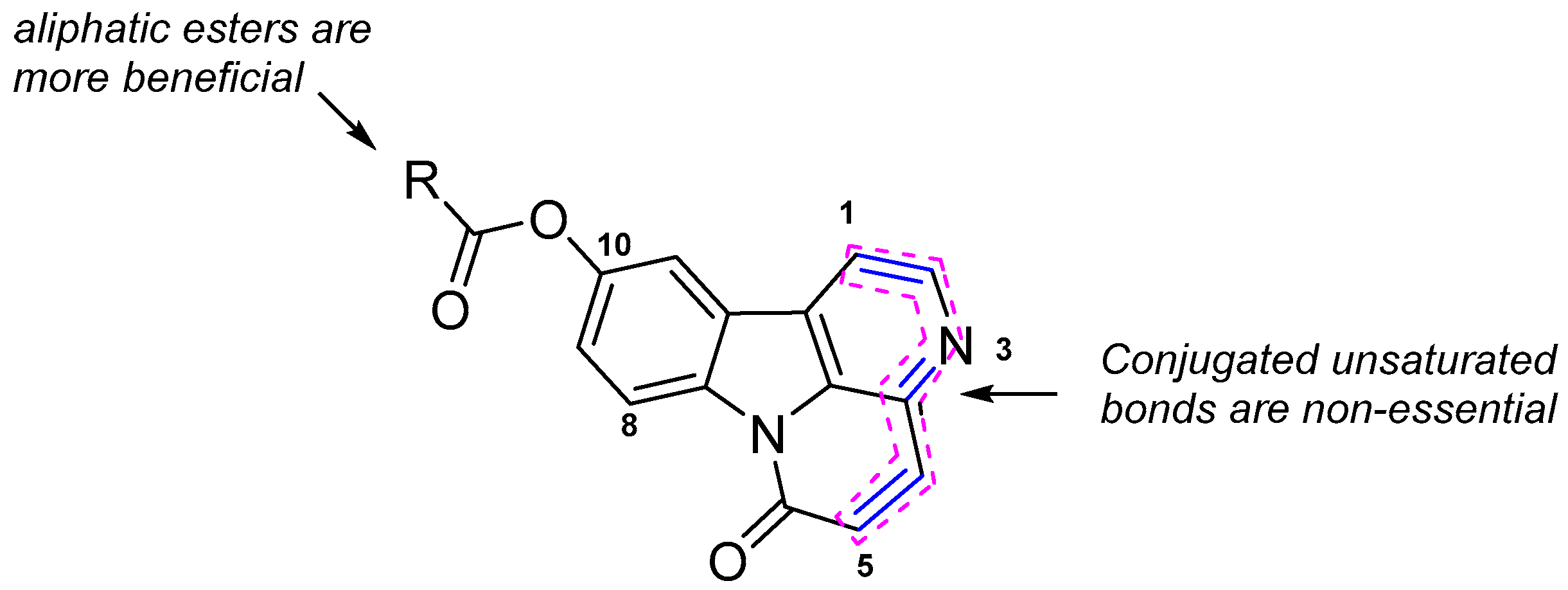
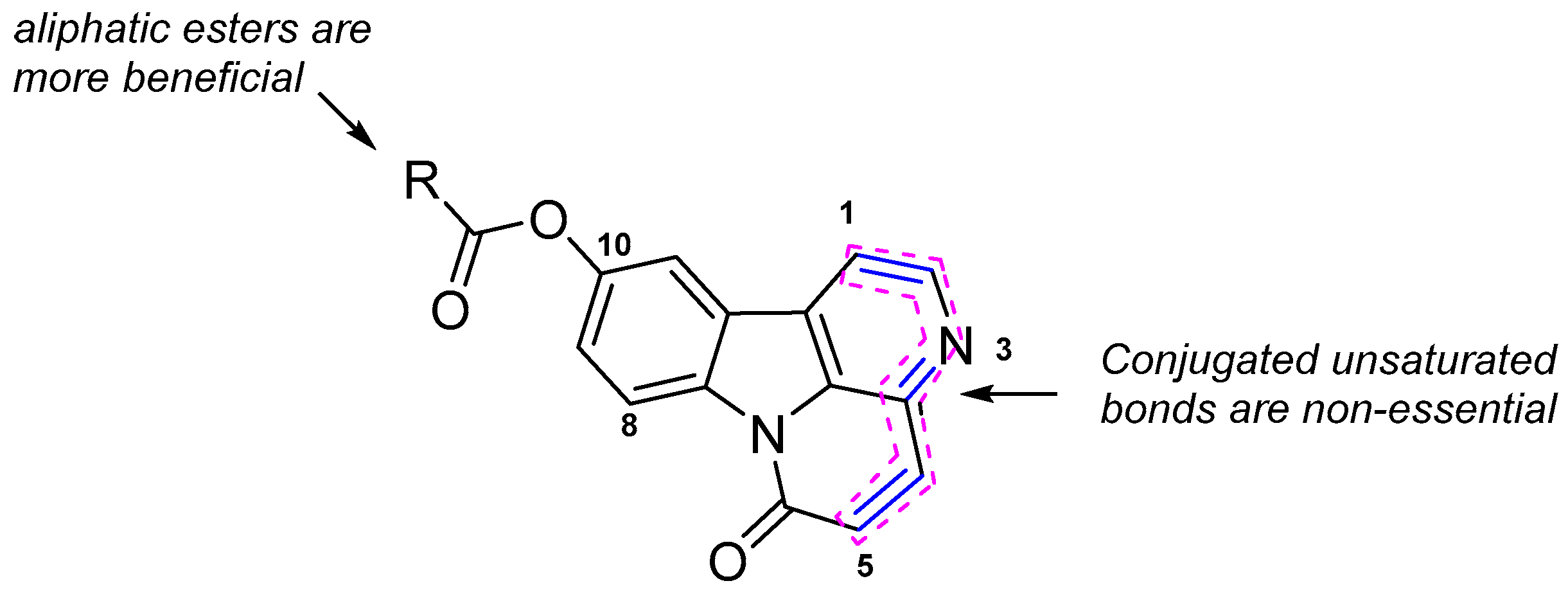
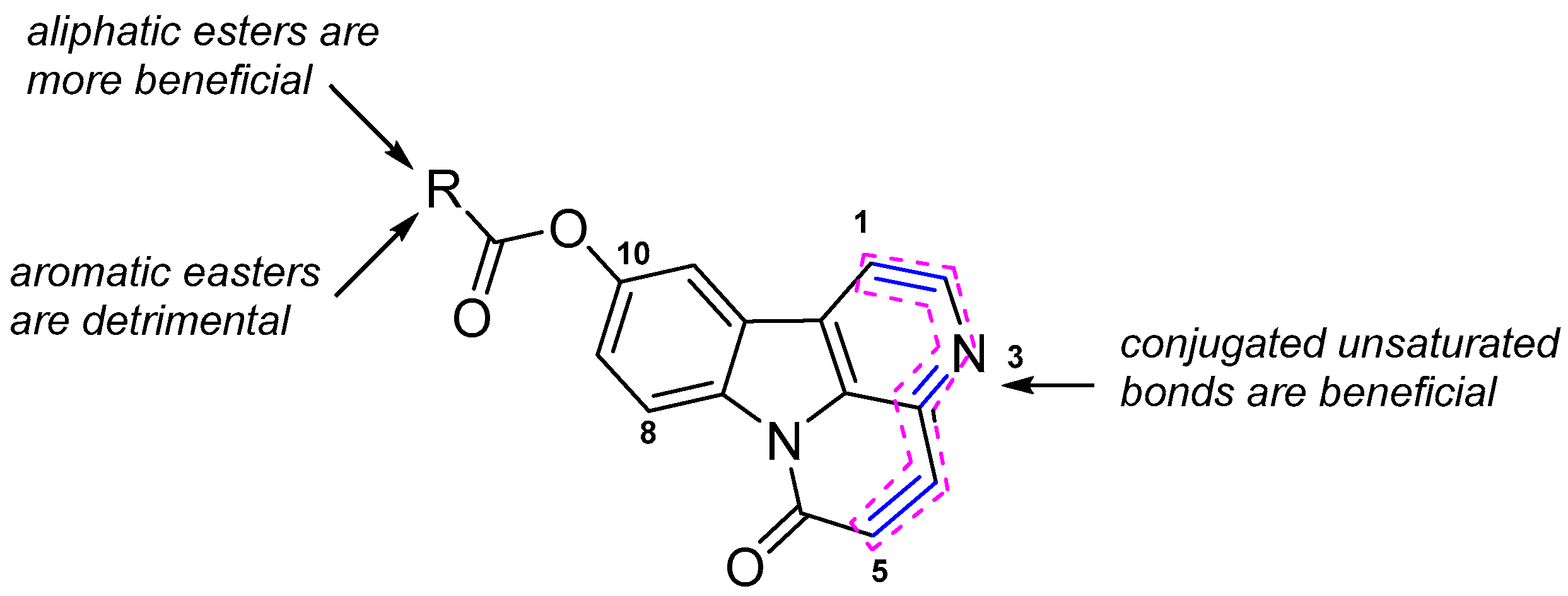
2.3. Structure-Activity Relationship
3. Materials and Methods
3.1. General Details
3.2. Synthesis of Target Compounds
3.2.1. Synthesis of Compound 2
3.2.2. Synthesis of Compound 3
3.2.3. Synthesis of Compound 4
3.2.4. Synthesis of Compound 5
3.2.5. Synthesis of Compound 6
3.2.6. General Procedure for the Synthesis of Compounds 7a–7x
3.3. Procedure for Determining Antifungal Activity
3.4. Procedure for Determining Antibacterial Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Haynes, H.F.; Nelson, E.R.; Price, J.R. Alkaloids of the australian rutaceae: Pentaceras australis hook. F. II. Identification of 5-methoxycanthinone. Aust. J. Sci. Res. Ser. B 1952, A5, 563–569. [Google Scholar]
- Showalter, H.D.H. Progress in the synthesis of canthine alkaloids and ring-truncated congeners. J. Nat. Prod. 2013, 76, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Brahmbhatt, K.G.; Ahmed, N.; Sabde, S.; Mitra, D.; Singh, I.P.; Bhutani, K.K. Synthesis and evaluation of β-carboline derivatives as inhibitors of human immunodeficiency virus. Bioorg. Med. Chem. Lett. 2010, 20, 4416–4419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.B.; Li, Y.; Ma, S.G.; Li, L.; Qu, J.; Zhang, D.; Chen, X.G.; Jiang, J.D.; Yu, S.S. Sesquiterpenes and alkaloids from the roots of alangium chinense. J. Nat. Prod. 2013, 76, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chang, F.R.; Wang, H.K.; Kashiwada, Y.; McPhail, A.T.; Bastow, K.F.; Tachibana, Y.; Cosentino, M.; Lee, K.H. Anti-HIV agents 45 and antitumor agents 205. Two new sesquiterpenes, leitneridanins A and B, and the cytotoxic and anti-HIV principles from Leitneria floridana. J. Nat. Prod. 2000, 63, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Bharitkar, Y.P.; Hazra, A.; Poduri, N.S.A.; Ash, A.; Maulik, P.R.; Mondal, N.B. Isolation, structural elucidation and cytotoxicity evaluation of a new pentahydroxy-pimarane diterpenoid along with other chemical constituents from Aerva lanata. Nat. Prod. Res. 2015, 29, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Dejos, C.; Voisin, P.; Bernard, M.; Regnacq, M.; Berges, T. Canthin-6-one displays antiproliferative activity and causes accumulation of cancer cells in the G2/M phase. J. Nat. Prod. 2014, 77, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Rivero-Cruz, J.F.; Lezutekong, R.; Lobo-Echeverri, T.; Ito, A.; Mi, Q.; Chai, H.B.; Soejarto, D.D.; Cordell, G.A.; Pezzuto, J.M.; Swanson, S.M.; et al. Cytotoxic constituents of the twigs of Simarouba glauca collected from a plot in Southern Florida. Phytother. Res. 2005, 19, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Al-Harrasi, A.; Al-Rawahi, A.; Green, I.R.; Gibbons, S. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem. Rev. 2014, 114, 10369–10428. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.E.; Nakayama, H.; Rojas de Arias, A.; Schinini, A.; Vera de Bilbao, N.; Serna, E.; Lagoutte, D.; Soriano-Agaton, F.; Poupon, E.; Hocquemiller, R.; et al. Effects of canthin-6-one alkaloids from Zanthoxylum chiloperone on trypanosoma cruzi-infected mice. J. Ethnopharmacol. 2007, 109, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.E.; de Rojas Arias, A.; de Torres Ortiz, S.; Inchausti, A.; Nakayama, H.; Thouvenel, C.; Hocquemiller, R.; Fournet, A. Leishmanicidal activity of two canthin-6-one alkaloids, two major constituents of Zanthoxylum chiloperone var. Angustifolium. J. Ethnopharmacol. 2002, 80, 199–202. [Google Scholar] [CrossRef]
- Jiao, W.H.; Gao, H.; Zhao, F.; Lin, H.W.; Pan, Y.M.; Zhou, G.X.; Yao, X.S. Anti-inflammatory alkaloids from the stems of Picrasma quassioides bennet. Chem. Pharm. Bull. 2011, 59, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Kuttan, G. Modulation of humoral immune responses and inhibition of proinflammatory cytokines and nitric oxide production by 10-methoxycanthin-6-one. Immunopharmacol. Immunotoxicol. 2012, 34, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.K.; Dan, W.J.; Li, N.; Du, H.T.; Zhang, J.W.; Wang, J.R. Synthesis, in vitro antibacterial activities of a series of 3-N-substituted canthin-6-ones. Bioorg. Med. Chem. Lett. 2016, 26, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Fournet, A.R.F.M.; Lagoutte, D.; Poupon, E.; Soriano-Agaton, F. Use of Canthin-6-one and Its Analogs in the Treatment of Mycobacteria-Linked Pathologier. U.S. Patent 0059977 A1, 10 March 2011. [Google Scholar]
- O’Donnell, G.; Gibbons, S. Antibacterial activity of two canthin-6-one alkaloids from Allium neapolitanum. Phytother. Res. 2007, 21, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Agaton, F.; Lagoutte, D.; Poupon, E.; Roblot, F. Extraction, hemisynthesis, and synthesis of canthin-6-one analogues. Evaluation of their antifungal activities. J. Nat. Prod. 2005, 68, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Lagoutte, D.; Nicolas, V.; Poupon, E.; Fournet, A.; Hocquemiller, R.; Libong, D.; Chaminade, P.; Loiseau, P.M. Antifungal canthin-6-one series accumulate in lipid droplets and affect fatty acid metabolism in Saccharomyces cerevisiae. Biomed. Pharmacother. 2008, 62, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.R.; Dai, J.K.; Zhao, F.; Dan, W.J.; Yin, D.Y.; Gao, Y.; Qin, W.J.; Zhang, J.W. Preparation of Canthin-6-one Quaternary Ammonium Salts as Antibacterial Agents. C.N. Patent 104530047A, 16 December 2014. [Google Scholar]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gao, Z.F.; Zhao, J.Y.; Li, W.B.; Zhou, L.; Miao, F. New class of 2-aryl-6-chloro-3, 4-dihydroisoquinolinium salts as potential antifungal agents for plant protection: Synthesis, bioactivity and structure-activity relationships. J. Agric. Food Chem. 2015, 63, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Czerwinski, K.M.; Zificsak, C.A.; Stevens, J.; Oberbeck, M. An improved synthesis of canthin-6-one. Synth. Commun. 2003, 33, 1225–1231. [Google Scholar] [CrossRef]
- Zhang, J.W.; Li, S.K.; Wu, W.J. The main chemical composition and in vitro antifungal activity of the essential oils of Ocimum basilicum linn. Var. Pilosum (willd.) benth. Molecules 2009, 14, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, H.; Yuan, M.; Zhou, J.; Tu, Q.; Liu, J.J.; Wang, J. Synthesis and biological evaluation of apigenin derivatives as antibacterial and antiproliferative agents. Molecules 2013, 18, 11496–11511. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.B.C.; Vanderwal, C.D. Efficient access to the core of the strychnos, aspidosperma and iboga alkaloids. A short synthesis of norfluorocurarine. J. Am. Chem. Soc. 2009, 131, 3472–3473. [Google Scholar] [CrossRef] [PubMed]
- Orbe, J.; Sanchez-Arias, J.A.; Rabal, O.; Rodriguez, J.A.; Salicio, A.; Ugarte, A.; Belzunce, M.; Xu, M.; Wu, W.; Tan, H.; et al. Design, synthesis, and biological evaluation of novel matrix metalloproteinase inhibitors as potent antihemorrhagic agents: From hit identification to an optimized lead. J. Med. Chem. 2015, 58, 2465–2488. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 5, 6, and 7a–7x are available from the authors.
| 7b: R = 2′-pyridyl | 7h: R = 4′-ClC6H4 | 7n: R = 2′-CF3C6H4 | 7t: R = CH3CH2 |
| 7c: R = 2′-CH3C6H4 | 7i: R = 2′,4′-Cl2C6H3 | 70: R = 3′-CF3C6H4 | 7u: R = CH3(CH2)2 |
| 7d: R = 3′-CH3C6H4 | 7j: R = 4′-CH3OC6H4 | 7p: R = 4′-CF3C6H4 | 7v: R = CH3(CH2)3 |
| 7e: R = 4′-CH3C6H4 | 7k: R = 2′-FC6H4 | 7q: R = 1′-naphthyl | 7w: R = CH3(CH2)4 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Average Inhibition Rate ± SD (%) | |||
|---|---|---|---|---|
| No. | R | A. solani | F. graminearum | F. solani |
| 4 | - | 61.2 ± 0.4 g | 100.0 ± 0.0 a | 53.4 ± 0.7 n |
| 5 | - | 71.8 ± 0.5 b | 77.7 ± 0.4 d | 92.9 ± 0.1 a |
| 6 | - | 2.0 ± 0.5 q | 74.5 ± 0.5 e | 57.9 ± 0.6 k |
| 7a | C6H5 | 53.1 ± 0.7 i | 48.8 ± 0.4 j | 41.7 ± 0.7 q |
| 7b | 2′-pyridyl | 66.6 ± 0.0 de | 47.0 ± 0.7 k | 79.8 ± 0.5 d |
| 7c | 2′-CH3C6H4 | 2.7 ± 0.7 q | 35.6 ± 0.4 n | 65.3 ± 1.5 i |
| 7d | 3′-CH3C6H4 | 45.8 ± 0.9 k | 43.6 ± 0.6 l | 64.1 ± 0.4 j |
| 7e | 4′-CH3C6H4 | 28.7 ± 0.8 l | 23.6 ± 0.2 q | 77.0 ± 0.6 e |
| 7f | 2′-ClC6H4 | 2.1 ± 0.8 q | 31.8 ± 0.4 p | 68.1 ± 0.4 h |
| 7g | 3′-ClC6H4 | 21.4 ± 0.8 n | 3.4 ± 0.3 s | 86.1 ± 0.6 c |
| 7h | 4′-ClC6H4 | 5.7 ± 0.9 p | 3.1 ± 0.6 s | 72.2 ± 0.5 g |
| 7i | 2′,4′-Cl2C6H3 | 50.1 ± 0.8 j | 7.1 ± 0.5 r | 22.8 ± 0.6 u |
| 7j | 4′-CH3OC6H4 | 50.6 ± 0.3 j | 6.3 ± 0.4 r | 41.9 ± 0.3 q |
| 7k | 2′-FC6H4 | 68.4 ± 0.7 c | 37.6 ± 0.5 m | 40.5 ± 0.4 r |
| 7l | 3′-FC6H4 | 54.0 ± 0.9 i | 33.0 ± 0.7 o | 49.4 ± 0.2 p |
| 7m | 4′-FC6H4 | 58.9 ± 1.3 h | 51.1 ± 0.6 h | 52.1 ± 0.9 o |
| 7n | 2′-CF3C6H4 | 64.4 ± 0.9 f | 80.5 ± 0.4 c | 13.5 ± 1.0 w |
| 7o | 3′-CF3C6H4 | 25.7 ± 0.7 m | 32.6 ± 0.8 po | 54.6 ± 0.3 m |
| 7p | 4′-CF3C6H4 | 8.3 ± 0.3 o | 80.1 ± 1.0 c | 22.9 ± 0.4 u |
| 7q | 1′-naphthyl | 65.1 ± 1.1 ef | 49.9 ± 0.6 i | 25.2 ± 0.7 t |
| 7r | 2′-naphthyl | 25.8 ± 0.2 m | 50.2 ± 0.6 hi | 20.3 ± 0.1 v |
| 7s | CH3 | 68.4 ± 0.6 c | 100.0 ± 0.0 a | 86.0 ± 0.2 c |
| 7t | CH3CH2 | 59.1 ± 0.9 h | 70.0 ± 0.3 f | 80.1 ± 0.3 d |
| 7u | CH3(CH2)2 | 66.9 ± 0.9 cd | 44.4 ± 0.7 l | 69.2 ± 0.9 h |
| 7v | CH3(CH2)3 | 49.9 ± 0.8 j | 85.4 ± 0.4 b | 74.2 ± 0.2 f |
| 7w | CH3(CH2)4 | 66.5 ± 0.9 de | 55.1 ± 0.2 g | 56.6 ± 0.3 l |
| 7x | CH3(CH2)10 | 72.2 ± 0.2 b | 85.6 ± 0.4 b | 29.8 ± 0.4 s |
| TBZ | - | 89.1 ± 0.6 a | 100.0 ± 0.0 a | 89.3 ± 0.4 b |
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | ||
|---|---|---|---|---|
| B. cereus | B. subtilis | R. solanacearum | P. syringae | |
| 5 | 11.2 ± 0.5 | 10.9 ± 0.8 | 12.6 ± 0.4 | 10.9 ± 1.0 |
| 6 | - * | 9.8 ± 0.4 | - | 8.5 ± 0.1 |
| 7s | 12.2 ± 0.1 | 16.9 ± 0.7 | 12.3 ± 0.2 | 18.4 ± 0.2 |
| 7t | 16.6 ± 0.2 | 17.4 ± 0.1 | 13.0 ± 0.2 | 21.0 ± 0.4 |
| 7u | 13.5 ± 0.3 | 13.9 ± 0.4 | - | 13.0 ± 0.8 |
| 7v | 12.7 ± 0.2 | 10.3 ± 1.1 | - | 15.4 ± 0.2 |
| 7w | 9.4 ± 0.1 | 8.5 ± 0.3 | - | 12.5 ± 0.2 |
| Penicillin Sodium | - | 22.5 ± 0.2 | 16.2 ± 0.2 | 20.4 ± 0.8 |
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | ||
|---|---|---|---|---|
| B. cereus | B. subtilis | R. solanacearum | P. syringae | |
| 5 | 3.91 | 7.81 | 15.62 | 7.81 |
| 6 | 15.62 | >125 | >125 | >125 |
| 7s | 15.62 | 15.62 | 31.25 | 7.81 |
| 7t | 15.62 | 7.81 | 15.62 | 7.81 |
| 7u | 31.25 | 31.25 | ND * | 15.62 |
| 7v | >125 | 125 | ND | 31.25 |
| 7w | >125 | 62.5 | ND | >125 |
| Penicillin Sodium | 3.91 | 7.81 | 7.81 | 0.98 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Dai, J.-K.; Liu, D.; Wang, S.-J.; Wang, J.-R. Synthesis and Evaluation of Ester Derivatives of 10-Hydroxycanthin-6-one as Potential Antimicrobial Agents. Molecules 2016, 21, 390. https://doi.org/10.3390/molecules21030390
Zhao F, Dai J-K, Liu D, Wang S-J, Wang J-R. Synthesis and Evaluation of Ester Derivatives of 10-Hydroxycanthin-6-one as Potential Antimicrobial Agents. Molecules. 2016; 21(3):390. https://doi.org/10.3390/molecules21030390
Chicago/Turabian StyleZhao, Fei, Jiang-Kun Dai, Dan Liu, Shi-Jun Wang, and Jun-Ru Wang. 2016. "Synthesis and Evaluation of Ester Derivatives of 10-Hydroxycanthin-6-one as Potential Antimicrobial Agents" Molecules 21, no. 3: 390. https://doi.org/10.3390/molecules21030390
APA StyleZhao, F., Dai, J.-K., Liu, D., Wang, S.-J., & Wang, J.-R. (2016). Synthesis and Evaluation of Ester Derivatives of 10-Hydroxycanthin-6-one as Potential Antimicrobial Agents. Molecules, 21(3), 390. https://doi.org/10.3390/molecules21030390