Design, Synthesis, and Biological Evaluation of Some Novel Pyrrolizine Derivatives as COX Inhibitors with Anti-Inflammatory/Analgesic Activities and Low Ulcerogenic Liability

 , , and
, , and
Abstract
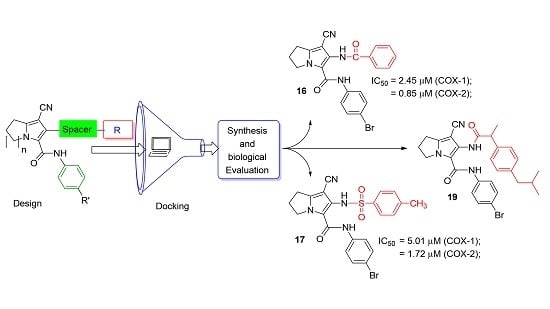
:
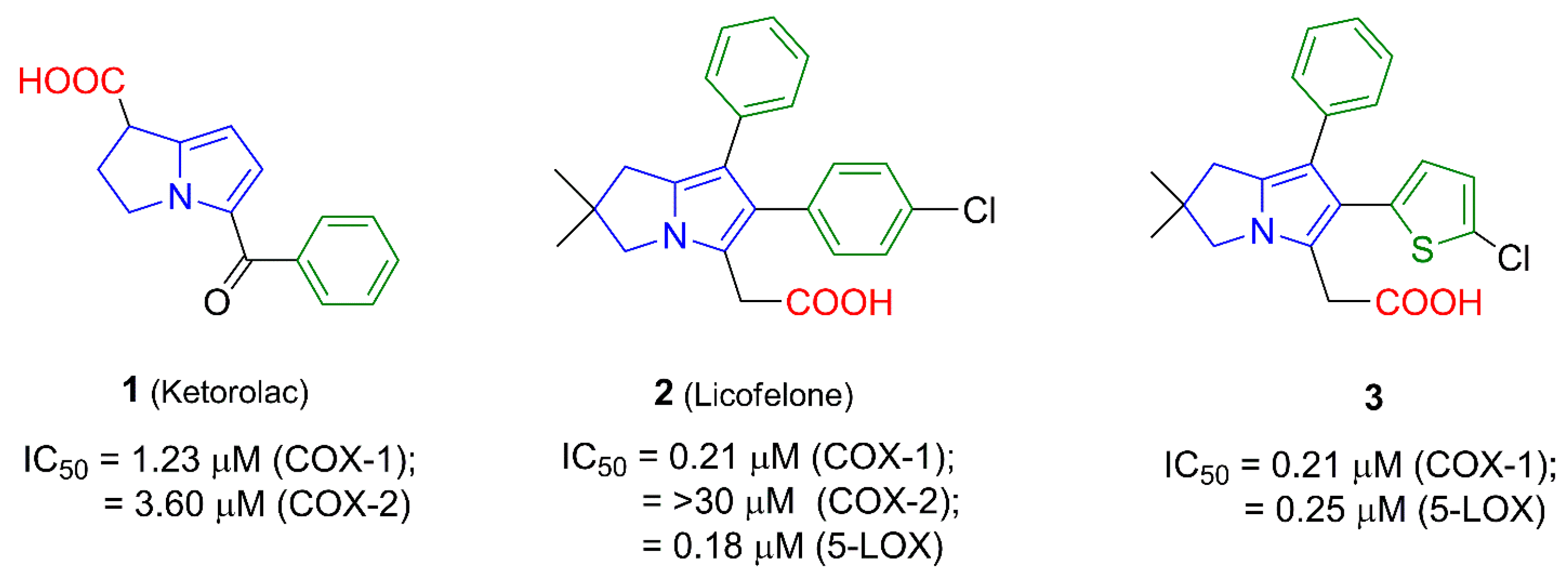
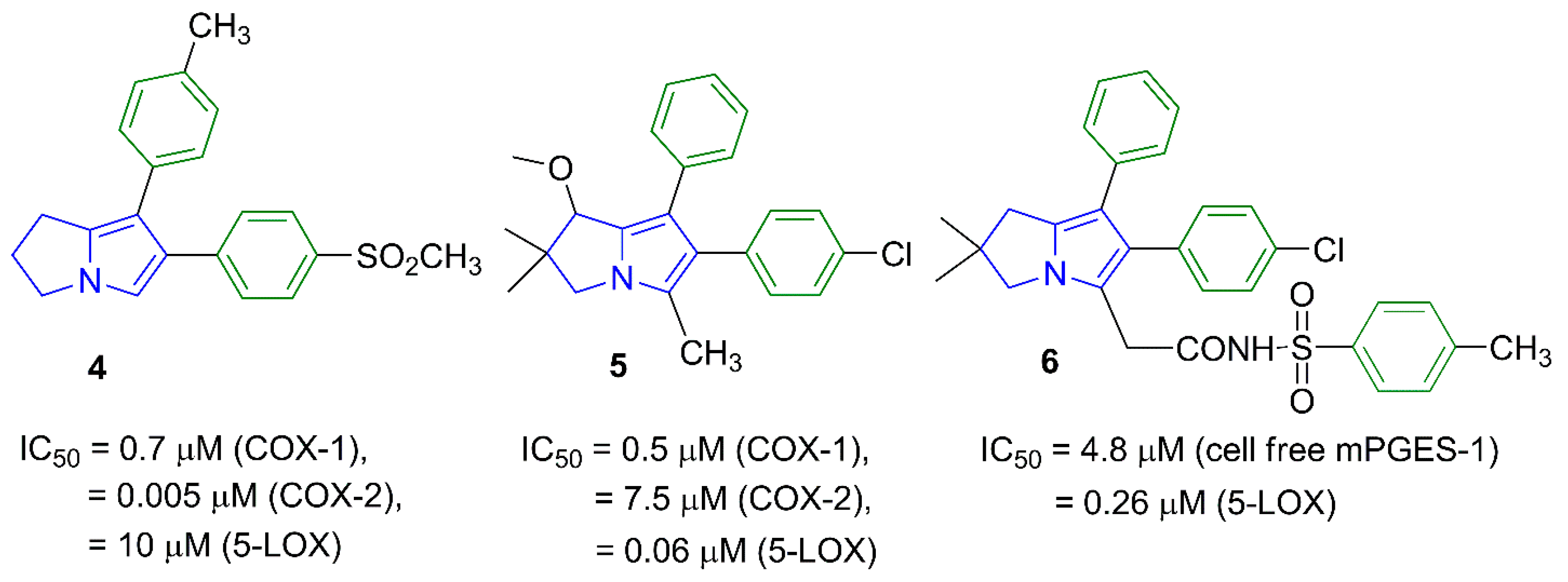
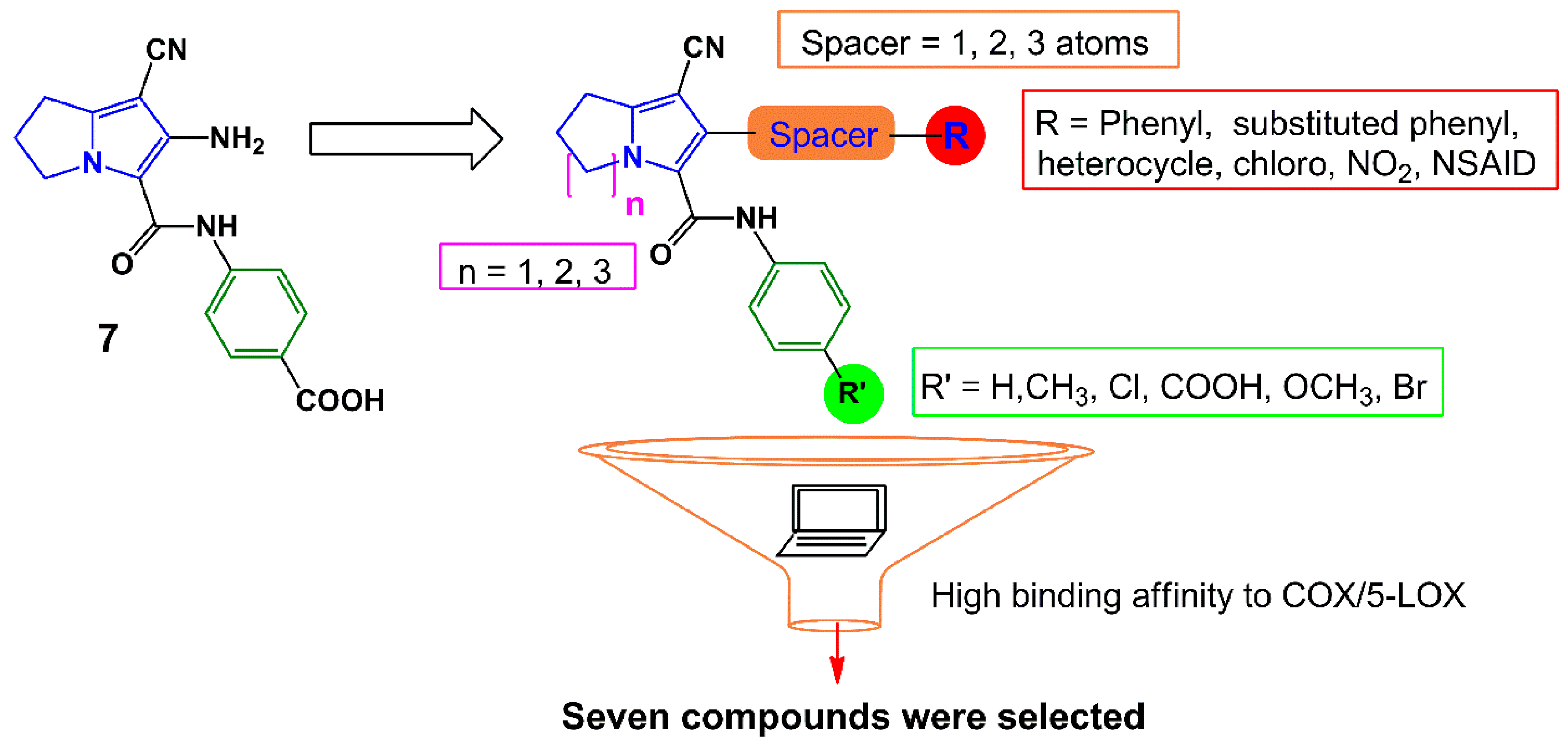
1. Introduction
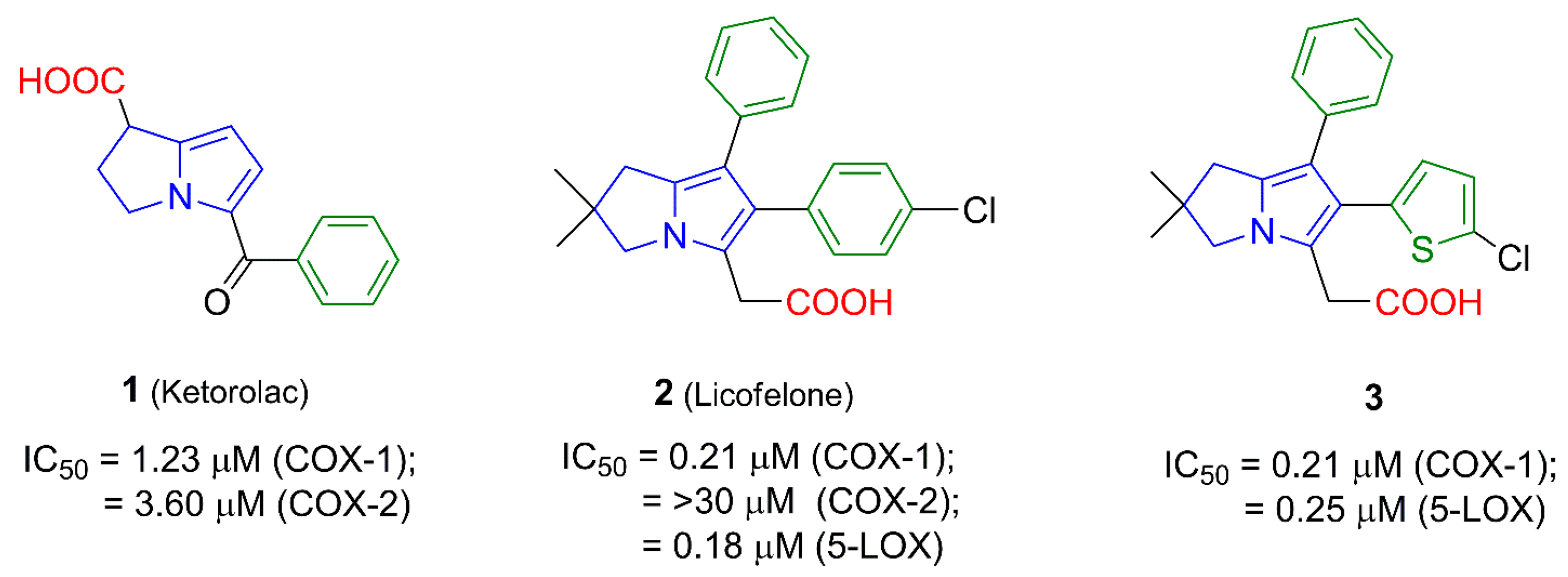
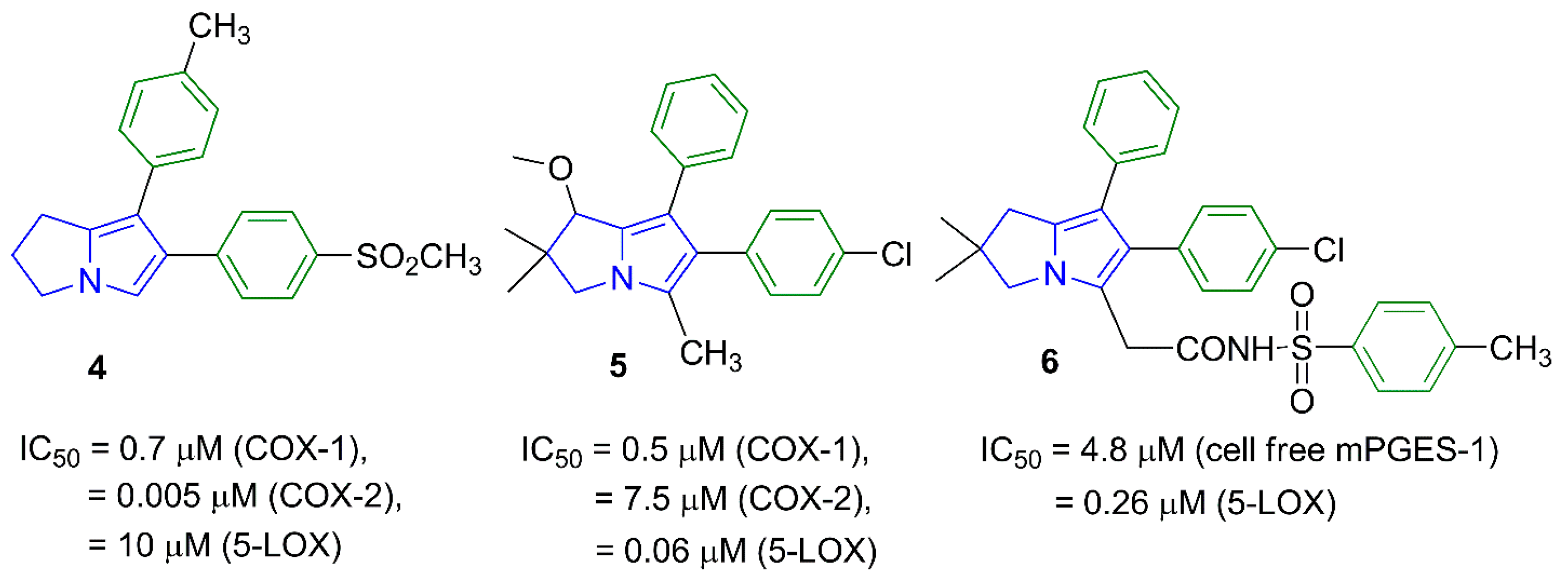

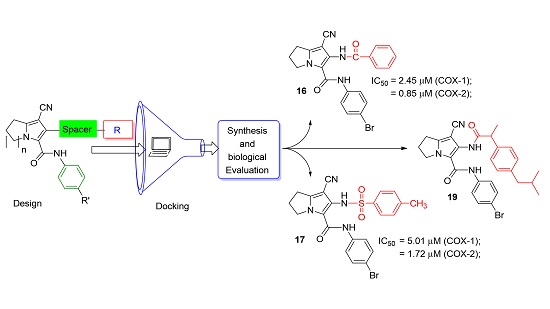
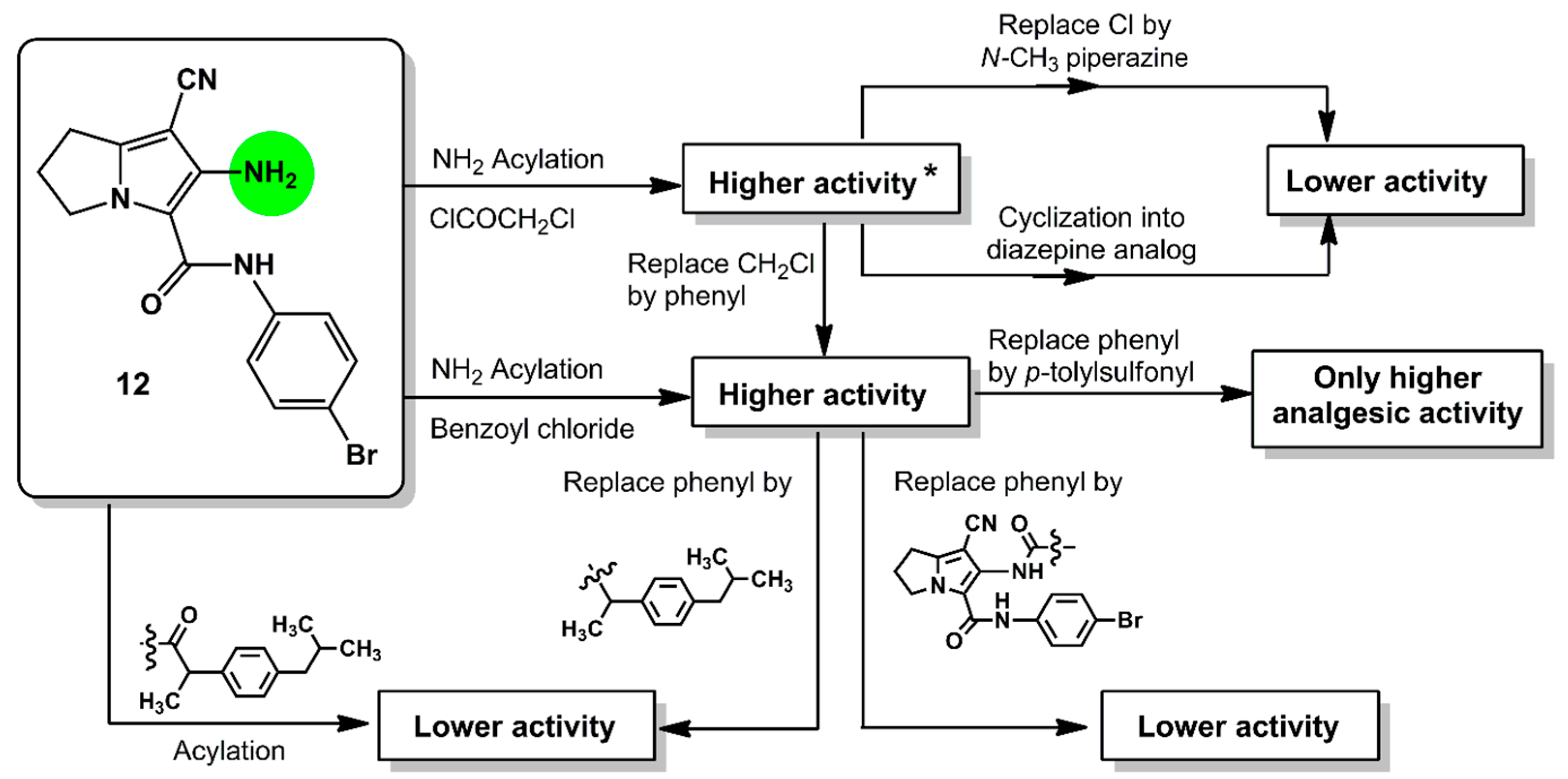
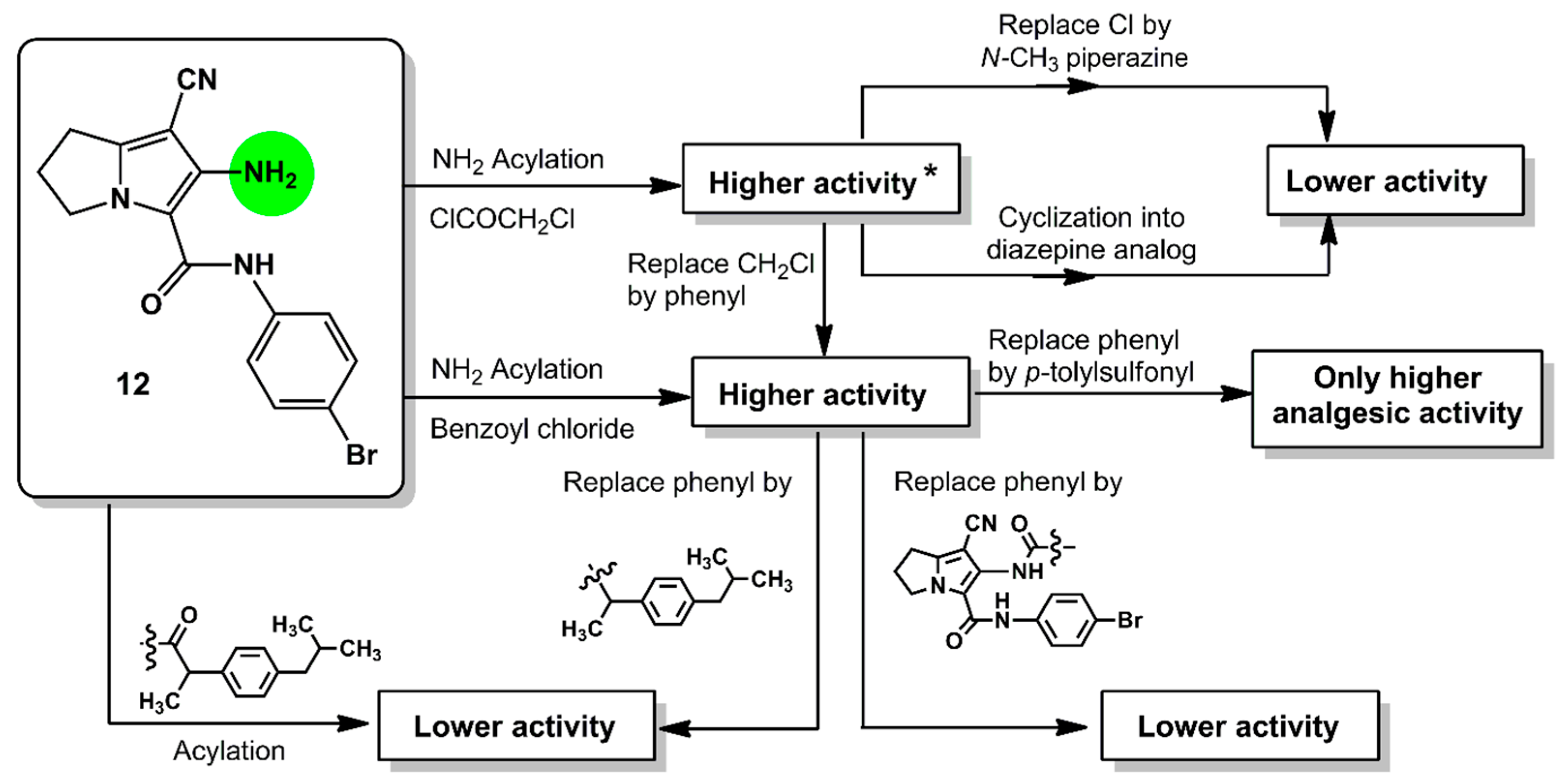
2. Results and Discussion
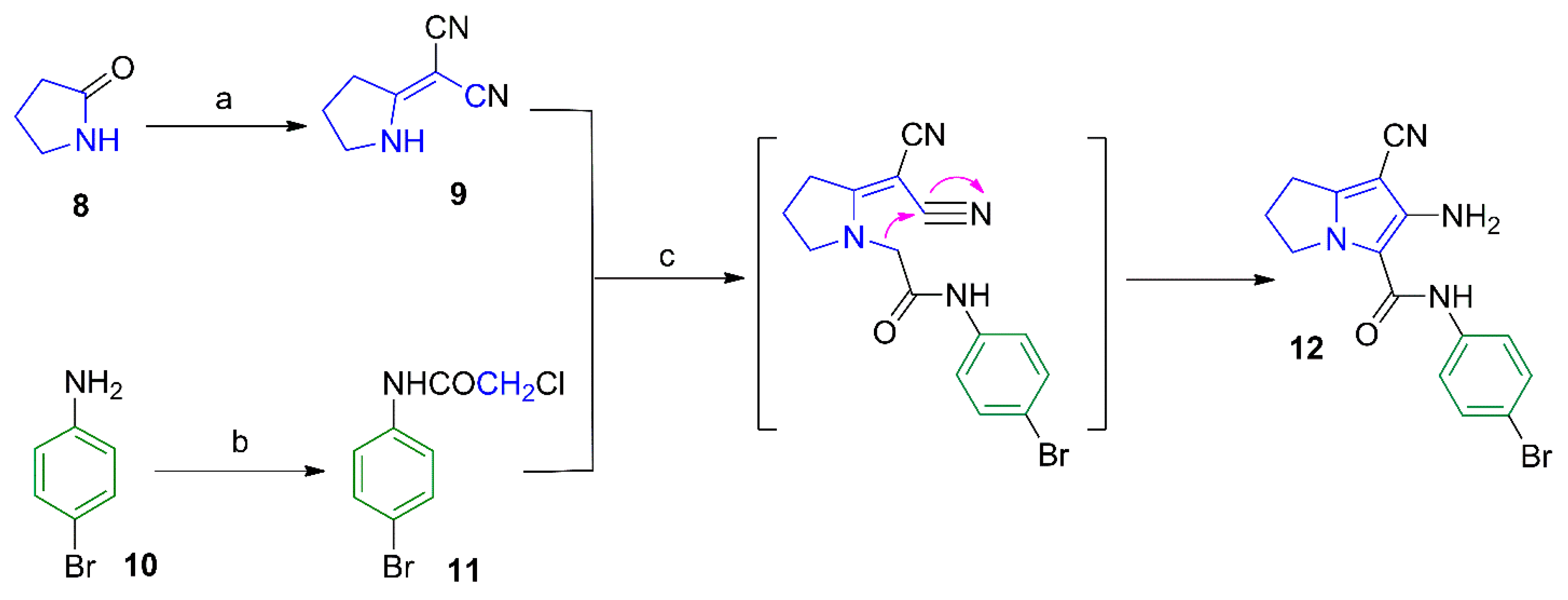
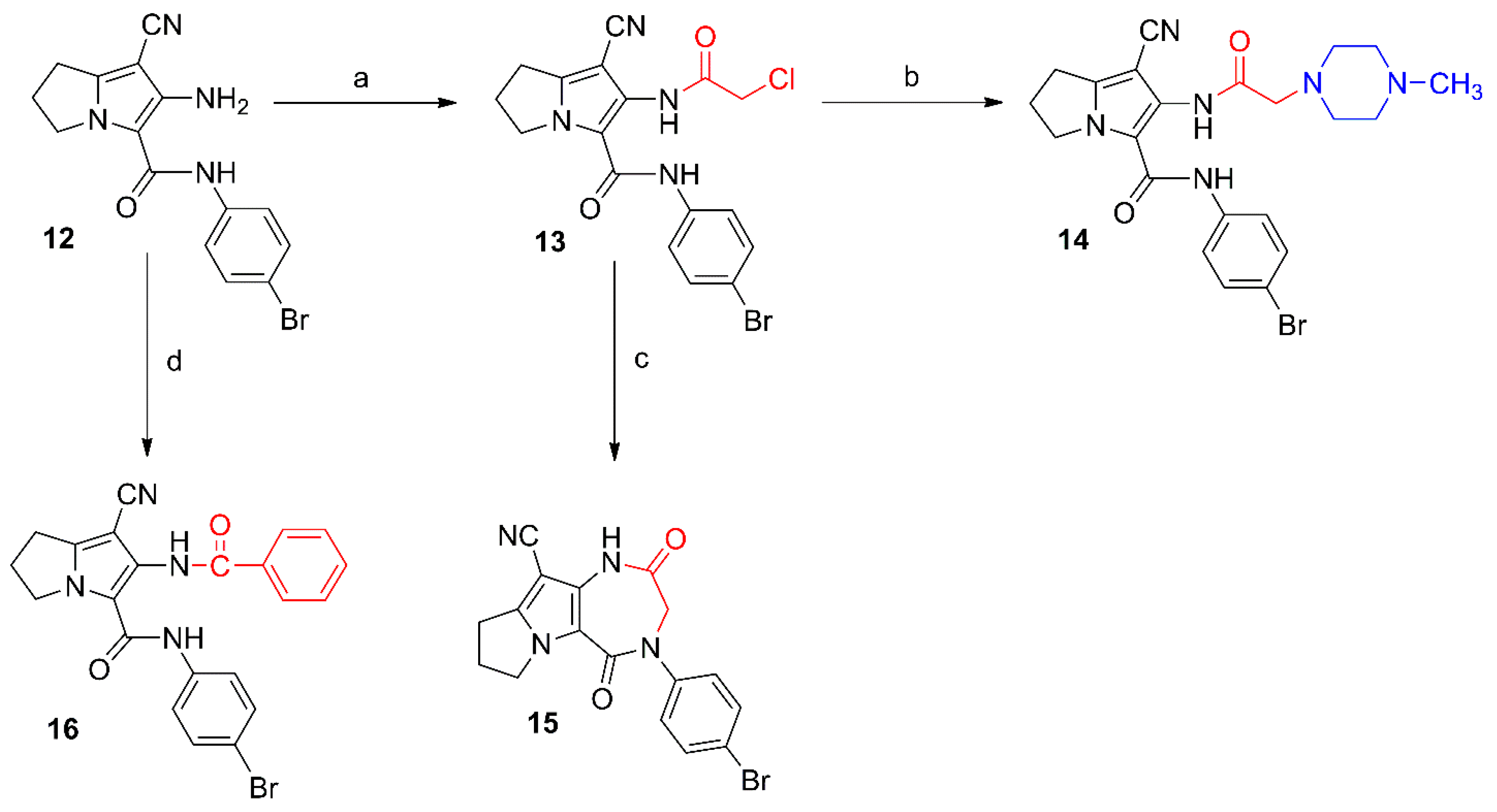
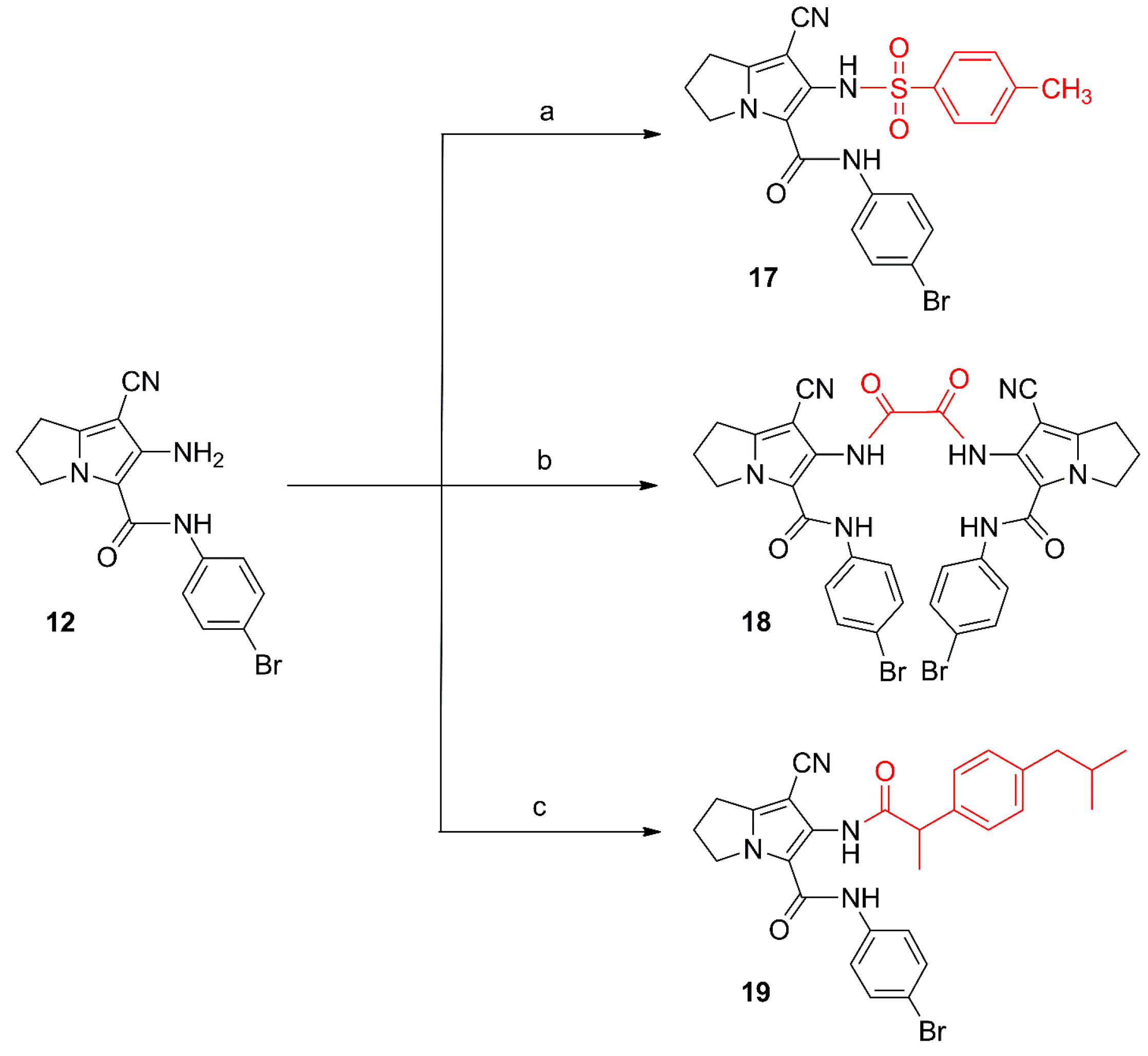
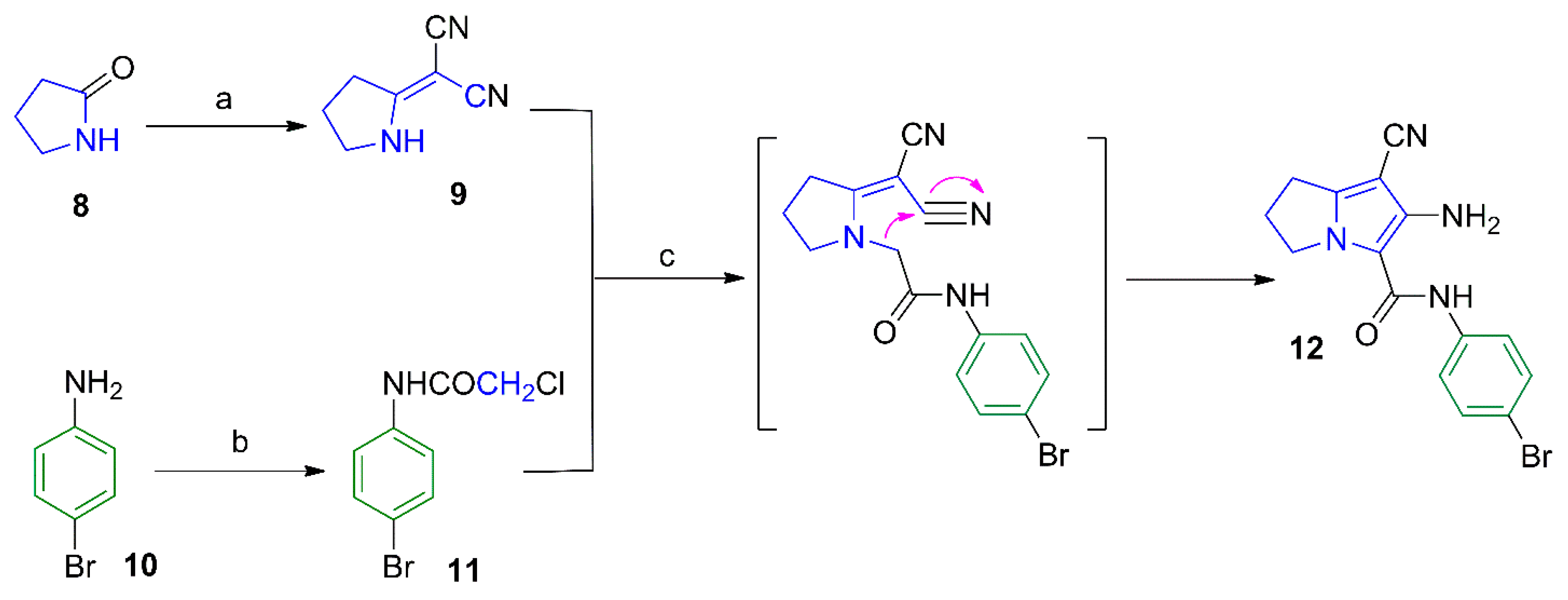
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro COX Inhibitory Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
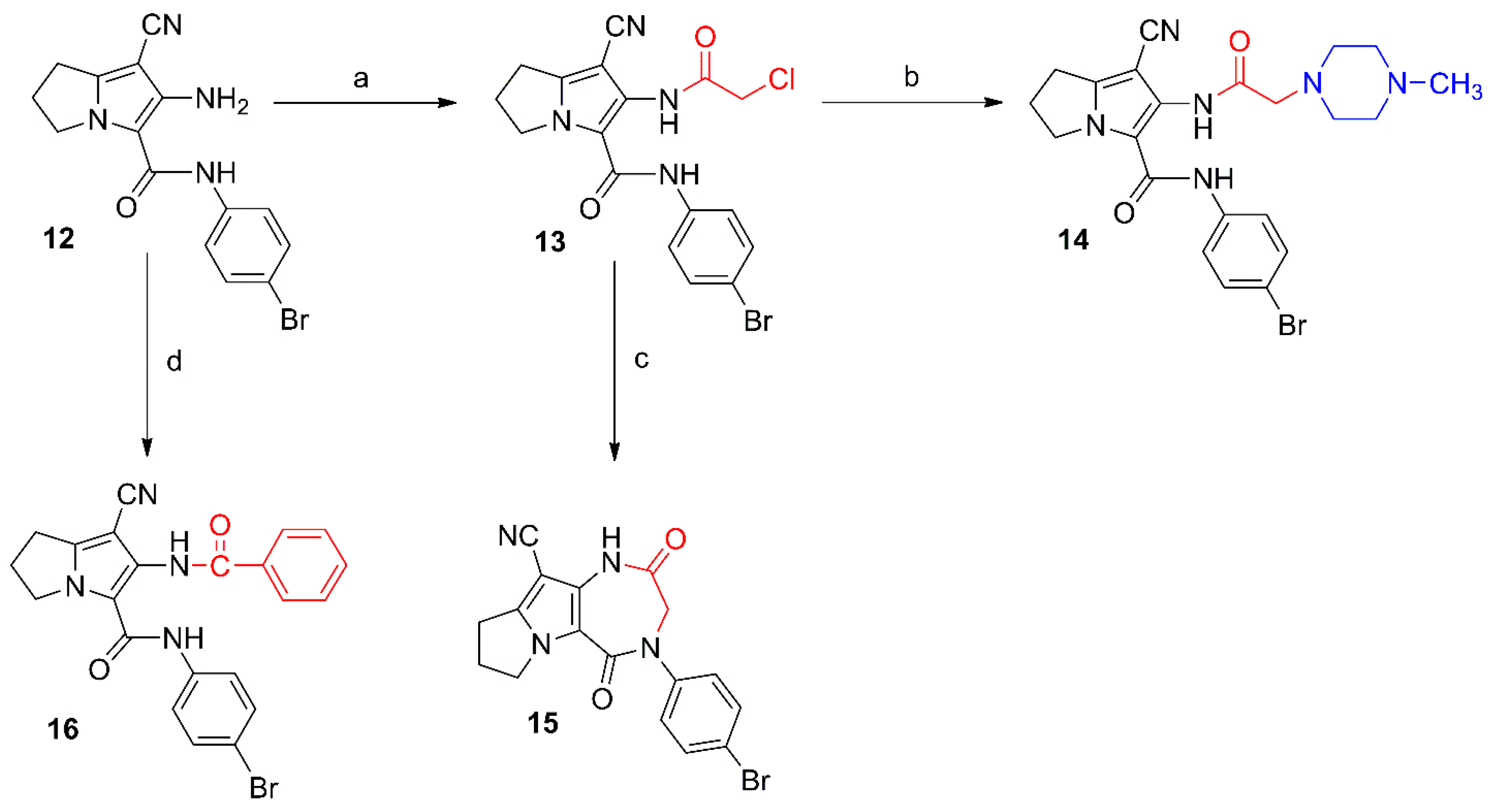{kind=link}
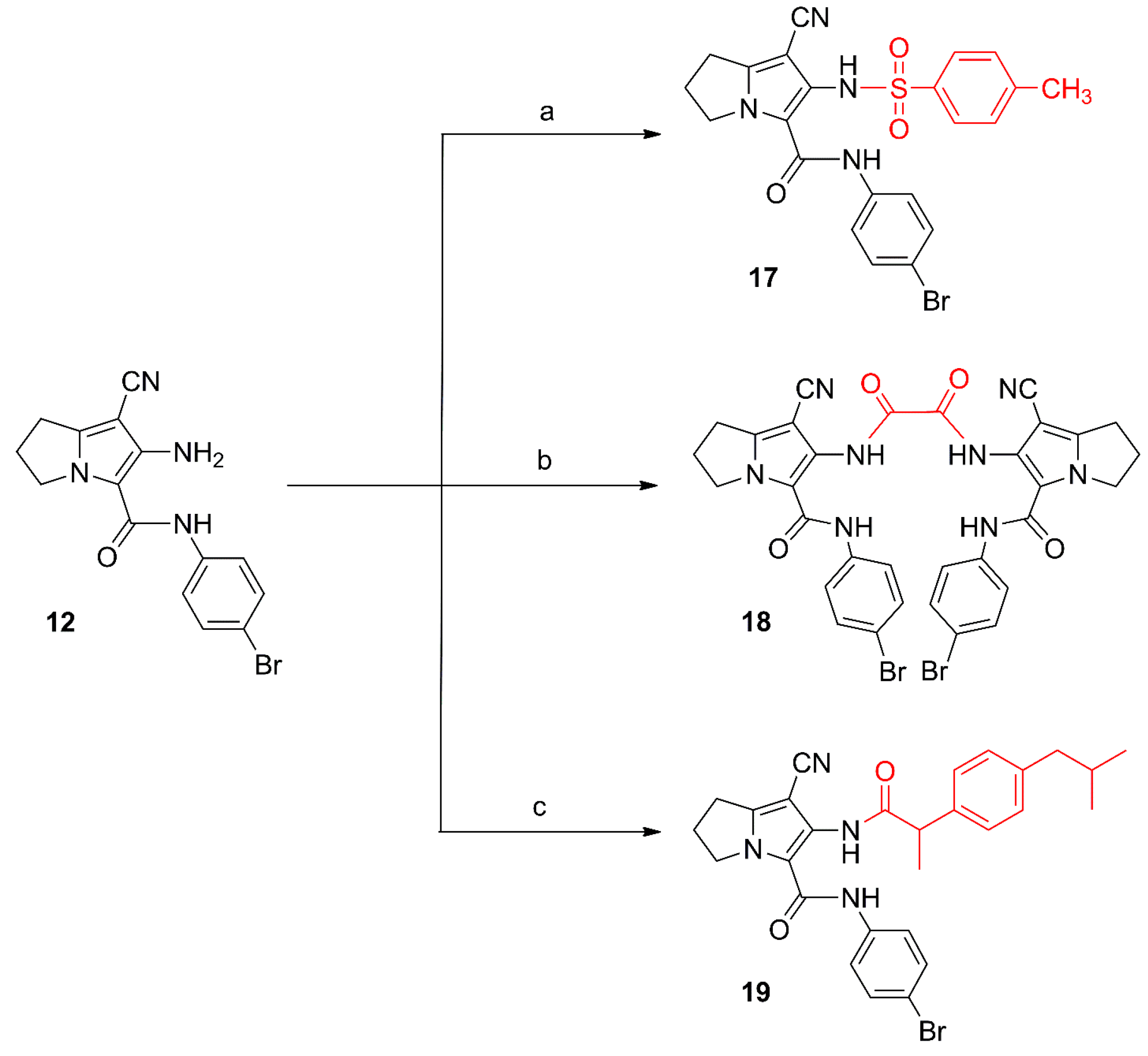{kind=link}
| Compd. No. | COX-1 | COX-2 | SI b |
|---|---|---|---|
| (IC50 µM) a | (IC50 µM) a | ||
| 12 | 4.64 | 1.27 | 3.64 |
| 13 | 5.69 | 1.64 | 3.48 |
| 14 | 3.50 | 1.09 | 3.21 |
| 15 | 3.37 | 1.06 | 3.17 |
| 16 | 2.45 | 0.85 | 2.89 |
| 17 | 5.01 | 1.72 | 2.91 |
| 18 | 5.10 | 0.85 | 6.03 |
| Indomethacin | 0.73 | 32.6 | 0.02 |
| Celecoxib | 15.6 | 0.32 | 48.75 |
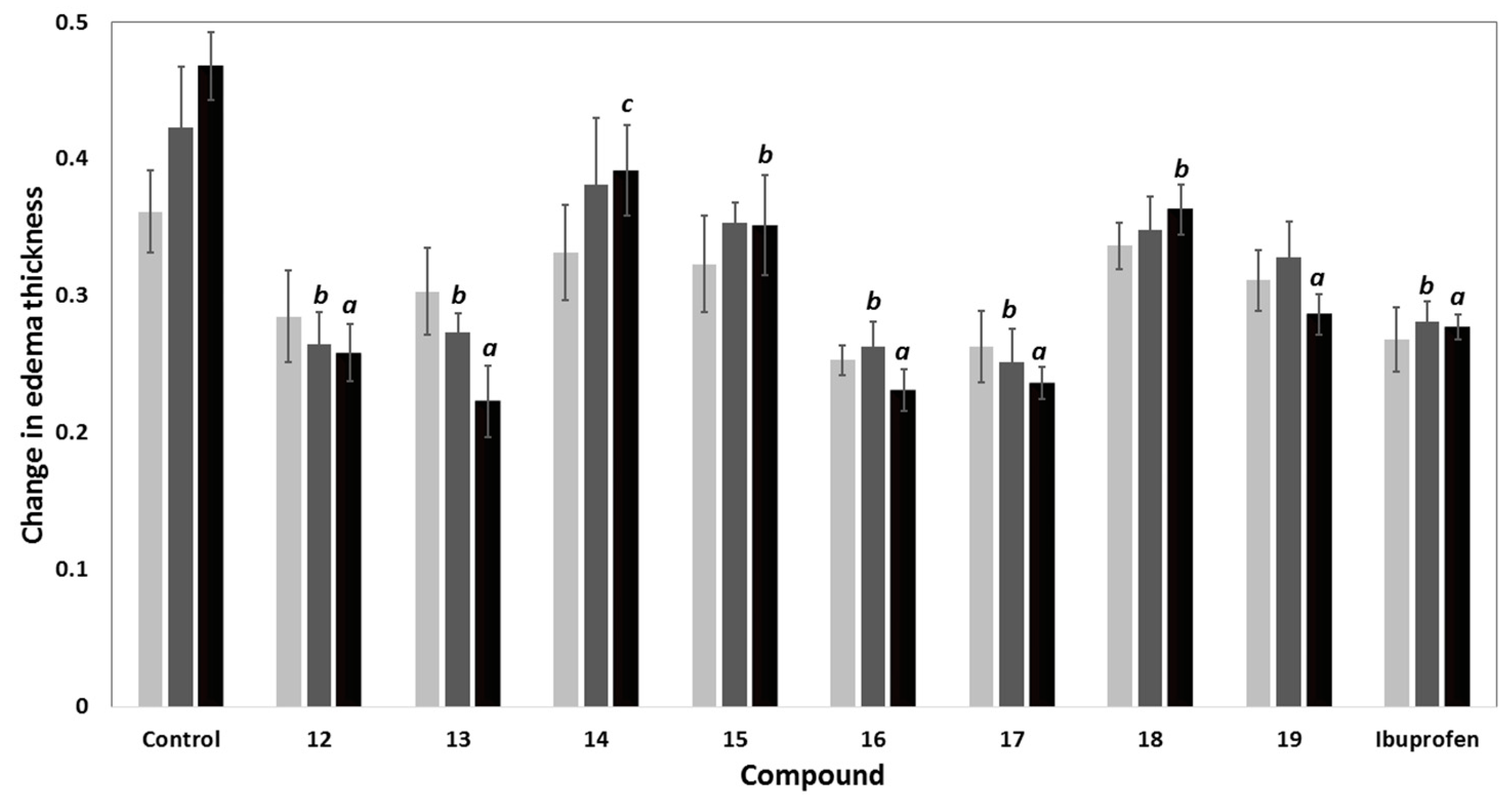
2.2.2. In Vivo Biological Evaluation
Anti-Inflammatory Activity

| Compd. | % Inhibition in Edema Thickness | Potency a | Ulcerogenicity | |||
|---|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | UI b | % Protection c | ||
| Control | - | - | - | 0 | 0 | 100 |
| 12 | 21.20 | 37.40 | 44.79 | 1.10 | 4.66 | 66.38 |
| 13 | 16.13 | 35.43 | 52.31 | 1.28 | 4.41 | 68.18 |
| 14 | 8.30 | 9.84 | 16.37 | 0.40 | 4.66 | 66.38 |
| 15 | 10.60 | 16.54 | 24.91 | 0.61 | 2.68 | 80.66 |
| 16 | 29.95 | 37.80 | 50.58 | 1.24 | 2.26 | 83.69 |
| 17 | 27.19 | 40.55 | 49.47 | 1.21 | 2.26 | 83.69 |
| 18 | 6.91 | 17.72 | 22.42 | 0.55 | 4.41 | 68.18 |
| 19 | 13.82 | 22.44 | 38.79 | 0.95 | 0 | 100 |
| Ibuprofen | 25.81 | 33.46 | 40.82 | 1.00 | 13.86 | 0 |
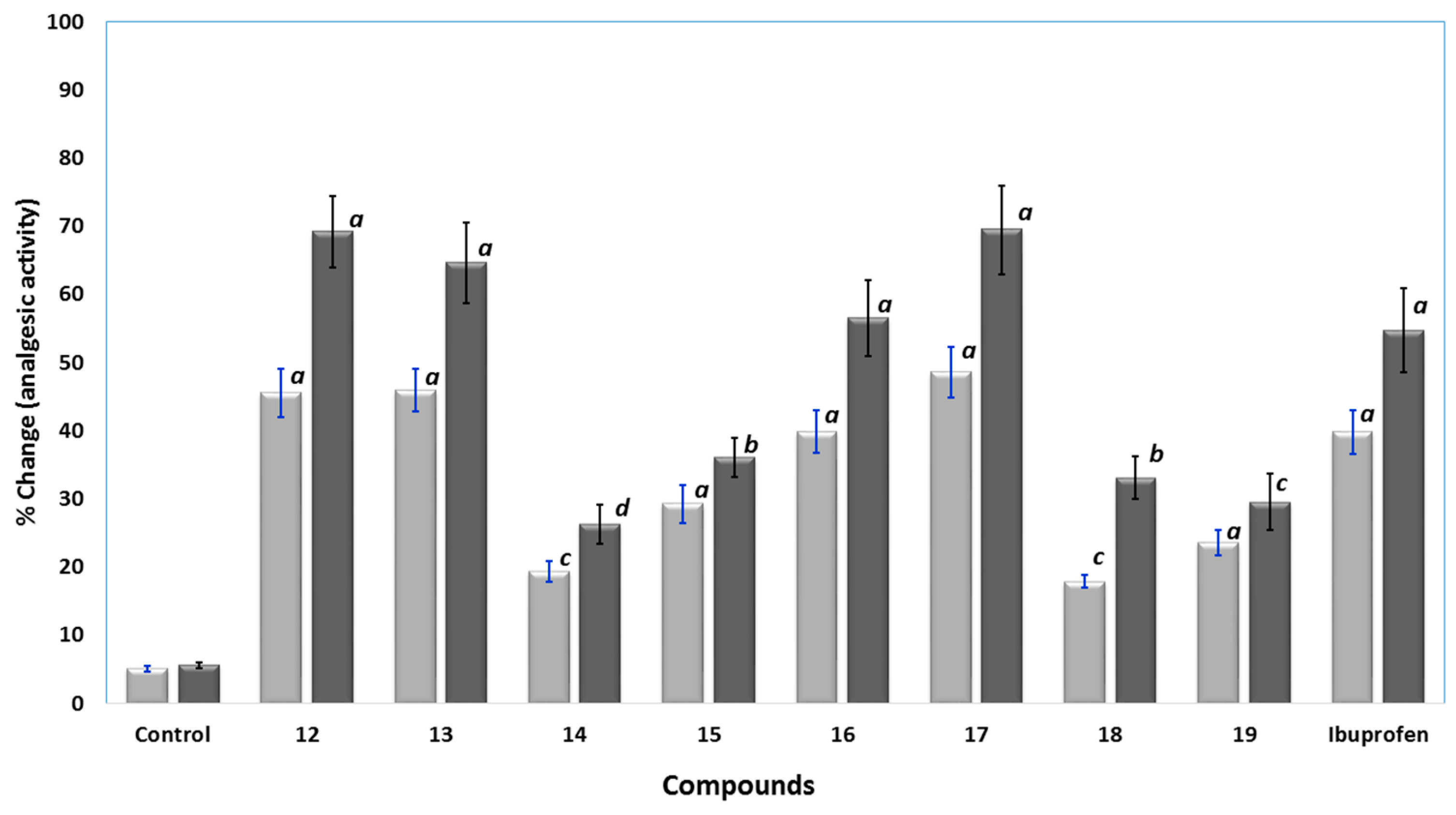
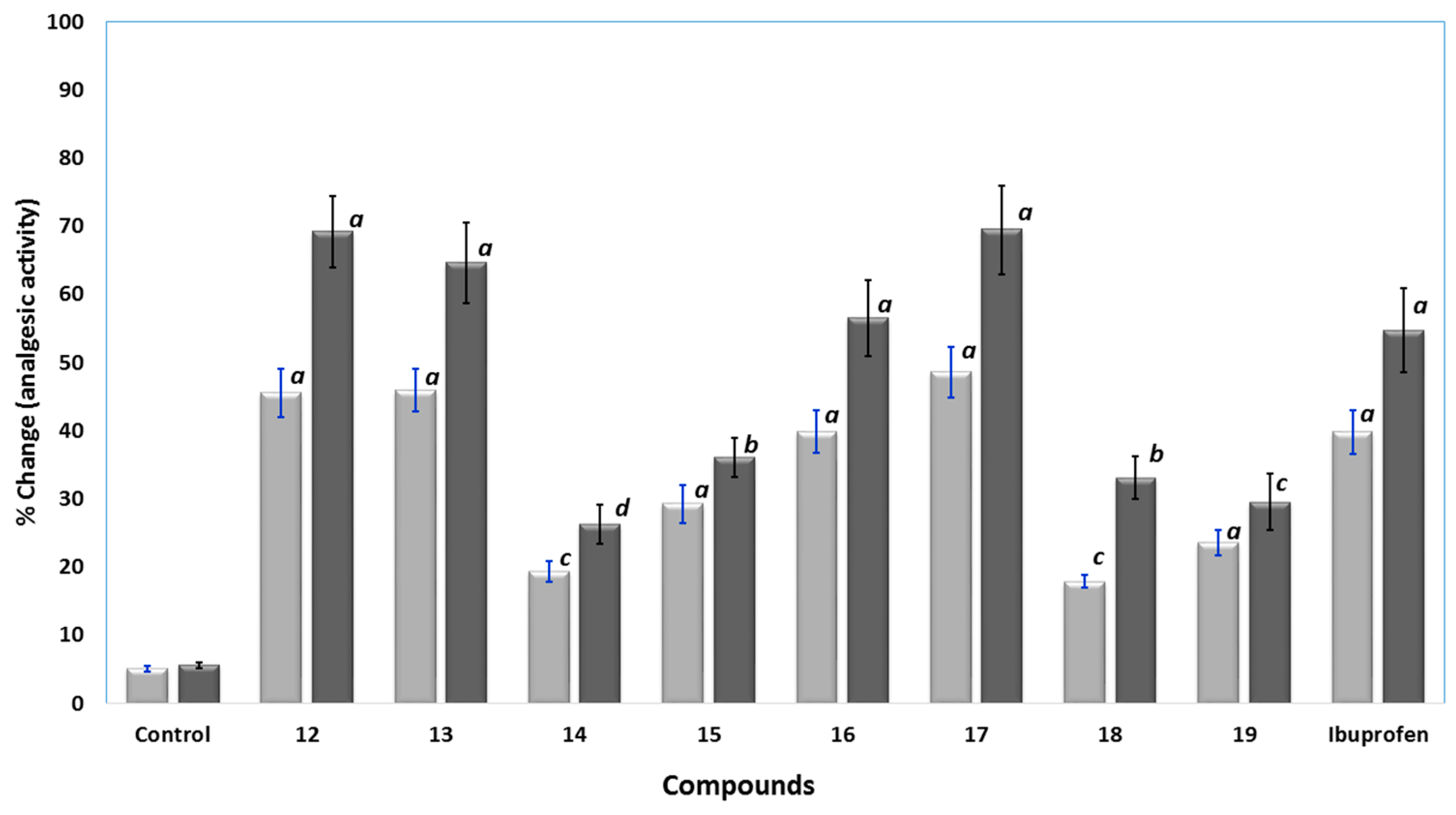
Analgesic Activity
Acute Ulcerogenicity Studies
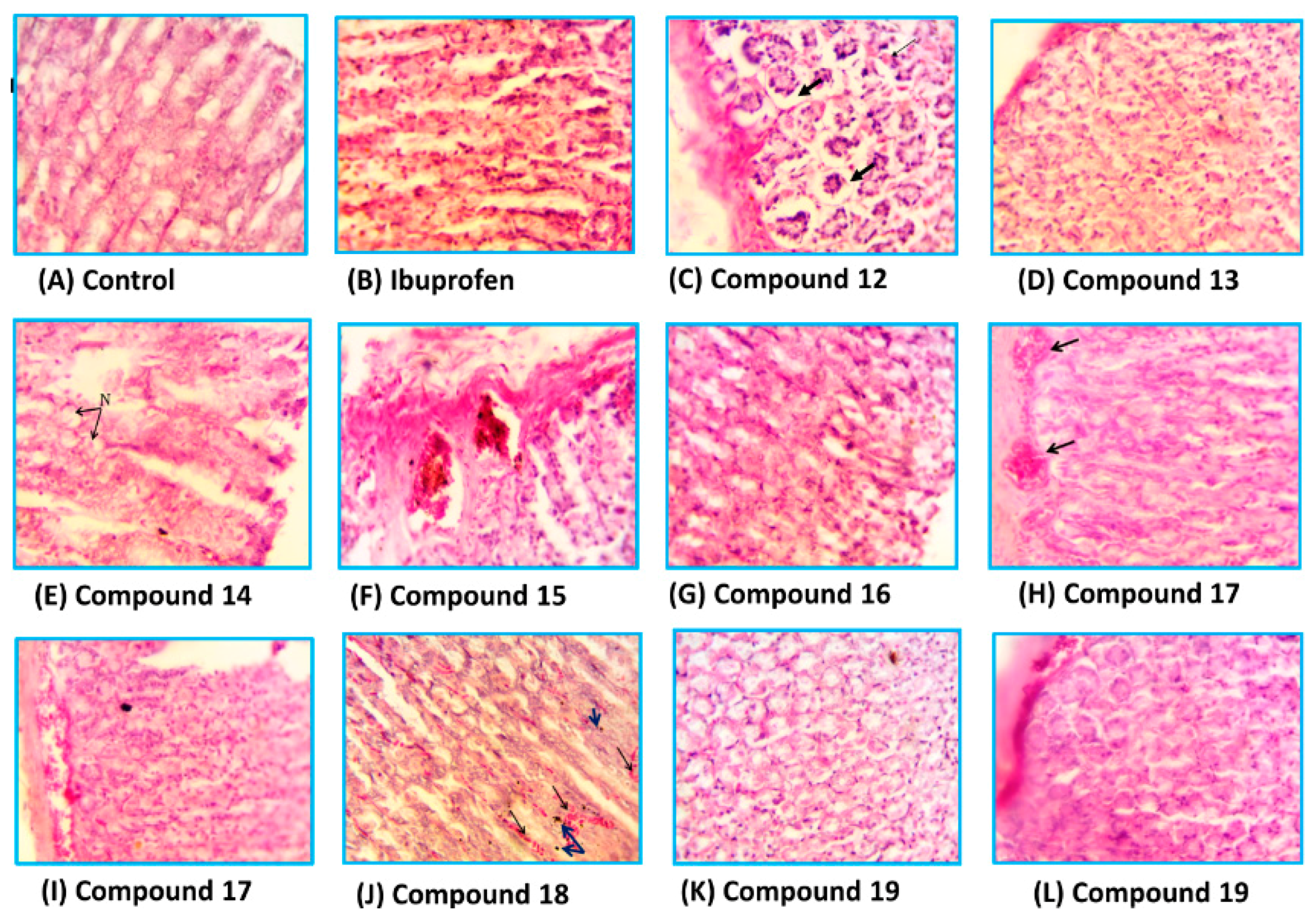
Histopathological Studies

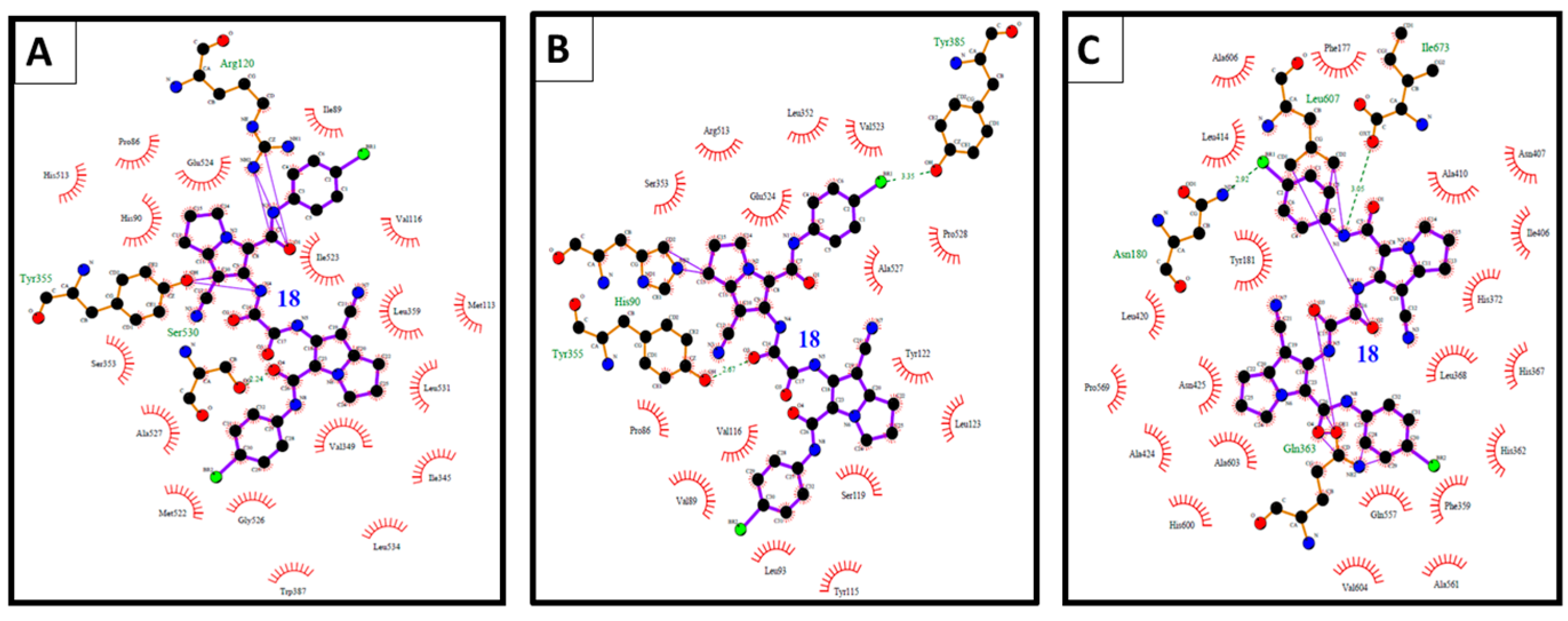
2.3. Molecular Docking Study
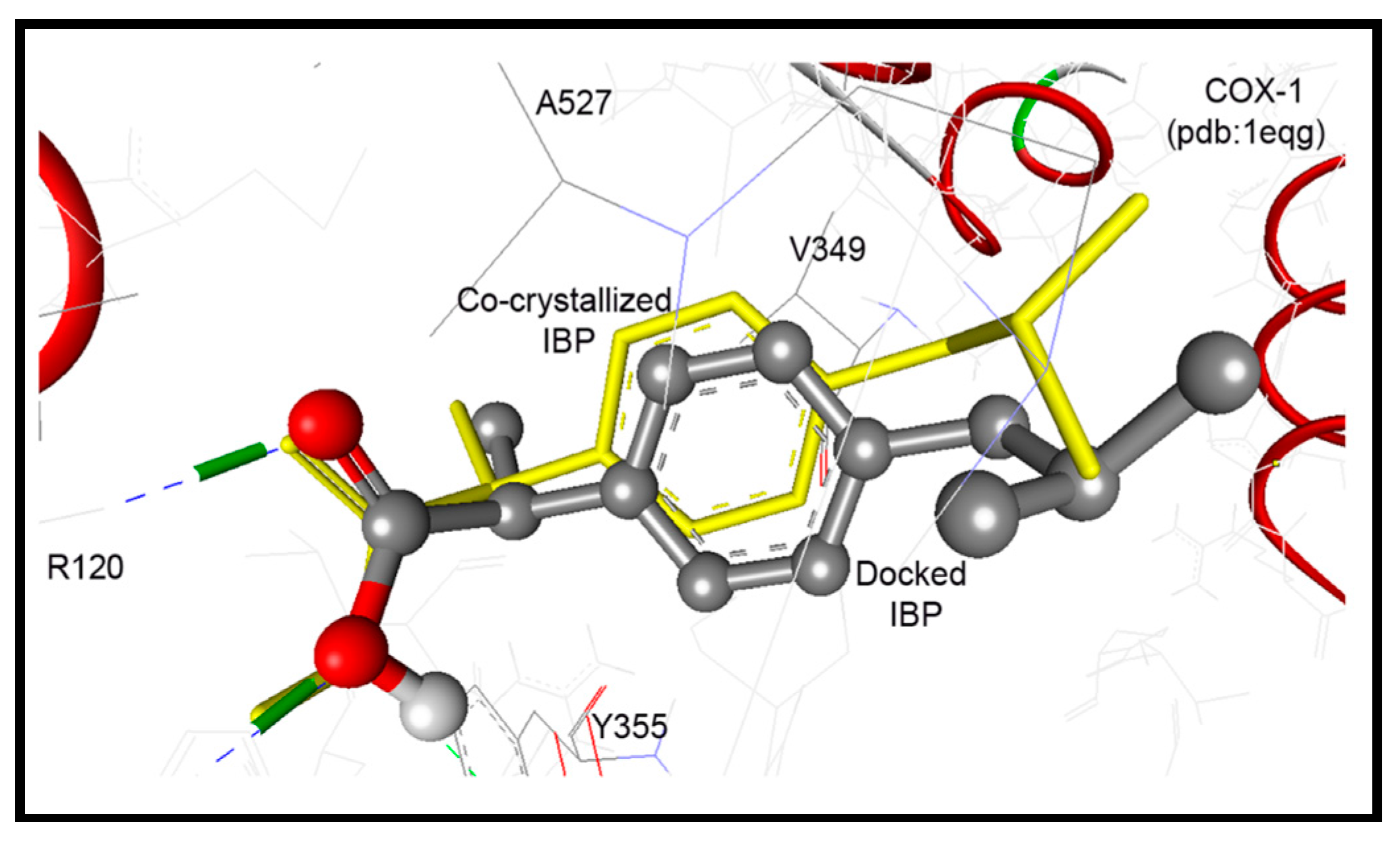
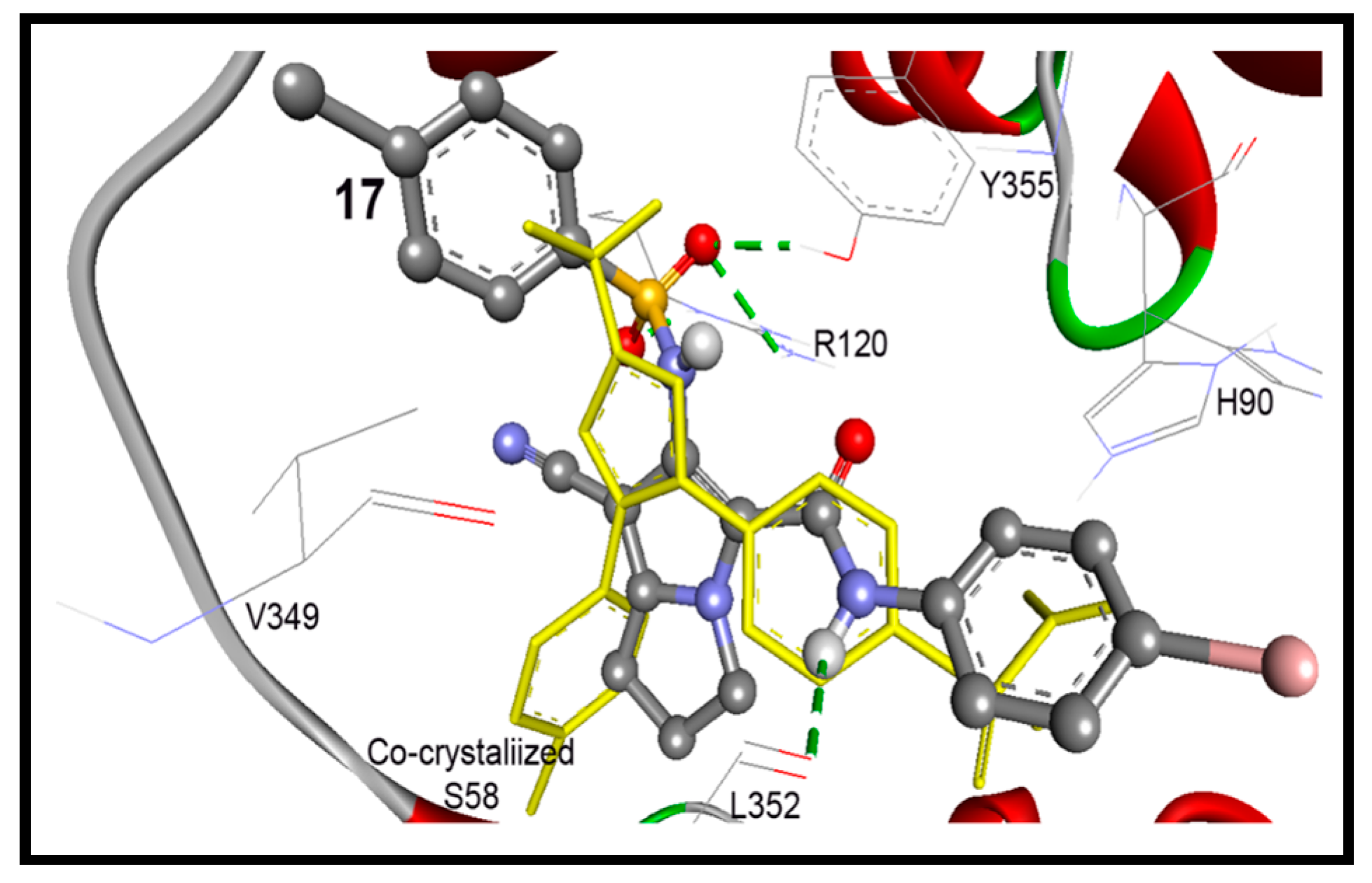
2.3.1. Docking Study into COX-1 Enzyme

| Compd. | ΔGb a (kcal/mol) | Ki b | Hydrogen Bonds between Atoms of Compounds and Amino Acids of COX-1 | RMSD c (Å) | |
|---|---|---|---|---|---|
| Atom of Compd. | Amino Acid | ||||
| 12 | −9.00 | 250.88 nM | 5-Ph-NH | OH of Tyr355 | 2.92 |
| 5-Ph-NHC=O | HN of Arg120 | ||||
| 13 | −9.34 | 142.76 nM | 7-CN | HN of Arg120 | 3.41 |
| 14 | −6.41 | 20.18 µM | 6-NH | OH of Tyr355 | 1.99 |
| 15 | −6.65 | 13.26 µM | 10-CN | HO of Tyr385 | 0.83 |
| 2-C=O | HO of Ser530 | ||||
| 2-C=O | HN of Leu531 | ||||
| 16 | −8.14 | 1.08 µM | 5-Ph-CONH | OH of Tyr355 | 2.27 |
| 17 | −10.31 | 27.77 nM | 6-NHS=O | HO of Tyr355 | 2.29 |
| 18 | −0.68 | 316.74 mM | 5-Ph-NHC=O | HN of Arg120 | 2.62 |
| 19 | −9.71 | 76.20 nM | 6-NHC=O | HN of Arg120 | 3.49 |
| Ibuprofen | −9.40 | 128.86 nM | COO | H1N of Arg120 | 0.62 |
| COOH | H2N of Arg120 | ||||
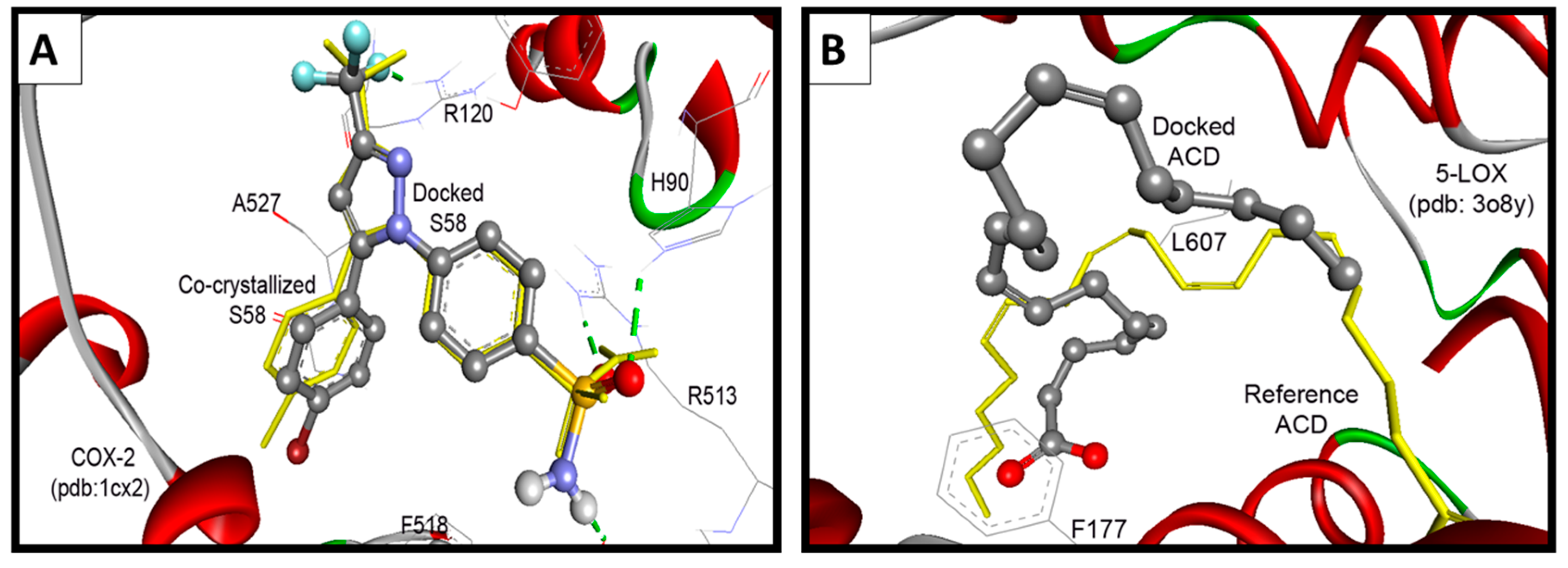
2.3.2. Docking Study into COX-2 Enzyme
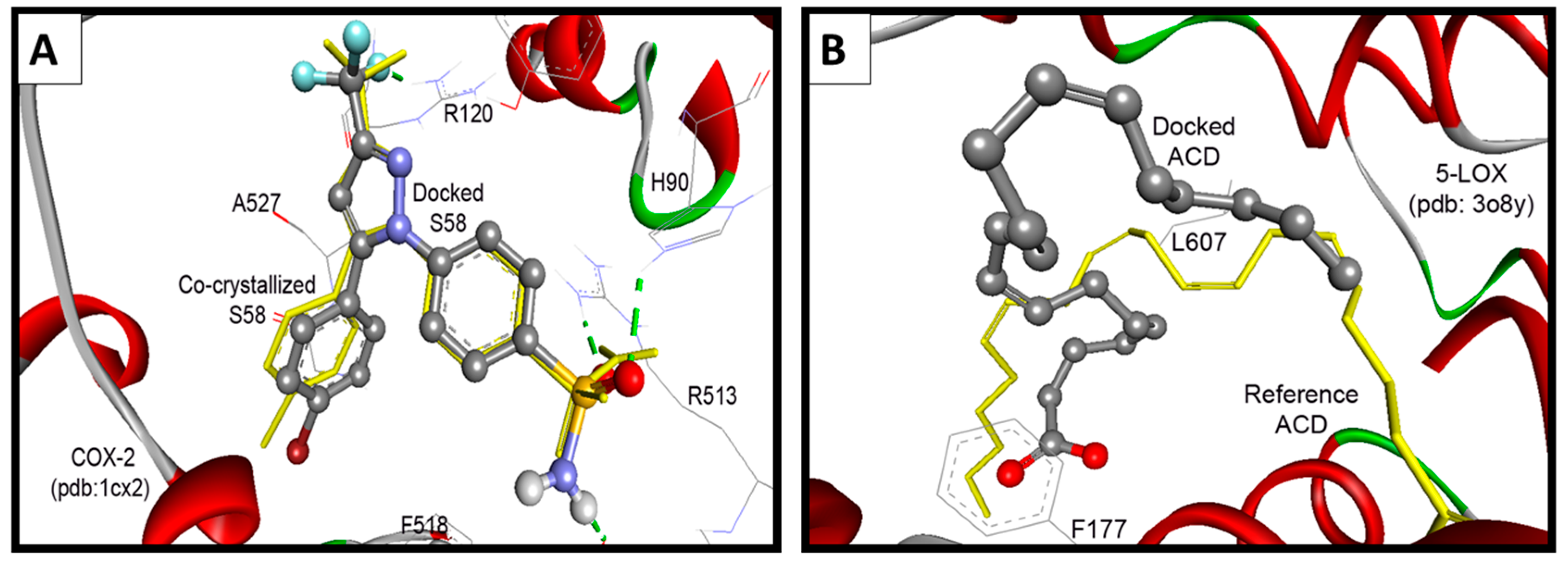
| Compd. | ΔGb a (kcal/mol) | Ki b | Hydrogen Bonds between Atoms of Compounds and Amino Acids of COX-2 | RMSD c (Å) | |
|---|---|---|---|---|---|
| Atom of Compd. | Amino Acid | ||||
| 12 | −8.79 | 362.12 nM | 5-Ph-NH | O=C of Glu524 | 3.39 |
| 5-Ph-NHC=O | H1N of Arg120 | ||||
| 5-Ph-NHC=O | H2N of Arg120 | ||||
| 5-NH | OH of Tyr355 | ||||
| 7-CN | HN of His90 | ||||
| 13 | −10.01 | 46.35 nM | 5-NHC=O | H1N of Arg120 | 0.49 |
| H2N of Arg120 | |||||
| 14 | −9.82 | 63.40 nM | -- d | 1.13 | |
| 15 | −8.73 | 400.2 nM | 10-CN | HO of Ser530 | 1.78 |
| 16 | −8.81 | 347.43 nM | 6-Ph-C=O | HN of Arg120 | 1.13 |
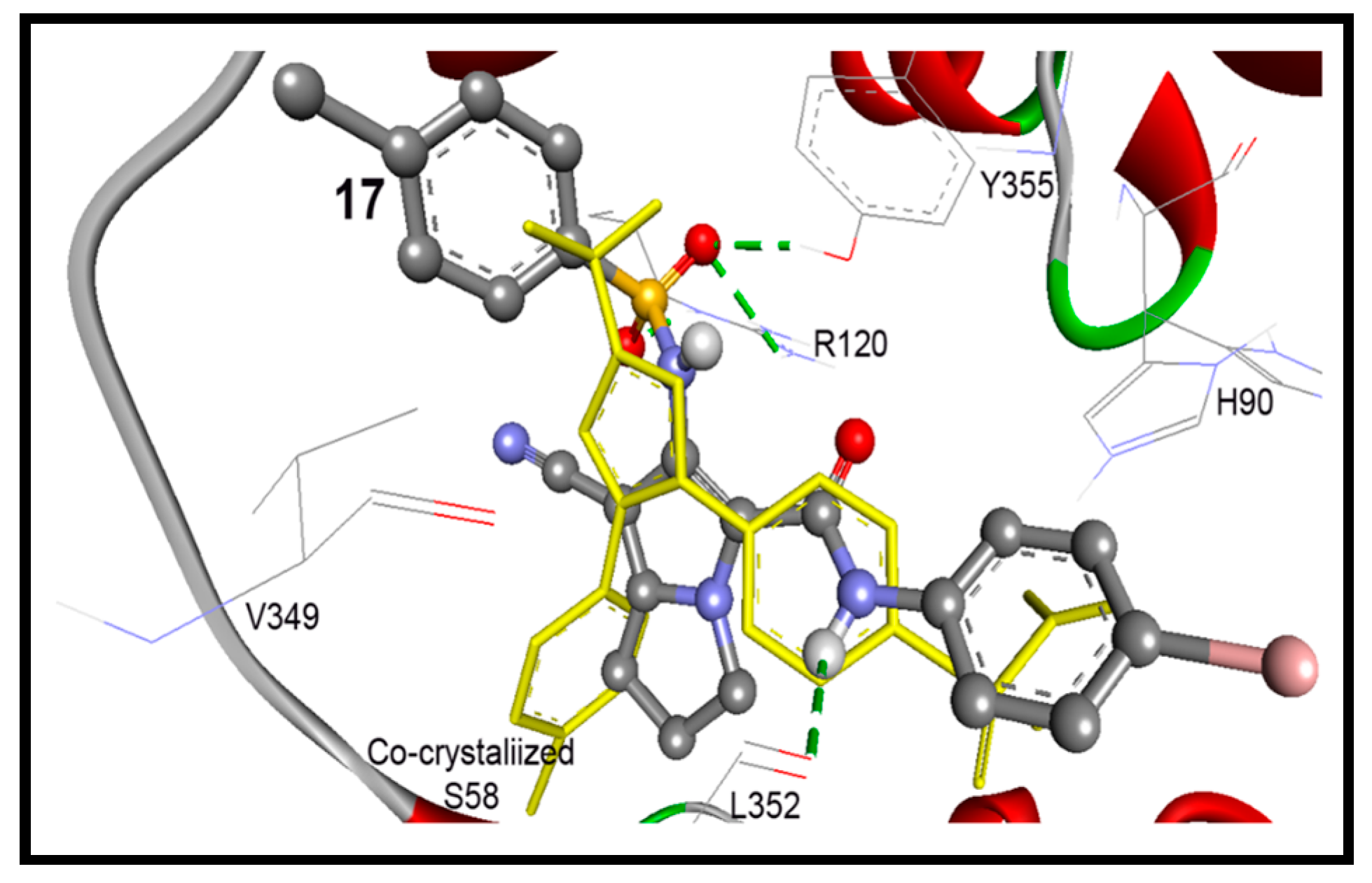
| 17 | −11.63 | 3.00 nM | 5-Ph-NH | O=C of Leu352 | 1.45 |
| 6-NHS=O1 | H1N of Arg120 | ||||
| 6-NHS=O1 | H2N of Arg120 | ||||
| 6-NHS=O2 | HO of Tyr355 | ||||
| 18 | −10.93 | 9.80 nM | 6-NH | O=C of Glu524 | 5.79 |
| 6-NHC=O | HO of Tyr355 | ||||
| 19 | −10.27 | 29.51 nM | 7-CN | HO of Ser530 | 1.59 |
| S58 Ligand e | −11.49 | 3.81 nM | 3-CF | HN of Arg120 | 0.38 |
| p-Ph-S=O1 | HN of His90 | ||||
| p-Ph-S=O2 | HN of Arg513 | ||||
| p-Ph-SONH | O=C of Phe518 | ||||
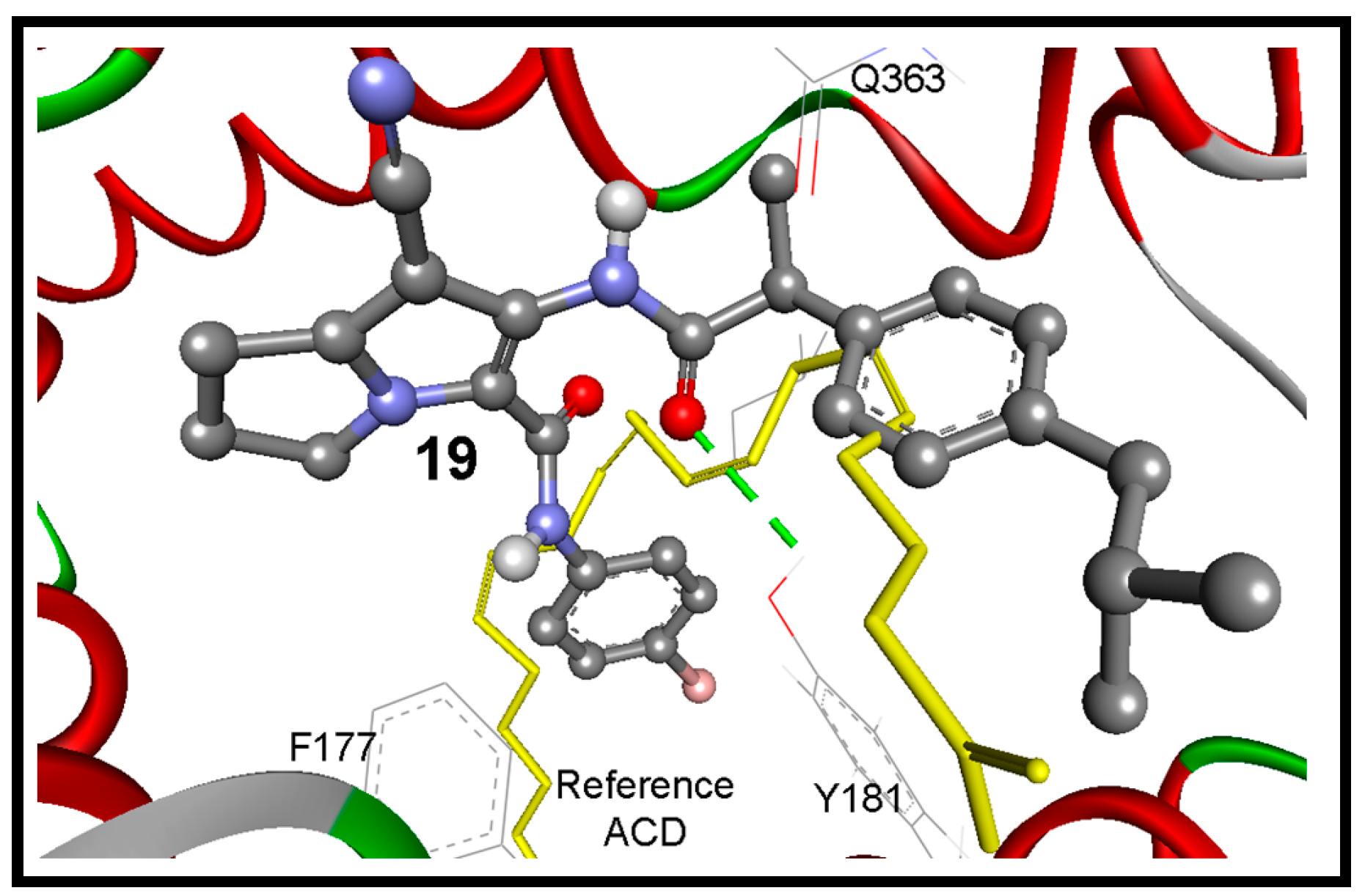
2.3.3. Docking Study into 5-LOX Enzyme
| Compd. | ΔGb a (kcal/mol) | Ki b | Hydrogen Bonds between Atoms of Compounds and Amino Acids of 5-LOX | RMSD c (Å) | |
|---|---|---|---|---|---|
| Atom of Compd. | Amino Acid | ||||
| 12 | −8.78 | 365.81 nM | 5-Ph-NH | O=C of Gln363 | 5.80 |
| 5-Ph-NHC=O | HO of Tyr181 | ||||
| 7-CN | HN of Ala424 | ||||
| 13 | −9.22 | 174.81 nM | 5-Ph-NH | O=C of Gln363 | 5.42 |
| 5-Ph-NHC=O | HO of Tyr181 | ||||
| 7-CN | HN of Asn425 | ||||
| 14 | −7.48 | 3.31 µM | 6-NH | OH of Tyr181 | 2.73 |
| 15 | −7.89 | 1.66 µM | 2-C=O | HO of Thr364 | 5.86 |
| 5-C=O | HN of His367 | ||||
| 16 | −11.06 | 7.78 nM | 5-Ph-NHC=O | HN of Gln363 | 2.68 |
| 6-NH | OH of Tyr181 | ||||
| 17 | −10.84 | 11.26 nM | 6-NHS=O1 | HN of Gln363 | 3.55 |
| 6--NHS=O2 | HN of His367 | ||||
| 18 | +6.69 | -- d | 5-Ph-NH | O=C of Ile673 | 4.25 |
| 7-CN | HN of Gln363 | ||||
| 19 | −12.16 | 1.23 nM | 6-NHC=O | HO of Tyr181 | 3.24 |
| ACD Ligand e | −4.50 | 502.87 µM | -- d | 2.27 | |
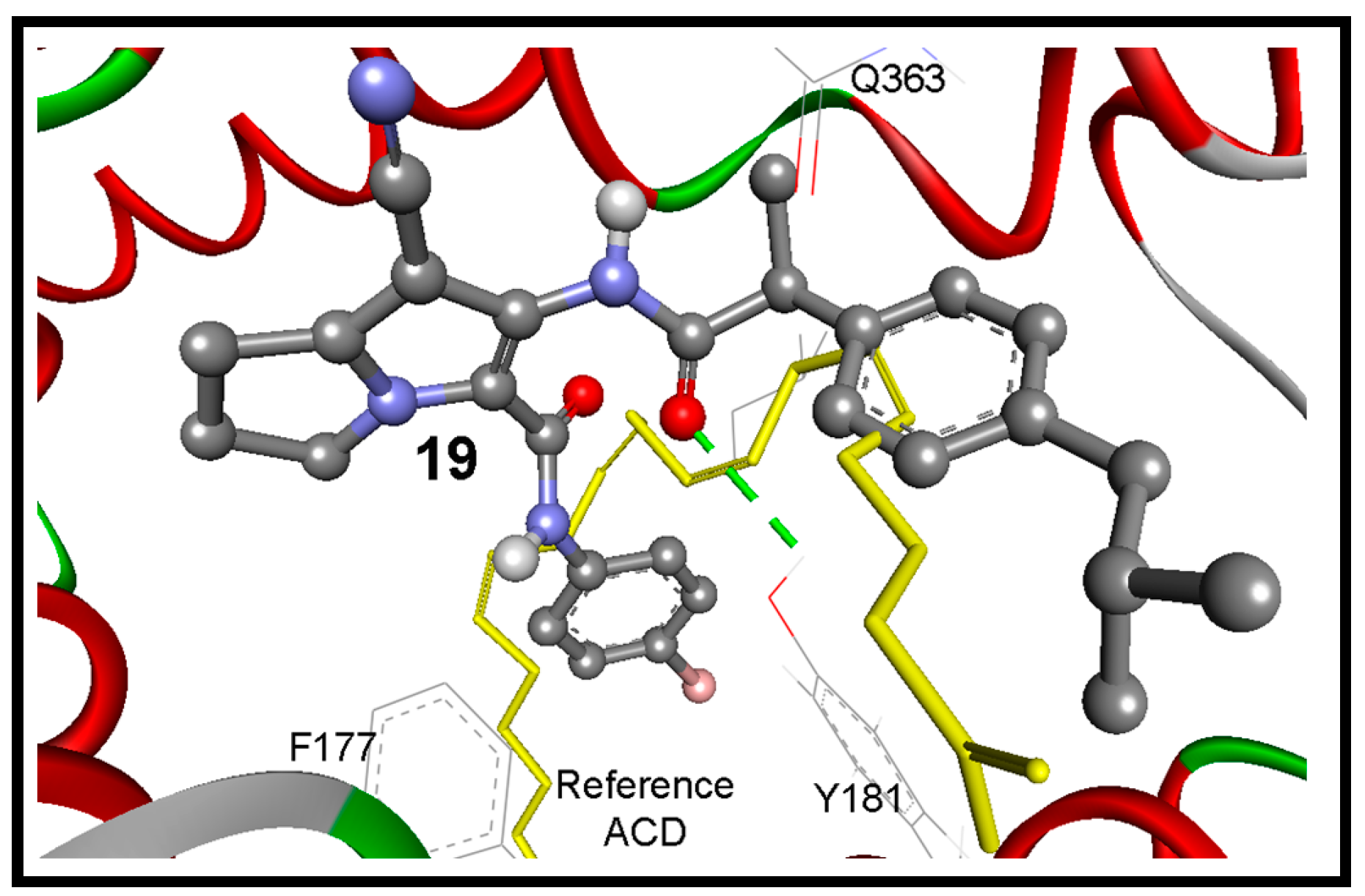

3. Experimental Section
3.1. General Information
3.2. Chemistry
3.3. Biological Evaluation
3.3.1. In Vitro COX-1/2 Inhibitory Assay
3.3.2. In Vivo Biological Evaluation
Animals
Anti-Inflammatory Activity
Analgesic Activity
Acute Ulcerogenicity Study
Histopathological Study
3.4. Molecular Docking Study
3.4.1. Preparation of the COX-1, COX-2, and 5-LOX Protein and Ligands (12–19)
3.4.2. Preparation of the Flexible Residue File
3.4.3. Calculation of Affinity Maps by Using AutoGrid
3.4.4. Defining the Docking Parameters and Running the Docking Simulation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roberts, L.J.; Morrow, J.D. The Pharmacological Basis of Therapeutics, 10th ed.; Goodman, L.S., Gilman, A.G., Hardman, J.G., Limbird, A.E., Eds.; McGraw Hill: New York, NY, USA, 2001; pp. 687–733. [Google Scholar]
- Mounier, G.; Guy, C.; Berthoux, F.; Beyens, M.N.; Ratrema, M.; Ollagnier, M. Severe renal adverse events with arylcarboxylic non-steroidal anti-inflammatory drugs: Results of a eight-year French national survey. Therapie 2006, 61, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Schneider, V.; Levesque, L.E.; Zhang, B.; Hutchinson, T.; Brophy, J.M. Association of selective and conventional nonsteroidal antiinflammatory drugs with acute renal failure: A population-based, nested case-control analysis. Am. J. Epidemiol. 2006, 164, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Cryer, B. NSAID-associated deaths: The rise and fall of NSAID-associated GI mortality. Am. J. Gastroenterol. 2005, 100, 1694–1695. [Google Scholar] [CrossRef] [PubMed]
- Singh, G. Recent considerations in nonsteroidal anti-inflammatory drug gastropathy. Am. J. Med. 1998, 105, 31S–38S. [Google Scholar] [CrossRef]
- Vane, J.R.; Botting, R.M. Mechanism of action of anti-inflammatory drugs. Scand. J. Rheumatol. Suppl. 1996, 102, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Tomisato, W.; Tsutsumi, S.; Rokutan, K.; Tsuchiya, T.; Mizushima, T. NSAIDs induce both necrosis and apoptosis in guinea pig gastric mucosal cells in primary culture. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1092–G1100. [Google Scholar] [PubMed]
- Tomisato, W.; Tsutsumi, S.; Hoshino, T.; Hwang, H.-J.; Mio, M.; Tsuchiya, T.; Mizushima, T. Role of direct cytotoxic effects of NSAIDs in the induction of gastric lesions. Biochem. Pharmacol. 2004, 67, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Yeomans, N.D.; Hawkey, C.J.; Brailsford, W.; Naesdal, J. Gastroduodenal toxicity of low-dose acetylsalicylic acid: A comparison with non-steroidal anti-inflammatory drugs. Curr. Med. Res. Opin. 2009, 25, 2785–2793. [Google Scholar] [CrossRef] [PubMed]
- Redasani, V.K.; Bari, S.B. Synthesis and evaluation of mutual prodrugs of ibuprofen with menthol, thymol and eugenol. Eur. J. Med. Chem. 2012, 56, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Masferrer, J.L.; Zweifel, B.S.; Manning, P.T.; Hauser, S.D.; Leahy, K.M.; Smith, W.G.; Isakson, P.C.; Seibert, K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. USA 1994, 91, 3228–3232. [Google Scholar] [CrossRef] [PubMed]
- Chiroli, V.; Benedini, F.; Ongini, E.; del Soldato, P. Nitric oxide-donating non-steroidal anti-inflammatory drugs: The case of nitro derivatives of aspirin. Eur. J. Med. Chem. 2003, 38, 441–446. [Google Scholar] [CrossRef]
- Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; et al. Novel antiarthritic agents with 1,2-isothiazolidine-1,1-dioxide (γ-sultam) skeleton: Cytokine suppressive dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase. J. Med. Chem. 2000, 43, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Jett, M.F.; Ramesha, C.S.; Brown, C.D.; Chiu, S.; Emmett, C.; Voronin, T.; Sun, T.; O’Yang, C.; Hunter, J.C.; Eglen, R.M.; et al. Characterization of the analgesic and anti-inflammatory activities of ketorolac and its enantiomers in the rat. J. Pharmacol. Exp. Ther. 1999, 288, 1288–1297. [Google Scholar] [PubMed]
- Lashbrook, J.M.; Ossipov, M.H.; Hunter, J.C.; Raffa, R.B.; Tallarida, R.J.; Porreca, F. Synergistic antiallodynic effects of spinal morphine with ketorolac and selective COX1- and COX2-inhibitors in nerve-injured rats. Pain 1999, 82, 65–72. [Google Scholar] [CrossRef]
- Laufer, S.; Augustin, J.; Dannhardt, G.; Kiefer, W. (6,7-Diaryldihydropyrrolizin-5-yl)acetic acids, a novel class of potent dual inhibitors of both cyclooxygenase and 5-lipoxygenase. J. Med. Chem. 1994, 37, 1894–1897. [Google Scholar] [CrossRef] [PubMed]
- Tries, S.; Laufer, S. The pharmacological profile of ML3000: A new pyrrolizine derivative inhibiting the enzymes cyclo-oxygenase and 5-lipoxygenase. Inflammopharmacology 2001, 9, 113–124. [Google Scholar] [CrossRef]
- Laufer, S.; Striegel, H.G.; Neher, K.; Zechmeister, P.; Donat, C.; Stolingwa, K.; Baur, S.; Tries, S.; Kammermeier, T.; Dannhardt, G.; et al. Synthesis and evaluation of a novel series of pyrrolizine derivatives as dual cyclooxygenase-1 and 5-lipoxygenase inhibitors. Arch. Pharm. 1997, 330, 307–312. [Google Scholar] [CrossRef]
- Ulbrich, H.; Fiebich, B.; Dannhardt, G. Cyclooxygenase-1/2 (COX-1/COX-2) and 5-lipoxygenase (5-LOX) inhibitors of the 6,7-diaryl-2,3–1H-dihydropyrrolizine type. Eur. J. Med. Chem. 2002, 37, 953–959. [Google Scholar] [CrossRef]
- Laufer, S.; Tollmann, K.; Striegel, H.-G. Anti-Inflammatory Oxo Derivatives and Hydroxy Derivatives of Pyrrolizines, and Their Pharmaceutical Use. U.S. Patent No. US6,878,738 B1, 12 April 2005. [Google Scholar]
- Liedtke, A.J.; Keck, P.R.; Lehmann, F.; Koeberle, A.; Werz, O.; Laufer, S.A. Arylpyrrolizines as inhibitors of microsomal prostaglandin E2 synthase-1 (mPGES-1) or as dual inhibitors of mPGES-1 and 5-lipoxygenase (5-LOX). J. Med. Chem. 2009, 52, 4968–4972. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.E.; Awadallah, F.M.; Ibrahim, N.A.; Gouda, A.M. Novel substituted and fused pyrrolizine derivatives: Synthesis, anti-inflammatory and ulcerogenecity studies. Eur. J. Med. Chem. 2010, 45, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Etienne, A.; Correia, Y. Derivatives of 2-pyrrolidone. Bull. Soc. Chem. 1969, 10, 3704–3712. [Google Scholar]
- Jacobs, W.A.; Heidelberger, M. The ferrous sulfate and ammonia method for the reduction of nitro to amino compounds. J. Am. Chem. Soc. 1917, 39, 1435–1439. [Google Scholar] [CrossRef] [Green Version]
- Gouda, A.M.; Abdelazeem, A.H.; Arafa, E.-S.; Abdellatif, K.R. Design, synthesis and pharmacological evaluation of novel pyrrolizine derivatives as potential anticancer agents. Bioorg. Chem. 2014, 53, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Praveen Rao, P.N.; Amini, M.; Li, H.; Habeeb, A.G.; Knaus, E.E. Design, synthesis, and biological evaluation of 6-substituted-3-(4-methanesulfonylphenyl)-4-phenylpyran-2-ones: A novel class of diarylheterocyclic selective cyclooxygenase-2 inhibitors. J. Med. Chem. 2003, 46, 4872–4882. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, J.; Bensdorf, K.; Zhang, H.; Liu, H.; Wang, Y.; Qian, H.; Zhang, Y.; Wellner, A.; Rubner, G.; et al. Investigations on cytotoxicity and anti-inflammatory potency of licofelone derivatives. Eur. J. Med. Chem. 2011, 46, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Abdelazeem, A.H.; Salama, S.A.; Maghrabi, I.A. Design, Synthesis, and Anti-Inflammatory Evaluation of Novel Diphenylthiazole-Thiazolidinone Hybrids. Arch. Pharm. 2015, 348, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenan-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Eddy, N.B.; Leimback, D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J. Pharmacol. Exp. Ther. 1953, 107, 385–393. [Google Scholar] [PubMed]
- Meshali, M.; El-Sabbagh, H.; Foda, A. Effect of encapsulation of flufenamic acid with acrylic resins on its bioavailability and gastric ulcerogenic activity in rats. Acta Pharm. Technol. 1983, 29, 217–219. [Google Scholar]
- Sivaraman, D.; Muralidharan, P. Anti-ulcerogenic evaluation of root extract of Ficus hispida Linn. in aspirin ulcerated rats. Afr. J. Pharm. Pharmacol. 2010, 4, 079–082. [Google Scholar]
- Bancroft, J.D.; Stevens, A.; Turmer, R. Theory and Practice of Histological Techniques, 4th ed.; Churchill Livingstone: Edinburgh, London, UK; Melbourne, Australia, 1996; pp. 47–67. [Google Scholar]
- Selinsky, B.S.; Gupta, K.; Sharkey, C.T.; Loll, P.J. Structural analysis of NSAID binding by prostaglandin H2 synthase: Time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 2001, 40, 5172–5180. [Google Scholar] [CrossRef] [PubMed]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.C.; Bartlett, S.G.; Waight, M.T.; Neau, D.B.; Boeglin, W.E.; Brash, A.R.; Newcomer, M.E. The structure of human 5-lipoxygenase. Science 2011, 331, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.C.; Rui, Z.; Neau, D.B.; Waight, M.T.; Bartlett, S.G.; Boeglin, W.E.; Brash, A.R.; Newcomer, M.E. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 2012, 26, 3222–3229. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 12–19 are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gouda, A.M.; Ali, H.I.; Almalki, W.H.; Azim, M.A.; Abourehab, M.A.S.; Abdelazeem, A.H. Design, Synthesis, and Biological Evaluation of Some Novel Pyrrolizine Derivatives as COX Inhibitors with Anti-Inflammatory/Analgesic Activities and Low Ulcerogenic Liability. Molecules 2016, 21, 201. https://doi.org/10.3390/molecules21020201
Gouda AM, Ali HI, Almalki WH, Azim MA, Abourehab MAS, Abdelazeem AH. Design, Synthesis, and Biological Evaluation of Some Novel Pyrrolizine Derivatives as COX Inhibitors with Anti-Inflammatory/Analgesic Activities and Low Ulcerogenic Liability. Molecules. 2016; 21(2):201. https://doi.org/10.3390/molecules21020201
Chicago/Turabian StyleGouda, Ahmed M., Hamed I. Ali, Waleed H. Almalki, Mohamed A. Azim, Mohammed A. S. Abourehab, and Ahmed H. Abdelazeem. 2016. "Design, Synthesis, and Biological Evaluation of Some Novel Pyrrolizine Derivatives as COX Inhibitors with Anti-Inflammatory/Analgesic Activities and Low Ulcerogenic Liability" Molecules 21, no. 2: 201. https://doi.org/10.3390/molecules21020201
APA StyleGouda, A. M., Ali, H. I., Almalki, W. H., Azim, M. A., Abourehab, M. A. S., & Abdelazeem, A. H. (2016). Design, Synthesis, and Biological Evaluation of Some Novel Pyrrolizine Derivatives as COX Inhibitors with Anti-Inflammatory/Analgesic Activities and Low Ulcerogenic Liability. Molecules, 21(2), 201. https://doi.org/10.3390/molecules21020201