
Utilization of Cyanoacetohydrazide and Oxadiazolyl Acetonitrile in the Synthesis of Some New Cytotoxic Heterocyclic Compounds

Abstract
:
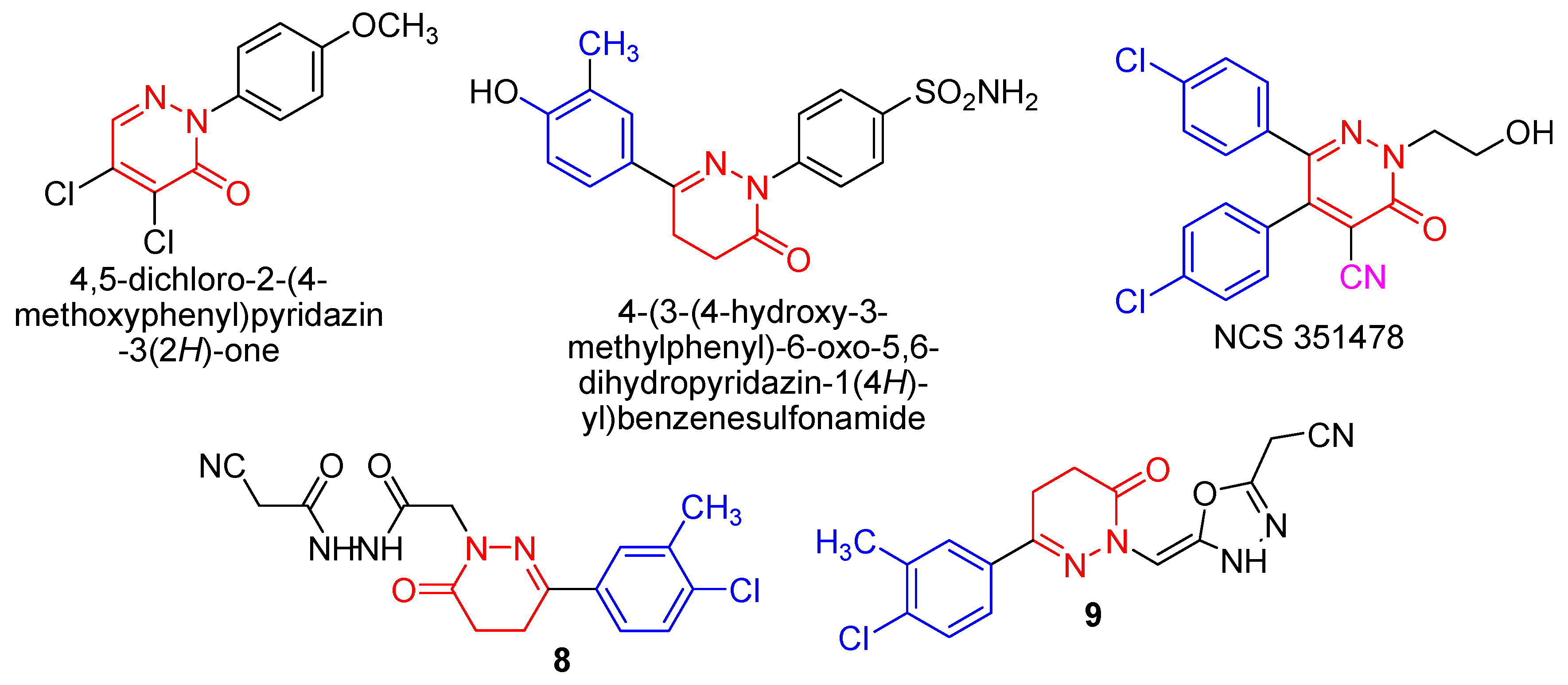
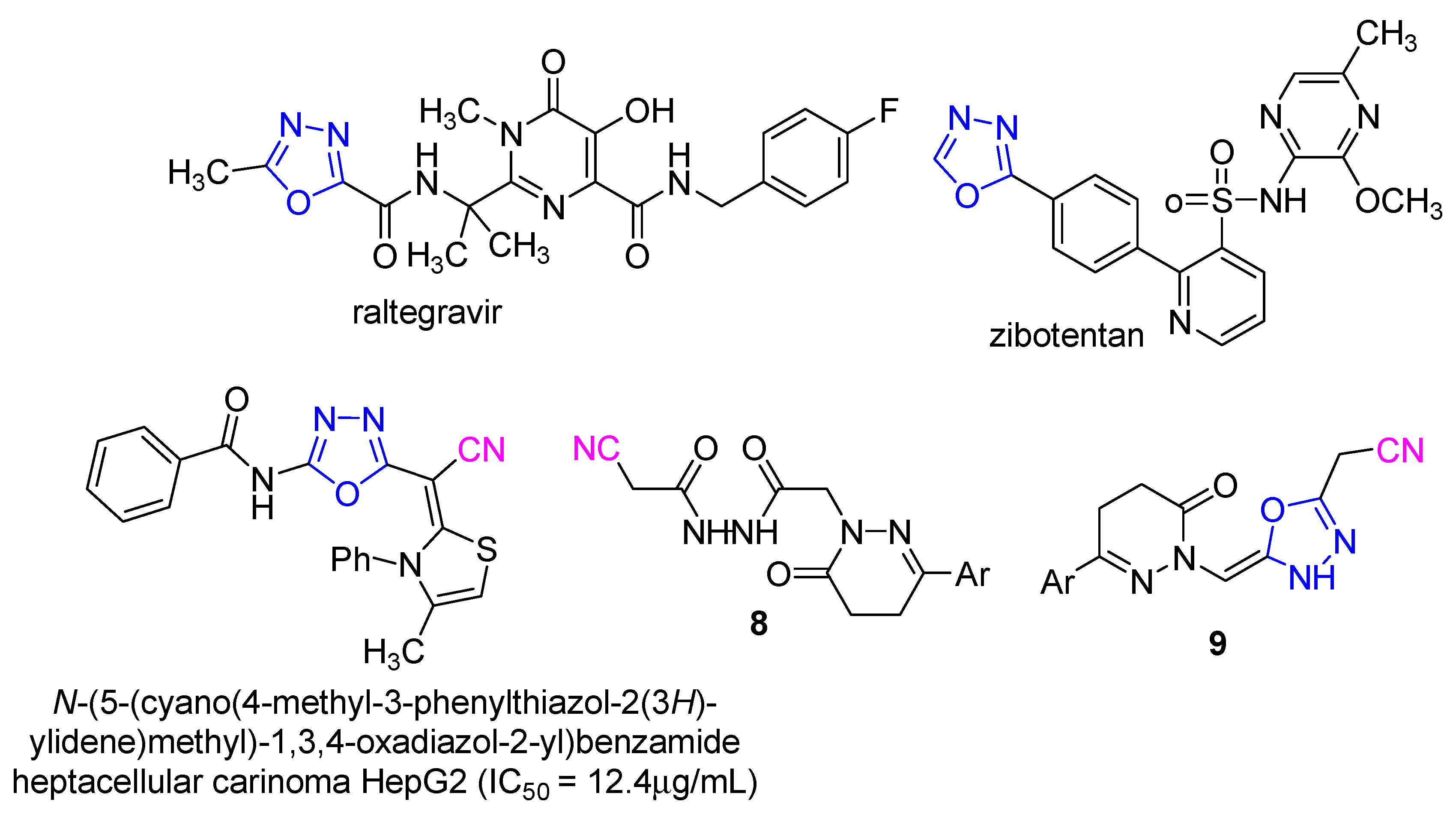
1. Introduction
2. Results and Discussion
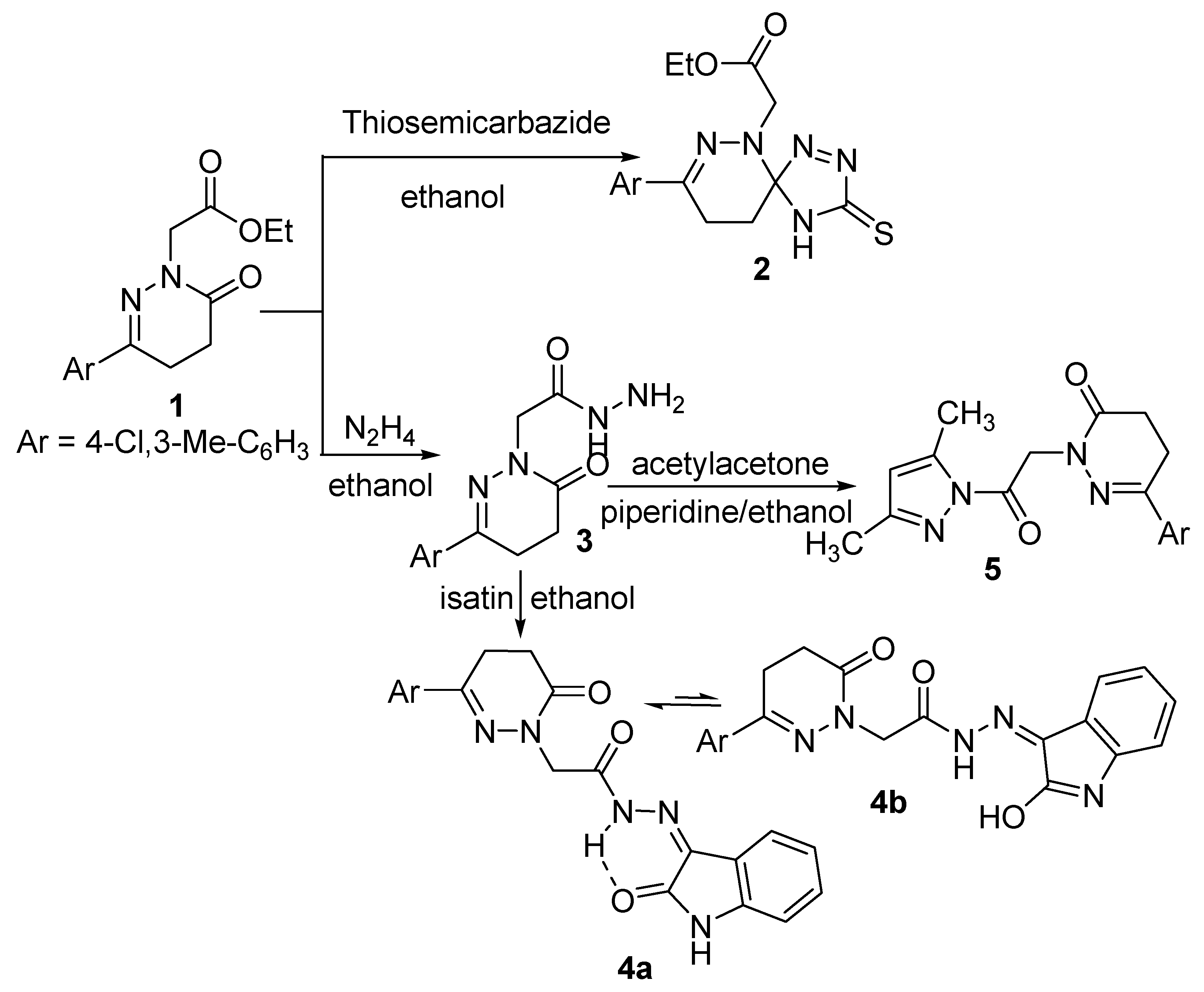
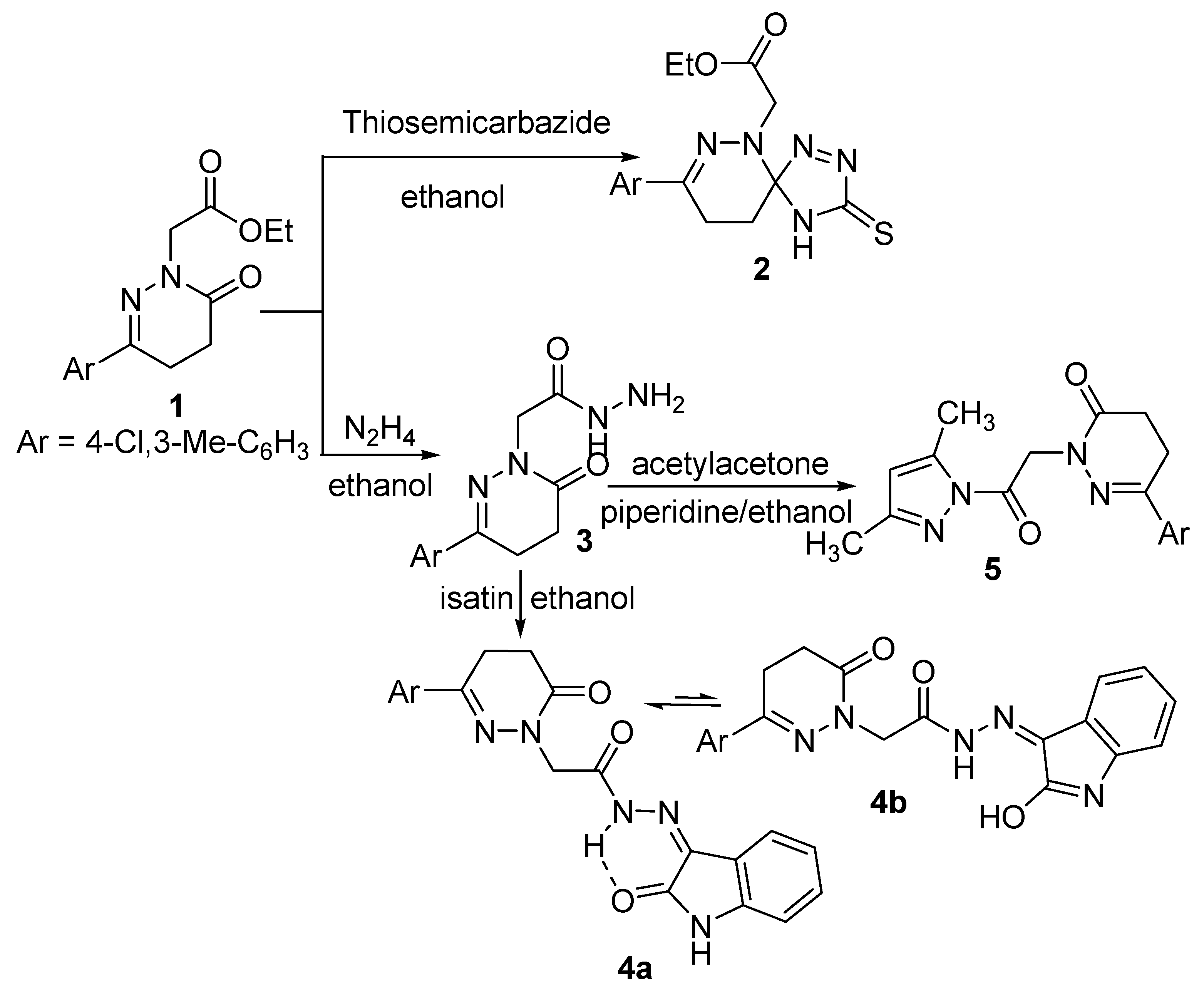
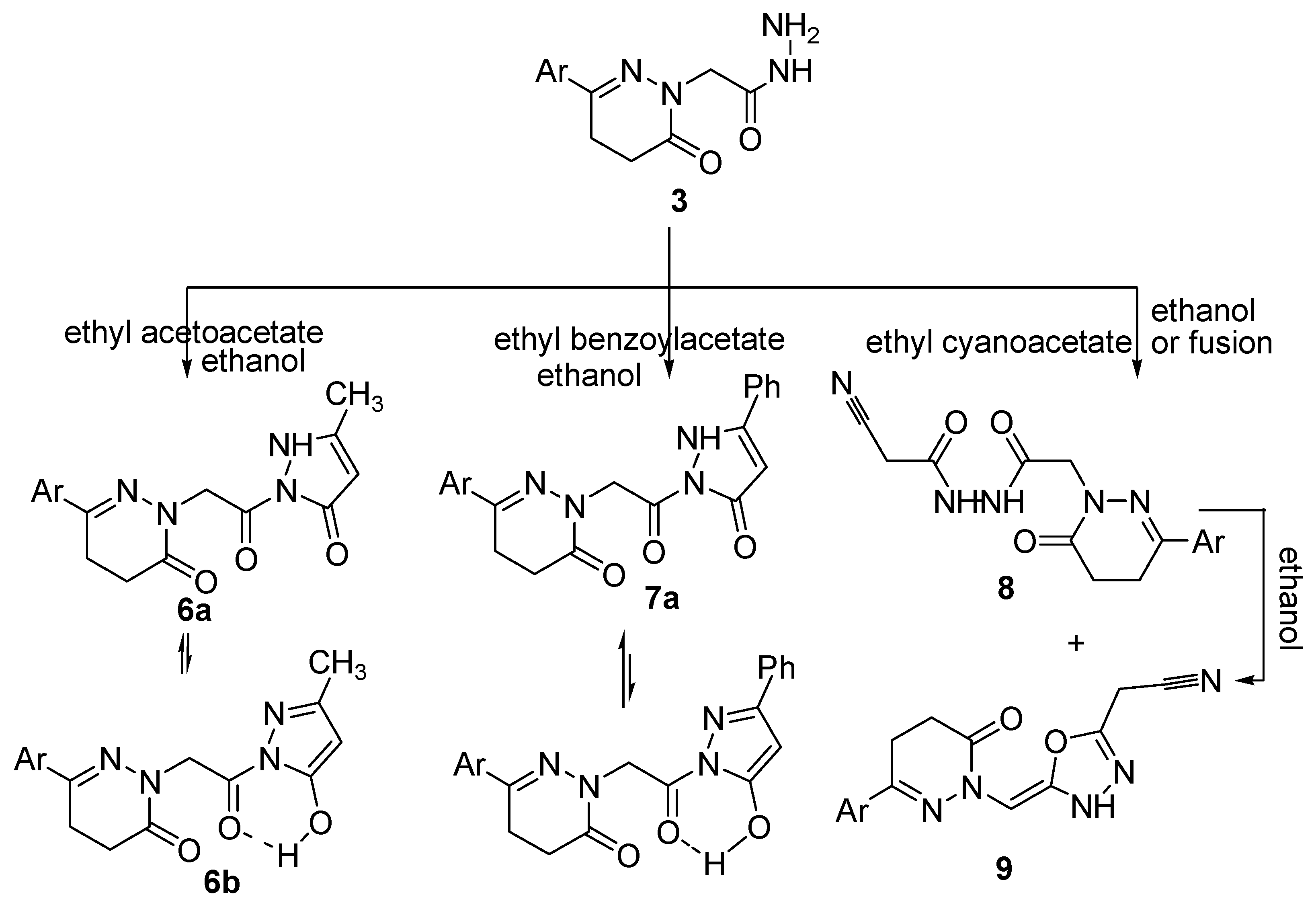
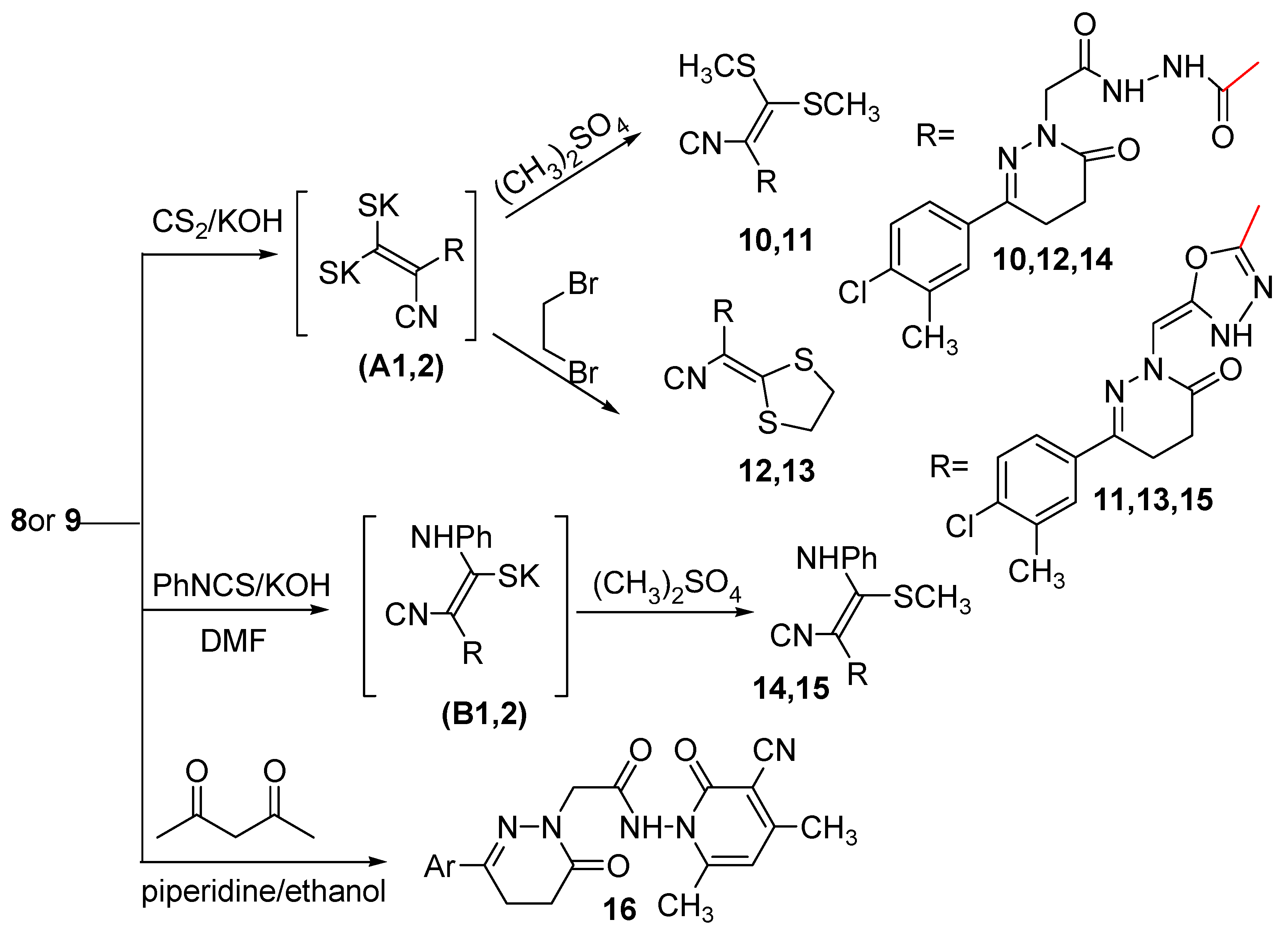
2.1. Chemistry
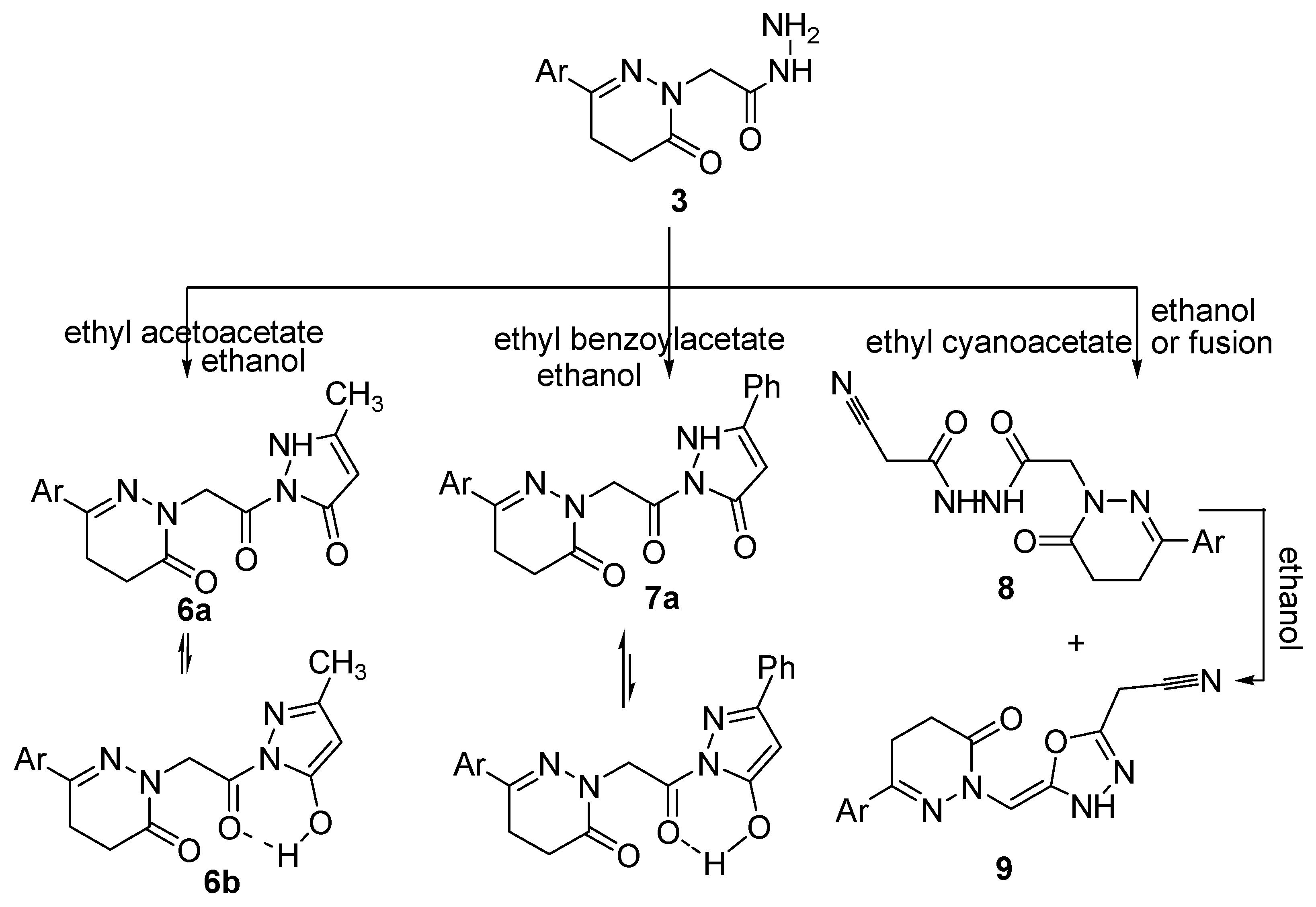
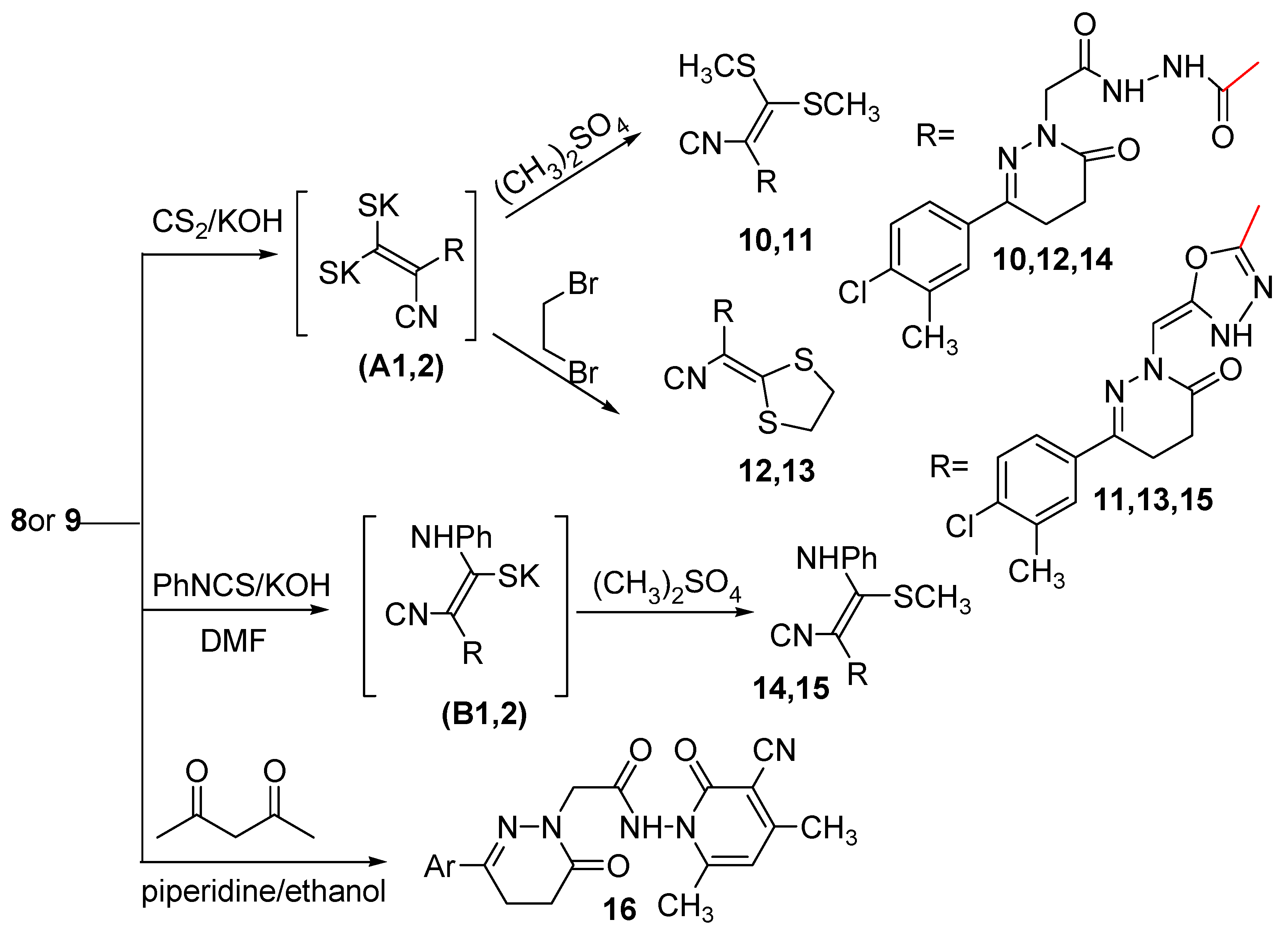
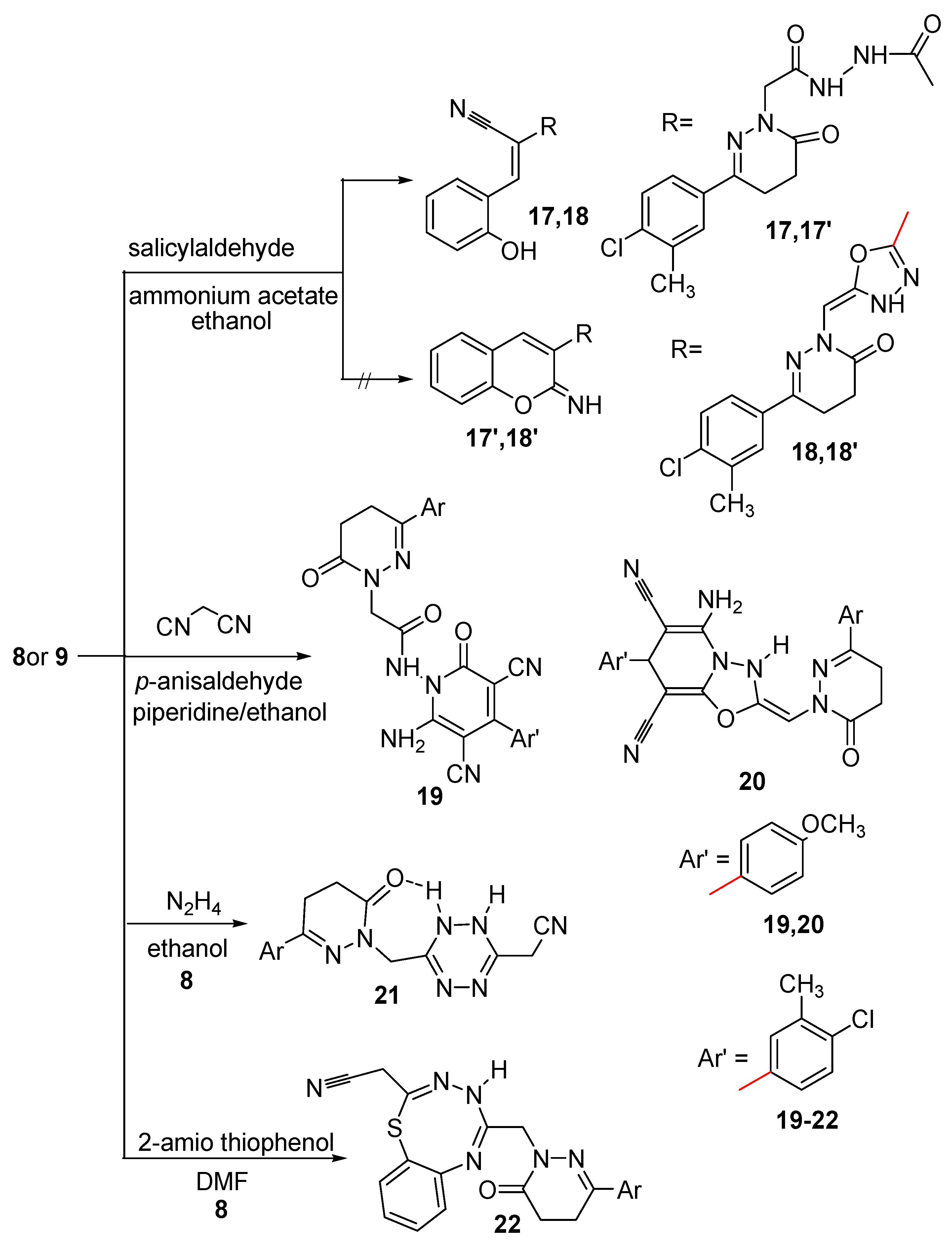
2.2. Pharmacological Activity
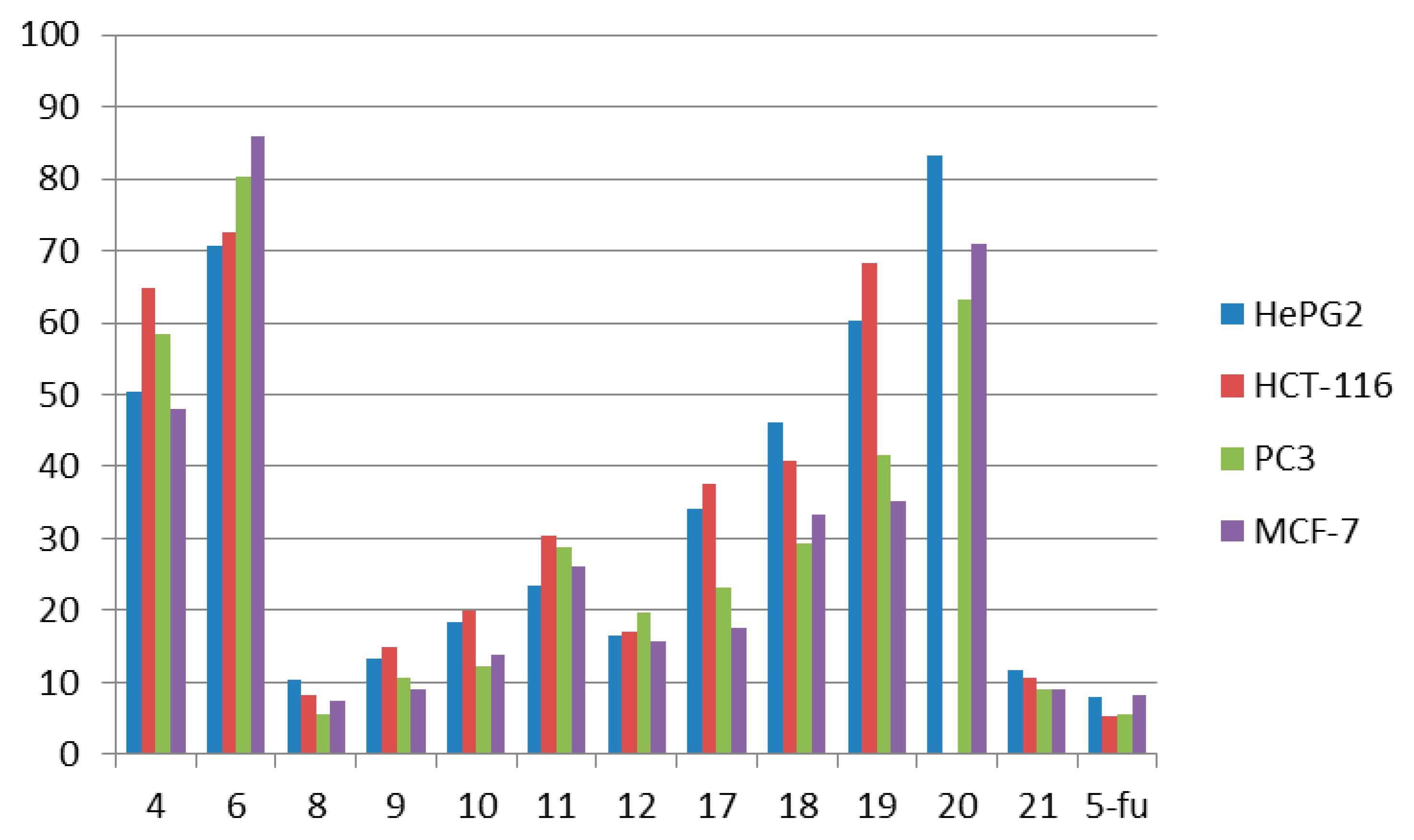
2.2.1. Antitumor Activity Using in Vitro Ehrlich Ascites Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
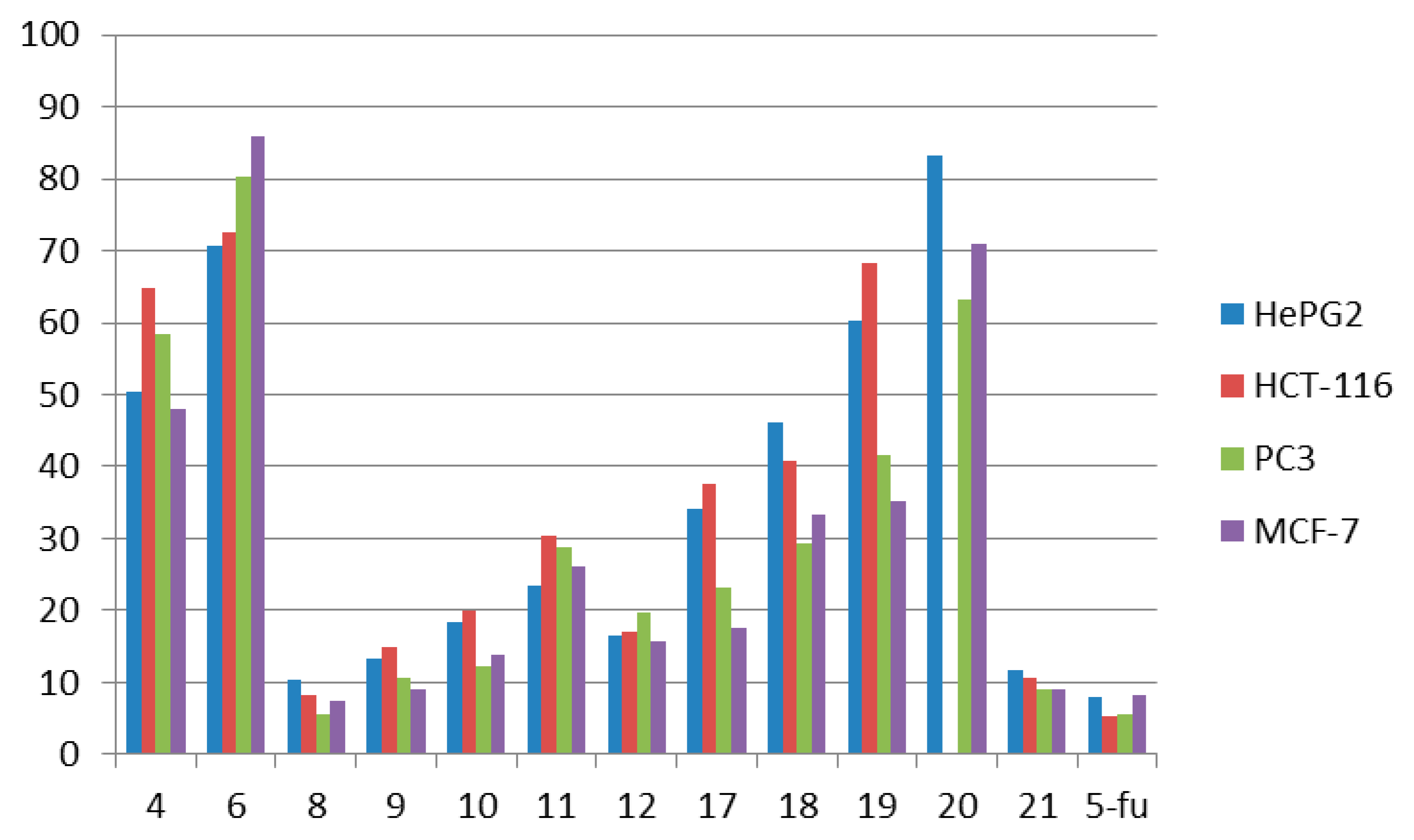
| Comp. No. | In Vitro Cytotoxicity | IC50 (μg/mL) a | ||
|---|---|---|---|---|
| HePG2 | HCT-116 | PC3 | MCF-7 | |
| 4 | 50.3 ± 4.22 | 64.7 ± 4.11 | 48.1 ± 3.64 | 58.4 ± 3.67 |
| 6 | 70.7 ± 4.65 | 72.6 ± 4.51 | 86.0 ± 4.63 | 80.4 ± 4.75 |
| 8 | 10.3 ± 0.81 | 8.1 ± 0.35 | 7.4 ± 0.34 | 5.6 ± 0.30 |
| 9 | 13.2 ± 1.31 | 14.8 ± 1.53 | 9.1 ± 0.86 | 10.5 ± 1.04 |
| 10 | 18.4 ± 1.06 | 20.0 ± 1.96 | 13.7 ± 1.37 | 12.3 ± 1.08 |
| 11 | 23.4 ± 1.46 | 30.3 ± 2.64 | 26.2 ± 1.60 | 28.7 ± 1.83 |
| 12 | 16.5 ± 1.35 | 16.9 ± 1.14 | 15.7 ± 1.56 | 19.7 ± 1.76 |
| 17 | 34.1 ± 2.30 | 37.5 ± 2.67 | 17.5 ± 1.42 | 23.1 ± 1.51 |
| 18 | 46.0 ± 3.61 | 40.7 ± 2.63 | 33.3 ± 2.07 | 29.4 ± 2.00 |
| 19 | 60.3 ± 3.97 | 68.3 ± 3.88 | 35.3 ± 2.94 | 41.5 ± 2.43 |
| 20 | 83.2 ± 4.83 | >100 | 70.9 ± 4.75 | 63.1 ± 3.89 |
| 21 | 11.8 ± 1.12 | 10.5 ± 0.89 | 8.9 ± 0.45 | 9.1 ± 0.87 |
| 5-fu | 7.9 ± 0.28 | 5.2 ± 0.14 | 8.3 ± 0.25 | 5.5 ± 0.21 |
| Compounds | Conc. (µg/mL) | HePG-2 | HCT-116 | PC3 | MCF-7 |
|---|---|---|---|---|---|
| 5-FU | 100 µg/mL | 8.6 | 7.2 | 8.1 | 7.7 |
| 50 µg/mL | 17.1 | 12.0 | 15.8 | 14.3 | |
| 25 µg/mL | 24.0 | 19.3 | 22.5 | 21.5 | |
| 12.5 µg/mL | 33.1 | 30.6 | 36.7 | 34.6 | |
| 6.25 µg/mL | 56.8 | 48.9 | 55.2 | 47.4 | |
| 3.125 µg/mL | 70.6 | 60.5 | 74.1 | 58.3 | |
| 1.56 µg/mL | 88.7 | 73.4 | 92.5 | 76.9 | |
| 4 | 100 µg/mL | 37.9 | 42.7 | 36.1 | 41.1 |
| 50 µg/mL | 48.8 | 55.5 | 48.4 | 52.9 | |
| 25 µg/mL | 63.0 | 67.2 | 62.5 | 64.6 | |
| 12.5 µg/mL | 75.6 | 81.3 | 74.2 | 78.2 | |
| 6.25 µg/mL | 93.1 | 94.9 | 95.6 | 99.3 | |
| 3.125 µg/mL | 100 | 100 | 100 | 100 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 6 | 100 µg/mL | 44.5 | 45.7 | 49.3 | 46.8 |
| 50 µg/mL | 57.2 | 56.6 | 60.6 | 61.2 | |
| 25 µg/mL | 70.4 | 71.3 | 78.1 | 73.3 | |
| 12.5 µg/mL | 82.9 | 83.4 | 91.7 | 86.9 | |
| 6.25 µg/mL | 98.8 | 96.5 | 100 | 100 | |
| 3.125 µg/mL | 100 | 100 | 100 | 100 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 8 | 100 µg/mL | 13.0 | 8.3 | 8.4 | 7.0 |
| 50 µg/mL | 18.2 | 17.9 | 14.3 | 12.3 | |
| 25 µg/mL | 25.8 | 25.1 | 22.5 | 19.5 | |
| 12.5 µg/mL | 36.9 | 33.7 | 31.9 | 31.9 | |
| 6.25 µg/mL | 68.5 | 56.8 | 52.6 | 50.6 | |
| 3.125 µg/mL | 75.6 | 71.4 | 70.8 | 62.2 | |
| 1.56 µg/mL | 97.4 | 89.3 | 91.1 | 74.4 | |
| 9 | 100 µg/mL | 18.4 | 16.2 | 8.1 | 13.1 |
| 50 µg/mL | 26.3 | 25.3 | 17.6 | 18.9 | |
| 25 µg/mL | 35.1 | 36.4 | 26.8 | 27.4 | |
| 12.5 µg/mL | 43.7 | 48.1 | 37.2 | 38.2 | |
| 6.25 µg/mL | 61.0 | 67.5 | 55.5 | 66.7 | |
| 3.125 µg/mL | 84.2 | 88.7 | 78.1 | 76.1 | |
| 1.56 µg/mL | 100 | 100 | 96.0 | 98.5 | |
| 10 | 100 µg/mL | 23.1 | 20.1 | 19.0 | 15.1 |
| 50 µg/mL | 30.0 | 32.4 | 26.4 | 24.2 | |
| 25 µg/mL | 41.5 | 42.8 | 35.9 | 35.1 | |
| 12.5 µg/mL | 53.8 | 55.5 | 43.2 | 42.8 | |
| 6.25 µg/mL | 67.3 | 71.2 | 65.3 | 59.9 | |
| 3.125 µg/mL | 89.7 | 95.3 | 83.6 | 81.7 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 11 | 100 µg/mL | 24.8 | 29.1 | 26.6 | 25.9 |
| 50 µg/mL | 35.7 | 39.7 | 37.3 | 38.8 | |
| 25 µg/mL | 47.4 | 52.5 | 49.2 | 50.1 | |
| 12.5 µg/mL | 58.1 | 63.8 | 60.5 | 65.9 | |
| 6.25 µg/mL | 72.3 | 77.9 | 74.1 | 78.6 | |
| 3.125 µg/mL | 91.8 | 96.2 | 97.2 | 96.7 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 |
| Compounds | Conc. (µg/mL) | HePG-2 | HCT-116 | PC3 | MCF-7 |
|---|---|---|---|---|---|
| 12 | 100 µg/mL | 19.9 | 18.3 | 21.0 | 22.9 |
| 50 µg/mL | 27.2 | 26.8 | 29.2 | 31.2 | |
| 25 µg/mL | 36.1 | 39.5 | 38.3 | 43.8 | |
| 12.5 µg/mL | 51.3 | 50.4 | 47.5 | 54.2 | |
| 6.25 µg/mL | 71.4 | 72.6 | 65.4 | 71.3 | |
| 3.125 µg/mL | 89.5 | 90.7 | 86.7 | 88.5 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 17 | 100 µg/mL | 29.1 | 31.5 | 19.7 | 24.2 |
| 50 µg/mL | 42.2 | 45.2 | 27.5 | 35.5 | |
| 25 µg/mL | 51.9 | 55.8 | 39.1 | 46.1 | |
| 12.5 µg/mL | 71.3 | 69.7 | 51.6 | 57.3 | |
| 6.25 µg/mL | 84.5 | 81.3 | 73.4 | 72.6 | |
| 3.125 µg/mL | 100 | 99.4 | 92.4 | 96.8 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 18 | 100 µg/mL | 35.9 | 33.6 | 31.1 | 28.6 |
| 50 µg/mL | 48.1 | 45.5 | 42.5 | 39.5 | |
| 25 µg/mL | 60.5 | 57.1 | 53.2 | 51.3 | |
| 12.5 µg/mL | 72.8 | 70.0 | 65.4 | 62.9 | |
| 6.25 µg/mL | 89.2 | 91.2 | 78.0 | 77.8 | |
| 3.125 µg/mL | 100 | 100 | 99.3 | 96.4 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 19 | 100 µg/mL | 38.7 | 43.7 | 29.5 | 31.9 |
| 50 µg/mL | 53.2 | 56.5 | 42.8 | 46.0 | |
| 25 µg/mL | 70.3 | 69.1 | 53.1 | 59.2 | |
| 12.5 µg/mL | 89.1 | 84.2 | 72.2 | 71.3 | |
| 6.25 µg/mL | 100 | 98.3 | 84.6 | 93.5 | |
| 3.125 µg/mL | 100 | 100 | 100 | 100 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 20 | 100 µg/mL | 48.1 | 53.7 | 45.0 | 39.8 |
| 50 µg/mL | 61.3 | 72.5 | 56.2 | 54.6 | |
| 25 µg/mL | 73.4 | 85.4 | 71.6 | 70.3 | |
| 12.5 µg/mL | 86.7 | 98.9 | 82.1 | 94.1 | |
| 6.25 µg/mL | 99.8 | 100 | 98.4 | 100 | |
| 3.125 µg/mL | 100 | 100 | 100 | 100 | |
| 1.56 µg/mL | 100 | 100 | 100 | 100 | |
| 21 | 100 µg/mL | 15.7 | 13.1 | 7.7 | 8.4 |
| 50 µg/mL | 22.1 | 19.2 | 16.1 | 17.3 | |
| 25 µg/mL | 27.4 | 27.6 | 24.5 | 26.5 | |
| 12.5 µg/mL | 42.9 | 38.5 | 38.2 | 37.0 | |
| 6.25 µg/mL | 65.8 | 66.8 | 55.3 | 55.4 | |
| 3.125 µg/mL | 83.2 | 75.7 | 78.1 | 78.8 | |
| 1.56 µg/mL | 99.0 | 97.1 | 94.4 | 95.9 |
2.2.2. Antioxidant Activity Using 2,2′-Azino-bis(3-ethylbenzthiazoline-6-sulfonic Acid (ABTS) Inhibition
| Comp. No. | Antioxidant Activity Absorbance | (ABTS Method) Inhibition (%) | Bleomycin Dependent DNA Damage |
|---|---|---|---|
| 4 | 0.261 | 48.4 | 0.116 |
| 6 | 0.271 | 46.4 | 0.129 |
| 8 | 0.060 | 88.1 | 0.069 |
| 9 | 0.127 | 74.9 | 0.077 |
| 10 | 0.143 | 71.7 | 0.088 |
| 11 | 0.204 | 59.7 | 0.094 |
| 12 | 0.162 | 68.0 | 0.081 |
| 17 | 0.201 | 60.3 | 0.104 |
| 18 | 0.230 | 54.5 | 0.074 |
| 19 | 0.257 | 49.2 | 0.122 |
| 20 | 0.268 | 47.0 | 0.143 |
| 21 | 0.071 | 86.0 | 0.074 |
| Control of ABTS | 0.506 | 0 | - |
| Ascorbic acid | 0.056 | 88.9 | 0.072 |
2.2.3. Bleomycin-Dependent Deoxyribonucleic Acid (DNA) Damage
2.2.4. Structure Activity Relationship
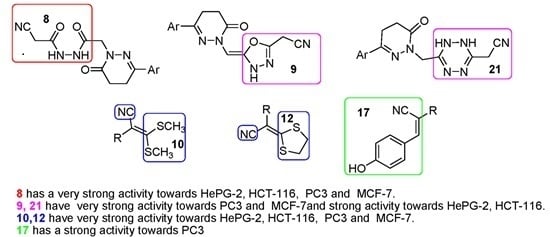
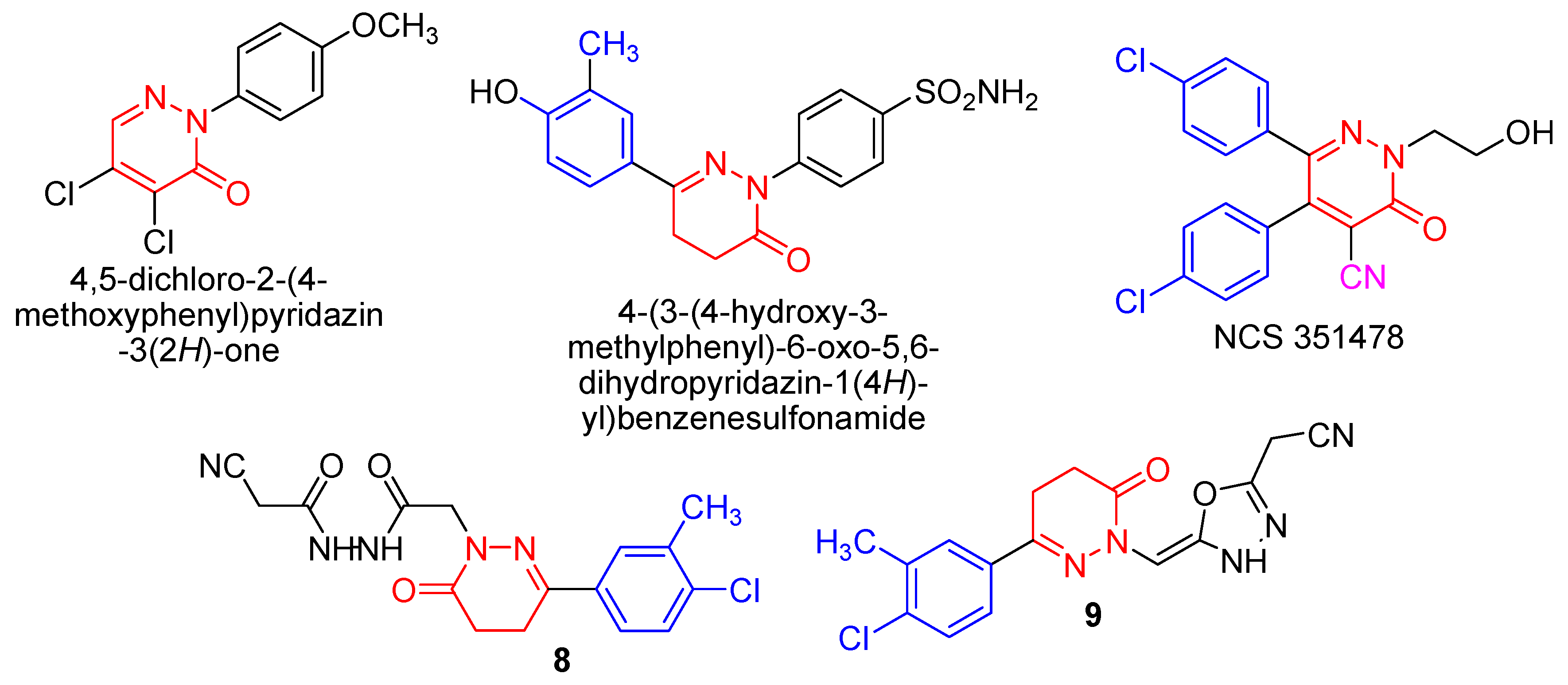
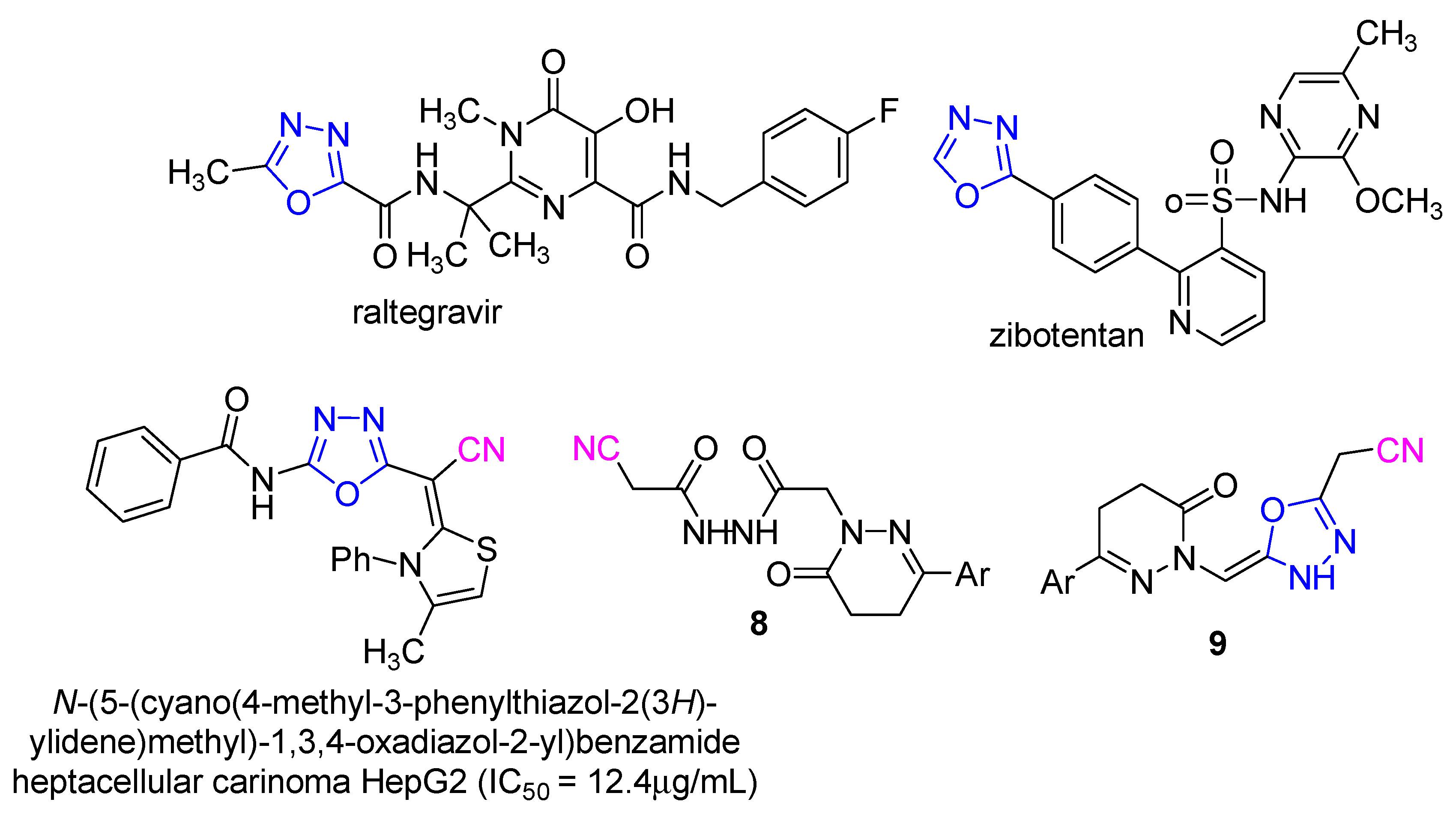
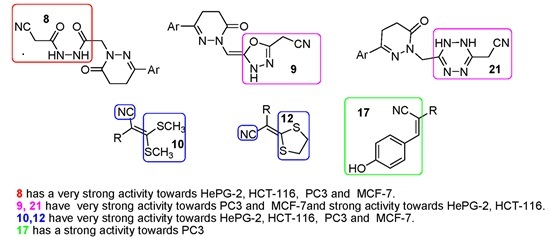
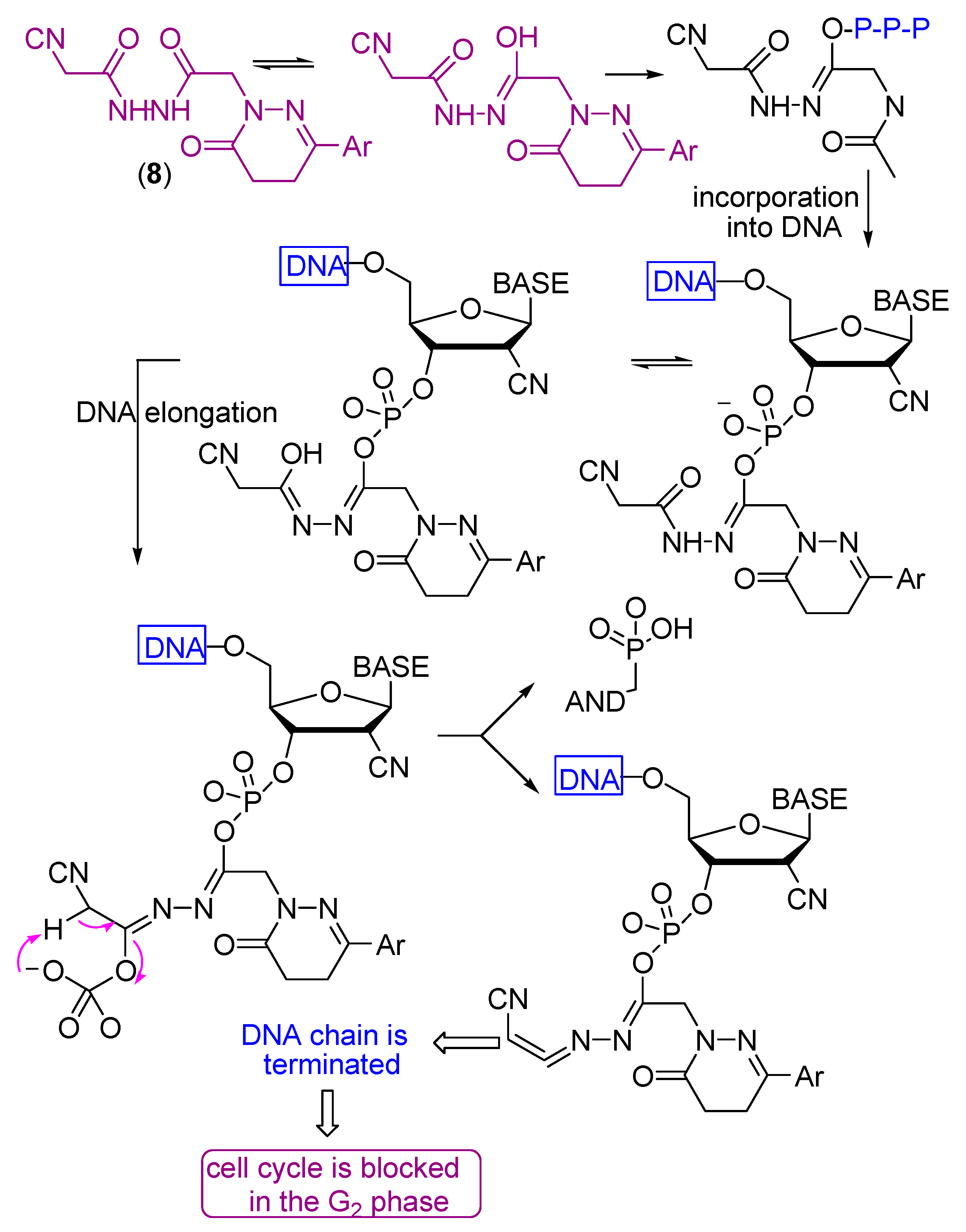
- Compound 8 showed very strong activities against the four cell lines, which may be due to the presence of two NH and C≡N groups.
- Compound 9 showed very strong activities against the PC-3 and MCF-7 cell lines, and strong activity against the HePG2 and HCT-116 cell lines which may be due to the presence of NH and the oxadiazole moiety.
- Compounds 10 and 12 showed strong activity against the four cell lines, which may be due to the presence of two NH groups and the sulfur atom, which has a vacant orbital that can accept electrons.
- Compound 17 showed strong and moderate activity due to the presence of NH and OH groups.
- Compound 21 showed very strong activities against the HCT-116, PC-3 and MCF-7 cell lines, and strong activity against the HePG2 cell line which may be due to the presence of NH of the tetrazine moiety and C≡N group with α-H and β-NH.
- Compounds 4, 6, 11, 18, 19 and 20 showed either weak or moderate activities because of the absence of C≡N as in compounds 4, 6 or the absence of α-H as in compounds 11, 18, 19 and 20.
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Ethyl 2-(8-(4-Chloro-3-methylphenyl)-3-thioxo-1,2,4,6,7-pentaazaspiro[4,5]deca-1,7-diene-6-yl)acetate (2)
3.2.2. 2-(3-(4-Chloro-3-methylphenyl)-6-oxo-5,6-dihydropyridazin-1(4H)-yl)-N′-(2-oxoindolin-3-ylidene)-acetohydrazide (4)
3.2.3. 6-(4-Chloro-3-methylphenyl)-2-(2-(3,5-dimethyl-1H-pyrazol-1-yl)-2-oxoethyl)-4,5-dihydropyridazin-3(2H)-one (5)
3.2.4. General Procedure for the Synthesis of Compounds 6 and 7
3.2.5. General Procedure for Synthesizing Compounds 8 and 9
3.2.6. Another Method for Synthesizing Compound 8
3.2.7. Another Method for Synthesizing Compound (9)
3.2.8. General Procedure for Synthesizing Compounds 10–13
3.2.9. General Procedure for Synthesizing Compounds 14 and 15
3.2.10. 2-(3-(4-Chloro-3-methylphenyl)-6-oxo-5,6-dihydropyridazin-1(4H)-yl)-N-(3-cyano-4,6-dimethyl-2-oxopyridin-1(2H)-yl)acetamide (16)
3.2.11. General Procedure for Synthesizing Compounds 17 and 18
3.2.12. General Procedure for Synthesizing Compounds 19 and 20
3.2.13. 2-(6-((3-(4-Chloro-3-methylphenyl)-6-oxo-5,6-dihydropyridazin-1(4H)-yl) methyl)-1,2-dihydro-1,2,4,5-tetrazin-3-yl)acetonitrile (21)
3.2.14. 2-(5-((3-(4-Chloro-3-methylphenyl)-6-oxo-5,6-dihydropyridazin-1(4H)-yl)methyl)-4H-benzo[g]- [1,3,4,6]-thiatriazocin-2-yl)acetonitrile (22)
3.3. Pharmacological Activity
3.3.1. Cytotoxicity Assay
MTT Assay
3.3.2. Antioxidant Assay
ABTS Method
Bleomycin—Dependent DNA Damage Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Youssef, A.S.; Marzouk, M.I.; Madkour, H.M.; El-Soll, A.M.; El-Hashash, M.A. Synthesis of some heterocyclic systems of anticipated biological activities via 6-aryl-4-pyrazol-1-yl-pyridazin-3-one. Can. J. Chem. 2005, 83, 251–259. [Google Scholar] [CrossRef]
- Takaya, M.; Sato, M.; Terashima, K.; Tanizawa, H.; Maki, Y. A new nonsteroidal analgesic-anti-inflammatory agent. Synthesis and activity of 4-ethoxy-2-methyl-5-morpholino-3(2H)-pyridazinone and related compounds. J. Med. Chem. 1979, 22, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Şüküroğlu, M.; Çalışkan-Ergun, B.; Ünlü, S.; Sahin, M.F.; Küpeli, E.; Yeşilada, E.; Banoğlu, E. Synthesis, analgesic, and anti-inflammatory activities of [6-(3,5-dimethyl-4-chloropyrazole-1-yl)-3(2H)-pyridazinon-2-yl]acetamides. Arch. Pharm. Res. 2005, 28, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Şüküroğlu, M.; Küpeli, E.; Banoğlu, E.; Ünlü, S.; Yeşilada, E.; Şahin, M.F. Synthesis and analgesic activity of some 4,6-disubstituted-3(2H)-pyridazinone derivatives. Arzneim. Forsch. Drug Res. 2006, 56, 337–345. [Google Scholar]
- Özdemir, Z.; Gökçe, M.; Karakurt, A. Synthesis and analgesic, anti-inflammatory and antimicrobial evaluation of 6-substituted-3(2H)-pyridazinone-2-acetyl-2-(substituted benzal) hydrazone derivatives. Fabad J. Pharm. Sci. 2012, 37, 111–122. [Google Scholar]
- Abouzid, K.A.M.; Khalil, N.A.; Ahmed, E.M.; Abd El-Latif, H.A.; El-Araby, M.E. Structure-based molecular design, synthesis, and in vivo anti-inflammatory activity of pyridazinone derivatives as nonclassic COX-2 inhibitors. Med. Chem. Res. 2010, 19, 629–642. [Google Scholar] [CrossRef]
- Sonmez, M.; Berber, I.; Akbas, E. Synthesis, antibacterial and antifungal activity of some new pyridazinone metal complexes. Eur. J. Med. Chem. 2006, 41, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Doğruer, D.S.; Őnkol, T.S.; Őzkan, S.; Őzgen, S.; Şahin, M.F. Synthesis and antimicrobial activity of some 3(2H)-pyridazinone and 1(2H)-phthalazinone derivatives. Turk. J. Chem. 2008, 32, 469–479. [Google Scholar]
- Kočar, T.; Rečnik, S.; Svete, J.; Stanovnik, B. Transformations of 3-aminopyridazines. Synthesis of 4-oxo-4H-pyrimido[1,2-b]pyridazine and 1-(substituted pyridazin-3-yl)-1H-1,2,3-triazole derivatives (AP-542HP). Arkivoc 2002, 2002, 143–156. [Google Scholar]
- Ahmad, S.; Rathish, I.G.; Bano, S.; Alam, M.S.; Javed, K. Synthesis and biological evaluation of some novel 6-aryl-2-(p-sulfamylphenyl)-4,5-dihydropyridazin-3(2H)-ones as anti-cancer, antimicrobial, and anti-inflammatory agents. J. Enzym. Inhib. Med. Chem. 2010, 25, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Lattmann, E.; Ayuko, W.O.; Kinchinaton, D.; Langley, C.A.; Singh, H.; Karimi, L.; Tisdale, M.J. Synthesis and evaluation of 5-arylated 2(5H)-furanones and 2-arylated pyridazin-3(2H)-ones as anti-cancer agents. J. Pharm. Pharmacol. 2003, 55, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E.; Lin, C.M. Guanosine 5′-O-(3-thiotriphosphate), a potent nucleotide inhibitor of microtubule assembly. J. Biol. Chem. 1984, 259, 11060–11069. [Google Scholar] [PubMed]
- Jha, K.K.; Samad, A.; Kumar, Y.; Shaharyar, M.; Khosa, R.L.; Jain, J.; Kumar, V.; Singh, P. Design, synthesis and biological evaluation of 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 4963–4967. [Google Scholar] [CrossRef] [PubMed]
- Mamolo, M.G.; Falagiani, V.; Zampieri, D.; Vio, L.; Banfi, E.; Scialino, G. Synthesis and antimycobacterial activity of (3,4-diaryl-3H-thiazol-2-ylidene)-hydrazide derivatives. IL Farmaco 2003, 58, 631–637. [Google Scholar] [CrossRef]
- Farshori, N.N.; Banday, M.R.; Ahmad, A.; Khan, A.U.; Rauf, A. Synthesis, characterization, and in vitro antimicrobial activities of 5-alkenyl/hydroxyalkenyl-2-phenylamine-1,3,4-oxadiazoles and thiadiazoles. Bioorg. Med. Chem. Lett. 2010, 20, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.-S.; Farkas, M.E.; Kadow, J.F.; Meanwell, N.A. A base-catalyzed, direct synthesis of 2,5-disubstituted 1,2,4-triazoles from nitriles and hydrazides. Tetrahedron Lett. 2005, 46, 3429–3432. [Google Scholar] [CrossRef]
- Almajan, G.L.; Barbuceanu, S.-F.; Bancescu, G.; Saramet, I.; Saramet, G.; Draghici, C. Synthesis and antimicrobial evaluation of some fused heterocyclic [1,2,4]triazolo[3,4-b][1,3,4]thiadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 6139–6146. [Google Scholar] [CrossRef] [PubMed]
- Skoumbourdis, A.P.; Huang, R.; Southall, N.; Leister, W.; Guo, V.; Cho, M.-H.; Inglese, J.; Nirenberg, M.; Austin, C.P.; Xia, M.; et al. Identification of a potent new chemotype for the selective inhibition of PDE4. Bioorg. Med. Chem. Lett. 2008, 18, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Attanasi, O.A.; Filippone, P.; Perrulli, F.R.; Santeusanio, S. Regioselective role of the hydrazide moiety in the formation of complex pyrrole-pyrazole systems. Tetrahedron 2001, 57, 1387–1394. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.; Abuo-Rahma, G.A.; Hassan, A.A. Synthesis of novel pyrazole derivatives and evaluation of their antidepressant and anticonvulsant activities. Eur. J. Med. Chem. 2009, 44, 3480–3487. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, S.; Mueller-Ibeler, J.; Kluge, J.; Wäss, L.; Burdinski, G.; Havens, J.R.; Onofrey, T.J.; Wang, D.; Schweitzer, M. Hydrazide oligonucleotides: New chemical modification for chip array attachment and conjugation. Nucleic Acids Res. 2002, 30, 4793–4802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Breslav, M.; Grimm, J.; Guan, K.; Huang, A.; Liu, F.; Maryanoff, C.A.; Palmer, D.; Patel, M.; Qian, Y.; et al. A new procedure for preparation of carboxylic acid hydrazides. J. Org. Chem. 2002, 67, 9471–9474. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kumar, R.; Kumar, R.; Devakumar, C. Development and assessment of green synthesis of hydrazides. Indian J. Chem. 2010, 49, 526–531. [Google Scholar]
- Aboul-Fadl, T.; Abdel-Aziz, H.A.; Kadi, A.; Bari, A.; Ahmad, P.; Al-Samani, T.; Ng, S.W. Microwave-assisted one-step synthesis of fenamic acid hydrazides from the corresponding acids. Molecules 2011, 16, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Marzouk, M.I. Microwave assisted reaction in synthesis of tetra- and hexa-cyclic spiro system. Bull. Chem. Commun. 2009, 41, 54–58. [Google Scholar]
- Bondock, S.; EL-Tarhoni, A.E.G.; Fadda, A.A. Utility of cyanoacetic acid hydrazide in heterocyclic synthesis. Arkivoc 2006, 2006, 113–156. [Google Scholar] [CrossRef]
- Marcos, M.; Dayse, N.M.; Clarissa, P.F.; Kelvis, L.; Nilo, Z.; Helio, G.B. Reaction of β-alkoxyvinyl halomethyl ketones with cyanoacetohydrazide. J. Braz. Chem. Soc. 2008, 19, 1361–1368. [Google Scholar]
- Şerban, G. 5-Arylamino-1,3,4-thiadiazol-2-yl acetic acid esters as intermediates for the synthesis of new bisheterocyclic compounds. Farmacia 2015, 63, 146–149. [Google Scholar]
- El-Emam, A.A.; Al-Deeb, O.A.; Al-Omar, M.; M. Lehmann, J. Synthesis, antimicrobial, and anti-HIV-1 activity of certain 5-(1-adamantyl)-2-substituted thio-1,3,4-oxadiazoles and 5-(1-adamntyl)-3-substituted aminomethyl-1,3,4-oxadiazoline-2-thiones. Bioorg. Med. Chem. 2004, 12, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Mohammed-Ali, M.A.; Majeed, N.N. Synthesis, characterization and study of antibacterial and antifungal activities of some 1,3,4-oxadiazole compounds. J. Chem. Pharm. Res. 2012, 4, 315–321. [Google Scholar]
- Kumar, V.; Singh, S. Synthesis of some new 1,3,4-oxadiazole derivatives and evaluation of their antibacterial effects. J. Chem. Pharm. Res. 2012, 4, 546–553. [Google Scholar]
- Sowjanya, C.; RamaBharathi, V.; Devi, G.K.; Rajitha, G. Synthesis and evaluation of some novel 3-[5-phenyl-1,3,4-oxadiazole- 2-yl]-2- (substituted styryl)-quinazoline-4(3H)-ones for antibacterial activity. J. Chem. Pharm. Res. 2011, 6, 212–216. [Google Scholar]
- Sumangalaa, V.; Poojary, B.; Chidananda, N.; Arulmoli, T.; Shenoy, S. Synthesis, characterization, antimicrobial and antioxidant activity of some disubstituted [1,3,4]-oxadiazoles carrying 4-(methylsulfonyl/sulfinyl)benzyl moieties. J. Chem. Pharm. Res. 2012, 4, 1661–1669. [Google Scholar]
- Kucukguzel, S.G.; Oruc, E.E.; Rollas, S.; Sahin, F.; Ozbek, A. Synthesis, characterisation and biological activity of novel 4-thiazolidinones, 1,3,4-oxadiazoles and some related compounds. Eur. J. Med. Chem. 2002, 37, 197–206. [Google Scholar] [CrossRef]
- Padmaja, A.; Rajasekhar, C.; Muralikrishna, A.; Padmavathi, V. Synthesis and antioxidant activity of disubstituted 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazoles. J. Chem. Pharm. Res. 2012, 4, 294–302. [Google Scholar]
- Kagthara, P.R.; Shah, N.S.; Doshi, R.K.; Parekh, H.H. Synthesis of 2,5-disubstituted 1,3,4-oxadiazoles as biologically active heterocycles. Ind. J. Chem. 1999, 38, 572–576. [Google Scholar]
- Akhter, M.; Husain, A.; Azad, B.; Ajmal, M. Aroylpropionic acid based 2,5-disubstituted-1,3,4-oxadiazoles: Synthesis and their anti-inflammatory and analgesic activities. Eur. J. Med. Chem. 2009, 44, 2372–2378. [Google Scholar] [CrossRef] [PubMed]
- Unangast, P.C.; Shrum, G.P.; Conner, D.T.; Dyer, C.D.; Schrier, D.J. Novel 1,2,4-oxadiazoles and 1,2,4-thiadiazoles as dual 5-lipoxygenase and cyclooxygenase inhibitors. J. Med. Chem. 1992, 35, 3691–3698. [Google Scholar]
- Sing, A.K.; Parthsarthy, R.; Lohani, M. Synthesis, characterization and anti-inflammatory activity of some 1, 3,4-oxadiazole derivatives. J. Chem. Pharm. Res. 2012, 4, 779–782. [Google Scholar]
- Khan, M.S.; Khan, R.M.; Drabu, S. Anticonvulsant and antibacterial activity of some new 1,3,4-oxadiazole derivatives. Ind. J. Heterocycl. Chem. 2001, 11, 119–122. [Google Scholar]
- O’Neal, J.B.; Rosen, H.; Russell, P.B.; Adams, A.C.; Blumenthal, A. Potential hypoglycemic agents: 1,3,4-Oxadiazoles and related compounds. J. Med. Chem. 1962, 5, 617–626. [Google Scholar] [CrossRef]
- Maslat, A.O.; Abussaud, M.; Tashtoush, H.; Al-Talib, M. Synthesis, antibacterial, antifungal and genotoxic activity of bis-1,3,4-oxadiazole derivatives. Pol. J. Pharmacol. 2002, 54, 55–59. [Google Scholar] [PubMed]
- Farghaly, A.A.; Bekhit, A.A.; Park, J.Y. Design and synthesis of some oxadiazolyl, thiadiazolyl, thiazolidinyl, and thiazolyl derivatives of 1H-pyrazole as anti-inflammatory antimicrobial agents. Arch. Pharm. 2000, 333, 53–57. [Google Scholar] [CrossRef]
- Savarino, A. A historical sketch of the discovery and development of HIV-1 integrase inhibitors. Expert Opin. Investig. Drugs 2006, 15, 1507–1522. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Growcott, J.W. Zibotentan. Drugs Future 2009, 34, 624–633. [Google Scholar] [CrossRef]
- Bondock, S.; Adel, S.; Etman, H.A.; Badria, F.A. Synthesis and antitumor evaluation of some new 1,3,4-oxadiazole-based heterocycles. Eur. J. Med. Chem. 2012, 48, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Koparır, M.; Çetin, A.; Cansız, A. 5-Furan-2yl[1,3,4]oxadiazole-2-thiol, 5-furan-2yl-4H[1,2,4]triazole-3-thiol and their thiol-thione tautomerism. Molecules 2005, 10, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Kumar, S. Synthesis and evaluation of anti-inflammatory, analgesic ulcerogenic and lipid peroxidation properties of ibuprofen derivatives. Acta Pharm. 2007, 57, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Mickevičius, V.; Vaickelionienė, R.; Sapijanskaitė, B. Synthesis of substituted 1,3,4-oxadiazolederivatives. Chem. Heterocycl. Comp. 2009, 45, 215–218. [Google Scholar] [CrossRef]
- Martin, P.J.; Bruce, D.W. Hydrogen-bonded oxadiazole mesogens. Liq. Cryst. 2007, 34, 767–774. [Google Scholar] [CrossRef]
- Hernández-Ainsa, S.; Barberá, J.; Marcos, M.; Serrano, J.L. Liquid crystalline ionic dendrimers containing luminescent oxadiazole moieties. Macromolecules 2012, 45, 1006–1015. [Google Scholar] [CrossRef]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in medicinal chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, A.; Mao, X.; Sun, X.; Gong, X. Silica-supported dichlorophosphate: A recoverable cyclodehydrant for the eco-friendly synthesis of 2,5-disubstituted 1,3,4-oxadiazoles under solvent-free and microwave irradiation conditions. J. Braz. Chem. Soc. 2008, 19, 1622–1626. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Rakesh, B.A.; Krishna, P.R. Zirconium(IV) chloride mediated cyclodehydration of 1,2-diacylhydrazines: A convenient synthesis of 2,5-diaryl 1,3,4-oxadiazoles. Synth. Commun. 2004, 34, 2387–2391. [Google Scholar] [CrossRef]
- Huisgen, R.; Szeimies, G.; Moebius, L. 1,3-Dipolar Cycloadditions. XXXII. Kinetics of the Addition of Organic Azides to Carbon-carbon Multiple Bonds. Chem. Ber. 1967, 100, 2494–2507. [Google Scholar] [CrossRef]
- Baranov, A.B.; Tsypin, V.G.; Malin, A.S.; Laskin, B.M. Synthesis of 2-chloromethyl-5-aryl-, 2-chloromethyl-5-(5-methyl-2-furyl)-, and 2-chloromethyl-5-(1,5-dimethyl-2-pyrrolyl)-1,3,4-oxadiazoles from tetrazole derivatives. Russ. J. Appl. Chem. 2005, 78, 773–775. [Google Scholar] [CrossRef]
- Efimova, Y.A.; Artamonova, T.V.; Koldobskii, G.I. Tetrazoles: LIII.* Microwave-activated acylation of 5-substituted tetrazoles. Russ. J. Org. Chem. 2008, 44, 1345–1347. [Google Scholar] [CrossRef]
- Salem, M.A.; Marzouk, M.I.; Mahmoud, N.F. Synthesis of various fused pyrimidine rings and their pharmacological and antimicrobial evaluation. J. Serbian Chem. Soc. 2014, 79, 1059–1073. [Google Scholar]
- Salem, M.A.I.; Marzouk, M.I.; Salem, M.S.; Alshibani, G.A. One-pot Synthesis of 1,2,3,4-tetrahydropyrimidin-2(1H)-thione derivatives and their biological activity. J. Heterocycl. Chem. 2015. [Google Scholar] [CrossRef]
- Salem, M.A.I.; Marzouk, M.I.; Mashaly, H.M. Synthesis of pharmacological dyes and their application on synthetic fabrics. Color. Technol. 2015, 131, 288–297. [Google Scholar] [CrossRef]
- Salem, M.A.I.; Marzouk, M.I.; Gaffer, H.E. Synthesis of 4-hydroxy coumarin dyes and their applications. Pigment Resin Technol. 2016, 45. in press. [Google Scholar]
- Marzouk, M.I.; Shaker, S.A.; Abdel-Hafiz, A.A.; El-Baghdady, K.Z. Design and synthesis of new phthalazinone derivatives containing benzyl moiety with anticipated antitumor activity. Biol. Pharm. Bull. 2016, 39. in press. [Google Scholar]
- El-Hashash, M.A.; Guirguis, D.B.; Kadhim, M.A. Synthesis and reaction of 2-(1-oxo-4(1,2,3,4-tetrahydronaphthalen-2-yl)phthalazin-1(2H)-yl)acetohydrazide. J. Am. Sc. 2013, 9, 180–185. [Google Scholar]
- Mohammed, A.A.; Abdelwahed, R.S.; Magdy, M.Y. Synthesis of novel triazoles, tetrazine, thiadiazoles and their biological activities. Molecules 2015, 20, 2591–2610. [Google Scholar]
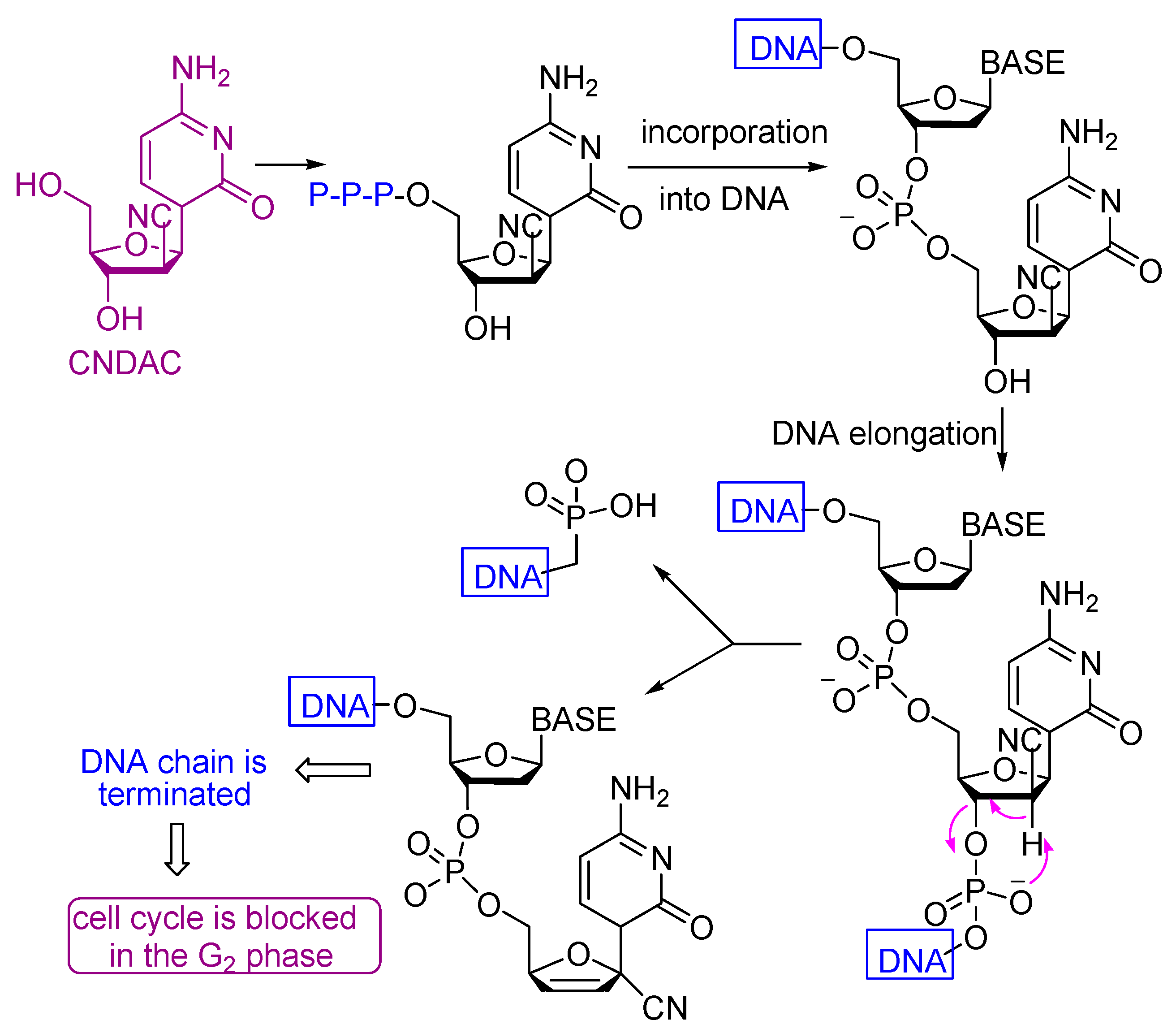
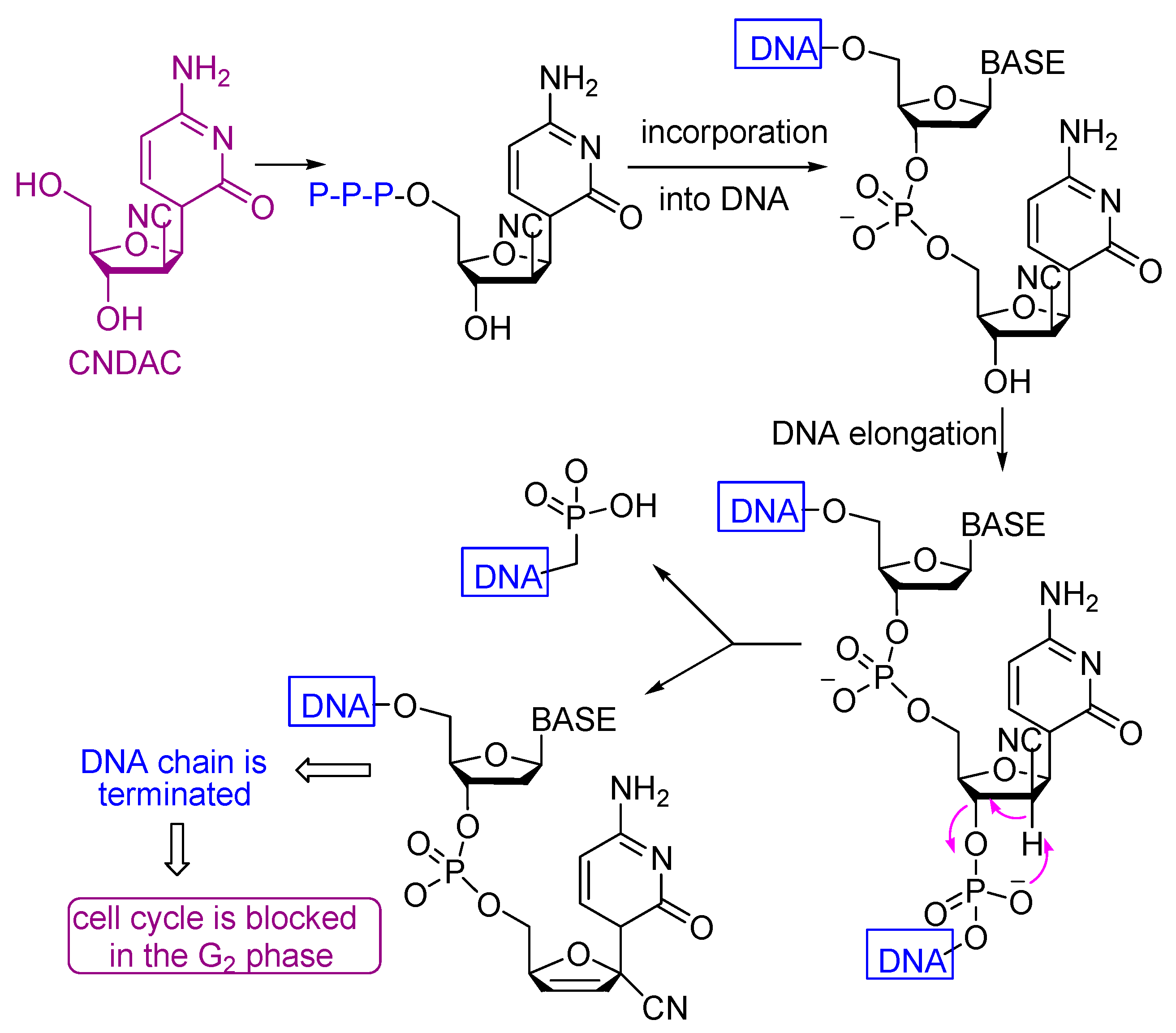
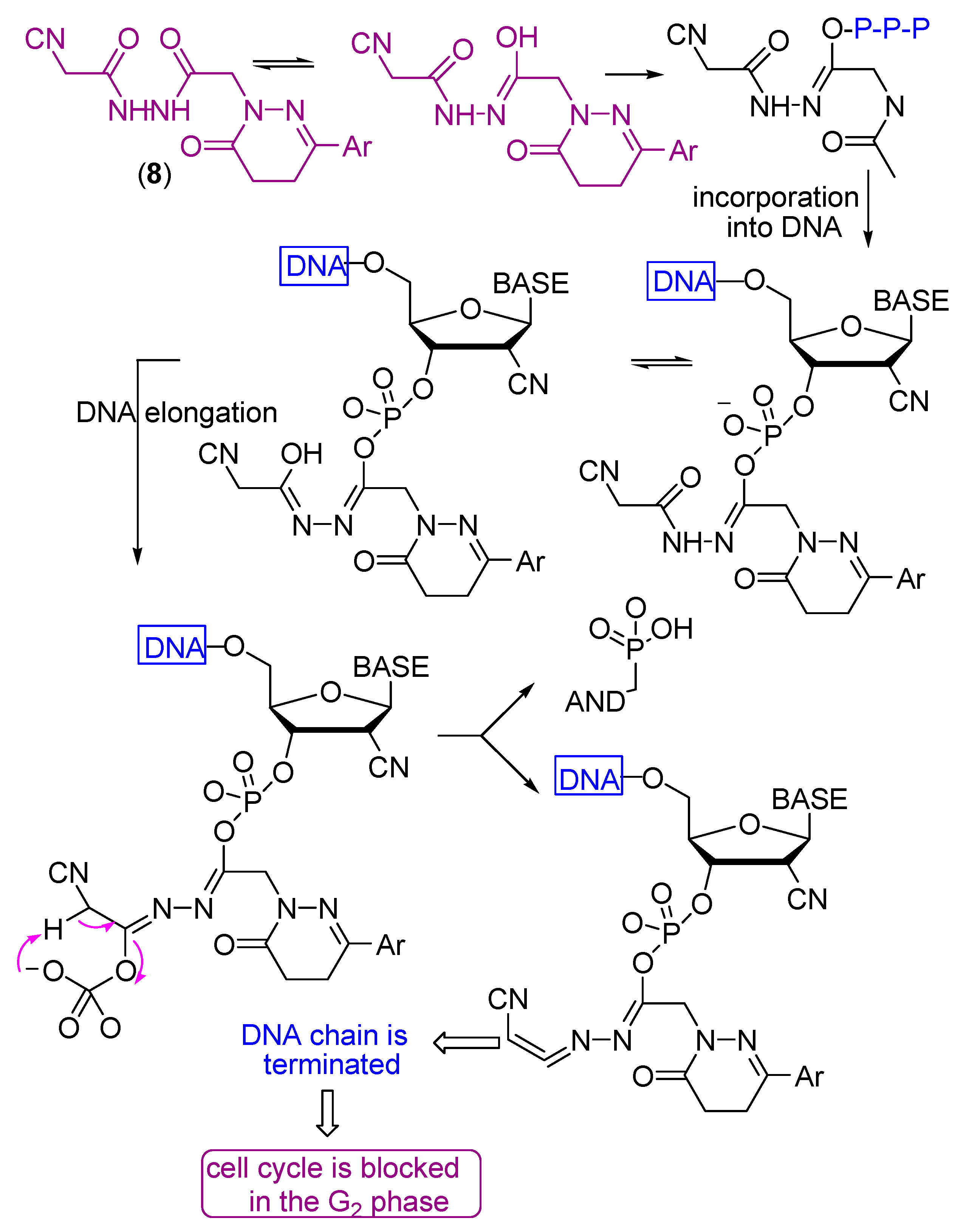
- Liu, X.; Guo, Y.; Li, Y.; Jiang, Y.; Chubb, S.; Azuma, A.; Huang, P.; Matsuda, A.; Hittelman, W.; Plunkett, W. Molecular basis for G2 arrest induced by 2′-C-cyano-2′-deoxy-1-β-d-arabino-pentofuran osylcytosine and consequences of checkpoint abrogation. Cancer Res. 2005, 65, 6874–6881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Avendaño, C.; Menendez, J.C. Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 73–74. [Google Scholar]
- Gutteridge, J.M.; Rowley, D.A.; Halliwell, B. Superoxide-dependent formation of hydroxyl radicals in the presence of iron salts. Detection of “free” iron in biological systems by using bleomycin-dependent degradation of DNA. Biochem. J. 1981, 199, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Mauceri, H.J.; Hanna, N.N.; Beckett, M.A.; Gorski, D.H.; Staba, M.; Stellato, K.A.; Bigelow, K.; Heimann, R.; Gately, S.; Dhanabal, M.; et al. Combined effects of angiostatin and ionizing radiation in antitumour therapy. Nature 1998, 394, 287–291. [Google Scholar] [PubMed]
- Lissi, E.A.; Modak, B.; Torres, R.; Escobar, J.; Urzua, A. Total antioxidant potential of resinous exudates from Heliotropium species, and a comparison of the ABTS and DPPH methods. Free Radic. Res. 1999, 30, 471–477. [Google Scholar] [CrossRef] [PubMed]
- El-Gazar, A.B.A.; Youssef, M.M.; Youssef, A.M.S.; Abu-Hashem, A.A.; Badria, F.A. Design and synthesis of azolopyrimidoquinolines, pyrimidoquinazolines as anti-oxidant, anti-inflammatory and analgesic activities. Eur. J. Med. Chem. 2009, 44, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Aeschlach, R.; Loliger, J.; Scott, B.C.; Murcia, A.; Butler, J.; Halliwell, B.; Aruoma, O.I. Antioxidant actions of thymol, carvacrol, 6-gingerol, zingerone and hydroxytyrosol. Food Chem. Toxicol. 1994, 32, 31–36. [Google Scholar] [CrossRef]
- Bakr, F.A.; Abdel-aziz, S.E.; Farid, A.B. Synthesis of new 2-naphthyl ethers and their protective activities against DNA damage induced by Bleomycin–Iron. Chem. Pharm. Bull. 2009, 57, 1348–1351. [Google Scholar]
- Badria, F.A.; Ameen, M.; Akl, M.R. Evaluation of cytotoxic compounds from Calligonum comosum L. growing in Egypt. Z. Naturforsch. C 2007, 62, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaker, S.A.; Marzouk, M.I. Utilization of Cyanoacetohydrazide and Oxadiazolyl Acetonitrile in the Synthesis of Some New Cytotoxic Heterocyclic Compounds. Molecules 2016, 21, 155. https://doi.org/10.3390/molecules21020155
Shaker SA, Marzouk MI. Utilization of Cyanoacetohydrazide and Oxadiazolyl Acetonitrile in the Synthesis of Some New Cytotoxic Heterocyclic Compounds. Molecules. 2016; 21(2):155. https://doi.org/10.3390/molecules21020155
Chicago/Turabian StyleShaker, Soheir A., and Magda I. Marzouk. 2016. "Utilization of Cyanoacetohydrazide and Oxadiazolyl Acetonitrile in the Synthesis of Some New Cytotoxic Heterocyclic Compounds" Molecules 21, no. 2: 155. https://doi.org/10.3390/molecules21020155
APA StyleShaker, S. A., & Marzouk, M. I. (2016). Utilization of Cyanoacetohydrazide and Oxadiazolyl Acetonitrile in the Synthesis of Some New Cytotoxic Heterocyclic Compounds. Molecules, 21(2), 155. https://doi.org/10.3390/molecules21020155