
Stereoselective Fluorescence Quenching in the Electron Transfer Photooxidation of Nucleobase-Related Azetidines by Cyanoaromatics




Abstract
:1. Introduction
2. Results
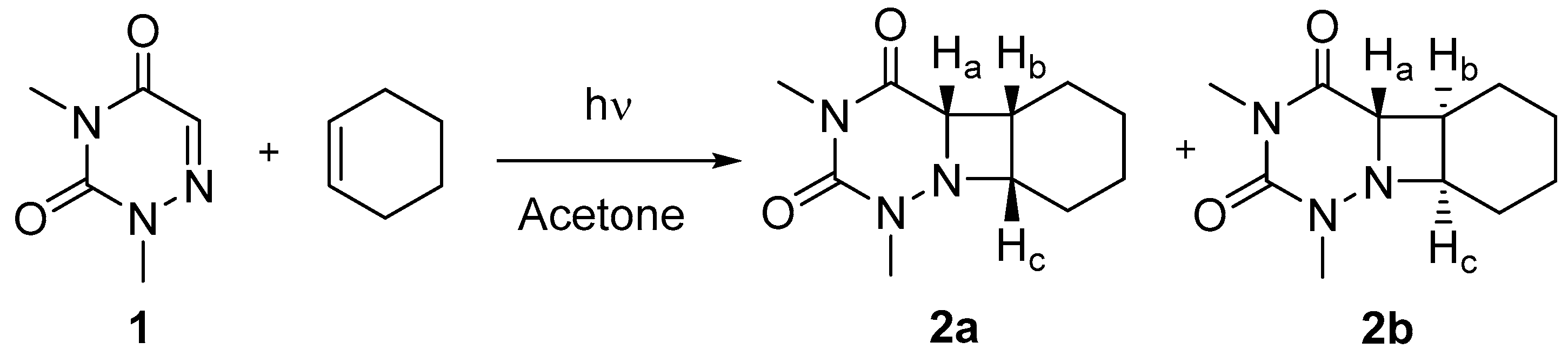
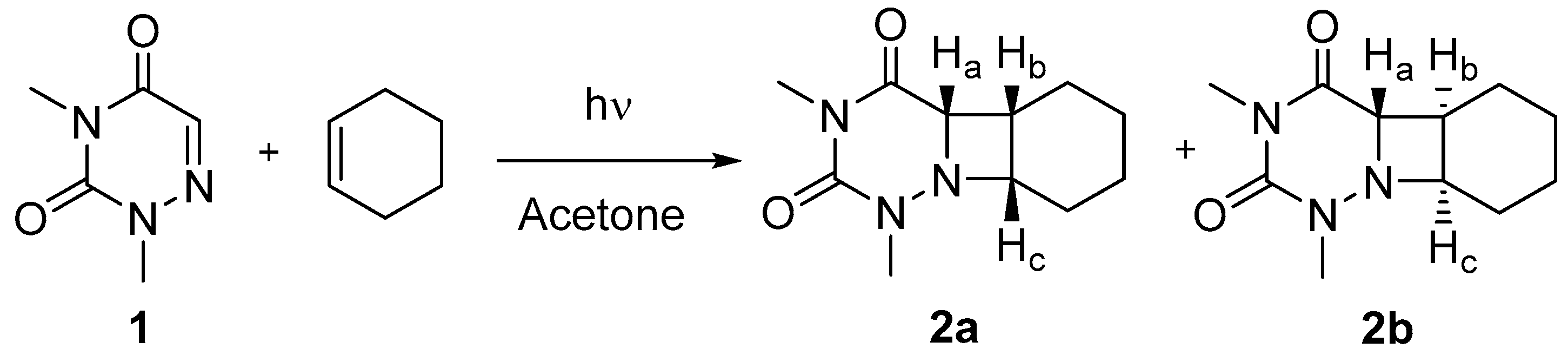
2.1. Synthesis
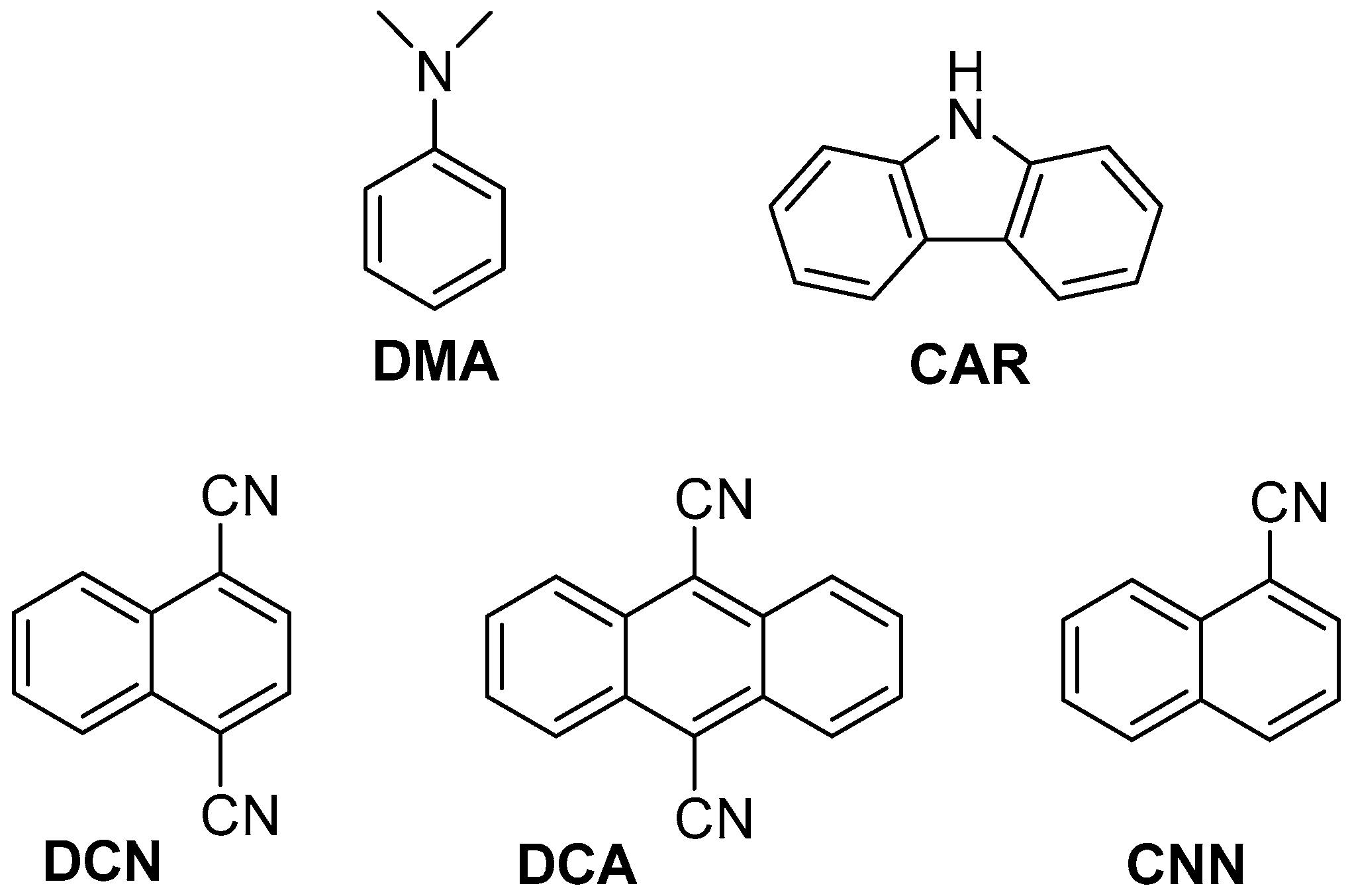
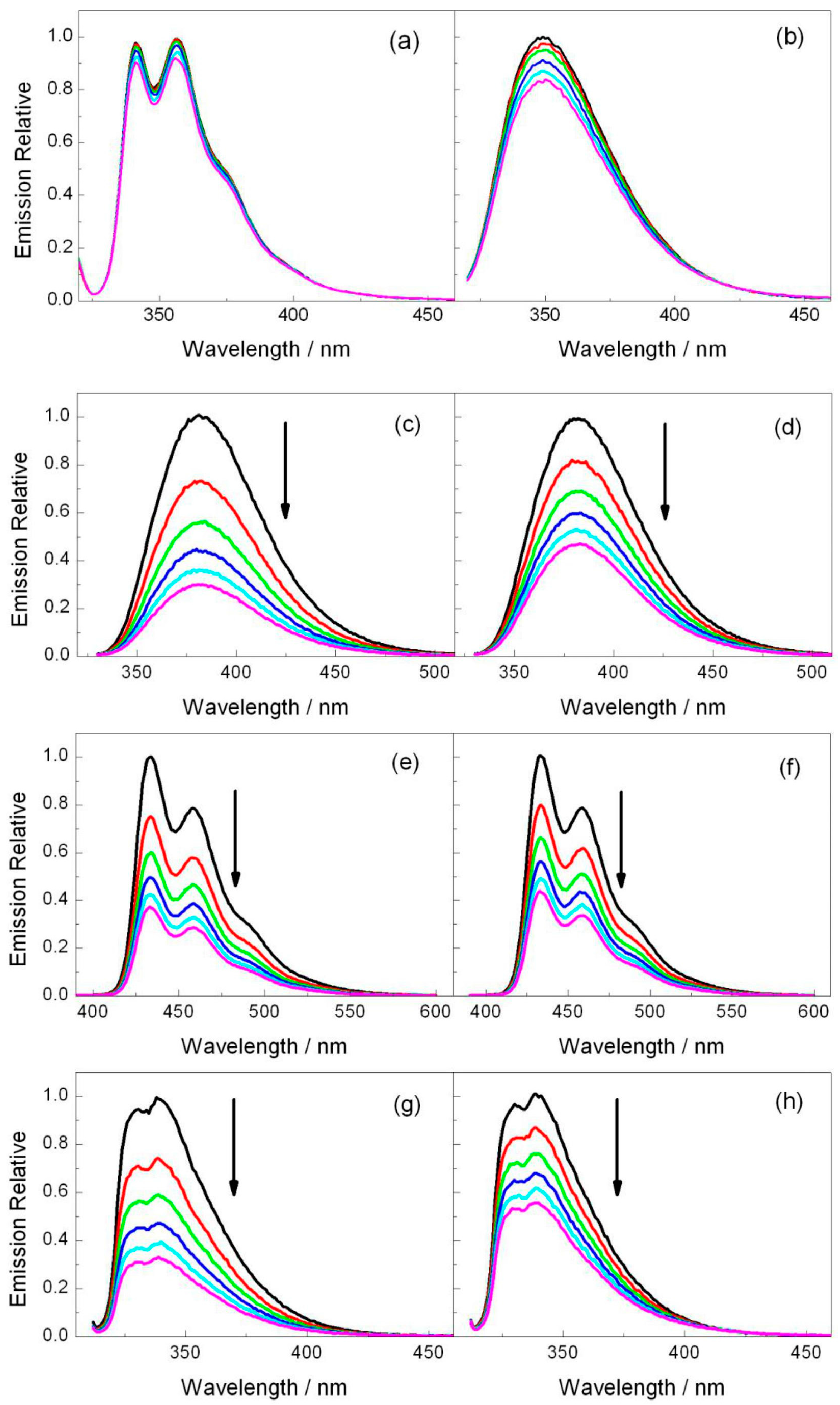
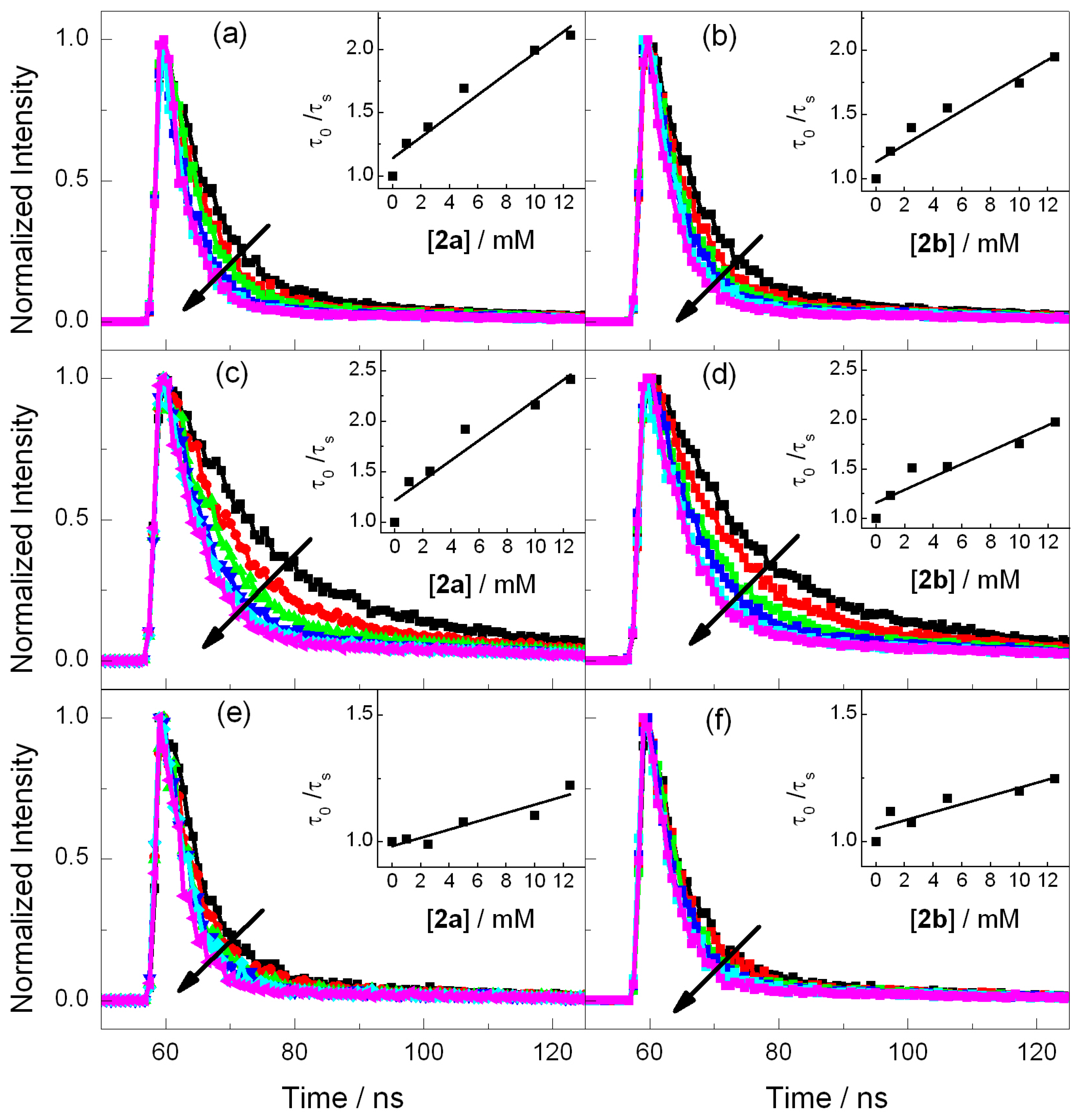
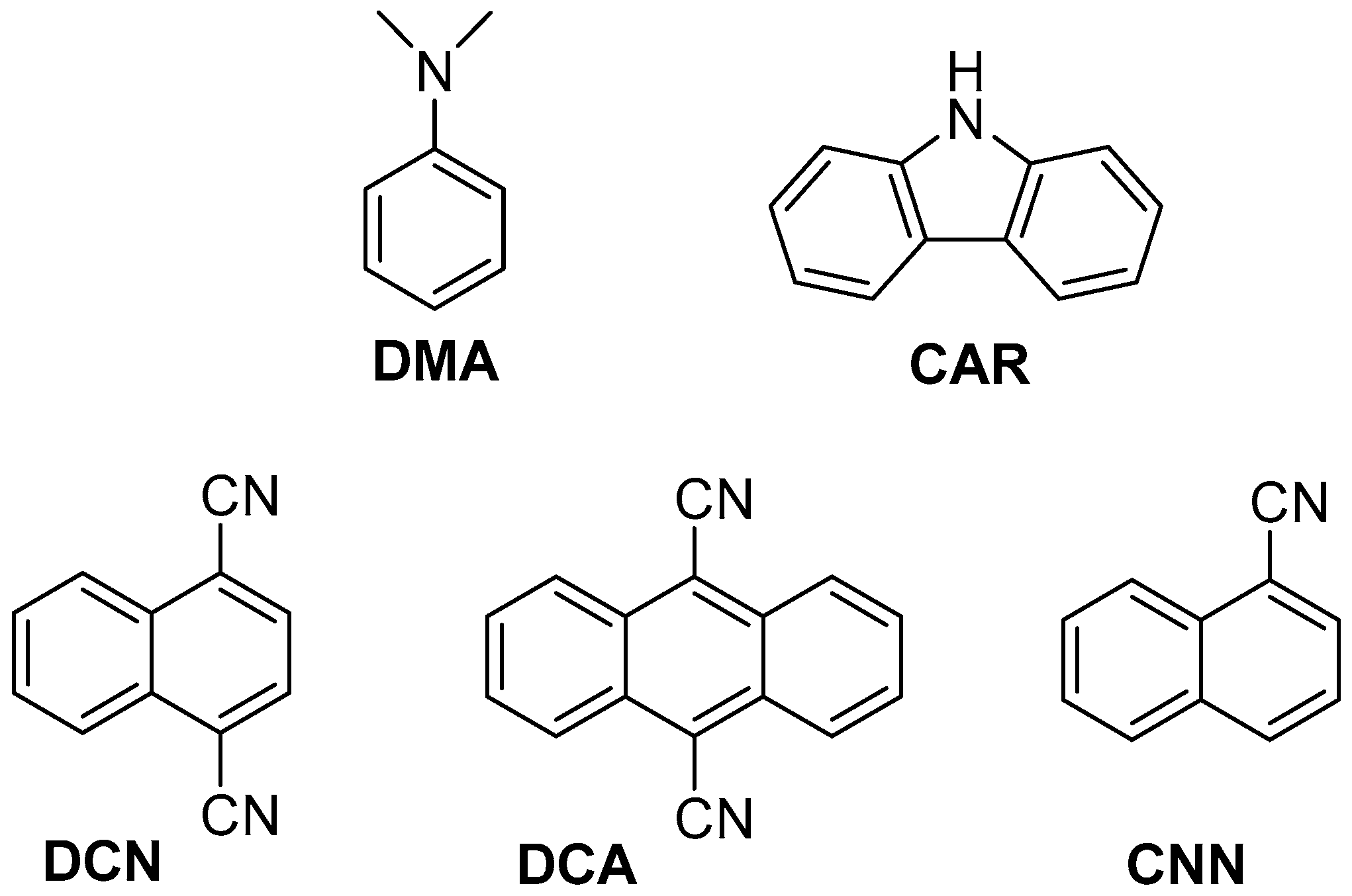
2.2. Fluorescence Experiments
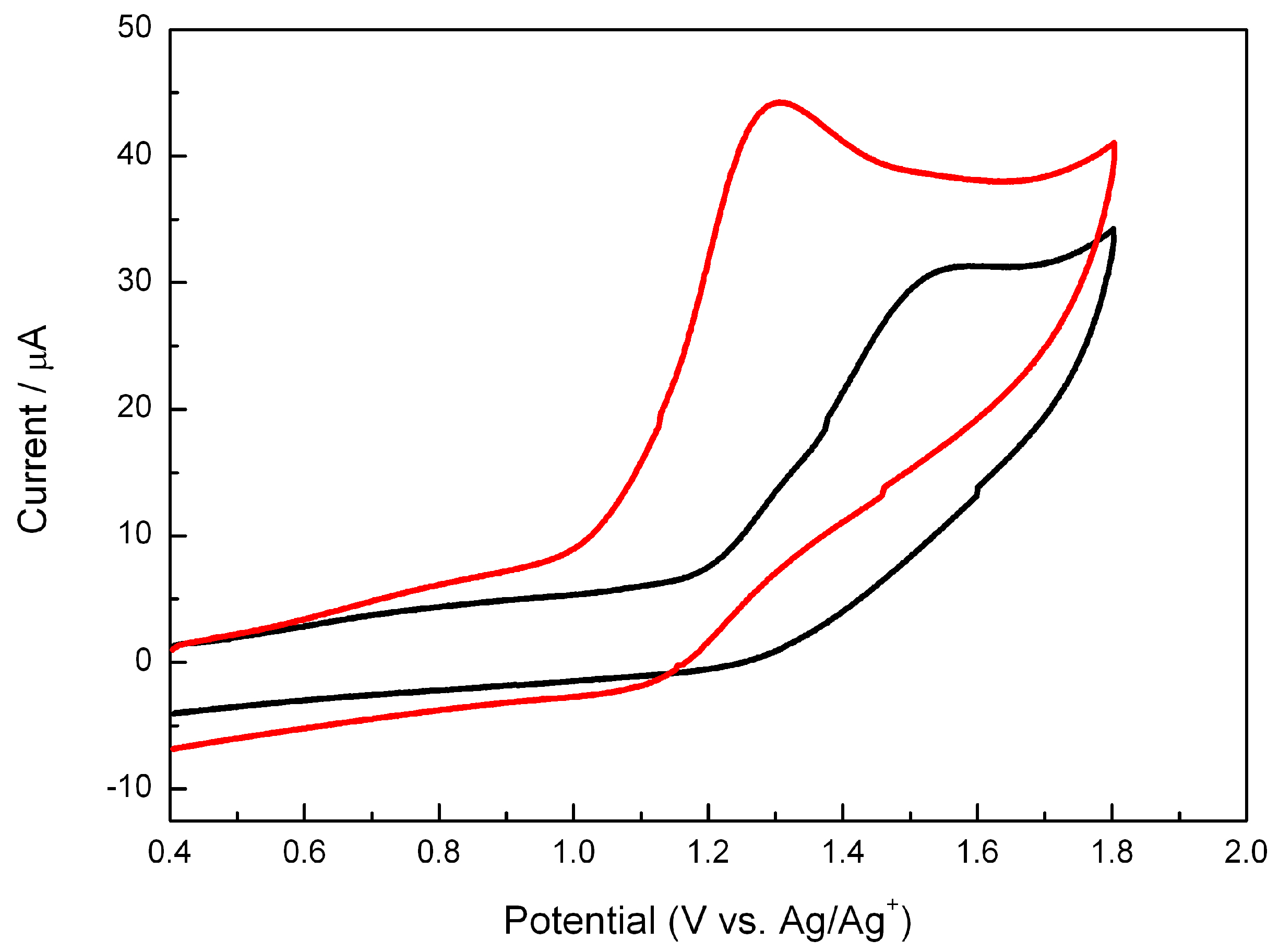
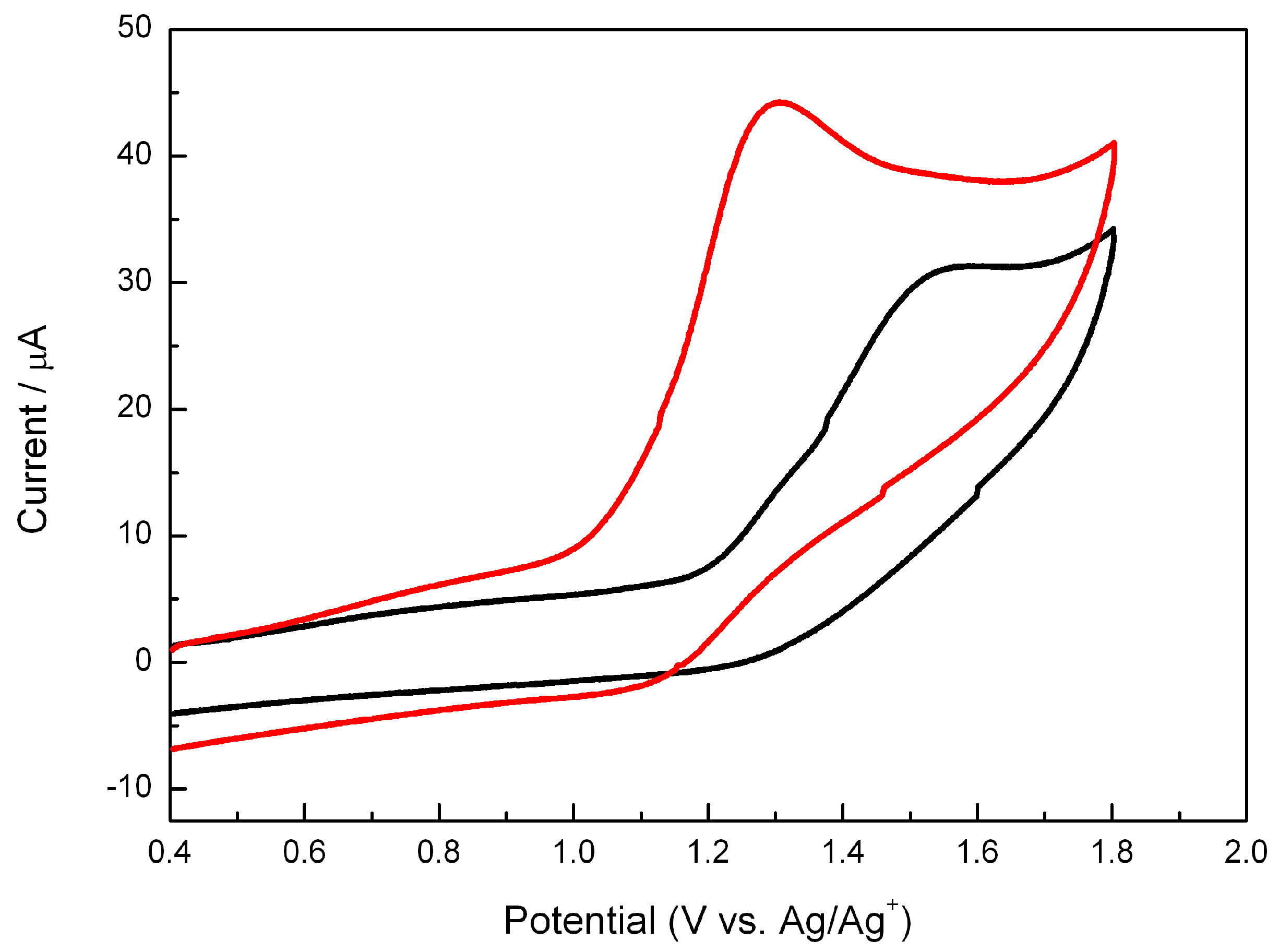
2.3. Electrochemistry
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Synthesis
3.4. Fluorescence Quenching
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. DNA charge transport: From chemical principles to the cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Ma, B.; Xin, N.; Guo, X. Carbon electrode–molecule junctions: A reliable platform for molecular electronics. Acc. Chem. Res. 2015, 48, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Beratan, D.N.; Liu, C.; Migliore, A.; Polizzi, N.F.; Skourtis, S.S.; Zhang, P.; Zhang, Y. Charge transfer in dynamical biosystems, or the treachery of (static) images. Acc. Chem. Res. 2015, 48, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Majima, T. Hole transfer kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 2003, 103, 2203–2238. [Google Scholar] [CrossRef] [PubMed]
- Kanvah, S.; Joseph, J.; Schuster, G.B.; Barnett, R.N.; Cleveland, C.L.; Landman, U. Oxidation of DNA: Damage to nucleobases. Acc. Chem. Res. 2010, 43, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.O.; Barton, J.K. Electron transfer between bases in double helical DNA. Science 1999, 283, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Breeger, S.; von Meltzer, M.; Hennecke, U.; Carell, T. Investigation of the pathways of excess electron transfer in DNA with flavin-donor and oxetane-acceptor modified DNA hairpins. Chem. Eur. J. 2006, 12, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Boussicault, F.; Robert, M. Electron transfer in DNA and in DNA-related biological processes. Electrochemical insights. Chem. Rev. 2008, 108, 2622–2645. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M. The Nobel Prize in Chemistry 2015—Advanced Information. Availabe online: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2015/advanced.html (accessed on 2 December 2016).
- Brettel, K.; Byrdin, M. Reaction mechanisms of DNA photolyase. Curr. Opin. Struct. Biol. 2010, 20, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Dandliker, P.J.; Holmlin, R.E.; Barton, J.K. Oxidative thymine dimer repair in the DNA helix. Science 1997, 275, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Vicic, D.A.; Odom, D.T.; Núñez, M.E.; Gianolio, D.A.; McLaughlin, L.W.; Barton, J.K. Oxidative repair of a thymine dimer in DNA from a distance by a covalently linked organic intercalator. J. Am. Chem. Soc. 2000, 122, 8603–8611. [Google Scholar] [CrossRef]
- Hartman, T.; Cibulka, R. Photocatalytic systems with flavinium salts: From photolyase models to synthetic tool for cyclobutane ring opening. Org. Lett. 2016, 18, 3710–3713. [Google Scholar] [CrossRef] [PubMed]
- Scannell, M.P.; Fenick, D.J.; Yeh, S.-R.; Falvey, D.E. Model studies of DNA photorepair: Reduction potentials of thymine and cytosine cyclobutane dimers measured by fluorescence quenching. J. Am. Chem. Soc. 1997, 119, 1971–1977. [Google Scholar] [CrossRef]
- Pérez-Ruiz, R.; Jiménez, M.C.; Miranda, M.A. Hetero-cycloreversions mediated by photoinduced electron transfer. Acc. Chem. Res. 2014, 47, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Boussicault, F.; Robert, M. Electrochemical approach to the repair of oxetanes mimicking DNA (6-4) photoproducts. J. Phys. Chem. B 2006, 110, 21987–21993. [Google Scholar] [CrossRef] [PubMed]
- Prakash, G.; Falvey, D.E. Model studies of the (6-4) photoproduct DNA photolyase: Synthesis and photosensitized splitting of a thymine-5,6-oxetane. J. Am. Chem. Soc. 1995, 117, 11375–11376. [Google Scholar] [CrossRef]
- Friedel, M.G.; Cichon, M.K.; Carell, T. Model compounds for (6-4) photolyases: A comparative flavin induced cleavage study of oxetanes and thietanes. Org. Biomol. Chem. 2005, 3, 1937–1941. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Timiraos, A.B.; Lhiaubet-Vallet, V.; Miranda, M.A. Repair of a dimeric azetidine related to the thymine–cytosine (6-4) photoproduct by electron transfer photoreduction. Angew. Chem. Int. Ed. 2016, 55, 6037–6040. [Google Scholar] [CrossRef] [PubMed]
- Andreu, I.; Delgado, J.; Espinós, A.; Pérez-Ruiz, R.; Jiménez, M.C.; Miranda, M.A. Cycloreversion of azetidines via oxidative electron transfer. Steady-state and time-resolved studies. Org. Lett. 2008, 10, 5207–5210. [Google Scholar] [CrossRef] [PubMed]
- Pac, C.; Ohtsuki, T.; Shiota, Y.; Yanagida, S.; Sakurai, H. Photochemical reactions of aromatic compounds. XLII. Photosensitized reactions of some selected diarylcyclobutanes by aromatic nitriles and chloranil. Implications of charge-transfer contributions on exciplex reactivities. Bull. Chem. Soc. Jpn. 1986, 59, 1133–1139. [Google Scholar] [CrossRef]
- Swenton, J.S.; Hyatt, J.A. Photosensitized cycloadditions to 1,3-dimethyl-6-azauracil and 1,3-dimethyl-6-azathymine. Imine linkage unusually reactive toward photocycloaddition. J. Am. Chem. Soc. 1974, 96, 4879–4885. [Google Scholar] [CrossRef] [PubMed]
- Stern, O.; Volmer, M. Über die abklingungszeit der fluoreszenz. Physik. Z. 1919, 20, 183–188. [Google Scholar]
- Scannell, M.P.; Prakash, G.; Falvey, D.E. Photoinduced electron transfer to pyrimidines and 5,6-dihydropyrimidine derivatives: Reduction potentials determined by fluorescence quenching kinetics. J. Phys. Chem. A 1997, 101, 4332–4337. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25 degrees C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2a or 2b are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PS | /ns | E*/V | ES (kJ·mol−1) | kq (2a)/109 M−1·s−1 | kq (2b)/109 M−1·s−1 |
|---|---|---|---|---|---|
| DMA | 1.5 | −3.0 (PS+●/PS*) 1 | 363 | N.D. 3 | N.D. 3 |
| CAR | 7.4 | −2.5 (PS+●/PS*) 1 | 338 | N.D. 3 | N.D. 3 |
| DCN | 7.5 | 2.4 (PS*/PS●−) 2 | 362 | 10 | 7.7 |
| DCA | 12.5 | 1.8 (PS*/PS●−) 2 | 276 | 6.4 | 4.6 |
| CNN | 5.5 | 1.4 (PS*/PS●−) 2 | 374 | 3.2 | 2.6 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraga-Timiraos, A.B.; Rodríguez-Muñiz, G.M.; Peiro-Penalba, V.; Miranda, M.A.; Lhiaubet-Vallet, V. Stereoselective Fluorescence Quenching in the Electron Transfer Photooxidation of Nucleobase-Related Azetidines by Cyanoaromatics. Molecules 2016, 21, 1683. https://doi.org/10.3390/molecules21121683
Fraga-Timiraos AB, Rodríguez-Muñiz GM, Peiro-Penalba V, Miranda MA, Lhiaubet-Vallet V. Stereoselective Fluorescence Quenching in the Electron Transfer Photooxidation of Nucleobase-Related Azetidines by Cyanoaromatics. Molecules. 2016; 21(12):1683. https://doi.org/10.3390/molecules21121683
Chicago/Turabian StyleFraga-Timiraos, Ana B., Gemma M. Rodríguez-Muñiz, Vicente Peiro-Penalba, Miguel A. Miranda, and Virginie Lhiaubet-Vallet. 2016. "Stereoselective Fluorescence Quenching in the Electron Transfer Photooxidation of Nucleobase-Related Azetidines by Cyanoaromatics" Molecules 21, no. 12: 1683. https://doi.org/10.3390/molecules21121683
APA StyleFraga-Timiraos, A. B., Rodríguez-Muñiz, G. M., Peiro-Penalba, V., Miranda, M. A., & Lhiaubet-Vallet, V. (2016). Stereoselective Fluorescence Quenching in the Electron Transfer Photooxidation of Nucleobase-Related Azetidines by Cyanoaromatics. Molecules, 21(12), 1683. https://doi.org/10.3390/molecules21121683