
Drug Release by Direct Jump from Poly(ethylene-glycol-b-ε-caprolactone) Nano-Vector to Cell Membrane

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
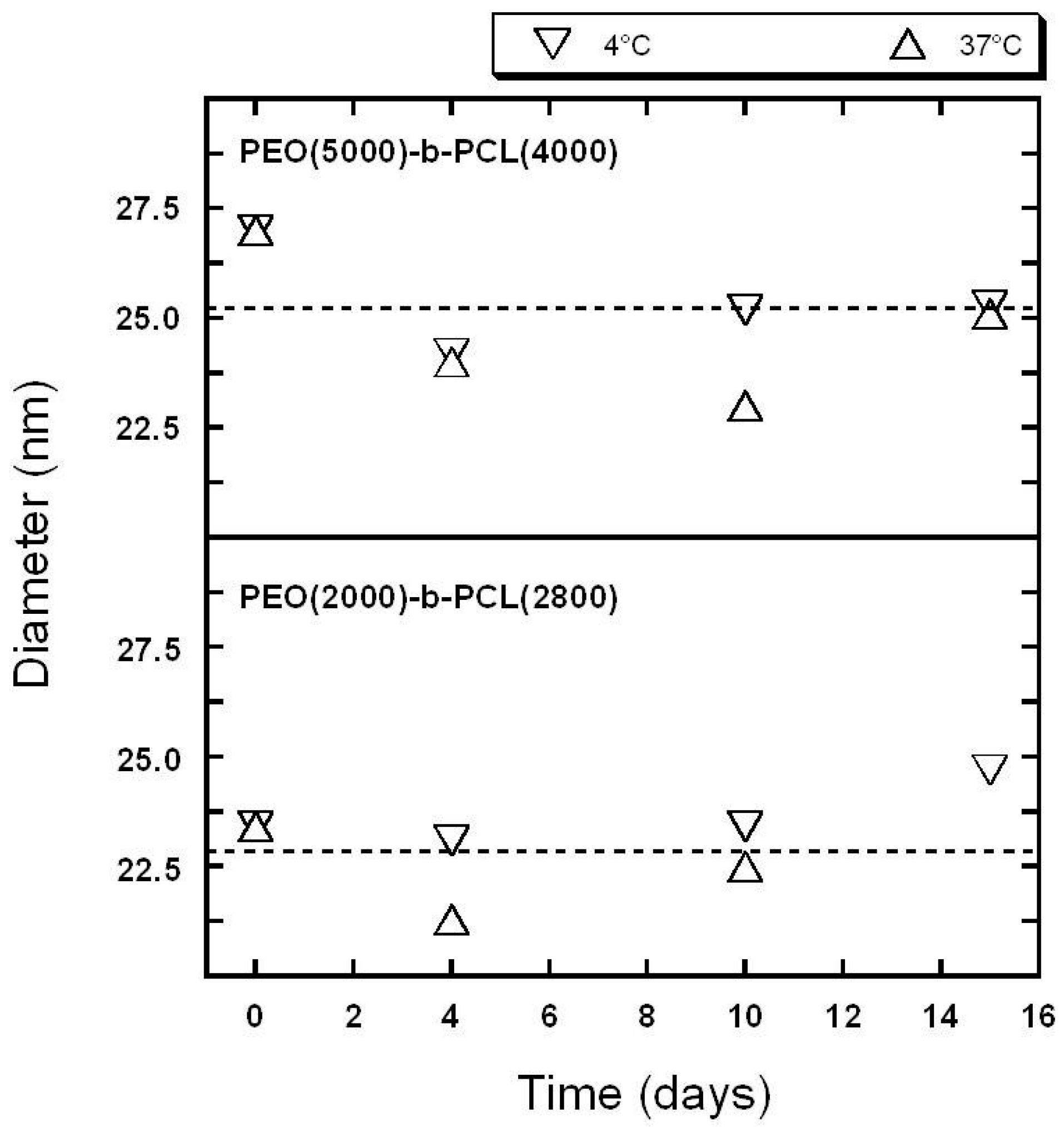
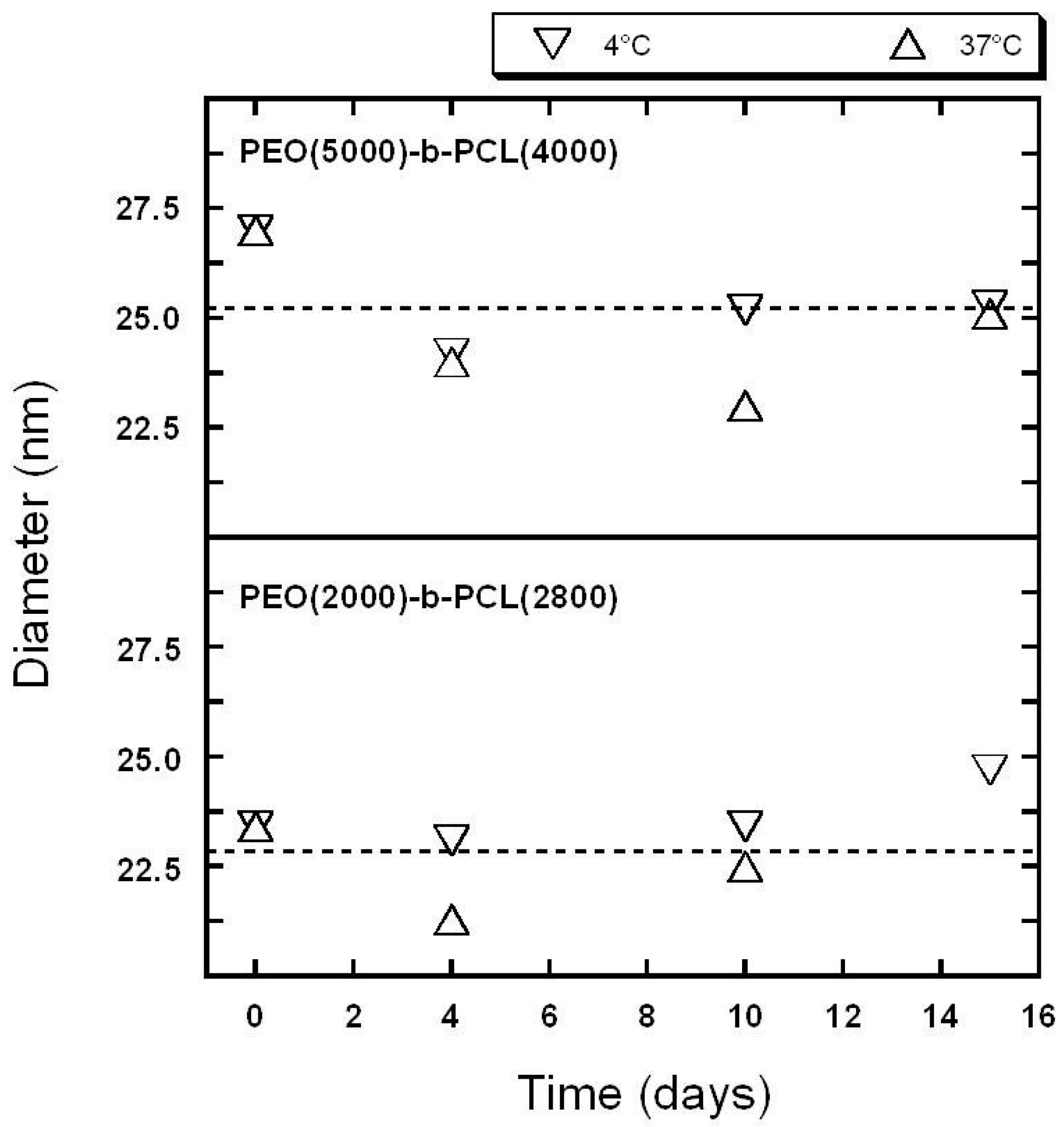
2.1. Characterization of Copolymer Micelles
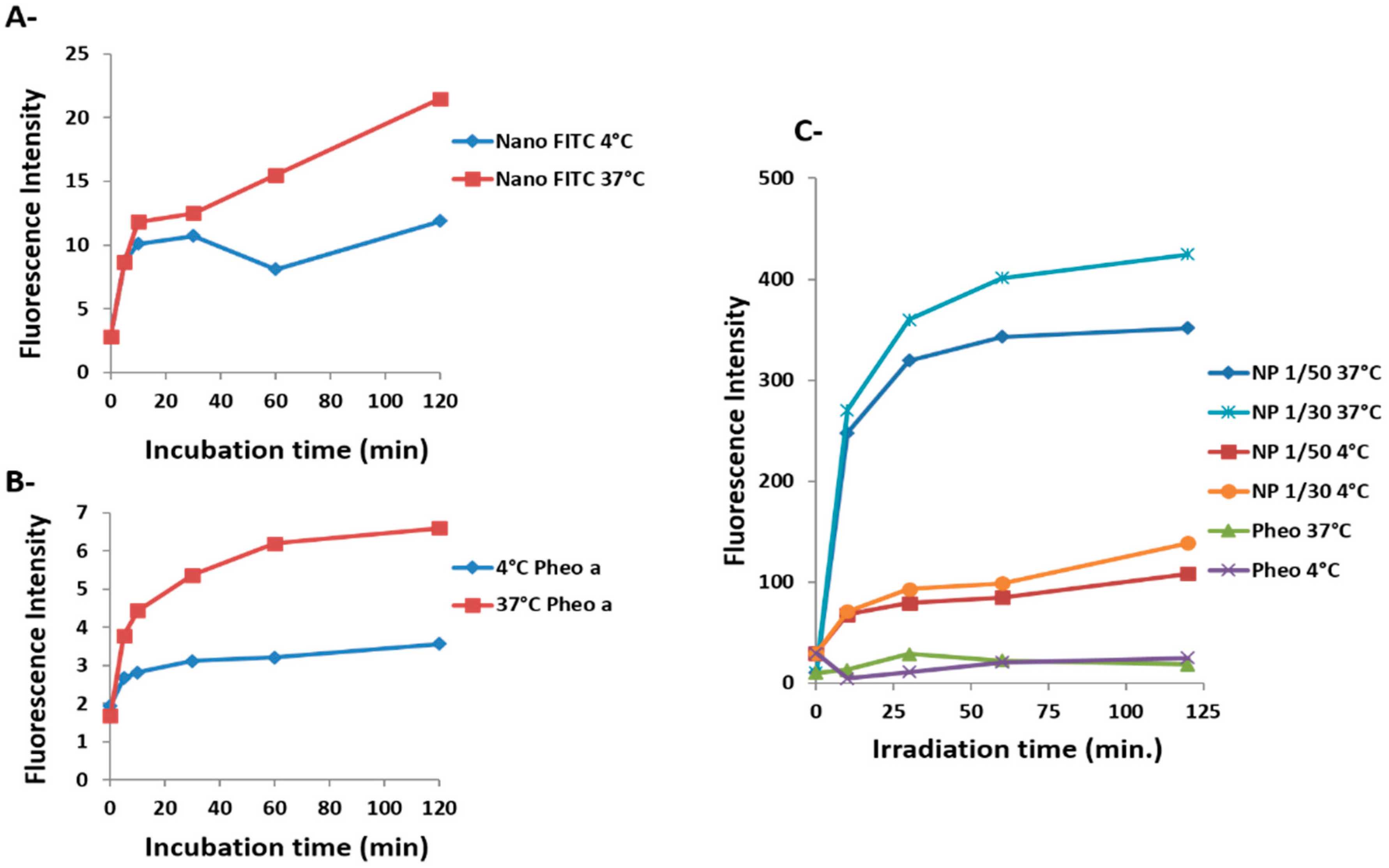
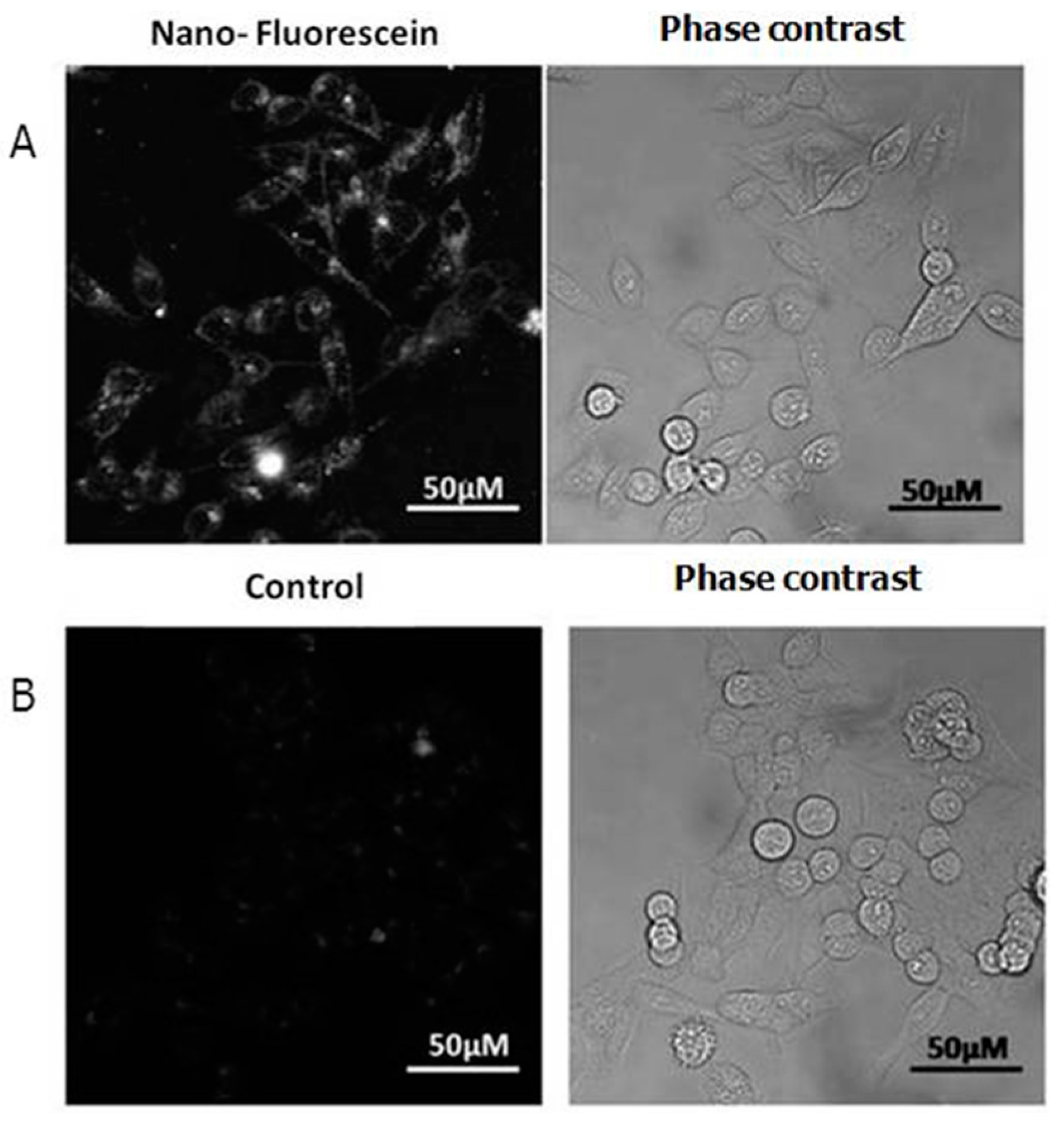
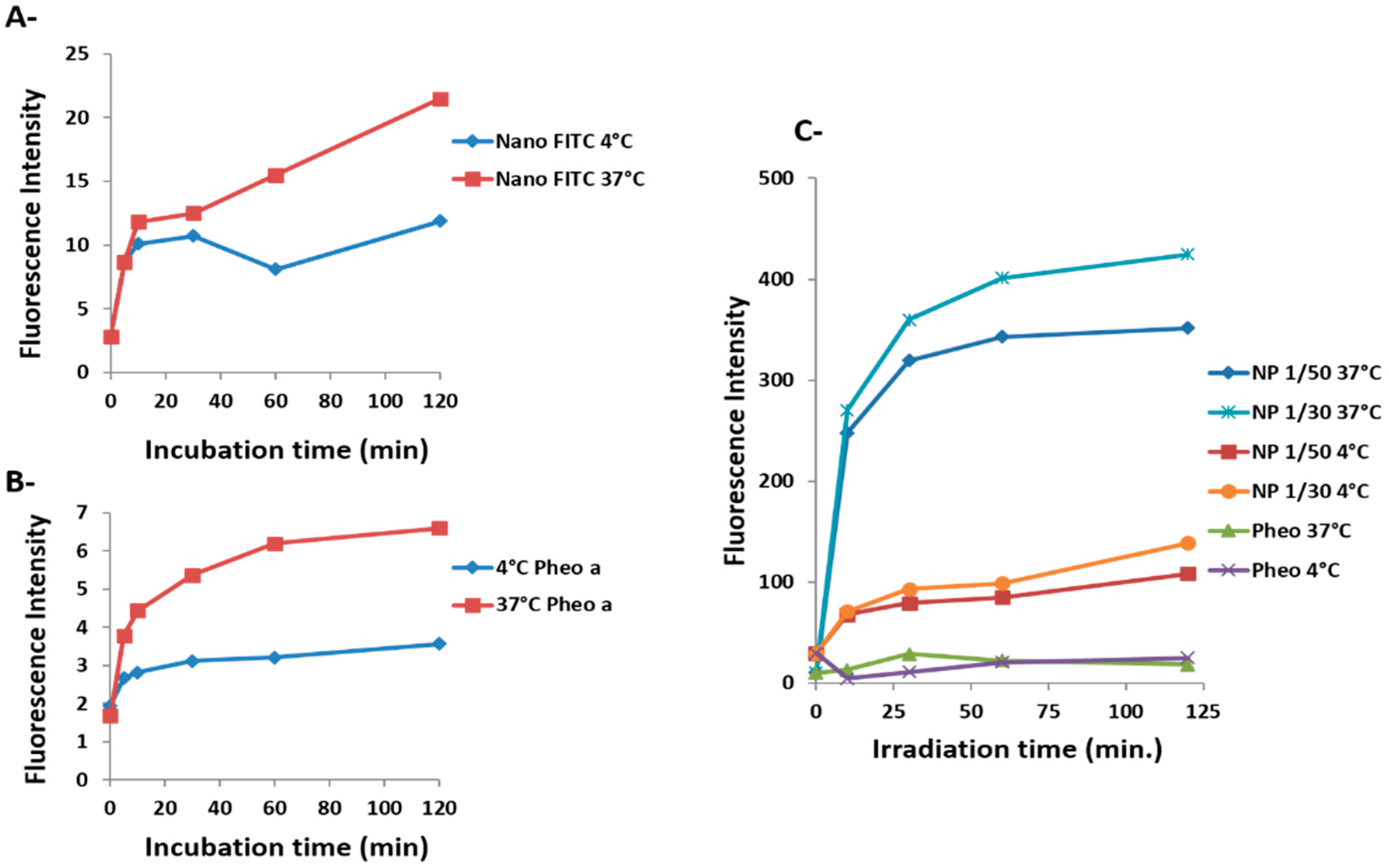
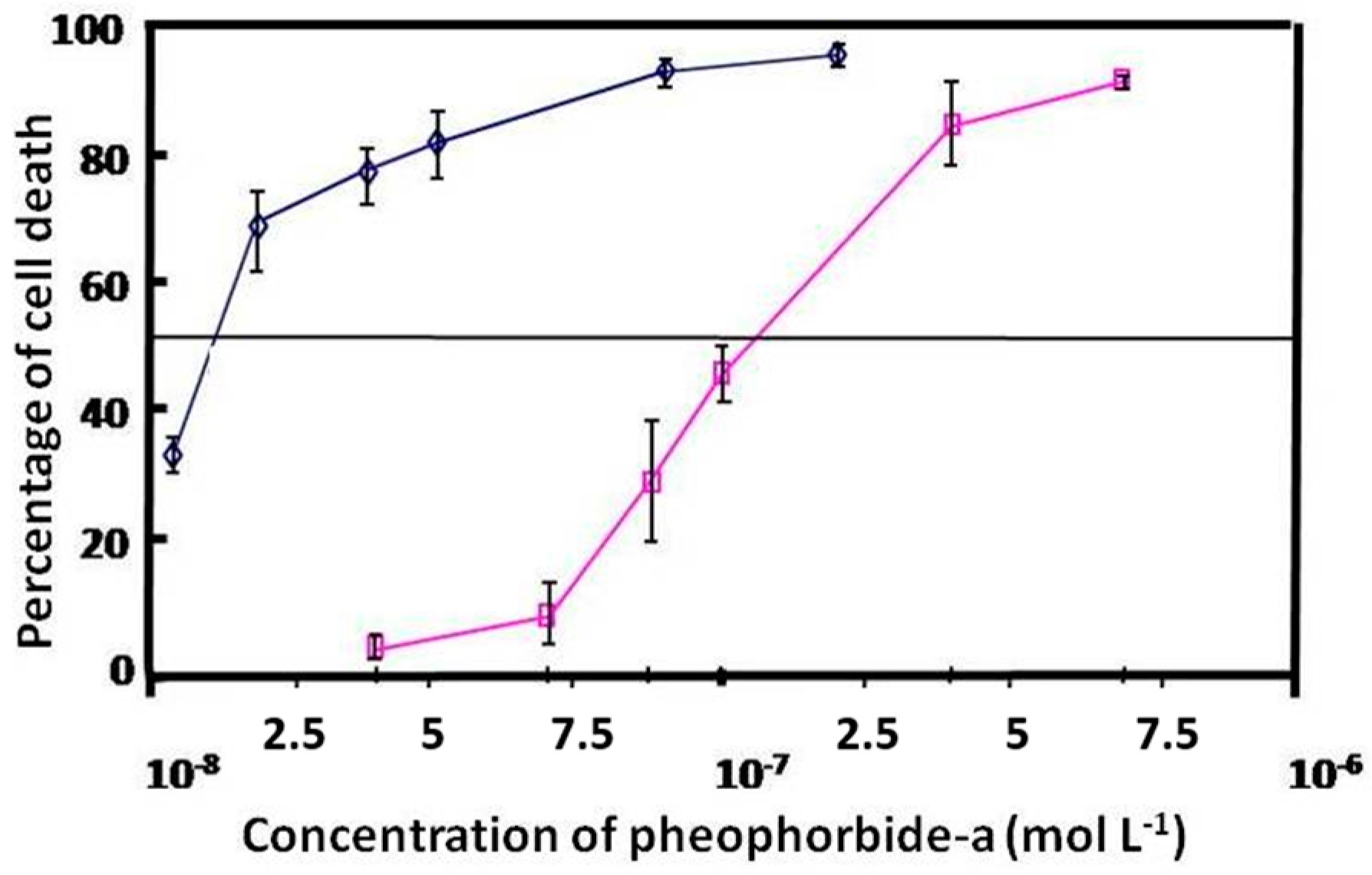
2.2. Cellular Uptake and Phototoxicity
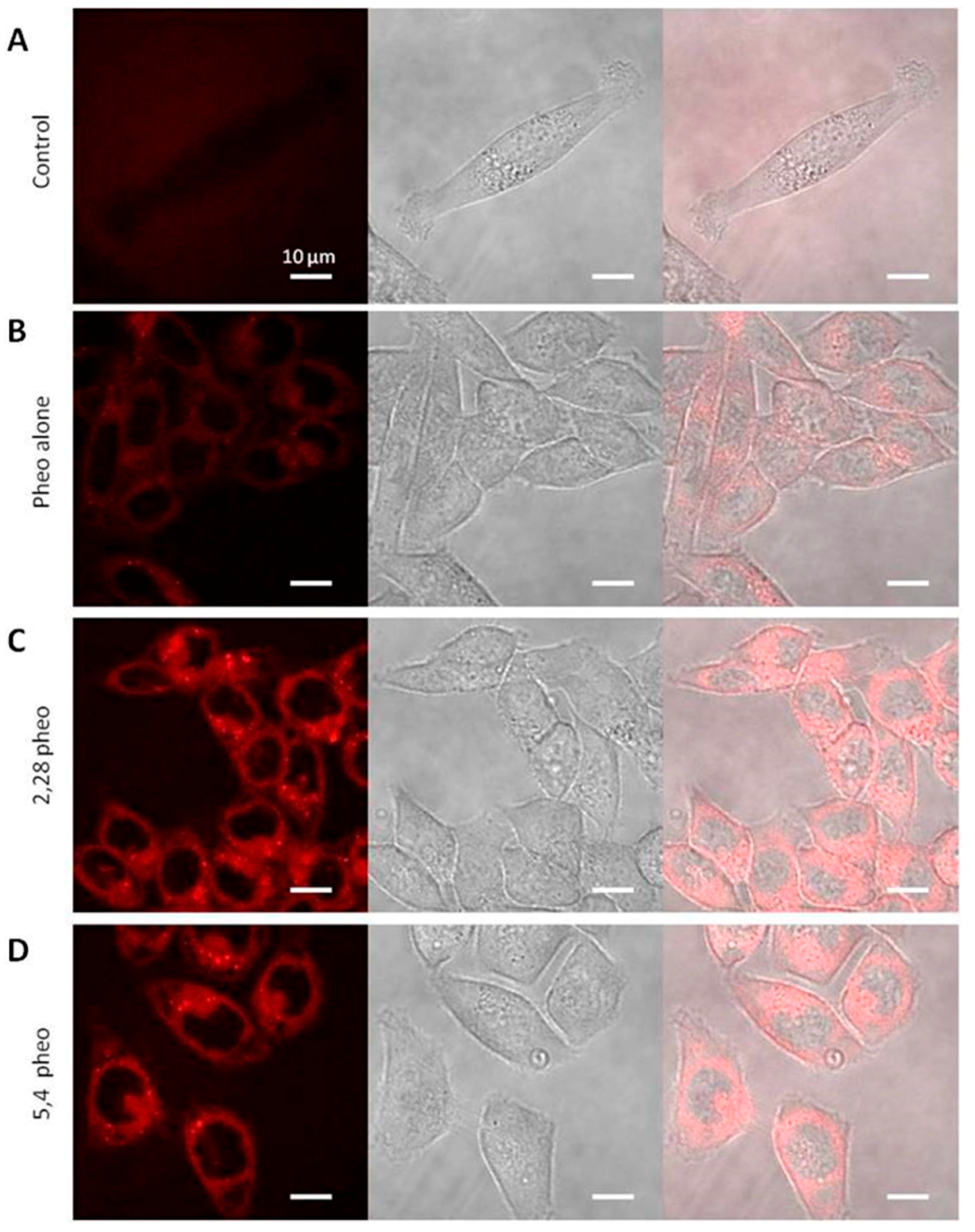
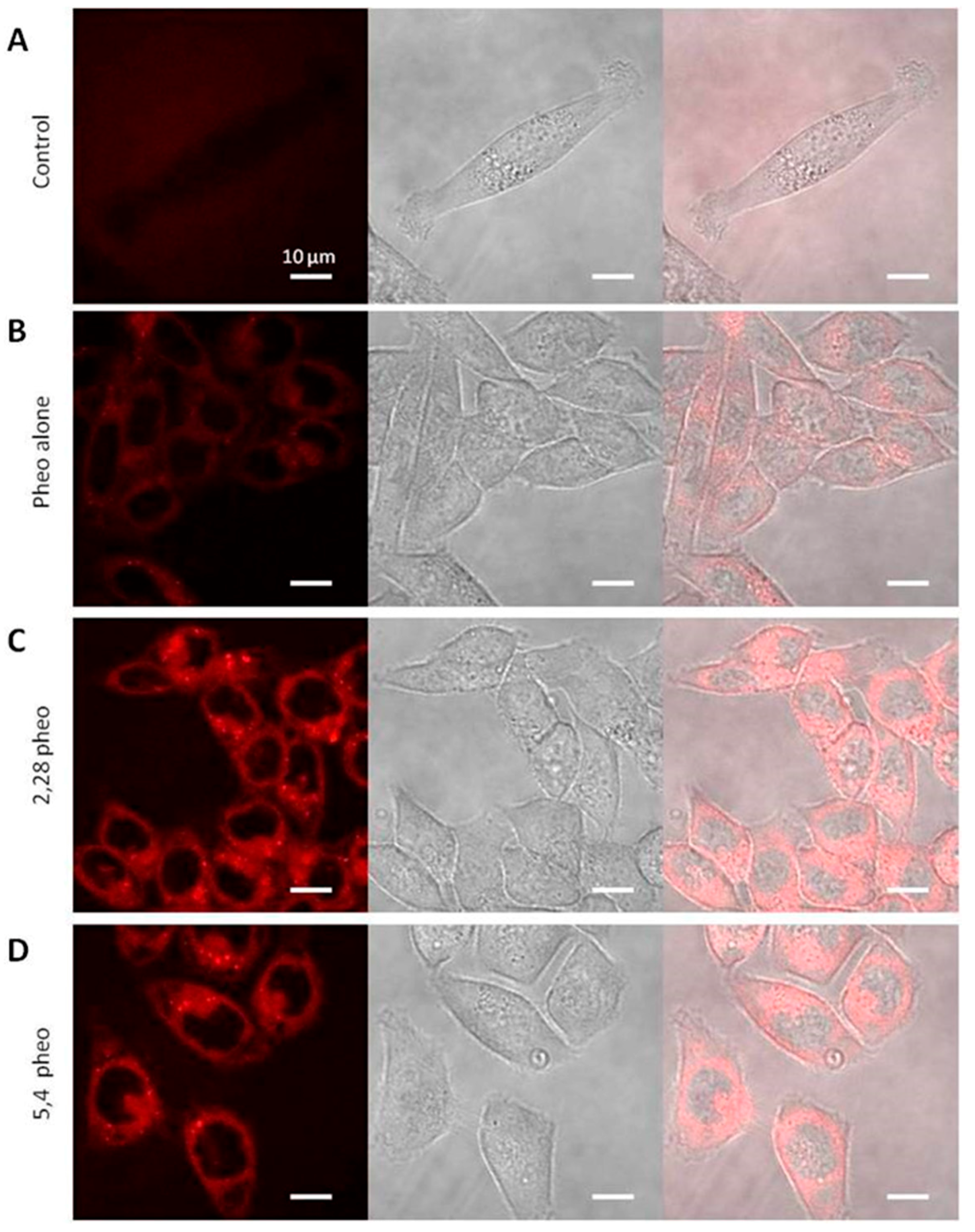
2.3. Confocal Microscopy Investigations
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Preparation of Polymer Micelles
3.3. Dynamic Light Scattering (DLS)
3.4. Differential Scanning Calorimetry (DSC)
3.5. Atomic Force Microscopy (AFM)
3.6. Small Angle Neutron Scattering (SANS)
3.7. Transmission Electron Microscopy (TEM)
3.8. The Asymmetric Flow Field Flow Fractionation
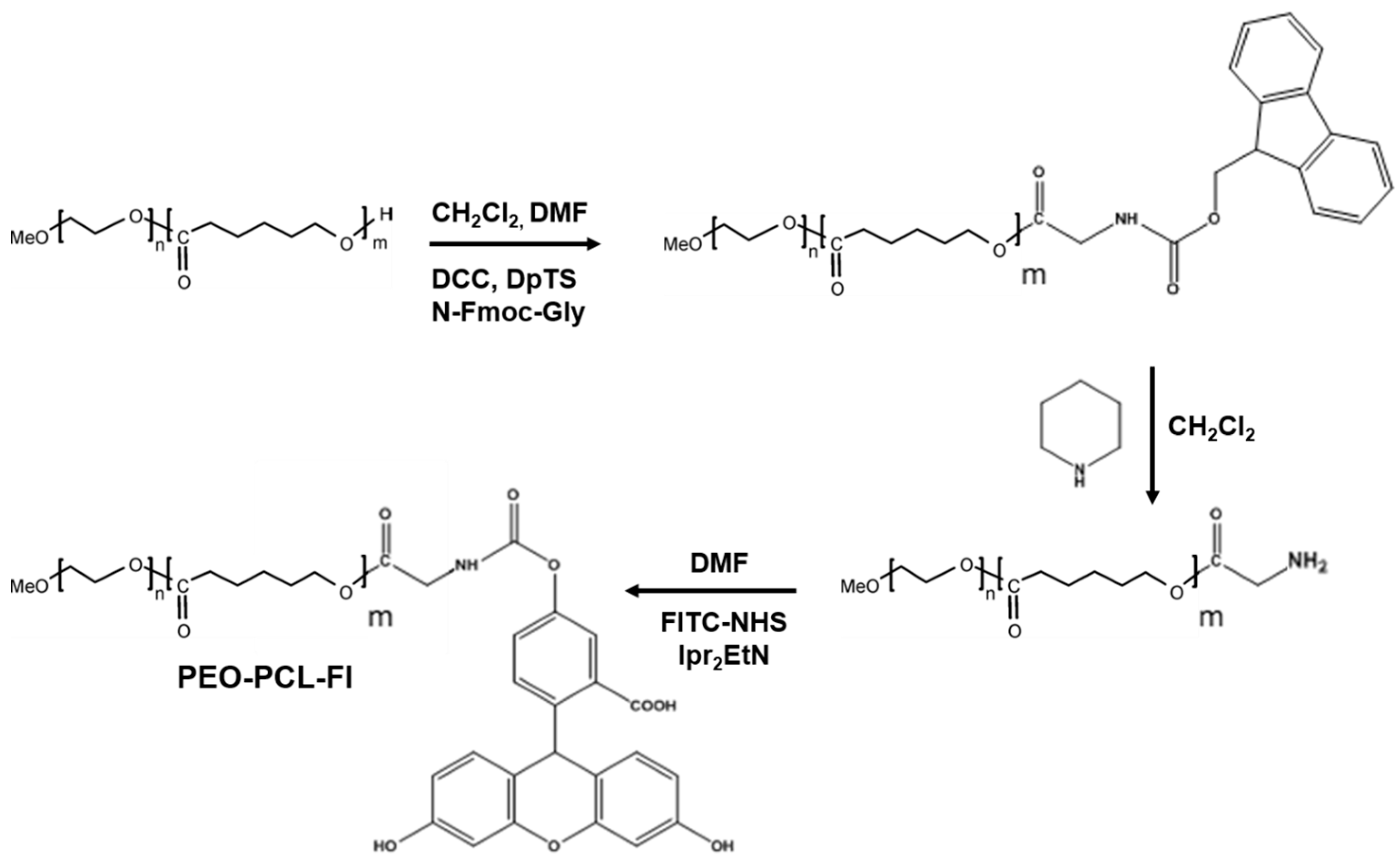
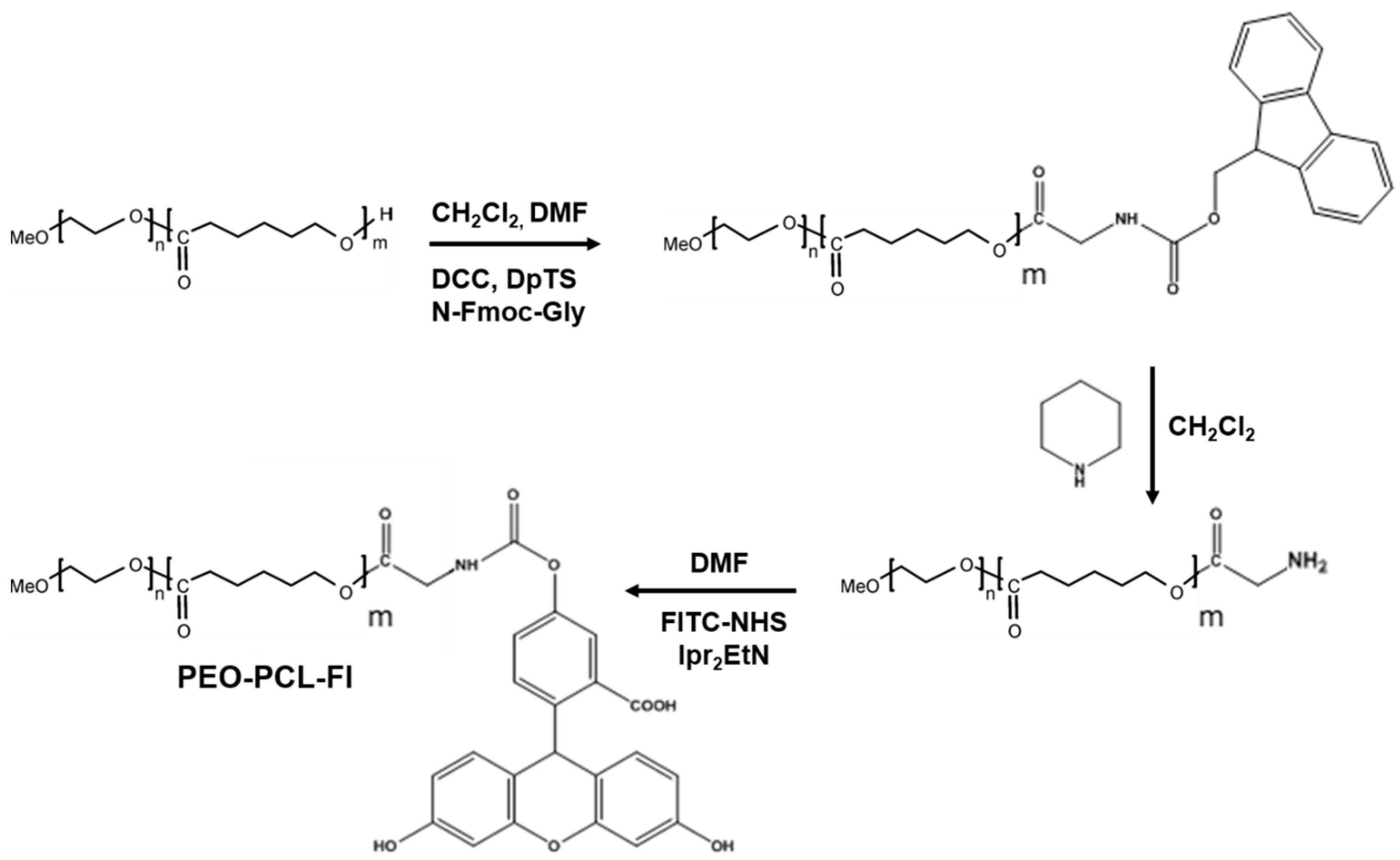
3.9. Synthesis of PEO-PCL-Fluorescein
3.9.1. Synthesis of PEO-PCL-Fmoc
3.9.2. Synthesis of PEO-PCL-NH2
3.9.3. PEO-PCL-Fl
3.10. Cell Line and Cell Culture
3.11. Measure of Uptake by Flow Cytometry
3.12. Cellular Imaging
3.13. Phototoxicity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kataoka, K.; Kwon, G.S.; Yokoyama, M.; Okano, T.; Sakurai, Y. Block Copolymer Micelles as Vehicles for Drug Delivery. J. Control. Release 1993, 24, 119–132. [Google Scholar]
- Kwon, G.S.; Kataoka, K. Block Copolymer Micelles as Long-Circulating Drug Vehicles. Adv. Drug Deliv. Rev. 1995, 16, 295–309. [Google Scholar] [CrossRef]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Y.; Allen, C. Polymer-Drug Compatibility: A Guide to the Development of Delivery Systems for the Anticancer Agent, Ellipticine. J. Pharm. Sci. 2004, 93, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. Supramolecular Assemblies of Block Copolymers in Aqueous Media as Nanocontainers Relevant to Biological Applications. Prog. Polym. Sci. 2006, 31, 949–982. [Google Scholar] [CrossRef]
- Stenzel, M.H. RAFT Polymerization: An Avenue to Functional Polymeric Micelles for Drug Delivery. Chem. Commun. 2008, 30, 3486–3503. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Pourgholami, M.H.; Morris, D.L.; Stenzel, M.H. An Optimized RGD-Decorated Micellar Drug Delivery System for Albendazole for the Treatment of Ovarian Cancer: From RAFT Polymer Synthesis to Cellular Uptake. Macromol. Biosci. 2011, 11, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-Engineering Block Copolymer Aggregates for Drug Delivery. Colloids Surf. B 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, H.B.; Andrade, J.D. Blood compatibility of Polyethylene Oxide Surfaces. Prog. Polym. Sci. 1995, 20, 1043–1079. [Google Scholar] [CrossRef]
- Letchford, K.; Zastre, R.; Burt, H. Synthesis and Micellar Characterization of Short Block Length Methoxy Poly(ethylene glycol)-block-poly(caprolactone) Diblock Copolymers. Colloids Surf. B 2004, 3, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.E.; Peppas, N.A. Opsonization, Biodistribution, and Pharmacokinetics of Polymeric Nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Tam, J.; Maysinger, D.; Eisenberg, A. Cellular Internalization of Poly(ethylene oxide)-b-poly(ε-caprolactone) Diblock Copolymer Micelles. Bioconjug. Chem. 2002, 13, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J. Controll. Release 2000, 6, 271–284. [Google Scholar] [CrossRef]
- Kreuter, J. Encyclopedia of Nanoscience and Nanotechnology; American Scientific Publishers: Valencia, CA, USA, 2004; Volume 7, pp. 161–180. [Google Scholar]
- Albertsson, A.C.; Varma, I.K. Recent Developments in Ring Opening Polymerization of Lactones for Biomedical Applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Kwon, G.S.; Forest, M.L. Amphiphilic Block Copolymer Micelles for Nanoscale Drug Delivery. Drug Dev. Res. 2006, 67, 15–22. [Google Scholar] [CrossRef]
- Wei, X.; Gong, C.; Gou, M.; Fu, S.; Guo, Q.; Shi, S.; Luo, F.; Guo, G.; Giu, L.; Qian, Z. Biodegradable Poly(ɛ-caprolactone)-poly(ethylene glycol) Copolymers as Drug Delivery System. Int. J. Pharm. 2009, 381, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Shi, Y.Z.; Kim, J.Y.; Park, K.; Cheng, J.X. Overcoming the Barriers in Micellar Drug Delivery: Loading Efficiency, in vivo Stability, and Micelle–Cell Interaction. Expert Opin. Drug Deliv. 2010, 7, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M. Polymeric Micelles as a New Drug Carrier System and their Required Considerations for Clinical. Expert Opin. Drug Deliv. 2010, 7, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Pourgholami, M.H.; Morris, D.L.; Stenzel, M.H. Effect of Cross Linking on the Performance of Micelles as Drug Delivery Carriers: A Cell Uptake Study. Biomacromolecules 2012, 13, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, R.; Xiao, H.; Cai, H.; Zhang, W.; Xie, Z.; Huang, Y.; Jing, X.; Liu, T. A cross-linked polymeric micellar delivery system for cisplatin(IV) complex. Eur. J. Pharm. Biopharm. 2013, 83, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Stefanick, J.F.; Jia, X.; Jing, X.; Kiziltepe, T.; Zhang, Y.; Bilgicer, B. Micellar nanoparticle formation via electrostatic interactions for delivering multinuclear platinum(II) drugs. Chem. Commun. 2013, 49, 4809–4811. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yan, L.; Xiao, H.; Li, X.; Jing, X. Application of microwave-assisted click chemistry in the preparation of functionalized copolymers for drug conjugation. J. Appl. Polym. Sci. 2013, 127, 3365–3373. [Google Scholar] [CrossRef]
- Qi, R.; Xiao, H.; Wu, S.; Li, Y.; Zhang, Y.; Jing, X. Design and delivery of camplatin to overcome cisplatin drug resistance. J. Mater. Chem. B, 2015, 3, 176–179. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Li, S.; Vinogradov, S.V.; Alakhov, V.Y.; Miller, D.W.; Kabanov, A.V. Mechanism of Pluronic Effect on P-glycoprotein Efflux System in Blood-Brain Barrier: Contribution of Energy Depletion and Membrane Fluidization. J. Pharmacol. Exp. Ther. 2001, 299, 483–493. [Google Scholar] [PubMed]
- Sahay, G.; Batrakova, E.V.; Kabanov, A.V. Different Internalization Pathways of Polymeric Micelles and Unimers and their Effects on Vesicular Transport. Bioconjug. Chem. 2008, 19, 2023–2029. [Google Scholar] [CrossRef] [PubMed]
- Savic, R.; Luo, L.; Eisenberg, A.; Maysinger, D. Micellar Nanocontainers Distribute to Defined Cytoplasmic Organelles. Science 2003, 300, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Savic, R.; Azzam, T.; Eisenberg, A.; Maysinger, D. Assessment of the Integrity of Poly(caprolactone)-b-poly(ethylene oxide) Micelles under Biological Conditions: A Fluorogenic-Based Approach. Langmuir 2006, 22, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Yu, Y.; Eisenberg, A.; Maysinger, D. Cellular internalization of PCL20-b-PEO44 Block Copolymer Micelles. Biochim. Biophys. Acta 1999, 1421, 32–38. [Google Scholar] [CrossRef]
- Hubell, J.A. Enhancing Drug Function. Science 2003, 300, 595–596. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kim, S.; Wang, S.; Park, K.; Cheng, J.-X. Release of Hydrophobic Molecules from Polymer Micelles into Cell Membranes Revealed by Förster Resonance Energy Transfer Imaging. Proc. Natl. Acad. Sci. USA 2008, 10, 6596–6601. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Lavasanifar, A. The Effect of Block Copolymer Structure on the Internalization of Polymeric Micelles by Human Breast Cancer Cells. Colloids Surf. B 2005, 4, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C.; Szewczyk, A. Cellular Distribution of Nonionic Micelles. Science 2004, 303, 626–627. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Mingotaud, A.-F.; Violleau, F. Asymmetrical Flow Field-Flow Fractionation with Multi-Angle Light Scattering and Quasi Elastic Light Scattering for Characterization of Poly(ethyleneglycol-b-ε-caprolactone) Block Copolymer Self-Assemblies Used as Drug Carriers for Photodynamic Therapy. J. Chromatogr. A 2011, 1218, 4249–4256. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Mingotaud, A.-F.; El-Akra, N.; Violleau, F.; Souchard, J.-P. Monomeric Pheophorbide(a)-containing Poly(ethyleneglycol-b-ε-caprolactone) Micelles for Photodynamic Therapy. Photochem. Photobiol. Sci. 2009, 8, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Gibot, L.; Lemelle, A.; Till, U.; Moukarzel, B.; Mingotaud, A.-F.; Pimienta, V.; Saint-Aguet, P.; Rols, M.-P.; Gaucher, M.; Violleau, F.; et al. Polymeric micelles encapsulating photosensitizer: Structure/photodynamic therapy efficiency relation. Biomacromolecules 2014, 1, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Maysinger, D.; Berezovska, O.; Savic, S.; Soo, R.; Eisenberg, L.P. Block Copolymers Modify the Internalization of Micelle-Incorporated of Probes into Neural Cells. Biomed. Biochim. Acta 2011, 139, 205–217. [Google Scholar] [CrossRef]
- Kerdous, R.; Sureau, F.; Bour, A.; Bonneau, S. Release kinetics of an amphiphilic photosensitizer by block-polymer nanoparticles. Int. J. Pharm. 2015, 49, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Tong, E.L.; Highley, C.B.; Errabelli, D.R.; Cheng, J.X.; Kohane, D.S.; Yeo, Y. Intracellular Drug Delivery by Poly(lactic-co-glycolic acid) Nanoparticles, revisited. Mol. Pharm. 2009, 6, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Han, J.; Yu, Y.; Maysinger, D.; Eisenberg, A. Polycaprolactone-b-poly(ethylene oxide) Copolymer Micelles as a Delivery Vehicle for Dihydrotestosterone. J. Control. Release 2000, 63, 275–286. [Google Scholar] [CrossRef]
- Allen, C.; Yu, Y.; Maysinger, D.; Eisenberg, A. Polycaprolactone-b-Poly(ethylene oxide) Block Copolymer Micelles as a Novel Drug Delivery Vehicle for Neurotrophic Agents FK506 and L-685,818. Bioconjug. Chem. 1998, 9, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Soo, L.P.; Luo, L.; Maysinger, D.; Eisenberg, A. Incorporation and Release of Hydrophobic Probes in Biocompatible Polycaprolactone-block-poly(ethylene oxide) Micelles: Implications for Drug Delivery. Langmuir 2002, 18, 9996–10004. [Google Scholar]
- Soo, L.P.; Sidorov, S.N.; Mui, J.; Bronstein, L.M.; Vali, H.; Eisenberg, A.; Maysinger, D. Gold-Labeled Block Copolymer Micelles Reveal Gold Aggregates at Multiple Subcellular Sites. Langmuir 2007, 23, 4830–4836. [Google Scholar] [PubMed]
- Maysinger, D.; Lovric, J.; Eisenberg, A.; Savic, R. Fate of Micelles and Quantum Dots in Cells. Eur. J. Pharm. Biopharm. 2007, 6, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Moriyama, E.H.; Li, F.; Jarvi, M.T.; Allen, C.; Wilson, B.C. Diblock Copolymer Micelles Deliver Hydrophobic Protoporphyrin IX for Photodynamic Therapy. Photochem. Photobiol. 2007, 83, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-W.; Yun, M.-H.; Jeong, S.W.; In, C.-H.; Kim, J.-Y.; Seo, M.-H.; Pai, C.-M.; Kim, S.-O. Development of docetaxel-loaded intravenous formulation, Nanoxel-PM™ using polymer-based delivery system. J. Control. Release 2011, 1, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Average Dh (Batch DLS) (nm) a | Average D (TEM) (nm) | Average Dh (DLS AF4) (nm) | Nagg b |
|---|---|---|---|---|
| {PEO(2000)-b-PCL(2600)} | 17.7 | 13.7 ± 2.7 | 20.2 | 200 |
| {PEO(2000)-b-PCL(2800)} | 22.0 | 13.7 ± 2.7 | 22.4 | N.D |
| {PEO(2000)-b-PCL(2800)} + Pheo 1/30 | 27.2 | 16.8 ± 2.7 | 27.4 | N.D. c |
| {PEO(5000)-b-PCL(4000)} | 24.6 | 12.6 ± 2.7 | 26.8 | 190 |
| {PEO(5000)-b-PCL(4000)} + Pheo 1/30 | 25.6 | N.D | N.D | N.D. |
| Sample | Guinier | Hairysphere Model | ||||
|---|---|---|---|---|---|---|
| Rg | R(sphere)eq | Nagg (Rsphere) | Rcore | Nagg (Rcore) | Total Diameter | |
| {PEO(2000)-b-PCL(2600)} | 5.6 nm | 7.3 nm | 396–222 | 5.0 nm | 127 | 14.8 nm |
| {PEO(2000)-b-PCL(2600)} + Pheo 1/10 | 6.4 nm | 8.3 nm | 582–326 | 5.2 nm | 143 | 15.2 nm |
| {PEO(5000)-b-PCL(4000)} + Pheo 1/20 | 7.1 nm | 9.2 nm | 515–227 | 6.0 nm | 143 | ca. 12 nm |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Till, U.; Gibot, L.; Mingotaud, A.-F.; Ehrhart, J.; Wasungu, L.; Mingotaud, C.; Souchard, J.-P.; Poinso, A.; Rols, M.-P.; Violleau, F.; et al. Drug Release by Direct Jump from Poly(ethylene-glycol-b-ε-caprolactone) Nano-Vector to Cell Membrane. Molecules 2016, 21, 1643. https://doi.org/10.3390/molecules21121643
Till U, Gibot L, Mingotaud A-F, Ehrhart J, Wasungu L, Mingotaud C, Souchard J-P, Poinso A, Rols M-P, Violleau F, et al. Drug Release by Direct Jump from Poly(ethylene-glycol-b-ε-caprolactone) Nano-Vector to Cell Membrane. Molecules. 2016; 21(12):1643. https://doi.org/10.3390/molecules21121643
Chicago/Turabian StyleTill, Ugo, Laure Gibot, Anne-Françoise Mingotaud, Jérôme Ehrhart, Luc Wasungu, Christophe Mingotaud, Jean-Pierre Souchard, Alix Poinso, Marie-Pierre Rols, Frédéric Violleau, and et al. 2016. "Drug Release by Direct Jump from Poly(ethylene-glycol-b-ε-caprolactone) Nano-Vector to Cell Membrane" Molecules 21, no. 12: 1643. https://doi.org/10.3390/molecules21121643
APA StyleTill, U., Gibot, L., Mingotaud, A.-F., Ehrhart, J., Wasungu, L., Mingotaud, C., Souchard, J.-P., Poinso, A., Rols, M.-P., Violleau, F., & Vicendo, P. (2016). Drug Release by Direct Jump from Poly(ethylene-glycol-b-ε-caprolactone) Nano-Vector to Cell Membrane. Molecules, 21(12), 1643. https://doi.org/10.3390/molecules21121643