
Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer



Abstract
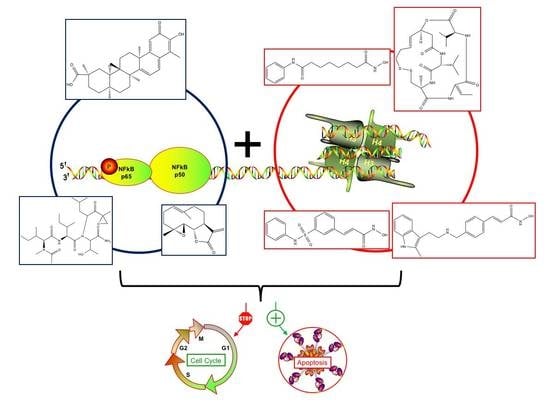
:
1. Introduction
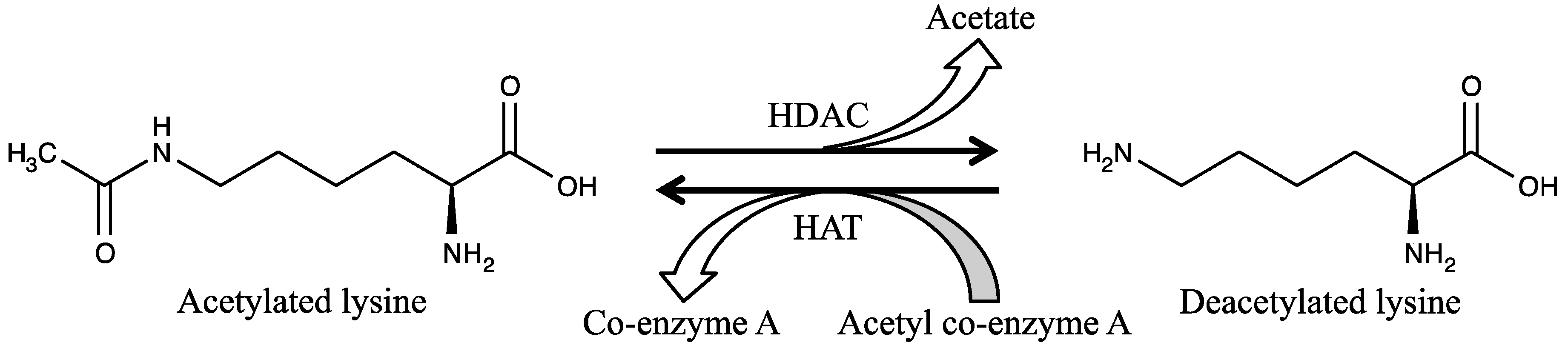
2. Histone Deacetylases and Their Classification
3. Histone Deacetylases and Cancer
3.1. Class I
3.2. Class II
3.3. Class III
3.4. Class IV
4. Natural Compound Histone Deacetylase Inhibitors
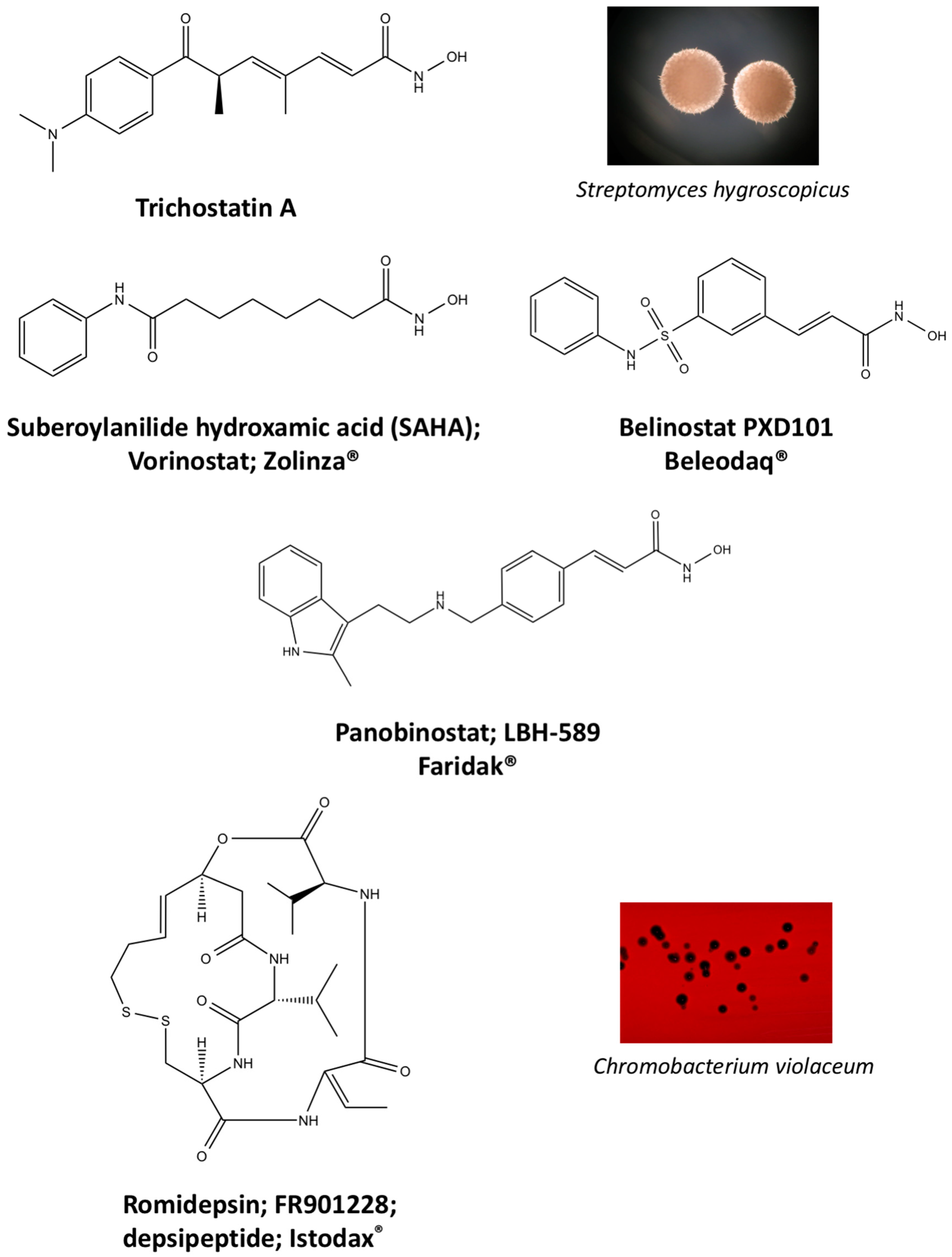
4.1. Natural Compound Scaffolds of Clinically Used HDAC Inhibitors
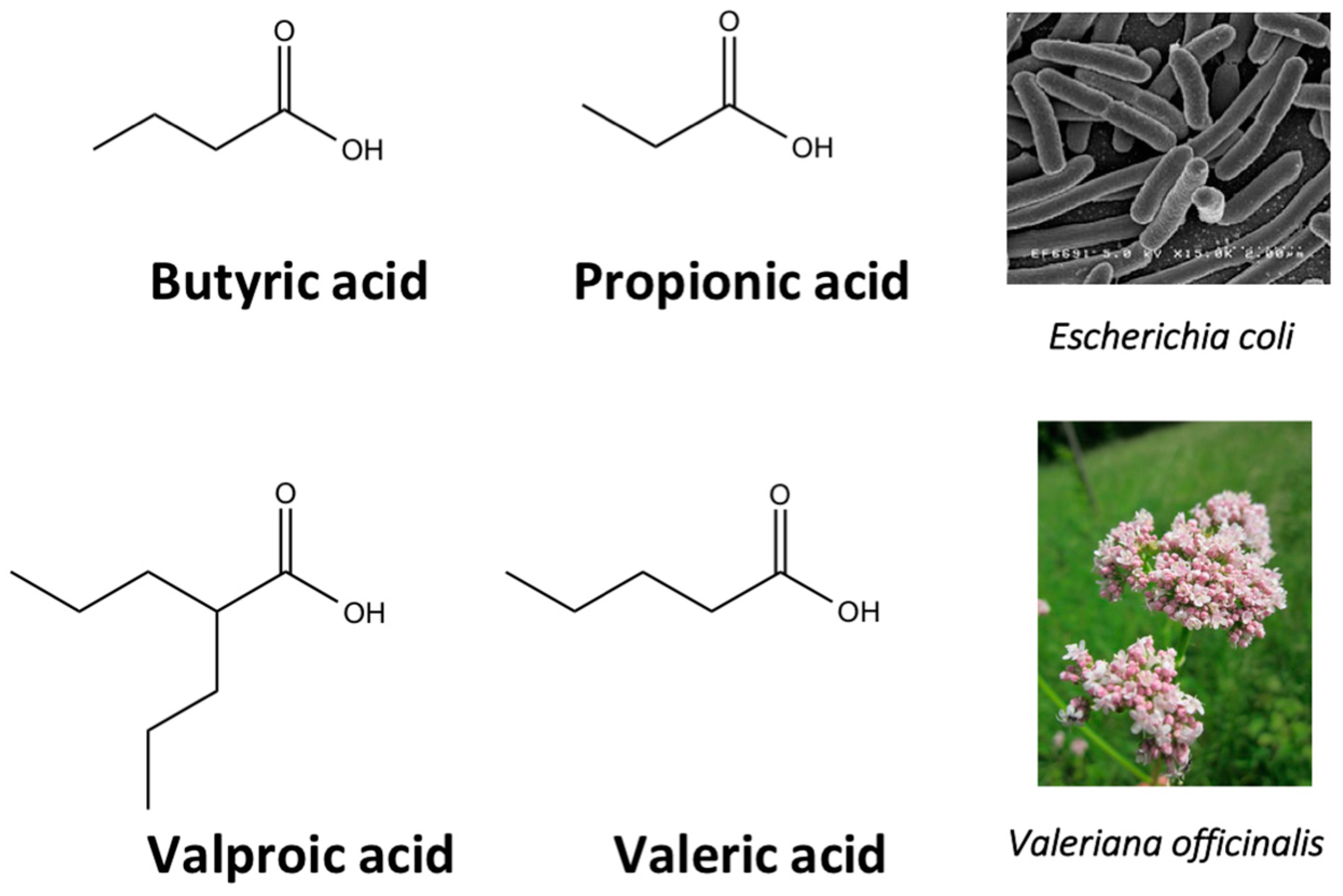
4.2. Experimentally Used Natural Compound Scaffolds without Clinical Utilization
5. Clinically Approved Histone Deacetylase Inhibitors
5.1. Vorinostat
5.2. Romidepsin
5.3. Belinostat
5.4. Panobinostat
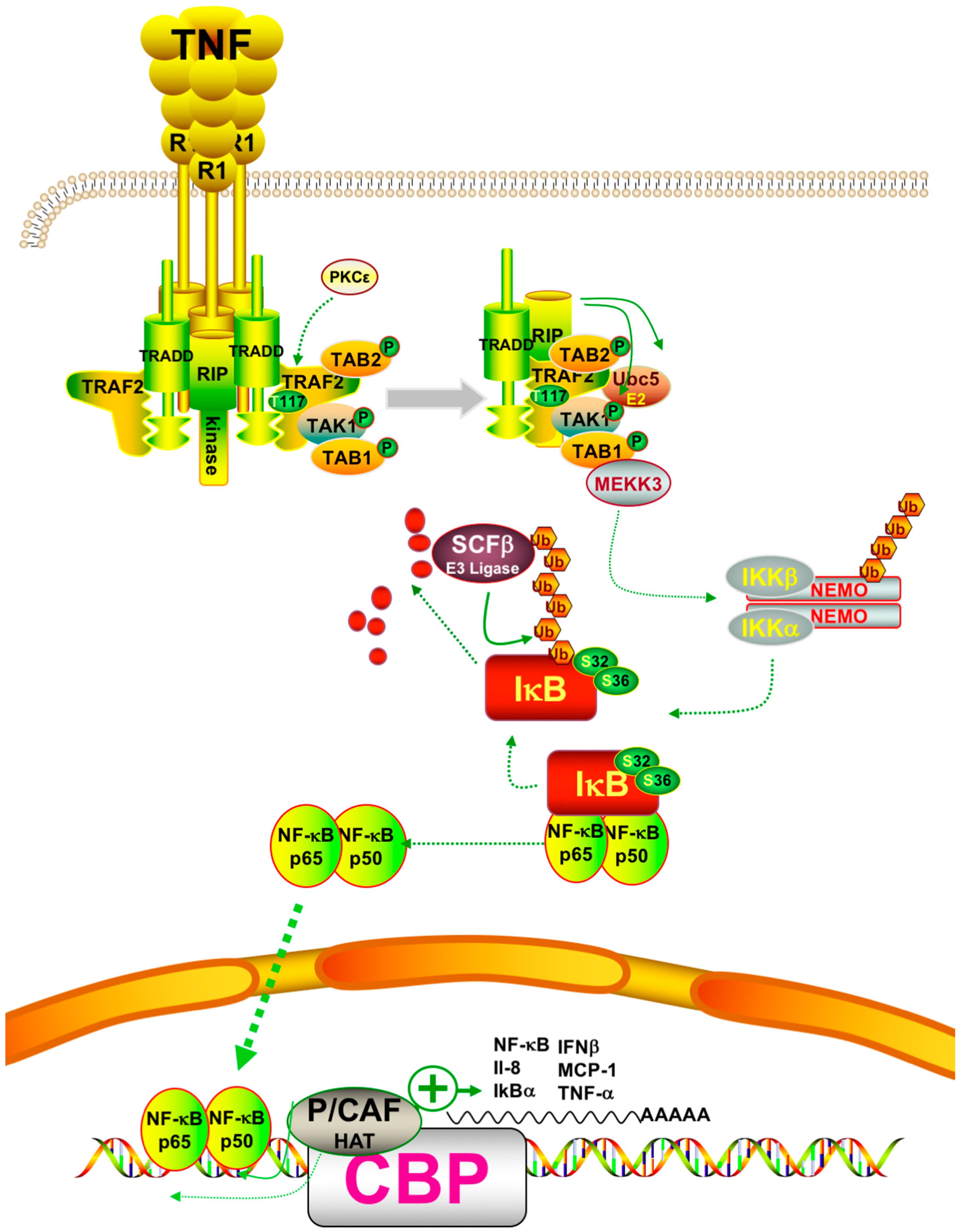
6. Combined Treatments of Histone Deacetylase Modulators and NF-κB Inhibitors
6.1. Post-Translational Modifications of NF-κB
6.2. Synergistic Treatments Involving HDAC and NF-κB inhibitors
7. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| CBP | CREB binding protein |
| cIAP | cellular inhibitor of apoptosis |
| CLL | chronic lymphocytic leukemia |
| CREB | cAMP-response element-binding protein |
| CSE | cystathionine-lyase |
| CTCL | cutaneous T-cell lymphoma |
| GLP | G9a-like protein |
| HAT | histone acetyltransferase |
| HAT | histone acetyltransferases |
| Hda1 | histone deacetylase 1 |
| HDAC | histone deacetylase |
| HDACi | histone deacetylase inhibitor |
| HMBA | hexamethylene bisacetamide |
| IKK | IκB kinase |
| IL | interleukin |
| INF | interferon |
| IκB | inhibitor of κB |
| MDS | myelodysplastic syndromes |
| MDS | myelodysplastic syndromes |
| MEKK | mitogen-activated protein kinase kinase kinase |
| NAD | nicotinamide adenine dinucleotide |
| NEMO | NF-κB essential modulator |
| NF-κB | nuclear factor-κB |
| P/CAF | P300/CBP-associated factor |
| PK | protein kinase |
| PTCL | peripheral T-cell lymphoma |
| RelA | REL associated |
| RHD | Rel homology domain |
| RIP | receptor-interacting protein |
| ROS | reactive oxygen species |
| Rpd3 | reduced potassium dependency-3 |
| SAHA | suberoylanilide hydroxamic acid |
| SCF | Skp, Cullin, F-box (containing complex) |
| SETD | SET domain containing |
| Sir2 | silent information regulator-2 |
| SIRT | sirtuin |
| TAB | TGF-β-activated kinase 1 and MAP3K7-binding protein |
| TAK | transforming growth factor β-activated kinase |
| TNF | tumor necrosis factor |
| TGF | tumor growth factor |
| TRADD | tumor necrosis factor receptor type 1-associated death domain |
| TRAFF | TNF receptor-associated factor |
| TSA | trichostatin A |
| Ubc | Ubiquitin-conjugating (enzyme) |
| USFDA | United States Food and Drug Administration |
| VPA | valproic acid |
| ZBD | Zn2+ binding domain |
References
- Seidel, C.; Florean, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Chromatin-modifying agents in anti-cancer therapy. Biochimie 2012, 94, 2264–2279. [Google Scholar] [CrossRef] [PubMed]
- Thurn, K.T.; Thomas, S.; Moore, A.; Munster, P.N. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011, 7, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase modulators provided by Mother Nature. Genes Nutr. 2012, 7, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.A.; Barnett, M.P. The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.; Uciechowska, U.; Schemies, J.; Rumpf, T.; Jung, M.; Sippl, W. Inhibitors to understand molecular mechanisms of NAD+-dependent deacetylases (sirtuins). Biochim. Biophys. Acta 2010, 1799, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kafeel, M.I.; Avezbakiyev, B.; Chen, C.; Sun, Y.; Rathnasabapathy, C.; Kalavar, M.; He, Z.; Burton, J.; Lichter, S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011, 81, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Nebbioso, A.; Altucci, L. Sirtuins and disease: The road ahead. Front. Pharmacol. 2012, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Ozdag, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Fischer, K.; Baus, K.; Kashyap, A.; Ma, S.; Krupp, M.; Linke, M.; Teufel, A.; Zechner, U.; Strand, D.; et al. Sirtuin-6-dependent genetic and epigenetic alterations are associated with poor clinical outcome in hepatocellular carcinoma patients. Hepatology 2013, 58, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.; Zino, S.; Macintyre, A.; Kingsmore, D.; Payne, A.P.; George, W.D.; Shiels, P.G. Altered sirtuin expression is associated with node-positive breast cancer. Br. J. Cancer 2006, 95, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- ME, L.L.; Vidal, F.; Gallardo, D.; Diaz-Fuertes, M.; Rojo, F.; Cuatrecasas, M.; Lopez-Vicente, L.; Kondoh, H.; Blanco, C.; Carnero, A.; et al. New p53 related genes in human tumors: Significant downregulation in colon and lung carcinomas. Oncol. Rep. 2006, 16, 603–608. [Google Scholar]
- Cerella, C.; Teiten, M.H.; Radogna, F.; Dicato, M.; Diederich, M. From nature to bedside: Pro-survival and cell death mechanisms as therapeutic targets in cancer treatment. Biotechnol. Adv. 2014, 32, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Morceau, F.; Chateauvieux, S.; Orsini, M.; Trecul, A.; Dicato, M.; Diederich, M. Natural compounds and pharmaceuticals reprogram leukemia cell differentiation pathways. Biotechnol. Adv. 2015, 33, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, M.; Dicato, M.; Diederich, M. Plant-derived epigenetic modulators for cancer treatment and prevention. Biotechnol. Adv. 2014, 32, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. Gold from the sea: Marine compounds as inhibitors of the hallmarks of cancer. Biotechnol. Adv. 2011, 29, 531–47. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Dicato, M.; Diederich, M. Cancer-type-specific crosstalk between autophagy, necroptosis and apoptosis as a pharmacological target. Biochem. Pharmacol. 2015, 94, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Diederich, M.; Cerella, C. Non-canonical programmed cell death mechanisms triggered by natural compounds. Semin. Cancer Biol. 2016, 40–41, 4–44. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Legrand, N.; Panning, J.; Dicato, M.; Diederich, M. Anti-inflammatory and anticancer drugs from nature. Cancer Treat. Res. 2014, 159, 123–143. [Google Scholar] [PubMed]
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Biol. 2016, 40–41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Duvoix, A.; Blasius, R.; Delhalle, S.; Schnekenburger, M.; Morceau, F.; Henry, E.; Dicato, M.; Diederich, M. Chemopreventive and therapeutic effects of curcumin. Cancer Lett. 2005, 223, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Schnekenburger, M.; Zloh, M.; Golais, F.; Diederich, M.; Tasdemir, D. Natural chalcones as dual inhibitors of HDACs and NF-κB. Oncol. Rep. 2012, 28, 797–805. [Google Scholar] [PubMed]
- Menezes, J.C.; Orlikova, B.; Morceau, F.; Diederich, M. Natural and Synthetic Flavonoids: Structure-Activity Relationship and Chemotherapeutic Potential for the Treatment of Leukemia. Crit. Rev. Food Sci. Nutr. 2016, 56, S4–S28. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Kobayashi, M.; Nagashima, K.; Wakisaka, Y.; Koizumi, K. A new antifungal antibiotic, trichostatin. J. Antibiot. 1976, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar] [PubMed]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Nakajima, H.; Hori, Y.; Goto, T.; Okuhara, M. Action of FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968, on Ha-ras transformed NIH3T3 cells. Biosci. Biotechnol. Biochem. 1994, 58, 1579–1583. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Kim, Y.B.; Terano, H.; Yoshida, M.; Horinouchi, S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp. Cell Res. 1998, 241, 126–133. [Google Scholar] [CrossRef] [PubMed]
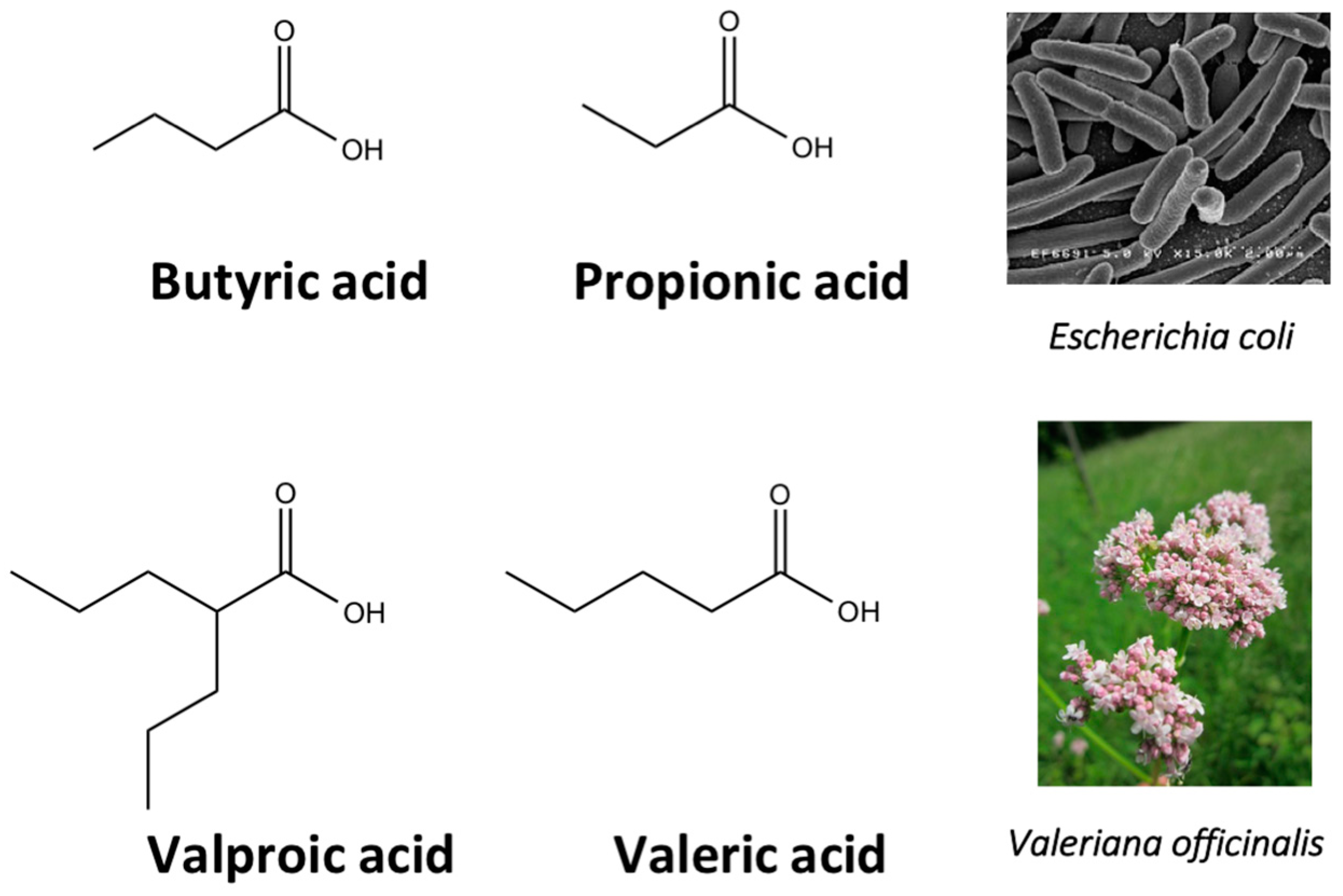
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- McKnight, G.S.; Hager, L.; Palmiter, R.D. Butyrate and related inhibitors of histone deacetylation block the induction of egg white genes by steroid hormones. Cell 1980, 22, 469–477. [Google Scholar] [CrossRef]
- Hassig, C.A.; Tong, J.K.; Schreiber, S.L. Fiber-derived butyrate and the prevention of colon cancer. Chem. Biol. 1997, 4, 783–789. [Google Scholar] [CrossRef]
- Chateauvieux, S.; Eifes, S.; Morceau, F.; Grigorakaki, C.; Schnekenburger, M.; Henry, E.; Dicato, M.; Diederich, M. Valproic acid perturbs hematopoietic homeostasis by inhibition of erythroid differentiation and activation of the myelo-monocytic pathway. Biochem. Pharmacol. 2011, 81, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dai, H.J.; Ye, M.; Wang, S.L.; Xiao, X.J.; Zheng, J.; Chen, H.Y.; Luo, Y.H.; Liu, J. Lycorine induces cell-cycle arrest in the G0/G1 phase in K562 cells via HDAC inhibition. Cancer Cell Int. 2012, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Biggins, J.B.; Gleber, C.D.; Brady, S.F. Acyldepsipeptide HDAC inhibitor production induced in Burkholderia thailandensis. Org. Lett. 2011, 13, 1536–1539. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Kulkarni, S. Enantioselective total synthesis of (+)-largazole, a potent inhibitor of histone deacetylase. Org. Lett. 2008, 10, 3907–3909. [Google Scholar] [CrossRef] [PubMed]
- Crabb, S.J.; Howell, M.; Rogers, H.; Ishfaq, M.; Yurek-George, A.; Carey, K.; Pickering, B.M.; East, P.; Mitter, R.; Maeda, S.; et al. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem. Pharmacol. 2008, 76, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Narita, K.; Matsuhara, K.; Itoh, J.; Akiyama, Y.; Dan, S.; Yamori, T.; Ito, A.; Yoshida, M.; Katoh, T. Synthesis and biological evaluation of novel FK228 analogues as potential isoform selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 592–609. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Henkes, L.M.; Doughty, L.B.; He, M.; Wang, D.; Meyer-Almes, F.J.; Cheng, Y.Q. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J. Nat. Prod. 2011, 74, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Parolin, C.; Calonghi, N.; Presta, E.; Boga, C.; Caruana, P.; Naldi, M.; Andrisano, V.; Masotti, L.; Sartor, G. Mechanism and stereoselectivity of HDAC I inhibition by (R)-9-hydroxystearic acid in colon cancer. Biochim. Biophys. Acta 2012, 1821, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Fennell, K.A.; Mollmann, U.; Miller, M.J. Syntheses and biological activity of amamistatin B and analogs. J. Org. Chem. 2008, 73, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duee, P.H.; Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Lea, M.A.; Rasheed, M.; Randolph, V.M.; Khan, F.; Shareef, A.; desBordes, C. Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutr. Cancer 2002, 43, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, S.E.; Rusche, J.J.; Bec, S.L.; Horn, D.J.; Janda, J.; Rim, S.H.; Smith, C.L.; Bowden, G.T. The effect of sulforaphane on histone deacetylase activity in keratinocytes: Differences between in vitro and in vivo analyses. Mol. Carcinog. 2015, 54, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.W.; Lee, D.H.; Shin, D.H.; Kim, S.H.; Kwon, S.H. Aceroside VIII is a new natural selective HDAC6 inhibitor that synergistically enhances the anticancer activity of HDAC inhibitor in HT29 cells. Planta Med. 2015, 81, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Venturelli, S.; Kallnischkies, M.; Bocker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Weiss, T.S.; Lauer, U.M.; et al. Kaempferol, a new nutrition-derived pan-inhibitor of human histone deacetylases. J. Nutr. Biochem. 2013, 24, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.B.; Lee, S.H. Protocatechualdehyde possesses anti-cancer activity through downregulating cyclin D1 and HDAC2 in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2013, 430, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Sim, C.J.; Lee, C.O. Cytotoxic compounds from a two-sponge association. J. Nat. Prod. 1995, 58, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Berger, A.; Bocker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS ONE 2013, 8, e73097. [Google Scholar] [CrossRef]
- Senawong, T.; Misuna, S.; Khaopha, S.; Nuchadomrong, S.; Sawatsitang, P.; Phaosiri, C.; Surapaitoon, A.; Sripa, B. Histone deacetylase (HDAC) inhibitory and antiproliferative activities of phenolic-rich extracts derived from the rhizome of Hydnophytum formicarum Jack.: Sinapinic acid acts as HDAC inhibitor. BMC Complement. Altern. Med. 2013, 13, 232. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Owa, T.; Hassig, C.A.; Shimada, J.; Schreiber, S.L. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. Proc. Natl. Acad. Sci. USA 1998, 95, 3356–3361. [Google Scholar] [CrossRef] [PubMed]
- El Amrani, M.; Lai, D.; Debbab, A.; Aly, A.H.; Siems, K.; Seidel, C.; Schnekenburger, M.; Gaigneaux, A.; Diederich, M.; et al. Protein kinase and HDAC inhibitors from the endophytic fungus Epicoccum nigrum. J. Nat. Prod. 2014, 77, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Li, J.; Xia, J.; Jiang, R.; Zuo, G.W.; Li, X.P.; Chen, Y.; Xiong, W.; Chen, D.L. Ginsenoside 20(s)-Rh2 as potent natural histone deacetylase inhibitors suppressing the growth of human leukemia cells. Chem. Biol. Interact. 2015, 242, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.M.; Kim, M.Y.; Park, W.H.; Moon, H.I. Histone deacetylase inhibitors from the rhizomes of Zingiber zerumbet. Pharmazie 2008, 63, 774–776. [Google Scholar] [PubMed]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones as lead-like natural products: The development of potent and selective histone deacetylase inhibitors. ACS Med. Chem. Lett. 2013, 4, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ahmad, M.; Bhat, J.A.; Kumar, A.; Kumar, M.; Zargar, M.A.; Hamid, A.; Shah, B.A. Design, synthesis and biological evaluation of beta-boswellic acid based HDAC inhibitors as inducers of cancer cell death. Bioorg. Med. Chem. Lett. 2014, 24, 4729–4734. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Altamura, S.; Chakravarty, P.K.; Cecchetti, O.; de Francesco, R.; Gallinari, P.; Ingenito, R.; Meinke, P.T.; Petrocchi, A.; Rowley, M.; et al. A series of novel, potent, and selective histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5948–5952. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, N.; Chini, M.G.; Micco, S.D.; Izzo, I.; Cafaro, E.; Russo, A.; Gallinari, P.; Paolini, C.; Nardi, M.C.; Casapullo, A.; et al. Molecular Insights into Azumamide E Histone Deacetylases Inhibitory Activity. J. Am. Chem. Soc. 2007, 129, 3007–3012. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Bruwiere, H.; Verhulst, T.; Steller, U.; Andries, L.; Wouters, W.; Janicot, M.; Arts, J.; van Heusden, J. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J. Pharmacol. Exp. Ther. 2003, 304, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [PubMed]
- Itazaki, H.; Nagashima, K.; Sugita, K.; Yoshida, H.; Kawamura, Y.; Yasuda, Y.; Matsumoto, K.; Ishii, K.; Uotani, N.; Nakai, H.; et al. Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J. Antibiot. 1990, 43, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov—A Service of the U.S. National Institutes of Health. Available online: https://clinicaltrials.gov/ (accessed on 18 November 2016).
- Chakraborty, A.R.; Robey, R.W.; Luchenko, V.L.; Zhan, Z.; Piekarz, R.L.; Gillet, J.P.; Kossenkov, A.V.; Wilkerson, J.; Showe, L.C.; Gottesman, M.M.; et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood 2013, 121, 4115–4125. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Wang, L.; Hao, J.; Ni, J.; Chang, L.; Graham, P.H.; Kearsley, J.H.; Li, Y. Low dose histone deacetylase inhibitor, LBH589, potentiates anticancer effect of docetaxel in epithelial ovarian cancer via PI3K/Akt pathway in vitro. Cancer Lett. 2013, 329, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Moore, D. Panobinostat (Farydak): A Novel Option for the Treatment of Relapsed Or Relapsed and Refractory Multiple Myeloma. P. T. 2016, 41, 296–300. [Google Scholar] [PubMed]
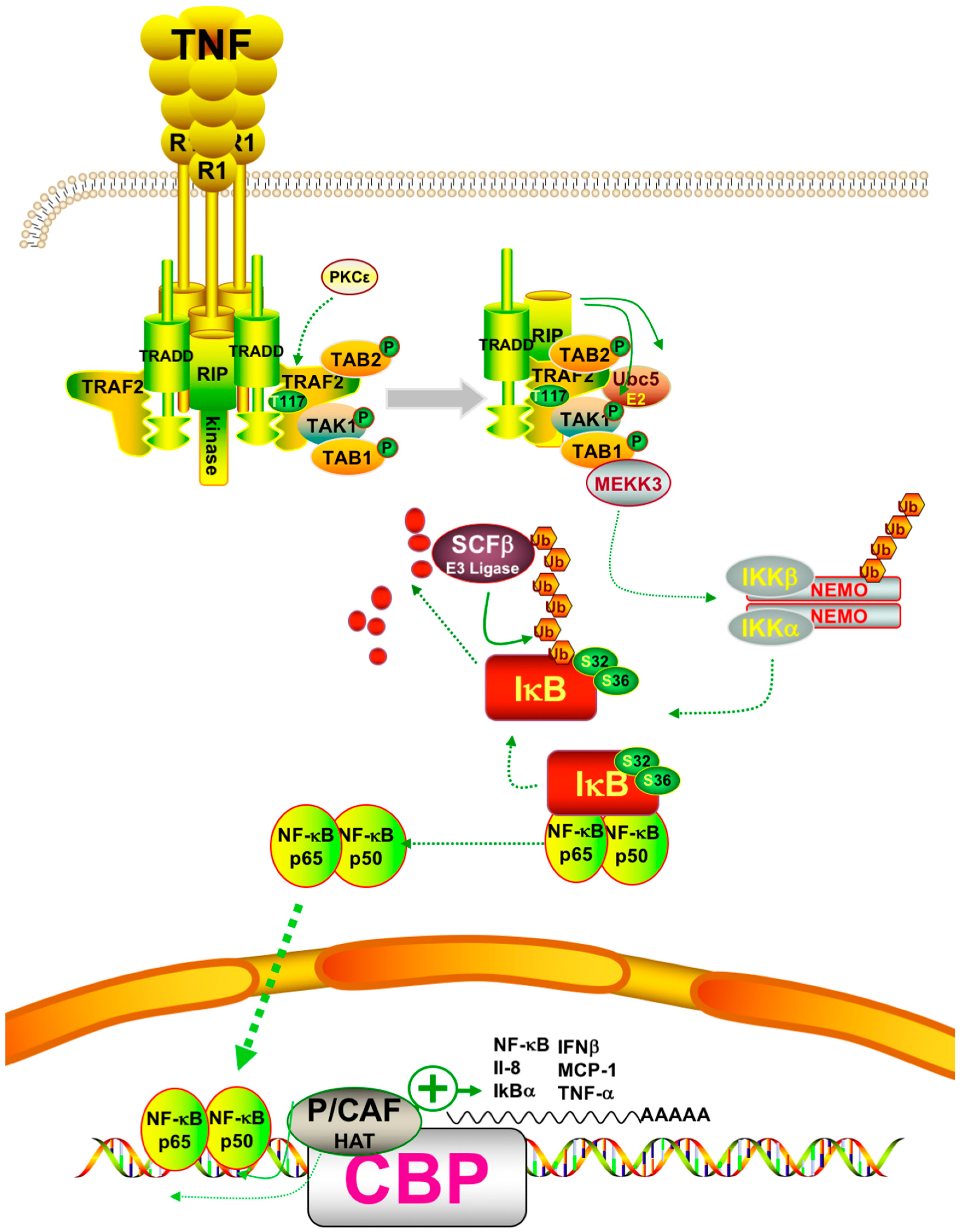
- Delhalle, S.; Blasius, R.; Dicato, M.; Diederich, M. A beginner’s guide to NF-κB signaling pathways. Ann. N. Y. Acad. Sci. 2004, 1030, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, Y.; Guo, Z.; Cheng, J.; Huang, J.; Deng, L.; Liao, W.; Chen, Z.; Liu, Z.; Su, B. The essential role of MEKK3 in TNF-induced NF-κB activation. Nat. Immunol. 2001, 2, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, R.; Kinnunen, L.; Kohler, M.; Julkunen, I.; Melen, K. NF-κB is transported into the nucleus by importin α3 and importin α4. J. Biol. Chem. 2005, 280, 15942–15951. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-κB: Another layer of regulation for NF-κB signaling pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, I.; Geng, H.; Sebald, A.; Hodel, M.; Bucher, C.; Kracht, M.; Schmitz, M.L. Inducible phosphorylation of NF-κB p65 at serine 468 by T cell costimulation is mediated by IKKε. J. Biol. Chem. 2006, 281, 6175–6183. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dorrie, A.; Schmitz, M.L.; Frank, R.; Livingstone, M.; Resch, K.; Kracht, M. Phosphorylation of serine 468 by GSK-3β negatively regulates basal p65 NF-κB activity. J. Biol. Chem. 2004, 279, 49571–49574. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Blonska, M.; Shambharkar, P.B.; Kobayashi, M.; Zhang, D.; Sakurai, H.; Su, B.; Lin, X. TAK1 is recruited to the tumor necrosis factor-α (TNF- α) receptor 1 complex in a receptor-interacting protein (RIP)-dependent manner and cooperates with MEKK3 leading to NF-κB activation. J. Biol. Chem. 2005, 280, 43056–43063. [Google Scholar] [CrossRef] [PubMed]
- Adli, M.; Baldwin, A.S. IKK-i/IKKε controls constitutive, cancer cell-associated NF-κB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 2006, 281, 26976–26984. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, Z.T.; Matsumoto, A.; Stamler, J.S.; Marshall, H.E. NOS2 regulation of NF-κB by S-nitrosylation of p65. J. Biol. Chem. 2007, 282, 30667–30672. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
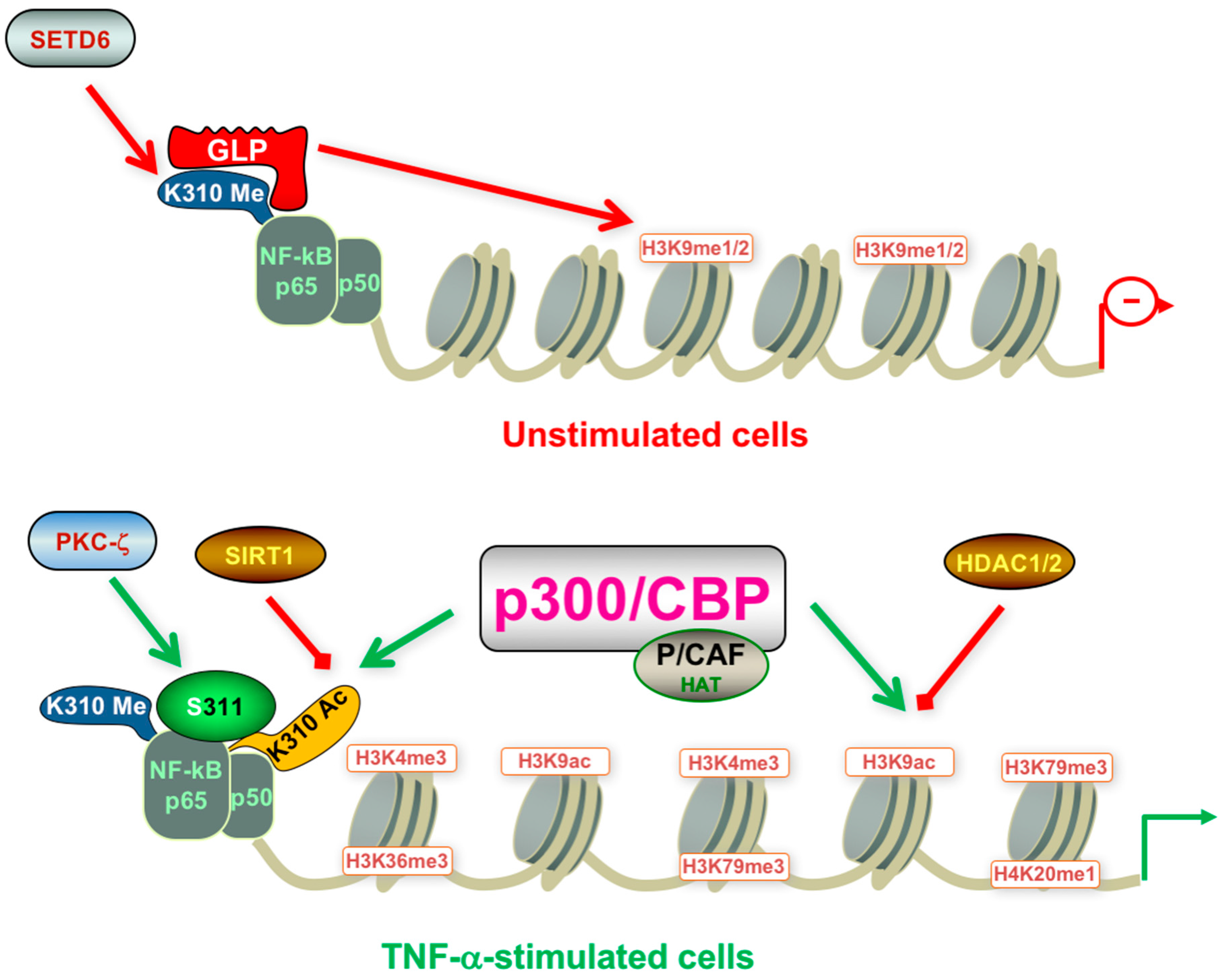
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Chen, L.f. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Cysteine 38 holds the key to NF-κB activation. Mol. Cell 2012, 45, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-κB-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tini, M.; Evans, R.M. HATs on and beyond chromatin. Curr. Opin. Cell Biol. 2001, 13, 218–224. [Google Scholar] [CrossRef]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-κB-dependent gene expression. BMC Genomics 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Buerki, C.; Rothgiesser, K.M.; Valovka, T.; Owen, H.R.; Rehrauer, H.; Fey, M.; Lane, W.S.; Hottiger, M.O. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 2008, 36, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. SIRT2 regulates NF-κB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Pandithage, R.; Lilischkis, R.; Harting, K.; Wolf, A.; Jedamzik, B.; Luscher-Firzlaff, J.; Vervoorts, J.; Lasonder, E.; Kremmer, E.; Knoll, B.; et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 2008, 180, 915–929. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.S.; Tainsky, M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Heltweg, B.; Gatbonton, T.; Schuler, A.D.; Posakony, J.; Li, H.; Goehle, S.; Kollipara, R.; Depinho, R.A.; Gu, Y.; Simon, J.A.; et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006, 66, 4368–4377. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Mosley, A.; Kitada, T.; Washburn, M.; Carey, M. The SIRT2 deacetylase regulates autoacetylation of p300. Mol. Cell 2008, 32, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Lee, E.K.; Kang, J.K.; Kim, J.A.; You, J.S.; Park, J.H.; Seo, D.W.; Hwang, J.W.; Kim, S.N.; Lee, H.Y.; et al. Activation of NF-κB by HDAC inhibitor apicidin through Sp1-dependent de novo protein synthesis: Its implication for resistance to apoptosis. Cell Death Differ. 2006, 13, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Chavey, C.; Muhlbauer, M.; Bossard, C.; Freund, A.; Durand, S.; Jorgensen, C.; Jobin, C.; Lazennec, G. Interleukin-8 expression is regulated by histone deacetylases through the nuclear factor-κB pathway in breast cancer. Mol. Pharmacol. 2008, 74, 1359–2066. [Google Scholar] [CrossRef] [PubMed]
- Abaza, M.S.; Bahman, A.M.; Al-Attiyah, R. Superior antimitogenic and chemosensitization activities of the combination treatment of the histone deacetylase inhibitor apicidin and proteasome inhibitors on human colorectal cancer cells. Int. J. Oncol. 2014, 44, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Friedman, J.; Nottingham, L.; Chen, Z.; Ara, G.; van Waes, C. Nuclear factor-κB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2007, 6, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; von Metzler, I.; Kaiser, M.; Rosche, M.; Sterz, J.; Rotzer, S.; Rademacher, J.; Jakob, C.; Fleissner, C.; Kuckelkorn, U.; et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur. J. Haematol. 2008, 80, 133–142. [Google Scholar] [CrossRef] [PubMed]
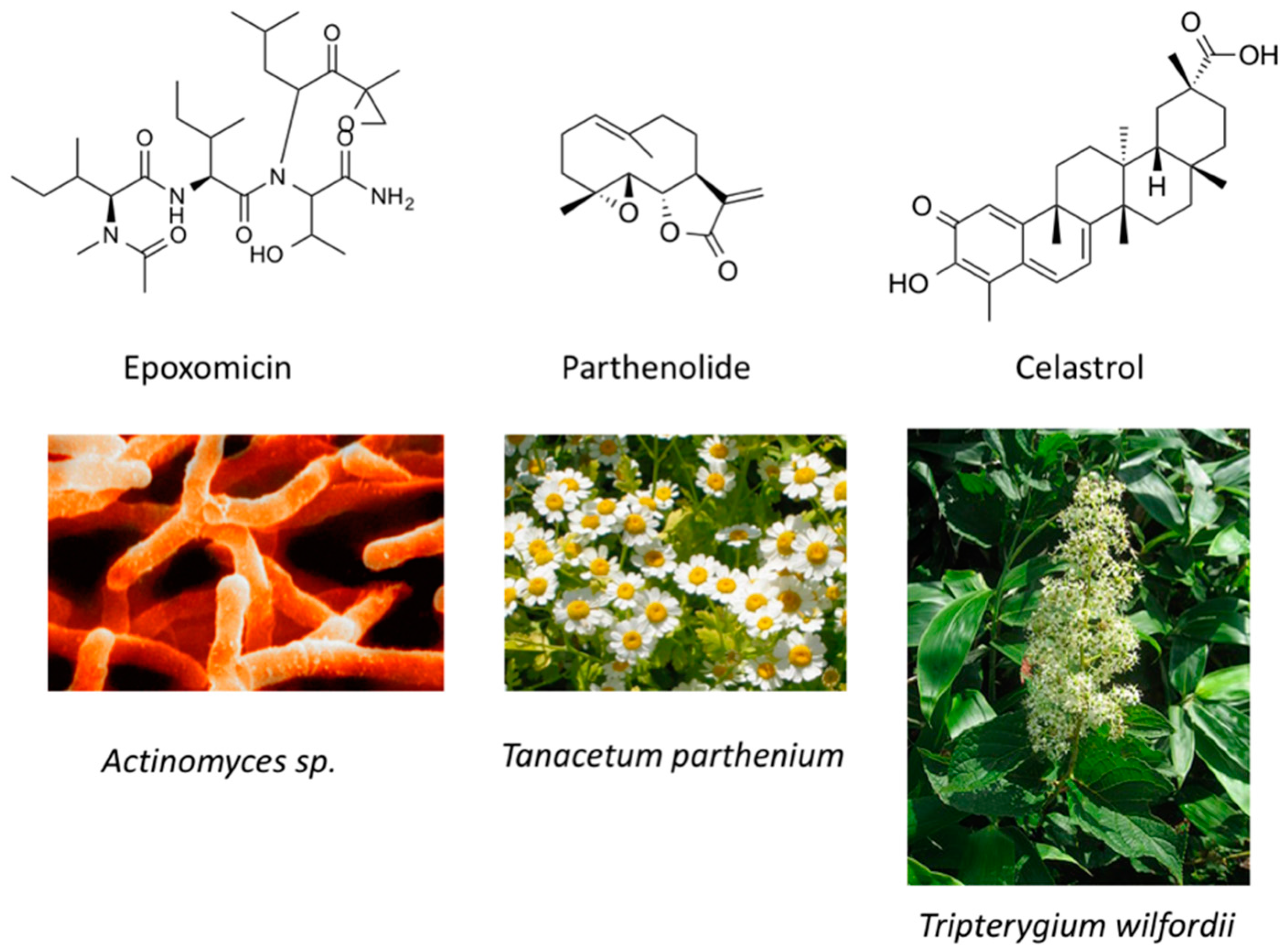
- Dai, Y.; Guzman, M.L.; Chen, S.; Wang, L.; Yeung, S.K.; Pei, X.Y.; Dent, P.; Jordan, C.T.; Grant, S. The NF (Nuclear factor)-κB inhibitor parthenolide interacts with histone deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in human acute myeloid leukaemia cells. Br. J. Haematol. 2010, 151, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Kanaya, N.; Petrossian, K.; Ye, J.; Warden, C.; Liu, Z.; Nishimura, R.; Osako, T.; Okido, M.; Shimada, K.; et al. Inhibition of the proliferation of acquired aromatase inhibitor-resistant breast cancer cells by histone deacetylase inhibitor LBH589 (panobinostat). Breast Cancer Res. Treat. 2013, 137, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Fu, Y.; Zhuang, L.; Gai, R.; Ma, J.; Lou, J.; Zhu, H.; He, Q.; Yang, B. Simultaneous NF-κB inhibition and E-cadherin upregulation mediate mutually synergistic anticancer activity of celastrol and SAHA in vitro and in vivo. Int. J. Cancer 2014, 135, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Kramer, O.H.; Baus, D.; Knauer, S.K.; Stein, S.; Jager, E.; Stauber, R.H.; Grez, M.; Pfitzner, E.; Heinzel, T. Acetylation of Stat1 modulates NF-κB activity. Genes Dev. 2006, 20, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, H.; Fujimura, T.; Mori, H.; Aramori, I.; Mutoh, S. Mechanism of HDAC inhibitor FR235222-mediated IL-2 transcriptional repression in Jurkat cells. Int. Immunopharmacol. 2007, 7, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
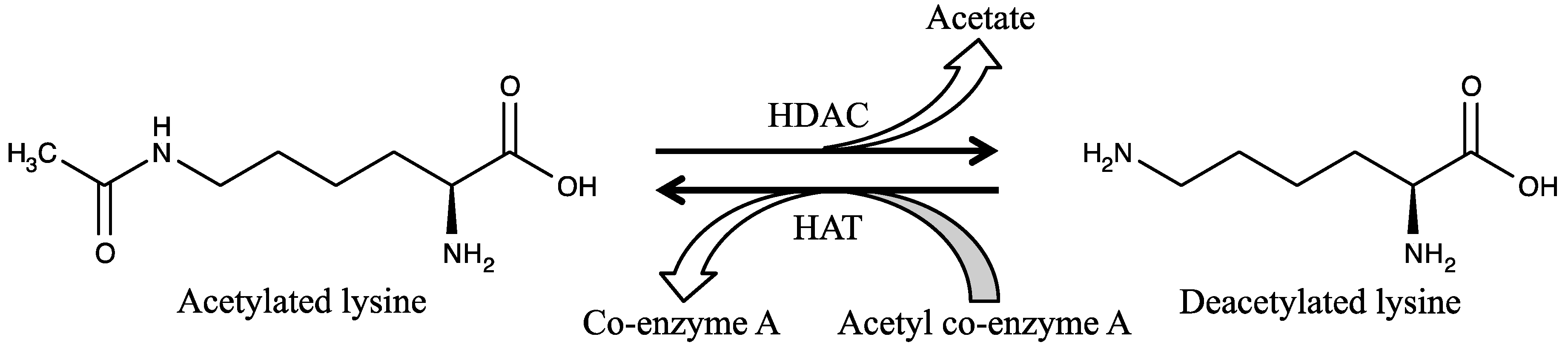
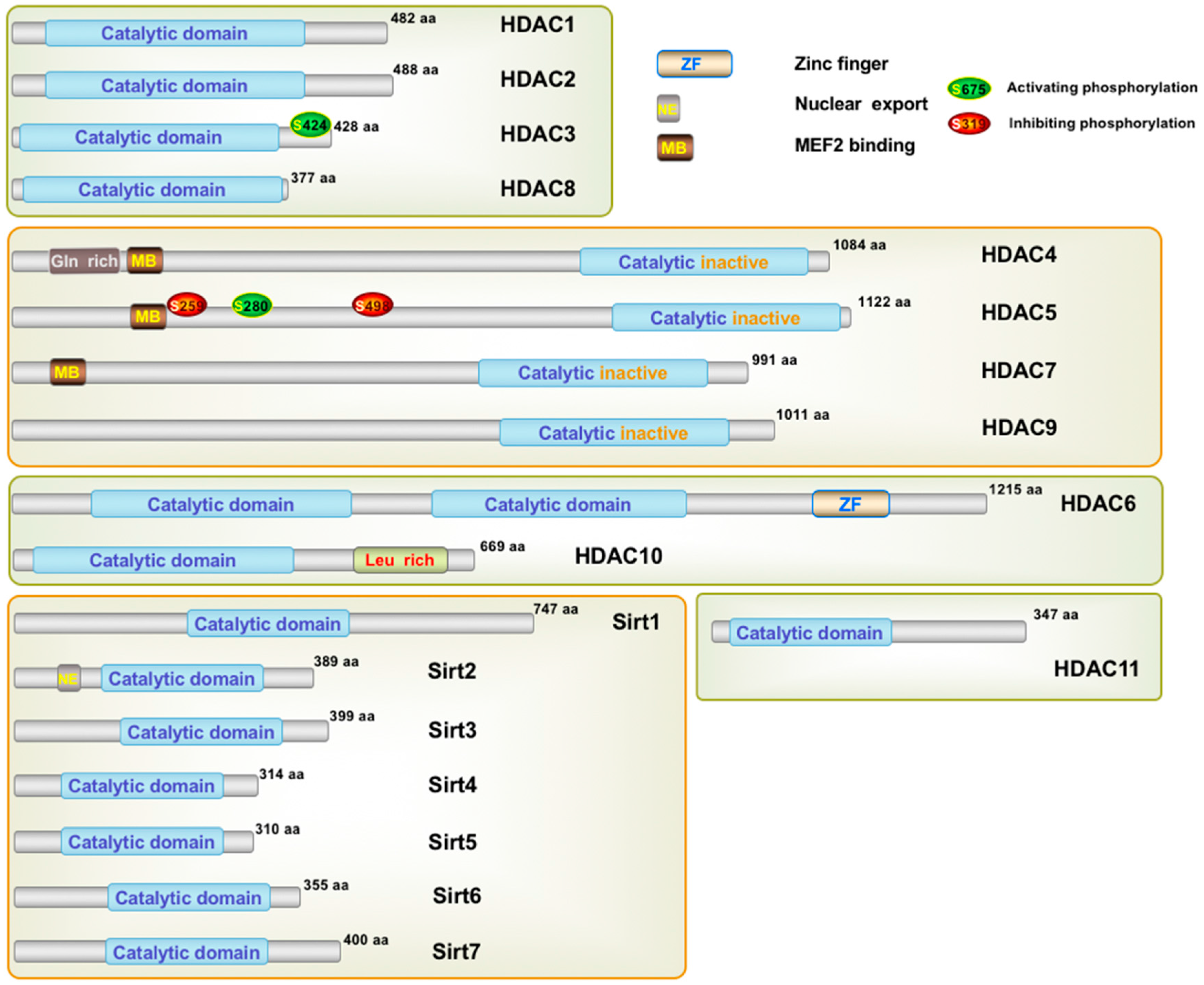
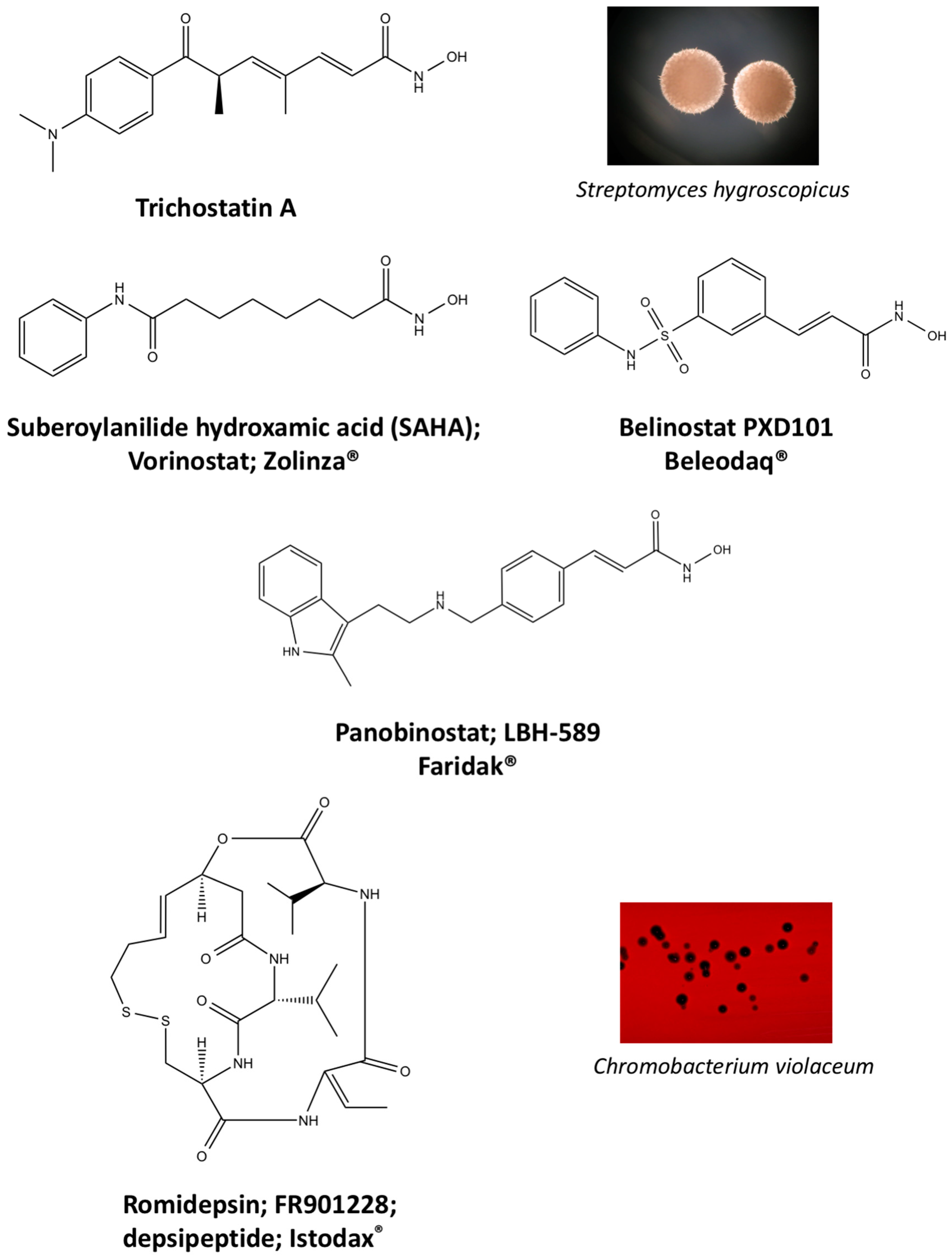

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Members | Size (Amino Acids) | Co-Factor | Cellular Localization | Physiological Function | Expression |
|---|---|---|---|---|---|---|
| I | HDAC1 | 483 | Zn2+ | Nucleus | Cell proliferation and survival | Ubiquitous |
| HDAC2 | 488 | Zn2+ | Nucleus | Cell proliferation and insulin resistance | ||
| HDAC3 | 428 | Zn2+ | Nucleus | Cell proliferation and survival | ||
| HDAC8 | 377 | Zn2+ | Nucleus | Cell proliferation | ||
| IIa | HDAC4 | 1084 | Zn2+ | Nucleus/Cytoplasm | Control of cytoskeletal dynamics and cell mobility | Tissue-specific |
| HDAC5 | 1122 | Zn2+ | Nucleus/Cytoplasm | Gluconeogenesis, cardiovascular growth, and function, and cardiac myocyte and endothelial cell function | ||
| HDAC7 | 912 | Zn2+ | Nucleus/Cytoplasm | Glycogenesis, endothelial function, and thymocyte differentiation | ||
| HDAC9 | 1069 | Zn2+ | Nucleus/Cytoplasm | Cardiovascular growth and function, homologous recombination, and thymocyte differentiation | ||
| IIb | HDAC6 | 1215 | Zn2+ | Cytoplasm | Control of cytoskeletal dynamics and cell mobility | Tissue-specific |
| HDAC10 | 669 | Zn2+ | Cytoplasm | Autophagy-mediated cell survival and homologous recombination | ||
| III | SIRT1 | 747 | NAD+ | Nucleus/Cytoplasm | Autoimmune system regulation, cell survival, aging, and redox regulation | Variable |
| SIRT2 | 389 | NAD+ | Nucleus | Cell survival, cell migration and invasion | ||
| SIRT3 | 399 | NAD+ | Mitochondria | Redox balance, ATP regulation and metabolism, urea cycle, apoptosis, and cell signaling | ||
| SIRT4 | 314 | NAD+ | Mitochondria | ATP regulation and metabolism, energy metabolism, apoptosis and cell signaling | ||
| SIRT5 | 310 | NAD+ | Mitochondria | ATP regulation, urea cycle, energy metabolism, apoptosis, and cell signaling | ||
| SIRT6 | 355 | NAD+ | Nucleus | Metabolic regulation | ||
| SIRT7 | 400 | NAD+ | Nucleus | Apoptosis | ||
| IV | HDAC11 | 347 | Zn2+ | Nucleus | Immunomodulation and DNA replication | Ubiquitous |
| Class | Members | Type of Variation | Cancer Involved | References |
|---|---|---|---|---|
| I | HDAC1 | Overexpression | Gastrointestinal carcinoma | [9] |
| Prostate carcinoma | [9] | |||
| Breast carcinoma | [9] | |||
| Colon adenocarcinoma | [7] | |||
| Chronic lymphocytic leukemia | [10] | |||
| HDAC1 overexpression | Ovarian cancer | [4] | ||
| Gastric cancer | [4] | |||
| Hodgkin’s lymphoma | [4] | |||
| Prostate cancer | [4] | |||
| HDAC1 downregulation | Colorectal cancer | [4] | ||
| HDAC2 | Overexpression | Uterine cancer | [9] | |
| Cervical cancer | [9] | |||
| Gastric cancer | [9] | |||
| HDAC2 overexpression | Ovarian cancer | [4] | ||
| Hodgkin’s lymphoma | [4] | |||
| Truncating HDAC2 mutation | Colon cancer | [4] | ||
| Endometrial cancer | [4] | |||
| Gastric cancer | [4] | |||
| HDAC3 | Overexpression | Colon cancer | [9] | |
| Hodgkin’s lymphoma | [4] | |||
| Chronic lymphocytic leukemia | [10] | |||
| HDAC3 overexpression | Ovarian cancer | [4] | ||
| Lung cancer | [4] | |||
| IIa | HDAC4 | HDAC4 mutations | Breast cancer | [4] |
| Colorectal cancer | [4] | |||
| HDAC4 overexpression | Prostate cancer | [4] | ||
| Breast cancer | [4] | |||
| HDAC4 downregulation | Colon cancer | [16] | ||
| Lung cancer | [16] | |||
| HDAC5 | HDAC5 downregulation | Colorectal cancer | [4] | |
| HDAC7 | Overexpression | Chronic lymphocytic leukemia | [10] | |
| HDAC7 overexpression | Colorectal cancer | [4] | ||
| Pancreatic cancer | [4] | |||
| HDAC9 | Overexpression | Chronic lymphocytic leukemia | [10] | |
| IIb | HDAC6 | Overexpression | Acute lymphoblastic leukemia | [11] |
| Acute myeloid leukemia | [11] | |||
| Breast cancer | [11] | |||
| Chronic lymphocytic leukemia | [11] | |||
| Cutaneous T-cell lymphoma | [11] | |||
| Hepatocellular carcinoma | [11] | |||
| Oral squamous cell carcinoma | [11] | |||
| Ovarian cancer | [11] | |||
| Urothelial cancer | [11] | |||
| HDAC10 | Overexpression | Chronic lymphocytic leukemia | [10] | |
| III | SIRT1 | Overexpression | Acute myeloid leukemia | [12] |
| Skin cancer | [12] | |||
| Colon cancer | [12] | |||
| Prostate cancer | [12] | |||
| Chronic lymphocytic leukemia | [10] | |||
| SIRT1 downregulation | Colorectal cancer | [13] | ||
| SIRT6 | Overexpression | Chronic lymphocytic leukemia | [10] | |
| Downregulation | Liver cancer | [14] | ||
| SIRT7 | Overexpression | Breast cancer | [15] |
| Compound Name | Chemical Category | HDAC Specificity | Origin of the Compound | Structure | Organism | Ref. |
|---|---|---|---|---|---|---|
| Lycorine | Alkaloid | Pan HDAC | Amaryllidaceae |  |  | [42] |
| Burkholdac A | Depsipeptide | Class I HDAC | Burkholderia thailandensis | 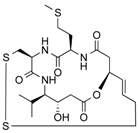 |  | [43] |
| Largazole | Depsipeptide | Class I HDAC HDAC1, 2, 3 | Cyanobacterium Symploca sp. | 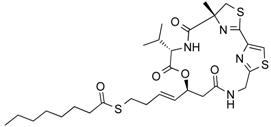 |  | [44] |
| Spiruchostatin A (YM753) | Depsipeptide | Class I HDAC | Pseudomonas sp. | 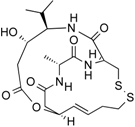 |  | [45] |
| Thailandepsin A (Burkholdac B) | Depsipeptide | Class I HDAC | Burkholderia thailandensis |  |  | [46,47] |
| 9-Hydroxystearic acid | Fatty acid | Class I HDACs | Endogenous lipid peroxidation by-product | 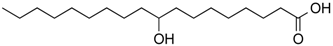 |  | [48] |
| Amamistatin B | Mycobacterial siderophore | Pan HDAC | Nocardia sp. |  |  | [49] |
| Diallyl disulfide | Organosulfur compound | Increased acetylation | Allium sativum | 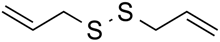 |  | [50] |
| (S)-allylmercaptocysteine | Organosulfur compound | Increased acetylation | Allium sativum | 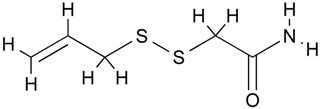 |  | [51] |
| Sulforaphane | Organosulfur compound | Increased acetylation | Brassica oleracea |  |  | [52] |
| Aceroside VIII | Phenolic | HDAC6 | Betula platyphylla | 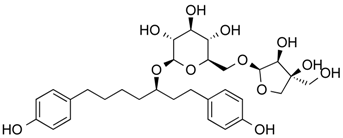 |  | [53] |
| Butein | Phenolic | HDAC classes I, II and IV | Toxicodendron vernicifluum | 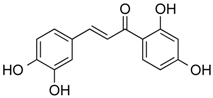 |  | [28] |
| Homobutein | Phenolic | HDAC classes I, II and IV | Butea frondosa | 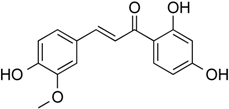 |  | [28] |
| Isoliquiritigenin | Phenolic | HDAC classes I, II and IV | Glycyrrhiza glabra |  |  | [28] |
| Kaempferol | Phenolic | HDACs of class I, II and IV | Aloe vera | 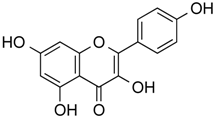 |  | [54] |
| Marein | Phenolic | HDAC classes I, II and IV | Coreopsis maritima | 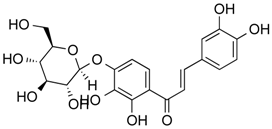 |  | [28] |
| Protocatechuic aldehyde | Phenolic | HDAC2 | Hordeum vulgare | 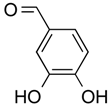 |  | [55] |
| Psammaplin A | Phenolic | Class I HDAC | Poecillastra sp. and Jaspis sp. |  |  | [56] |
| Resveratrol | Phenolic | HDACs of class I, II and IV | Vitis vinifera | 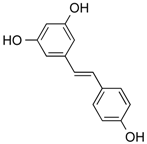 |  | [57] |
| Sinapinic acid | Phenolic | Pan HDAC | Hydnophytum formicarum Jack. | 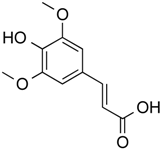 |  | [58] |
| Depudecin | Polyketide | HDAC1 | Alternaria brassicicola | 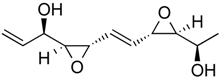 | 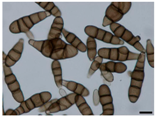 | [59] |
| Epicocconigrone A | Polyketide | HDACs of class I, IIb and IV | Epicoccum nigrum | 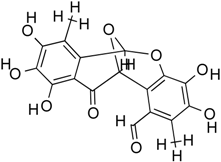 |  | [60] |
| 20(S)-Ginsenoside Rh2 | Steroid glycoside | HDAC1, HDAC2, HDAC6 | Panax ginseng | 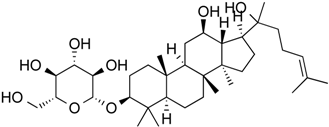 |  | [61] |
| 6-methoxy-2E,9E-humuladien-8-one | Terpenoid (sesqui-) | Pan HDAC | Zingiber zerumbet (L.) J. E. Smith | 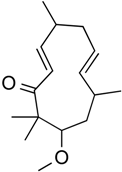 |  | [62] |
| Zerumbone | Terpenoid (sesqui-) | Pan HDAC | Zingiber zerumbet (L.) J. E. Smith | 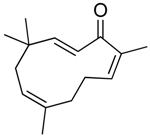 |  | [62] |
| β-Thujaplicin (hinokitiol) | Terpenoid (mono) | HDAC2 | Cupressaceae sp. |  |  | [63] |
| β-boswellic acid derivatives | Terpenoid (pentacyclic tri-) | HDAC8 | Boswellia sacra | 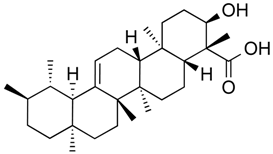 |  | [64] |
| Apicidin | Tetrapeptide | Class I HDAC HDAC1, 2, 3, 8 | Fusarium sp. | 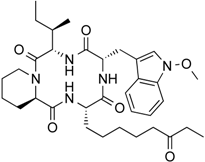 |  | [65] |
| Azumamide E | Tetrapeptide | Class I HDAC HDAC1, 2, 3 | Mycale izuensis |  |  | [66] |
| Chlamydocin | Tetrapeptide | HDAC1, 6 | Diheterospora chlamydosporia | 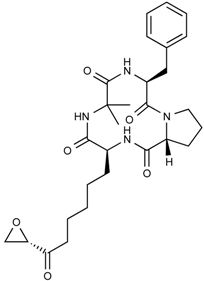 |  | [67] |
| Trapoxin A | Tetrapeptide | Class I HDAC | Helicoma ambiens RF-1023 | 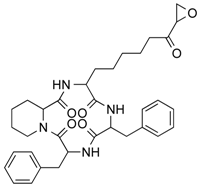 |  | [68,69] |
| Trapoxin B | Tetrapeptide | Class I HDAC | Helicoma ambiens RF-1023 |  |  | [68,69] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608. https://doi.org/10.3390/molecules21111608
Losson H, Schnekenburger M, Dicato M, Diederich M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules. 2016; 21(11):1608. https://doi.org/10.3390/molecules21111608
Chicago/Turabian StyleLosson, Hélène, Michael Schnekenburger, Mario Dicato, and Marc Diederich. 2016. "Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer" Molecules 21, no. 11: 1608. https://doi.org/10.3390/molecules21111608
APA StyleLosson, H., Schnekenburger, M., Dicato, M., & Diederich, M. (2016). Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules, 21(11), 1608. https://doi.org/10.3390/molecules21111608