
Deprotection Reagents in Fmoc Solid Phase Peptide Synthesis: Moving Away from Piperidine?

 ,
,  ,
,
Abstract
: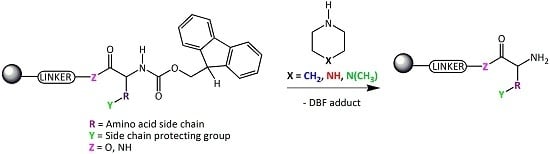
1. Introduction
2. Results
2.1. Yield and Purity of Synthesized Peptides
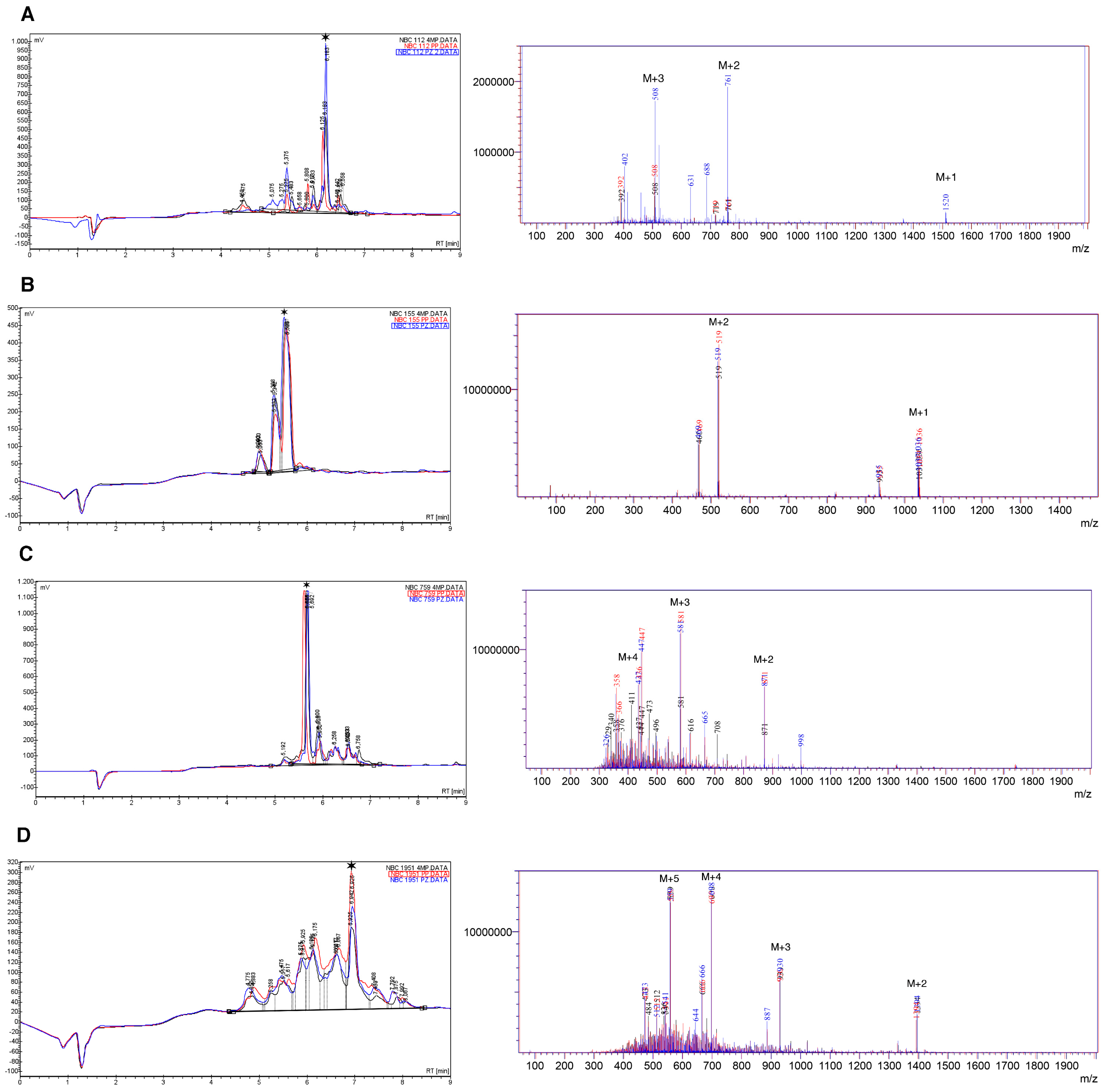
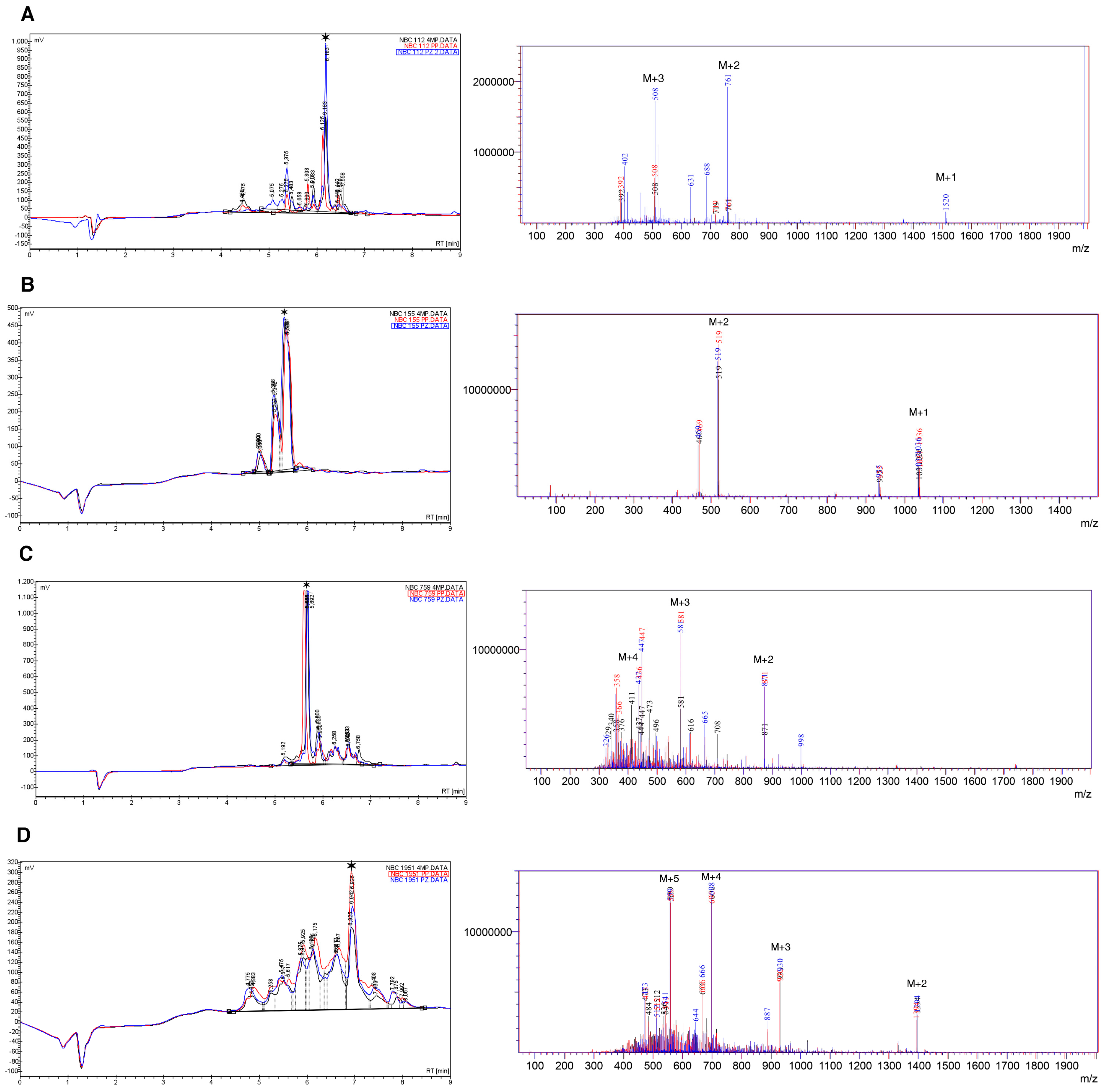
2.2. HPLC and Mass Spectrometry
- (1)
- NBC112: Alanine and histidine deletion with 4MP and PZ with a %area higher than 10%.
- (2)
- NBC759: Lysine deletion with a %area higher than 10% in all cases.
- (3)
- NBC1951: Glutamine/lysine deletion with a %area higher than 10% in all cases, the two amino acids co-eluted in the chromatogram (Figure S4 peak 1).
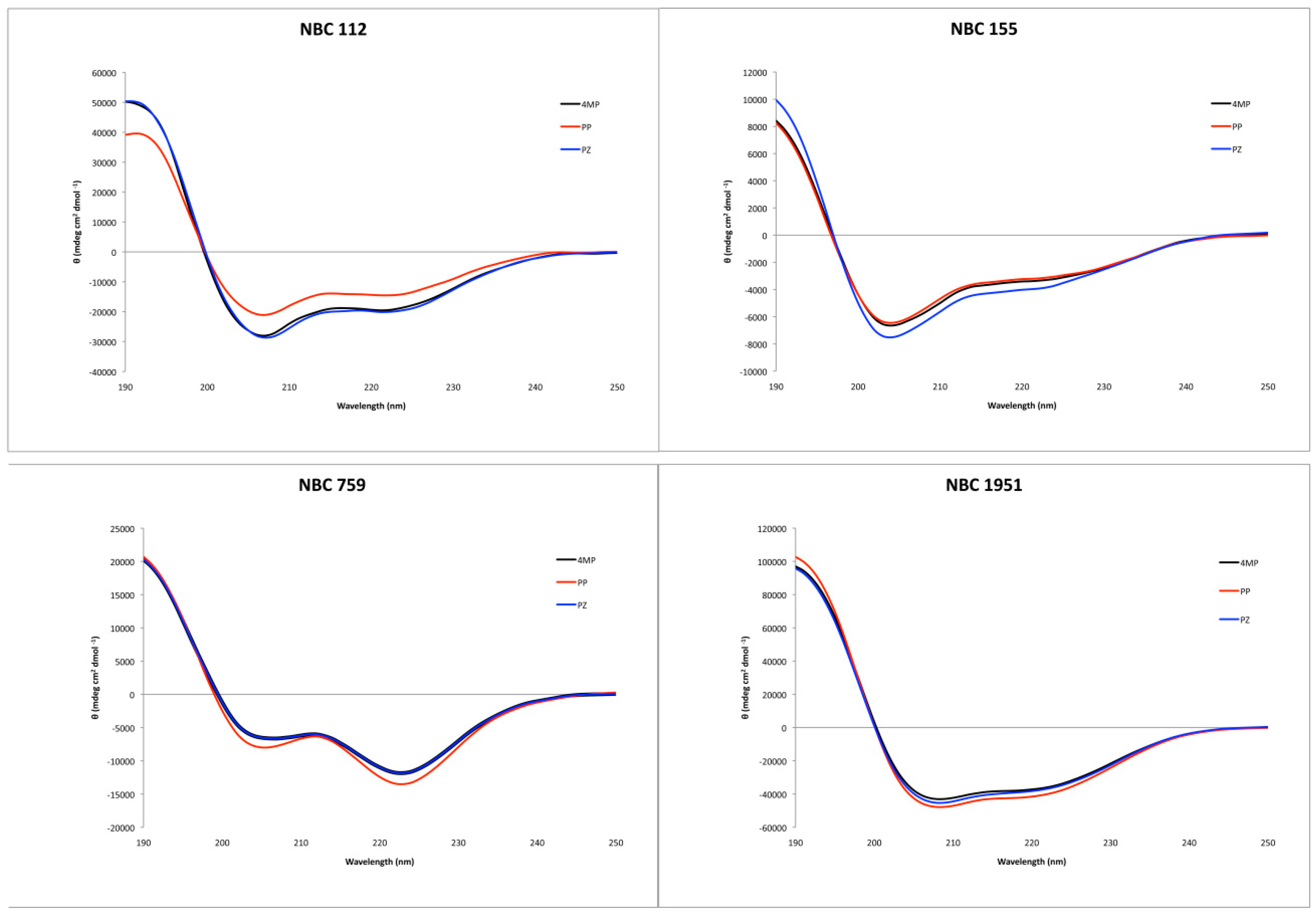
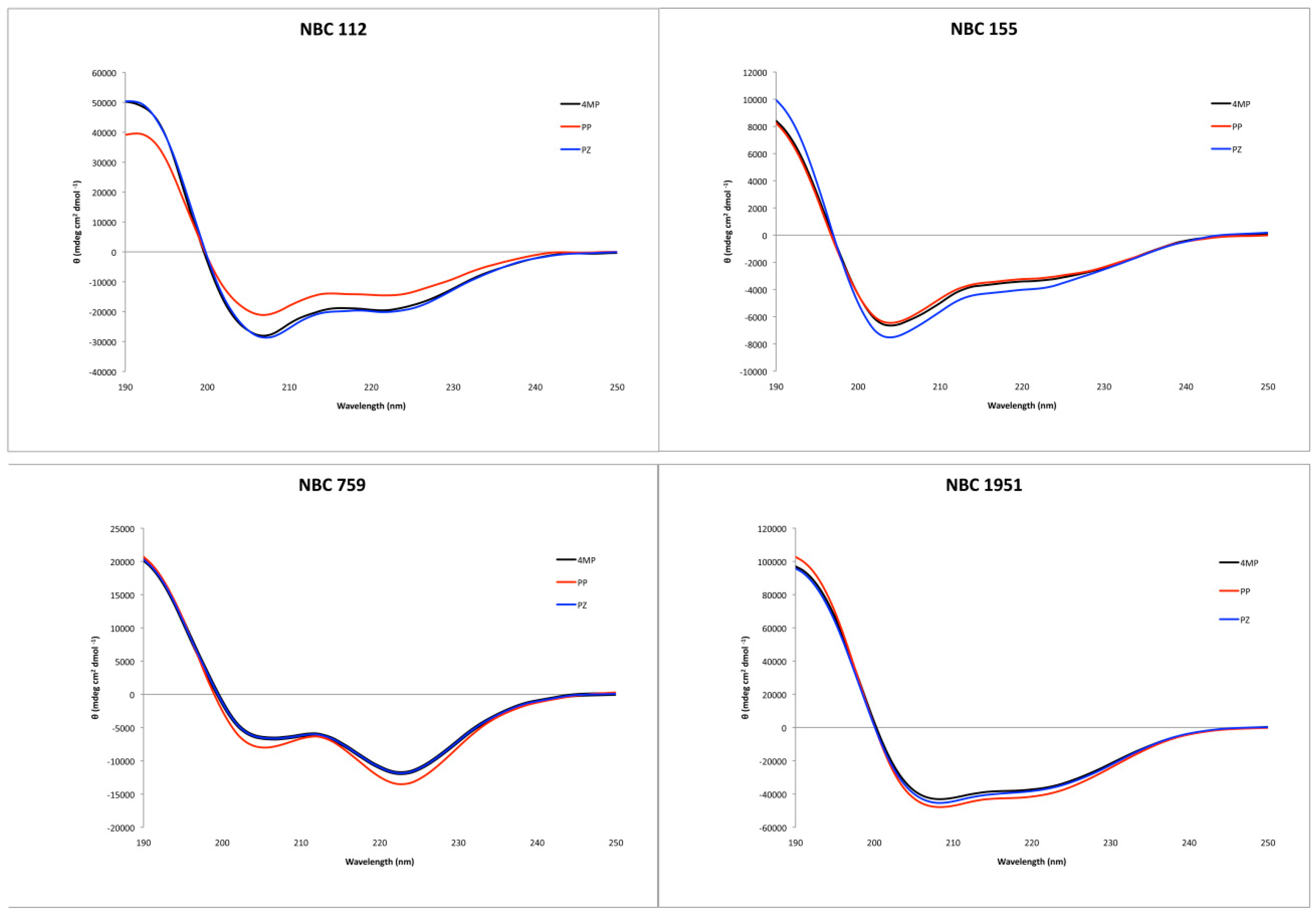
2.3. Secondary Structure Characterization
2.4. Physicochemical Properties
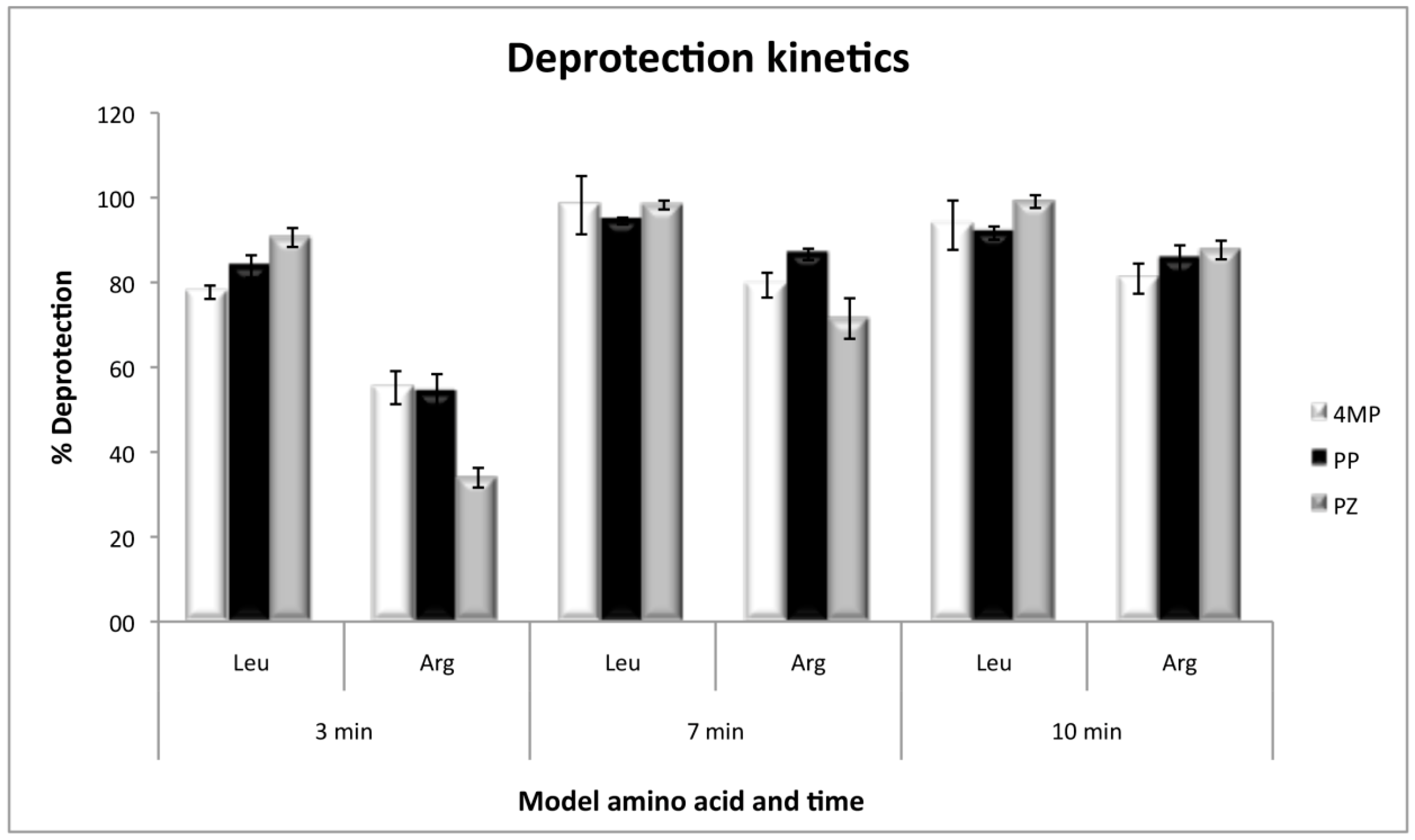
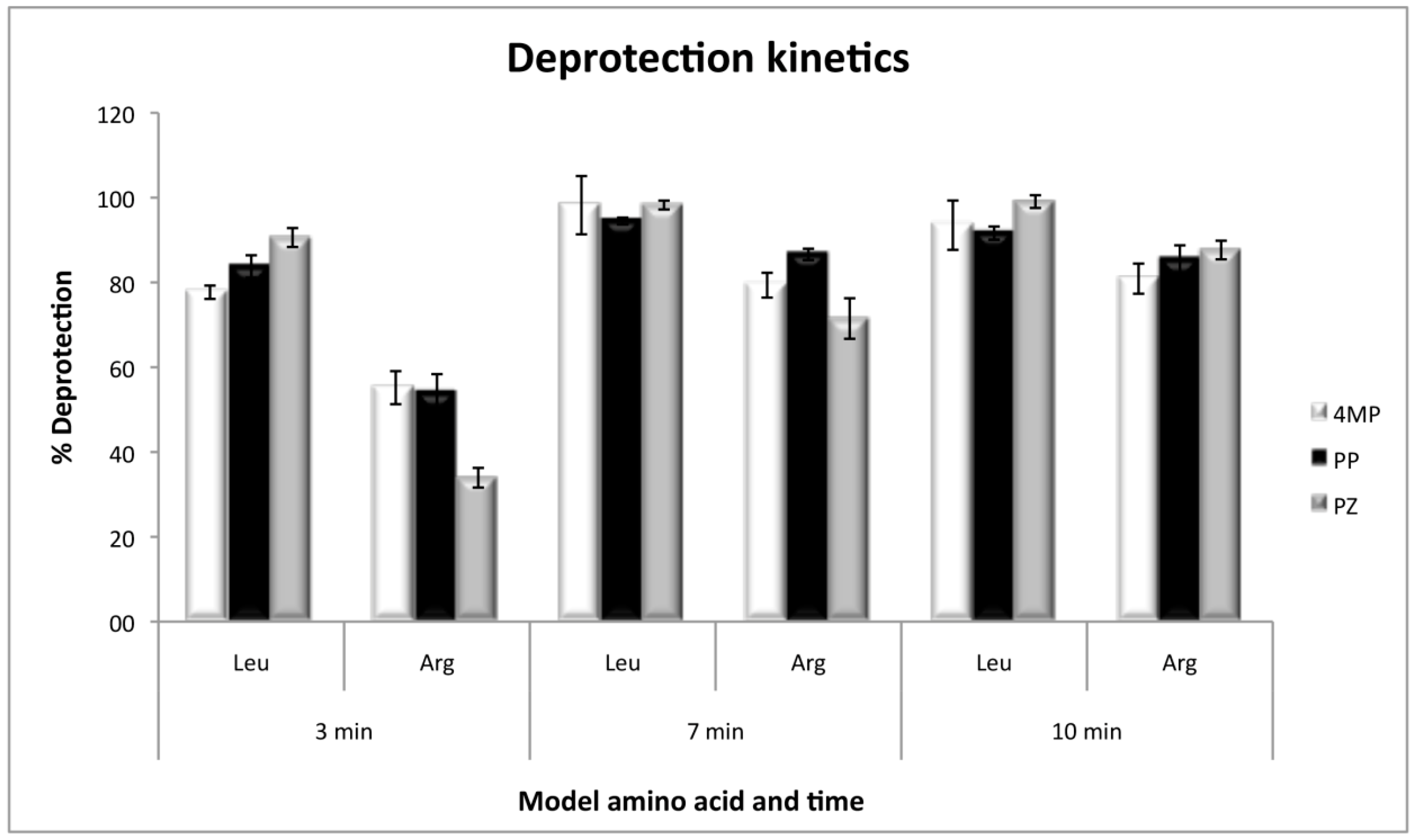
2.5. Deprotection Kinetics
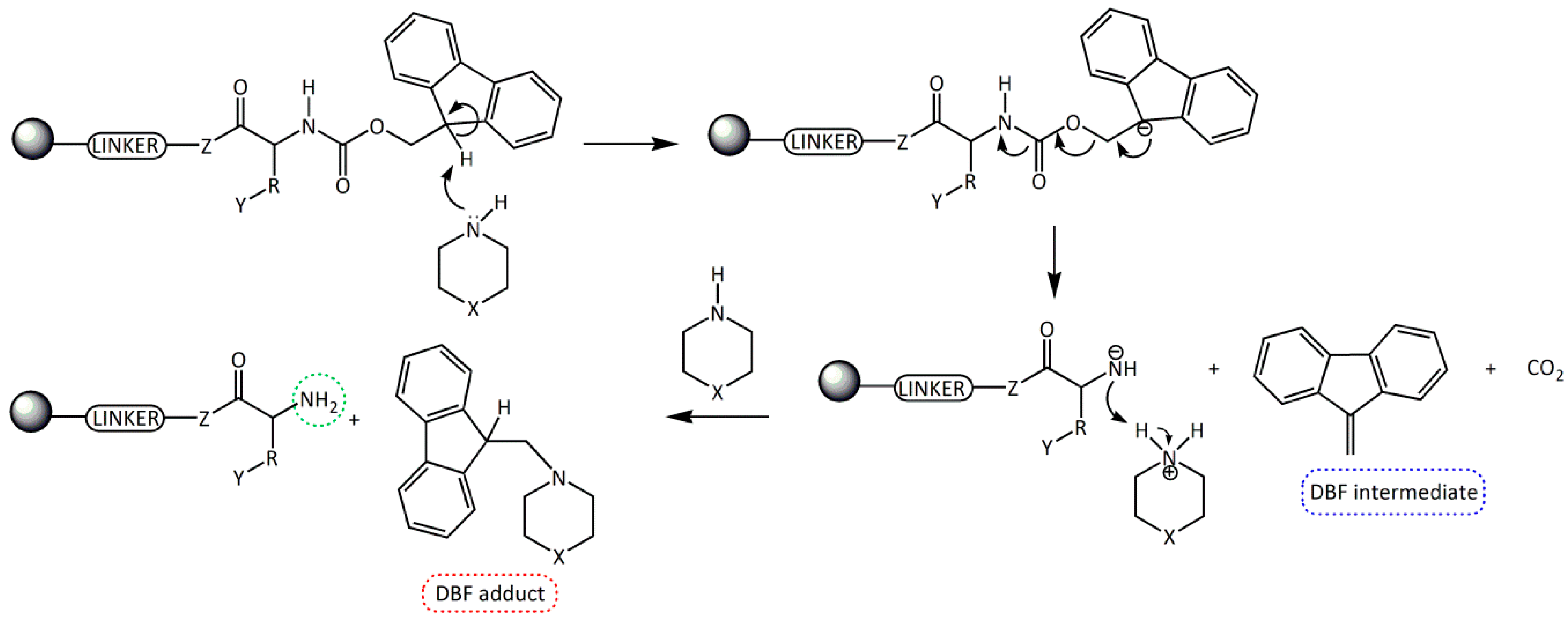
3. Discussion
4. Materials and Methods
4.1. Reagents and Solvents
4.2. Peptide Synthesis
- (1)
- Total crude yield calculated as the percentage of total crude obtained versus the theoretical maximum attainable (0.1 mmol according the scale used in the Liberty Blue synthesizer).
- (2)
- Purity calculated automatically by integrating the area under the curve for the main peak, assigned upon identification by mass spectrometry, in the chromatogram (Tables S1–S4).
- (3)
- Peptide-specific yield calculated as the ratio between the obtained weight in mg with the theoretical one, multiplied by the purity (%area) of the main peak.
4.3. Enrichment of the Main Product
4.4. Characterization
4.4.1. HPLC
4.4.2. Mass Spectrometry
4.4.3. Circular Dichroism Spectroscopy
4.4.4. Theoretical Physicochemical Properties
4.5. Deprotection Kinetics Assay
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carpino, L.; Han, G. The 9-Fluorenylmethoxycarbonyl Amino-Protecting Group. J. Org. Chem. 1979, 44, 3739–3739. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide coupling reagents, more than a letter soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef] [PubMed]
- Kimmerlin, T.; Seebach, D. “100 years of peptide synthesis”: Ligation methods for peptide and protein synthesis with applications to β-peptide assemblies*: 100 Years of peptide synthesis. J. Pept. Res. 2008, 65, 229–260. [Google Scholar] [CrossRef] [PubMed]
- Guzman, F.; Barberis, S.; Illanes, A. Peptide synthesis: Chemical or enzymatic. Electron. J. Biotechnol. 2007, 10, 279–314. [Google Scholar] [CrossRef]
- Fields, G.B.; Noble, R.L. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.A.T.; Bemquerer, M.P.; do Nascimento, C.J. Some Mechanistic Aspects on Fmoc Solid Phase Peptide Synthesis. Int. J. Pept. Res. Ther. 2014, 20, 53–69. [Google Scholar] [CrossRef]
- Zinieris, N.; Leondiadis, L.; Ferderigos, N. N α-Fmoc Removal from Resin-Bound Amino Acids by 5% Piperidine Solution. J. Comb. Chem. 2005, 7, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, J.; Lebl, M. Alternative to piperidine in Fmoc solid-phase synthesis. J. Comb. Chem. 2006, 8, 149. [Google Scholar] [CrossRef] [PubMed]
- Vergel Galeano, C.F.; Rivera Monroy, Z.J.; Rosas Pérez, J.E.; García Castañeda, J.E. Efficient Synthesis of Peptides with 4-Methylpiperidine as Fmoc Removal Reagent by Solid Phase Synthesis. J. Mex. Chem. Soc. 2014, 58, 386–392. [Google Scholar]
- Ralhan, K.; KrishnaKumar, V.G.; Gupta, S. Piperazine and DBU: A safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Adv. 2015, 5, 104417–104425. [Google Scholar] [CrossRef]
- Wade, J.D.; Mathieu, M.N.; Macris, M.; Tregear, G.W. Base-induced side reactions in Fmoc-solid phase peptide synthesis: Minimization by use of piperazine as N-α-deprotection reagent. Lett. Pept. Sci. 2000, 7, 107–112. [Google Scholar] [CrossRef]
- Palasek, S.A.; Cox, Z.J.; Collins, J.M. Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. J. Pept. Sci. 2007, 13, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Prieto, I.; Hervás-Stubbs, S.; García-Granero, M.; Berasain, C.; Riezu-Boj, J.I.; Lasarte, J.J.; Sarobe, P.; Prieto, J.; Borrás-Cuesta, F. Simple strategy to induce antibodies of distinct specificity: Application to the mapping of gp120 and inhibition of HIV-1 infectivity. Eur. J. Immunol. 1995, 25, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Santana, P.A.; Álvarez, C.A.; Guzmán, F.; Mercado, L. Development of a sandwich ELISA for quantifying hepcidin in Rainbow trout. Fish Shellfish Immunol. 2013, 35, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Diez, H.; Lopez, M.C.; Del Carmen Thomas, M.; Guzman, F.; Rosas, F.; Velazco, V.; Gonzalez, J.M.; Puerta, C. Evaluation of IFN-gamma production by CD8+ T lymphocytes in response to the K1 peptide from KMP-11 protein in patients infected with Trypanosoma cruzi. Parasite Immunol. 2006, 28, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Díez, H.; Guzmán, F.; Alba, M.P.; Cuéllar, A.; Thomas, M.C.; López, M.C.; Rosas, F.; Velasco, V.; González, J.M.; Patarroyo, M.E.; et al. Immunological and structural characterization of an epitope from the Trypanosoma cruzi KMP-11 protein. Peptides 2007, 28, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Murillo, L.A.; Lan, C.-Y.; Agabian, N.M.; Larios, S.; Lomonte, B. Fungicidal activity of a phospholipase-A2-derived synthetic peptide variant against Candida albicans. Rev. Esp. Quimioter. 2007, 20, 330–333. [Google Scholar] [PubMed]
- Segura, C.; Guzmán, F.; Salazar, L.M.; Patarroyo, M.E.; Orduz, S.; Lemeshko, V. BTM-P1 polycationic peptide biological activity and 3D-dimensional structure. Biochem. Biophys. Res. Commun. 2007, 353, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Hopp, T.P.; Woods, K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3824–3828. [Google Scholar] [CrossRef] [PubMed]
- Bachem Peptide Calculator. Available online: http://www.bachem.com/service-support/peptide-calculator/ (accessed on 5 September 2016).
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Cabrele, C.; Vanejews, M.; Schreiner, P.R. γ-Aminoadamantanecarboxylic Acids through Direct C–H Bond Amidations. Eur. J. Org. Chem. 2007, 2007, 1474–1490. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the synthetic peptides are available by request from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | MW (Da) | Internal ID | Reference |
|---|---|---|---|
| H-FISEAIIHVLHSR-NH2 | 1520.8 | NBC112 | [14,15] |
| H-TLEEFSAKL-NH2 | 1036.2 | NBC155 | [16,17] |
| H-KKWRWWLKALAKK-NH2 | 1741.2 | NBC759 | [18] |
| H-VAPIAKYLATALAKWALKQGFAKLKS-NH2 | 2787.4 | NBC1951 | [19] |
| Peptide | Total Crude Yield (%) | Purity (%) | Peptide-Specific Yield (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 4MP | PP | PZ | 4MP | PP | PZ | 4MP | PP | PZ | |
| NBC112 | 73.2 | 72.0 | 53.6 | 47.6 | 43.6 | 57.7 | 34.9 | 31.3 | 31.0 |
| NBC155 | 99.1 | 98.4 | 99.4 | 65.1 | 83.0 | 74.8 | 64.5 | 81.7 | 74.3 |
| NBC759 | 93.6 | 81.6 | 74.7 | 50.4 | 59.1 | 55.6 | 44.4 | 48.2 | 41.5 |
| NBC1951 | 68.5 | 79.3 | 68.2 | 20.6 | 29.0 | 21.4 | 14.1 | 23.8 | 14.6 |
| m/z | NBC112 | NBC155 | NBC759 | NBC1951 | ||||
|---|---|---|---|---|---|---|---|---|
| Theoretical | Obtained | Theoretical | Obtained | Theoretical | Obtained | Theoretical | Obtained | |
| M+1 | 1521 | 1521 * | 1036 | 1036 | 1741 | 2787 | ||
| M+2 | 761 | 761 | 519 | 519 | 872 | 871 | 1395 | 1394 |
| M+3 | 508 | 508 | 346 | 581 | 581 | 930 | 930 | |
| M+4 | 381 | 260 | 436 | 436 | 698 | 698 | ||
| M+5 | 305 | 208 | 349 | 559 | 559 | |||
| Peptide | m/z | ID | 4MP | PP | PZ |
|---|---|---|---|---|---|
| NBC112 | 1520.9 | Molecular ion | X | X | X |
| 1449.8 | Alanine deletion | X | X | ||
| 1383.7 | Histidine deletion | X | X | ||
| NBC155 | 1036.6 | Molecular ion | X | X | X |
| NBC759 | 1742.1 | Molecular ion | X | X | X |
| 1613.9 | Lysine deletion | X | X | X | |
| NBC1951 | 2787.4 | Molecular ion | X | X | X |
| 2659.3 | Glutamine/Lysine deletion | X | X | X |
| Peptide | #Res | Hopp and Woods Hydrophobicity | Hydrophilic Res/Total Res Ratio |
|---|---|---|---|
| NBC112 | 13 | 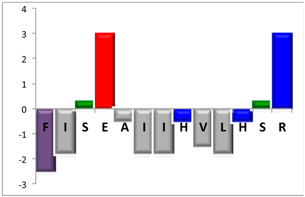 | 31% [2.38] |
| NBC155 | 9 | 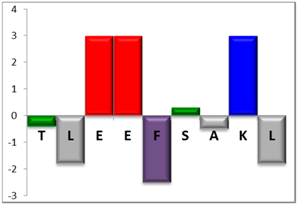 | 44% [4.89] |
| NBC759 | 13 | 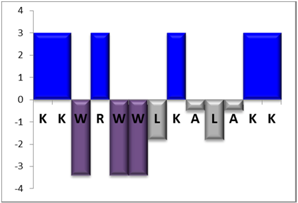 | 46% [3.54] |
| NBC1951 | 26 | 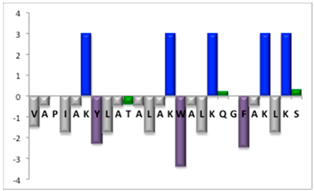 | 27% [1.04] |
| Step | Temperature (°C) | Power (W) | Hold Time (s) |
|---|---|---|---|
| Deprotection | 75 | 155 | 15 |
| 90 | 30 | 50 | |
| Coupling | 75 | 170 | 15 |
| 90 | 30 | 110 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna, O.F.; Gomez, J.; Cárdenas, C.; Albericio, F.; Marshall, S.H.; Guzmán, F. Deprotection Reagents in Fmoc Solid Phase Peptide Synthesis: Moving Away from Piperidine? Molecules 2016, 21, 1542. https://doi.org/10.3390/molecules21111542
Luna OF, Gomez J, Cárdenas C, Albericio F, Marshall SH, Guzmán F. Deprotection Reagents in Fmoc Solid Phase Peptide Synthesis: Moving Away from Piperidine? Molecules. 2016; 21(11):1542. https://doi.org/10.3390/molecules21111542
Chicago/Turabian StyleLuna, Omar F., Johana Gomez, Constanza Cárdenas, Fernando Albericio, Sergio H. Marshall, and Fanny Guzmán. 2016. "Deprotection Reagents in Fmoc Solid Phase Peptide Synthesis: Moving Away from Piperidine?" Molecules 21, no. 11: 1542. https://doi.org/10.3390/molecules21111542
APA StyleLuna, O. F., Gomez, J., Cárdenas, C., Albericio, F., Marshall, S. H., & Guzmán, F. (2016). Deprotection Reagents in Fmoc Solid Phase Peptide Synthesis: Moving Away from Piperidine? Molecules, 21(11), 1542. https://doi.org/10.3390/molecules21111542