
Simultaneous Determination of Coumarin and Its Derivatives in Tobacco Products by Liquid Chromatography-Tandem Mass Spectrometry

Abstract
:1. Introduction
2. Results and Discussion
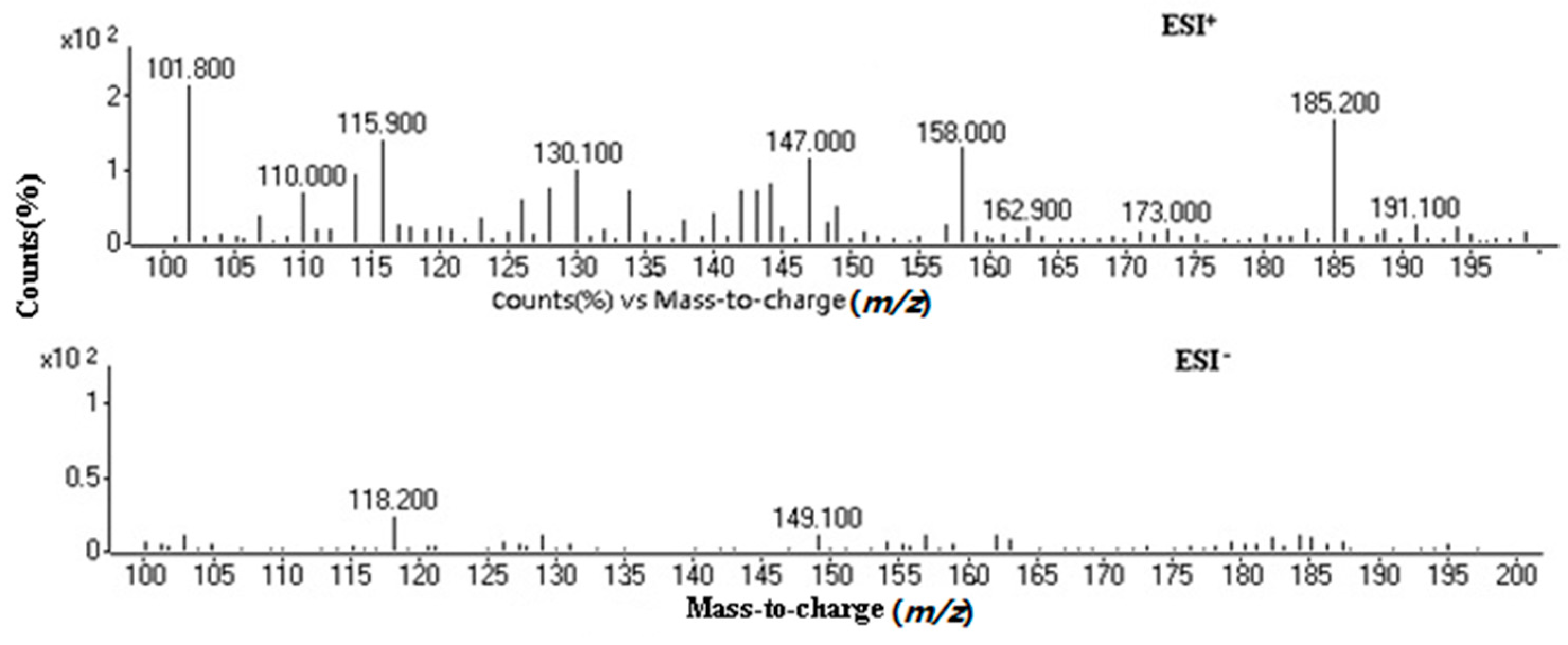
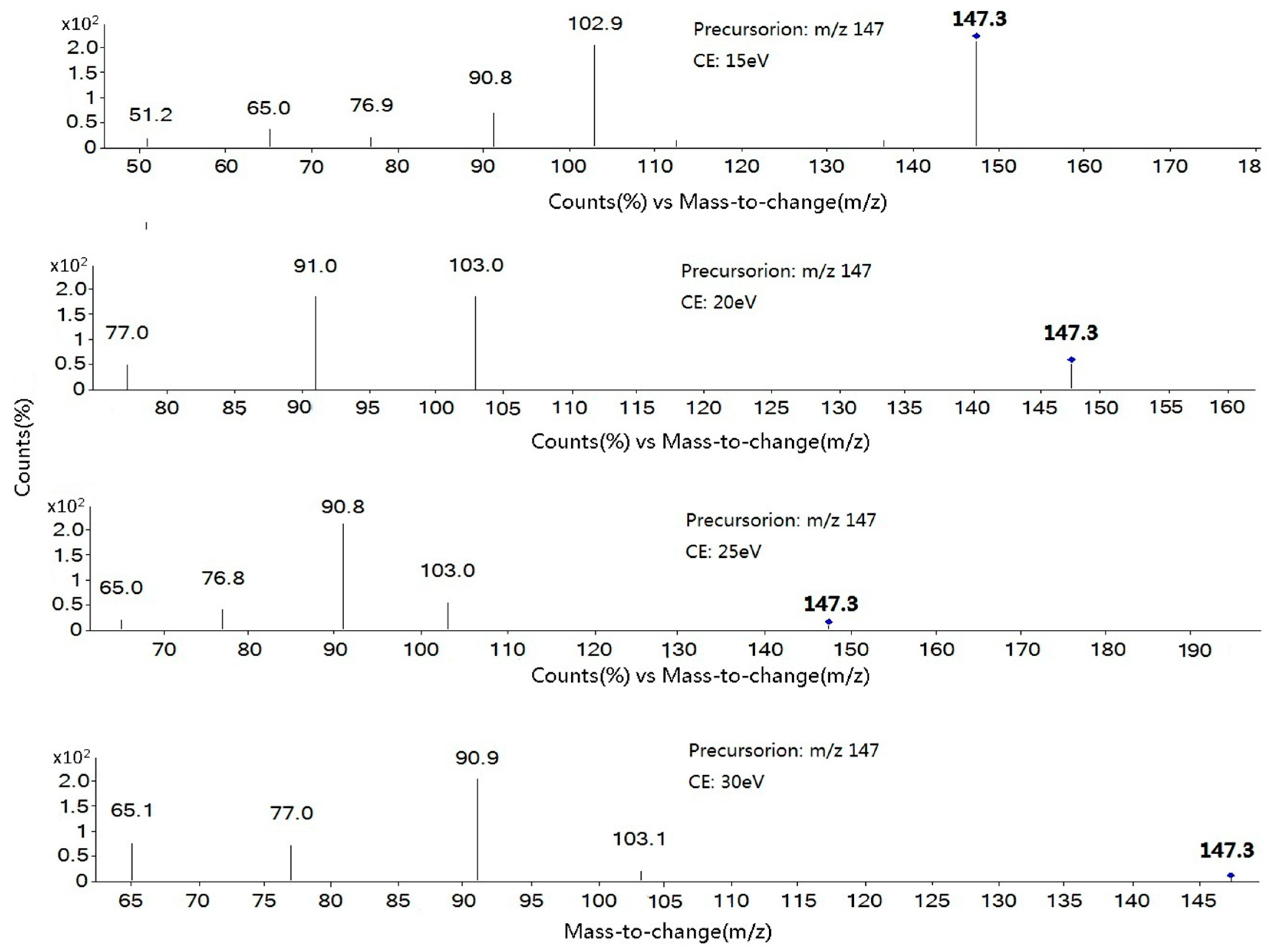
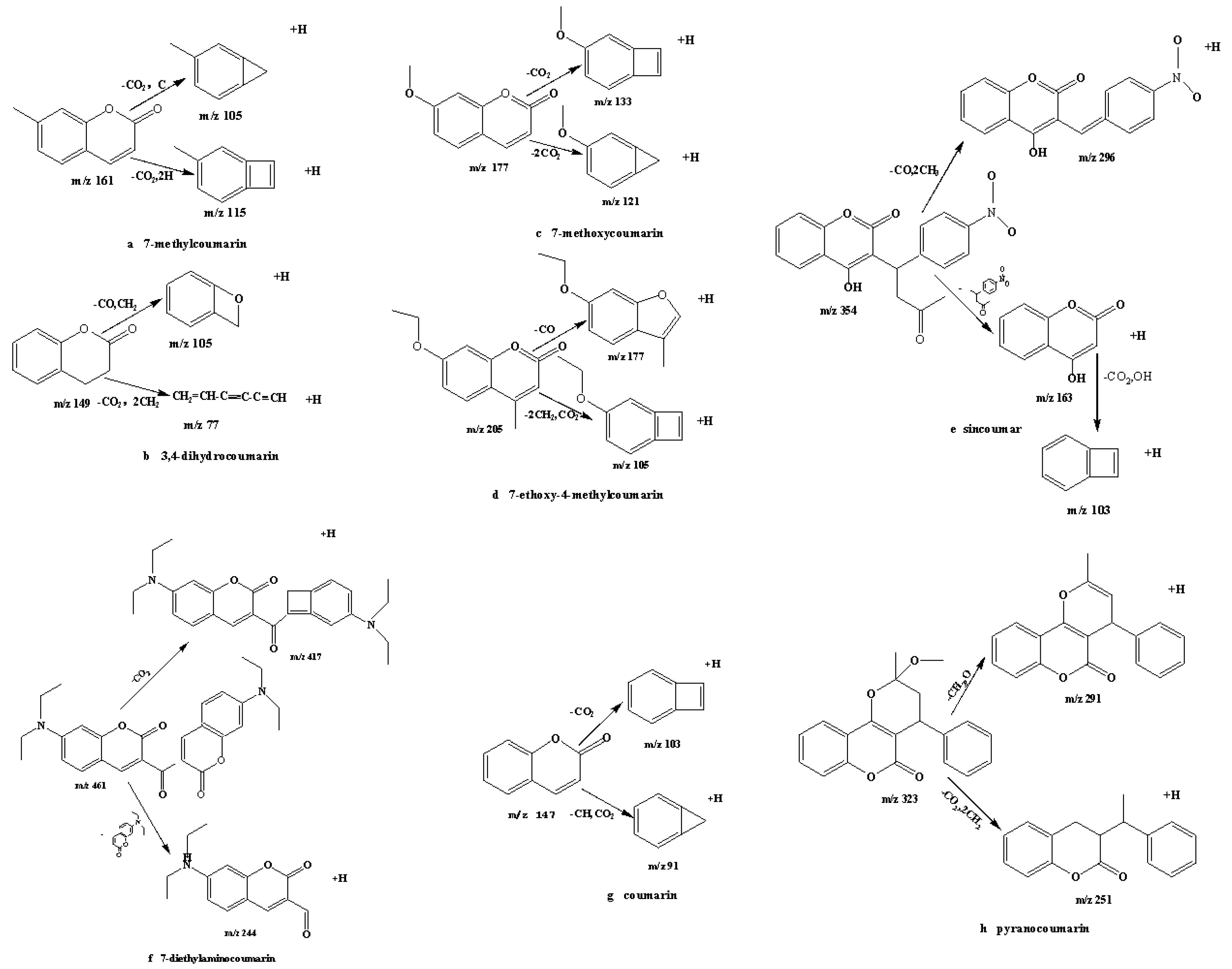
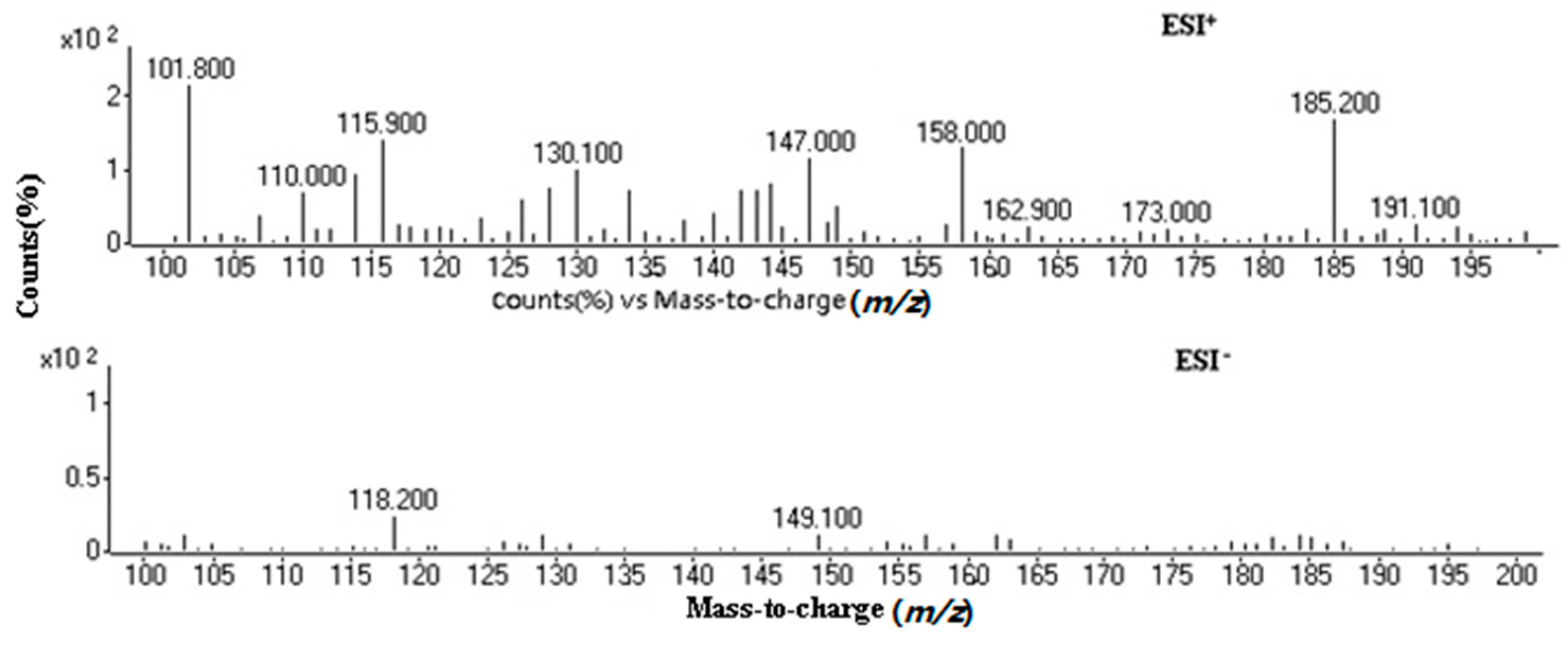
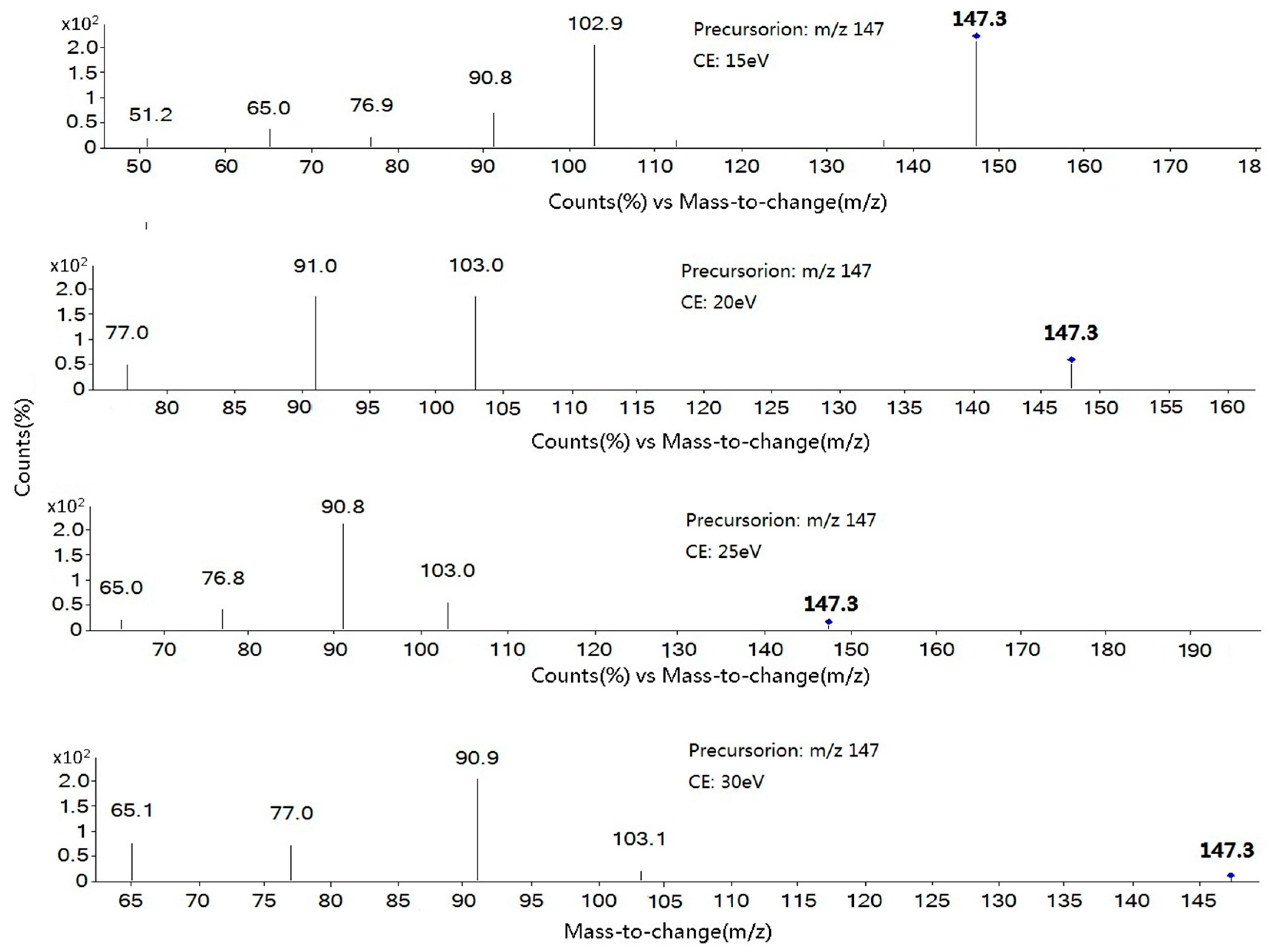
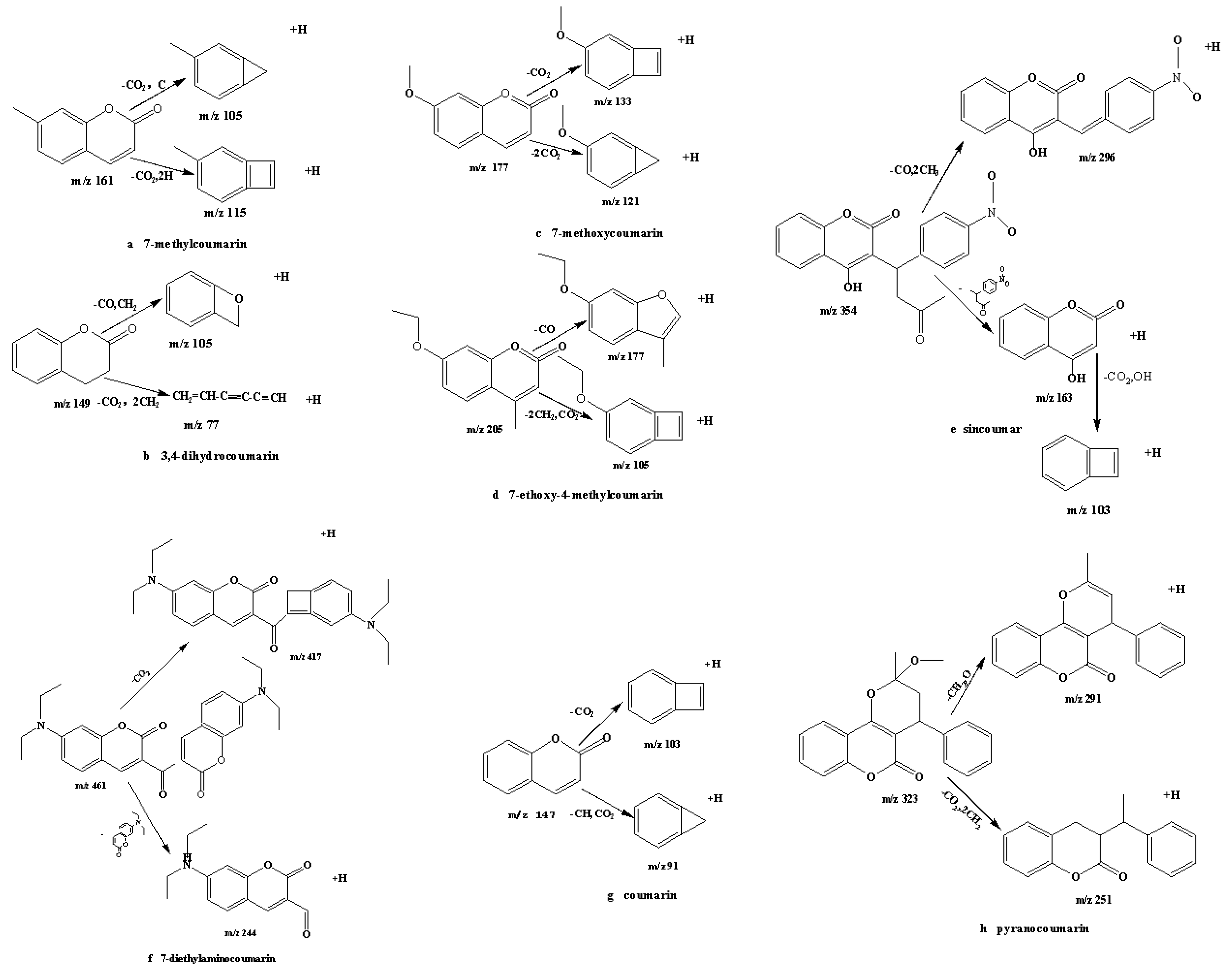
2.1. Optimization of Mass Spectrometry
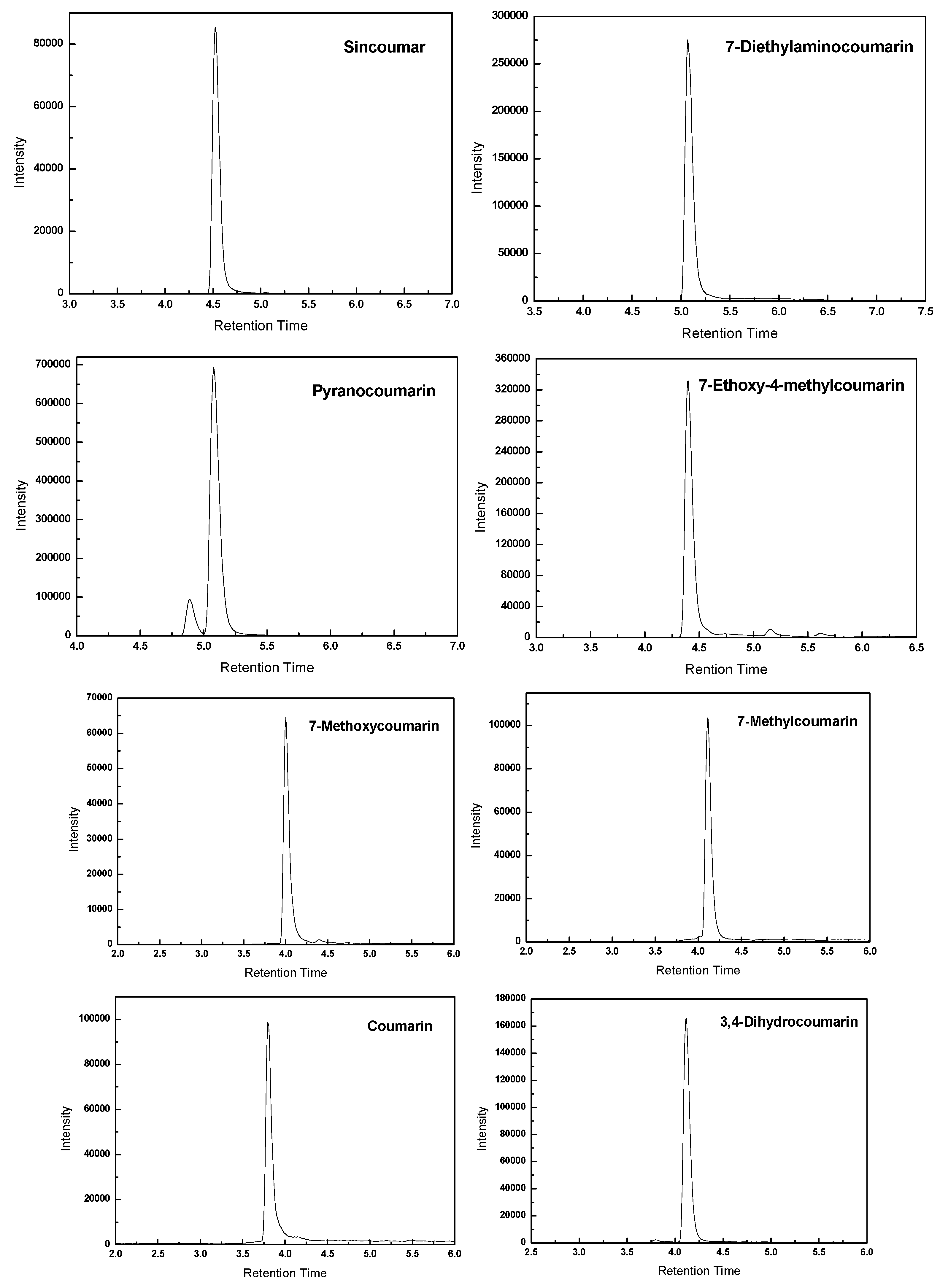
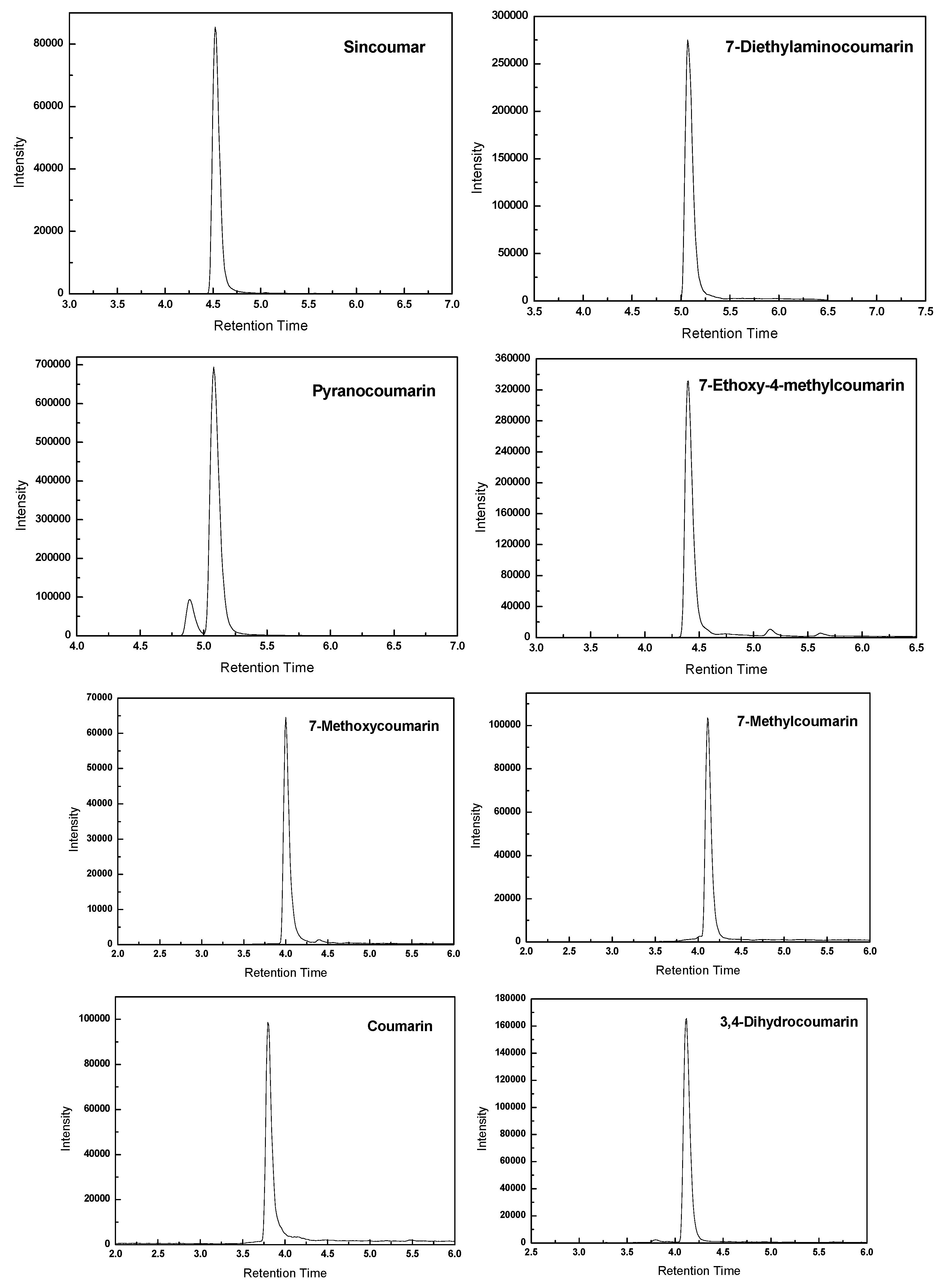
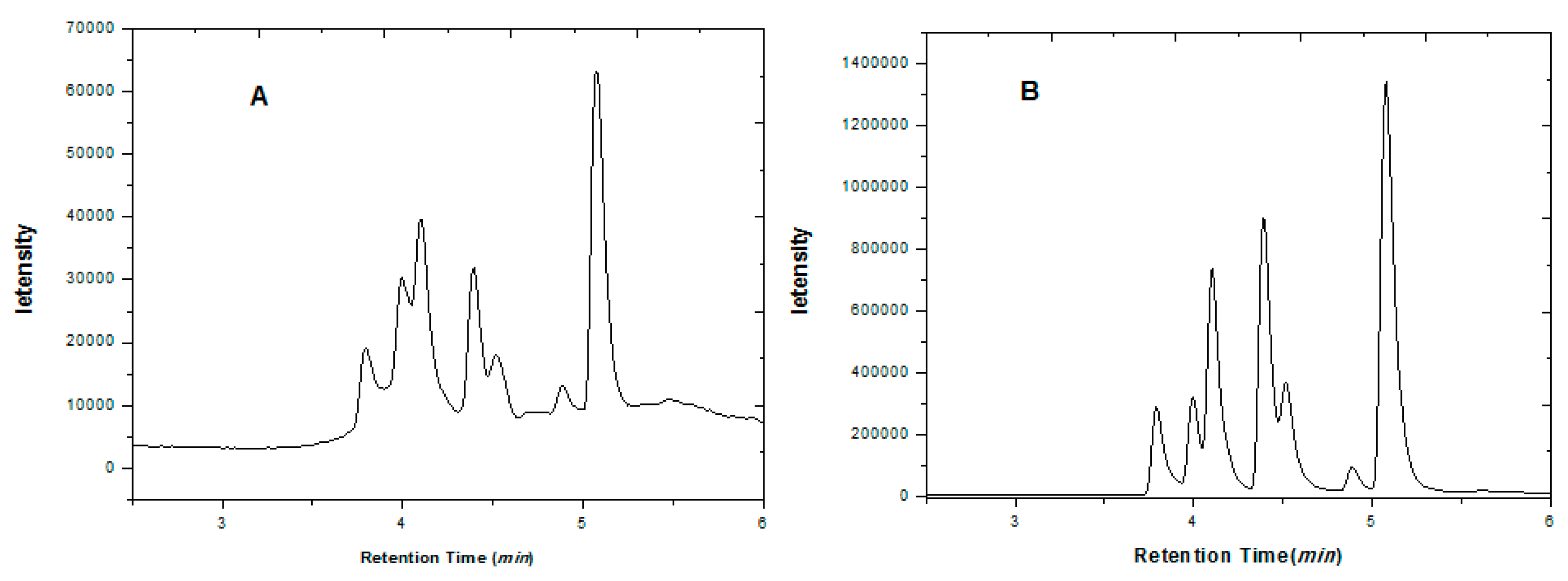
2.2. Optimization of HPLC Analysis
2.3. Optimization of Extraction and Purification Methods
2.3.1. Optimization of Extraction Solvents
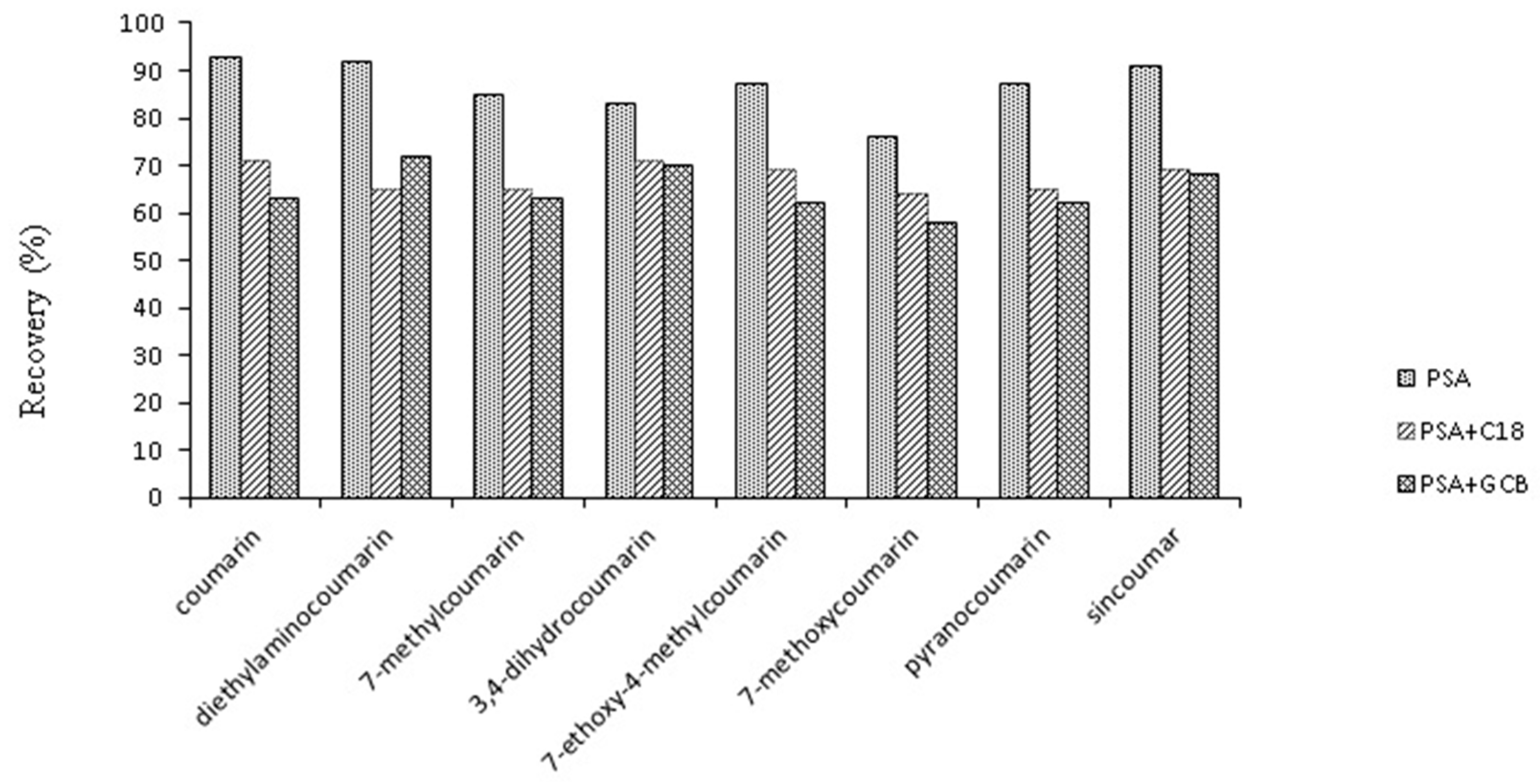
2.3.2. Optimization of Purification Material
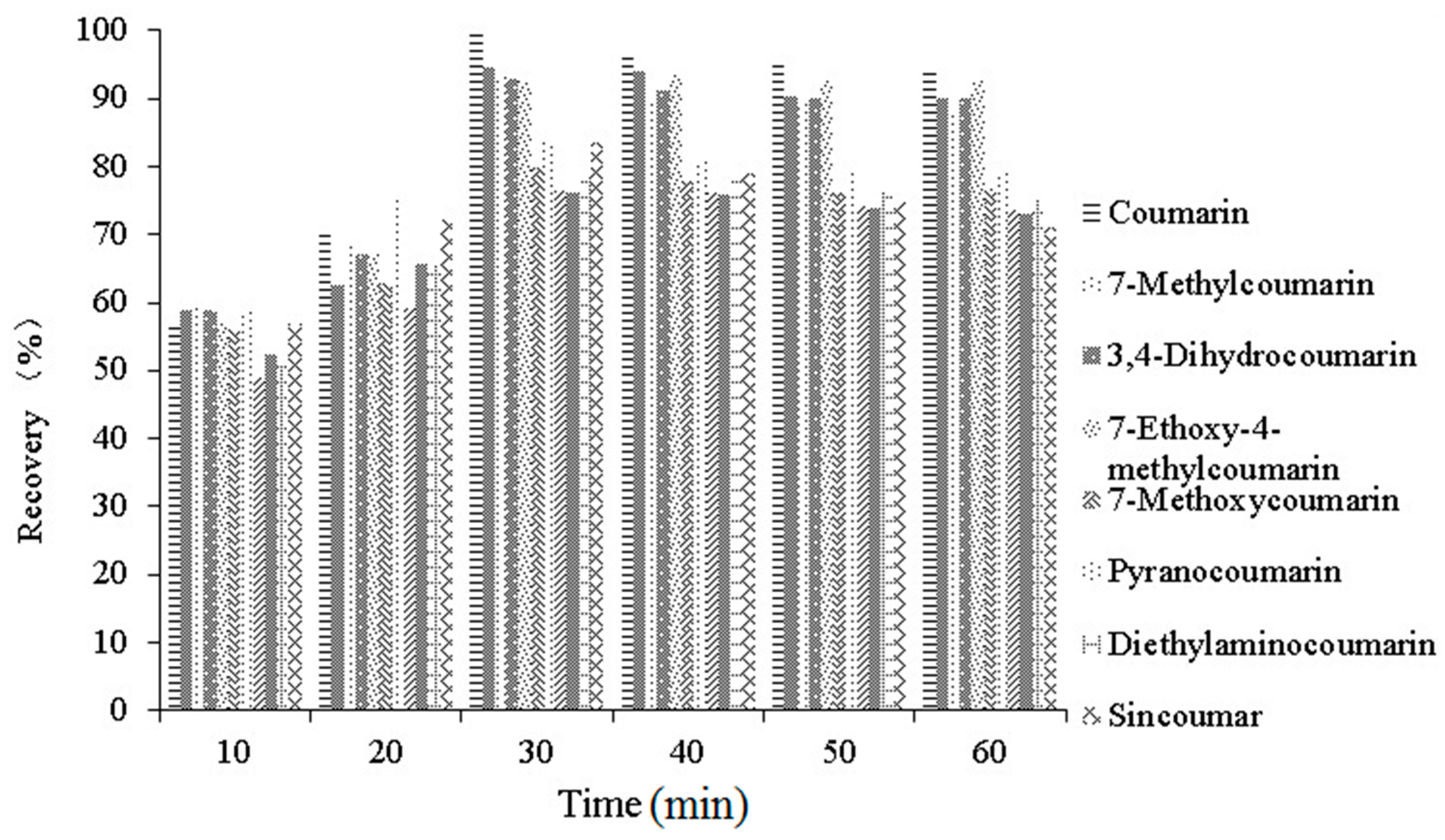
2.3.3. Optimization of Extraction Time
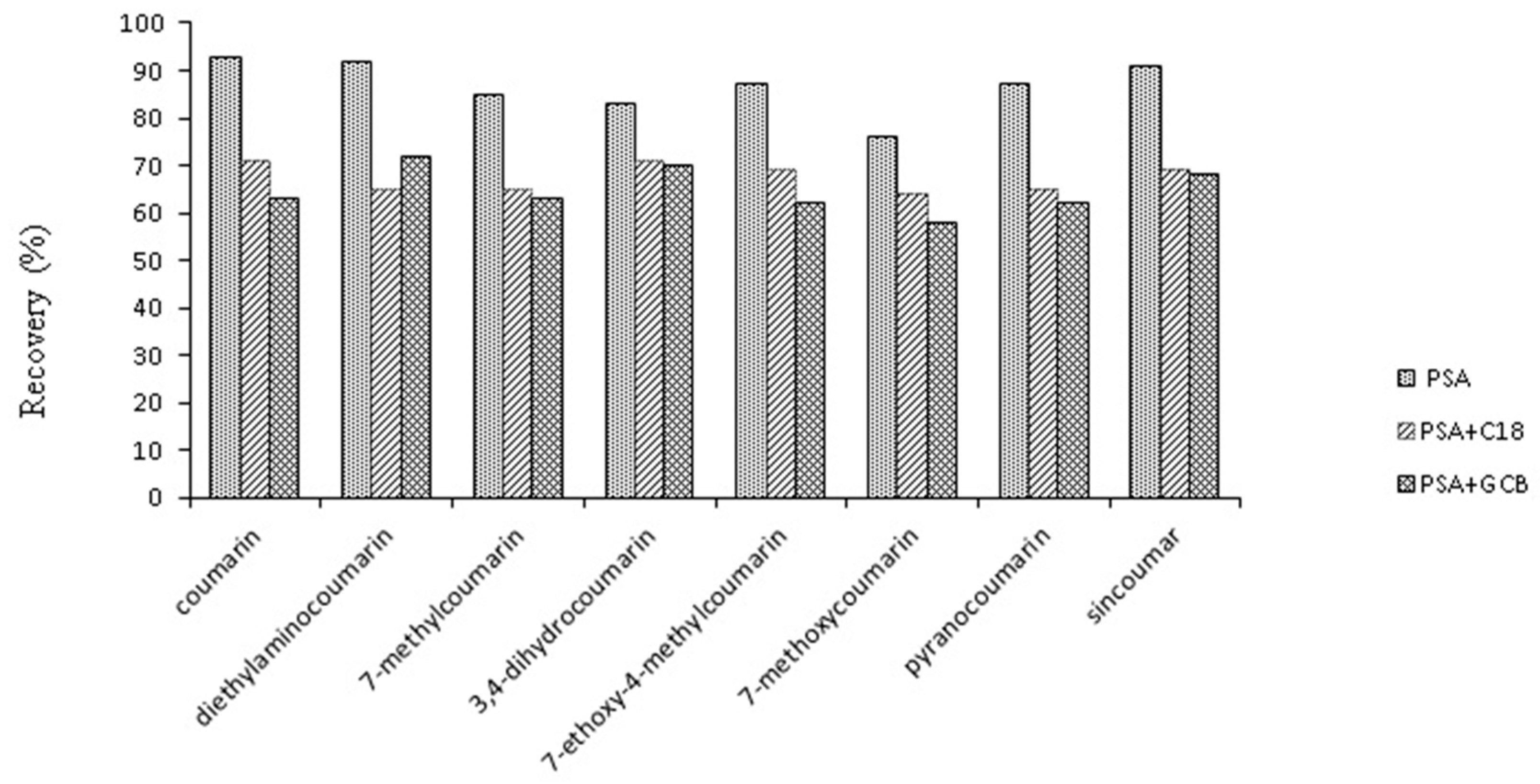
2.3.4. Optimization of Purification Method
2.4. Validation
2.4.1. Calibration and Sensitivity
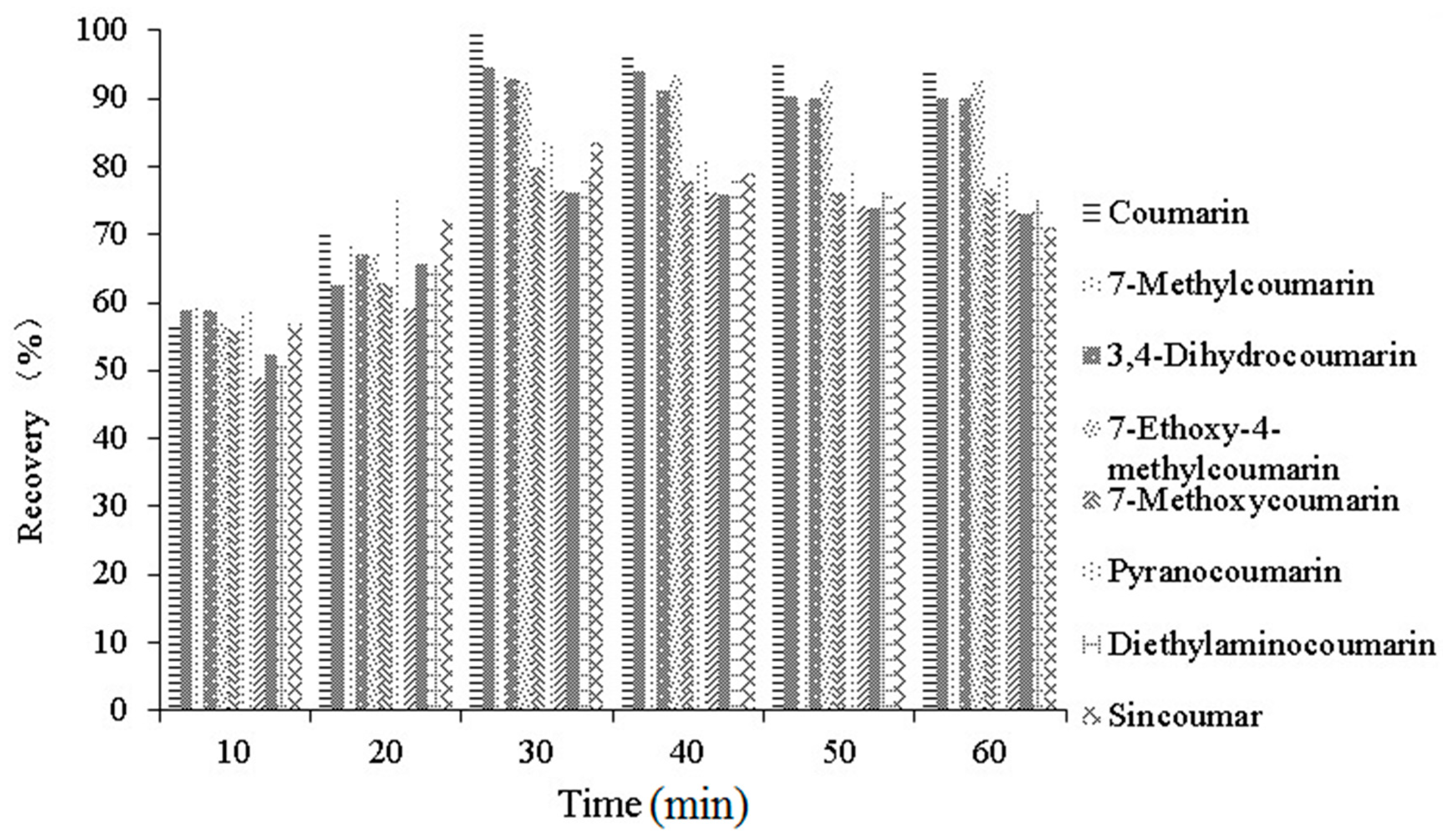
2.4.2. Recoveries and Precision
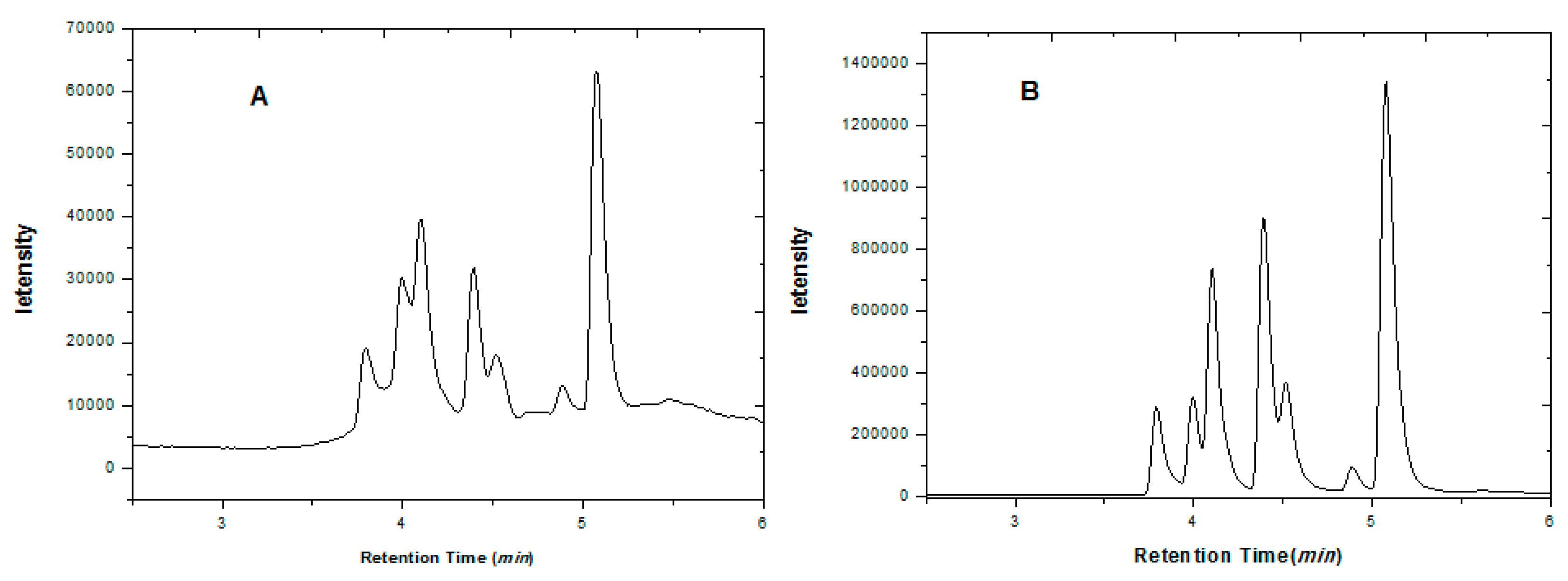
2.5. Analysis of Real Tobacco Samples
3. Experimental
3.1. Reagents and Materials
3.2. Sample Treatment
3.3. HPLC-MS/MS Instrumentation and Conditions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. WHO Global Report on Trends in Prevalence of Tobacco Smoking 2015; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Baker, R.R.; da Silva, J.R.P.; Smith, G. The effect of tobacco ingredients on smoke chemistry. Part II: Casing ingredients. Food Chem. Toxicol. 2004, 42, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Loscos, N.; Hernandez-Orte, P.; Cacho, J.; Ferreira, V. Current awareness in flavour and fragrance. Food Chem. 2009, 116, 59–65. [Google Scholar]
- Mcdonald, J.W.; Heffner, J.E. Eugenol Causes Oxidant-mediated Edema in Isolated Perfused Rabbit Lungs 1–4. Am. Rev. Respir. Dis. 1991, 143, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Stanfill, S.B.; Ashley, D.L. Solid phase microextraction of alkenylbenzenes and other flavor-related compounds from tobacco for analysis by selected ion monitoring gas chromatography-mass spectrometry. J. Chromatogr. A 1999, 858, 79–89. [Google Scholar] [CrossRef]
- Stanfill, S.; Calafat, A.; Brown, C.; Polzin, G.; Chiang, J.; Watson, C.; Ashley, D. Concentrations of nine alkenylbenzenes, coumarin, piperonal and pulegone in Indian bidi cigarette tobacco. Food Chem. Toxicol. 2003, 41, 303–317. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Wang, X.; Han, S.G.; Zhuang, H. A case-control study of risk factors for hepatocellular carcinoma in Henan, China. Am. J. Trop. Med. Hyg. 1998, 59, 947–951. [Google Scholar] [PubMed]
- Rychlik, M. Quantification of free coumarin and its liberation from glucosylated precursors by stable isotope dilution assays based on liquid chromatography− tandem mass spectrometric detection. J. Agric. Food Chem. 2008, 56, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Liu, B.; Ling, P.; Su, Q. Analysis of free and bound volatiles by gas chromatography and gas chromatography-mass spectrometry in uncased and cased tobaccos. J. Chromatogr. A 2002, 947, 267–275. [Google Scholar] [CrossRef]
- Umegaki, T.; Imamura, S.; Toyama, N.; Kojima, Y. Influence of preparation conditions on the morphology of hollow silica-alumina composite spheres and their activity for hydrolytic dehydrogenation of ammonia borane. Microporous Mesoporous Mater. 2014, 196, 349–353. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Li, L.; Li, W.; Zhang, F.; Du, T.; Chu, X. Simultaneous determination of 23 flavor additives in tobacco products using gas chromatography-triple quadrupole mass spectrometry. J. Chromatogr. A 2013, 1306, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Stange, K.C.; Nutting, P.A.; Miller, W.L.; Jaén, C.R.; Crabtree, B.F.; Flocke, S.A.; Gill, J.M. Defining and measuring the patient-centered medical home. J. Gen. Int. Med. 2010, 25, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Clancy, L. Progress in tobacco control. Healthy Policy 2009, 91, S3–S14. [Google Scholar] [CrossRef]
- Hahn, J.; Schaub, J. Influence of tobacco additives on the chemical composition of mainstream smoke. J. BTFI GmbH 2010, 24, 100–116. [Google Scholar] [CrossRef]
- Walorczyk, S. Development of a multi-residue screening method for the determination of pesticides in cereals and dry animal feed using gas chromatography-triple quadrupole tandem mass spectrometry. J. Chromatogr. A 2007, 1165, 200–212. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Some Naturally Occurring and Synthetic Food Components, Furocoumarins and Ultraviolet Radiation. In Proceedings of Apresentado em: IARC Working Group on the Evaluation of the Carcinogenic Risk of Chemicals to Humans: Some Naturally Occurring and Synthetic Food Components, Lyon, France, 1986.
- Scordino, M.; Sabatino, L.; Belligno, A.; Gagliano, G. Flavonoids and furocoumarins distribution of unripe chinotto (Citrus × myrtifolia Rafinesque) fruit: Beverage processing homogenate and juice characterization. Eur. Food Res. Technol. 2011, 233, 759–767. [Google Scholar] [CrossRef]
- Turin, L. The Secret of Scent; Harper Collins: New York, NY, USA, 2006. [Google Scholar]
- Evans, J.G.; Gaunt, I.F.; Lake, B.G. Two-year toxicity study on coumarin in the baboon. Food Cosmet. Toxicol. 1979, 17, 187–193. [Google Scholar] [CrossRef]
- Meineke, I.; Desel, H.; Kahl, R.; Kahl, G.; Gundert-Remy, U. Determination of 2-hydroxyphenylacetic acid (2HPAA) in urine after oral and parenteral administration of coumarin by gas-liquid chromatography with flame-ionization detection. J. Pharm. Biomed. Anal. 1998, 17, 487–492. [Google Scholar] [CrossRef]
- Rahim, A.A.; Saad, B.; Osman, H.; Hashim, N.; Yahya, S.; Talib, K.M. Simultaneous determination of diethylene glycol, diethylene glycol monoethyl ether, coumarin and caffeine in food items by gas chromatography. Food Chem. 2011, 126, 1412–1416. [Google Scholar] [CrossRef]
- Mondello, L.; Casilli, A.; Tranchida, P.Q.; Dugo, G.; Dugo, P. Comprehensive two-dimensional gas chromatography in combination with rapid scanning quadrupole mass spectrometry in perfume analysis. J. Chromatogr. A 2005, 1067, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.; Seabra, R.; Valentao, P.; Areias, F. Simultaneous determination of flavonoids, phenolic acids, and coumarins in seven medicinal species by HPLC/diode-array detector. J. Liq. Chromatogr. Related Technol. 1998, 21, 2813–2820. [Google Scholar] [CrossRef]
- Russo, M.; Torre, G.; Carnovale, C.; Bonaccorsi, I.; Dugo, P.; Mondello, L. A new HPLC method developed for the analysis of oxygen heterocyclic compounds in Citrus essential oils. J. Essent. Oil Res. 2012, 24, 119–129. [Google Scholar] [CrossRef]
- Russo, M.; Bonaccorsi, I.; Costa, R.; Dugo, P.; Mondello, L. Reduced time HPLC analyses for fast quality control of Citrus essential oils. J. Essent. Oil Res. 2015, 27, 307–315. [Google Scholar] [CrossRef]
- Celeghini, R.; Vilegas, J.H.; Lanças, F.M. Extraction and quantitative HPLC analysis of coumarin in hydroalcoholic extracts of Mikania glomerata Spreng: (“guaco”) leaves. J. Braz. Chem. Soc. 2001, 12, 706–709. [Google Scholar] [CrossRef]
- Sproll, C.; Ruge, W.; Andlauer, C.; Godelmann, R.; Lachenmeier, D.W. HPLC analysis and safety assessment of coumarin in foods. Food Chem. 2008, 109, 462–469. [Google Scholar] [CrossRef] [PubMed]
- De Jager, L.S.; Perfetti, G.A.; Diachenko, G.W. Determination of coumarin, vanillin, and ethyl vanillin in vanilla extract products: Liquid chromatography mass spectrometry method development and validation studies. J. Chromatogr. A, 2007, 1145, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Han, C.; Liu, B.; Lin, Z.; Zhou, X.; Wang, C.; Zhu, Z. Determination of vanillin, ethyl vanillin, and coumarin in infant formula by liquid chromatography-quadrupole linear ion trap mass spectrometry. J. Dairy Sci. 2014, 97, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhao, W.; Zhou, T.; Fan, G.; Wu, Y. An efficient strategy based on MAE, HPLC-DAD-ESI-MS/MS and 2D-prep-HPLC-DAD for the rapid extraction, separation, identification and purification of five active coumarin components from radix angelicae dahuricae. Phytochem. Anal. 2010, 21, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Li, K.P.; Gao, C.K.; Li, W.M. Analysis of coumarins in extract of Cnidium monnieri by ultra-performance liquid chromatographic coupled to electrospray ionization time of flight mass/mass spectrometry. Chin. Tradit. Pat. Med. 2009, 4, 584–587. [Google Scholar]
- Yang, R.; Wei, B.; Gao, H.; Yu, W. Determination of five coumarins in toys by high performance liquid chromatography-tandem mass spectrometry. Chin. J. Chromatogr. 2012, 30, 160–164. [Google Scholar] [CrossRef]
- Polzin, G.M.; Stanfill, S.B.; Brown, C.R.; Ashley, D.L.; Watson, C.H. Determination of eugenol, anethole, and coumarin in the mainstream cigarette smoke of Indonesian clove cigarettes. Food Chem. Toxicol. 2007, 45, 1948–1953. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Zhu, H.F.; Zhao, Y.B.; Gang, L.T.; Qing, Y.H. Dihydrogen coumarin in cigarette flavoring and GC/MS analysis of 6-methyl coumarin. J. Inf. Sci. Technol. 2011, 24, 345–346. [Google Scholar]
- Chen, Y.; Wang, C.; Xue, Y.M.; Chen, W.; Wang, X.; Bai, H.; Cai, T.P.; Hu, K.X. Determination of dicumarol and cyclocoumarol in cosmetics by HPLC/DAD. J. Instrum. Anal. 2008, 27, 196–199. [Google Scholar]
- Li, J.; Wang, C.; Wu, T.; Li, N. Determination of three coumarins-coumarin, 6-methylcoumarin, 7-methoxy coumarins- in cosmetics by gas chromatography and mass spectrometry. J. Life Sci. Instrum. 2006, 4, 33–36. [Google Scholar]
- Christakopoulos, A.; Feldhusen, K.; Norin, H.; Palmqvist, A.; Wahlberg, I. Determination of natural levels of coumarin in different types of tobacco using a mass fragmentographic method. J. Agric. Food Chem. 1992, 40, 1358–1361. [Google Scholar] [CrossRef]
- Sample Availability: Not available

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CAS | Elemental Composition | Retention Time (min) | Ionization Mode | Precursor (m/z) | Production (m/z) (Collision Energy/eV) |
|---|---|---|---|---|---|---|
| Coumarin | 91-64-5 | C9H6O2 | 3.79 | [M + H]+ | 147.3 | 91.0 * (25); 103.1 (15) |
| 7-Methylcoumarin | 2445-83-2 | C10H8O2 | 4.10 | [M + H]+ | 161.1 | 105.0 * (25); 115.1 (20) |
| 3,4-Dihydrocoumarin | 119-84-6 | C9H8O2 | 4.10 | [M + H]+ | 149.1 | 107.0 * (10); 7 7.0 (15) |
| 7-Ethoxy-4-methyl-coumarin | 87-05-8 | C12H12O3 | 4.39 | [M + H]+ | 205.1 | 177.2 * (20); 105.0 (25) |
| 7-Methoxycoumarin | 531-59-9 | C10H8O3 | 3.99 | [M + H]+ | 177.1 | 121.0 * (25); 132.9 (15) |
| Pyranocoumarin | 518-20-7 | C20H18O4 | 5.08 | [M + H]+ | 323.1 | 251.0 * (15); 291.0 * (10) |
| 7-Diethylaminocoumarin | 63226-13-1 | C27H28N2O5 | 5.08 | [M + H]+ | 461.0 | 244.0 * (25); 417.0 (35) |
| Sincoumar | 152-72-7 | C19H15NO6 | 4.52 | [M + H]+ | 354.1 | 296.1 * (20); 162.9 (15) |
| Analytes | Linear Range (μg·kg−1) | Regression Equation | Correlation Coefficient (r2) | LOD | LOQ |
|---|---|---|---|---|---|
| (μg·kg−1) | (μg·kg−1) | ||||
| Coumarin | 2–500 | Y = 10493X + 26.32 | 0.9987 | 0.5 | 2.0 |
| 7-Methylcoumarin | 5–500 | Y = 16661X + 24.99 | 0.9989 | 0.9 | 3.0 |
| 3,4-Dihydrocoumarin | 5–500 | Y = 17936X + 152.4 | 0.9996 | 1.5 | 5.0 |
| 7-Ethoxy-4-methylcoumarin | 2–500 | Y = 37567X + 94.14 | 0.9995 | 0.5 | 1.7 |
| 7-Methoxycoumarin | 5–500 | Y = 19204X + 86.41 | 0.9993 | 1.2 | 3.5 |
| Pyranocoumarin | 5–500 | Y = 71867X + 21.56 | 0.9992 | 0.6 | 2.1 |
| 7-Diethylaminocoumarin | 5–00 | Y = 32635X + 44.76 | 0.9994 | 1.5 | 5.0 |
| Sincoumar | 5–500 | Y = 10161X + 45.72 | 0.9995 | 0.9 | 3.1 |
| NO. | Analytes | Spiked Level (μg·kg−1) | Average Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 1 | Coumarin | 2.0 | 76.1 | 3.2 |
| 3.0 | 85.0 | 2.1 | ||
| 4.0 | 88.3 | 1.5 | ||
| 2 | 7-Methylcoumarin | 3.0 | 75.1 | 5.3 |
| 4.5 | 78.4 | 2.4 | ||
| 6.0 | 76.3 | 2.0 | ||
| 3 | 3,4-Dihydrocoumarin | 5.2 | 69.8 | 5.3 |
| 7.8 | 70.9 | 4.4 | ||
| 10.4 | 72.4 | 2.5 | ||
| 4 | 7-Ethoxy-4-methylcoumarin | 1.7 | 86.8 | 2.2 |
| 2.5 | 91.3 | 2.1 | ||
| 3.4 | 94.2 | 2.2 | ||
| 5 | 7-Methoxycoumarin | 3.5 | 90.5 | 3.7 |
| 5.3 | 93.4 | 2.7 | ||
| 7.0 | 95.1 | 1.9 | ||
| 6 | Pyranocoumarin | 2.1 | 81.5 | 4.3 |
| 3.2 | 90.0 | 3.3 | ||
| 4.2 | 94.3 | 2.3 | ||
| 7 | 7-Diethylaminocoumarin | 5.0 | 73.2 | 3.4 |
| 7.5 | 76.5 | 3.8 | ||
| 10.0 | 81.2 | 2.9 | ||
| 8 | Sincoumar | 3.1 | 80.5 | 5.3 |
| 4.7 | 84.3 | 4.2 | ||
| 6.2 | 83.5 | 3.7 |
| Compounds | T1 | T2 | T3 | T4 | T5 | T6 | T7 | T8 | T9 | T10 | T11 | T12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coumarin | 5.4 | 3.51 | 5.6 | 5.21 | 4.72 | 2.35 | 5.67 | 3.2 | - | 4.38 | 5.16 | 2.77 |
| 7-Methylcoumarin | 4.9 | 1.87 | 3.71 | 2.89 | 3.01 | 1.61 | 4.32 | - | 1.93 | - | 1.89 | 2.17 |
| 3,4-Dihydrocoumarin | - | - | - | - | - | - | 5.21 | - | - | - | - | - |
| 7-Ethoxy-4-methylcoumarin | 3.72 | 3.61 | 3.95 | 2.99 | 3.14 | 2.13 | 1.81 | 3.91 | 2.07 | 2.64 | 3.45 | 3.05 |
| 7-Methoxycoumarin | - | 3.53 | - | 3.75 | - | - | 3.53 | - | 3.55 | - | 3.25 | - |
| Pyranocoumarin | - | 2.29 | - | 2.23 | - | 2.45 | - | 2.53 | - | 2.42 | 2.31 | - |
| Diethylaminocoumarin | - | 5.48 | - | - | - | - | - | - | - | - | - | - |
| Sincoumar | - | - | - | - | - | - | 3.34 | - | - | - | - | - |
| Total coumarins content | 14.02 | 20.29 | 13.26 | 17.07 | 10.87 | 8.54 | 23.88 | 9.64 | 7.55 | 9.44 | 16.06 | 4.94 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Z.; Nie, B.; Liu, T.; Yuan, F.; Feng, F.; Zhang, Y.; Zhou, W.; Xu, X.; Yao, M.; Zhang, F. Simultaneous Determination of Coumarin and Its Derivatives in Tobacco Products by Liquid Chromatography-Tandem Mass Spectrometry. Molecules 2016, 21, 1511. https://doi.org/10.3390/molecules21111511
Ren Z, Nie B, Liu T, Yuan F, Feng F, Zhang Y, Zhou W, Xu X, Yao M, Zhang F. Simultaneous Determination of Coumarin and Its Derivatives in Tobacco Products by Liquid Chromatography-Tandem Mass Spectrometry. Molecules. 2016; 21(11):1511. https://doi.org/10.3390/molecules21111511
Chicago/Turabian StyleRen, Zhiqin, Bo Nie, Tong Liu, Fei Yuan, Feng Feng, Yuan Zhang, Weie Zhou, Xiuli Xu, Meiyi Yao, and Feng Zhang. 2016. "Simultaneous Determination of Coumarin and Its Derivatives in Tobacco Products by Liquid Chromatography-Tandem Mass Spectrometry" Molecules 21, no. 11: 1511. https://doi.org/10.3390/molecules21111511
APA StyleRen, Z., Nie, B., Liu, T., Yuan, F., Feng, F., Zhang, Y., Zhou, W., Xu, X., Yao, M., & Zhang, F. (2016). Simultaneous Determination of Coumarin and Its Derivatives in Tobacco Products by Liquid Chromatography-Tandem Mass Spectrometry. Molecules, 21(11), 1511. https://doi.org/10.3390/molecules21111511