
Developing HIV-1 Protease Inhibitors through Stereospecific Reactions in Protein Crystals



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
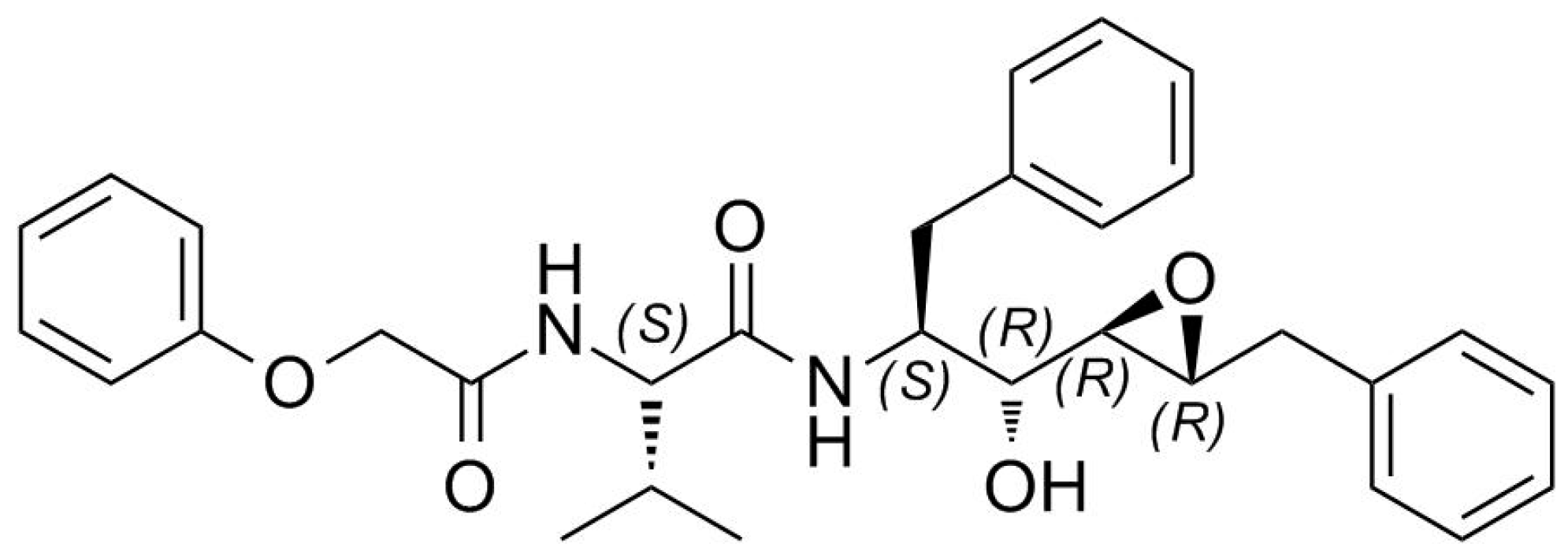
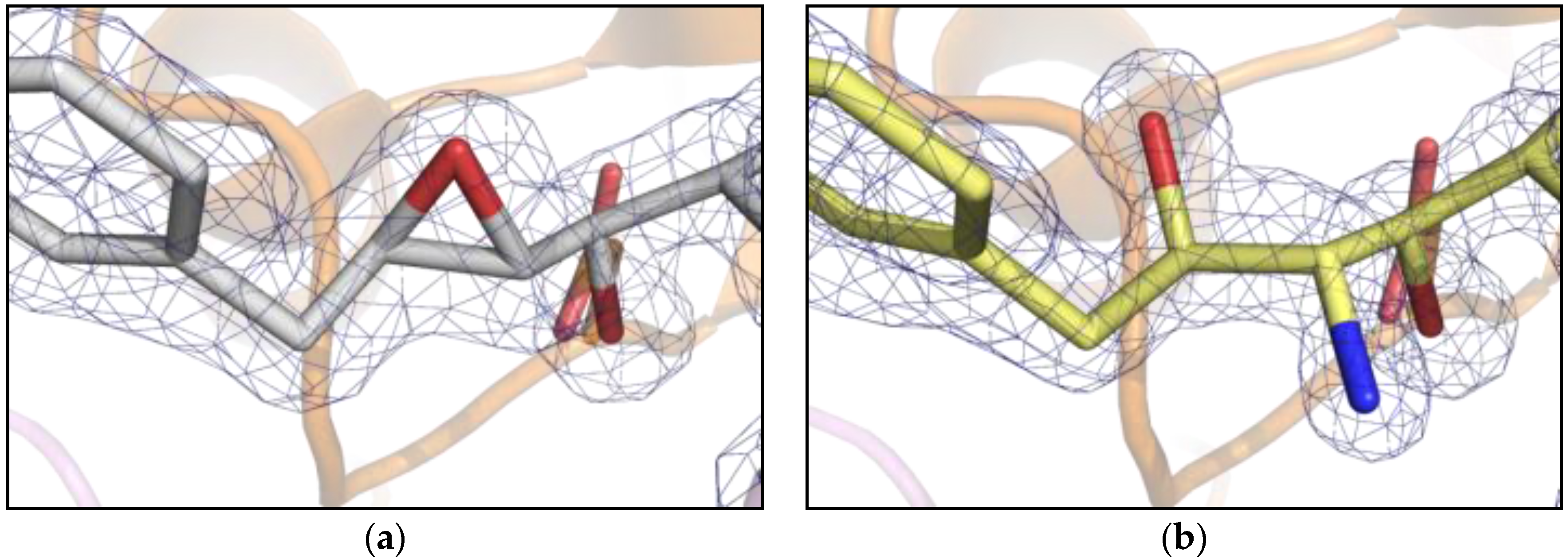
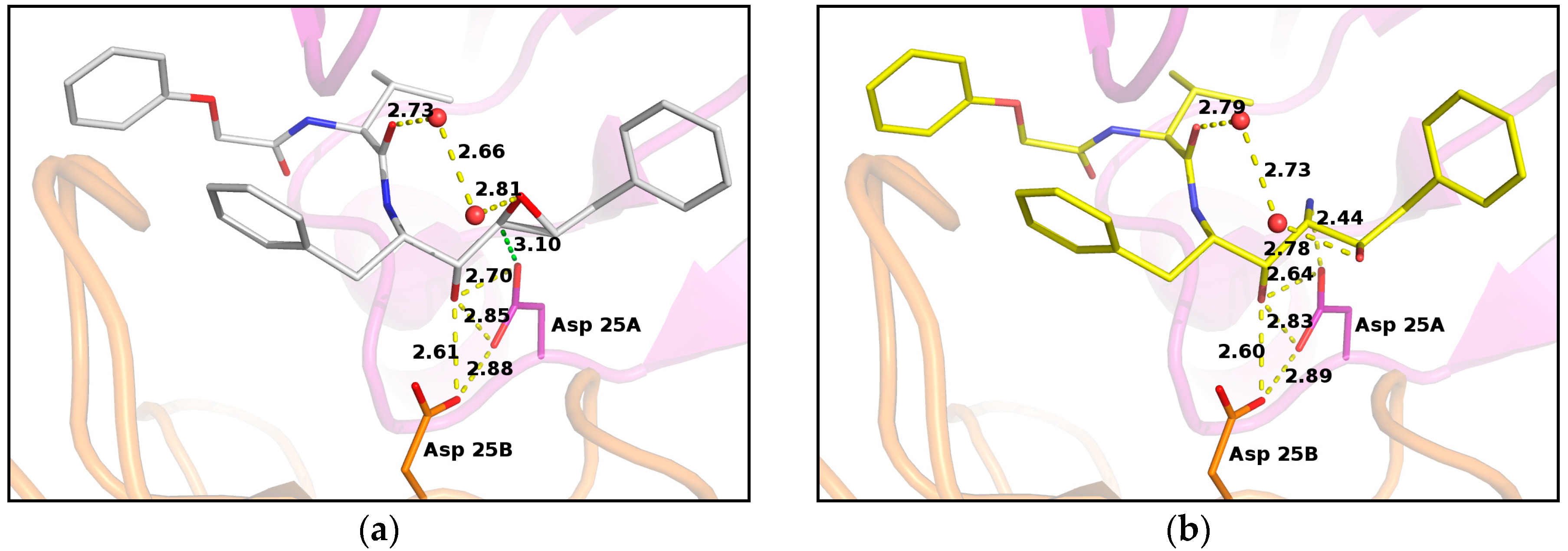
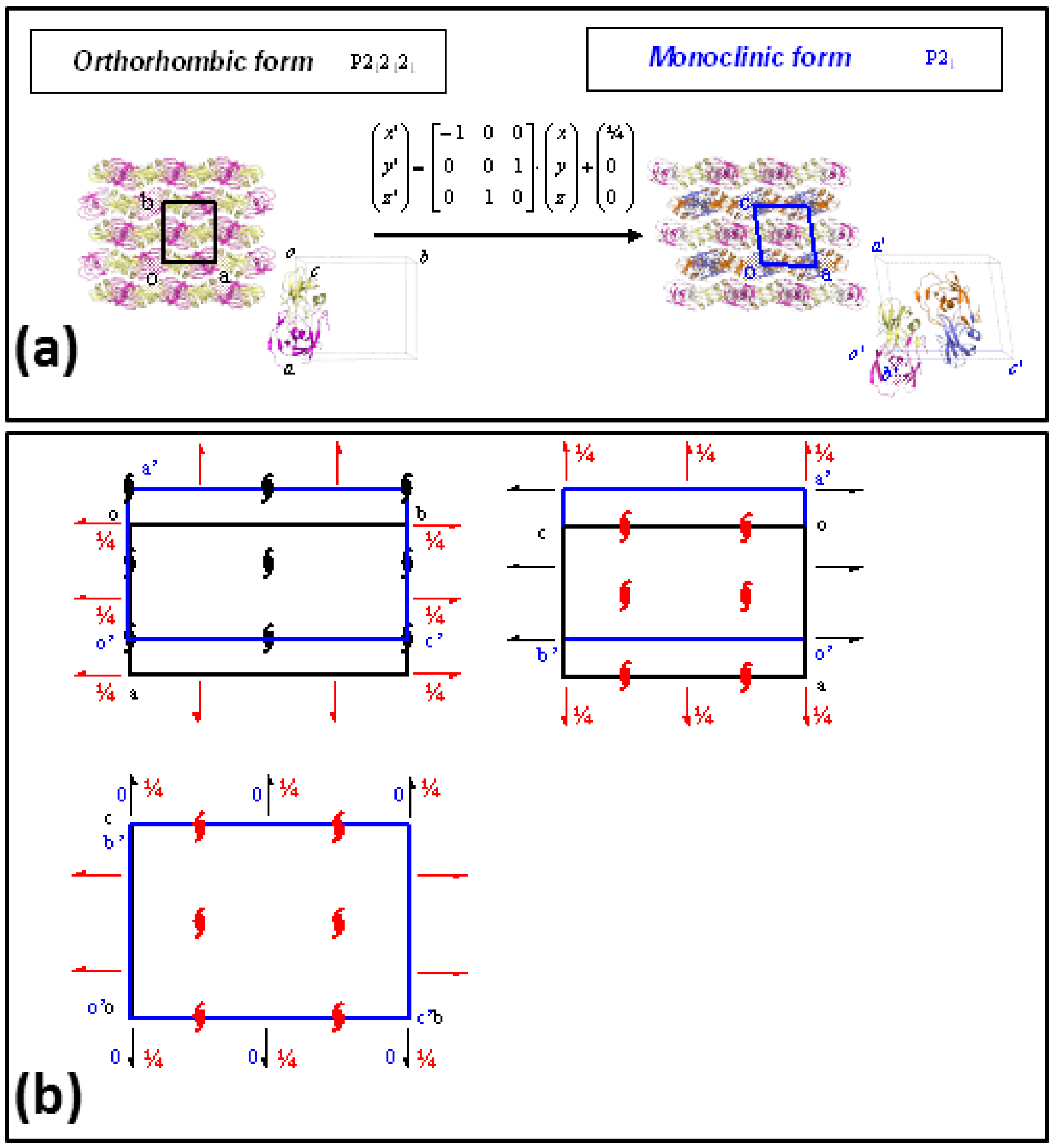
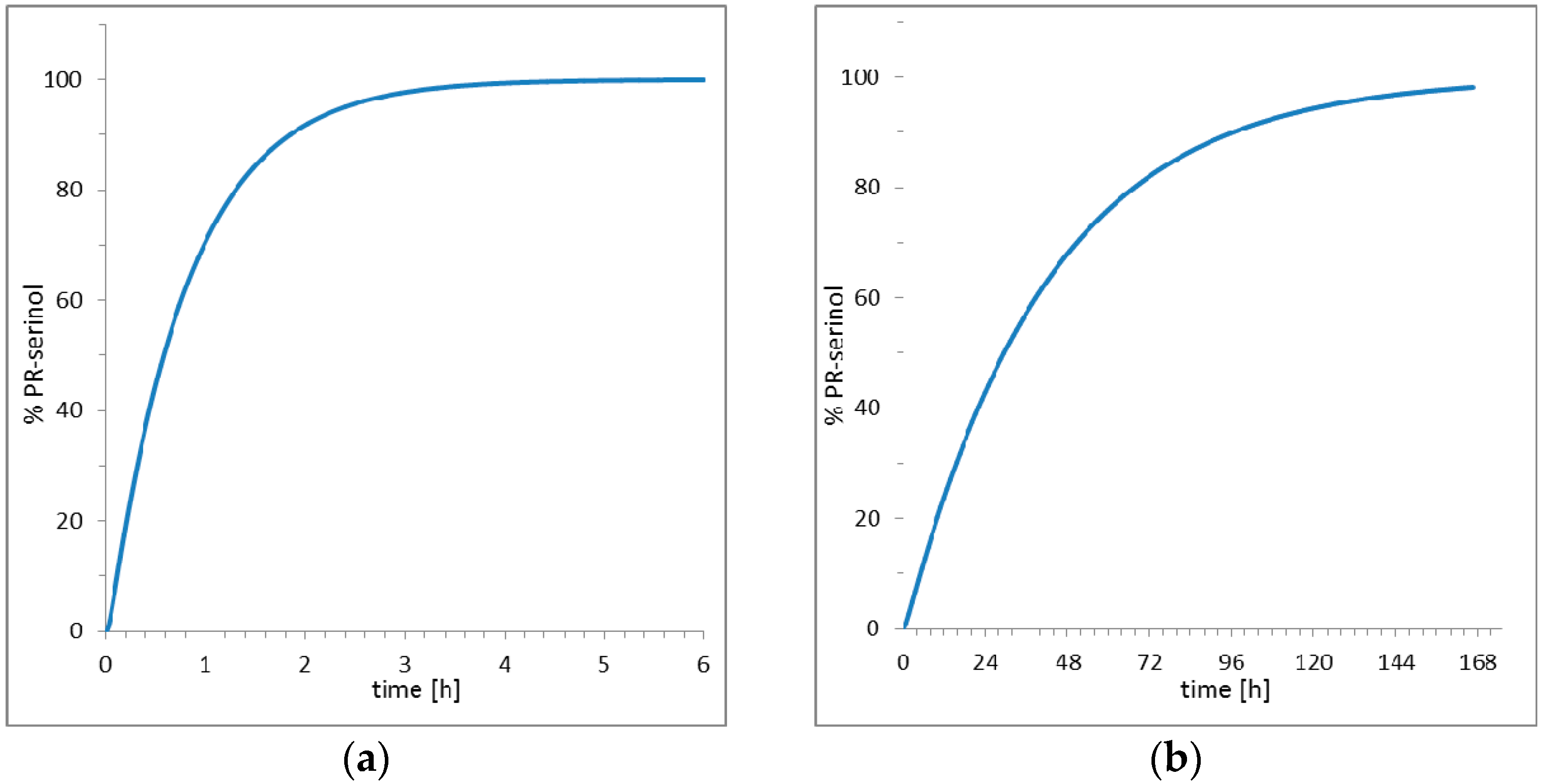
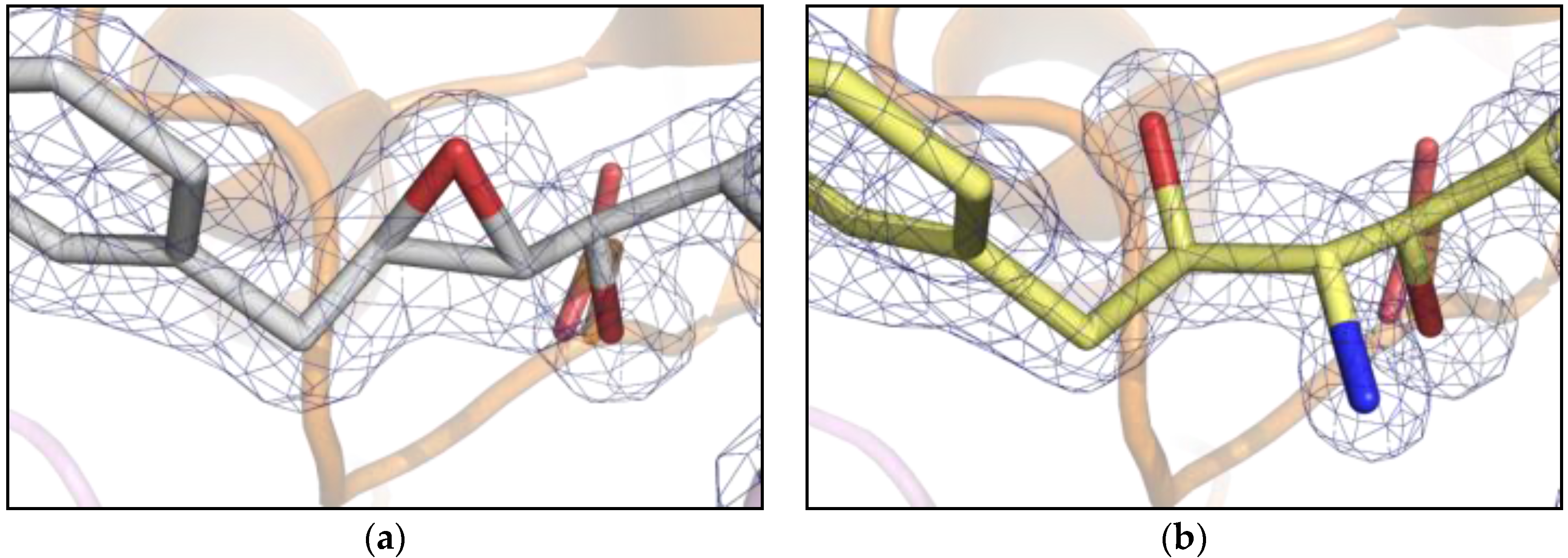
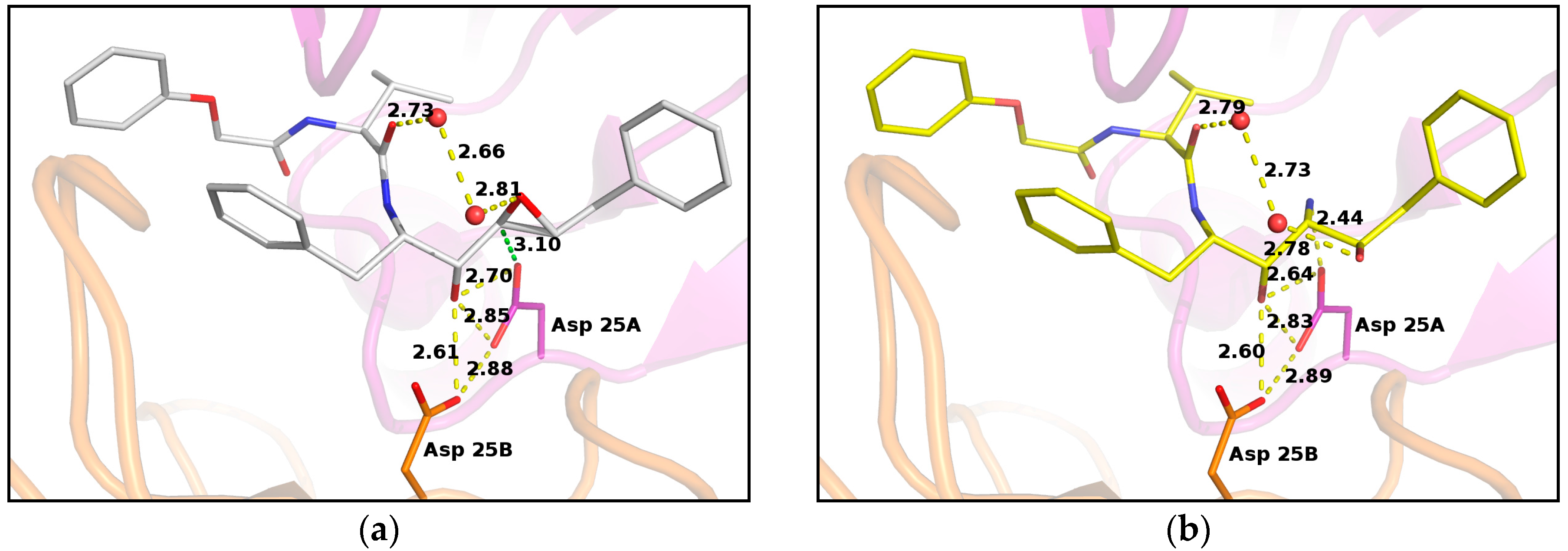
2. Results
3. Discussion
4. Materials and Methods
4.1. Expression, Purification and Refolding of HIV-1 Protease
4.2. Crystallization and Triggering of Reactions in Crystals of PR/EPX
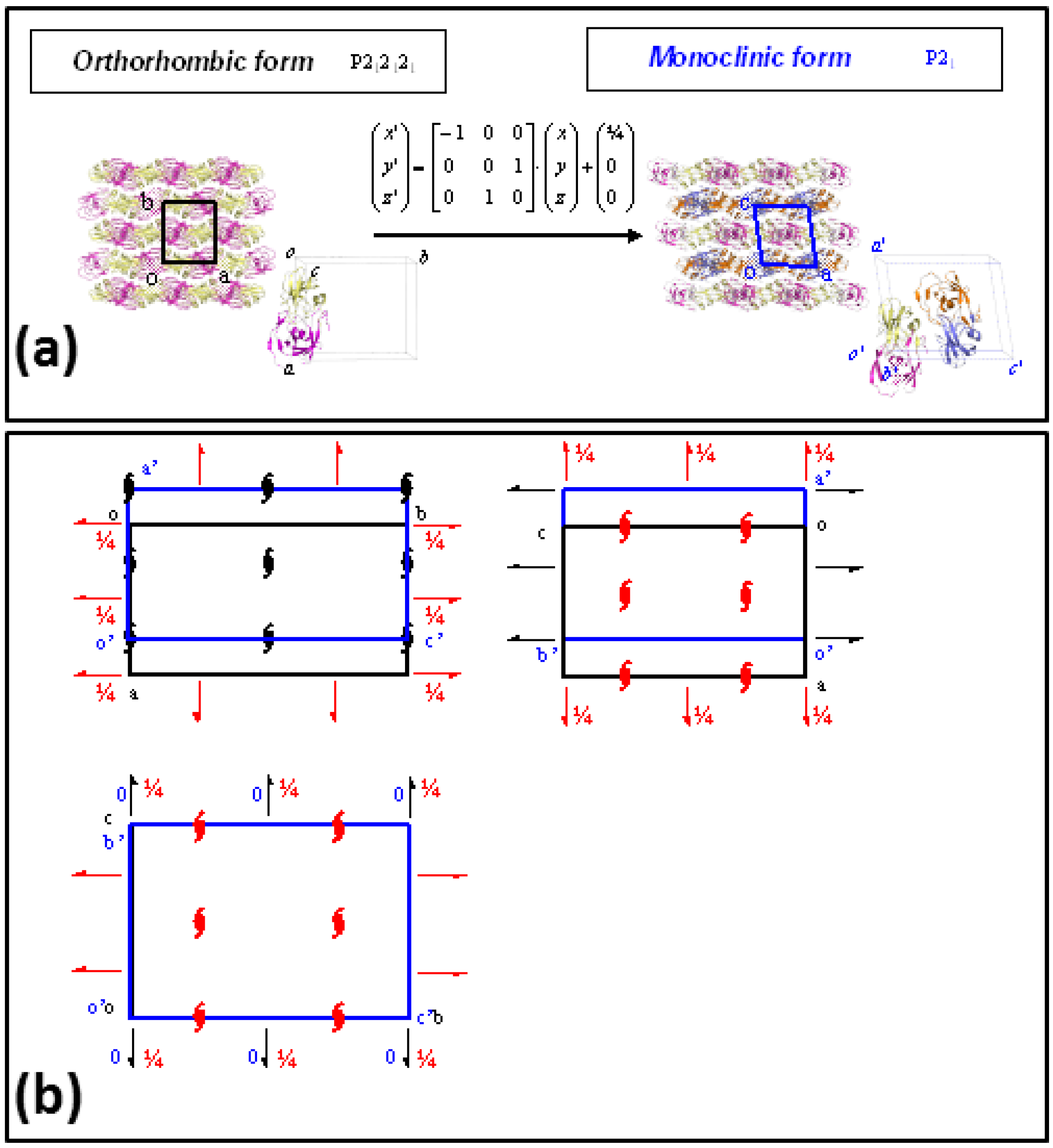
4.3. Crystallographic Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mehellou, Y.; De Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Liu, Z.P. Recent developments of peptidomimetic HIV-1 protease inhibitors. Curr. Med. Chem. 2011, 18, 4513–4537. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salveson, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Pokorná, J.; Machala, L.; Řezáčová, P.; Konvalinka, J. Current and Novel Inhibitors of HIV Protease. Viruses 2009, 1, 1209–1239. [Google Scholar] [CrossRef] [PubMed]
- Hagel, M.; Niu, D.; St Martin, T.; Sheets, M.P.; Qiao, L.; Bernard, H.; Karp, R.M.; Zhu, Z.; Labenski, M.T.; Chaturvedi, P.; et al. Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat. Chem. Biol. 2011, 7, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Lebon, F.; Ledecq, M. Approaches to the design of effective HIV-1 protease inhibitors. Curr. Med. Chem. 2000, 7, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Parham, G.L.; Martyr, C.D.; Nyalapatla, P.R.; Osswald, H.L.; Agniswamy, J.; Wang, Y.F.; Amano, M.; Weberand, I.T.; Mitsuya, H. Highly potent HIV-1 protease inhibitors with novel tricyclic P2 ligands: Design, synthesis, and protein-ligand X-ray studies. J. Med. Chem. 2013, 56, 6792–6802. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbrunn, T.; Forli, S.; Happer, M.; Gonzalez, A.; Tsai, Y.; Soltis, M.; Elder, J.H.; Olson, A.J.; Stout, C.D. Crystallographic fragment-based drug discovery: Use of a brominated fragment library targeting HIV protease. Chem. Biol. Drug Des. 2014, 83, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, Z.; Usmani, S.; Shamshad, H.; Mahmood, U.; Halim, S.A. A combined 3D-QSAR and docking studies for the In-silico prediction of HIV-protease inhibitors. Chem. Cent. J. 2013, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.G.; Zhang, X.; Yuan, J.F. Anti-HIV drug development through computational methods. AAPS J. 2014, 16, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Aneja, R.; Chaiken, I. Click chemistry in peptide-based drug design. Molecules 2013, 18, 9797. [Google Scholar] [CrossRef] [PubMed]
- Paton, N.I.; Stöhr, W.; Oddershede, L.; Arenas-Pinto, A.; Walker, S.; Sculpher, M.; Dunn, D.T. The protease inhibitor monotherapy versus ongoing triple therapy (PIVOT) trial: A randomised controlled trial of a protease inhibitor monotherapy strategy for long-term management of human immunodeficiency virus infection. Health Technol. Assess. 2016, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, M.; Ehret, R.; Wienbreyer, A.; Walter, H.; Berg, T.; Baumgarten, A. Resistance remains a problem in treatment failure. J. Int. AIDS Soc. 2014, 17, 19756. [Google Scholar] [CrossRef] [PubMed]
- Rabi, S.A.; Laird, G.M.; Durand, C.M.; Laskey, S.; Shan, L.; Bailey, J.R.; Chioma, S.; Moore, R.D.; Siliciano, R.F. Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J. Clin. Investig. 2013, 123, 3848–3860. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Yu, X.; Zhang, Y.; Tie, Y.; Wang, Y.F.; Yashchuk, S.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. Potent antiviral HIV-1 protease inhibitor GRL-02031 adapts to the structures of drug resistant mutants with its P1’-pyrrolidinone ring. J. Med. Chem. 2012, 55, 3387–3397. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, F.; Berti, F.; Campaner, P.; Fanfoni, L.; Demitri, N.; Olajuyigbe, F.M.; De March, M.; Geremia, S. Impact of Stereochemistry on Ligand Binding: X-ray Crystallographic Analysis of an Epoxide-Based HIV Protease Inhibitor. ACS Med. Chem. Lett. 2014, 5, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.B.; Rosé, J.R.; Salto, R.; Craik, C.S.; Stroud, R.M. Structure of the protease from simian immunodeficiency virus: Complex with an irreversible non peptide inhibitor. Biochemistry 1993, 32, 12498–12507. [Google Scholar] [CrossRef] [PubMed]
- Salto, R.; Babé, L.M.; Li, J.; Rosé, J.R.; Yu, Z.; Burlingame, A.; de Voss, J.J.; Sui, Z.; Ortiz-de-Montellano, P.; Craik, C.S. In vitro characterization of non peptide irreversible inhibitors of HIV proteases. J. Biol. Chem. 1994, 269, 10691–10698. [Google Scholar] [PubMed]
- Blacker, A.J.; Roy, M.; Hariharan, S.; Headley, C.; Upare, A.; Jagtap, A.; Kadam, N. Convenient Method for Synthesis of N-Protected α-Amino Epoxides: Key Intermediates for HIV Protease Inhibitors. Org. Process Res. Dev. 2011, 15, 331–338. [Google Scholar] [CrossRef]
- Miller, M. The early years of retroviral protease crystal structures. Biopolymers 2010, 94, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, B.; Louis, J.M.; Hung, J.; Harrison, R.W.; Weber, I.J. Structural implications of drug-resistant mutants of HIV-1 protease: High-resolution crystal structures of the mutant protease/substrate analogue complexes. Proteins 2001, 43, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Olajuyigbe, F.M.; Demitri, N.; Geremia, S. Investigation of 2-Fold Disorder of Inhibitors and Relative Potency by Crystallizations of HIV-1 Protease in Ritonavir and Saquinavir Mixtures. Cryst. Growth Des. 2011, 11, 4378–4385. [Google Scholar] [CrossRef]
- Olajuyigbe, F.M.; Demitri, N.; Ajele, J.O.; Maurizio, E.; Randaccio, L.; Geremia, S. Carbamylation of N-terminal proline. ACS Med. Chem. Lett. 2010, 1, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Geremia, S.; Demitri, N.; Wuerges, J.; Benedetti, F.; Berti, F.; Tell, G.; Randaccio, L. A potent HIV protease inhibitor identified in an epimeric mixture by high-resolution protein crystallography. ChemMedChem 2006, 1, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Gelin, M.; Poncet-Montange, G.; Assairi, L.; Morellato, L.; Huteau, V.; Dugué, L.; Dussurget, O.; Pochet, S.; Labesse, G. Screening and in situ synthesis using crystals of a NAD kinase lead to a potent antistaphylococcal compound. Structure 2012, 20, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Mavri, J. Irreversible inhibition of the HIV-1 protease: A theoretical study. Int. J. Quantum Chem. 1998, 69, 753–759. [Google Scholar] [CrossRef]
- Geremia, S.; Campagnolo, M.; Demitri, N.; Johnson, L.N. Simulation of diffusion time of small molecules in protein crystals. Structure 2006, 14, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Andreeßen, B.; Steinbüchel, A. Serinol: Small molecule—Big impact. AMB Express 2011, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.E.; Trylska, J.; Tozzini, V.; McCammon, J.A. Binding pathways of ligands to HIV-1 protease: Coarse-grained and atomistic simulations. Chem. Biol. Drug Des. 2007, 69, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Heaslet, H.; Rosenfeld, R.; Giffin, M.; Lin, Y.-C.; Tam, K.; Torbett, B.E.; Elder, J.H.; McRee, D.E.; Stout, C.D. Conformational flexibility in the flap domains of ligand-free HIV protease. Acta Cryst. 2007, D63, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A Complex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Di Russo, N.V.; Estrin, D.A.; Martí, M.A.; Roitberg, A.E. pH-Dependent conformational changes in proteins and their effect on experimental pK(a)s: The case of Nitrophorin 4. PLoS Comput. Biol. 2012, 8, e1002761. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Srajer, V.; Purwar, N.; Henning, R.; Schmidt, M. pH dependence of the photoactive yellow protein photocycle investigated by time-resolved crystallography. Biophys. J. 2012, 102, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.E.; Majumdar, A.; García-Moreno, E.B. pH dependence of conformational fluctuations of the protein backbone. Proteins 2014, 82, 3132–3143. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Talukdar, A.; Cushman, M. Regioselective synthesis of N-β-hydroxyethylaziridines by the ring-opening reaction of epoxides with aziridine generated in situ. Org. Lett. 2006, 8, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Azizi, N.; Gholibeglo, E.; Maryami, M.; Nayeri, S.D.; Bolourtchian, S.M. Ultrasound mediated efficient ring opening of epoxides by in situ generated dithiocarbamates in green reaction media. C. R. Chim. 2013, 16, 412–418. [Google Scholar] [CrossRef]
- Geremia, S.; Campagnolo, M.; Schinzel, R.; Johnson, L.N. Enzymatic catalysis in crystals of Escherichia coli maltodextrin phosphorylase. J. Mol. Biol. 2002, 322, 413–423. [Google Scholar] [CrossRef]
- Shen, C.H.; Tie, Y.; Yu, X.; Wang, Y.F.; Kovalevsky, A.Y.; Harrison, R.W.; Weber, I.T. Capturing the reaction pathway in near-atomic-resolution crystal structures of HIV-1 protease. Biochemistry 2012, 51, 7726–7732. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Mahale, S.; Prashar, V.; Bihani, S.; Ferrer, J.L.; Hosur, M.V. X-ray snapshot of HIV-1 protease in action: Observation of tetrahedral intermediate and short ionic hydrogen bond SIHB with catalytic aspartate. J. Am. Chem. Soc. 2010, 132, 6366–6373. [Google Scholar] [CrossRef] [PubMed]
- Kóňa, J. Theoretical study on the mechanism of a ring-opening reaction of oxirane by the active-site aspartic dyad of HIV-1 protease. Org. Biomol. Chem. 2008, 6, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Ohhara, T.; Kurihara, K.; Tamada, T.; Honjo, E.; Okazaki, N.; Arai, S.; Shoyama, Y.; Kimura, K.; Matsumura, H.; et al. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc. Natl. Acad. Sci. USA 2009, 106, 4641–4646. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Anderson, D.D.; Weber, I.T.; Mitsuya, H. Enhancing protein backbone binding—A fruitful concept for combating drug-resistant HIV. Angew. Chem. Int. Ed. Engl. 2012, 51, 1778–1802. [Google Scholar] [CrossRef] [PubMed]
- Geremia, S.; di Costanzo, L.; Randaccio, L.; Engel, D.E.; Lombardi, A.; Nastri, F.; DeGrado, W.F. Response of a designed metalloprotein to changes in metal ion coordination, exogenous ligands, and active site volume determined by X-ray crystallography. J. Am. Chem. Soc. 2005, 127, 17266–17276. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, L.; Forneris, F.; Geremia, S.; Randaccio, L. Phasing protein structures using the group-subgroup relation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1435–1439. [Google Scholar] [CrossRef]
- Louis, J.M.; Wondrak, E.M.; Kimmel, A.R.; Wingfield, P.T.; Nashed, N.T. Proteolytic processing of HIV-1 protease precursor, kinetics and mechanism. J. Biol. Chem. 1999, 274, 23437–23442. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.G.W. Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography No. 26; Daresbury Laboratory: Warrington, UK, 1992. [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. Available online: http://www.pymol.org (accessed on 12 January 2005).
- Sample Availability: Samples of the amino diol derivative are not available.


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olajuyigbe, F.M.; Demitri, N.; De Zorzi, R.; Geremia, S. Developing HIV-1 Protease Inhibitors through Stereospecific Reactions in Protein Crystals. Molecules 2016, 21, 1458. https://doi.org/10.3390/molecules21111458
Olajuyigbe FM, Demitri N, De Zorzi R, Geremia S. Developing HIV-1 Protease Inhibitors through Stereospecific Reactions in Protein Crystals. Molecules. 2016; 21(11):1458. https://doi.org/10.3390/molecules21111458
Chicago/Turabian StyleOlajuyigbe, Folasade M., Nicola Demitri, Rita De Zorzi, and Silvano Geremia. 2016. "Developing HIV-1 Protease Inhibitors through Stereospecific Reactions in Protein Crystals" Molecules 21, no. 11: 1458. https://doi.org/10.3390/molecules21111458
APA StyleOlajuyigbe, F. M., Demitri, N., De Zorzi, R., & Geremia, S. (2016). Developing HIV-1 Protease Inhibitors through Stereospecific Reactions in Protein Crystals. Molecules, 21(11), 1458. https://doi.org/10.3390/molecules21111458