
Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn-Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts

Abstract
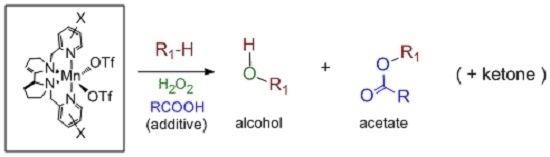
:
1. Introduction
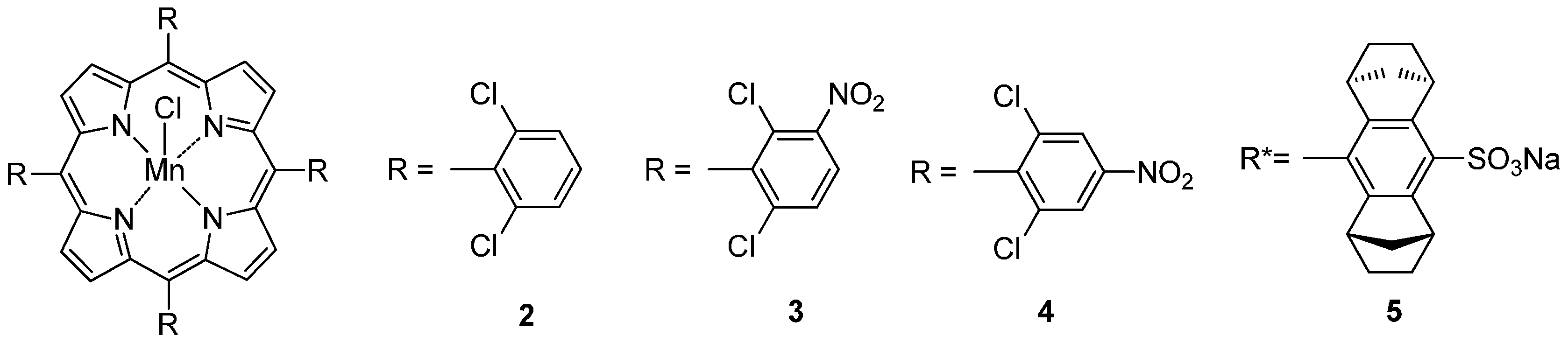
2. C–H Oxidations with H2O2 in the Presence of Mn Porphyrins
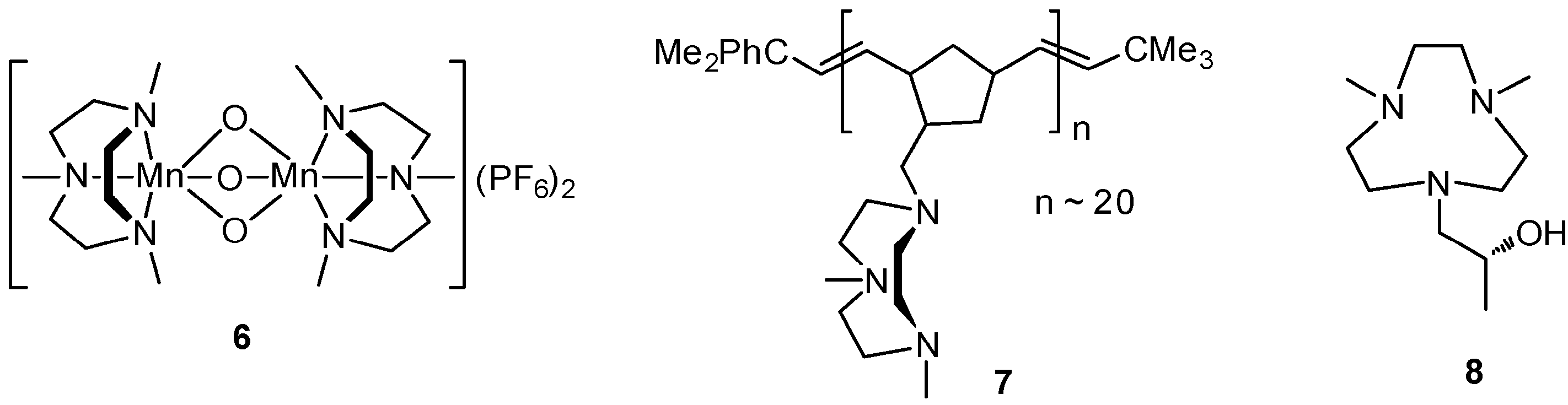
3. C–H Oxidations with H2O2 in the Presence of Mn Complexes with Me3tacn Derived Ligands
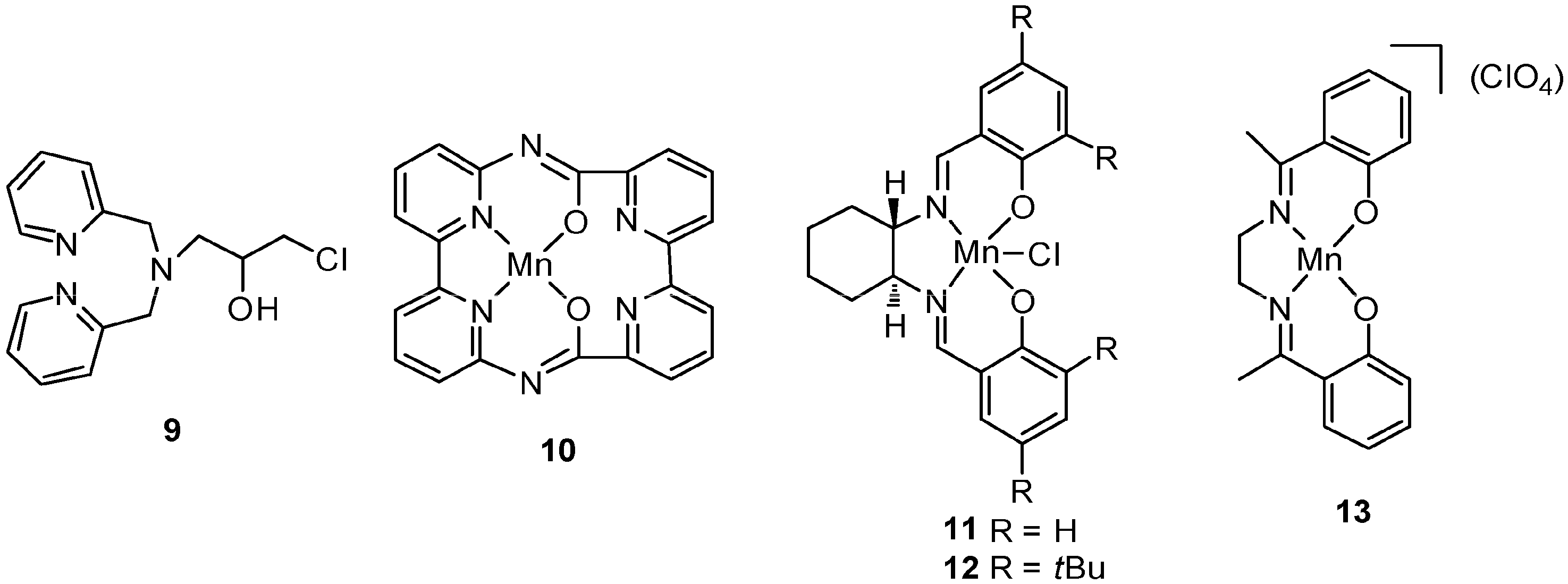
4. C–H Oxidations with H2O2 in the Presence of Mn Complexes with N,O Donor Ligands
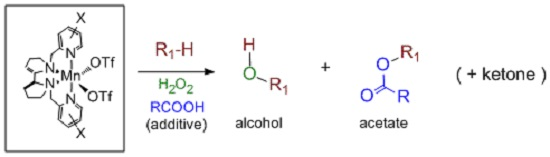
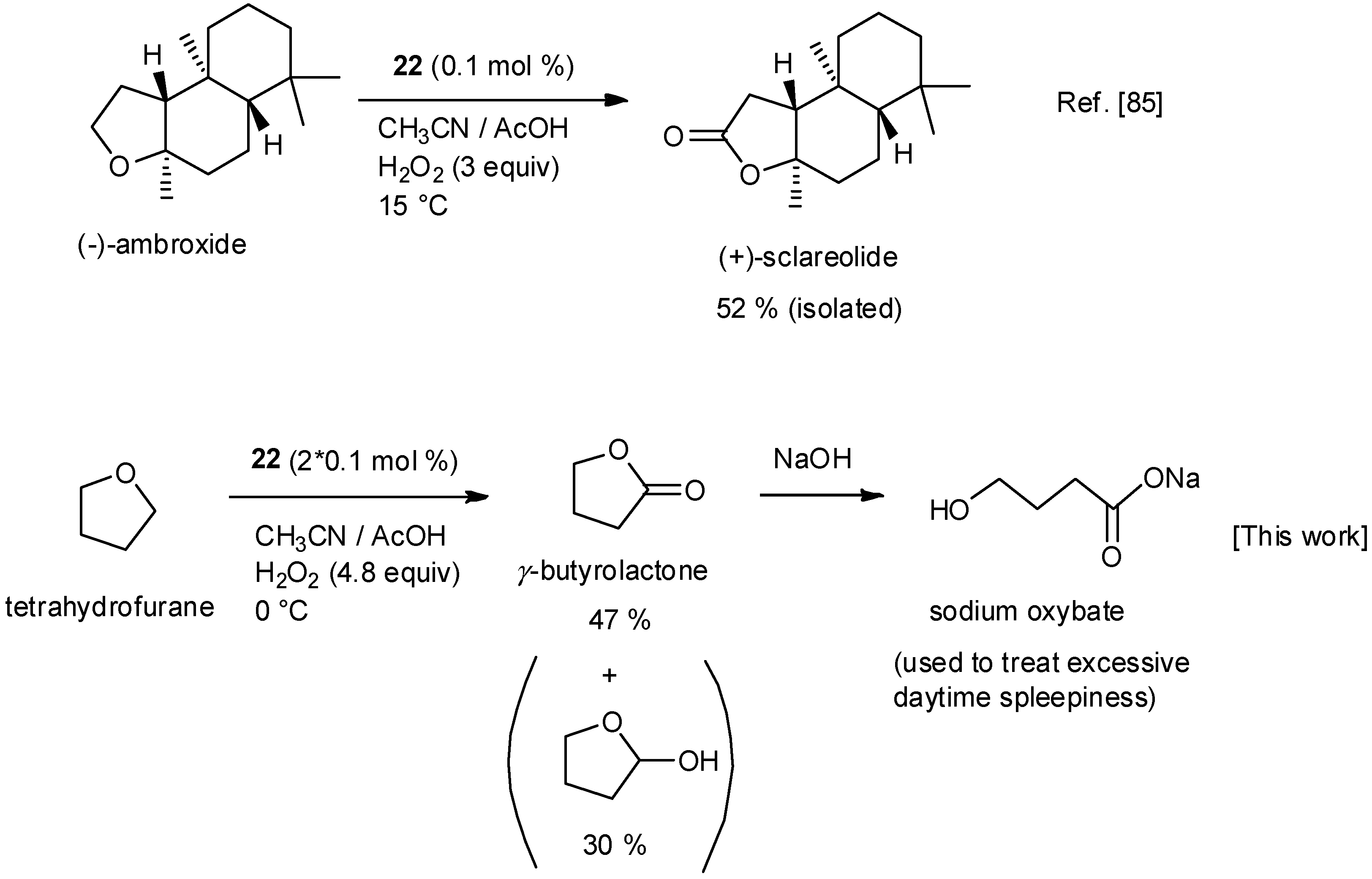
5. C–H Oxidations with H2O2 in the Presence of Mn Complexes with Aminopyridine Ligands
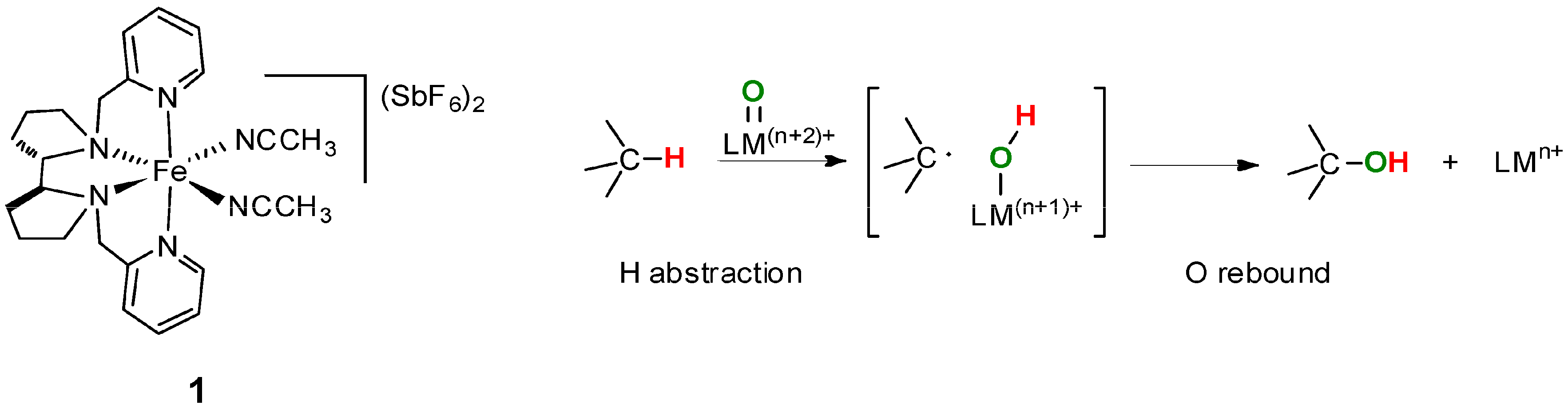
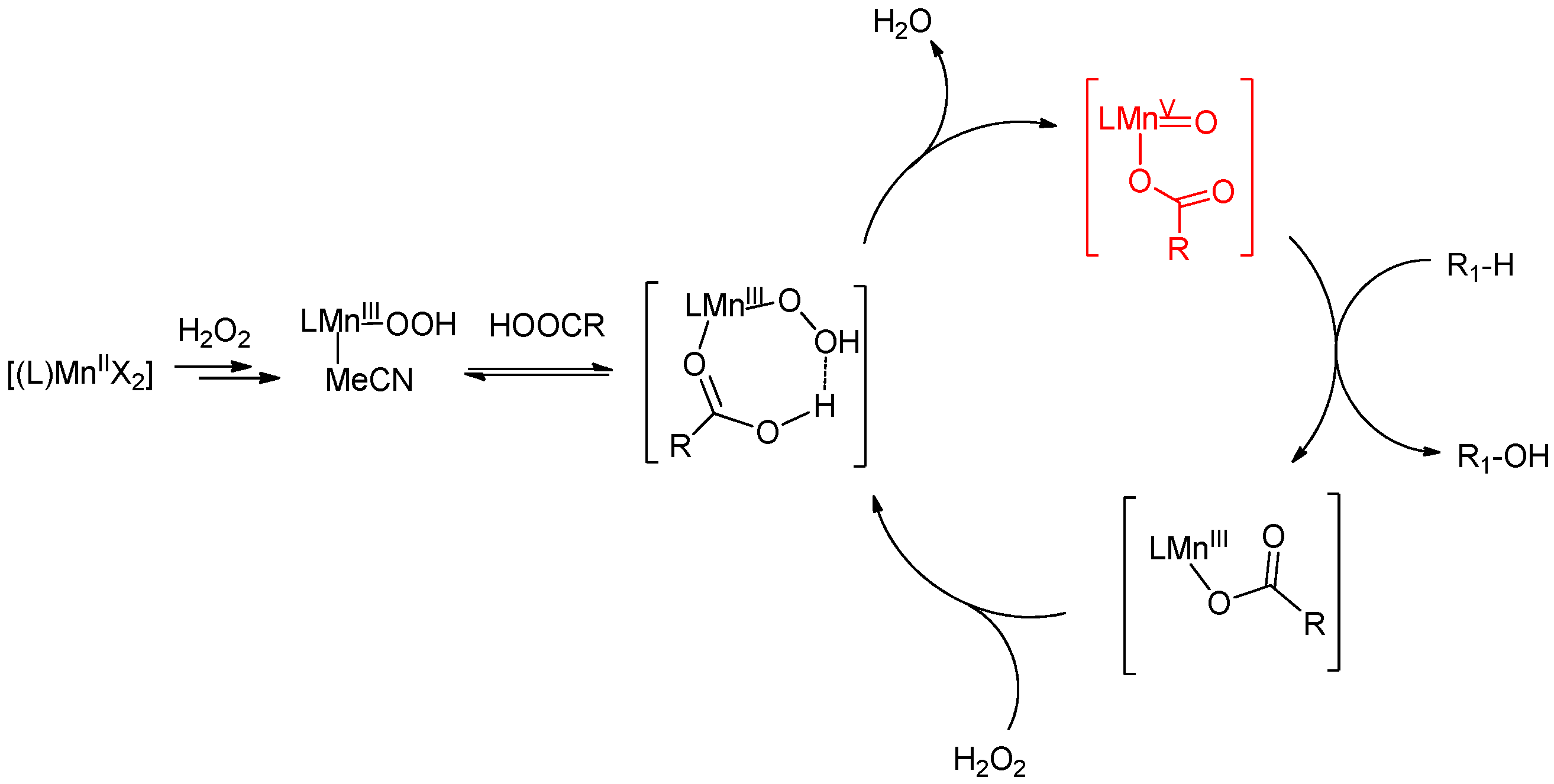
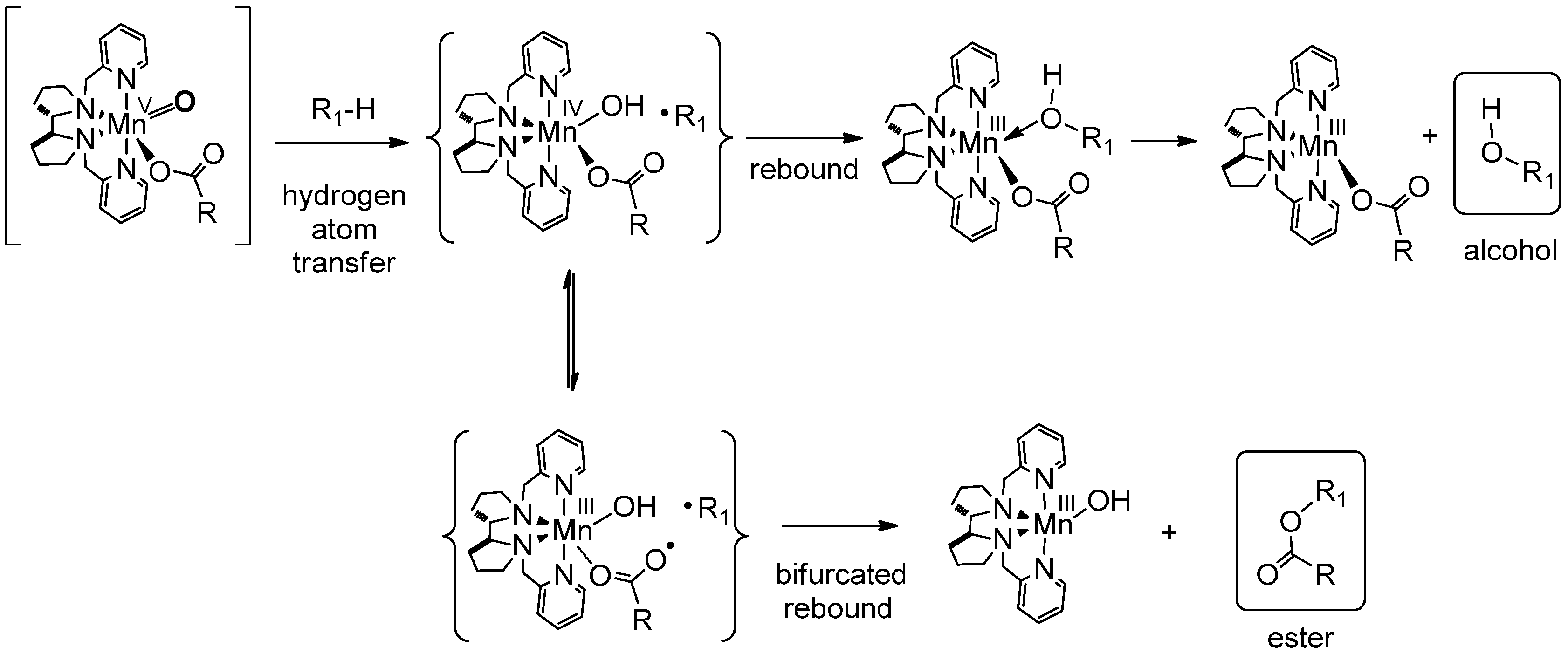
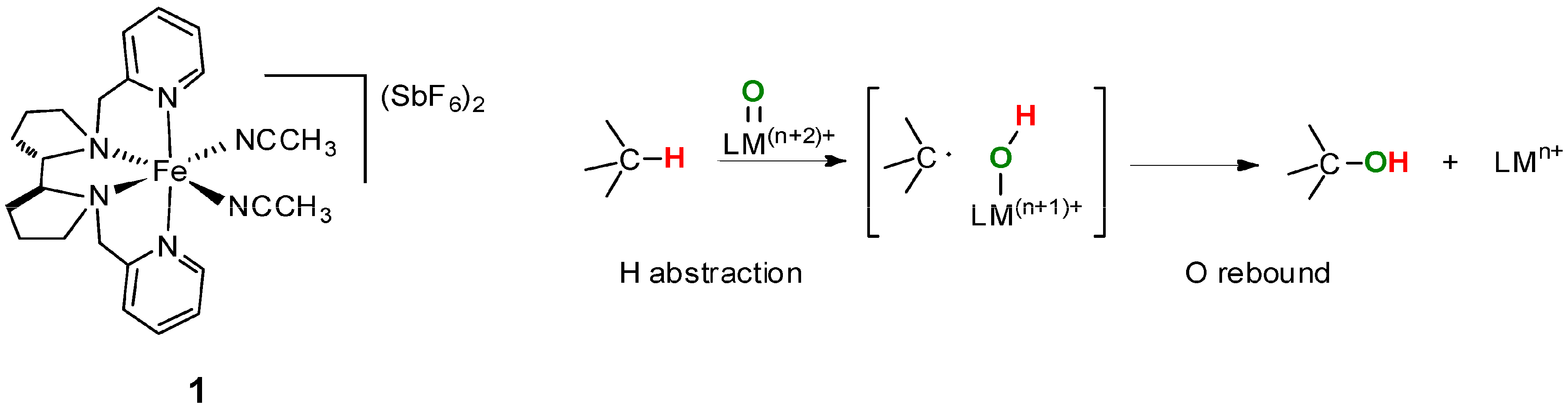
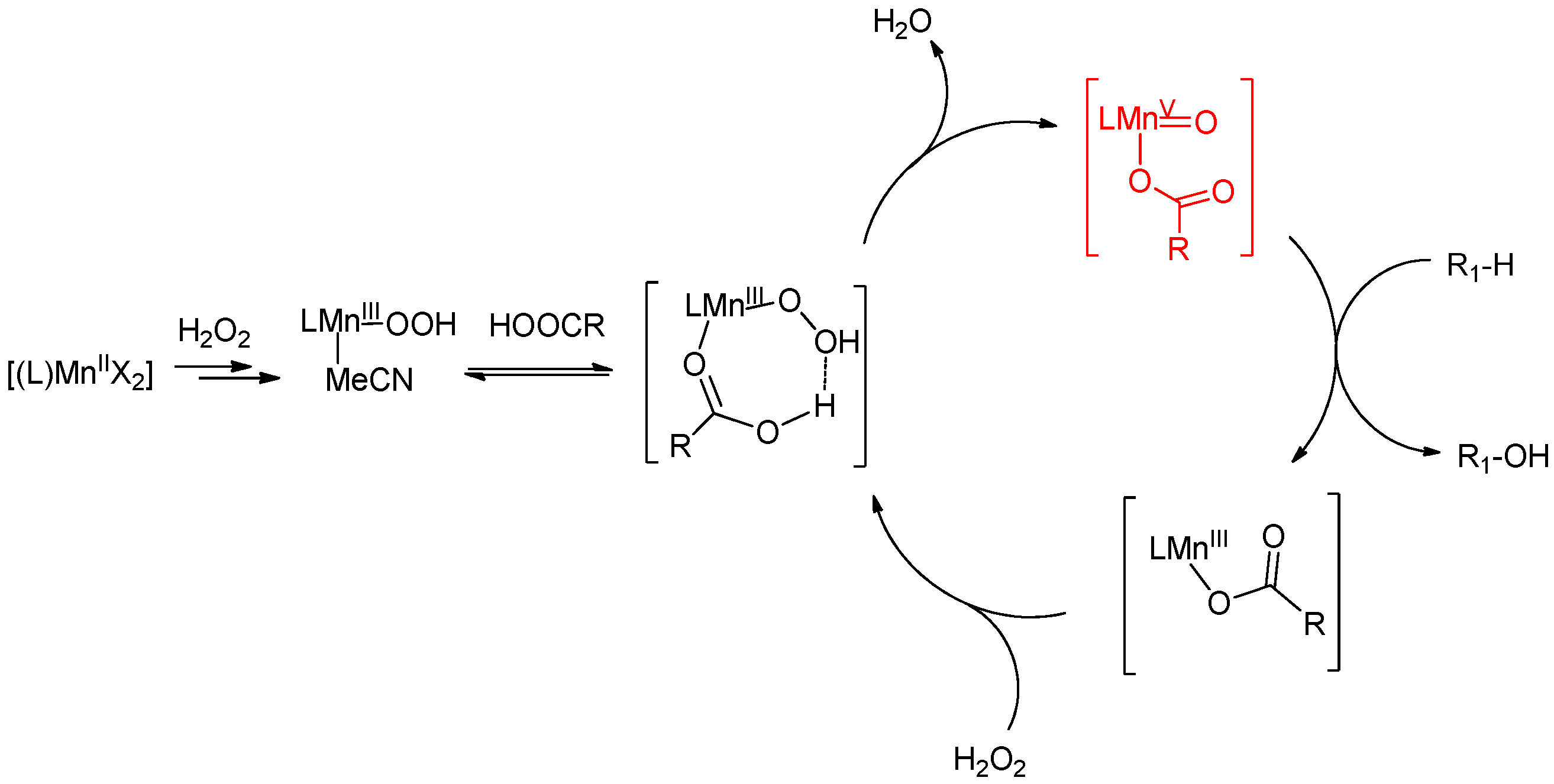
6. Elucidation of the Mechanism of C–H Oxidations with H2O2 in the Presence of Mn Complexes
7. Experimental Conditions
8. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shilov, A.E.; Shul’pin, G.B. Activation of C–H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E.; Shteinman, A.A. Oxygen atom transfer into C–H bond in biological and model chemical systems. Mechanistic aspects. Acc. Chem. Res. 1999, 32, 763–771. [Google Scholar] [CrossRef]
- Christmann, M. Selective Oxidation of Aliphatic C–H Bonds in the Synthesis of Complex Molecules. Angew. Chem. Int. Ed. 2008, 47, 2740–2742. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, T.; Baran, P.S. If C–H Bonds Could Talk: Selective C–H Bond Oxidation. Angew. Chem. Int. Ed. 2011, 50, 3362–3374. [Google Scholar] [CrossRef] [PubMed]
- Che, C.M.; Kar-Yan Lo, V.; Zhou, C.Y.; Huang, J.S. Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 2011, 40, 1950–1975. [Google Scholar] [CrossRef] [PubMed]
- McMurray, L.; O’Hara, F.; Gaunt, M.J. Recent developments in natural product synthesis using metal-catalysed C–H bond functionalisation. Chem. Soc. Rev. 2011, 40, 1885–1898. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Yamaguchi, A.D.; Itami, K. C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2012, 51, 8960–9009. [Google Scholar] [CrossRef] [PubMed]
- Gunnoe, T.B. Alkane C–H Activation by Single-Site Metal Catalysis; Pérez, P.J., Ed.; Springer: Dordrecht, Germany, 2012; pp. 1–15. [Google Scholar]
- Roduner, E.; Kaim, W.; Sarkar, B.; Urlacher, V.B.; Pleiss, J.; Gläser, R.; Einicke, W.D.; Sprenger, G.A.; Beifuß, U.; Klemm, E.; Liebner, C.; Hieronymus, H.; Hsu, S.F.; Plietker, B.; Laschat, S. Selective Catalytic Oxidation of C–H Bond with Molecular Oxygen. ChemCatChem 2013, 5, 82–112. [Google Scholar] [CrossRef]
- Qiu, Y.; Gao, S. Trends in applying C–H oxidation to the total synthesis of natural products. Nat. Prod. Rep. 2016, 33, 562–581. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. New Trends in Oxidative Functionalization of Carbon-Hydrogen Bonds: A Review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef]
- Gol’dshleger, N.F.; Tyabin, M.B.; Shilov, A.E.; Shteinman, A.A. Activation of Saturated Hydrocarbons. H/D Exchange in Solutions of Transition Metal Complexes. Zh. Fiz. Khim. 1969, 43, 2174–2175. (In Russian) [Google Scholar]
- Periana, R.A.; Bergman, R.G. Oxidative Addition of Rhodium to Alkane C–H Bonds: Enhancement in Selectivity and Alkyl Group Functionalization. Organometallics 1984, 3, 508–510. [Google Scholar] [CrossRef]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.S.; Zhou, J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–350. [Google Scholar] [CrossRef] [PubMed]
- Costas, M.; Chen, K.; Que, L., Jr. Biomimetic nonheme iron catalysts for alkane hydroxylation. Coord. Chem. Rev. 2000, 200, 517–544. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L., Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef] [PubMed]
- Tanase, S.; Bowman, E. Advances in Inorganic Chemistry; van Eldik, R., Reedijk, J., Eds.; Academic Press: Amsterdam, The Netherlands, 2006; pp. 29–75. [Google Scholar]
- Battaini, G.; Granata, A.; Monzani, E.; Gullotti, M.; Casella, L. Advances in Inorganic Chemistry; van Eldik, R., Reedijk, J., Eds.; Academic Press: Amsterdam, The Netherlands, 2006; pp. 185–233. [Google Scholar]
- Shteinman, A.A. Iron oxygenases: Structure, mechanism and modeling. Russ. Chem. Rev. 2008, 77, 945–966. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Hydrocarbon Oxygenations with Peroxides Catalyzed by Metal Compounds. Mini-Rev. Org. Chem. 2009, 6, 95–104. [Google Scholar] [CrossRef]
- Costas, M. Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Company, A.; Gómez, L.; Costas, M. Iron Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; de Visser, S.P., Kumar, D., Eds.; RCS Publishing: Cambridge, UK, 2011; pp. 148–208. [Google Scholar]
- Talsi, E.P.; Bryliakov, K.P. Chemo- and stereoselective C–H oxidations and epoxidations/cis-dihydroxylations with H2O2, catalyzed by non-heme iron and manganese complexes. Coord. Chem. Rev. 2012, 256, 1418–1434. [Google Scholar] [CrossRef]
- Bryliakov, K.P. Environmentally Sustainable Catalytic Asymmetric Oxidations; CRC Press: Boca Raton, FL, USA, 2014; pp. 109–119. [Google Scholar]
- Canta, M.; Rodriguez, M.; Costas, M. Recent Advances in the Selective Oxidation of Alkyl C–H Bonds Catalyzed by Iron Coordination Complexes. Top. Curr. Chem. 2016, 372, 27–54. [Google Scholar] [PubMed]
- Afanasiev, P.; Sorokin, A.B. μ-Nitrido Diiron Macrocyclic Platform: Particular Structure for Particular Catalysis. Acc. Chem. Res. 2016, 49, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Olivo, G.; Lanzalunga, O.; di Stefano, S. Non-Heme Imine-Based Iron Complexes as Catalysts for Oxidative Processes. Adv. Synth. Catal. 2016, 358, 843–863. [Google Scholar] [CrossRef]
- Sheu, C.; Reichert, S.A.; Cofré, P.; Ross, B., Jr.; Sobkowiak, A.; Sawyer, D.T.; Kanofski, J.R. Iron-Induced Activation of Hydrogen Peroxide for the Direct Ketonization of Methylenic Carbon [c-C6H12→c-C6H10(O)] and the Dioxygenation of Acetylenes and Arylolefins. J. Am. Chem. Soc. 1990, 112, 1936–1942. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; White, M.C. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Bigi, M.A.; Reed, S.A.; White, M.C. Diverting non-haem iron catalysed aliphatic C–H hydroxylations towards desaturations. Nat. Chem. 2011, 3, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Bigi, M.A.; Reed, S.A.; White, M.C. Directed Metal (Oxo) Aliphatic C–H Hydroxylations: Overriding Substrate Bias. J. Am. Chem. Soc. 2012, 134, 9721–9726. [Google Scholar] [CrossRef] [PubMed]
- Gormisky, P.; White, M.C. Catalyst-Controlled Aliphatic C–H Oxidations with a Predictive Model for Site-Selectivity. J. Am. Chem. Soc. 2013, 135, 14052–14055. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.M.; Feng, K.; Clark, J.R.; Trzepkowski, L.J.; White, M.C. Remote Oxidation of Aliphatic C-H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2015, 137, 14590–14593. [Google Scholar] [CrossRef] [PubMed]
- Prat, I.; Gomez, L.; Canta, M.; Ribas, X.; Costas, M. An Iron Catalyst for Oxidation of Alkyl CH Bonds Showing Enhanced Selectivity for Methylenic Sites. Chem. Eur. J. 2013, 19, 1908–1913. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Canta, M.; Font, D.; Prat, I.; Ribas, X.; Costas, M. Regioselective Oxidation of Nonactivated Alkyl C–H Groups Using Highly Structured Non-Heme Iron Catalysts. J. Org. Chem. 2013, 78, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Canta, M.; Font, D.; Gomez, L.; Ribas, X.; Costas, M. The Iron(II) Complex [Fe(CF3SO3)2(mcp)] as a Convenient, Readily Available Catalyst for the Selective Oxidation of Methylenic Sites in Alkanes. Adv. Synth. Catal. 2014, 356, 818–830. [Google Scholar] [CrossRef]
- Font, D.; Canta, M.; Milan, M.; Cusso, O.; Ribas, X.; Gebbink, R.J.M.K.; Costas, M. Readily Accessible Bulky Iron Catalysts Exhibiting Site Selectivity in the Oxidation of Steroidal Substrates. Angew. Chem. Int. Ed. 2016, 55, 5776–5779. [Google Scholar] [CrossRef] [PubMed]
- Bryliakov, K.P.; Talsi, E.P. Active sites and mechanisms of bioinspired oxidation with H2O2, catalyzed by non-heme Fe and related Mn complexes. Coord. Chem. Rev. 2014, 276, 73–96. [Google Scholar] [CrossRef]
- Groves, J.T. High-valent iron in chemical and biological oxidations. J. Inorg. Biochem. 2006, 100, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Lyakin, O.Y.; Bryliakov, K.P.; Britovsek, G.J.P.; Talsi, E.P. EPR Spectroscopic Trapping of the Active Species of Nonheme Iron-Catalyzed Oxidation. J. Am. Chem. Soc. 2009, 131, 10798–10799. [Google Scholar] [CrossRef] [PubMed]
- Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. EPR, 1H and 2H NMR, and Reactivity Studies of the Iron–Oxygen Intermediates in Bioinspired Catalyst Systems. Inorg. Chem. 2011, 50, 5526–5538. [Google Scholar] [CrossRef] [PubMed]
- Prat, I.; Mathieson, J.S.; Güell, M.; Ribas, X.; Luis, J.M.; Cronin, L.; Costas, M. Observation of Fe(V)=O using variable-temperature mass spectrometry and its enzyme-like C–H and C=C oxidation reactions. Nat. Chem. 2011, 3, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Asymmetric Epoxidations with H2O2 on Fe and Mn Aminopyridine Catalysts: Probing the Nature of Active Species by Combined Electron Paramagnetic Resonance and Enantioselectivity Study. ACS Catal. 2012, 2, 1196–1202. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Prat, I.; Bryliakov, K.P.; Costas, M.; Talsi, E.P. EPR detection of Fe(V)=O active species in nonheme iron-catalyzed oxidations. Catal. Commun. 2012, 29, 105–108. [Google Scholar] [CrossRef]
- Oloo, W.N.; Meier, K.K.; Wang, Y.; Shaik, S.; Münck, E.; Que, L. Identification of a low-spin acylperoxoiron(III) intermediate in bio-inspired non-heme iron-catalysed oxidations. Nat. Commun. 2014, 5, 3046. [Google Scholar] [CrossRef] [PubMed]
- Lyakin, O.Y.; Zima, A.M.; Samsonenko, D.G.; Bryliakov, K.P.; Talsi, E.P. EPR Spectroscopic Detection of the Elusive FeV=O Intermediates in Selective Catalytic Oxofunctionalizations of Hydrocarbons Mediated by Biomimetic Ferric Complexes. ACS Catal. 2015, 5, 2702–2707. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; García-España, E.; Basallote, M.G.; Münck, E.; Que, L., Jr.; Company, A.; Costas, M. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C–H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.M.; Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Dramatic Effect of Carboxylic Acid on the Electronic Structure of the Active Species in Fe(PDP)-Catalyzed Asymmetric Epoxidation. ACS Catal. 2016, 6, 5399–5404. [Google Scholar] [CrossRef]
- Battioni, P.; Renaud, J.P.; Bartoli, J.F.; Mansuy, D. Hydroxylation of alkanes by hydrogen peroxide: And efficient system using manganese porphyrins and imidazole as catalysts. J. Chem. Soc. Chem. Commun. 1986, 341–343. [Google Scholar] [CrossRef]
- Talsi, E.P.; Ottenbacher, R.V.; Bryliakov, K.P. Bioinspired oxidations of aliphatic C–H groups with H2O2 in the presence of manganese complexes. J. Organomet. Chem. 2015, 793, 102–107. [Google Scholar] [CrossRef]
- Battioni, P.; Renaud, J.P.; Bartoli, J.F.; Reina-Artiles, M.; Fort, M.; Mansuy, D. Monooxygenase-like Oxidation of Hydrocarbons by H2O2 Catalyzed by Manganese Porphyrins and Imidazole: Selection of the Best Catalytic System and Nature of the Active Oxygen Species. J. Am. Chem. Soc. 1988, 110, 8462–8470. [Google Scholar] [CrossRef]
- Banfi, S.; Maiocchi, A.; Moggi, A.; Montanari, F.; Quici, S. Hydrogen peroxide oxygenation of alkanes catalysed by manganese(III)-tetraarylporphyrins: The remarkable co-catalytic effect of lipophilic carboxylic acids and heterocyclic bases. J. Chem. Soc. Chem. Commun. 1990, 24, 1794–1796. [Google Scholar] [CrossRef]
- Banfi, S.; Legramandi, F.; Montanari, F.; Pozzi, G.; Quici, S. Biomimetic models of cytochrome P-450. A doubly tailed manganese(III)–tetraaryl porphyrin; an extremely efficient catalyst for hydrocarbon oxygenations promoted by 30% H2O2. J. Chem. Soc. Chem. Commun. 1991, 18, 1285–1287. [Google Scholar] [CrossRef]
- Carrier, M.N.; Scheer, C.; Gouvine, P.; Bartoli, J.F.; Battioni, P.; Mansuy, D. Biomimetic hydroxylation of aromatic compounds: Hydrogen peroxide and manganese-polyhalogenated porphyrins as a particularly good system. Tetrahedron Lett. 1990, 31, 6645–6648. [Google Scholar] [CrossRef]
- Thellend, A.; Battioni, P.; Mansuy, D. Ammonium acetate as a very simple and efficient cocatalyst for manganese porphyrin-catalysed oxygenation of hydrocarbons by hydrogen peroxide. J. Chem. Soc. Chem. Commun. 1994, 1035–1036. [Google Scholar] [CrossRef]
- Srour, H.; Le Maux, P.; Simonneaux, G. Enantioselective Manganese-Porphyrin-Catalyzed Epoxidation and C–H Hydroxylation with Hydrogen Peroxide in Water/Methanol Solutions. Inorg. Chem. 2012, 51, 5850–5856. [Google Scholar] [CrossRef] [PubMed]
- Amiri, N.; Le Maux, P.; Srour, H.; Nasri, H.; Simonneaux, G. Nitration of Halterman porphyrin: A new route for fine tuning chiral iron and manganese porphyrins with application in epoxidation and hydroxylation reactions using hydrogen peroxide as oxidant. Tetrahedron 2014, 70, 8836–8842. [Google Scholar] [CrossRef]
- Lindsay Smith, J.R.; Shul’pin, G.B. Efficient stereoselective oxygenation of alkanes by peroxyacetic acid or hydrogen peroxide and acetic acid catalysed by a manganese(IV) 1,4,7-trimethyl-1,4,7-triazacyclononane complex. Tetrahedron Lett. 1998, 39, 4909–4912. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Lindsay Smith, J.R. Oxidation with the “H2O2-managanese(IV) complex-carboxylic acid” reagent. 1. Oxidation of saturated hydrocarbons with peroxy acids and hydrogen peroxide. Russ. Chem. Bull. 1998, 47, 2379–2386. [Google Scholar] [CrossRef]
- Wieghardt, K.; Bossek, U.; Nuber, B.; Weiss, J.; Bonvoisin, J.; Corbella, M.; Vitols, S.E.; Girerd, J.J. Synthesis, Crystal Structures, Reactivity, and Magnetochemistry of a Series of Binuclear Complexes of Manganese(II), -(III), and -(IV) of Biological Relevance. The Crystal Structure of [L’MIV(μ-O)3MIVL’](PF6)2·H2O Containing an Unprecedented Short Mn···Mn Distance of 2.296 Å. J. Am. Chem. Soc. 1998, 110, 7398–7411. [Google Scholar]
- Shul’pin, G.B.; Nesterov, D.S.; Shul’pina, L.S.; Pombeiro, A.J.L. A hydroperoxo-rebound mechanism of alkane oxidation with hydrogen peroxide catalyzed by binuclear manganese(IV) complex in the presence of an acid with involvement of atmospheric dioxygen. Inorg. Chim. Acta 2016. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Süss-Fink, G.; Lindsay Smith, J.R. Oxidations by the system “hydrogen peroxide—manganese(IV) complex—acetic acid”—Part II. Hydroperoxidation and hydroxylation of alkanes in acetonitrile. Tetrahedron 1999, 55, 5345–5358. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Süss-Fink, G.; Shul’pina, L.S. Oxidations by the system “hydrogen peroxide–manganese(IV) complex–carboxylic acid”: Part 3. Oxygenation of ethane, higher alkanes, alcohols, olefins and sulfides. J. Mol. Catal. A Chem. 2001, 170, 17–34. [Google Scholar] [CrossRef]
- Nizova, G.V.; Bolm, C.; Ceccarelli, S.; Pavan, C.; Shul’pin, G.B. Hydrocarbon Oxidations with Hydrogen Peroxide Catalyzed by a Soluble Polymer-Bound Manganese(IV) Complex with 1,4,7-Triazacyclononane. Adv. Synth. Catal. 2002, 344, 899–905. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V.; Kozlov, Y.N.; Pechenkina, I.G. Oxidations by the “hydrogen peroxide–manganese(IV) complex–carboxylic acid” system. Part 4. Efficient acid-base switching between catalase and oxygenase activities of a dinuclear manganese(IV) complex in the reaction with H2O2 and an alkane. New J. Chem. 2002, 26, 1238–1245. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Metal-catalysed hydrocarbon oxidations. C. R. Chim. 2003, 6, 163–178. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Nizova, G.V.; Kozlov, Y.N.; Arutyunov, V.S.; dos Santos, A.C.M.; Ferreira, A.C.T.; Mandelli, D. Oxidations by the system “hydrogen peroxide–[Mn2L2O3][PF6]2(L = 1,4,7-trimethyl-1,4,7-triazacyclononane)–oxalic acid”. Part 6. Oxidation of methane and other alkanes and olefins in water. J. Organomet. Chem. 2005, 690, 4498–4504. [Google Scholar] [CrossRef]
- Nizova, G.V.; Shul’pin, G.B. A unique rate-accelerating effect of certain amino acids in the H2O2 oxidation of alkanes catalyzed by a dinuclear manganese complex containing 1,4,7-trimethyl-1,4,7-triazacyclononane. Tetrahedron 2007, 63, 7997–8001. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Matthes, M.G.; Romakh, V.B.; Barbosa, M.I.F.; Aoyagi, J.L.T.; Mandelli, D. Oxidations by the system ‘hydrogen peroxide–[Mn2L2O3][PF6]2(L=1,4,7-trimethyl-1,4,7-triazacyclononane)–carboxylic acid’. Part 10: Co-catalytic effect of different carboxylic acids in the oxidation of cyclohexane, cyclohexanol, and acetone. Tetrahedron 2008, 64, 2143–2152. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Kim, S.O.; Nam, W.; Heo, S.; Kim, J. Alkane Oxidation Catalyzed by Manganese-tmtacn Complexes with H2O2. Bull. Korean Chem. Soc. 2003, 24, 1835–1837. [Google Scholar] [CrossRef]
- Bennur, T.H.; Sabne, S.; Desphande, S.S.; Srinivas, D.; Sivasanker, S. Benzylic oxidation with H2O2 catalyzed by Mn complexes of N,N′,N′′-trimethyl-1,4,7-triazacyclononane: Spectroscopic investigations of the active Mn species. J. Mol. Catal. A Chem. 2002, 185, 71–80. [Google Scholar] [CrossRef]
- Saisaha, P.; Buettner, L.; van der Meer, M.; Hage, R.; Feringa, B.L.; Browne, W.R.; de Boer, J.W. Selective Catalytic Oxidation of Alcohols, Aldehydes, Alkanes and Alkenes Employing Manganese Catalysts and Hydrogen Peroxide. Adv. Synth. Catal. 2013, 355, 2591–2603. [Google Scholar] [CrossRef]
- Romakh, V.B.; Therrien, B.; Süss-Fink, G.; Shul’pin, G.B. Dinuclear Manganese Complexes Containing Chiral 1,4,7-Triazacyclononane-Derived Ligands and Their Catalytic Potential for the Oxidation of Olefins, Alkanes, and Alcohols. Inorg. Chem. 2007, 46, 1315–1331. [Google Scholar] [CrossRef] [PubMed]
- Lessa, J.A.; Horn, A.; Bull, É.S.; Rocha, M.R.; Benassi, M.; Catharino, R.R.; Eberlin, M.N.; Casellato, A.; Noble, C.J.; Hanson, G.R.; Schenk, G.; Giselle, C.; Silva, O.A.; Antunes, O.A.C.; Fernandes, C. Catalase vs. Peroxidase Activity of a Manganese(II) Compound: Identification of a Mn(III)–(μ-O)2–Mn(IV) Reaction Intermediate by Electrospray Ionization Mass Spectrometry and Electron Paramagnetic Resonance Spectroscopy. Inorg. Chem. 2009, 48, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, N.; Tsuchiya, S.; Ogawa, S. Hydrocarbon oxidation with hydrogen peroxide and pentafluoroiodosylbenzene catalyzed by unusually distorted macrocycle manganese complexes. J. Mol Catal. A Chem. 2007, 277, 61–71. [Google Scholar] [CrossRef]
- Lee, N.H.; Lee, C.S.; Jung, D.S. Selective Oxidation of Benzylic Hydrocarbons to Carbonyl Compounds Catalyzed by Mn(III) Salen Complexes. Tetrahedron Lett. 1998, 39, 1385–1388. [Google Scholar] [CrossRef]
- Mardani, H.P.; Golchoubian, H. Selective and efficient C–H oxidation of alkanes with hydrogen peroxide catalyzed by a manganese(III) Schiff base complex. J. Mol. Catal. A. Chem. 2006, 259, 197–200. [Google Scholar] [CrossRef]
- Golchoubian, H.; Ghasemi, N. Diphenylmethane Oxidation Catalyzed by Mononuclear Mn(III) Complexes. J. Chin. Chem. Soc. 2011, 58, 470–473. [Google Scholar] [CrossRef]
- Gómez, I.; Garcia-Bosch, A.; Company, J.; Benet-Buchholz, A.; Polo, X.; Sala, X.; Ribas, M.; Costas, M. Stereospecific C–H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem. Int. Ed. 2009, 48, 5720–5723. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Efficient, Regioselective, and Stereospecific Oxidation of Aliphatic C–H Groups with H2O2, Catalyzed by Aminopyridine Manganese Complexes. Org. Lett. 2012, 14, 4310–4313. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Miao, C.; Wang, S.; Xia, C.; Sun, W. Efficient Benzylic and Aliphatic C–H Oxidation with Selectivity for Methylenic Sites Catalyzed by a Bioinspired Manganese Complex. Org. Lett. 2014, 16, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Miao, C.; Xu, D.; Xia, C.; Sun, W. Highly Efficient Oxidation of Secondary Alcohols to Ketones Catalyzed by Manganese Complexes of N4 Ligands with H2O2. Org. Lett. 2015, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Lv, Y.; Wang, L.; Shang, S.; Chen, B.; Li, G.; Gao, S. Highly efficient oxidation of alcohols ca4alyzed by a porphyrin-inspired manganese complex. Chem. Commun. 2015, 51, 11268–11271. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Mechanism of Selective C–H Hydroxylation Mediated by Manganese Aminopyridine Enzyme Models. ACS Catal. 2015, 5, 39–44. [Google Scholar] [CrossRef]
- Adams, A.M.; Du Bois, J.; Malik, H.A. Comparative Study of the Limitations and Challenges in AtomTransfer C–H Oxidations. Org. Lett. 2015, 17, 6066–6069. [Google Scholar] [CrossRef] [PubMed]
- Costas, M.; Que, L., Jr. Ligand Topology Tuning of Iron-Catalyzed Hydrocarbon Oxidations. Angew. Chem. Int. Ed. 2002, 41, 2179–2181. [Google Scholar] [CrossRef]
- Chen, K.; Costas, M.; Que, L., Jr. Spin state tuning of non-heme iron-catalyzed hydrocarbon oxidations: Participation of FeIII–OOH and FeV=O intermediates. J. Chem. Soc. Dalton Trans. 2002, 672–679. [Google Scholar] [CrossRef]
- Que, L., Jr. The Road to Non-Heme Oxoferryls and Beyond. Acc. Chem. Res. 2007, 40, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Mas-Ballesté, R.; Que, L., Jr. Iron-Catalyzed Olefin Epoxidation in the Presence of Acetic Acid: Insights into the Nature of the Metal-Based Oxidant. J. Am. Chem. Soc. 2007, 129, 15964–15972. [Google Scholar] [CrossRef] [PubMed]
- Mas-Ballesté, R.; Fujita, M.; Que, L., Jr. High-valent iron-mediated cis-hydroxyacetoxylation of olefins. Dalton Trans. 2008, 1828–1830. [Google Scholar] [CrossRef] [PubMed]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Enantioselective Bioinspired Epoxidation of Electron-Deficient Olefins with H2O2 on Aminopyridine Mn Catalysts. ACS Catal. 2014, 4, 1599–1606. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Shubin, A.A.; Shashkov, M.V.; Talsi, E.P.; Bryliakov, K.P. Evidence for “Bifurcated Rebound Mechanism” in Biomimetic C–H Hydroxylation Mediated by Manganese-Aminopyridine Complexes. 2016; Submitted. [Google Scholar]
- Hartwig, J.F.; Larsen, M.A. Undirected, Homogeneous C–H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci. 2016, 2, 281–292. [Google Scholar] [CrossRef] [PubMed]
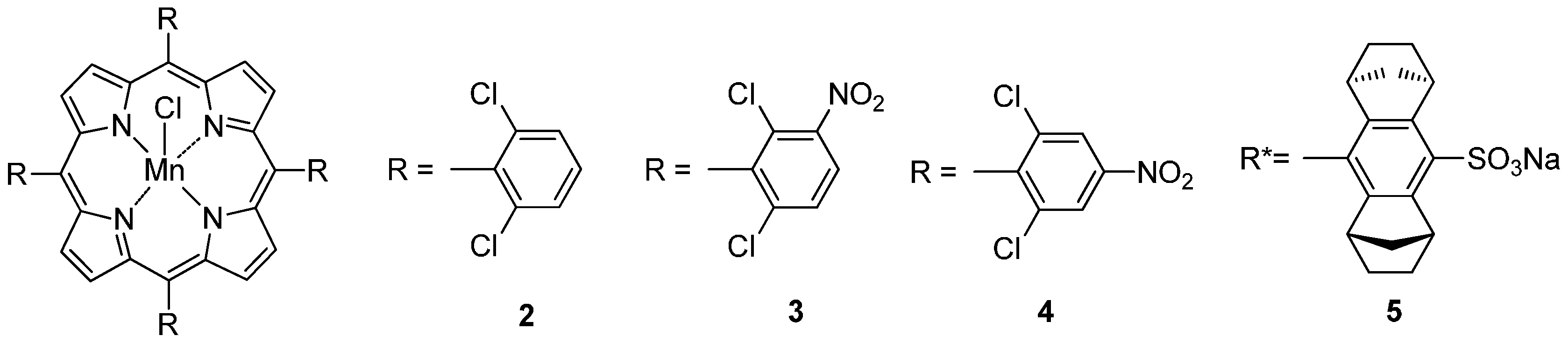
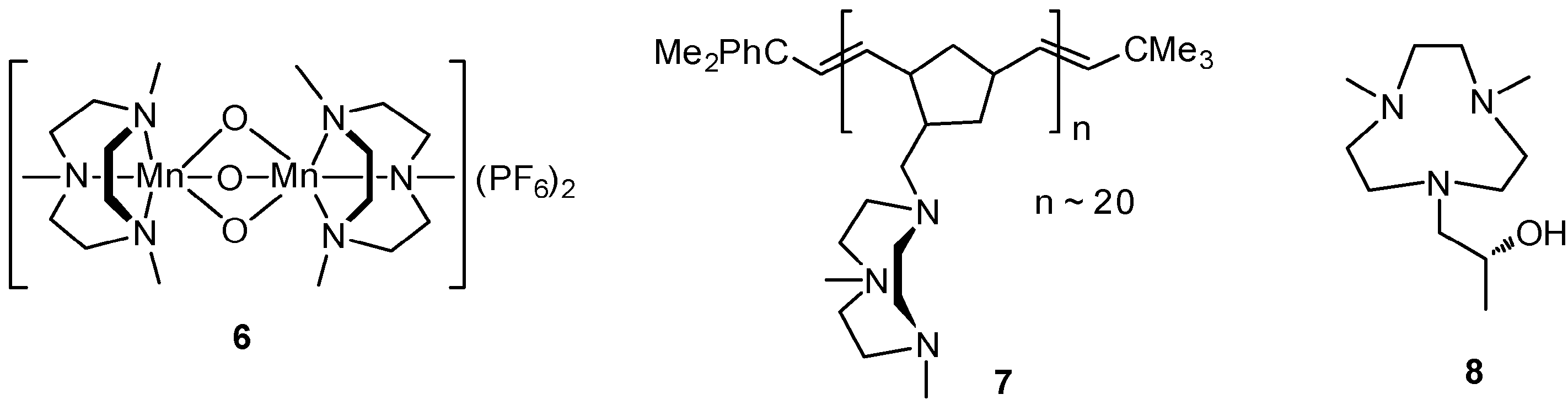
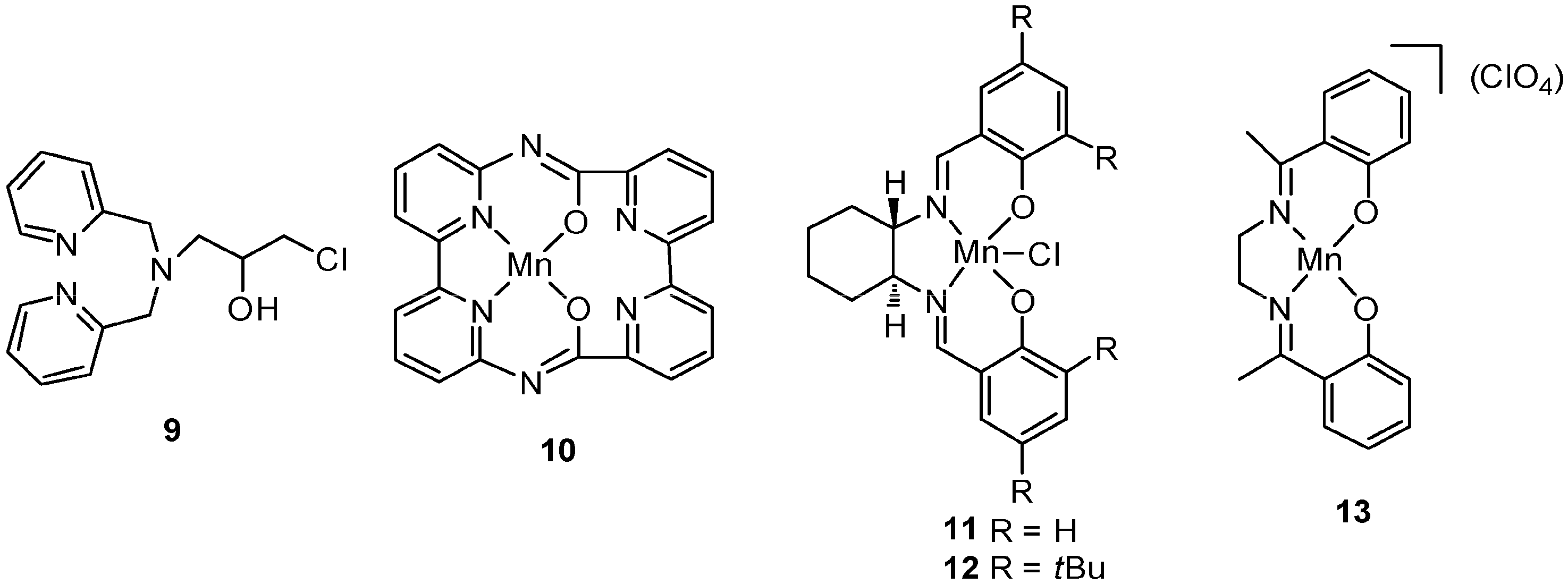
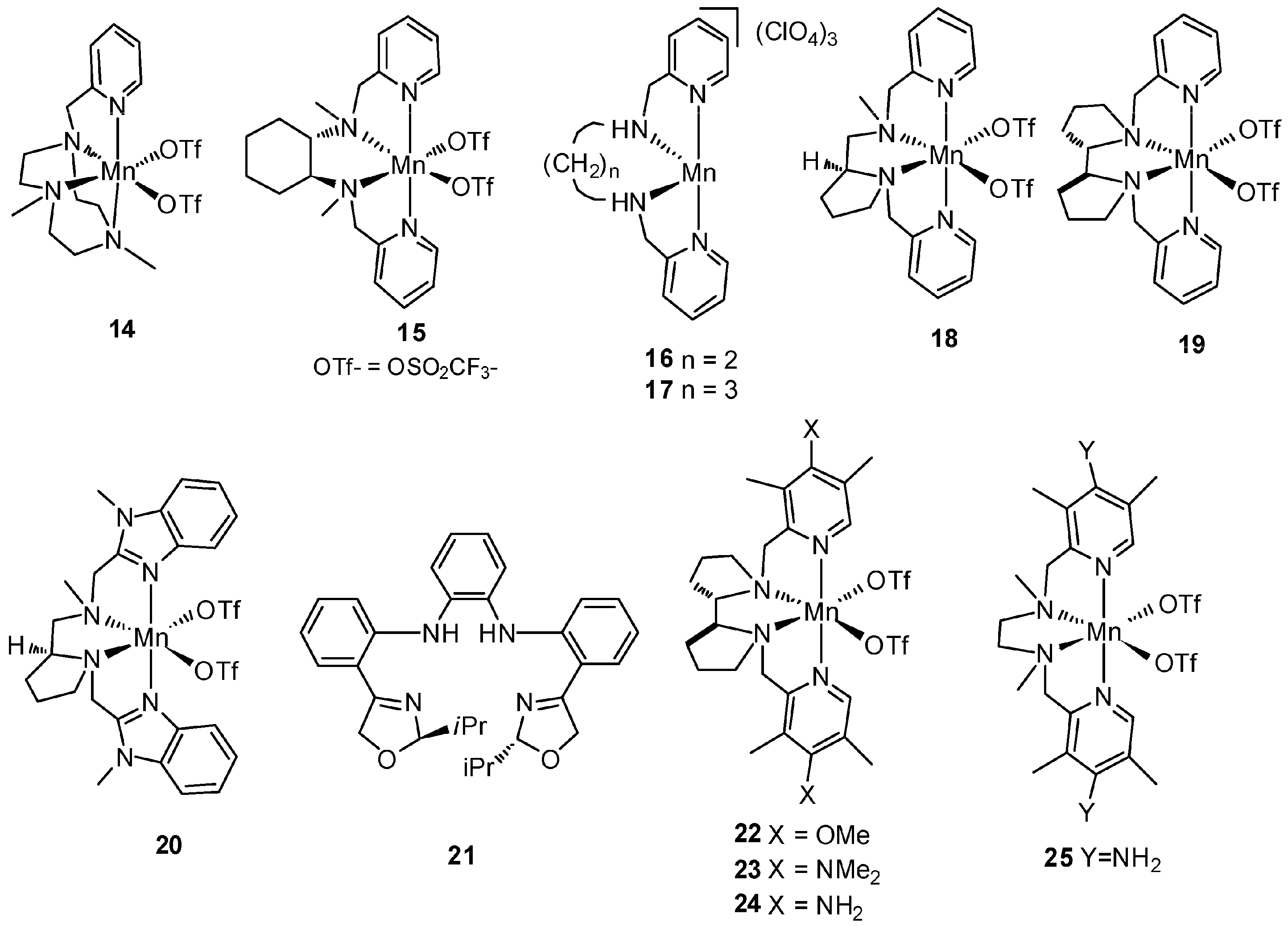
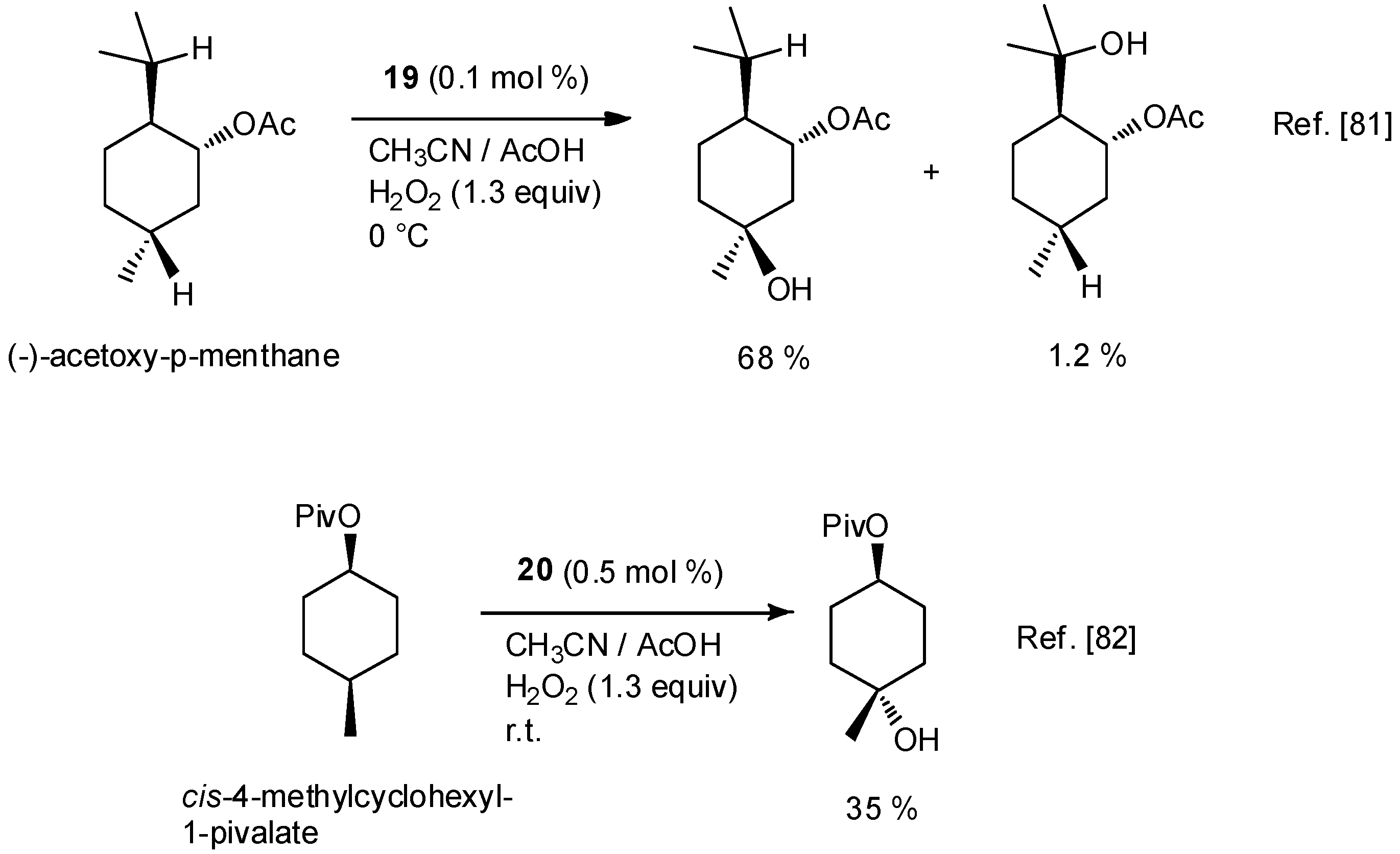
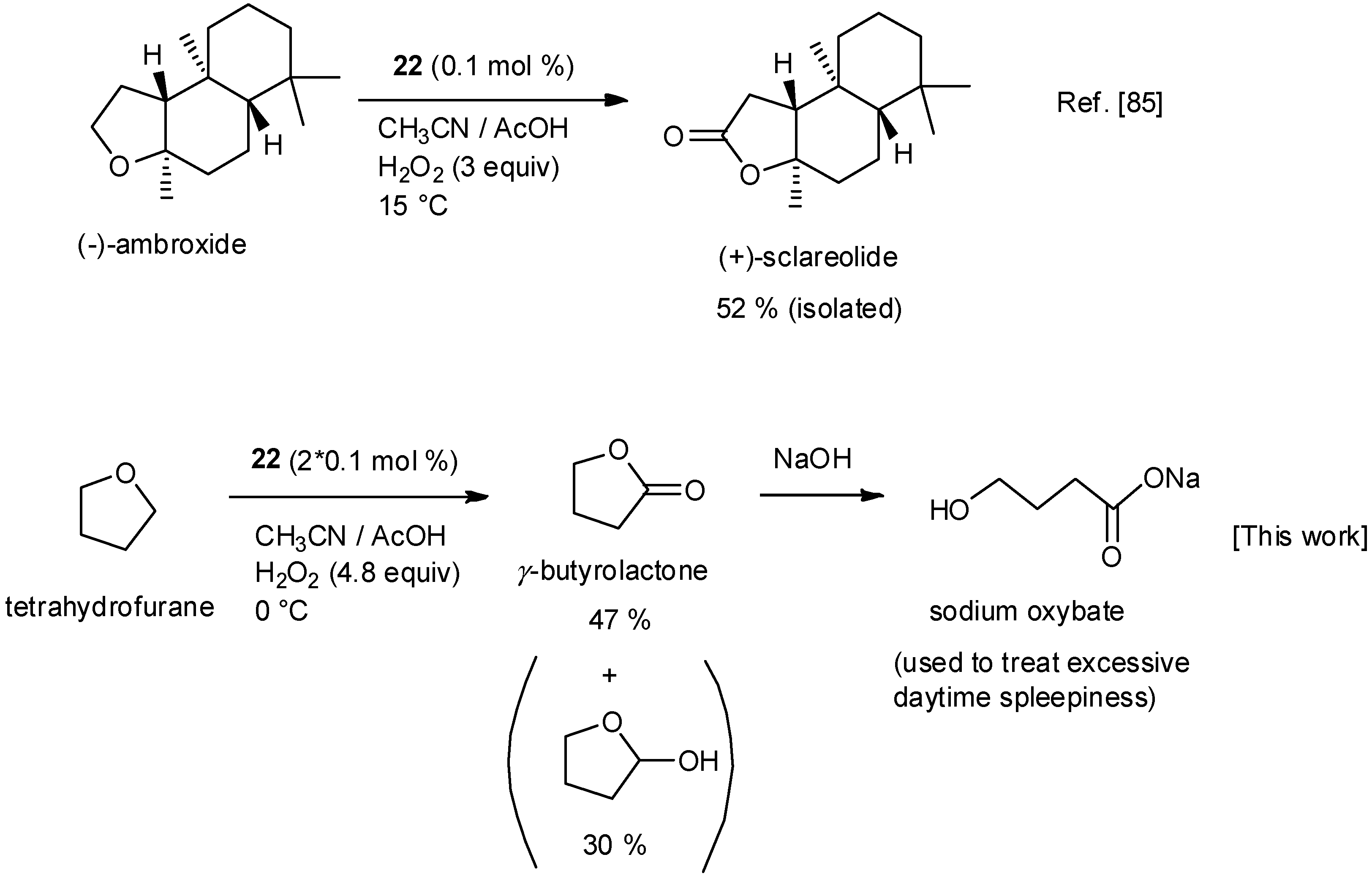

| No. | Cat. | Substrate | Additive | Products (yield a) | Ref. |
|---|---|---|---|---|---|
| 1 | 2 | cyclohexane | imidazole | cyclohexanol (12), cyclohexanone (4) | [50] |
| 2 | 3 | cyclohexane | p-tBu-pyridine/benzoic acid | cyclohexanol (233 b), cyclohexanone (47 b) | [53] |
| 3 | 4 | cyclooctane | p-tBu-pyridine/benzoic acid | cyclooctanol (565 b), cyclooctanone (235 b) | [53] |
| 4 | 2 | naphthalene | imidazole | 1-naphthol (3.5), 2-naphthol (0.4) | [55] |
| 5 | 2 | anisole | imidazole | p-OH-anisole (4.7), o-OH-anisole (0.35), phenol (0.2) | [55] |
| 6 | 2 | cyclohexane | imidazole | cyclohexanol (12.4), cyclohexanone (1.6) | [56] |
| 7 | 2 | cyclohexane | CH3COONH4 | cyclohexanol (20), cyclohexanone (5.2) | [56] |
| 8 | 5 | p-Et-toluene | imidazole | p-acetyltoluene (2.8), 1-(p-tolyl)ethanol (37.2, 57% ee) | [57] |
| No. | Cat. | Substrate | Additive | Products (yield a) | Ref. |
|---|---|---|---|---|---|
| 1 | 6 | hexane | AcOH | hexanones (775), hexanols (575) | [59] |
| 2 | 6 | ethane (20 bar) | AcOH | EtOOH (260), EtOH (120), MeCHO (20) | [64] |
| 3 | 6 | hexane | Na oxalate | 2-hexanol (4.5), 3-hexanol (4.5), 2-hexanone (29), 3-hexanone (29) | [71] |
| 4 | 6 | ethylbenzene | oxalate buffer | 1-phenylethanol (48.5), acetophenone (102) | [72] |
| 6 | 13 | ethylbenzene | none | 1-phenylethanol (7), acetophenone (683) | [78] |
| No. | Cat. | Substrate | Additive | Products (yield a) | Ref. |
|---|---|---|---|---|---|
| 1 | 17 | diphenylmethane | none | benzophenone (250), diphenylmethanol (530) | [79] |
| 2 | 19 | cyclohexane | AcOH | cyclohexanol (28), cyclohexanone (842) | [81] |
| 3 | 20 | cyclohexane | AcOH | cyclohexanone (122) | [82] |
| 4 | 20 | 1-Ph-ethanol | AcOH | acetophenone (4700) | [83] |
| 5 | 21/Mn(OTf)2 | cyclohexanol | Ada-COOH | cyclohexanone (38) | [84] |
| 6 | 19 | cumene | AcOH | cumyl alcohol (209), cumyl acetate (139) | [93] |
| 7 | 23 | cumene | AcOH | cumyl alcohol (560), cumyl acetate (75), acetophenone (30) | [85] |
| 8 | 24 | cumene | AcOH | cumyl alcohol (492), cumyl acetate (61), acetophenone (110) | [93] |
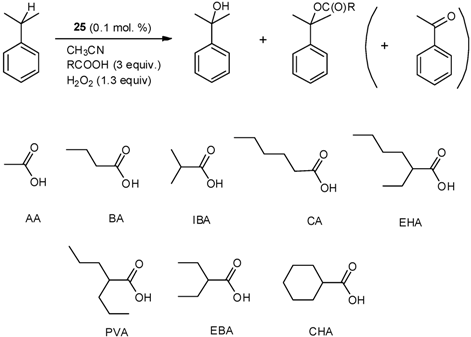
| No | Additive | Conversion (%) | Yield of Alcohol/Acetate/Other a (%) | Alcohol/Ester Ratio |
|---|---|---|---|---|
| 1 | AA | 82.9 | 62.9/12.1/7.9 | 5.2/1 |
| 2 | BA | 74.7 | 60.2/7.3/7.2 | 8.2/1 |
| 3 | CA | 56.5 | 46.6/4.3/5.6 | 10.8/1 |
| 4 | CHA | 34.3 | 30.3/2.0/2.0 | 15.4/1 |
| 5 | IBA | 61.8 | 51.7/3.2/6.9 | 16.2/1 |
| 6 | EHA | 29.8 | 26.1/1.6/2.1 | 16.3/1 |
| 7 | PVA | 38.6 | 34.5/0.9/3.2 | 36.7/1 |
| 8 | EBA | 39.2 | 35.4/0.8/3.0 | 44.7/1 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottenbacher, R.V.; Talsi, E.P.; Bryliakov, K.P. Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn-Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts. Molecules 2016, 21, 1454. https://doi.org/10.3390/molecules21111454
Ottenbacher RV, Talsi EP, Bryliakov KP. Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn-Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts. Molecules. 2016; 21(11):1454. https://doi.org/10.3390/molecules21111454
Chicago/Turabian StyleOttenbacher, Roman V., Evgenii P. Talsi, and Konstantin P. Bryliakov. 2016. "Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn-Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts" Molecules 21, no. 11: 1454. https://doi.org/10.3390/molecules21111454
APA StyleOttenbacher, R. V., Talsi, E. P., & Bryliakov, K. P. (2016). Direct Selective Oxidative Functionalization of C–H Bonds with H2O2: Mn-Aminopyridine Complexes Challenge the Dominance of Non-Heme Fe Catalysts. Molecules, 21(11), 1454. https://doi.org/10.3390/molecules21111454