
Accumulation of Stable Full-Length Circular Group I Intron RNAs during Heat-Shock


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
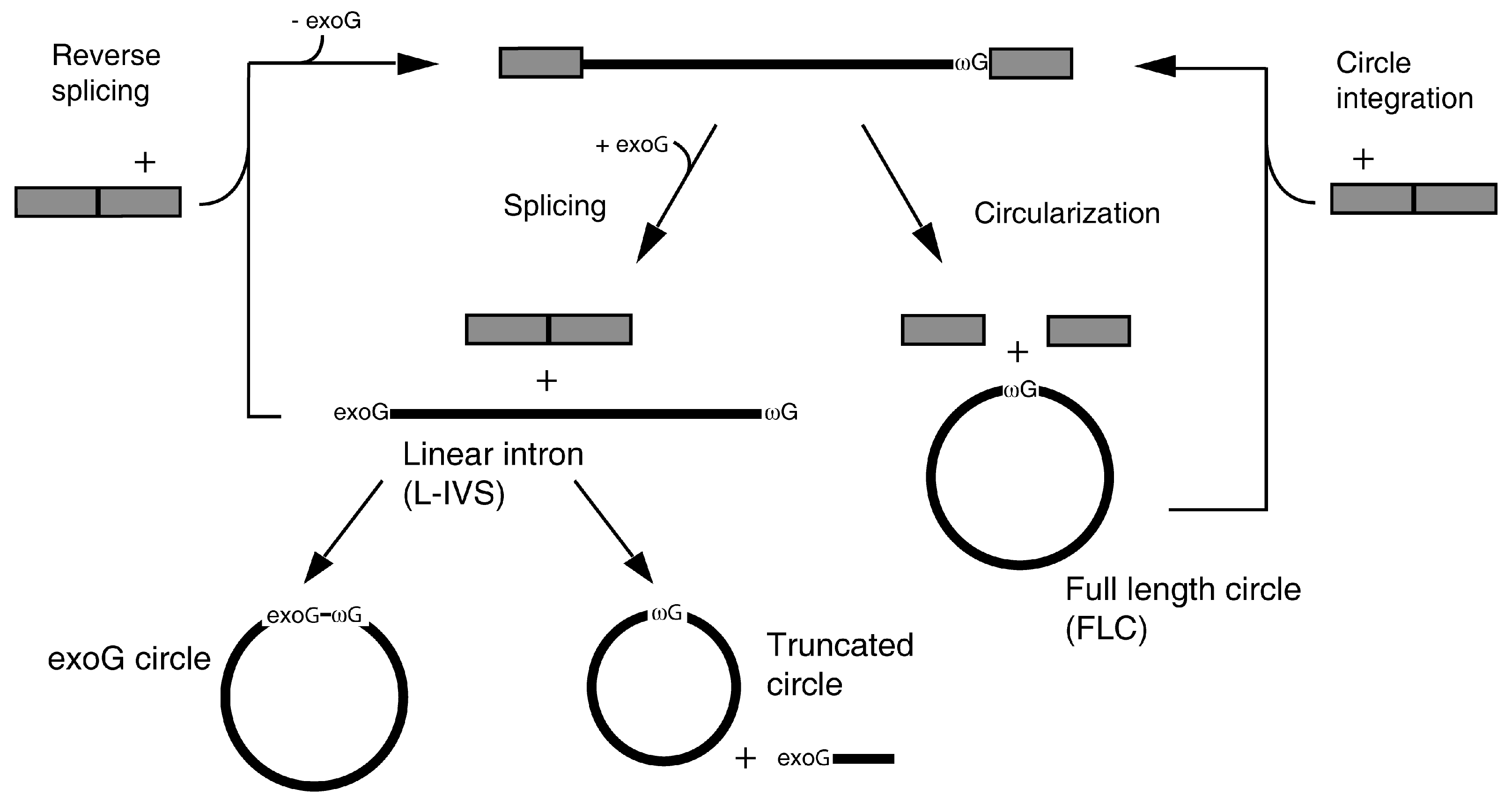
:1. Introduction
2. Results
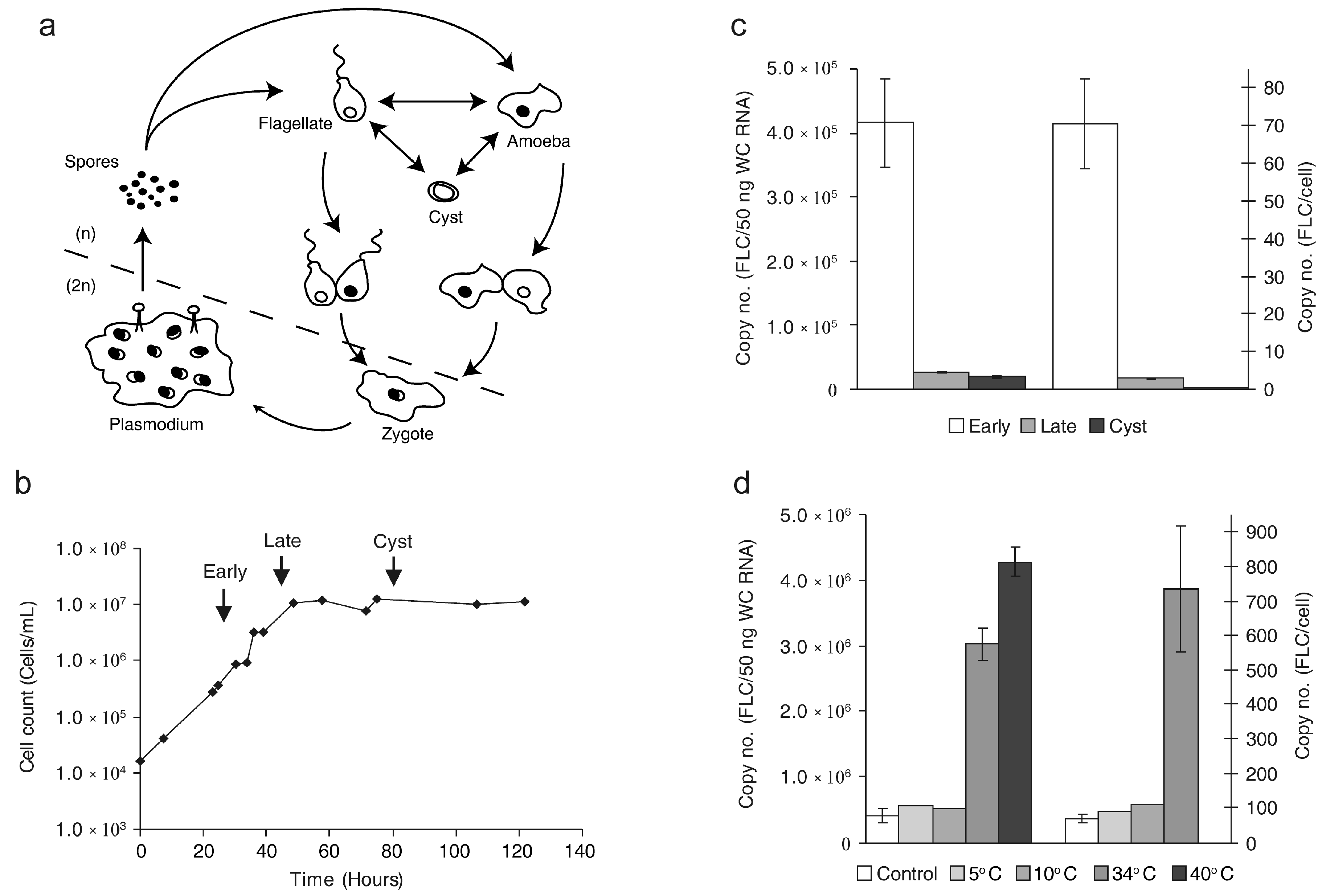
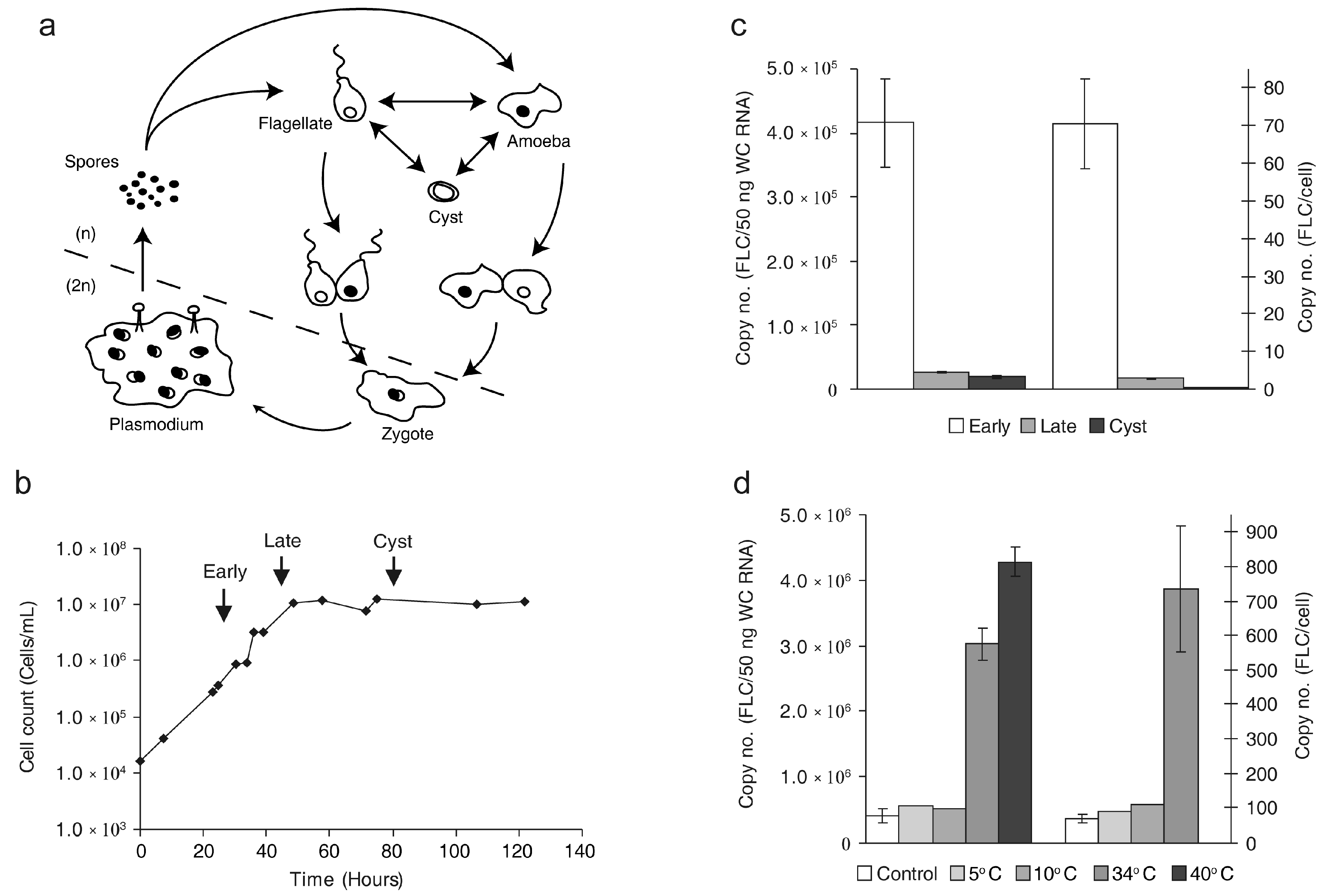
2.1. The Copy Number of FLC Is Sensitive to External Stimuli
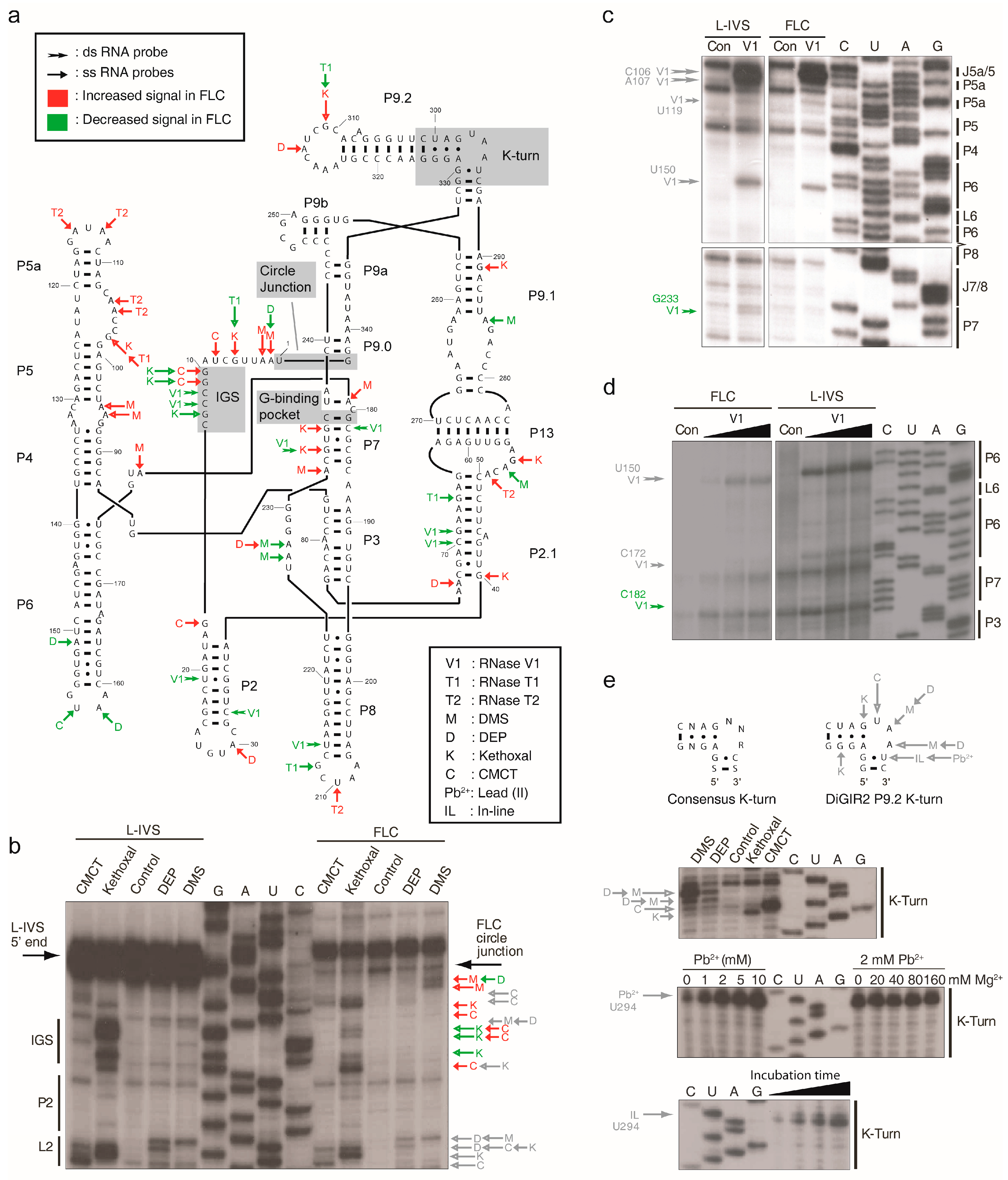
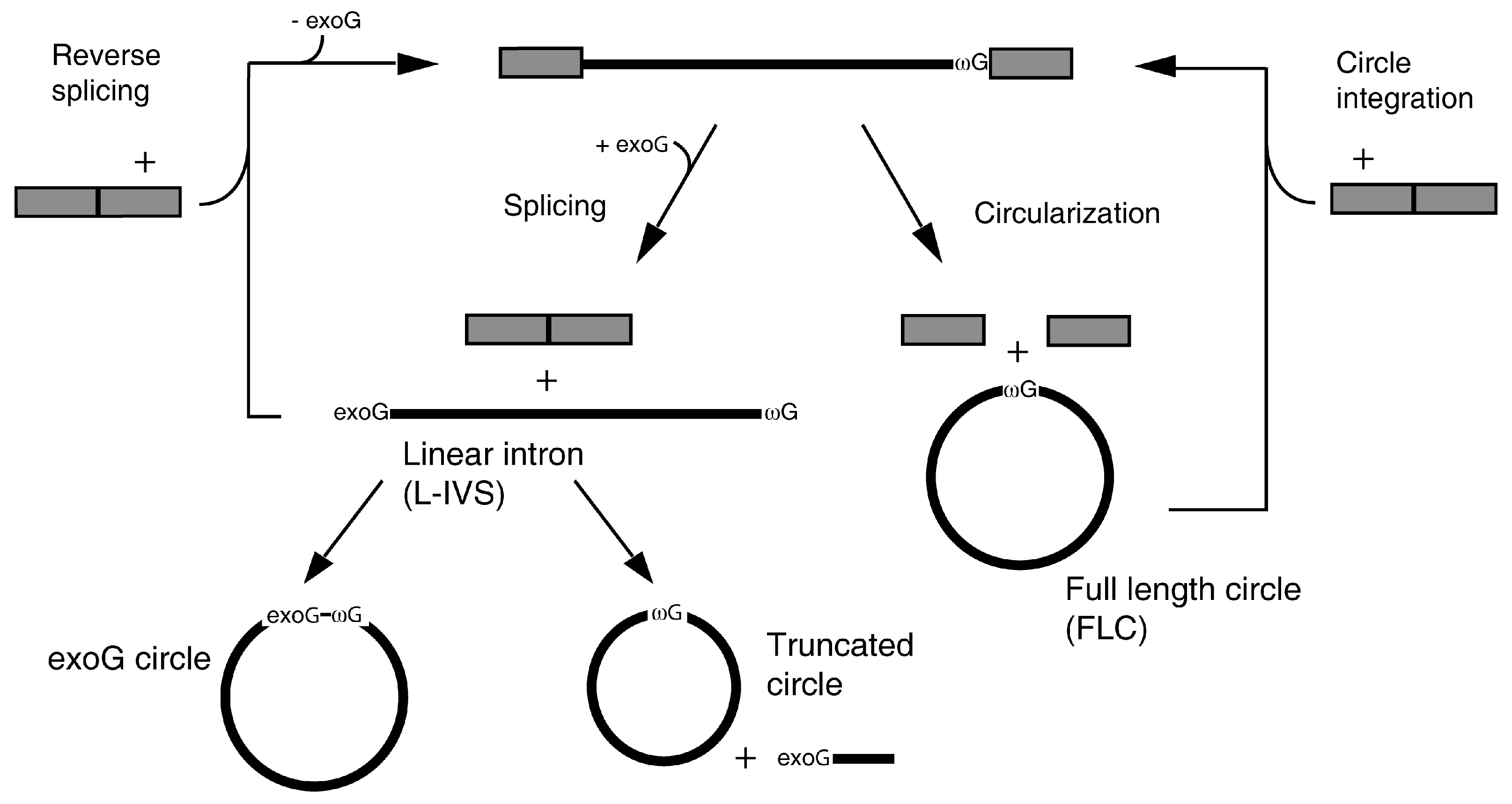
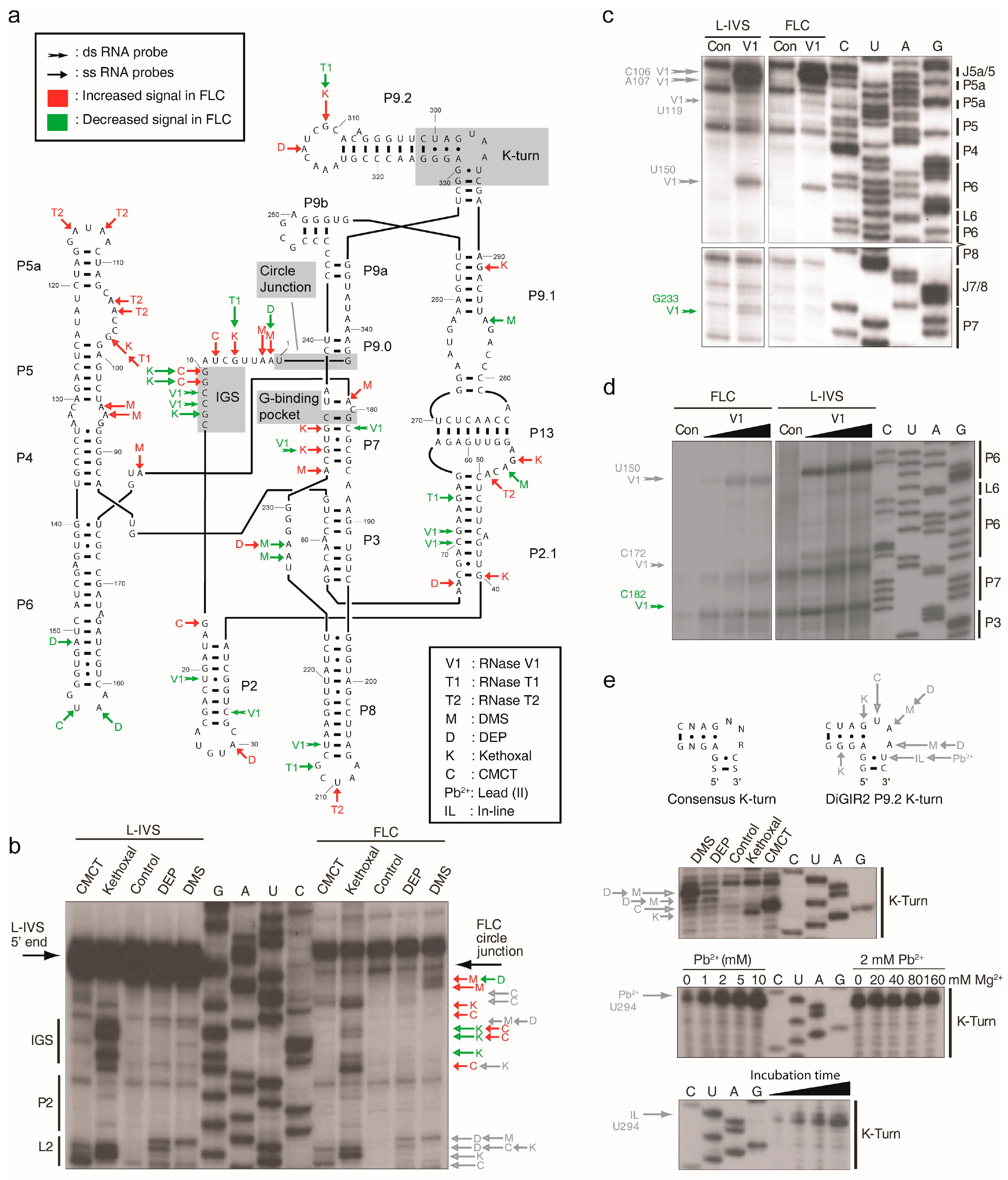
2.2. FLC Is Very Similar in Structure to L-IVS But Has an Unstable Active Site
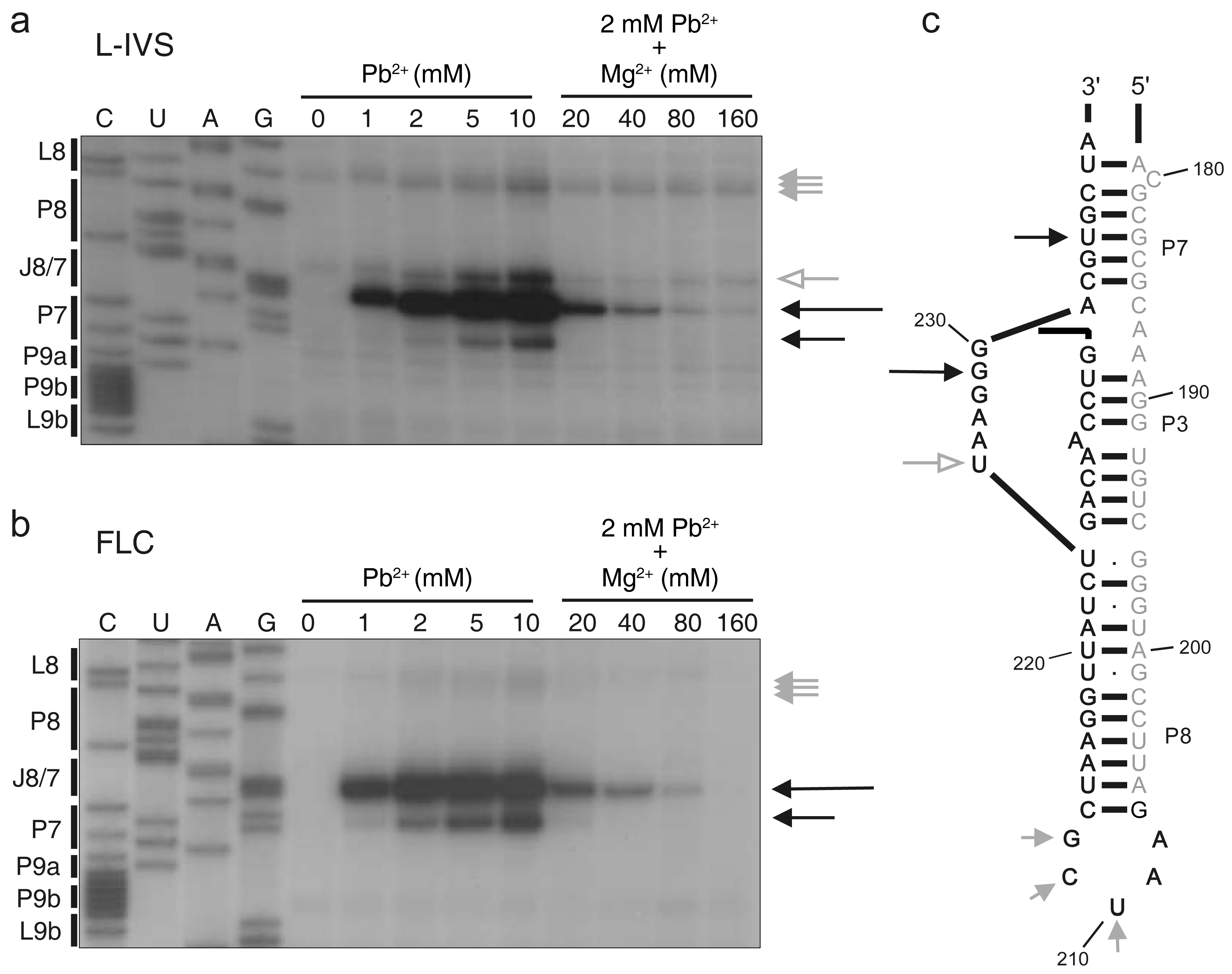
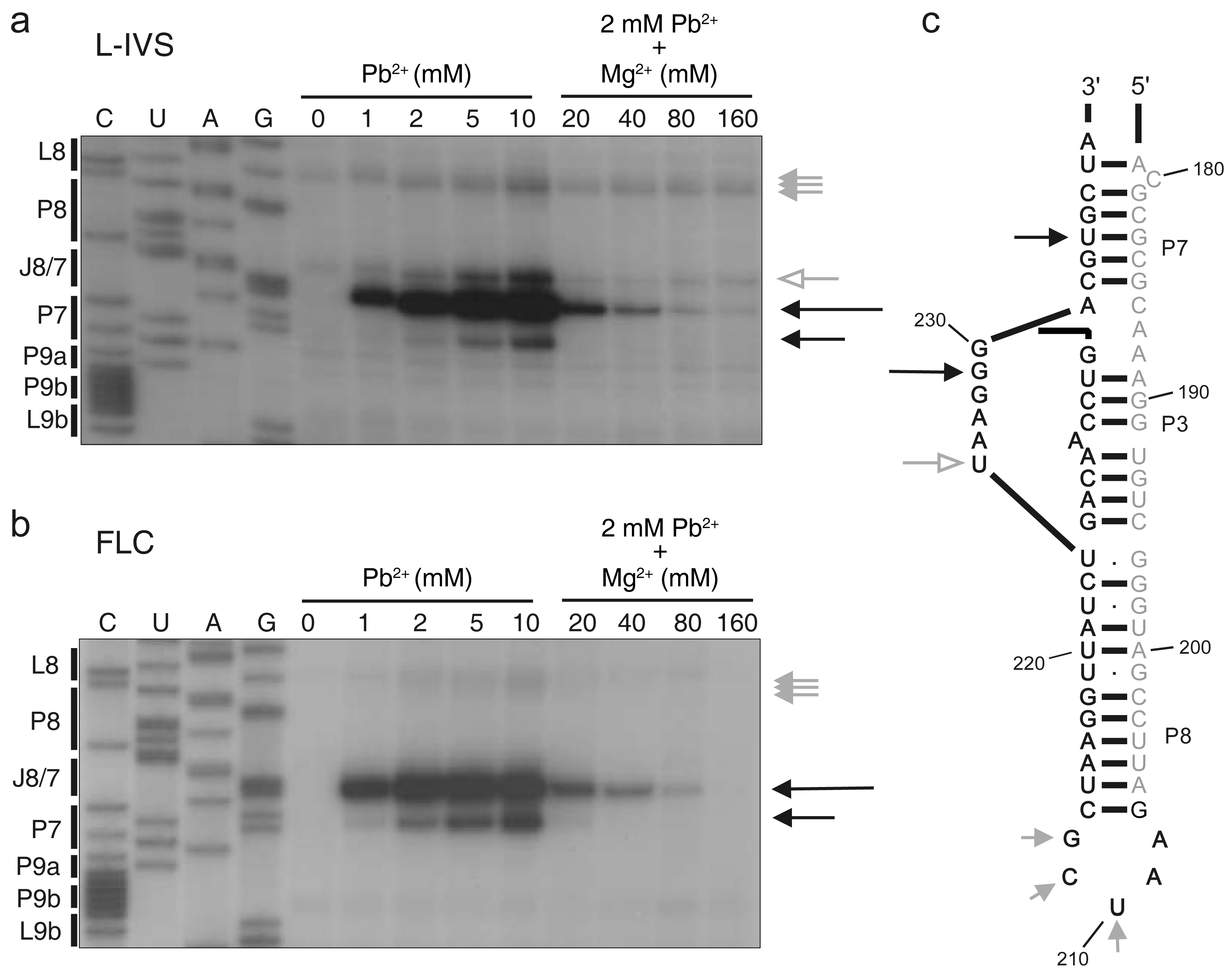
2.3. Pb2+ Cleavage Sites in the Catalytic Core
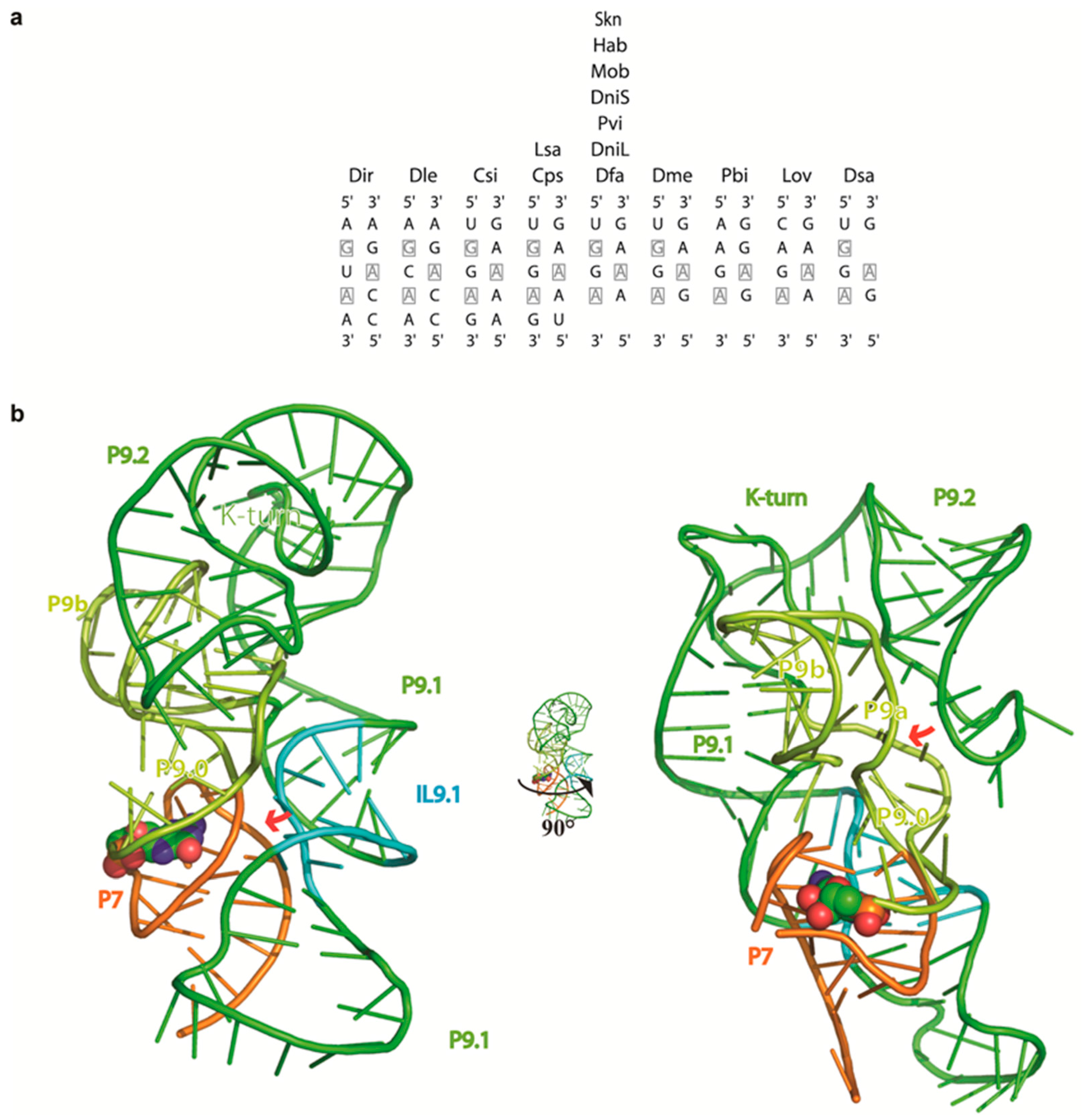
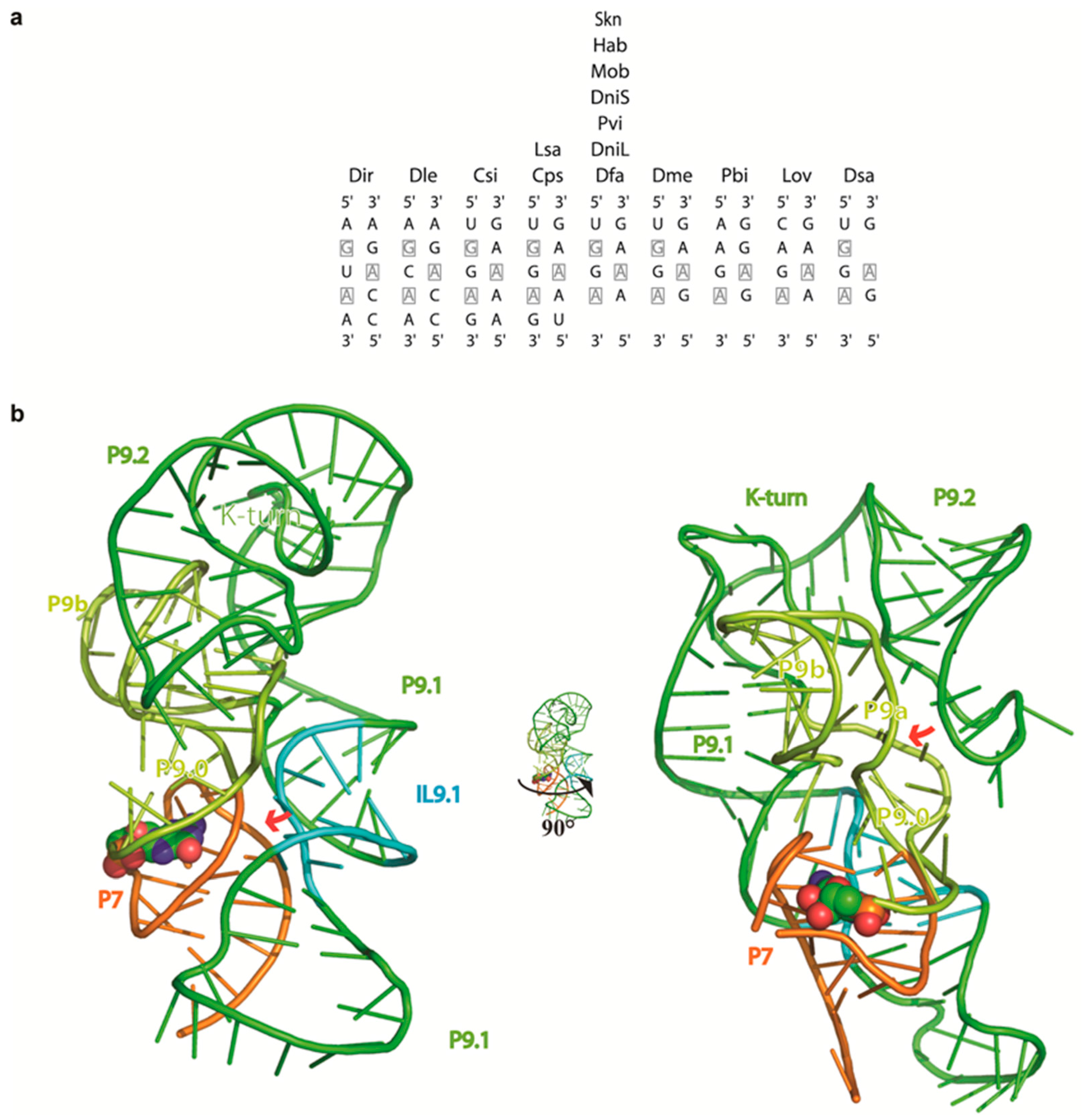
2.4. 3D Modeling Supports the Involvement of the P9 Appendages in Circle Formation
3. Discussion
3.1. The FLC Copy Number Varies in Response to Internal and External Factors
3.2. Structural Differences between L-IVS and FLC
3.3. The Structure of the P9 Domain Suggests a Role in Promoting Circularization
3.4. A Role for FLC in Intron Mobility?
4. Materials and Methods
4.1. Strains and Sequences
4.2. Growth Conditions
4.3. RNA Isolation from Cells
4.4. Cell Fractionation
4.5. Reverse Transcription and Quantitative RT-PCR (qRT-PCR)
4.6. In Vitro Transcription and RNA Processing
4.7. Chemical and Enzymatic Structure Probing, RNA Purification and Primer Extension
4.8. Molecular Modeling
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chan, W.K.; Belfort, G.; Belfort, M. Stability of group I intron RNA in Escherichia coli and its potential application in a novel expression vector. Gene 1988, 73, 295–304. [Google Scholar] [CrossRef]
- Harland, R.; Misher, L. Stability of RNA in developing Xenopus embryos and identification of a destabilizing sequence in TFIIIA messenger RNA. Development 1988, 102, 837–852. [Google Scholar] [PubMed]
- Branch, A.D.; Robertson, H.D. A replication cycle for viroids and other small infectious RNA’s. Science 1984, 223, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.L.; Mikheeva, S.; Coljee, V.W.; Turczyk, B.M.; Donahue, W.F.; Bar-Shalom, A.; Jarrell, K.A. Excision of group II introns as circles. Mol. Cell 2001, 8, 201–211. [Google Scholar] [CrossRef]
- Cote, F.; Levesque, D.; Perreault, J.P. Natural 2′,5′-phosphodiester bonds found at the ligation sites of peach latent mosaic viroid. J. Virol. 2001, 75, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Vicens, Q.; Westhof, E. Biogenesis of circular RNAs. Cell 2014, 159, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J. Circular RNA expression: Its potential regulation and function. Trends Genet. 2016, 32, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, K.K.; Kjems, J.; Hansen, T.B. Circular RNAs: Identification, biogenesis and function. Biochim. Biophys. Acta 2016, 1859, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Grammatikakis, I.; Munk, R.; Gorospe, M.; Abdelmohsen, K. Emerging roles and context of circular RNAs. Wiley Interdiscip. Rev. RNA 2016. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Johansen, S.D. Group I introns: Moving in new directions. RNA Biol. 2009, 6, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, A.; Johansen, S.D. Nuclear group I introns in self-splicing and beyond. Mob. DNA 2013, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Fiskaa, T.; Birgisdottir, A.B.; Haugen, P.; Einvik, C.; Johansen, S. The ability to form full-length intron RNA circles is a general property of nuclear group I introns. RNA 2003, 9, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Vicens, Q.; Cech, T.R. A natural ribozyme with 3′,5′ RNA ligase activity. Nat. Chem. Biol. 2009, 5, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Been, M.D.; Cech, T.R. Sites of circularization of the Tetrahymena rRNA IVS are determined by sequence and influenced by position and secondary structure. Nucleic Acids Res. 1985, 13, 8389–8408. [Google Scholar] [CrossRef] [PubMed]
- Engberg, J.; Zaug, A.J.; Nielsen, H. Circularization site choice in the self-splicing reaction of the ribosomal RNA intervening sequence of Tetrahymena silvana. Mol. Genet. Life Sci. Adv. 1988, 1, 50–55. [Google Scholar]
- Grabowski, P.J.; Zaug, A.J.; Cech, T.R. The intervening sequence of the ribosomal RNA precursor is converted to a circular RNA in isolated nuclei of Tetrahymena. Cell 1981, 23, 467–476. [Google Scholar] [CrossRef]
- Brehm, S.L.; Cech, T.R. Fate of an intervening sequence ribonucleic acid: Excision and cyclization of the Tetrahymena ribosomal ribonucleic acid intervening sequence in vivo. Biochemistry 1983, 22, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, E.W.; Einvik, C.; Ronning, S.; Haugli, K.; Johansen, S. Twelve Group I introns in the same pre-rRNA transcript of the myxomycete Fuligo septica: RNA processing and evolution. Mol. Biol. Evol. 2004, 21, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Johansen, S. Site-specific reverse splicing of a HEG-containing group I intron in ribosomal RNA. Nucleic Acids Res. 2005, 33, 2042–2051. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, B.L. Homing endonuclease structure and function. Q. Rev. Biophys. 2005, 38, 49–95. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.; Woodson, S.A. Integration of the Tetrahymena group I intron into bacterial rRNA by reverse splicing in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Sullivan, F.X.; Cech, T.R. New reactions of the ribosomal RNA precursor of Tetrahymena and the mechanism of self-splicing. J. Mol. Biol. 1986, 189, 143–165. [Google Scholar] [CrossRef]
- Nielsen, H.; Beckert, B.D.; Masquida, B.; Johansen, S.D. Ribozymes and RNA Catalysis; Lilley, D.M.J., Eckstein, F., Eds.; Royal Society of Chemistry: Cambridge, UK, 2008; Volume 12, pp. 229–252. [Google Scholar]
- Decatur, W.A.; Einvik, C.; Johansen, S.; Vogt, V.M. Two group I ribozymes with different functions in a nuclear rDNA intron. EMBO J. 1995, 14, 4558–4568. [Google Scholar] [PubMed]
- Meyer, M.; Nielsen, H.; Olieric, V.; Roblin, P.; Johansen, S.D.; Westhof, E.; Masquida, B. Speciation of a group I intron into a lariat capping ribozyme. Proc. Natl. Acad. Sci. USA 2014, 111, 7659–7664. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.; Westhof, E.; Johansen, S. An mRNA is capped by a 2′,5′ lariat catalyzed by a group I-like ribozyme. Science 2005, 309, 1584–1587. [Google Scholar] [CrossRef] [PubMed]
- Vader, A.; Johansen, S.; Nielsen, H. The group I-like ribozyme DiGIR1 mediates alternative processing of pre-rRNA transcripts in Didymium iridis. Eur. J. Biochem. 2002, 269, 5804–5812. [Google Scholar] [CrossRef] [PubMed]
- Raub, T.J.; Aldrich, H.C. Cell Biology of Physarum and Didymium; Aldrich, H.C., Daniel, J.W., Eds.; Academic Press: New York, NY, USA, 1982; Volume 2, pp. 21–75. [Google Scholar]
- Wright, M.; Tollon, Y. Induction of heat-shock proteins at permissive growth temperatures in the plasmodium of the myxomycete Physarum polycephalum. Eur. J. Biochem. 1982, 127, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Johansen, S.; Vogt, V.M. An intron in the nuclear ribosomal DNA of Didymium iridis codes for a group I ribozyme and a novel ribozyme that cooperate in self-splicing. Cell 1994, 76, 725–734. [Google Scholar] [CrossRef]
- Streicher, B.; von Ahsen, U.; Schroeder, R. Lead cleavage sites in the core structure of group I intron-RNA. Nucleic Acids Res. 1993, 21, 311–317. [Google Scholar] [PubMed]
- Streicher, B.; Westhof, E.; Schroeder, R. The environment of two metal ions surrounding the splice site of a group I intron. EMBO J. 1996, 15, 2556–2564. [Google Scholar] [PubMed]
- Waldsich, C.; Masquida, B.; Westhof, E.; Schroeder, R. Monitoring intermediate folding states of the td group I intron in vivo. EMBO J. 2002, 21, 5281–5291. [Google Scholar] [CrossRef] [PubMed]
- Golden, B.L.; Kim, H.; Chase, E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat. Struct. Mol. Biol. 2005, 12, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Leontis, N.B.; Westhof, E. A common motif organizes the structure of multi-helix loops in 16 S and 23 S ribosomal RNAs. J. Mol. Biol. 1998, 283, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Lescoute, A.; Westhof, E. Topology of three-way junctions in folded RNAs. RNA 2006, 12, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Westhof, E.; Masquida, B. A structural module in RNase P expands the variety of RNA kinks. RNA Biol. 2012, 9, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Haugen, P.; Andreassen, M.; Birgisdottir, A.B.; Johansen, S. Hydrolytic cleavage by a group I intron ribozyme is dependent on RNA structures not important for splicing. Eur. J. Biochem. 2004, 271, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J.; Aagaard, C.; Semionenkov, M.; Garrett, R.A. Archaeal introns: Splicing, intercellular mobility and evolution. Trends Biochem. Sci. 1997, 22, 326–331. [Google Scholar] [CrossRef]
- Vader, A.; Nielsen, H.; Johansen, S. In vivo expression of the nucleolar group I intron-encoded I-DirI homing endonuclease involves the removal of a spliceosomal intron. EMBO J. 1999, 18, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Sanger, H.L.; Riesner, D. Subcellular localization of viroids in highly purified nuclei from tomato leaf tissue. EMBO J. 1983, 2, 1549–1555. [Google Scholar] [PubMed]
- Bell, J.; Neilson, L.; Pellegrini, M. Effect of heat shock on ribosome synthesis in Drosophila melanogaster. Mol. Cell. Biol. 1988, 8, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Nover, L.; Munsche, D.; Neumann, D.; Ohme, K.; Scharf, K.D. Control of ribosome biosynthesis in plant cell cultures under heat-shock conditions. Ribosomal RNA. Eur. J. Biochem. 1986, 160, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.L.; Stahley, M.R.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a self-splicing group I intron with both exons. Nature 2004, 430, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Gooding, A.R.; Cech, T.R. Structure of the Tetrahymena ribozyme: Base triple sandwich and metal ion at the active site. Mol. Cell 2004, 16, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Michel, F.; Ellington, A.D.; Couture, S.; Szostak, J.W. Phylogenetic and genetic evidence for base-triples in the catalytic domain of group I introns. Nature 1990, 347, 578–580. [Google Scholar] [CrossRef] [PubMed]
- Lipchock, S.V.; Strobel, S.A. A relaxed active site after exon ligation by the group I intron. Proc. Natl. Acad. Sci. USA 2008, 105, 5699–5704. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Rieder, R.; Micura, R. Ligand-induced folding of the thiM TPP riboswitch investigated by a structure-based fluorescence spectroscopic approach. Nucleic Acids Res. 2007, 35, 5370–5378. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.; Lang, K.; Graber, D.; Micura, R. Ligand-induced folding of the adenosine deaminase A-riboswitch and implications on riboswitch translational control. Chembiochem 2007, 8, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Stahley, M.R.; Strobel, S.A. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science 2005, 309, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S.; Dewan, J.C.; Klug, A. Crystallographic and biochemical investigation of the lead(II)-catalyzed hydrolysis of yeast phenylalanine tRNA. Biochemistry 1985, 24, 4785–4801. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, V.; Jaeger, L.; Michel, F.; Westhof, E. New loop-loop tertiary interactions in self-splicing introns of subgroup IC and ID: A complete 3D model of the Tetrahymena thermophila ribozyme. Chem. Biol. 1996, 3, 993–1009. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y. Predicting the secondary structures and tertiary interactions of 211 group I introns in IE subgroup. Nucleic Acids Res. 2005, 33, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.J.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A. The kink-turn: A new RNA secondary structure motif. EMBO J. 2001, 20, 4214–4221. [Google Scholar] [CrossRef] [PubMed]
- Johansen, S.; Elde, M.; Vader, A.; Haugen, P.; Haugli, K.; Haugli, F. In vivo mobility of a group I twintron in nuclear ribosomal DNA of the myxomycete Didymium iridis. Mol. Microbiol. 1997, 24, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Haugen, P.; Wikmark, O.G.; Vader, A.; Coucheron, D.H.; Sjottem, E.; Johansen, S.D. The recent transfer of a homing endonuclease gene. Nucleic Acids Res. 2005, 33, 2734–2741. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.; Egebjerg, J.; Larsen, N.; Garrett, R.A. Ribosomes and Protein Synthesis: A Practical Appoach; Spedding, G., Ed.; Oxford University Press: Oxford, UK, 1990; pp. 229–252. [Google Scholar]
- Brunel, C.; Romby, P. Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol. 2000, 318, 3–21. [Google Scholar] [PubMed]
- Winkler, W.C.; Nahvi, A.; Sudarsan, N.; Barrick, J.E.; Breaker, R.R. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol. 2003, 10, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Massire, C.; Jaeger, L.; Westhof, E. Derivation of the three-dimensional architecture of bacterial ribonuclease P RNAs from comparative sequence analysis. J. Mol. Biol. 1998, 279, 773–793. [Google Scholar] [CrossRef] [PubMed]
- Jossinet, F.; Westhof, E. Sequence to Structure (S2S): Display, manipulate and interconnect RNA data from sequence to structure. Bioinformatics 2005, 21, 3320–3321. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.L.; Stahley, M.R.; Gill, M.L.; Kosek, A.B.; Wang, J.; Strobel, S.A. Crystal structure of a group I intron splicing intermediate. RNA 2004, 10, 1867–1887. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of plasmids and specialized chemicals (e.g. PNA) are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersen, K.L.; Beckert, B.; Masquida, B.; Johansen, S.D.; Nielsen, H. Accumulation of Stable Full-Length Circular Group I Intron RNAs during Heat-Shock. Molecules 2016, 21, 1451. https://doi.org/10.3390/molecules21111451
Andersen KL, Beckert B, Masquida B, Johansen SD, Nielsen H. Accumulation of Stable Full-Length Circular Group I Intron RNAs during Heat-Shock. Molecules. 2016; 21(11):1451. https://doi.org/10.3390/molecules21111451
Chicago/Turabian StyleAndersen, Kasper L., Bertrand Beckert, Benoit Masquida, Steinar D. Johansen, and Henrik Nielsen. 2016. "Accumulation of Stable Full-Length Circular Group I Intron RNAs during Heat-Shock" Molecules 21, no. 11: 1451. https://doi.org/10.3390/molecules21111451
APA StyleAndersen, K. L., Beckert, B., Masquida, B., Johansen, S. D., & Nielsen, H. (2016). Accumulation of Stable Full-Length Circular Group I Intron RNAs during Heat-Shock. Molecules, 21(11), 1451. https://doi.org/10.3390/molecules21111451