
Comparative Transcriptomic Analysis of Grape Berry in Response to Root Restriction during Developmental Stages


Abstract
:1. Introduction
2. Results and Discussion
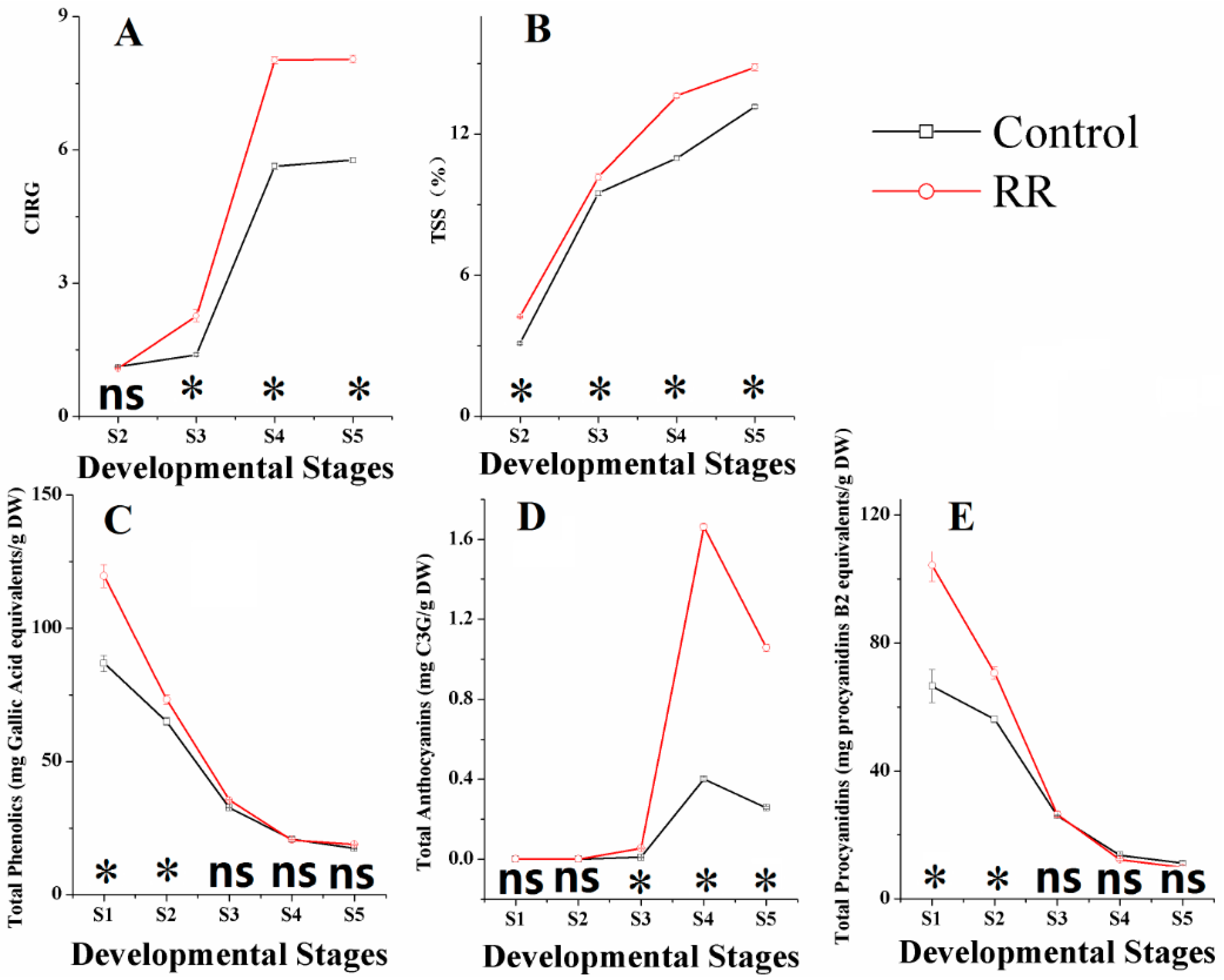
2.1. Physical Properties of Grapes
2.2. Evaluation of RNA Sequencing Data
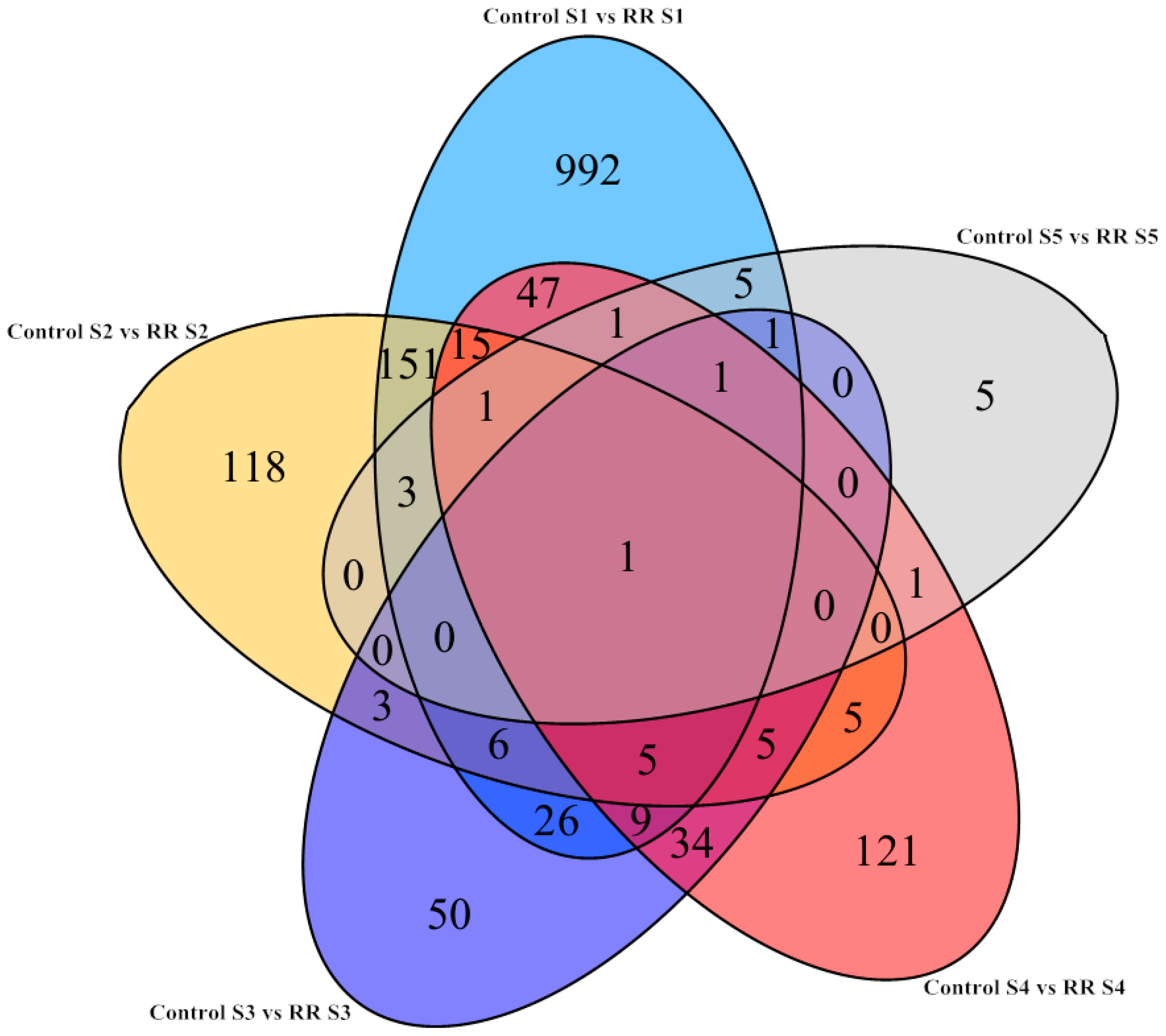
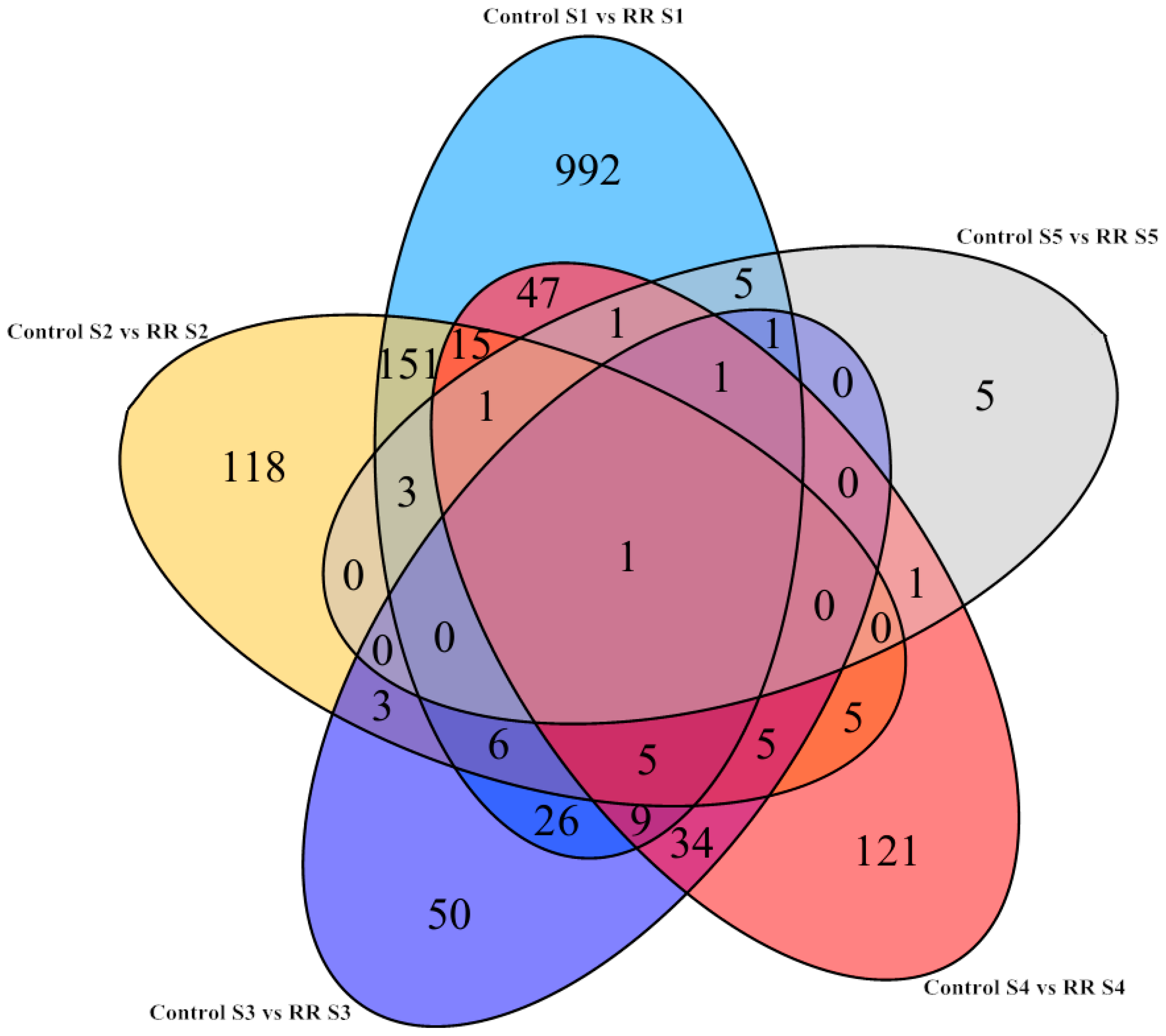
2.3. Differential Gene Expression Triggered by Root Restriction
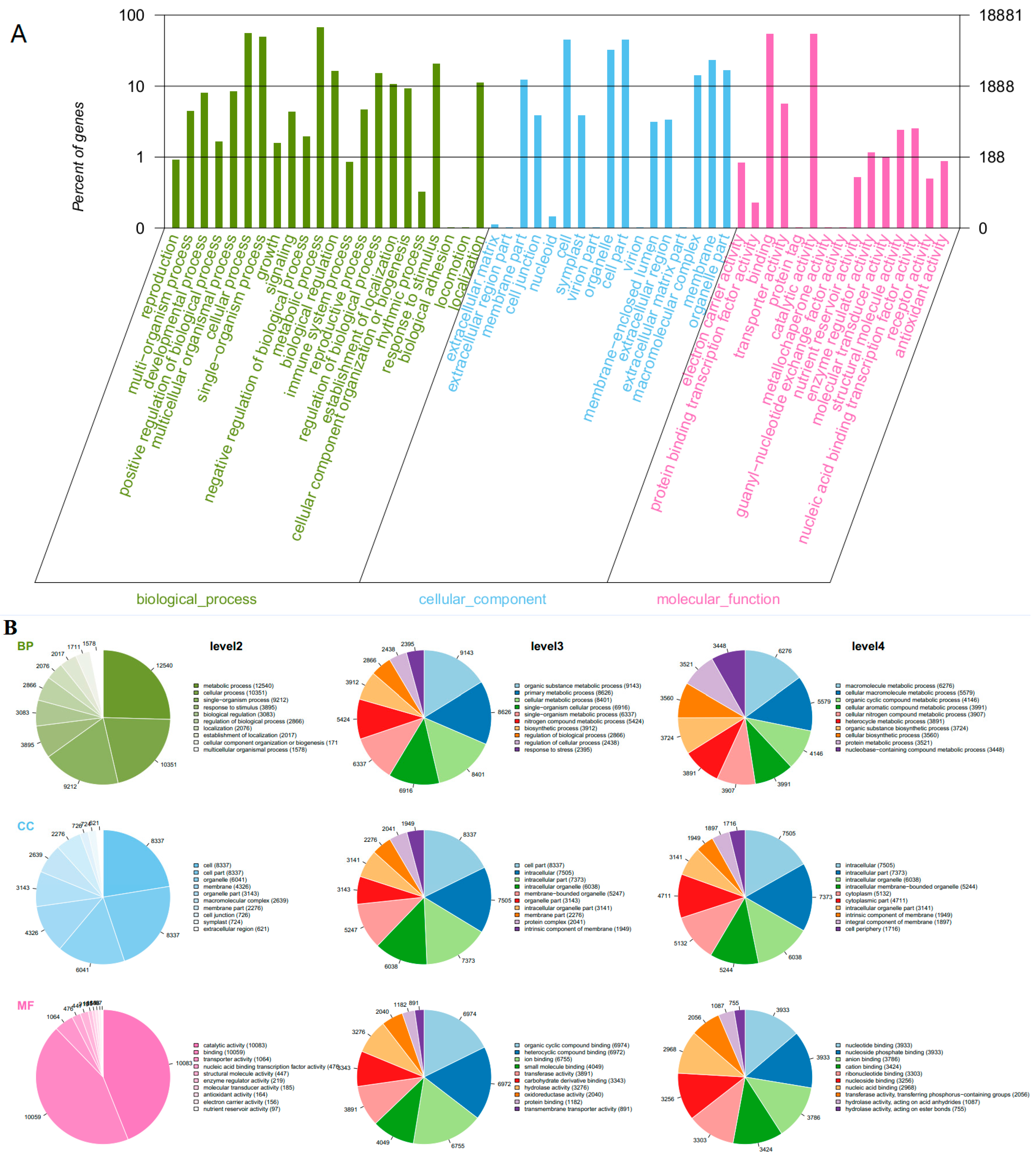
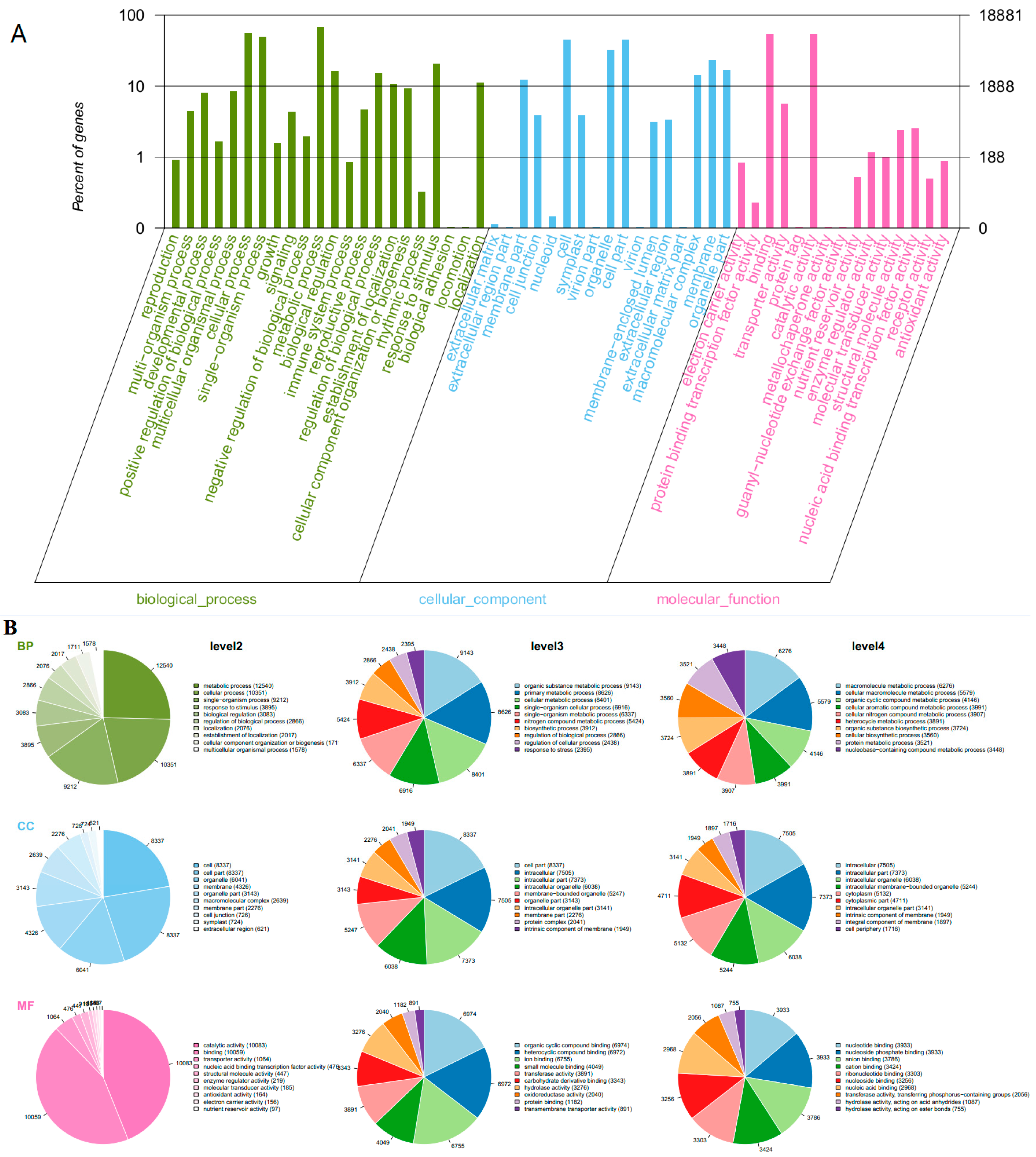
2.4. GO Functional Annotation and KEGG Analysis
2.5. Validation of Gene Expression Using qRT-PCR
3. Materials and Methods
3.1. Color and TSS Measurement
3.2. Total Soluble Phenolics, Total Anthocyanins and Total Procyanidins
3.3. RNA Extraction and RNA-Seq
3.4. qRT-PCR Validation of RNA-Seq Data
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vivier, M.A.; Pretorius, I.S. Genetically tailored grapevines for the wine industry. Trends Biotechnol. 2002, 20, 472–478. [Google Scholar] [CrossRef]
- Coombe, B.G. Research on development and ripening of the grape berry. Am. J. Enol. Viticult. 1992, 43, 101–110. [Google Scholar]
- Deluc, L.; Bogs, J.; Walker, A.R.; Ferrier, T.; Decendit, A.; Merillon, J.M.; Robinson, S.P.; Barrieu, F. The transcription factor vvmyb5b contributes to the regulation of anthocyanin and proanthocyanidin biosynthesis in developing grape berries. Plant Physiol. 2008, 147, 2041–2053. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Ergul, A.; Grimplet, J.; Tillett, R.L.; Tattersall, E.A.R.; Bohlman, M.C.; Vincent, D.; Sonderegger, J.; Evans, J.; Osborne, C.; et al. Water and salinity stress in grapevines: Early and late changes in transcript and metabolite profiles. Funct. Integr. Genom. 2007, 7, 111–134. [Google Scholar] [CrossRef] [PubMed]
- Grimplet, J.; Deluc, L.G.; Tillett, R.L.; Wheatley, M.D.; Schlauch, K.A.; Cramer, G.R.; Cushman, J.C. Tissue-specific mrna expression profiling in grape berry tissues. BMC Genom. 2007, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Deluc, L.G.; Quilici, D.R.; Decendit, A.; Grimplet, J.; Wheatley, M.D.; Schlauch, K.A.; Merillon, J.M.; Cushman, J.C.; Cramer, G.R. Water deficit alters differentially metabolic pathways affecting important flavor and quality traits in grape berries of cabernet sauvignon and chardonnay. BMC Genom. 2009, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, M.; Nicolas, P.; Kappel, C.; Dai, Z.W.; Hilbert, G.; Peccoux, A.; Lafontaine, M.; Ollat, N.; Gomes, E.; Delrot, S. Water limitation and rootstock genotype interact to alter grape berry metabolism through transcriptome reprogramming. Hortic. Res. 2015, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.Y.; Zhu, L.N.; Wang, S.P.; Gu, W.J.; Huang, D.F.; Xu, W.P.; Jiang, A.L.; Li, S.C. Nitrate uptake kinetics of grapevine under root restriction. Sci. Hortic. 2007, 111, 358–364. [Google Scholar] [CrossRef]
- Xie, Z.S.; Li, B.; Forney, C.F.; Xu, W.P.; Wang, S.P. Changes in sugar content and relative enzyme activity in grape berry in response to root restriction. Sci. Hortic. 2009, 123, 39–45. [Google Scholar] [CrossRef]
- Wang, S.P.; Okamoto, G.; Hirano, K.; Lu, J.; Zhang, C.X. Effects of restricted rooting volume on vine growth and berry development of kyoho grapevines. Am. J. Enol. Viticult. 2001, 52, 248–253. [Google Scholar]
- Baryosef, B.; Schwartz, S.; Markovich, T.; Lucas, B.; Assaf, R. Effect of root volume and nitrate solution concentration on growth, fruit yield, and temporal n and water-uptake rates by apple-trees. Plant Soil 1988, 107, 49–56. [Google Scholar] [CrossRef]
- Boland, A.M.; Jerie, P.H.; Mitchell, P.D.; Goodwin, I. Long-term effects of restricted root volume and regulated deficit irrigation on peach: I. Growth and mineral nutrition. J. Am. Soc. Hortic. Sci. 2000, 125, 135–142. [Google Scholar]
- Wang, B.; He, J.J.; Bai, Y.; Yu, X.M.; Li, J.F.; Zhang, C.X.; Xu, W.P.; Bai, X.J.; Cao, X.J.; Wang, S.P. Root restriction affected anthocyanin composition and upregulated the transcription of their biosynthetic genes during berry development in ‘summer black’ grape. Acta Physiol. Plant 2013, 35, 2205–2217. [Google Scholar] [CrossRef]
- Wang, B.; He, J.J.; Duan, C.Q.; Yu, X.M.; Zhu, L.N.; Xie, Z.S.; Zhang, C.X.; Xu, W.P.; Wang, S.P. Root restriction affects anthocyanin accumulation and composition in berry skin of ‘kyoho’ grape (Vitis vinifera L. × Vitis labrusca L.) during ripening. Sci. Hortic. 2012, 137, 20–28. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, U463–U465. [Google Scholar] [CrossRef] [PubMed]
- Hoen, P.A.C.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.A.M.; de Menezes, R.X.; Boer, J.M.; van Ommen, G.J.B.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. Rna-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genom. Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Zenoni, S.; Ferrarini, A.; Giacomelli, E.; Xumerle, L.; Fasoli, M.; Malerba, G.; Bellin, D.; Pezzotti, M.; Delledonne, M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-seq. Plant Physiol. 2010, 152, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Fasoli, M.; Dal Santo, S.; Zenoni, S.; Tornielli, G.B.; Farina, L.; Zamboni, A.; Porceddu, A.; Venturini, L.; Bicego, M.; Murino, V.; et al. The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell 2012, 24, 3489–3505. [Google Scholar] [CrossRef] [PubMed]
- Sweetman, C.; Wong, D.C.J.; Ford, C.M.; Drew, D.P. Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genom. 2012, 13, 691. [Google Scholar] [CrossRef] [PubMed]
- Fortes, A.M.; Agudelo-Romero, P.; Silva, M.S.; Ali, K.; Sousa, L.; Maltese, F.; Choi, Y.H.; Grimplet, J.; Martinez-Zapater, J.M.; Verpoorte, R.; et al. Transcript and metabolite analysis in trincadeira cultivar reveals novel information regarding the dynamics of grape ripening. BMC Plant Biol. 2011, 11, 149. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Nakabayashi, R.; Ogata, Y.; Sakurai, N.; Tokimatsu, T.; Goto, S.; Suzuki, M.; Jasinski, M.; Martinoia, E.; Otagaki, S.; et al. Multiomics in grape berry skin revealed specific induction of the stilbene synthetic pathway by ultraviolet-c irradiation. Plant Physiol. 2015, 168, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Deluc, L.G.; Grimplet, J.; Wheatley, M.D.; Tillett, R.L.; Quilici, D.R.; Osborne, C.; Schooley, D.A.; Schlauch, K.A.; Cushman, J.C.; Cramer, G.R. Transcriptomic and metabolite analyses of cabernet sauvignon grape berry development. BMC Genom. 2007, 8, 429. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wei, G.C.; Zhou, H.; Gu, C.; Vimolmangkang, S.; Liao, L.; Han, Y.P. Unraveling the mechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 2014, 166, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.A.; Matthews, M.A.; Waterhouse, A.L. Effect of maturity and vine water status on grape skin and wine flavonoids. Am. J. Enol. Viticult. 2002, 53, 268–274. [Google Scholar]
- Ryan, J.M.; Revilla, E. Anthocyanin composition of cabernet sauvignon and tempranillo grapes at different stages of ripening. J. Agric. Food Chem. 2003, 51, 3372–3378. [Google Scholar] [CrossRef] [PubMed]
- Canals, R.; Llaudy, M.C.; Valls, J.; Canals, J.M.; Zamora, F. Influence of ethanol concentration on the extraction of color and phenolic compounds from the skin and seeds of tempranillo grapes at different stages of ripening. J. Agric. Food Chem. 2005, 53, 4019–4025. [Google Scholar] [CrossRef] [PubMed]
- Fournand, D.; Vicens, A.; Sidhoum, L.; Souquet, J.M.; Moutounet, M.; Cheynier, V. Accumulation and extractability of grape skin tannins and anthocyanins at different advanced physiological stages. J. Agric. Food Chem. 2006, 54, 7331–7338. [Google Scholar] [CrossRef] [PubMed]
- Hellin, P.; Manso, A.; Flores, P.; Fenoll, J. Evolution of aroma and phenolic compounds during ripening of ‘superior seedless’ grapes. J. Agric. Food Chem. 2010, 58, 6334–6340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tao, X.; Tang, X.M.; Xiao, L.; Sun, J.L.; Yan, X.F.; Li, D.; Deng, H.Y.; Ma, X.R. Comparative transcriptome analysis of tomato (solanum lycopersicum) in response to exogenous abscisic acid. BMC Genom. 2013, 14, 841. [Google Scholar] [CrossRef] [PubMed]
- Palliotti, A.; Silvestroni, O.; Petoumenou, D. Seasonal patterns of growth rate and morphophysiological features in green organs of cabernet sauvignon grapevines. Am. J. Enol. Viticult. 2010, 61, 74–82. [Google Scholar]
- Deng, W.; Yan, F.; Zhang, X.L.; Tang, Y.W.; Yuan, Y.J. Transcriptional profiling of canola developing embryo and identification of the important roles of bndof5.6 in embryo development and fatty acids synthesis. Plant Cell Physiol. 2015, 56, 1624–1640. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.P.; Zhang, Y.; Song, J.; Zhao, L.J.; Wang, Z.Z. De novo transcriptome sequencing in salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 2011, 98, 272–279. [Google Scholar]
- Sweetman, C.; Deluc, L.G.; Cramer, G.R.; Ford, C.M.; Soole, K.L. Regulation of malate metabolism in grape berry and other developing fruits. Phytochemistry 2009, 70, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.F.; Wu, B.H.; Fan, P.G.; Li, S.H.; Li, L.S. Sugar and acid concentrations in 98 grape cultivars analyzed by principal component analysis. J. Sci. Food Agric. 2006, 86, 1526–1536. [Google Scholar] [CrossRef]
- Lund, S.T.; Bohlmann, J. The molecular basis for wine grape quality—A volatile subject. Science 2006, 311, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Carreno, J.; Martinez, A.; Almela, L.; Fernandezlopez, J.A. Proposal of an index for the objective evaluation of the color of red table grapes. Food Res. Int. 1995, 28, 373–377. [Google Scholar] [CrossRef]
- Feng, C.; Chen, M.; Xu, C.J.; Bai, L.; Yin, X.R.; Li, X.; Allan, A.C.; Ferguson, I.B.; Chen, K.S. Transcriptomic analysis of chinese bayberry (myrica rubra) fruit development and ripening using Rna-seq. BMC Genom. 2012, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.P.; Jiang, Q.; Lin, J.Y.; Li, X.; Sun, C.D.; Chen, K.S. Physicochemical characterisation of four cherry species (prunus spp.) grown in China. Food Chem. 2015, 173, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Singleton, V.L. Citation classic-colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. CC/Agric. Biol. Environ. 1985, 18. [Google Scholar]
- Payne, M.J.; Hurst, W.J.; Stuart, D.A.; Ou, B.X.; Fan, E.; Ji, H.P.; Kou, Y. Determination of total procyanidins in selected chocolate and confectionery products using dmac. J. AOAC Int. 2010, 93, 89–96. [Google Scholar] [PubMed]
- Shan, L.L.; Li, X.; Wang, P.; Cai, C.; Zhang, B.; Sun, C.D.; Zhang, W.S.; Xu, C.J.; Ferguson, I.; Chen, K.S. Characterization of cdnas associated with lignification and their expression profiles in loquat fruit with different lignin accumulation. Planta 2008, 227, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Wang, S.Q.; Li, W. Rseqc: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.B.; Wang, X.Y.; Bowers, J.E.; Ming, R.; Alam, M.; Paterson, A.H. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genom. Res. 2008, 18, 1944–1954. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. Venndiagram: A package for the generation of highly-customizable venn and euler diagrams in r. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.G.; Peatman, E.; Tang, H.B.; Lewis, J.; Liu, Z.J. Profiling of gene duplication patterns of sequenced teleost genomes: Evidence for rapid lineage-specific genome expansion mediated by recent tandem duplications. BMC Genom. 2012, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Osada, T.; Glass, O.; Yang, X.Y.; Lei, G.J.; Lyerly, H.K.; Clay, T.M. Ligand-independent toll-like receptor signals generated by ectopic overexpression of myd88 generate local and systemic antitumor immunity. Cancer Res. 2010, 70, 7209–7220. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Sequence data of grape are available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Analysis | Treatments | Developmental Stages | ||||
|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | ||
| Raw bases (bp) | Control | 6043206288 | 6388028052 | 5629860684 | 5937329496 | 6421364544 |
| RR | 5863704000 | 6208024116 | 5503911840 | 5821091052 | 5730388524 | |
| Raw reads (No.) | Control | 47961955 | 50698635 | 44681434 | 47121663 | 50963211 |
| RR | 46537333 | 49270033 | 43681840 | 46199135 | 45479274 | |
| Clean bases (bp) | Control | 5507614346 | 5804699908 | 5088328621 | 5360779171 | 5781234462 |
| RR | 5326390300 | 5642227230 | 4920575938 | 5245451851 | 5189109353 | |
| Clean reads (No.) | Control | 44822587 | 47465685 | 41663959 | 44128070 | 47813283 |
| RR | 43519598 | 45949313 | 40551247 | 43479523 | 42667291 | |
| ≥Q30 | Control | 93.62 | 93.31 | 93.22 | 93.11 | 92.91 |
| RR | 94.09 | 93.76 | 92.81 | 93.08 | 93.12 | |
| Mapped sequences (No.) | Control | 33064934 | 31668177 | 27537423 | 30466585 | 31557453 |
| RR | 31883094 | 28392274 | 29022488 | 25252114 | 26946449 | |
| Mapped percentage (%) | Control | 73.74 | 66.94 | 66.27 | 68.99 | 67.01 |
| RR | 73.34 | 61.45 | 71.51 | 59.55 | 63.06 | |
| Log2FC | Upregulated Genes | Downregulated Genes | Not Differentially Expressed |
|---|---|---|---|
| RRS1/ControlS1 | 987 | 277 | 28707 |
| RRS2/ControlS2 | 72 | 241 | 29658 |
| RRS3/ControlS3 | 112 | 29 | 29830 |
| RRS4/ControlS4 | 158 | 88 | 29725 |
| RRS5/ControlS5 | 11 | 8 | 29952 |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| GAPDH | TGGAGCTGAATTTGTTGT | GTGGAGTTCTGGCTTGTA |
| VIT_04s0023g02290 | TTTGTTTGCGGTCTTGGA | GAACAGCCTGCCGTAGAA |
| VIT_05s0049g00770 | CCACCATCTCCCACCCAT | TGTCACAATACTCATCACCC |
| VIT_07s0197g00240 | AGCCATTTATCAGAGCGAACAG | GCACCAGCTTGAGGAGAACAT |
| VIT_09s0002g06590 | ATGAATACAACTTCGTCCTT | GCTTTGAGTTCAGCCATT |
| VIT_14s0068g00920 | TCCCAGGGTTGATTTCCA | TGCTGCCTTTCCCTTCTT |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leng, F.; Lin, Q.; Wu, D.; Wang, S.; Wang, D.; Sun, C. Comparative Transcriptomic Analysis of Grape Berry in Response to Root Restriction during Developmental Stages. Molecules 2016, 21, 1431. https://doi.org/10.3390/molecules21111431
Leng F, Lin Q, Wu D, Wang S, Wang D, Sun C. Comparative Transcriptomic Analysis of Grape Berry in Response to Root Restriction during Developmental Stages. Molecules. 2016; 21(11):1431. https://doi.org/10.3390/molecules21111431
Chicago/Turabian StyleLeng, Feng, Qiong Lin, Di Wu, Shiping Wang, Dengliang Wang, and Chongde Sun. 2016. "Comparative Transcriptomic Analysis of Grape Berry in Response to Root Restriction during Developmental Stages" Molecules 21, no. 11: 1431. https://doi.org/10.3390/molecules21111431
APA StyleLeng, F., Lin, Q., Wu, D., Wang, S., Wang, D., & Sun, C. (2016). Comparative Transcriptomic Analysis of Grape Berry in Response to Root Restriction during Developmental Stages. Molecules, 21(11), 1431. https://doi.org/10.3390/molecules21111431