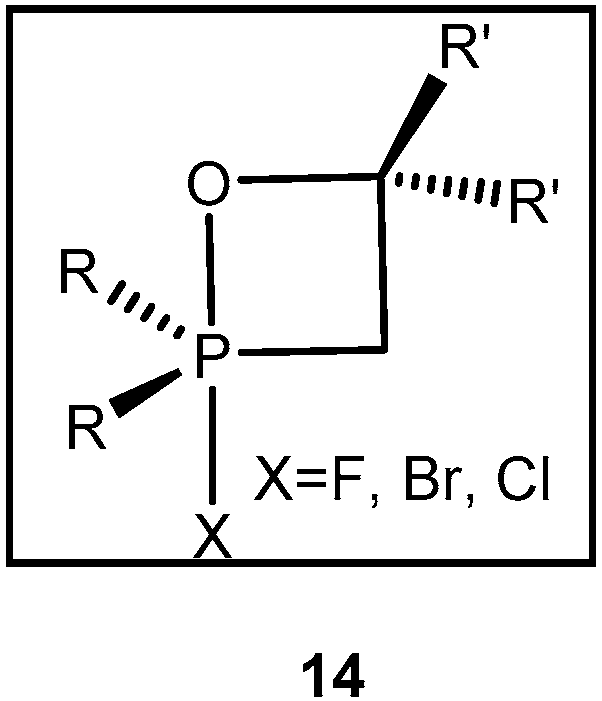
2.2. Synthesis of 2-Halo-1,2λ5-oxaphosphetane
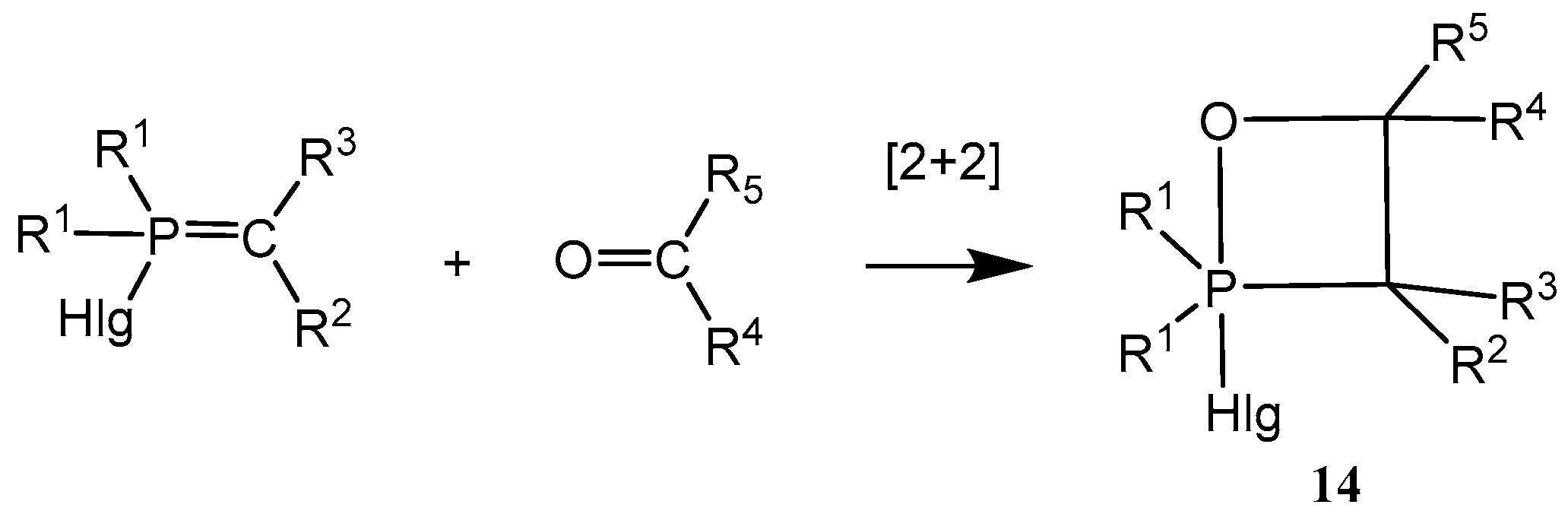
Available methods for the synthesis of 2-halo-1,2λ
5-oxaphosphetanes can be used for investigation of the reaction mechanism of phosphorus ylides with carbonyl compounds as well as for preparing stable oxaphosphetanes that can be used as reactants for organic synthesis. The 2-halo-1,2λ
5-oxaphosphetanes were prepared by reaction of
P-fluoro-, chloro- or bromoylides with carbonyl compounds.
P-Chloro- and
P-bromoylides react with active ketones, containing a trifluoromethyl group, with the formation of stable [2+2]-cycloaddition products, 2-chloro- or 2-bromo-1,2λ
5-oxaphosphetenes
14 were isolated in yields close to quantitative as crystalline substances or as liquids distillable in vacuum (
Scheme 8,
Table 1) [
29,
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40,
41,
42].
The addition of
P-halogen-ylides to ketones proceeded stereoselectively and led predominantly to the formation of one of the possible 2-halo-1,2λ
5-oxaphosphetane diastereomers. 2-Halo-1,2λ
5-oxaphosphetanes dissociate at the
P-halogen bond in solution with the formation of cyclic phosphonium salts, as a result of which an equilibrium is established between the forms with five- and four-coordinate phosphorus atoms. Dissociation of 2-halo-1,2λ
5-oxaphosphetanes is enhanced by reducing the electron-accepting properties of substituents and also by increasing priority solvent. The 2-halo-1,2λ
5-oxaphosphetanes containing electron-accepting groups at C(4) are distinctly stabler than oxaphosphetanes with alkyl groups in this position [
25,
26,
27].
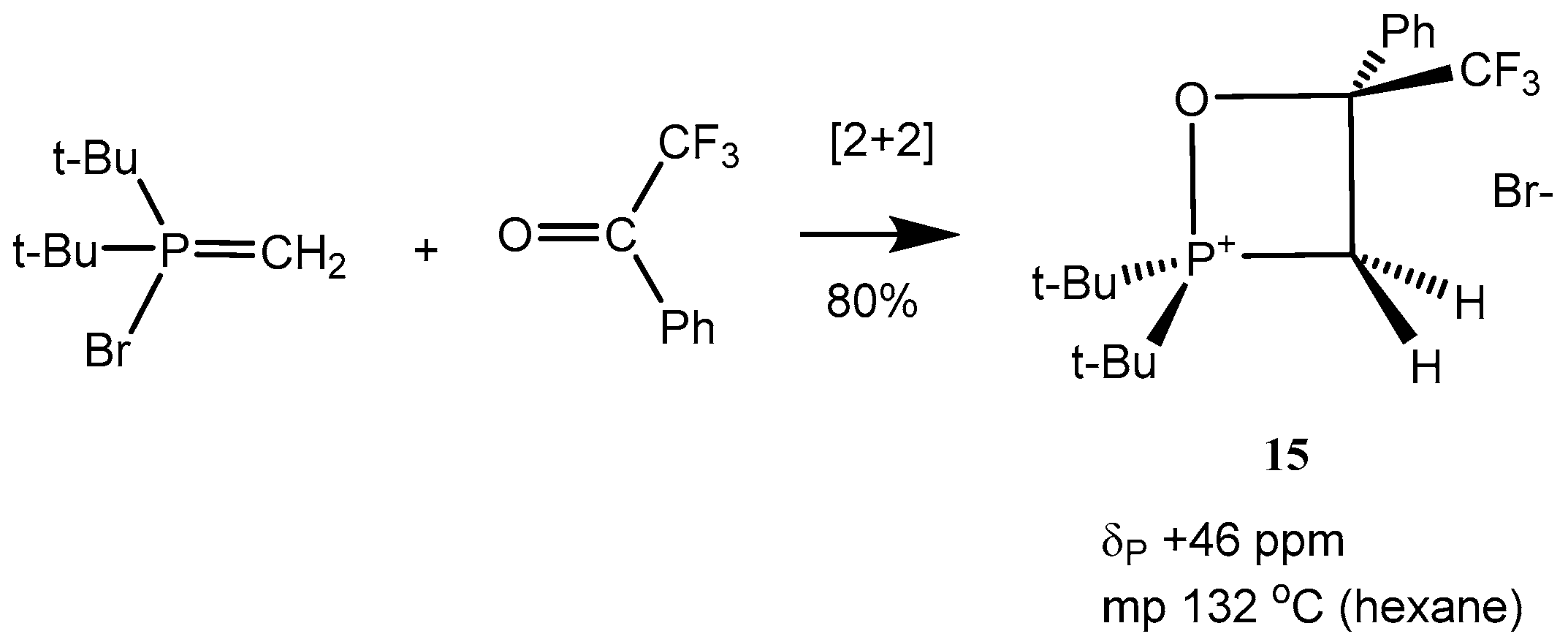
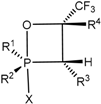
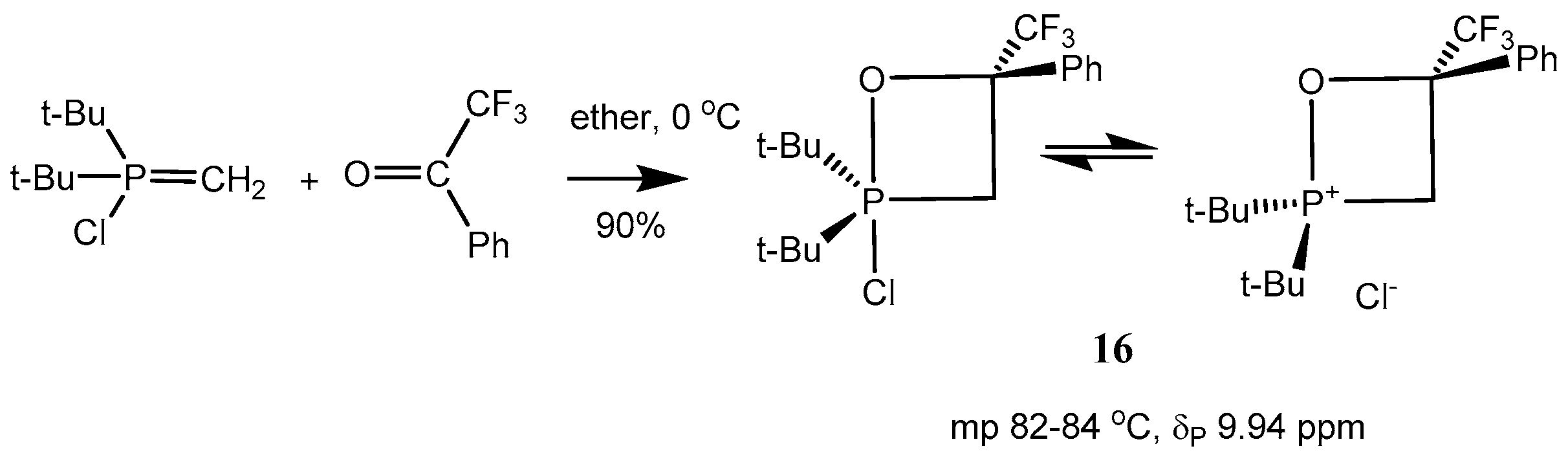
The reaction of
P-bromomethylides with fluorinated acetophenone afforded in high yield oxaphosphetanes
15, which were isolated as crystalline compounds (
Scheme 9 and
Table 1, entries 10–13). The compounds
15 exist in solution as cyclic phosphonium salt.
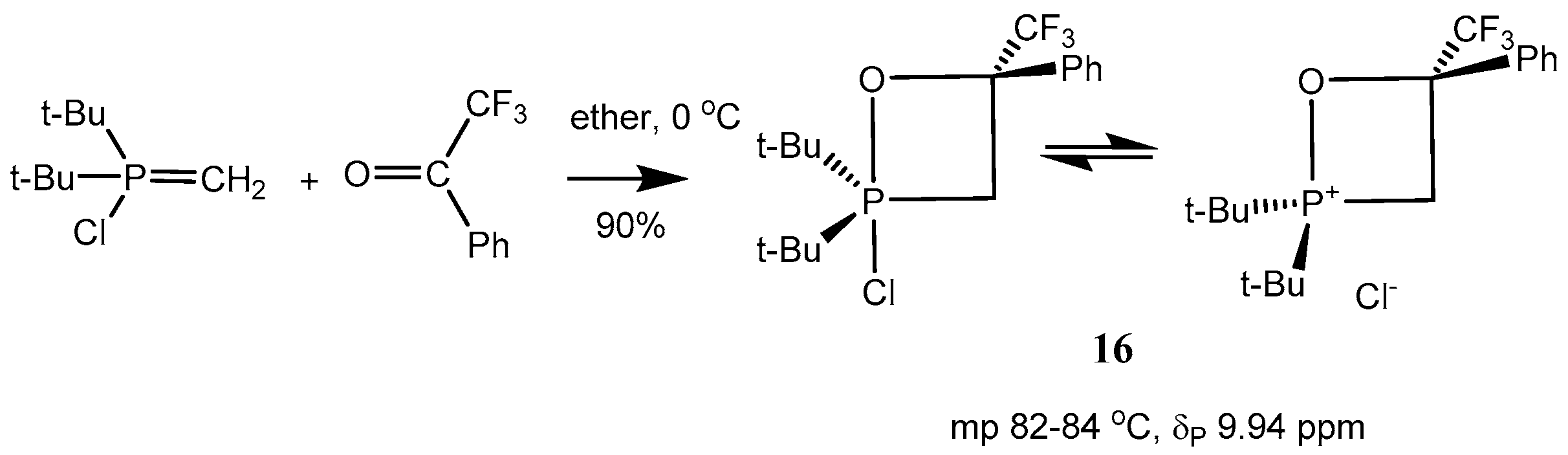
At the same time the 2-chloro-1,2λ
5-oxaphosphetanes
16 exist as mixture of P(IV) and P(V)-forms. These compounds can be distilled under vacuum and dissolved in non-polar solvents (benzene) (
Scheme 10 and
Table 1, entries 1–9). Reaction of
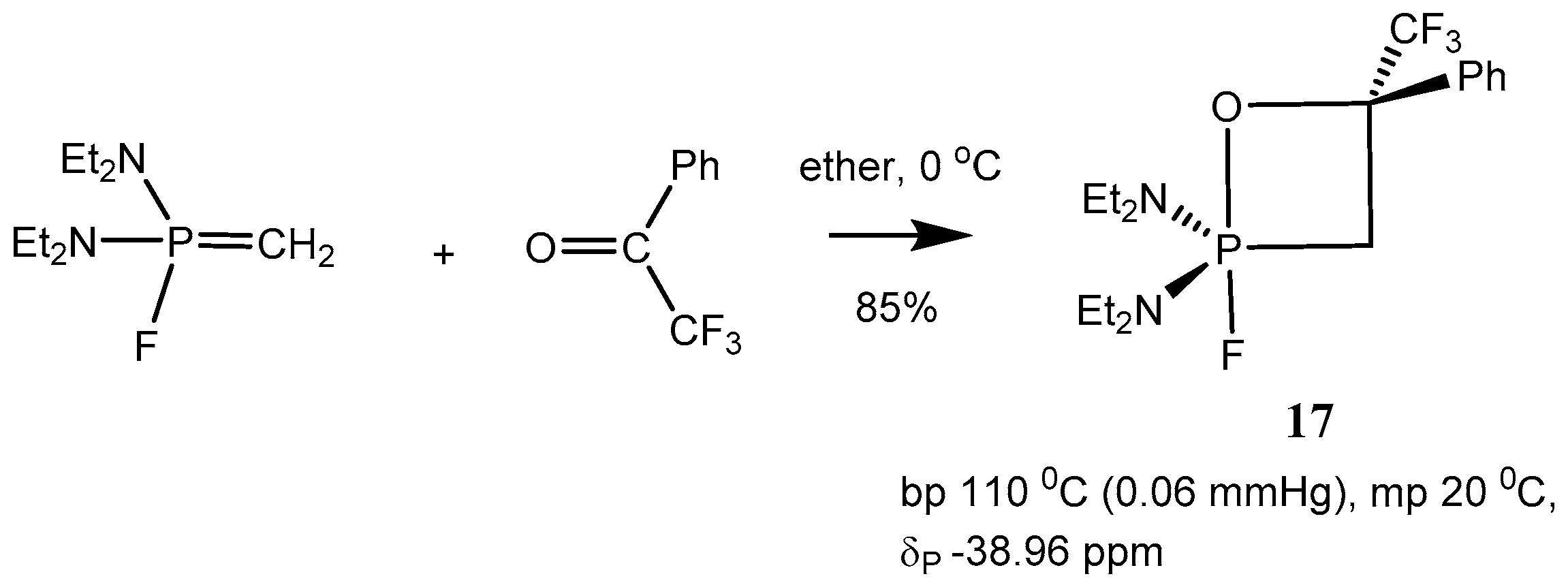
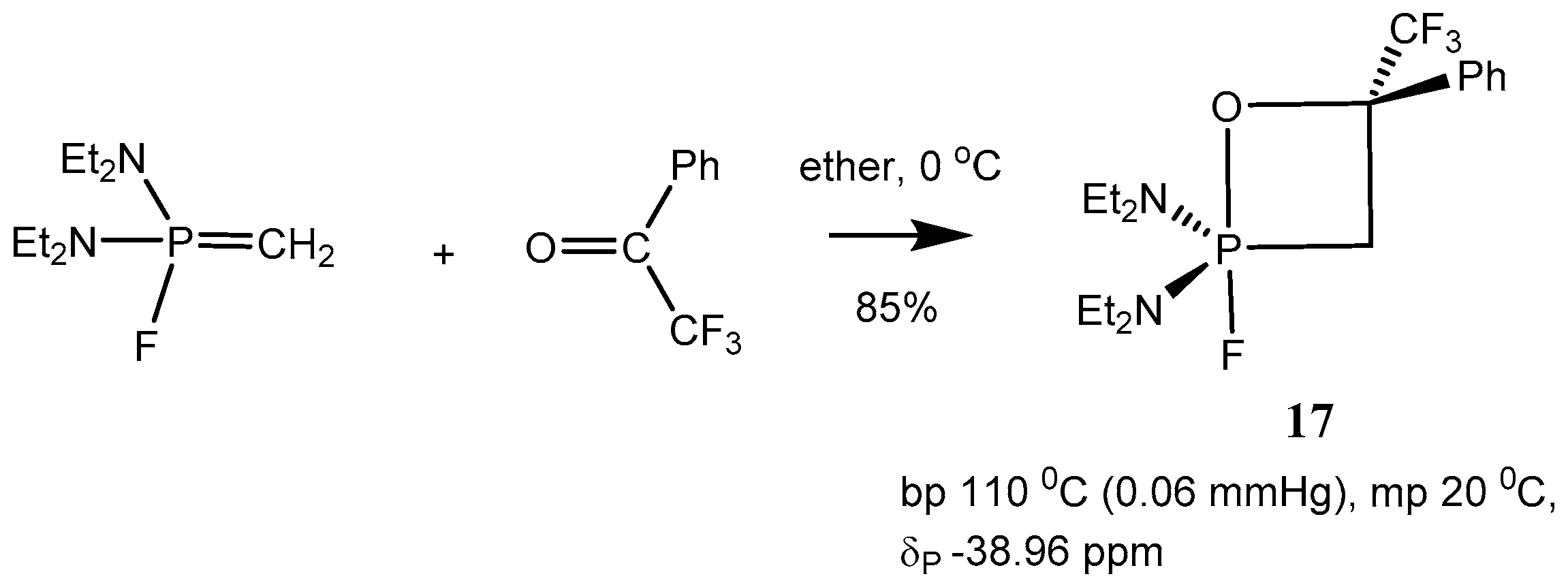
P-Fluoroylids with aldehydes and ketones proceeds in ether or pentane at −40–−20 °С and leads to the formation of stable 2-fluoro-1,2λ
5-oxaphosphetanes
17 (
Scheme 11 and
Table 2).
The compounds
17 are liquids distilling in vacuo, the structure of which was proved by means of mass and NMR spectra. The
31Р-NMR spectra of 2-fluoro-oxaphosphetanes
17 present doublets with 800 Hz
1JРF constants in the high magnetic field of a NMR spectrum at −37–−8 ppm. This corresponds to a pentacoordinate state of compounds
17 [
24,
26,
27,
28,
29,
30,
31]. Tetracoordinated forms of 2-fluoro-oxaphosphetanes
17 were not registered by
19F- and
31P-NMR spectroscopy (
Scheme 11).
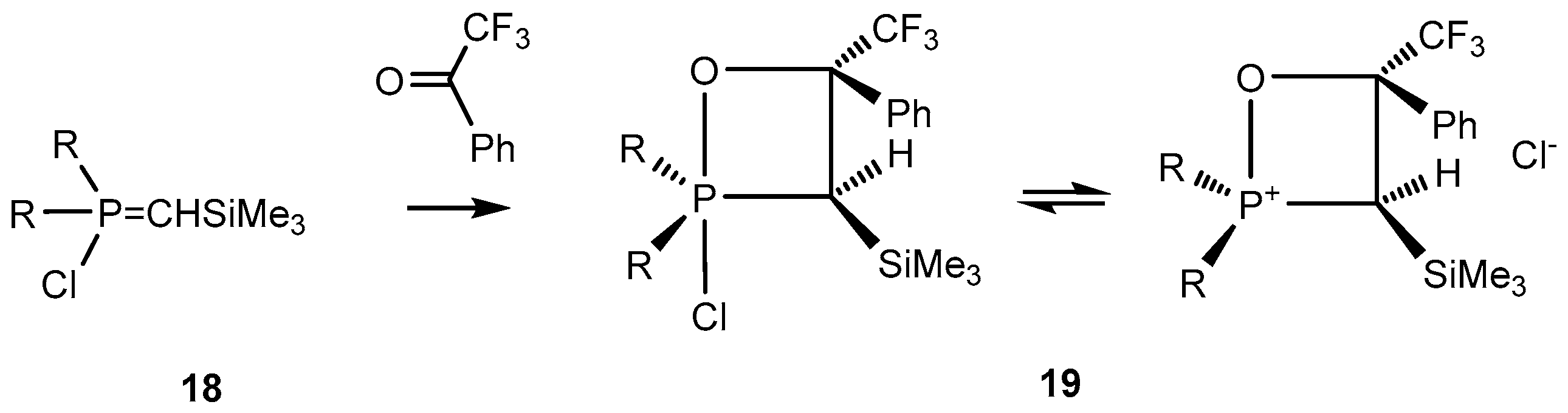
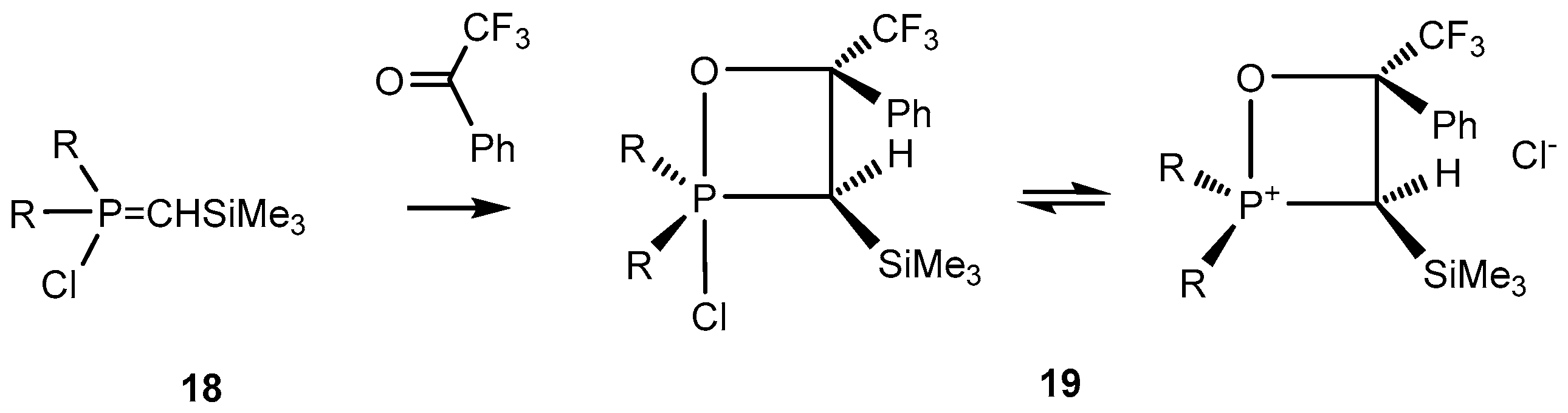
C-Silyl-
P-chloroylides
18 react with carbonyl compounds to afford 2-chloro-1,2λ
5-oxaphosphetanes
19. The oxaphosphetanes
19 bearing an electronegative CF
3 substituent at С-4 are relatively stable and can be isolated and analyzed by NMR (
Scheme 12). The NMR spectra of these compounds reveal signals at 0.2 ppm, singlet (Me
3Si), at 2 ppm, doublet,
3JPH 18.0 Hz (С
3Н), and at 4.5 ppm (С
4Н).
13С-NMR signals of С-3 and С-4 carbons were found at 30 and 90 ppm, correspondingly. The
31P-NMR signals of 2-chloro-1,2λ
5-oxaphosphetanes
19 at δ
P +48 ppm (R
1 =
i-PrO) and at δ
P = 60 ppm (R
1 = Et
2N) correspond to tetracoordinate phosphorus included in four-membered phosphetane cycle (
Table 1) [
25,
32].
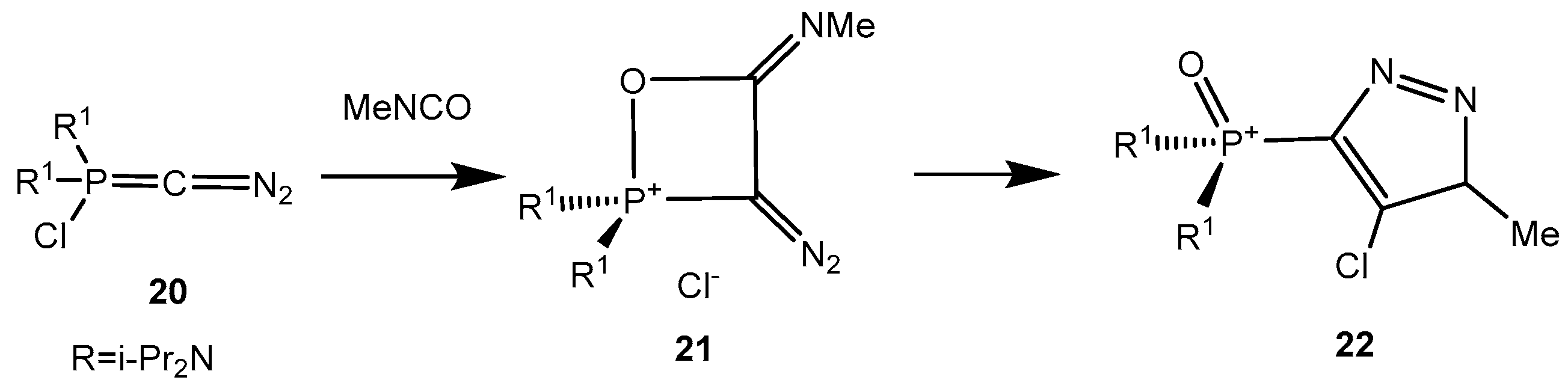
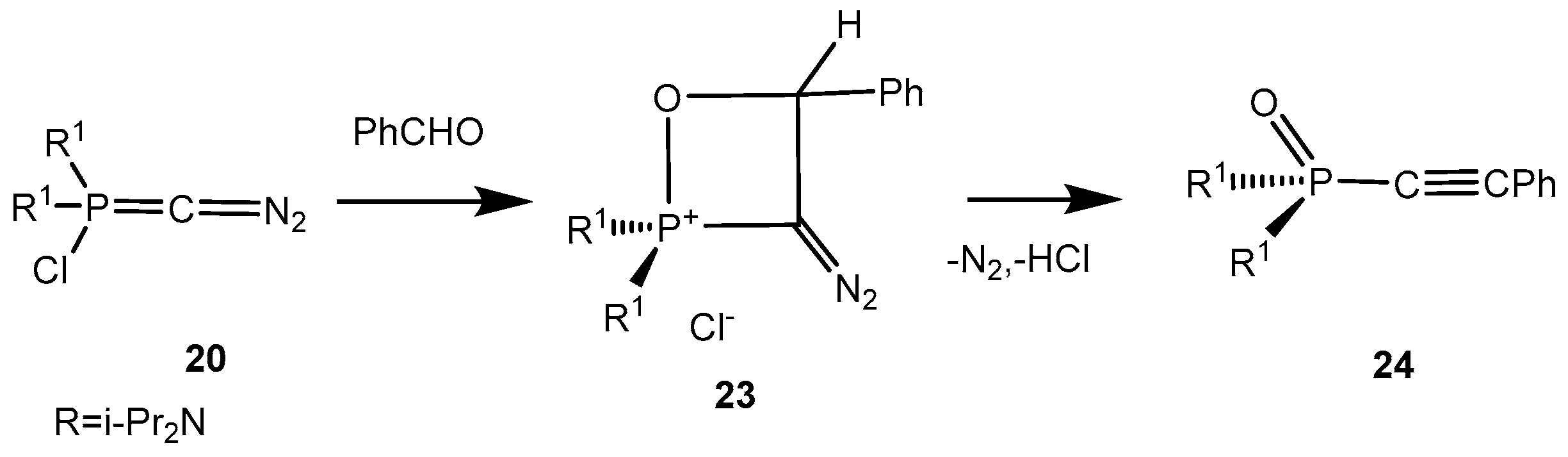
Sotiropulos and Bertrand [
33] reported the addition of phosphacumulene ylides
20 bearing the diazo group to isocyanates, leading to the formation of products
22. It was proposed that initial nucleophilic attack of the ylide carbon atom on the carbonyl carbon gives a oxaphosphetane
21, which depending on the relationship of the oxygen atom (or NR group) to nitrogen or phosphorus rearranges into products of 1,4- or 1,5-cyclisation
22 (
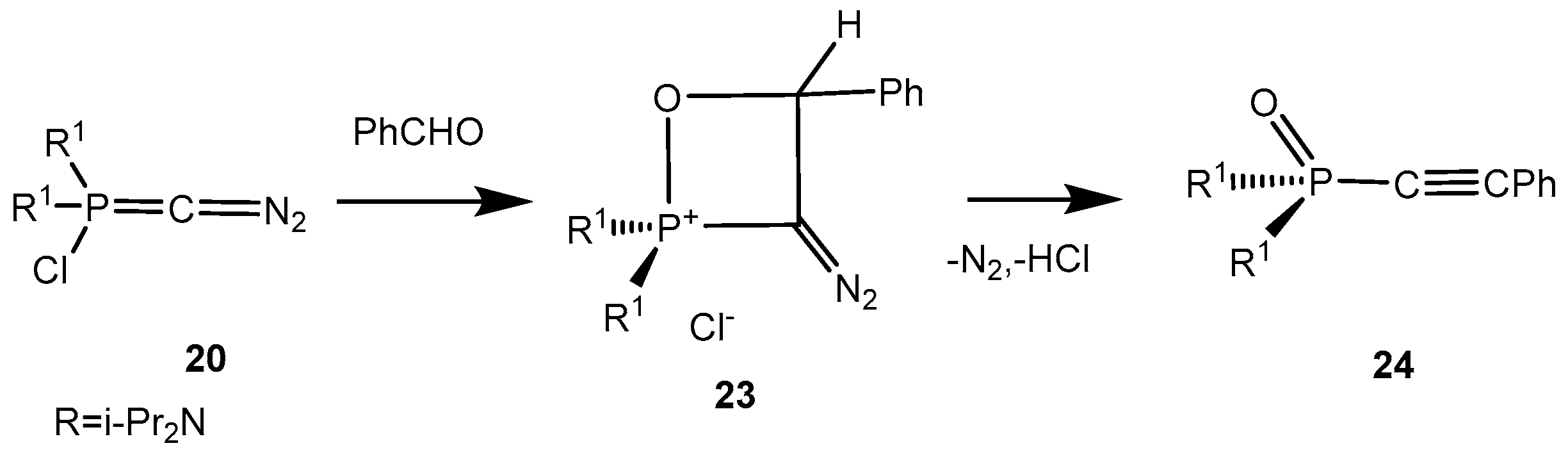
Scheme 13). Benzaldehyde gives the 2-chlorooxaphosphetane
23 with the ylide
20 which readily eliminates hydrogen chloride and a nitrogen molecule being converted into an acetylene phosphonate
24 (
Scheme 14).
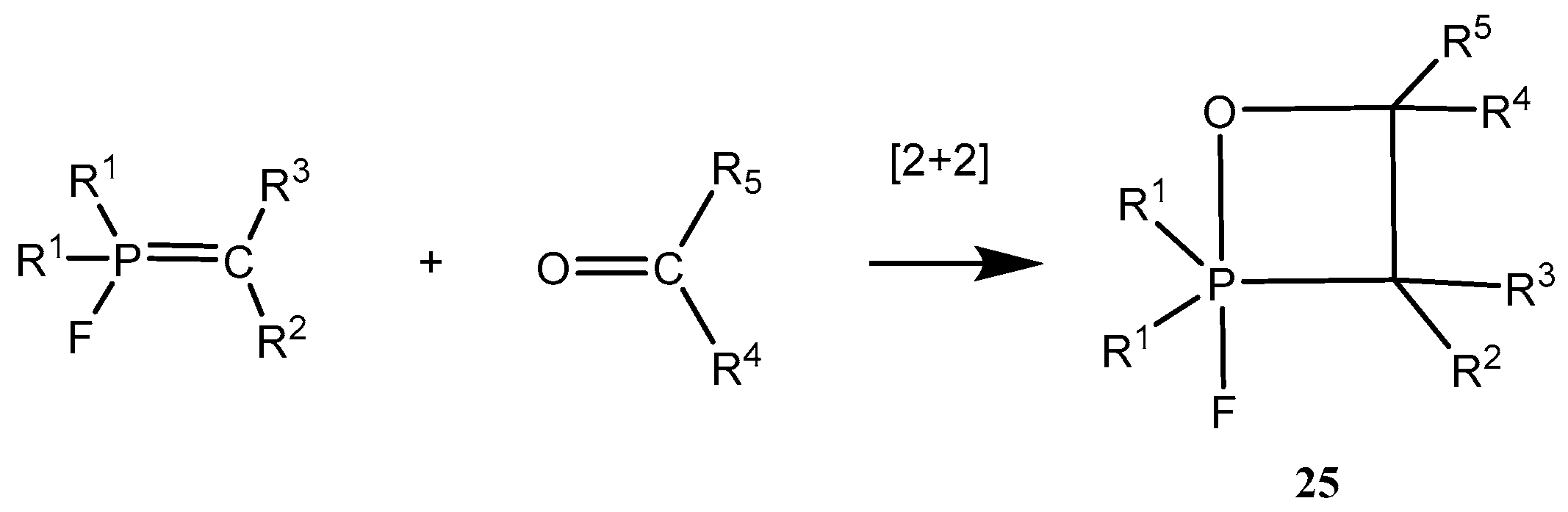
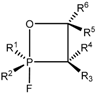
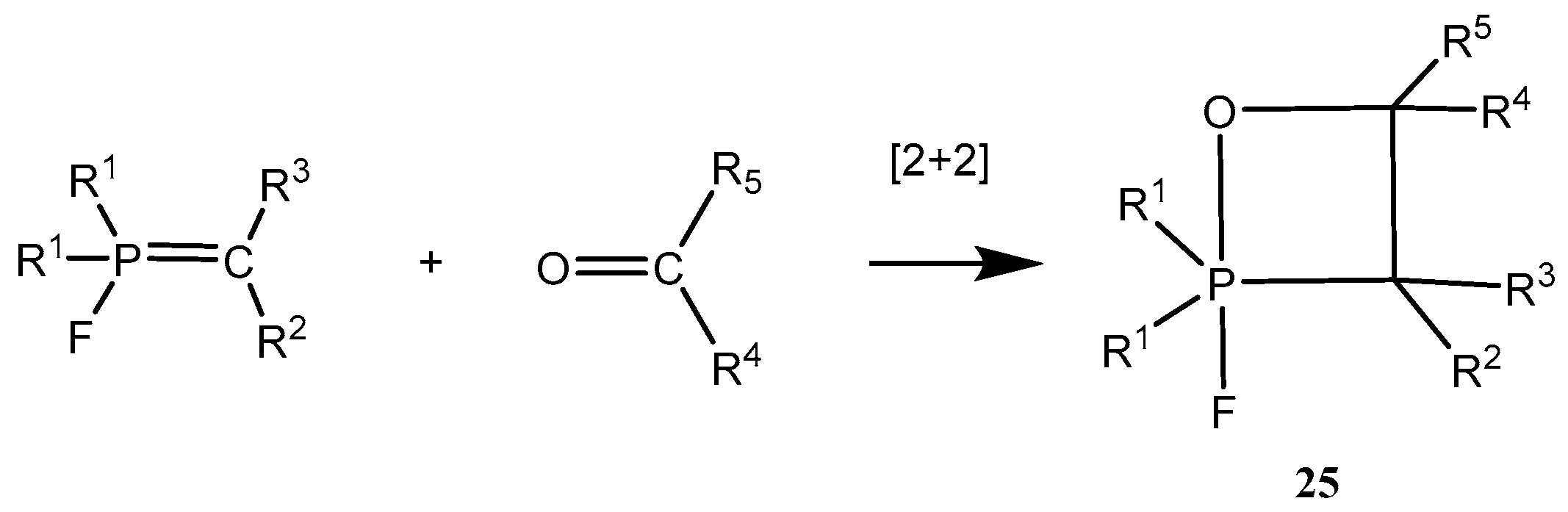
A number of 2-fluoro-1,2λ
5-oxaphosphetanes
25 were prepared by reaction of
P-fluoroylides with aldehydes and ketones (
Scheme 15,
Table 2). The 2-fluoro-1,2λ
5-oxaphosphetanes
25 are stable compounds, which can be isolated and purified by distillation under vacuum or by crystallization from non-polar solvents. These compounds are much distinguished from unstable adducts of carbonyl compounds with triphenylphosphonium ylides. Cycloadducts of
P-fluoroylides with carbonyl compounds, 2-fluoro-1,2λ
5-oxaphosphetanes, are also much more stable than 2-chloro- or 2-bromo-1,2λ
5-oxaphosphetanes [
25,
26,
27,
28,
29,
30,
31].
The stability of 2-fluoro-oxaphosphetanes is explained by the high electronegativity of the fluorine atom, compared to the electronegativities of chlorine and bromine. The P-F bond in 2-fluoro-1,2λ
5-oxaphosphetanes is very strong, and, therefore, these compounds do not dissociate with formation of cyclic phosphonium salts, what is observed, for example, with 2-chloro-1,2λ
5-oxaphosphetanes. Various stable 2-fluoro-1,2λ
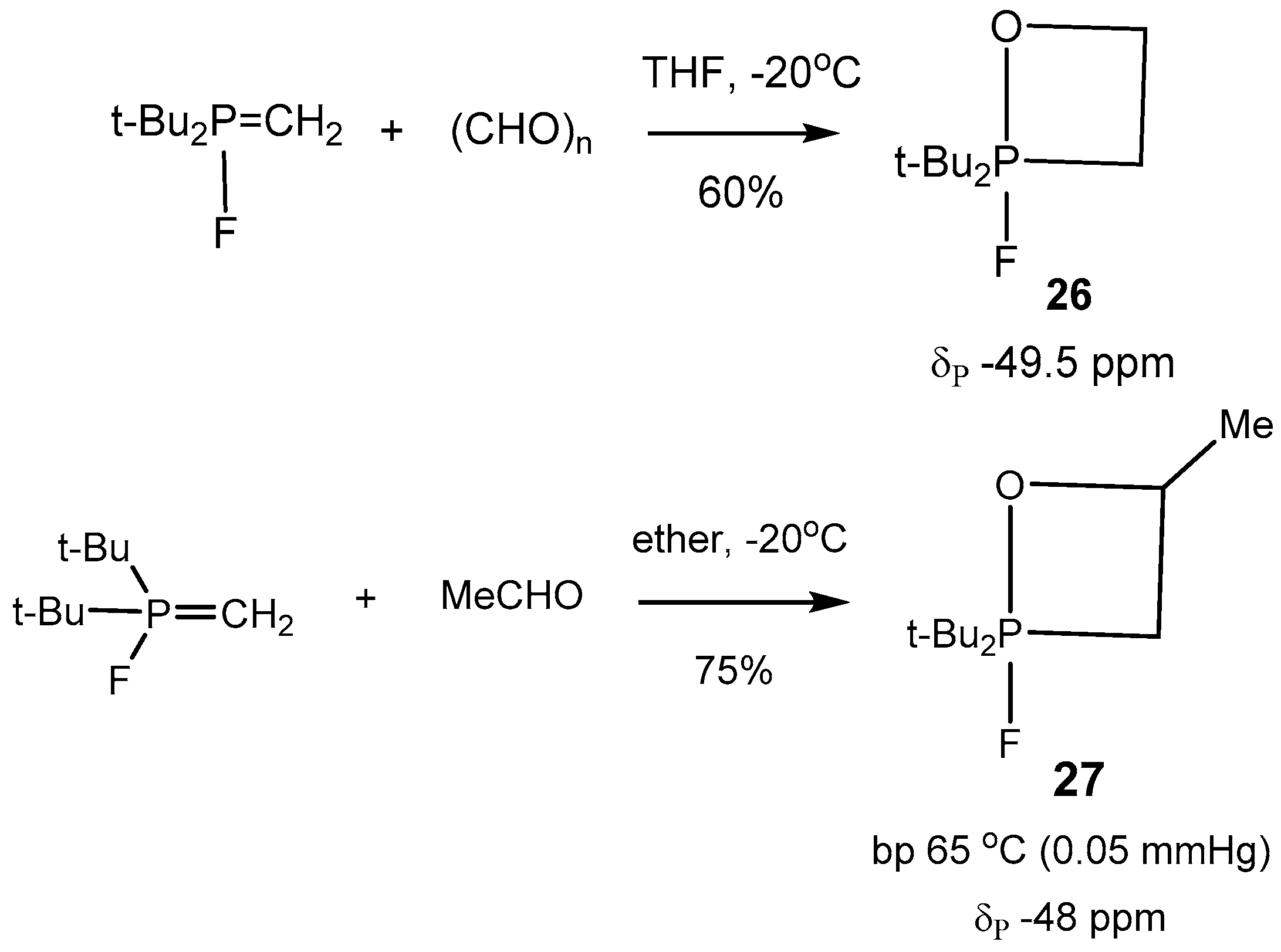
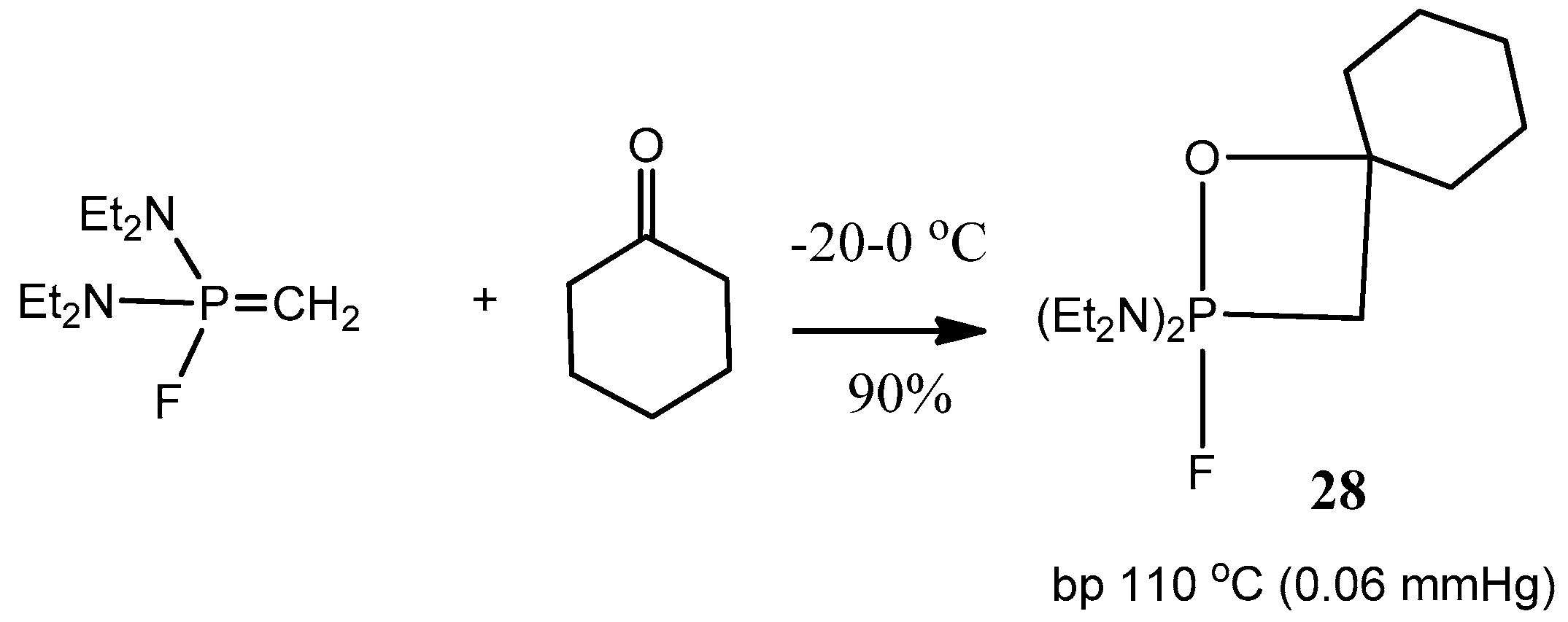
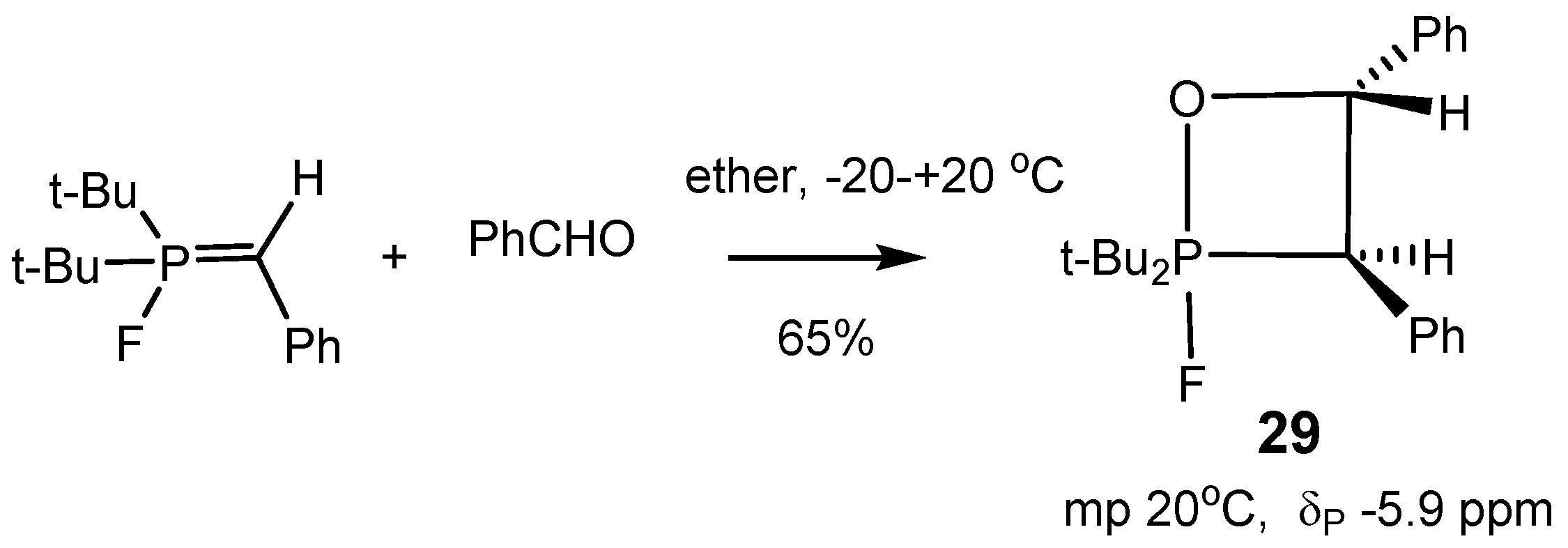
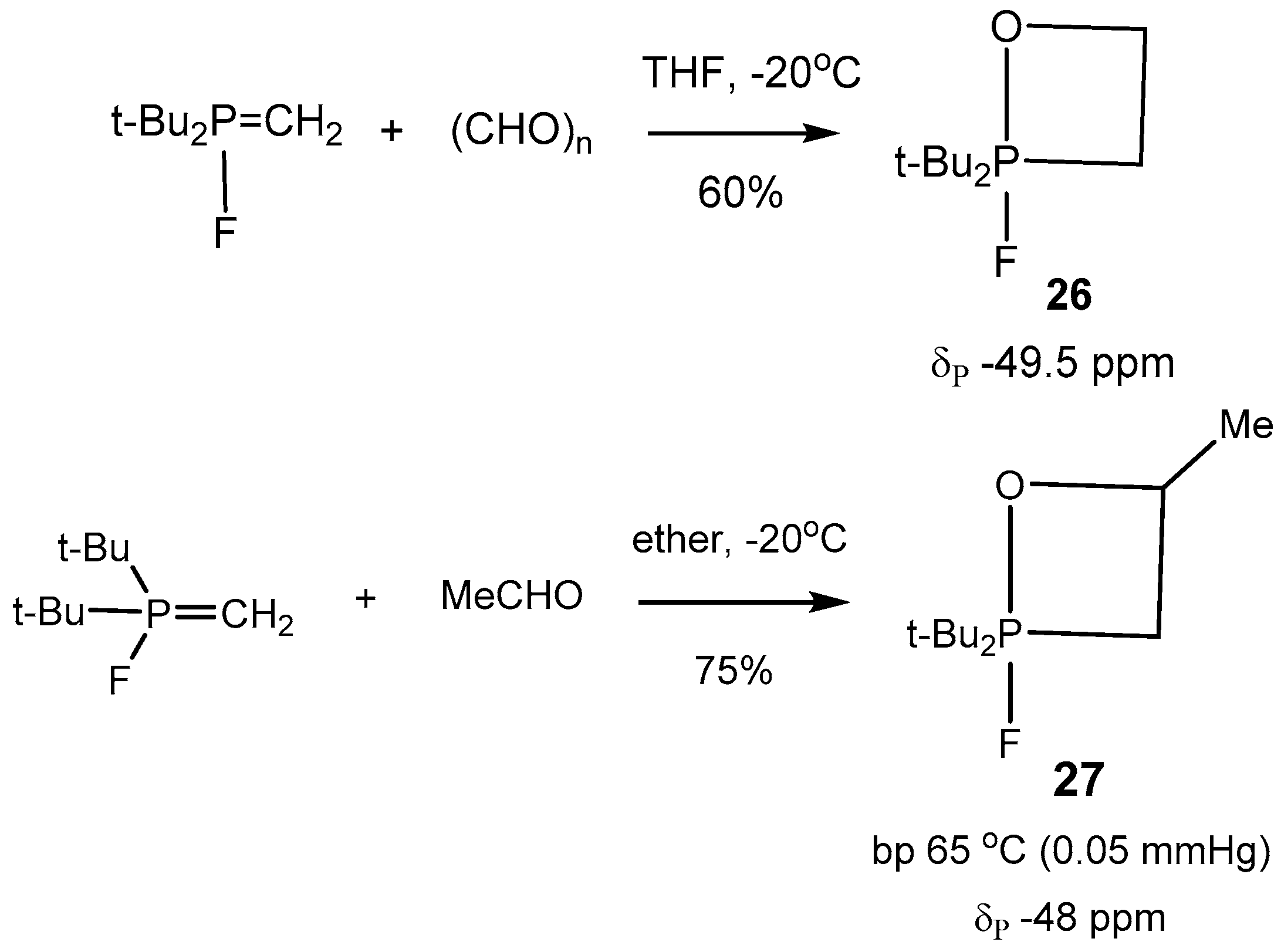
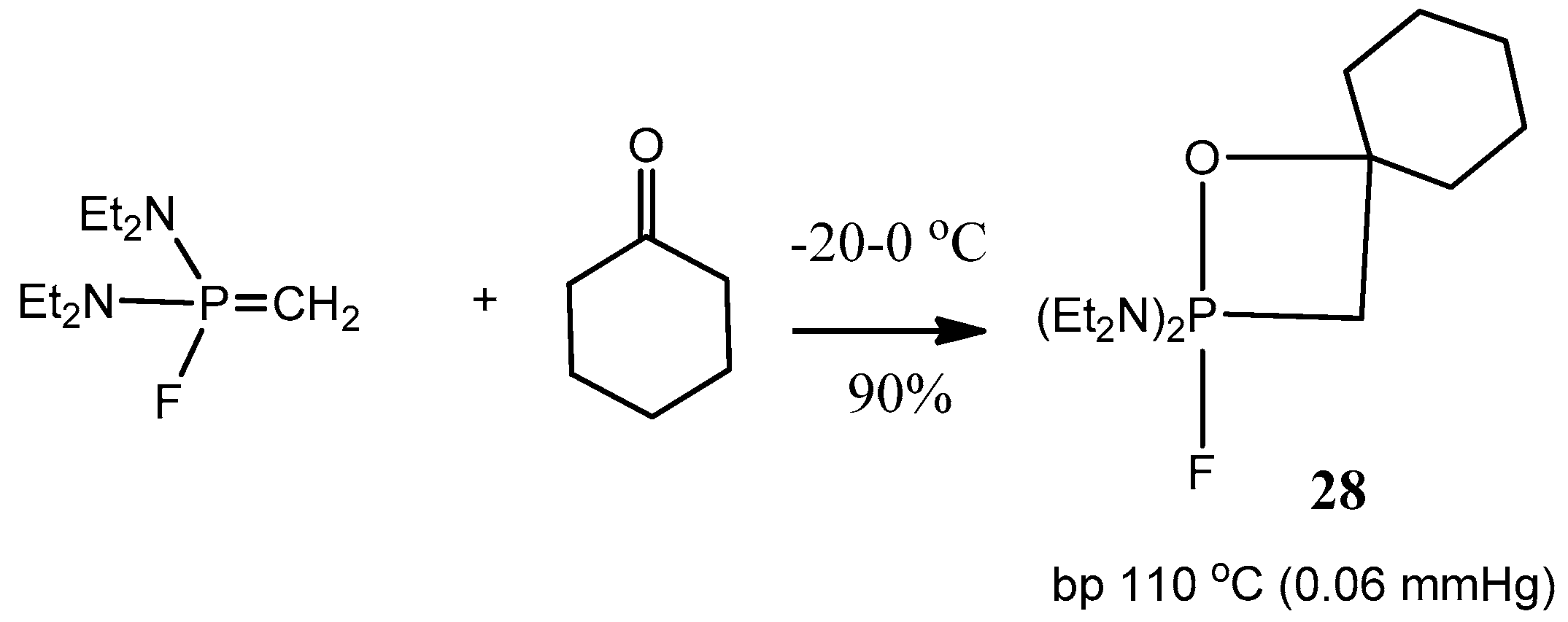
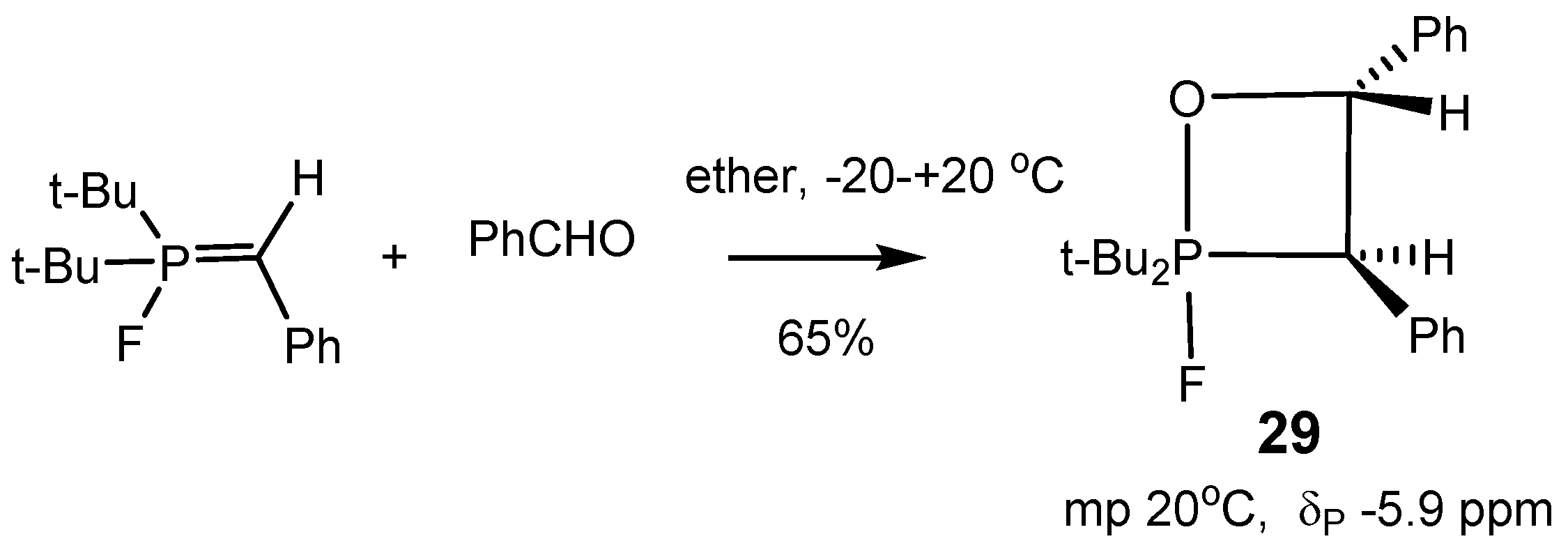
5-oxaphosphetanes were synthesized, isolated as pure specimens, and characterized (
Table 2). Typical representatives of such compounds
26–
29 are shown in
Scheme 16,
Scheme 17 and
Scheme 18 [
26,
29].
The 31P-NMR spectra of compounds 25 show a doublet at −6 to −45 ppm with 760–850 Hz PF coupling constants appropriate for axial fluorine atoms. The 31P-NMR spectra of 2-fluoro-1,2λ5-oxaphosphetanes exhibit a doublet in the range −6 to −45 ppm, belonging to the five-coordinate phosphorus with corresponding coupling constants on axial fluorine atoms (760–850 Hz). The 19F-NMR spectra of 2-fluoro-1,2λ5-oxaphosphetanes exhibit doublets with the same PF coupling constants.
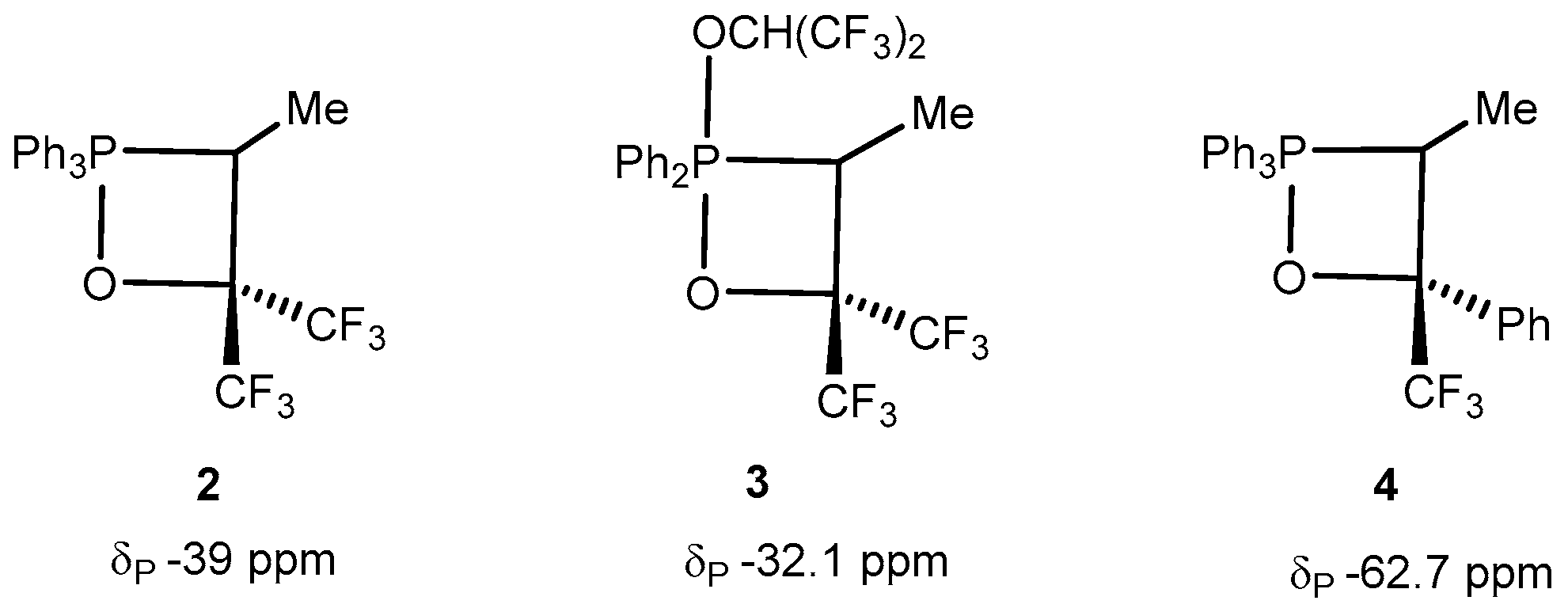
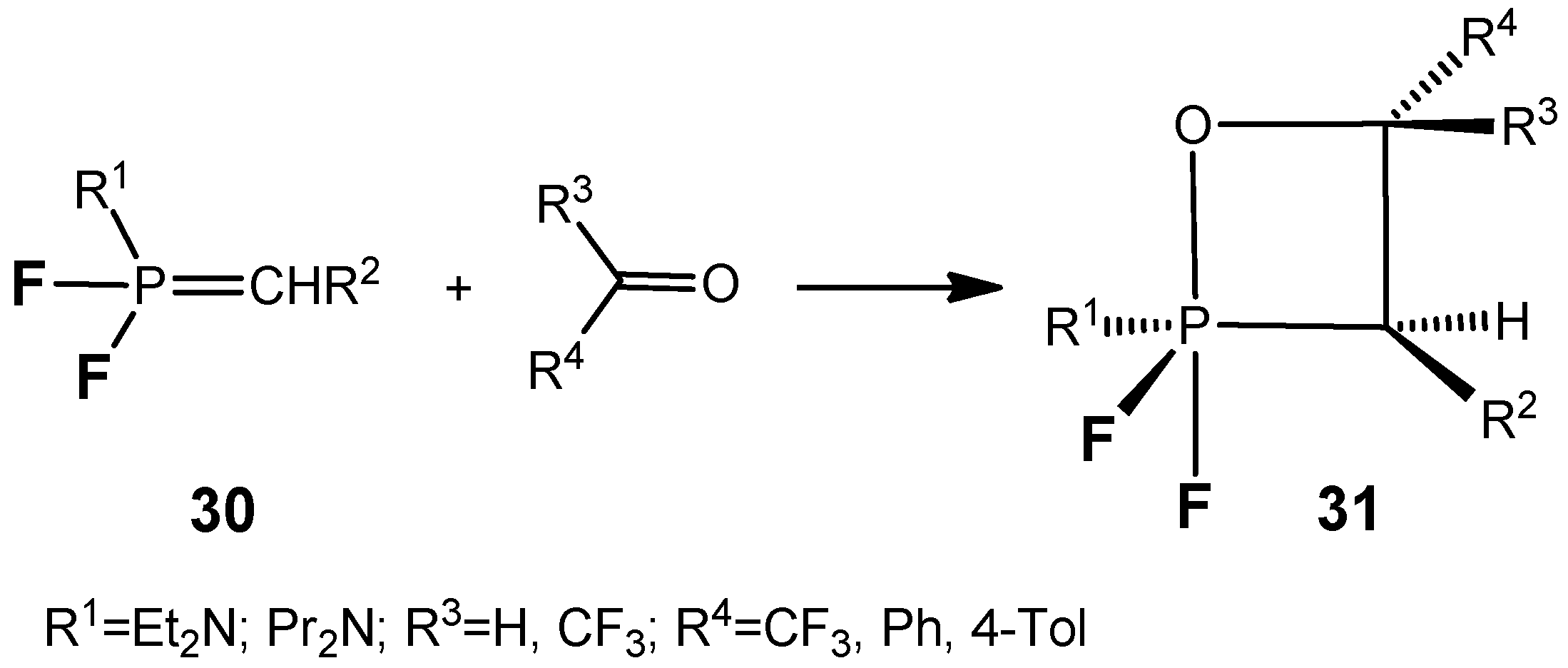
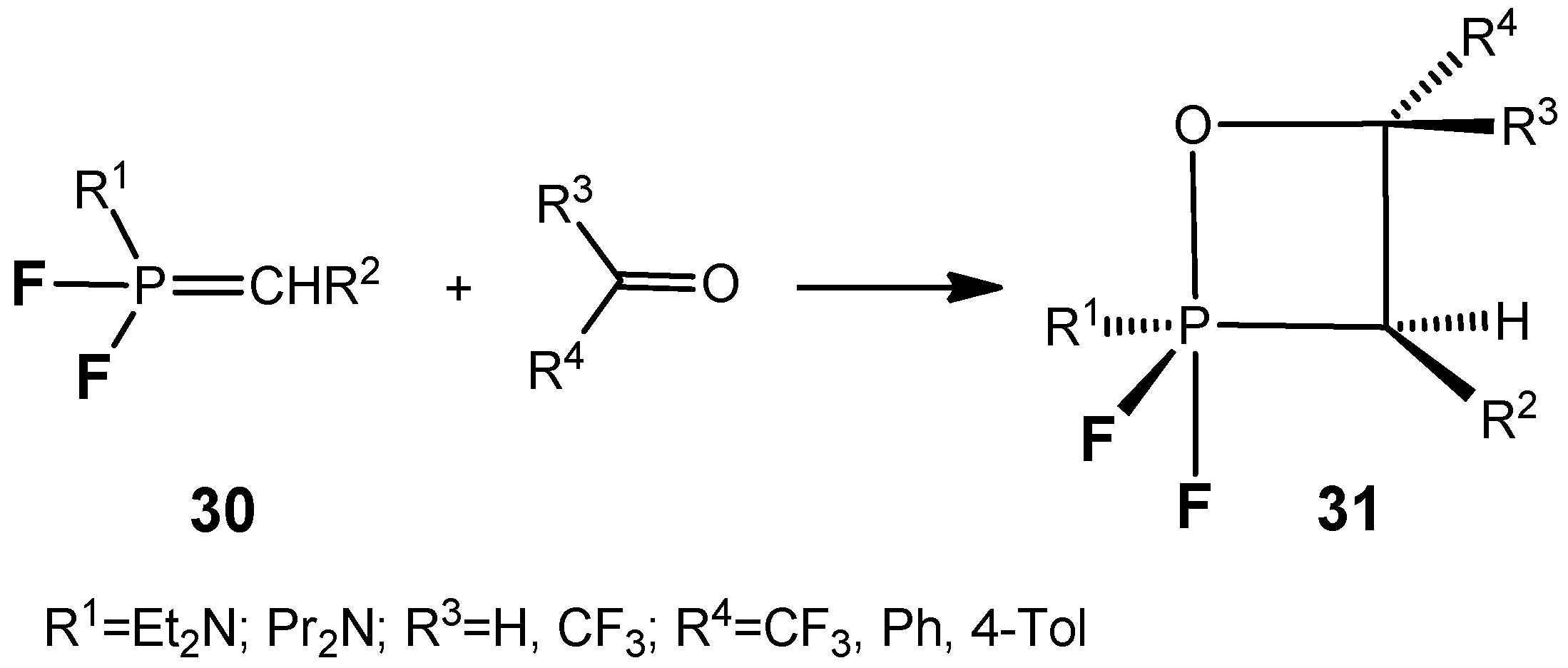
The
P,P-difluoroylides
30 react with aldehydes and active ketones to afford the stable oxaphosphetanes
31 bearing two fluorine atoms at the phosphorus (
Scheme 19) [
34,
35,
36]. The compounds
31 (R=CF
3) were distilled under reduced pressure without decomposition and were characterized by means of NMR spectroscopy.
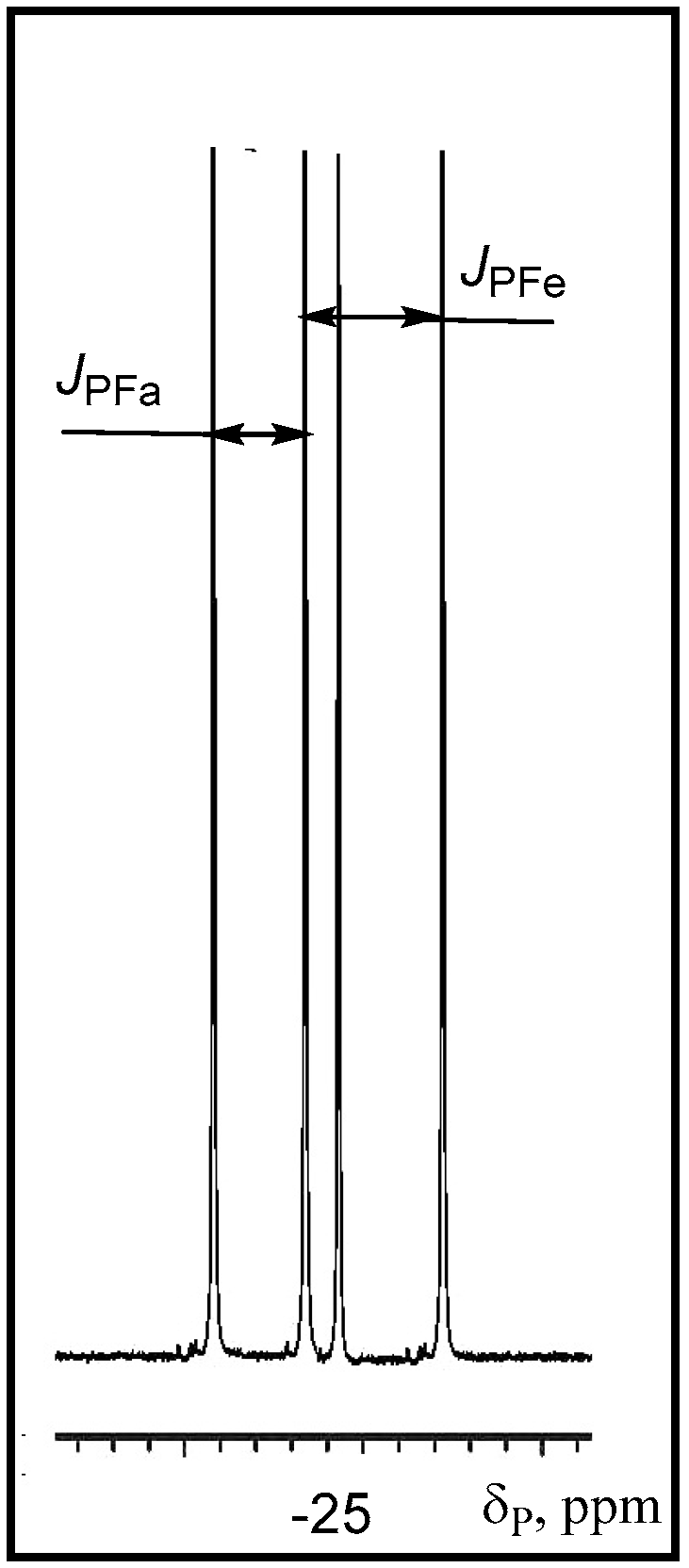
The
19F-NMR spectra contain two double doublets at −47 and −65 ppm with expected coupling constants for the axial and equatorial fluorine atoms:
1JPFa = 915 Hz,
1JPFe = 1025 Hz, and
2JFaFe = 62 Hz. Apparently [2+2]-cycloaddition of the C=O group to the ylide
15 proceeds with high stereoselectivity, because the
19F- and
31P-NMR spectra show the signals belonging to the single diastereomer of the compounds
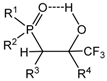
31. The signals of the second diastereomer, the existence of which one can suppose as a consequence of the presence of two asymmetric endocyclic C-3 and C-4 carbon atoms are absent. The
13C-NMR spectra reveal the presence of the signals at 62.5 ppm (
1JCP = 150 Hz,
2JCF = 53 Hz) and 75.6 ppm due to the C-3 and C-4 carbon atoms in complete accordance with the assigned structure of oxaphosphetane (
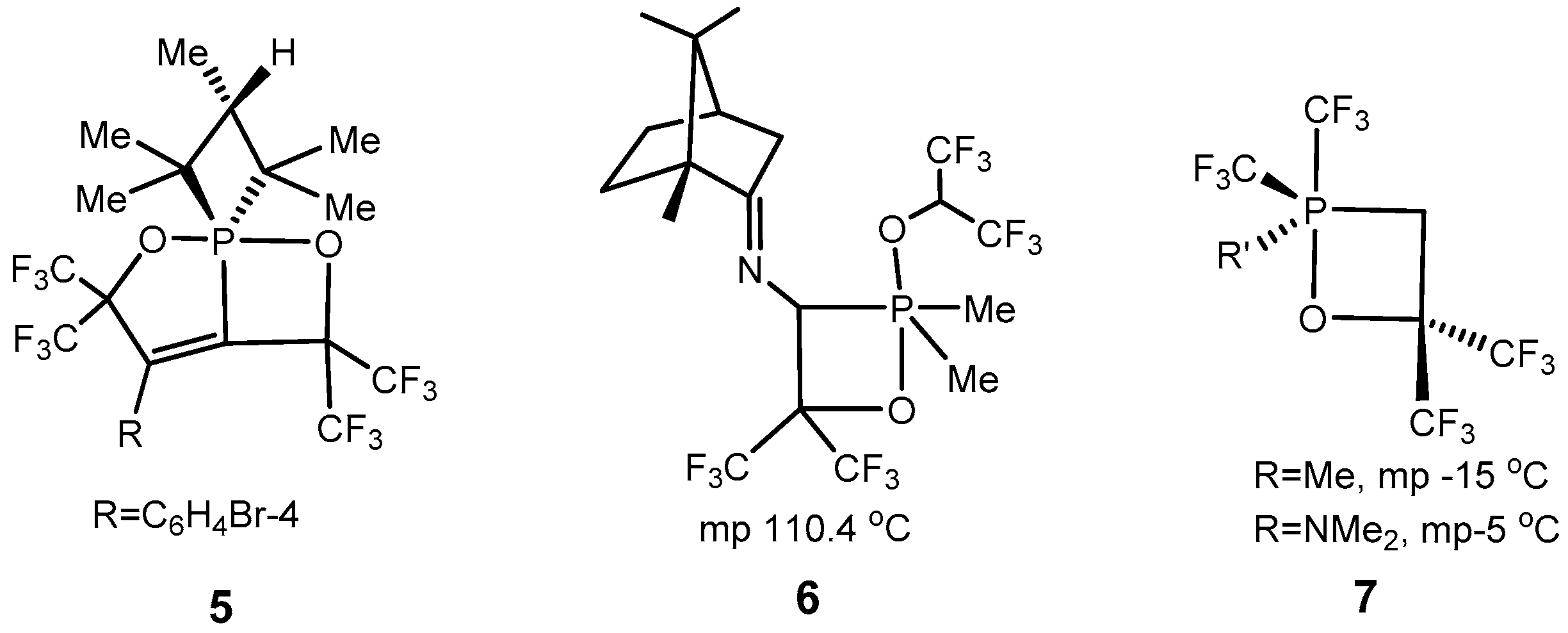
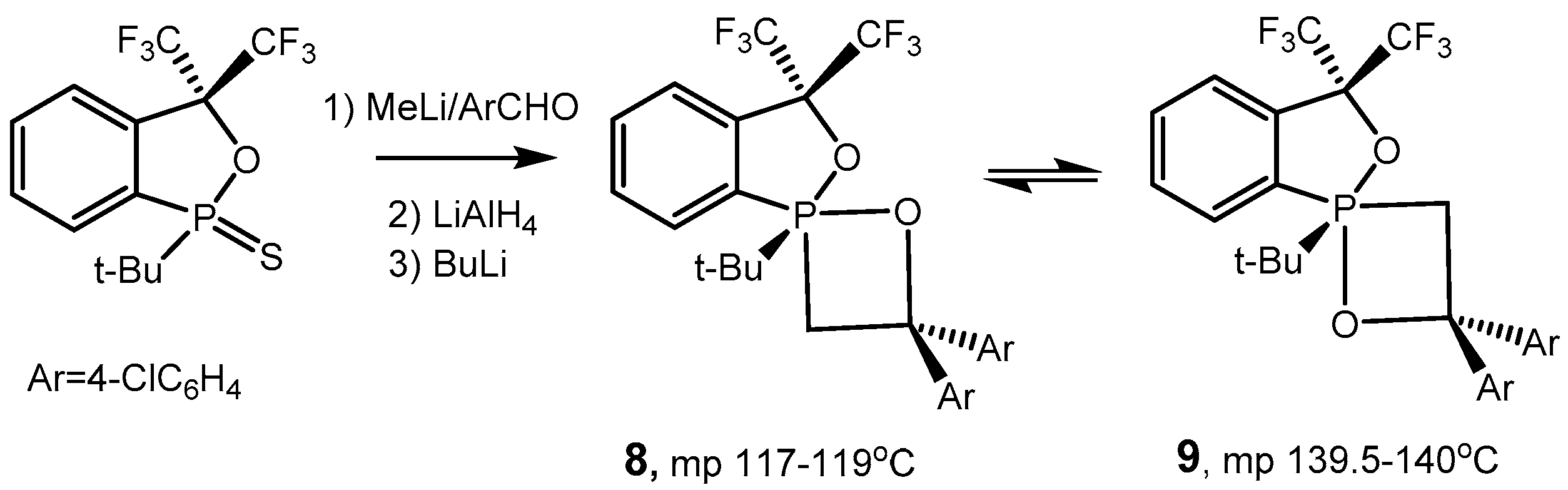
Figure 1 and
Figure 2).
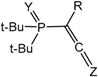
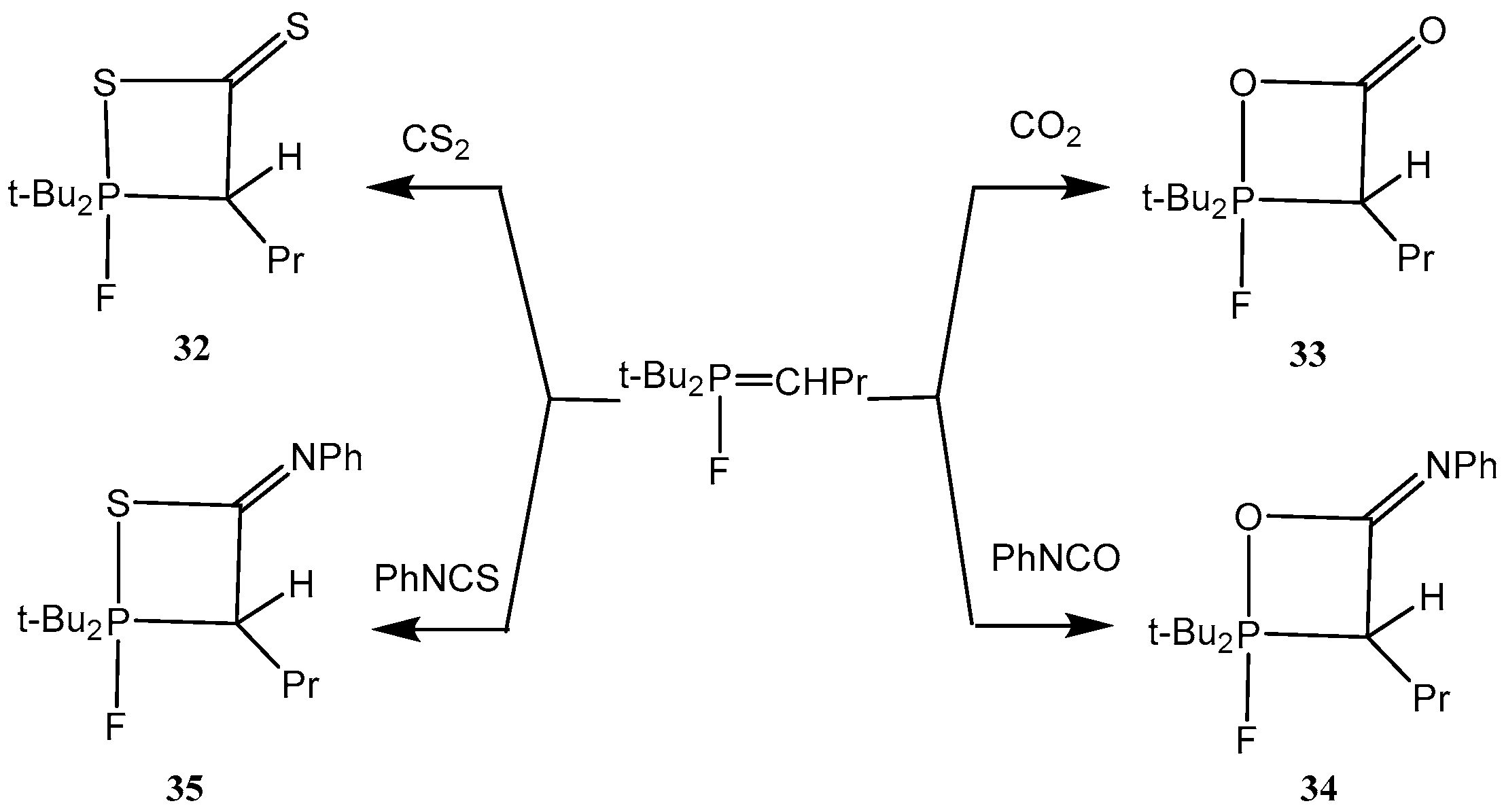
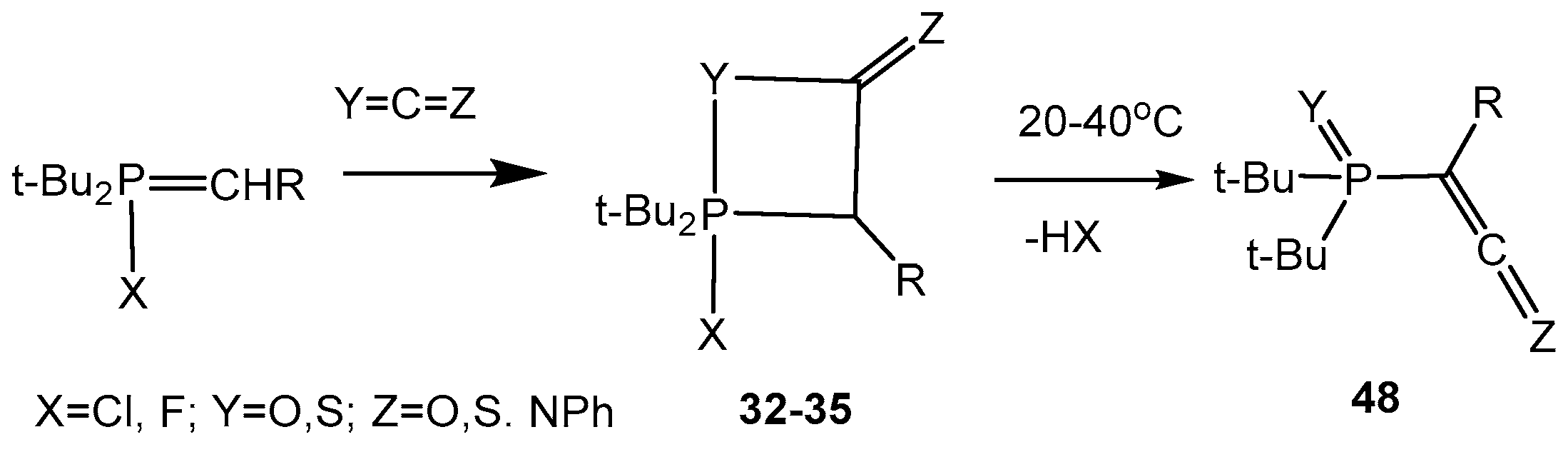
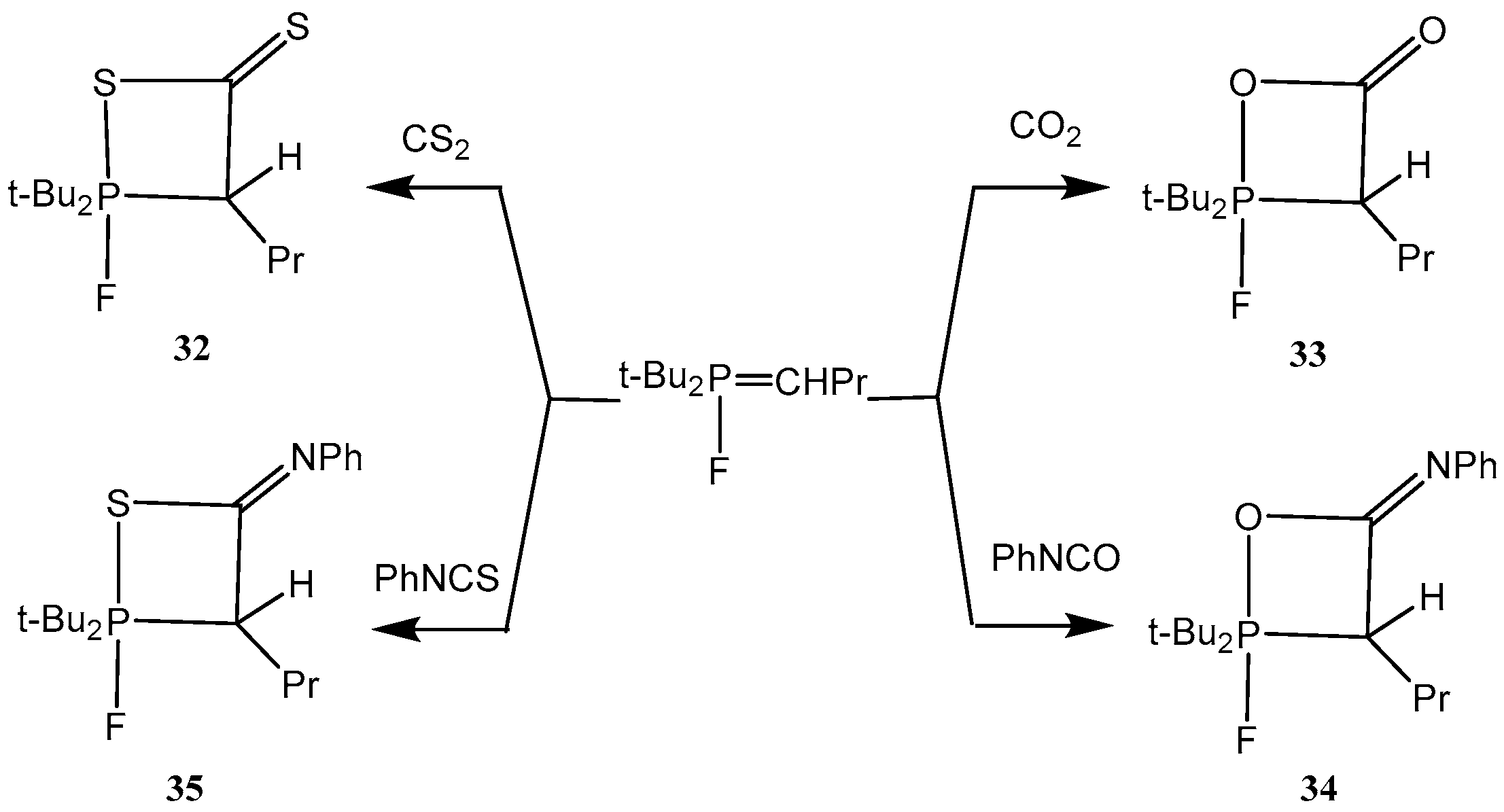
P-Fluoroylides add easily isocyanates, carbon dioxide and carbon disulfide with formation of oxaphosphetanes
32–
35. For example, the reaction of
P-fluoroylides with phenyl isocyanate resulted in the formation of 2-fluorooxaphosphetane 34 which was stable during several hours at ambient temperature. Compound
34 was purified by crystallization in pentane and isolated as colorless crystalline matter. In IR spectra of compounds was found the strong band at 1730 cm
−1 belonging to the C=O bond in a four-membered cycle. The
31Р-NMR spectra disclosed a doublet with constant
1JPF = 780 Hz and a doublet with the same constant in
19F-NMR spectra. The oxaphosphetanes
32–
35 convert at room temperature slowly and at heating quickly into phosphorylated heterocumulenes (ketenes, thioketenes, ketenimines, see
Scheme 20) [
29,
37,
38,
39].
2.3. Properties of Oxaphosphetanes
The cycloaddition of
P-halogenylides to aldehydes and ketones proceeds stereoselectively and leads predominantly to the formation of one of the possible diastereomers of 2-halo-1,2λ
5-oxaphosphetanes (
Table 1 and
Table 2).
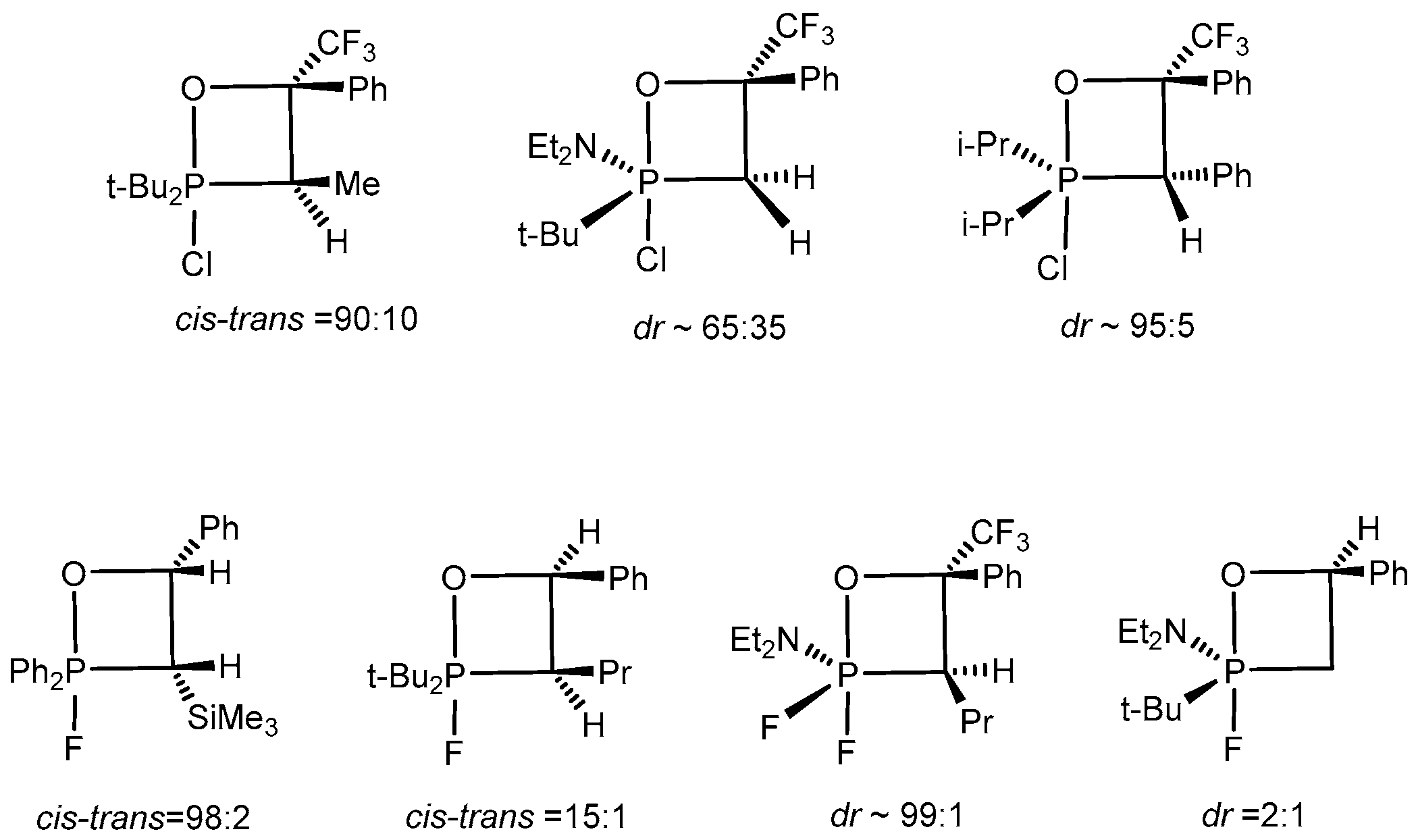
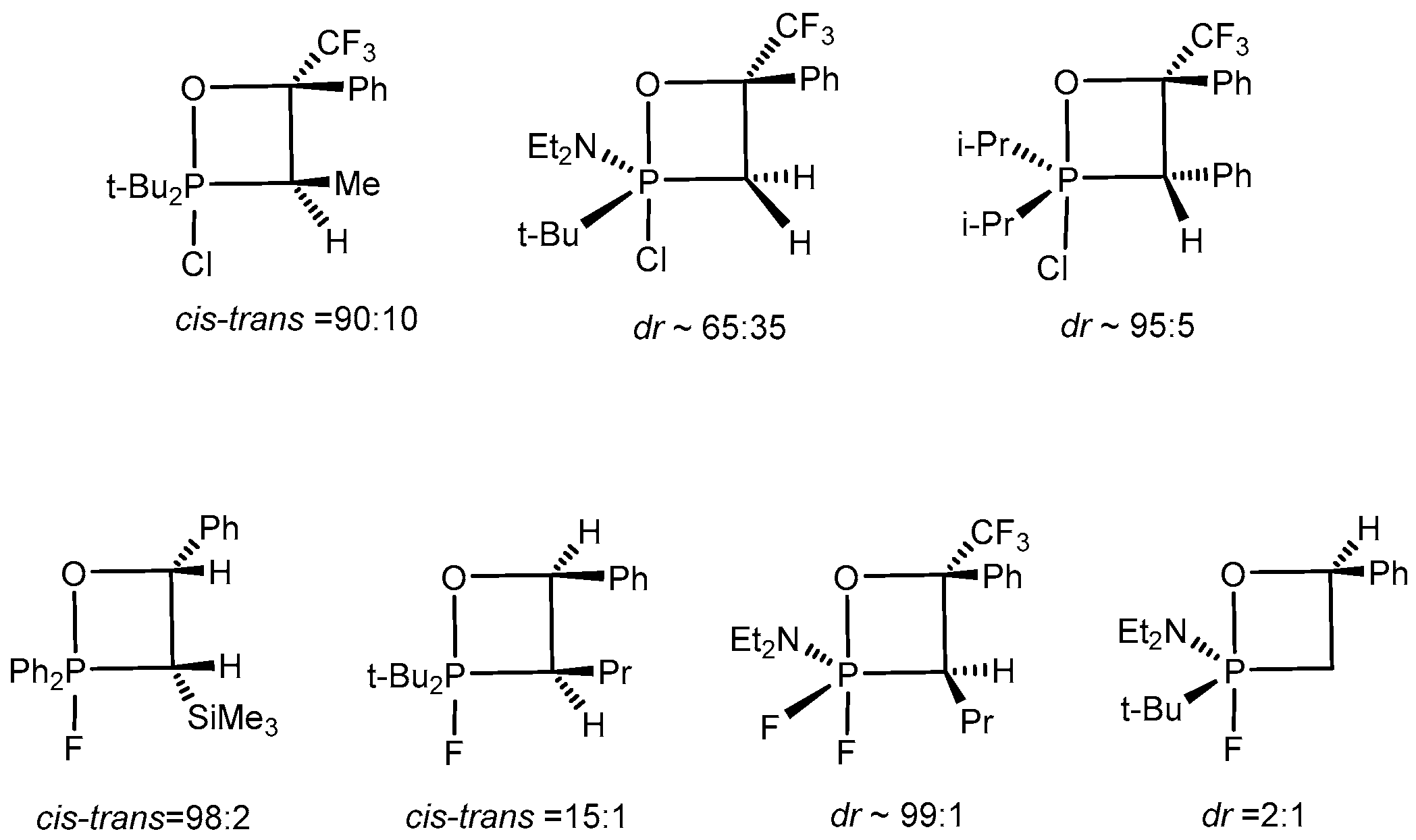
P-Haloylides (chloro- and bromo) react stereoselectively with aldehydes to give predominantly single diastereoisomers of 2-halo-oxaphosphetanes, bearing asymmetric endocyclic C-3 and C-4 carbon atoms (dr ~7:1–15:1). It was established by NMR that the ratio of diastereoisomers of 2-halooxaphosphetanes containing asymmetric atoms at C(3) and C(4) was within the limits ~99:1–90:10. The ratio of diastereomers after completion of the reaction was approximately 6:4–9:1. However on heating, as a result of permutational changes in the molecule, the ratio of diastereomers grew in favour of the thermodynamically more stable diastereomer. 2-Fluoro- and 2-chloroxaphosphetanes containing chiral phosphorus or endocyclic carbon atoms exist as mixtures of diastereomers whose ratio depends on the nature of the starting reagents (
Scheme 21). The diastereomeric purity of the compounds, assessed by NMR spectroscopy, was 80%–96%. The reaction of α,α,α-trifluoroacetophenone with
P,P-difluoroylides proceeded with very high stereo-selectivity to furnish only one diastereomer of compound. At the same time 2-halo-1,2λ
5-oxa-phosphetanes bearing asymmetric phosphorus atom are formed with low stereoselectivity (dr~ 3:1).
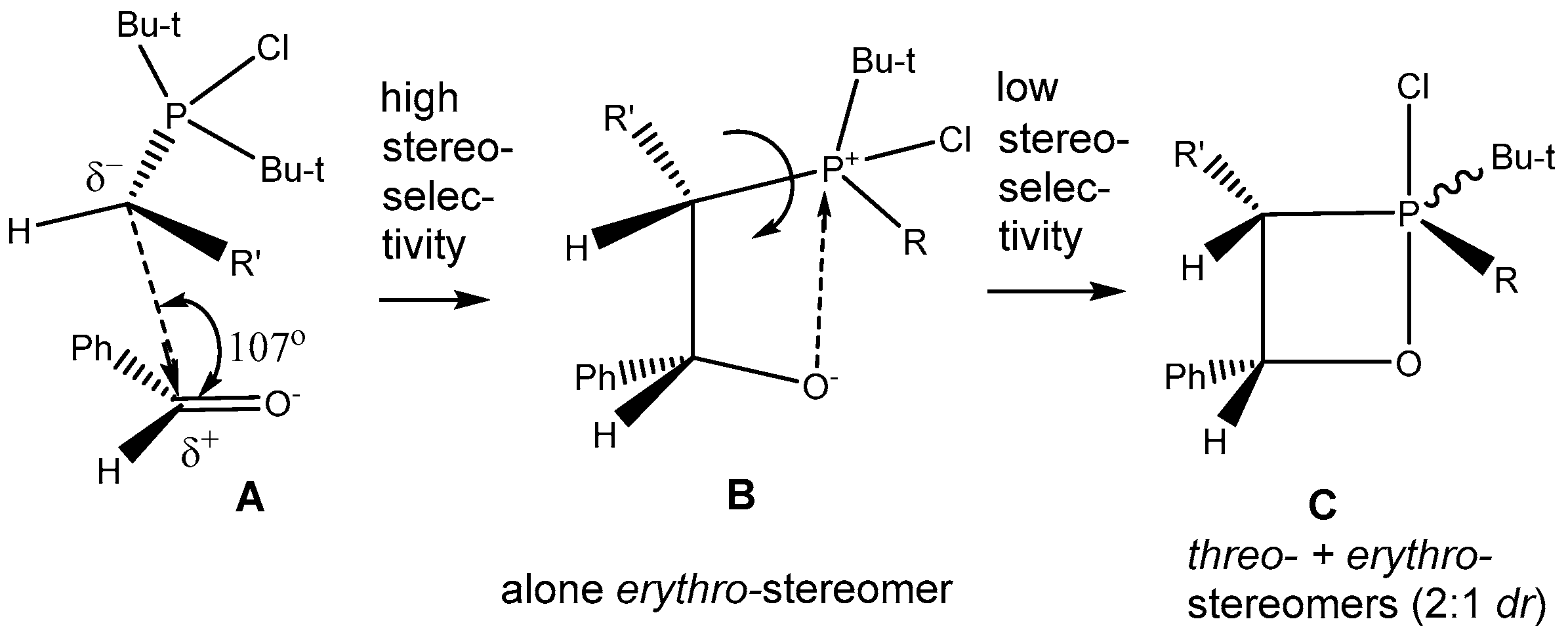
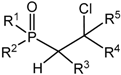
The reaction of
P-halogenylides with aldehydes leads to the formation of an
erythro-oxaphosphetanes with the oxygen in the apical position, because of the tendency of electron-acceptor atoms to occupy this position [
40]. The preferred orientation of the transition state leading to the
erythro-oxaphosphetane is stereoselective approach of the ylide nucleophilic center to the carbonyl group at an angle of 107°, with the double bonds of both reagents arranged in one plane. This was associated with the Burgi-Dunitz trajectory concept (
Scheme 22) [
41]. The first step of reaction stereoselectively leads to the formation of the betaine
B. The conversion of reagents into oxaphosphetane requires minimum energy when it proceeds with the non-synchronous formation of bonds between the carbonyl and ylidic carbon atoms. On the second step the betaine converts into oxaphosphetane
C with low stereoselectivity, because of free rotation around of the P-C bond [
28,
42]. This mechanism of asynchronous addition of
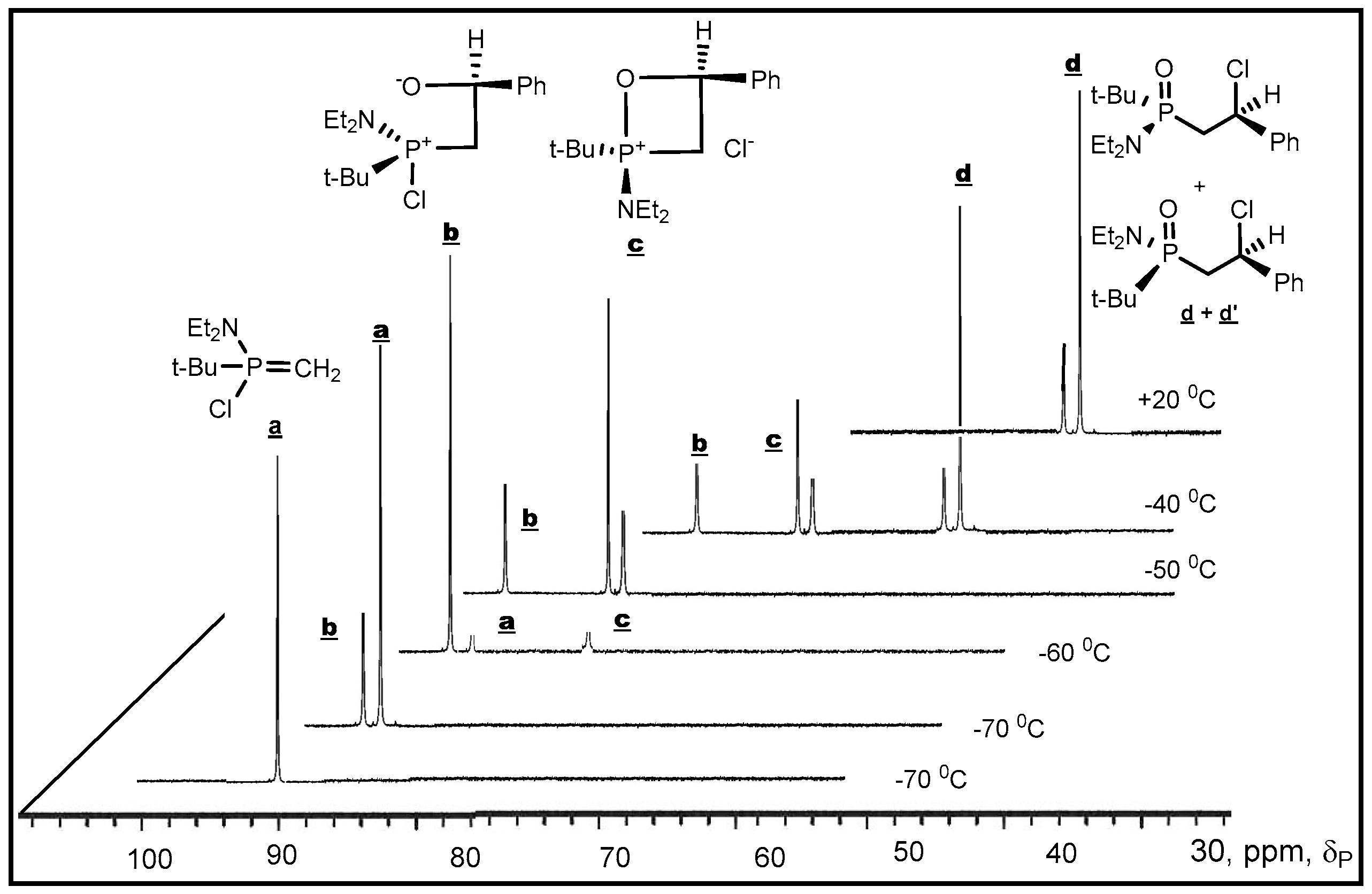
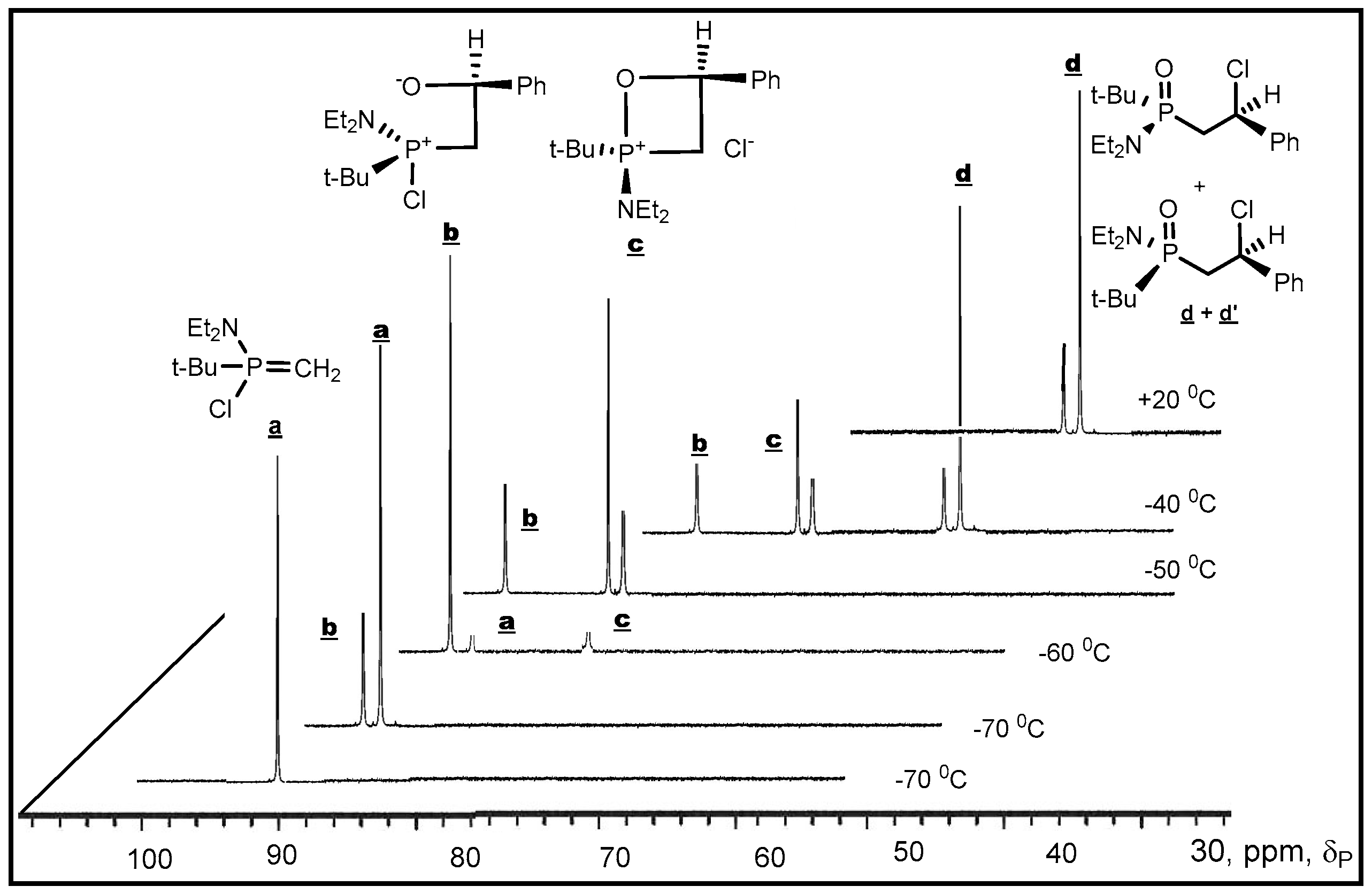
P-haloylides to carbonyl compounds was confirmed by low temperature NMR investigations. The reaction of di-
tert-butylсhlorphosphonium methylide with Ph(CF
3)C=O was studied by
31Р-NMR at low temperature (−70–0 °C) in diethyl ether solution. At −70 °С was found only a decreasing signal of
P-chloroylide (+114 ppm) and a growing signal of 2-chloro-1,2λ
5-oxaphosphetane (+9.9 ppm). The signal which could be referred to betaine was not found, probably, because of the high speed of betaine cyclisation into 2-chloroxaphosphetane. However the reaction of
tert-butyl(diethylamino)chlorophosphonium methylide with benzaldehyde began at −70 °С and led to the formation of
erythro-betaine which was registered by increasing signal δ
P +106 ppm, located in more weaker field than the signal of initial
P-chloroylide (+102 ppm) (
Figure 3). The betaine formation was proceeded stereoselectively as in the
31P-(
1Н-)NMR spectrum only one diastereomer signal was registered. The rise of temperature to −50 °С led to the formation of two signals of
threo and
erythro-oxaphosphetane diastereomers δ
Р + 92.6 and +84.6 ppm in the ratio of 2:1. Evidently, the formation of diastereomers proceeded via S
N2@P substitution with inversion of configuration at the phosphorus atom [
43]. The formation of isomers indicates on the nucleophilic substitution at asymmetric tetracoordinate phosphorus atom, proceeding with partial inversion of configuration. At −50–−40 °С the oxaphosphetane was converted into 2-chloralkylphosphine oxide, which was also formed as two
threo- and
erythro-diastereomers in the ratio of 2:1 (δ
Р +48.7 and +49.2 ppm). The conversion was completed at −20 °С. The
threo- and
erythro diastereomers were separated and isolated in pure state by chromatography and crystallization (m.p. 100 and 123 °С, see
Table 3, entries 13, 14). Although at present many of the puzzling features about the reaction mechanism of ylides phosphorus with carbonyl compounds have been clarified, it seems there is no general mechanism, which could explain the progress of the reaction, transition states and stereochemistry. Nevertheless in case of
P-chloroylide addition to aldehydes, on the basis of presented above stereochemical researches the asynchronous [2+2]-сycloaddition seems to be the most likely mechanism.
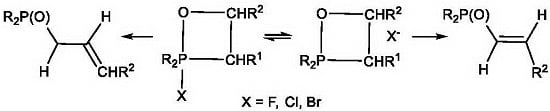
2-Halo-oxaphosphetanes exist in pentacoordinate form, but ionize under certain conditions with formation of tetracoordinate forms. In the mass spectra of 2-chlorooxaphosphetanes the peak of molecular ion was observed that indicated the covalent character of the P–Cl bond. All oxaphosphetanes show methyl carbons with lJPC values characteristic of equatorial placement in the trigonal bipyramid. The resonances of the methylene carbons of the phosphetane ring system lay downfield of the methyl carbons and the large measured lJPC values indicate equatorial placement. The four-membered ring is occupying an apical-equatorial plane where the O-P-C angle is 90°, and never a diequatorial plane or a diapical plane where the O-P-C angle would have to be 120 or 180°, respectively.
In the
1H-NMR spectra of the 2-halooxaphosphetanes possessing asymmetric carbon atom C
4 the magnetic nonequivalence of protons СН
aН
b in the four-membered ring became apparent because the geminal spin-spin interaction arose between them. Each of the signals СН
a and СН
b was the double doublet with the coupling constant with phosphorus nucleus
2JPH = 20–22 Hz and a constant of geminal interaction
2JHH = 16–17 Hz. The chemical shifts
31P of 2-halogenoxaphosphetanes depended on the polarity of the solvent. Thus, in nonpolar solvents (diethyl ether, pentane) the signals of δ
P are located in the strong field of the
31P-NMR spectrum (from −3 to 10 ppm) that corresponds to the pentacoordinated state of the phosphorus atom. In polar solvents and especially in the presence of Lewis acids, like AlCl
3, the values of δ
P were shifted downfield. For example, for 2-chloro-oxaphosphetanes the chemical shift of phosphorus, δ
P was as follows (ppm): 9 (pentane), 22 (СН
2С1
2), 30 (СНС1
3), 35 (СН
3СN), 45 (СНС1
3 + А1С1
3 (traces)). With the increase in amount of aluminum chloride up to equimolar level the value of δ
P 100 ppm was registered for 2-chlorooxaphosphetanes that was in accordance with the values of chemical shifts for the known alkoxyphosphonium salts and apparently indicated complete ionization of chlorophosphorane with the formation of phosphonium salt. The dependence of the chemical shift value of phosphorus in 2-chlorooxaphosphetanes on the solvent polarity and on the presence of Lewis acid is in accordance with the published data showing that chlorophosphoranes can be ionized with the formation of phosphonium structures. Herewith in the
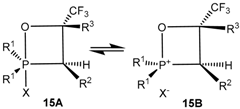
31P-NMR spectrum the resultant signal is registered due to fast exchange in the phosphorus coordination
15A ![Molecules 21 01371 i011]() 15B
15B. The value of δ
P is shifted downfield proportionally to the increase in the phosphonium structure content, which in its turn depends on the solvent polarity. Substitution of electron withdrawing CF
3 group at C4 atom by hydrogen atom destabilizes the oxaphosphetane cycle and considerably reinforces the ionization of Р-С1 bond. The stability of 2-halogenoxaphosphetanes decreases, and positive values δ
Р and ionization of Р-С1 bond increases in the sequence of substituents at С4 atom: СF
3 > С
6Н
4F-4 >-C
6N
5 > С
6Н
4ОMе > H (
Table 4) [
25,
30,
31]. Chemical shifts of phosphorus in 2-bromo-1,2λ
5-oxaphosphetanes (δ
Р = +20–+75 ppm) were in the weaker fields relatively to chemical shifts of chloro-oxaphosphetanes. Evidently high positive values δ
Р of 2-bromoxaphosphetanes is undoubtedly explained by bigger, than in case of 2-chlorooxaphosphetanes, contribution of phosphonium forms
15B in the equilibrium 15
A ![Molecules 21 01371 i011]()
15
B.
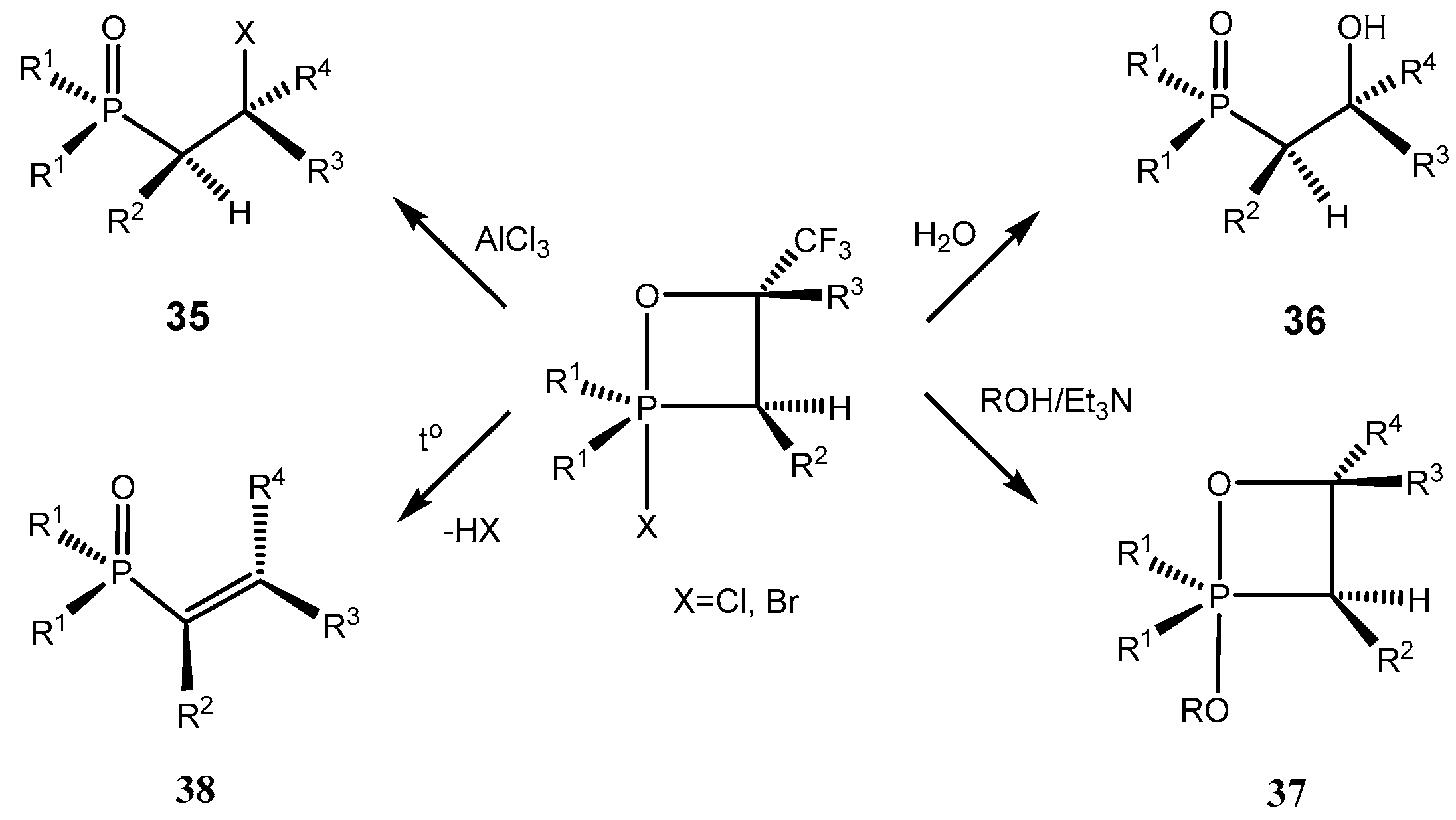
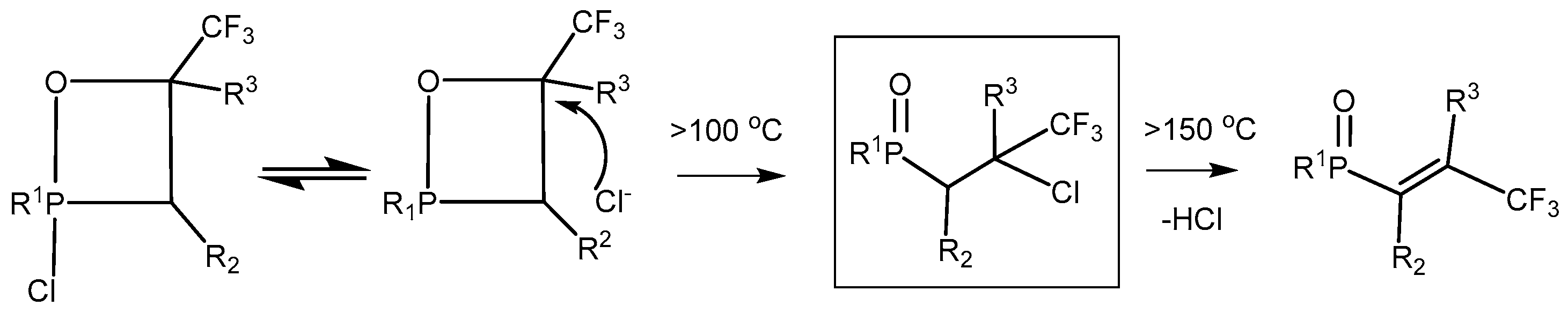
2-Chloro-1,2λ
5-oxaphosphetanes enter into a number of interesting chemical conversions. Thus, 2- chloro-1,2λ
5-oxaphosphetanes as a result of [
1,
3]-migration of chlorine atom to carbon atom underwent 2-chlorooxaphosphetane-2-chlorooxyphosphine oxide rearrangement to convert into the 2-chlorooxyphosphine oxides
35. Thermal stability of 2-chloro-1,2λ
5-oxaphosphetanes was decreased in case of oxaphosphetanes which not contain at С4 atom strong acceptor substituents, which rearranged into chloroalkylphosphonates at room temperature (
Table 3). At heating 2-chloralkylphosphine oxides yield vinylphosphine oxides
38. Hydrolysis of 2-chloro- and bromo-oxaphosphetanes led to the formation of 2-hydroxyalkylphosphine oxides
36 (
Table 5)
. The chlorine atom of 2-сhloro-1,2λ
5-oxaphosphetanes is easily substituted on methoxy- or phenoxy groups by reaction with methanol or phenol in the presence of triethylamine with formation of 2-alkoxyoxaphosphetanes
37, which at heating were converted into alkenes (
Scheme 23,
Table 1, entries 14, 15) [
27,
42].
The 2-chloro-1,2λ
5-oxaphosphetanes not containing electron accepting groups at C-3 are unstable and rearrange easily into 2-cloroalkylphosphonates at temperature below +20 °C. The oxaphophetanes bearing at C4 electronegative CF
3 groups convert into vinylphosphonates with elimination of hydrogen chloride at heading up to 150–160 °C. 2-Bromooxaphosphetanes are less stable than 2-chlorooxaphosphetanes and are converted into vinylphosphine oxides at room temperature. At heating (160–190 °С) 2-chloralkylphosphine oxides eliminate HCl to convert into vinylphosphine oxides (
Scheme 24) [
27,
28,
29,
42].
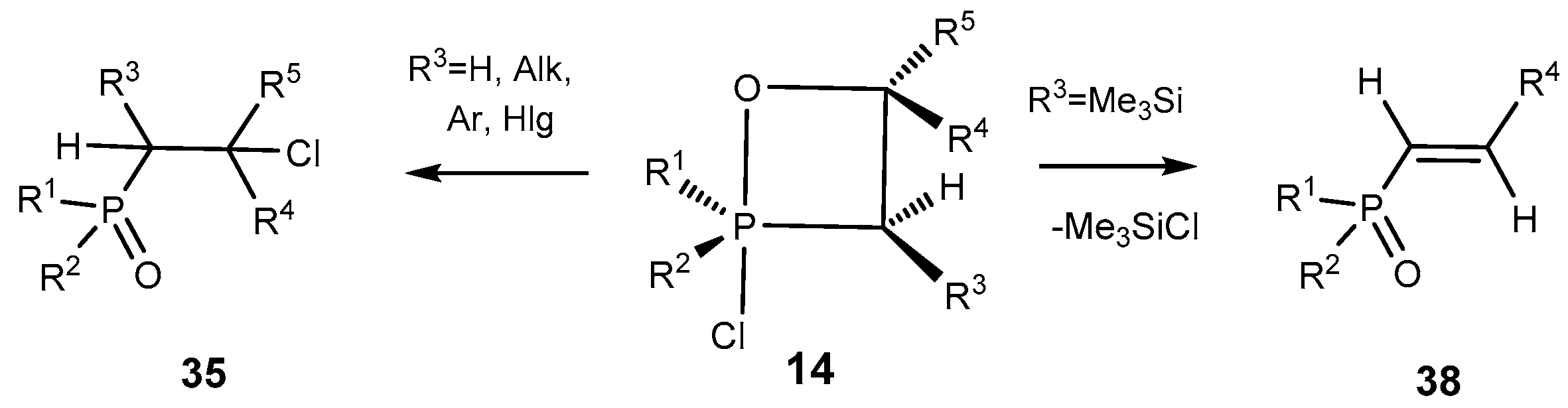
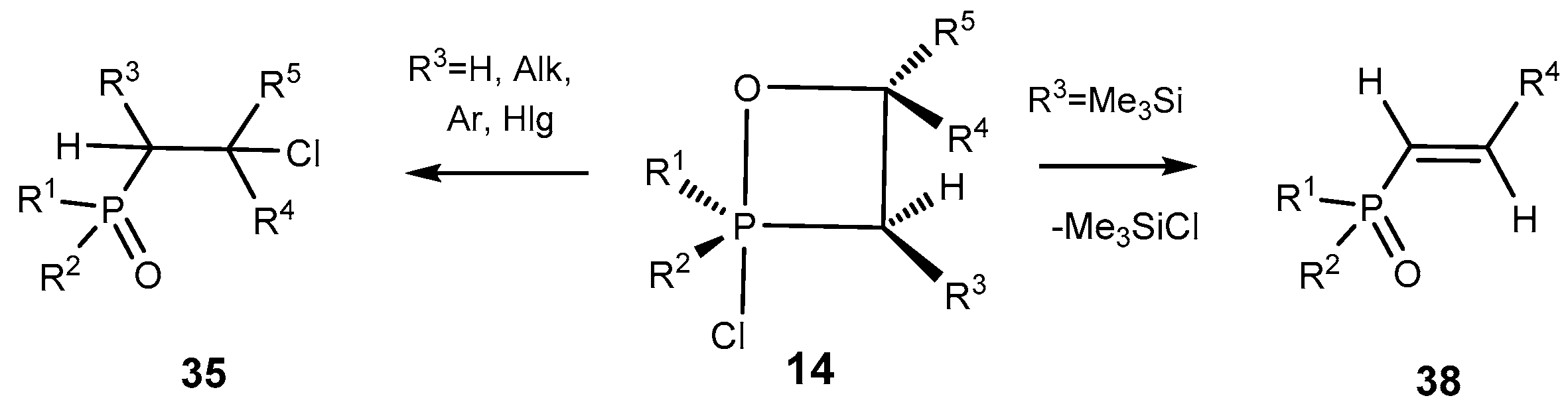
The oxaphosphetanes bearing a trimethylsilyl group at C-3 atom eliminate the trimethylchlorosilane moiety and convert into the phosphorylated alkenes
38. The conversion of oxaphosphetanes into alkenephosphonates proceeds at room temperature slowly, and at heating faster to give the alkenes
38 in good yields (
Scheme 25). The phosphorylated alkenes
38 were purified by distillation under vacuum and isolated as pure compounds. The reaction of ylides
14 with aldehydes is regioselective. For example, the oxaphosphetanes obtained by reaction of silylated
P-chloroylide
5 with terephthalic aldehyde depending on a ratio of initial reactants led to the formation of 1,4-bis-vinylphosphonobenzene or phosphonovinylbenzaldehyde, that represent interest as reactants for organic synthesis (See
Table 6, entries 10, 11) [
13,
18,
19,
20].
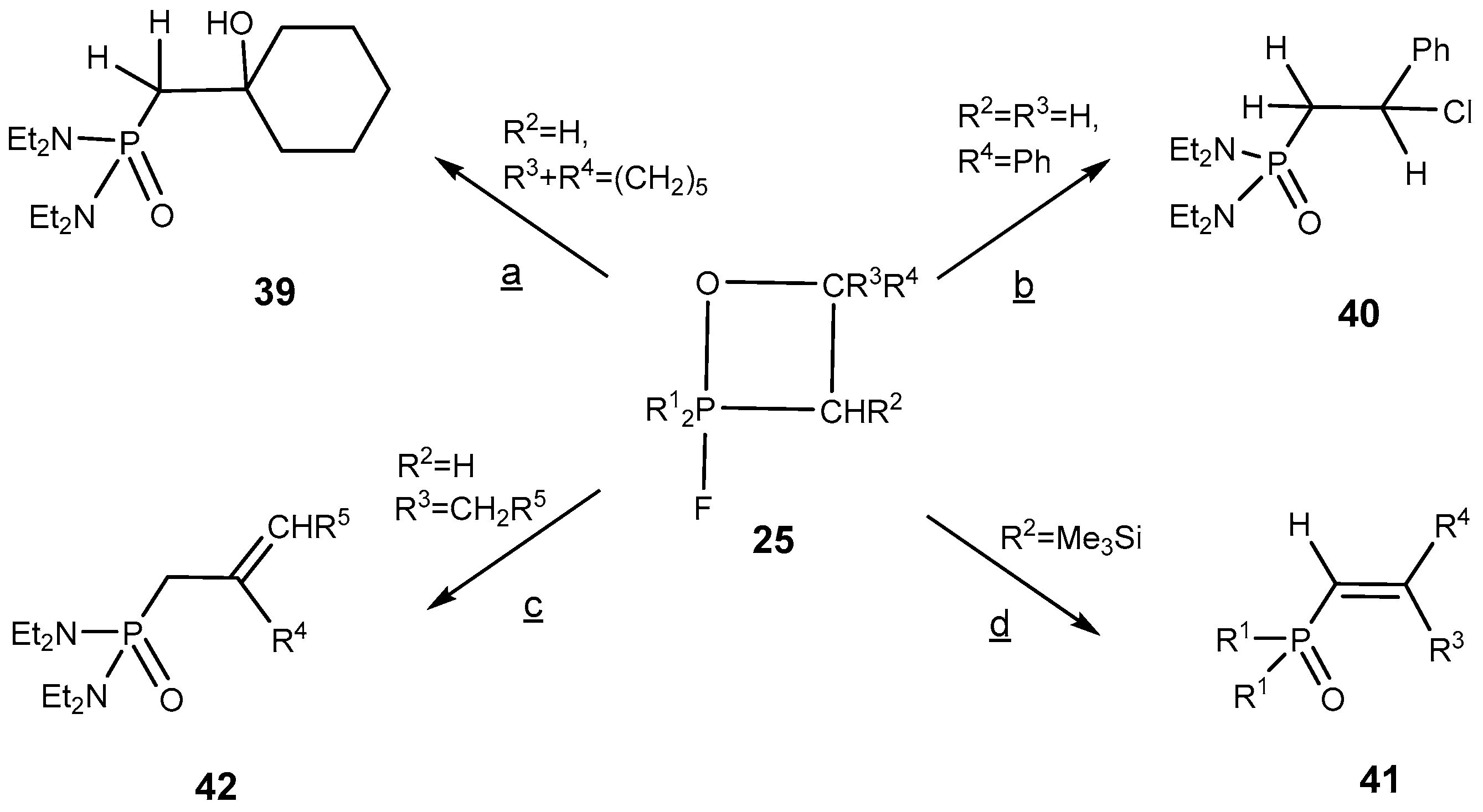
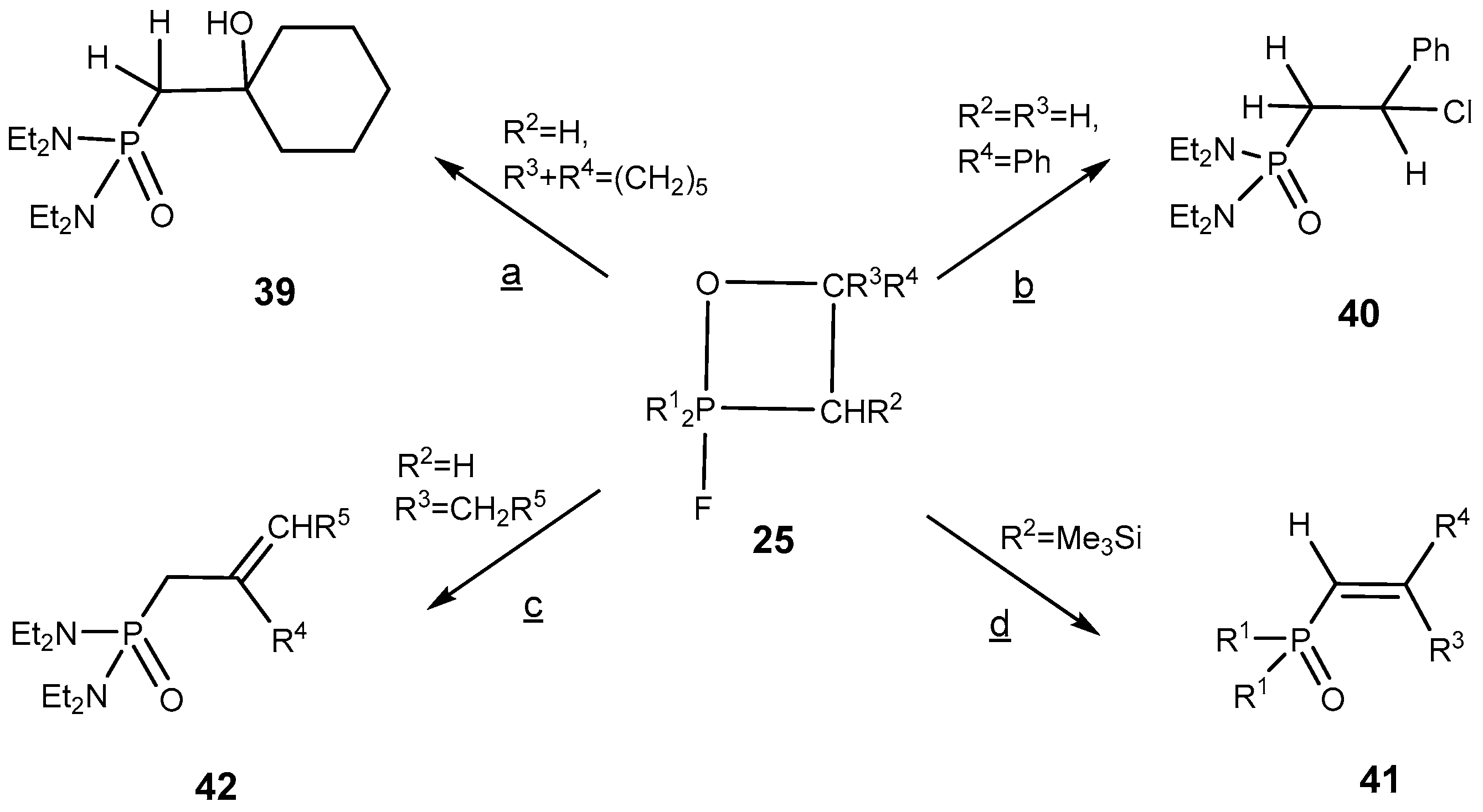
The 2-fluoro-1,2λ
5-oxaphosphetanes enter readily into a number of interesting organophosphorus compounds proceeding without Р—С bond cleavage [
26]. Thus, the treatment of 2-fluorooxaphosphetanes
25 with ether solution of HCl led to the formation of 2-chloro-oxaphosphetanes
40, which was isolated in good yield. At heating the 2-fluorooxaphosphetanes
25, in contrast to triphenyloxaphosphetanes, convert into phosphorylated alkenes: vinylphosphonates or allylphosphonates. The direction of reaction depended on substituents R
2 and R
3 at C-3 and C-4 of oxaphosphetane cycle. The 2-fluorooxaphosphetanes bearing at C-3 R
2 = H, Alk, Аr, and at C-4 R
3 = Alkyl eliminated HF to convert into the allylphosphonates
42 [
39,
44,
45]. The reaction was catalyzed by boron trifluoride etherate. At the same time the 2-fluorooxaphosphetanes
25 bearing at C-3 substituent R = Me
3Si eliminated Me
3SiF and afforded the vinylphosphonates
41 (
Scheme 26,
Table 7). This reaction represent a convenient method for the preparation of phosphorylated alkenes that are versatile building blocks for organic synthesis [
45,
46,
47,
48]. 2-Fluoro-1,2λ
5-oxaphosphetanes containing two alkyl groups at C3 as well eliminated hydrogen fluoride and afforded the allylphosphonates
42 (
Scheme 26,
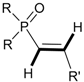
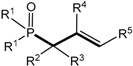
Table 8).
The reaction occurs by the 1,4-elimination as shown in
Scheme 26 [
25]. Opposite to 2,2,2-triphenyloxaphosphetanes, upon heating, the decyclization of 2-fluorooxaphosphetanes led to the formation of phosphorylated alkenes
41,
42.
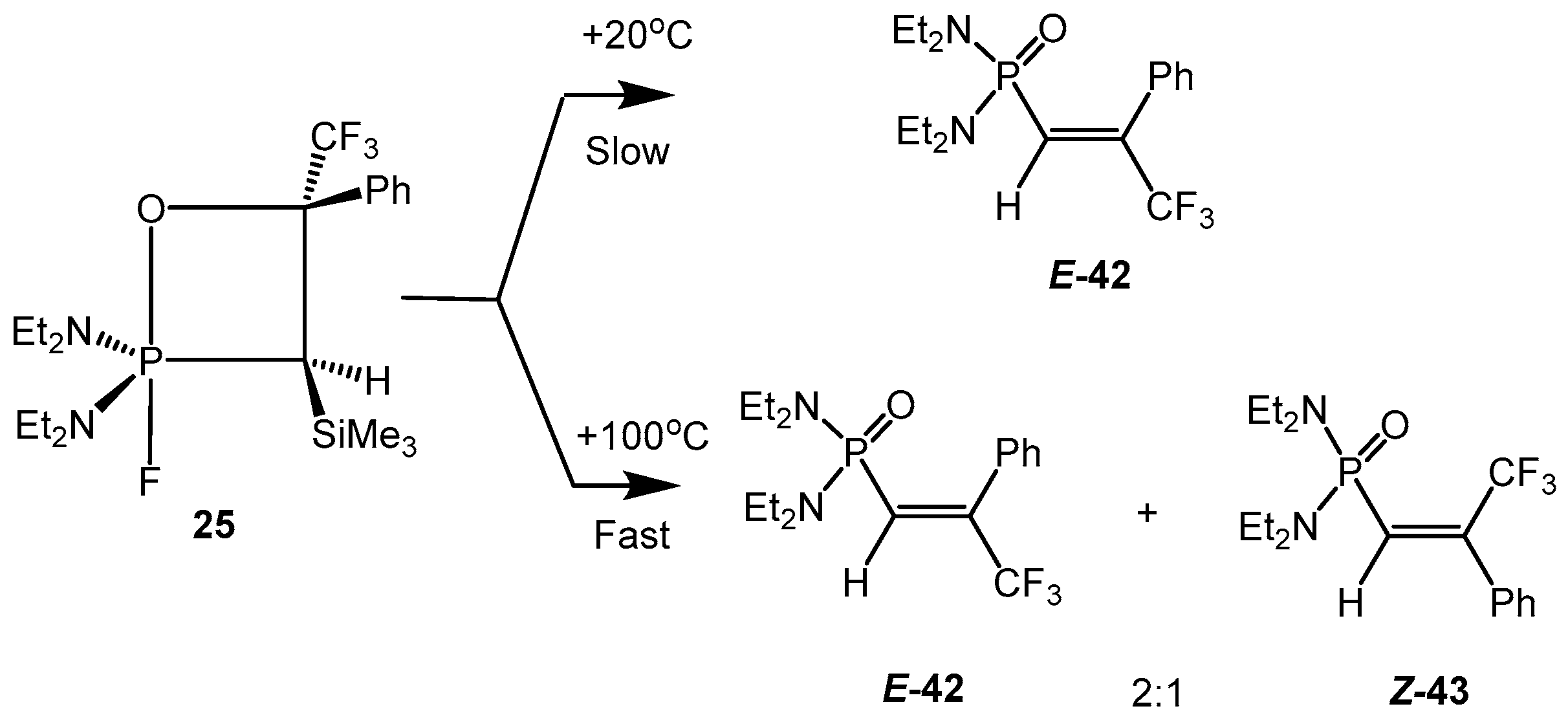
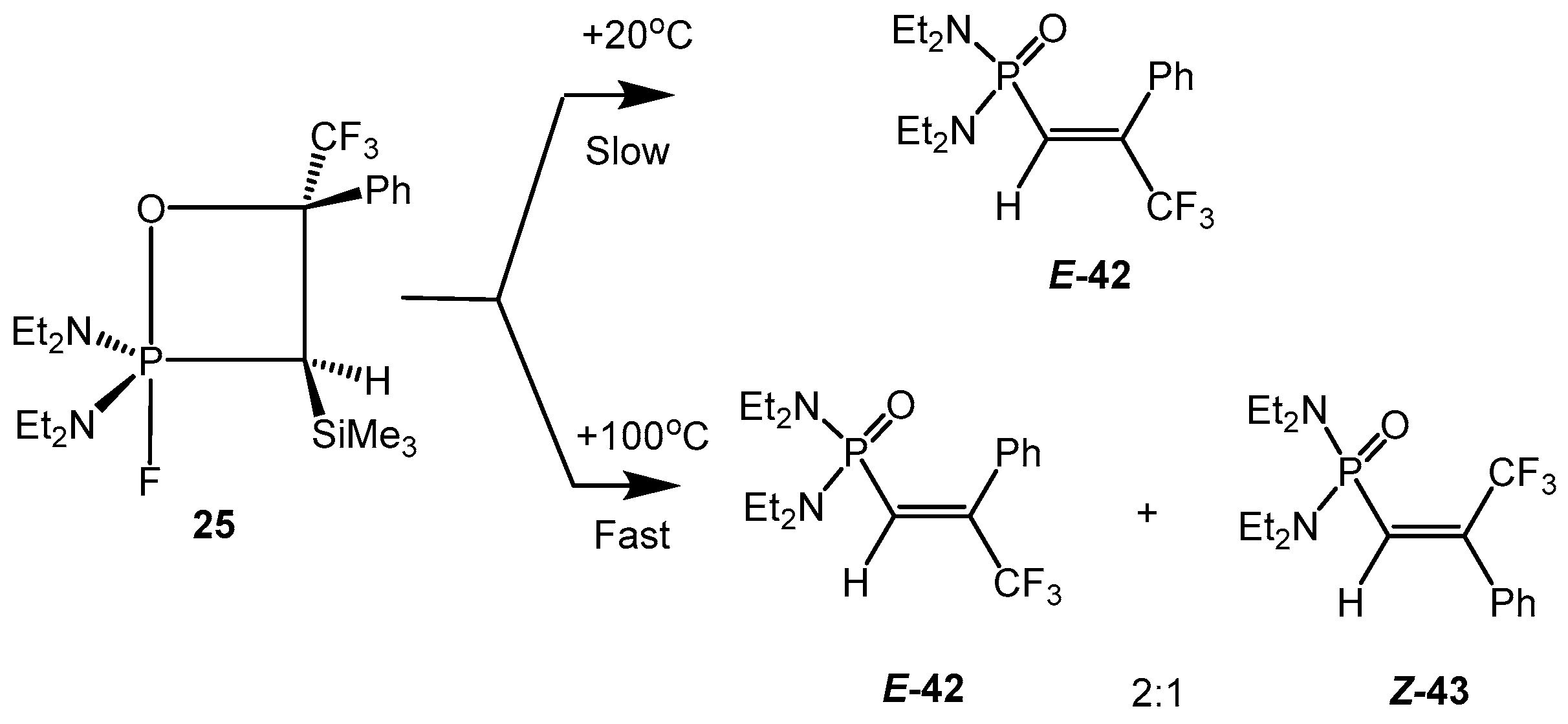
At heating to +100 °C the 2-fluoro-3-silyloxaphosphetanes
25, bearing CF
3 group at С-4, afforded a mixture of
E- and
Z-vinylphosphonates
29,
30 in the ratio 2:1 with elimination of Me
3SiF. However the slow conversion of oxaphosphetane at +20 °С during several days provided almost pure vinylphosphonates
E-
29, containing only 2%–3% of
Z-isomer (
Scheme 27) [
26]. This effect was explained by formation of carbocation intermediate and rotation of substituents around the C-C bond (
Scheme 28).
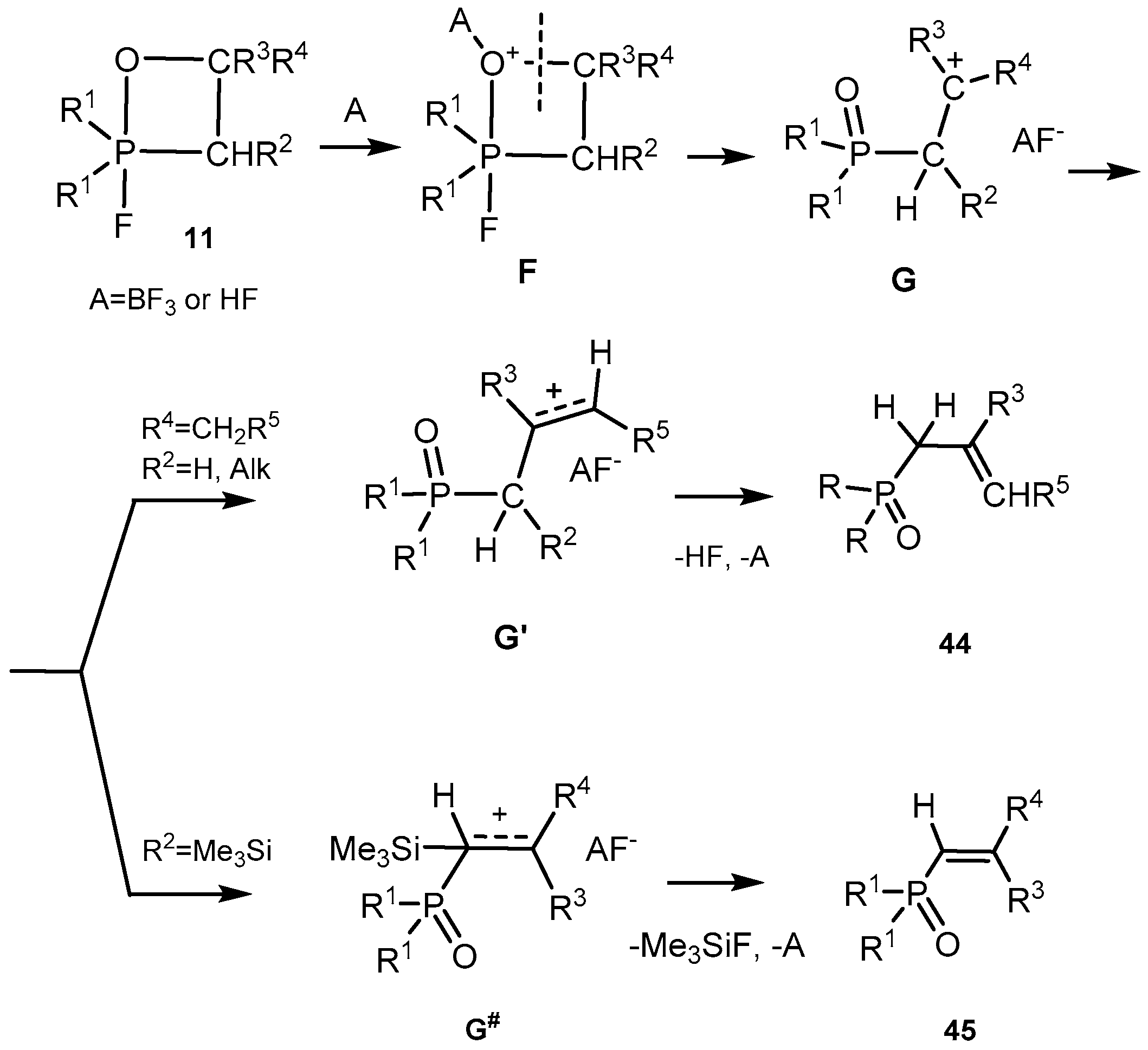
The conversion of 2-fluoro-1,2λ
5-oxaphosphetanes bearing alkyl groups at C-3 into allylphosphonates
42 represents an interesting example of 1,4-elimination as
Scheme 28 and
Table 8 show [
45]. The study of the reaction mechanism showed that Lewis and Broensted acids actively catalyze the conversion of 2-fluorooxaphosphetanes into allylphosphine oxides. The reaction is autocatalytic because the evolving hydrogen fluoride catalyzes the transition of 2-fluorooxaphosphetanes into allylphosphine oxides. The decomposition of protonated 2-fluorooxaphosphetanes leads to the formation of oxonium salts and carbocation intermediates under conditions of E
N1 elimination. We suppose that the 2-fluorooxaphosphetanes under condition of acid catalysis (with HF or BF
3) via the formation of an oxonium intermediate F, convert to carbocation intermediate
G which has a planar configuration [
46]. The removal of a proton from the carbocation intermediate
G depends on electronic effects of the substituents. Alkyl groups possessing the +
I-effect and the effect of hyperconjugation stabilize the positive charge and reduce the energy of intermediate
G formation. Therefore, the intermediate
G′ leading to allylphosphonates is energetically more favorable than this one that is converted into vinylphosphonates.
In addition, even in case when the initial 2-fluorooxaphosphetanes
25 exist as a mixture of
threo- and
erythro-diastereomers, they converted into pure
E-vinylphosphonates. Probably, this effect can be explained by rotation of substituents around the C-C bond in carbocation intermediate G. However, the presence of Me
3Si at С-3 in
G” leads to the elimination of Me
3SiF, which has a high energy of formation, that creates a preference for the formation of vinylphosphonates
45 (
Scheme 28) [
26,
40].
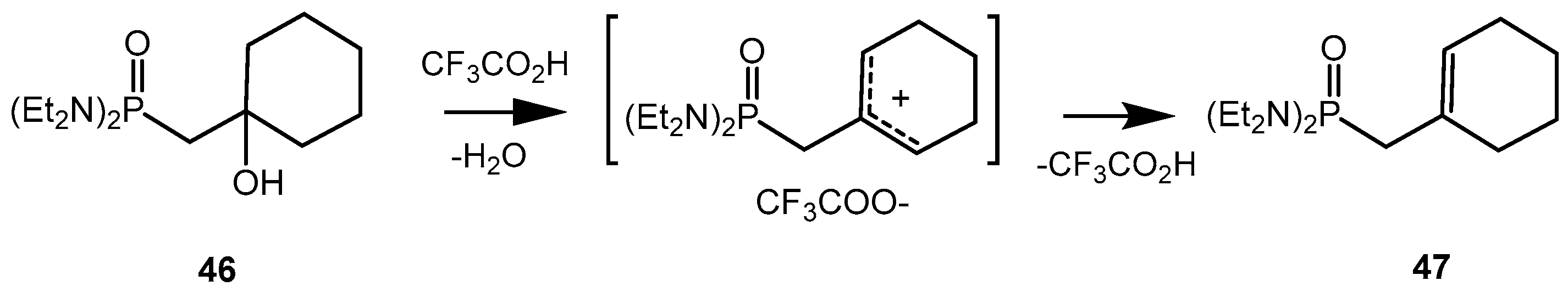
The formation of carbocation intermediate
G was experimentally confirmed (
Scheme 29). The treatment of 2-hydroxyphosphonate
46 with trifluoracetic acid and refluxing for several hours, generated the carbocation intermediate, as a result of acid-catalyzed dehydration of alcohol.
After that the carbocation intermediate is converted into allylphosphonate
47, which is identical to the one obtained from 2-fluorooxaphosphetane
25 [
26,
40,
45].
The reaction of
P-fluoroylides with carbonyl compounds is a convenient method for the synthesis of allylphosphonates having various applications in the synthesis of naturally occurring compounds (
Scheme 30) [
45,
46,
47]. In this case, the carbonyl compounds can be used twice for the constructing of diene structures: first in reaction with
P-fluoroylide and then in the Wittig reaction with allylphosphonate. This reaction was used for preparing analogs of the juvenile hormone.
The cycloadducts of
P-fluoroylides with carbon dioxide or with carbon disulfide were isolated as colorless liquids or crystalline substances. Their structure was confirmed by the NMR spectra. Upon gentle heating or at room temperature these cycloadducts
32–
35 (
Table 2, entries 21, 22) are converted to phosphorylated ketenes or thioketenes
48 (
Scheme 31). Stable [2+2]-cycloadducts of 2-fluoroylides with alkyl and aryl isothiocyanates were also synthesized and converted into phosphorylated ketenimines. The reaction of
P-fluoroylides with carbon disulfide gives unstable cycloadducts that at temperatures above 0 °C converted completely into phosphorylated thioketenes in high yields. Phosphorylated thioketenes are red liquids distillable in vacuum and susceptible for various transformations [
29,
36,
37]. The same phosphorylated ketenes or thioketenes
29 were prepared by reaction of
P-cloroylides corresponding with CO
2 and CS
2 (See
Scheme 31 and
Table 9) [
37,
38,
39].


 15B. The value of δP is shifted downfield proportionally to the increase in the phosphonium structure content, which in its turn depends on the solvent polarity. Substitution of electron withdrawing CF3 group at C4 atom by hydrogen atom destabilizes the oxaphosphetane cycle and considerably reinforces the ionization of Р-С1 bond. The stability of 2-halogenoxaphosphetanes decreases, and positive values δР and ionization of Р-С1 bond increases in the sequence of substituents at С4 atom: СF3 > С6Н4F-4 >-C6N5 > С6Н4ОMе > H (Table 4) [25,30,31]. Chemical shifts of phosphorus in 2-bromo-1,2λ5-oxaphosphetanes (δР = +20–+75 ppm) were in the weaker fields relatively to chemical shifts of chloro-oxaphosphetanes. Evidently high positive values δР of 2-bromoxaphosphetanes is undoubtedly explained by bigger, than in case of 2-chlorooxaphosphetanes, contribution of phosphonium forms 15B in the equilibrium 15A
15B. The value of δP is shifted downfield proportionally to the increase in the phosphonium structure content, which in its turn depends on the solvent polarity. Substitution of electron withdrawing CF3 group at C4 atom by hydrogen atom destabilizes the oxaphosphetane cycle and considerably reinforces the ionization of Р-С1 bond. The stability of 2-halogenoxaphosphetanes decreases, and positive values δР and ionization of Р-С1 bond increases in the sequence of substituents at С4 atom: СF3 > С6Н4F-4 >-C6N5 > С6Н4ОMе > H (Table 4) [25,30,31]. Chemical shifts of phosphorus in 2-bromo-1,2λ5-oxaphosphetanes (δР = +20–+75 ppm) were in the weaker fields relatively to chemical shifts of chloro-oxaphosphetanes. Evidently high positive values δР of 2-bromoxaphosphetanes is undoubtedly explained by bigger, than in case of 2-chlorooxaphosphetanes, contribution of phosphonium forms 15B in the equilibrium 15A 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}