
Three New Cytotoxic ent-Kaurane Diterpenes from Isodon excisoides

Abstract
:1. Introduction
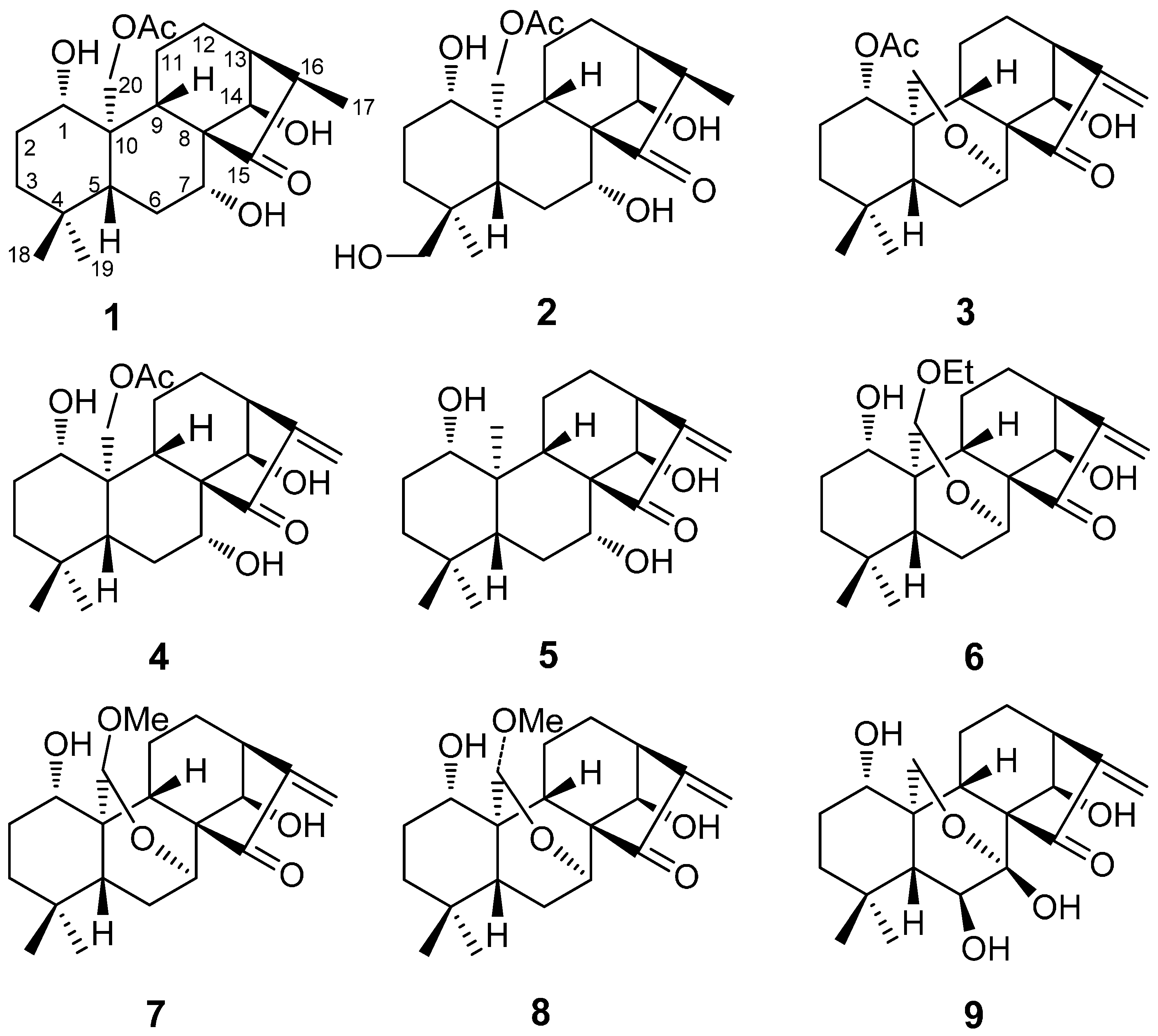
2. Results and Discussion
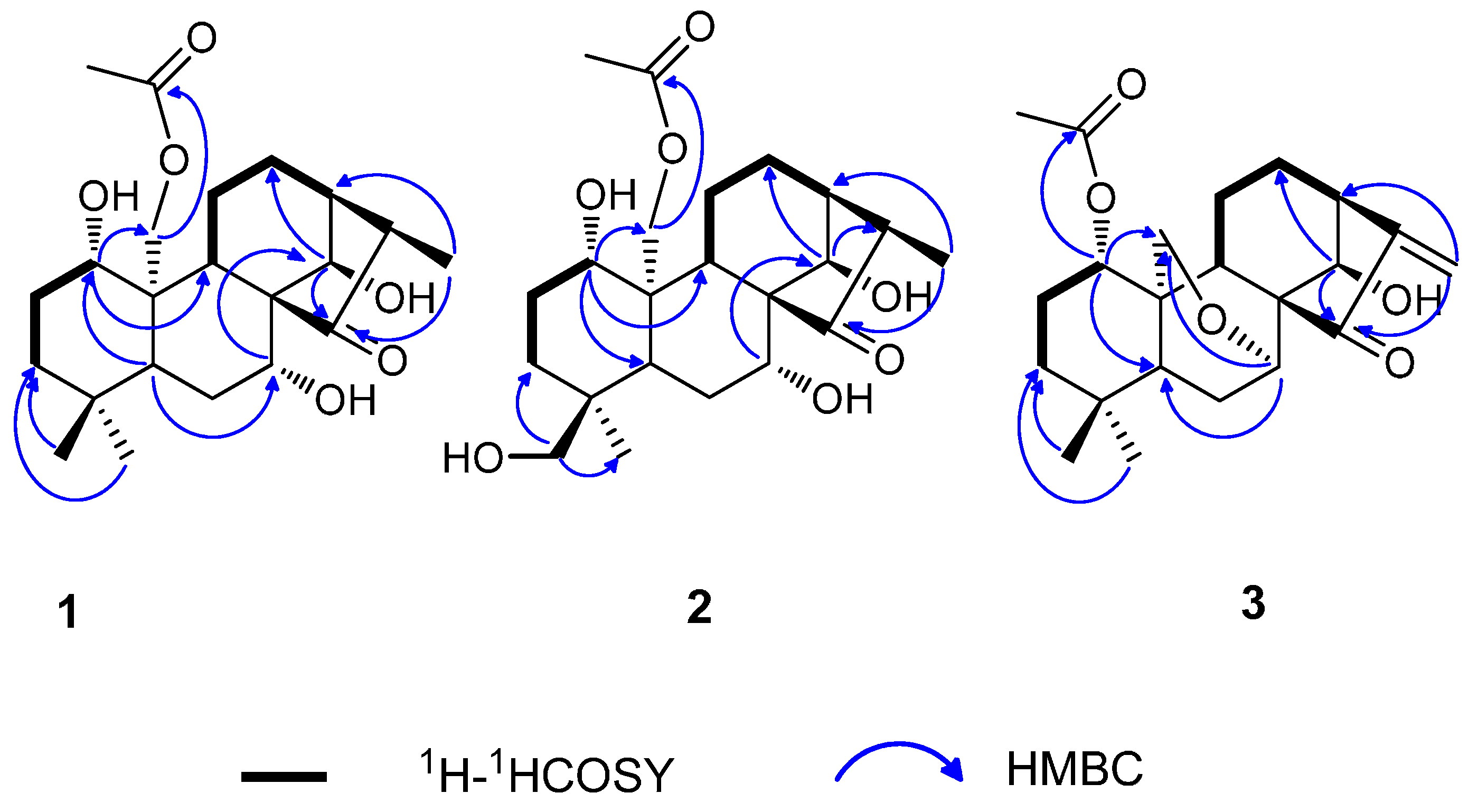
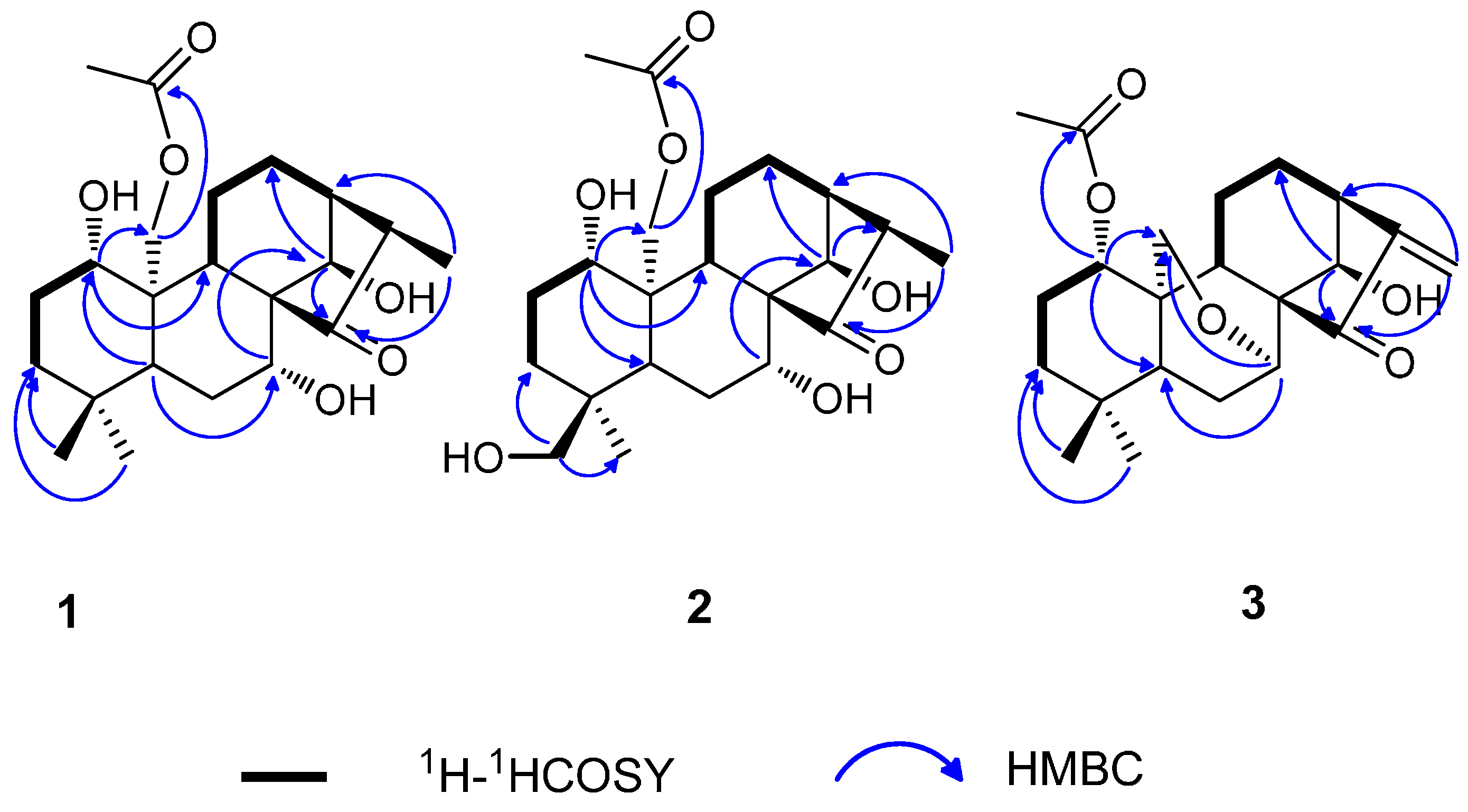
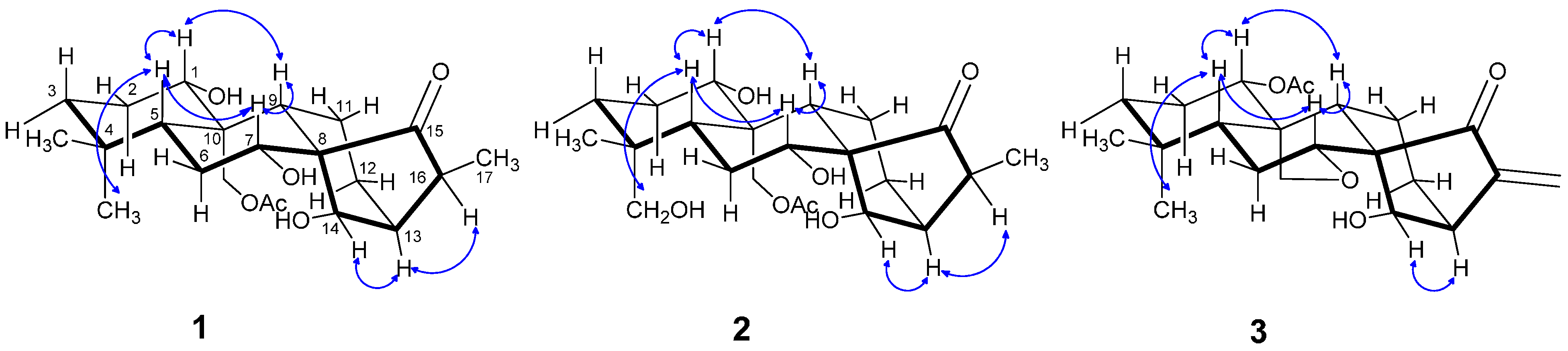
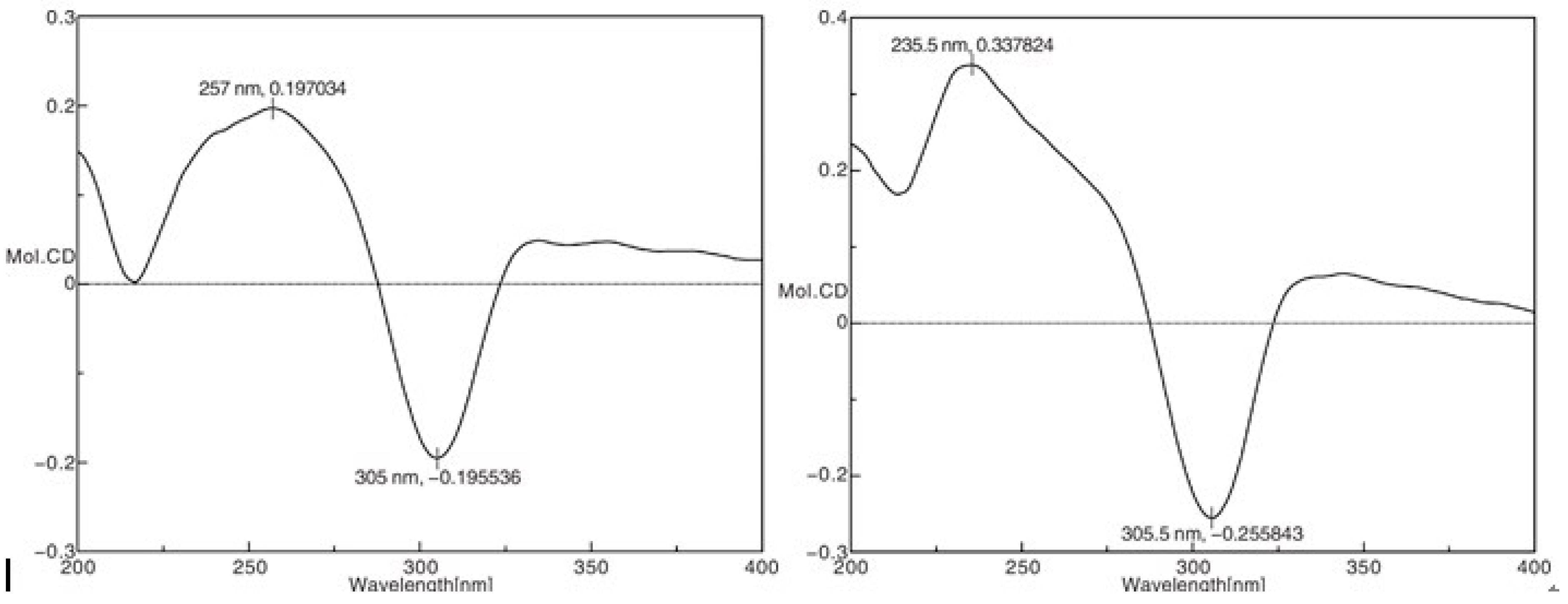
2.1. Structure Elucidation

{kind=link}
{kind=link}
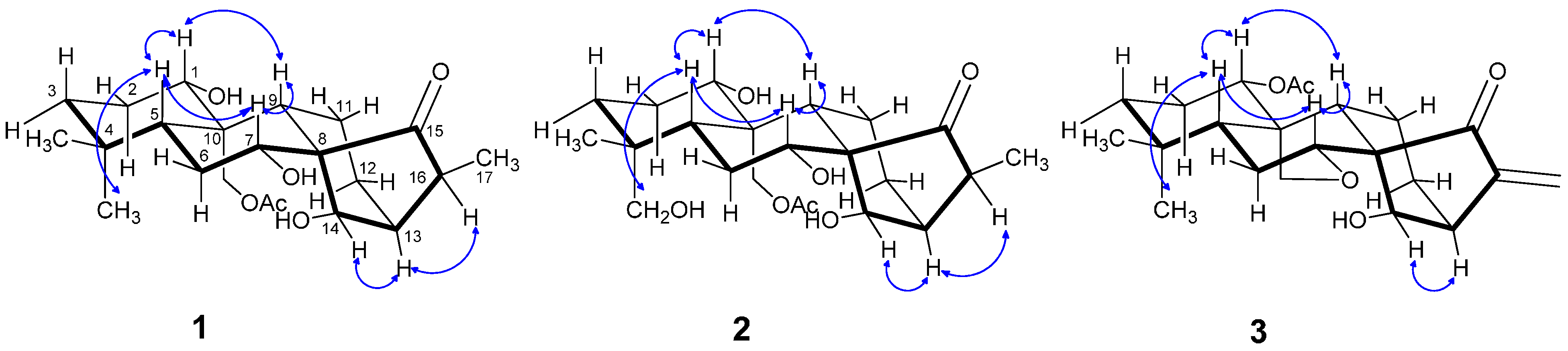{kind=link}
{kind=link}
| No. | 1 (In CDCl3) | 2 (In DMSO-d6 and D2O) | 3 (In CDCl3) | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | β 3.31, dd, (11.0, 4.6) | 81.6 | β 3.09, dd, (10.8, 4.5) | 80.5 | β 4.57, dd, (11.2, 5.3) | 76.6 |
| 2 | α 1.82, m | 30.5 | α 1.63, overlapped | 29.2 | α 1.69, overlapped | 25.1 |
| β 1.65, overlapped | β 1.53, overlapped | β 1.47, m | ||||
| 3 | α 1.47, dt, (13.8, 3.7) | 39.4 | α 1.53, overlapped | 32.9 | α 1.44, m | 37.9 |
| β 1.29, dt, (13.8, 4.4) | β 1.05, m | β 1.24, m | ||||
| 4 | – | 32.9 | – | 36.6 | – | 33.6 |
| 5 | β 0.98, dd, (11.6, 3.5) | 52.5 | β 1.21, d, (12.2) | 42.0 | β 1.33, ddd, (11.8, 7.4, 1.5) | 47.1 |
| 6 | 1.93, m | 28.6 | β 1.80, d, (12.2) | 28.6 | β 2.85, ddd, (14.0, 11.8, 1.9) | 25.1 |
| α 1.63, overlapped | α 1.84, overlapped | |||||
| 7 | β 4.20, dd (11.4, 6.0) | 75.4 | β 3.77, dd, (11.8, 4.3) | 73.8 | β 3.95, dd, (3.8, 1.9) | 64.5 |
| 8 | – | 61.0 | – | 59.8 | – | 58.7 |
| 9 | β 1.55, d, (8.8) | 55.5 | β 1.37, d, (8.6) | 55.7 | β 1.69, overlapped | 50.8 |
| 10 | – | 46.0 | – | 44.1 | – | 39.2 |
| 11 | α 2.77, q, (6.1) | 19.9 | α 2.82, dd, (15.9, 5.6) | 19.3 | α 1.84, overlapped | 17.8 |
| β 1.20, m | β 0.98, m | β 1.15, q, (6.5) | ||||
| 12 | α 1.88, m | 24.2 | α 1.63, overlapped | 24.3 | α 2.42, dt, (14.1, 9.0) | 30.9 |
| β 1.65, overlapped | β 1.53, overlapped | β 1.54, m | ||||
| 13 | α 2.42, m | 42.4 | α 2.24, m | 42.5 | α 2.99, br d, (9.9) | 42.0 |
| 14 | α 4.82, d, (1.1) | 76.1 | α 4.72, br. s | 75.2 | α 4.61, br s | 71.5 |
| 15 | – | 222.8 | – | 221.2 | – | 204.5 |
| 16 | 2.88, m | 43.2 | 2.67, m | 44.6 | – | 151.1 |
| 17 | 1.12, d, (7.1) | 9.2 | 1.00, d, (7.1) | 9.1 | 6.00, br s; 5.38, br.s | 117.5 |
| 18 | 0.91, s | 33.3 | 3.19, d, (10.6) | 69.5 | 0.88, s | 31.4 |
| 2.84, d, (10.6) | ||||||
| 19 | 0.85, s | 21.5 | 0.62, s | 17.3 | 1.08, s | 20.3 |
| 20 | α 4.74, d, (13.2) | 63.7 | α 4.35, d, (13.3) | 64.1 | α 4.09, dd, (10.3, 1.5) | 61.0 |
| β 4.37, d, (13.2) | β 4.28, d, (13.3) | β 4.03, dd, (10.3, 1.6) | ||||
| OAc | – | 170.8 | – | 170.2 | – | 170.1 |
| OAc | 2.13, s | 21.5 | 2.07, s | 21.2 | 1.95, s | 21.5 |
2.2. Cytotoxicity Assay
| Sample | IC50 (μM) | ||||
|---|---|---|---|---|---|
| HCT-116 | HepG2 | BGC-823 | NCI-H1650 | A2780 | |
| 1 | 2.94 ± 0.06 | 3.07 ± 0.02 | 5.59 ± 0.19 | >10 | 6.33 ± 0.34 |
| 2 | 2.45 ± 0.12 | 3.21 ± 0.09 | 4.17 ± 0.25 | >10 | 5.61 ± 0.19 |
| 3 | 2.13 ± 0.81 | 2.20 ± 1.12 | >10 | 5.68 ± 0.73 | 1.09 ± 0.13 |
| 4 | 1.77 ± 0.22 | 1.54 ± 0.32 | 1.31 ± 0.76 | 2.07 ± 0.36 | 1.42 ± 0.20 |
| 5 | 4.85 ± 0.33 | 7.88 ± 1.02 | 2.99 ± 0.76 | >10 | 1.56 ± 0.34 |
| 6 | 4.81 ± 0.01 | 7.45 ± 0.22 | 5.17 ± 0.61 | >10 | 1.98 ± 0.13 |
| 7 | 4.87 ± 1.12 | 1.09 ± 0.06 | 5.31 ± 0.14 | 2.58 ± 0.23 | 1.44 ± 0.07 |
| 8 | 4.55 ± 0.02 | 7.35 ± 0.48 | 4.97 ± 0.84 | 2.38 ± 0.31 | 4.88 ± 0.22 |
| 9 | 4.85 ± 0.33 | 7.88 ± 1.02 | 2.99 ± 0.76 | >10 | 1.56 ± 0.34 |
| DDP | 7.81 ± 0.14 | >10 | 8.56 ± 1.05 | >10 | 8.65 ± 0.59 |
| Taxol | (3.07 ± 0.12) × 10−2 | (1.31 ± 0.44) × 10−2 | (4.06 ± 0.35) × 10−3 | (2.61 ± 1.02) × 10−2 | (7.13 ± 0.51) × 10−3 |
2.3. Analysis of Structure-Activity Relationships
3. Experimental Section
3.1. General Information
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sun, H.D.; Xu, Y.L.; Jiang, B. Diterpenoids from Isodon Species; Science Press: Beijing, China, 2001; p. 2. [Google Scholar]
- Li, H.; PU, J.X.; Li, J. Diterpenoids chemodiversity of the genus Isodon spach from Lamiaceae. Plant Divers. Resour. 2013, 35, 81–88. [Google Scholar]
- Jiao, K.; Li, H.Y.; Zhang, P.; Pi, H.F.; Ruan, H.L.; Wu, J.Z. Two new ent-kaurane diterpenoids from the aerial parts of Isodon excisoides. Chin. Chem. Lett. 2014, 25, 131–133. [Google Scholar] [CrossRef]
- Wang, T.; Tang, F.M.; Zhang, Y.H.; Chen, Z. A natural diterpenoid kamebacetal with anti-tumor activity. J. Mol. Struct. 2010, 97, 317–322. [Google Scholar] [CrossRef]
- Wang, Y.J.; Kim, J.Y.; Dong, S.P.; Wang, K.Y. Study on the immunomodulation effect of Isodon japonicus extract via splenocyte function and NK anti-tumor activity. Int. J. Mol. Sci. 2012, 13, 4880–4888. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.X.; Zhang, W.; Li, J.C.; Liu, N. Chemical constituents of flowers and fruits of Rabdosia excisa. Chin. J. Nat. Med. 2012, 10, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.S.; Yamashiro, A.; Dohzono, I.M.; Maki, M. Development of microsatellite markers for Isodon Longitubus (lamiaceae). Appl. Plant Sci. 2013, 1, 128–130. [Google Scholar]
- Liao, Y.J.; Bai, H.Y.; Li, Z.H.; Zou, J. Longikaurin A, a natural ent-kaurane, induces G2/M phase arrest via downregulation of Skp2 and apoptosis induction through ROS/JNK/c-Jun pathway in hepatocellular carcinoma cells. Cell. Death Dis. 2014, 5, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.M.; Cheng, P.Y. Structure elucidation of a bis-ent-kauranoid isolated from Isodon pharicus. Acta Pharm. Sin. 1994, 29, 532–538. [Google Scholar]
- Wang, Z.M.; Cheng, P.Y. Structure elucidation of a new bis-ent-kaurane compound, isodopharicin E, isolated from Isodon pharicus (Prain) Murata. Chin. Chem Lett. 1991, 2B, 847. [Google Scholar]
- Wang, Z.M.; Feng, H.; Zhang, Q.; Liu, F.S.; Jin, W.S.; Mu, M.; Fang, Q.H.; Kong, M.; He, W.Y. The structures elucidation of isodopharicin D and F. Acta Pharm. Sin. 1998, 33, 207–211. [Google Scholar]
- Jiang, H.L.; Zhu, M.; Chen, Y.; Li, W.T. Simultaneous determination of glaucocalyxin A, glaucocalyxin B and glaucocalyxin D in weifuchun tablets by HPLC. Chin. Tradit. Patent Med. 2013, 35, 86–89. [Google Scholar]
- Wu, M.J.; Fu, J.G.; Dong, J.J.; Li, J.C.; Zhao, T.Z. Advances on the plants of Isodon of the chemical constituents and pharmacological effects. World Latest Med. Inf. 2004, 3, 1214–1218. [Google Scholar]
- Ding, B.Z.; Wang, S.Y.; Gao, Z.Y. Flora of Henan; Henan People Publishing Press: Zhengzhou, China, 1981; Volume 3, p. 76. [Google Scholar]
- Li, H.Y.; Jiao, K.; Zhang, P.; Gong, S.Z.; Sun, Y.; Wu, J.Z. Chemical constituents from Isodon excisoides. Chin. Tradit. Herb. Drug 2014, 45, 154–160. [Google Scholar]
- Wang, Y.X.; Zhu, L.L.; He, Z.A.; Zhang, J.X. New diterpenoids from Isodon excisoides. Chin. Chem. Lett. 2010, 21, 610–612. [Google Scholar] [CrossRef]
- Li, J.C.; Liu, C.J.; An, X.Z.; Sun, H.D.; Lin, Z.W. The structure of Henryin. Acta Bot. Yunnan 1984, 6, 453–456. [Google Scholar]
- Takeda, Y.; Ichihara, T.; Takaishi, Y. Structure elucidation of new diterpenoids Rabdosia umbrosa var. leucantha f. Kameba. J. Chem. Soc. Perkin Trans. 1987, 1, 2403–2409. [Google Scholar] [CrossRef]
- Lan, D.; Liu, G.A.; Yang, D.J.; Wang, H.; Wang, L.; Sun, K. Cytotoxic ent-kaurane diterpenoids from Isodon weisiensis. Pharmazie 2005, 60, 458–460. [Google Scholar]
- Wang, X.R.; Wang, Z.Q.; Dong, J.G. Chemical structures of Reniformin A, B and C. Acta Bot. Sin. 1986, 28, 292–298. [Google Scholar]
- Sun, H.D.; Li, J.C. Excisanin A and B, new diterpenoids from Rabdosia excisa. Chem. Lett. 1981, 753–756. [Google Scholar]
- Zhang, H.B.; PU, J.X.; Wang, Y.Y.; He, F.; Zhao, Y.; Li, X.N.; Luo, X.; Xiao, W.L.; Li, Y.; Sun, H.D. Four New ent-Kauranoids from Isodon rubescens var. lushanensis and Data Reassignment of Dayecrystal B. Chem. Pharm. Bull. 2010, 58, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.M.; Kuo, C.Y.; Lin, C.Y.; Hung, M.F.; Shen, J.J.; Wang, T.L. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules 2014, 19, 3327–3344. [Google Scholar] [CrossRef] [PubMed]
- Zhan, R.; Li, X.N.; Du, X.; Wang, W.G.; Dong, K.; Su, J.; Li, Y.; Pu, J.X.; Sun, H.D. ent-Atisane and ent-kaurane diterpenoids from Isodon rosthornii. Fitoterapia 2013, 88, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Y.; Zhan, R.; Wang, W.G.; Jiang, H.Y.; Du, X.; Li, X.N.; Li, Y.; Pu, J.X.; Sun, H.D. Cytotoxic ent-kaurane diterpenoids from Isodon wikstroemioides. J. Nat. Prod. 2014, 77, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Gui, Z.; Li, S.; Liu, X.; Ji, A.; Xu, B.; Xu, J. Oridonin alters the expression profiles of microRNAs in BxPC-3 human pancreatic cancer cells. BMC Complement. Altern. Med. 2015, 115, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Wang, S.; Gao, Y.; Zhang, X.; Lu, C. Oridonin phosphate-induced autophagy effectively enhances cell apoptosis of human breast cancer cells. Med. Oncol. 2015, 32, 365. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Xie, H.H.; Hao, J.; Jiang, Y.M.; Wei, X.Y. Eudesmane sesquiterpene glucosides from lycheeseed and their cytotoxic activity. Food Chem. 2010, 123, 1123–1126. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–3 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, L.-P.; Li, C.; Yang, H.-Z.; Lu, Y.-Q.; Yu, H.-Y.; Gao, H.-M.; Wang, Z.-M. Three New Cytotoxic ent-Kaurane Diterpenes from Isodon excisoides. Molecules 2015, 20, 17544-17556. https://doi.org/10.3390/molecules200917544
Dai L-P, Li C, Yang H-Z, Lu Y-Q, Yu H-Y, Gao H-M, Wang Z-M. Three New Cytotoxic ent-Kaurane Diterpenes from Isodon excisoides. Molecules. 2015; 20(9):17544-17556. https://doi.org/10.3390/molecules200917544
Chicago/Turabian StyleDai, Li-Ping, Chun Li, Han-Ze Yang, Yan-Qing Lu, Hong-Yan Yu, Hui-Min Gao, and Zhi-Min Wang. 2015. "Three New Cytotoxic ent-Kaurane Diterpenes from Isodon excisoides" Molecules 20, no. 9: 17544-17556. https://doi.org/10.3390/molecules200917544
APA StyleDai, L.-P., Li, C., Yang, H.-Z., Lu, Y.-Q., Yu, H.-Y., Gao, H.-M., & Wang, Z.-M. (2015). Three New Cytotoxic ent-Kaurane Diterpenes from Isodon excisoides. Molecules, 20(9), 17544-17556. https://doi.org/10.3390/molecules200917544