
Asymmetric Synthesis of 1,3-Oxazolidine Derivatives with Multi-Component Reaction and Research of Kinetic Resolution

Abstract
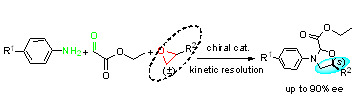
:
1. Introduction
2. Results and Discussion
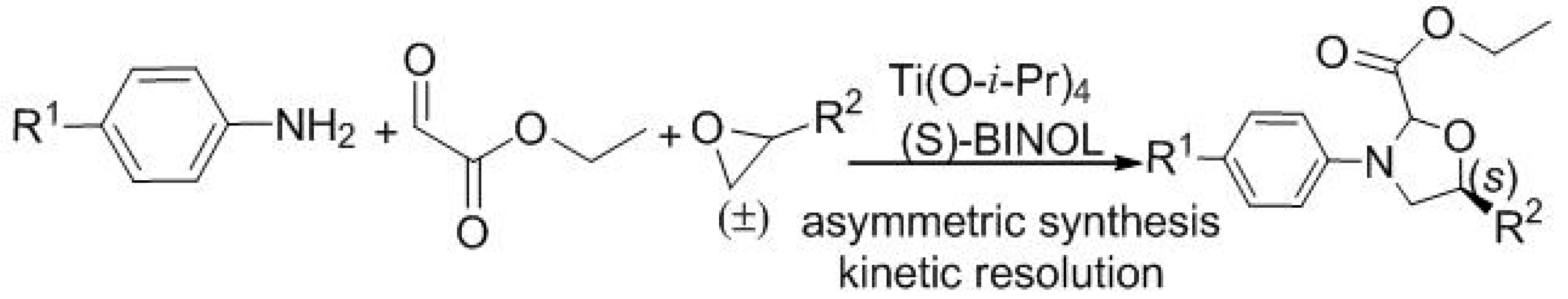
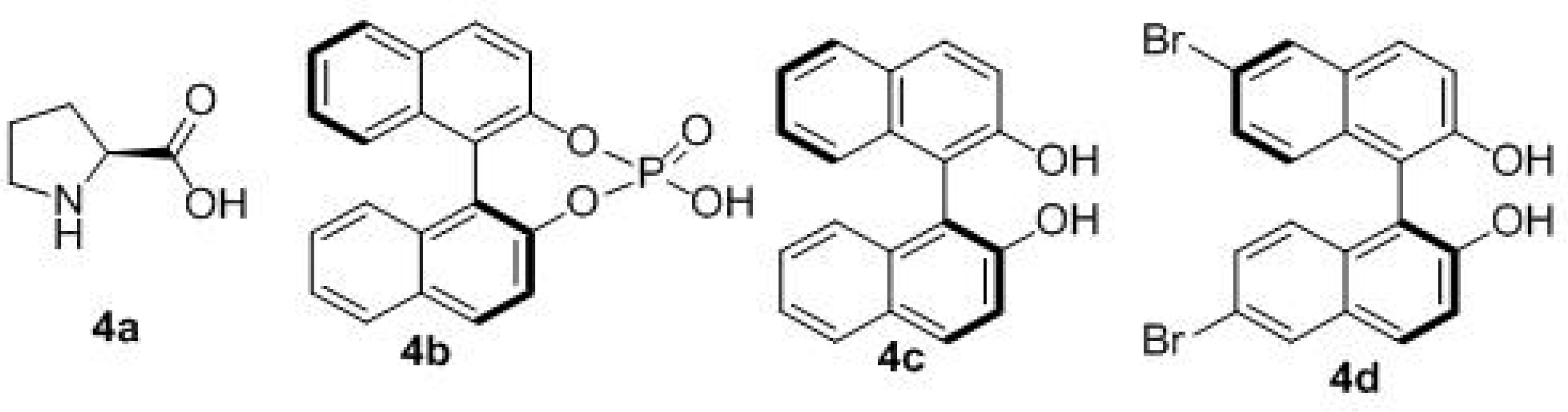

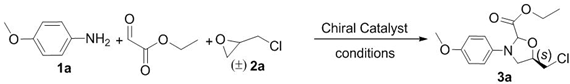
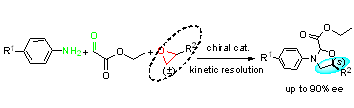
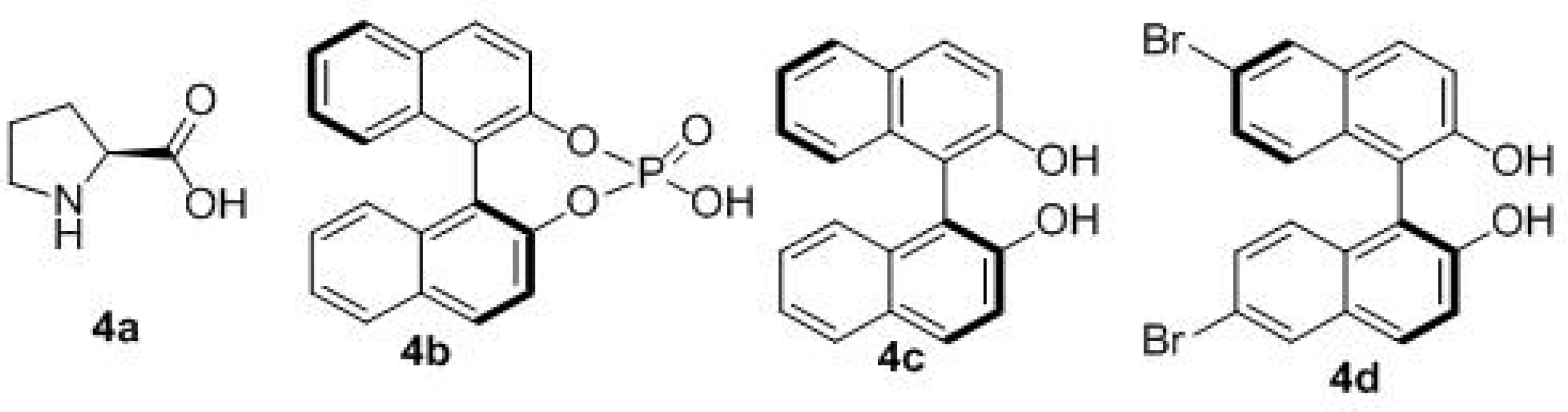
| Entry | Solvent | Catal. | T (°C) | Yield (%) b | d.r. c | ee (%) d |
|---|---|---|---|---|---|---|
| 1 | CH2Cl2 | 4a | 18 | 5 | 2:1 | 10 |
| 2 | CH2Cl2 | 4b | 18 | 13 | 3:1 | 15 |
| 3 | CH2Cl2 | 4b | −10 | 13 | 2:1 | 30 |
| 4 e | CH2Cl2 | 4c/Ti(IV) | −40 | 13 | 3:1 | 41 |
| 5 e | PhCH3 | 4c/Ti(IV) | −40 | 15 | 4:1 | 60 |
| 6 e | PhCH3 | 4c/Ti(IV) | −40 | 50 | 4:1 | 61 |
| 7 f | PhCH3 | 4c/Ti(IV) | −40 | 53 | 11:1 | 72 |
| 8 f | PhCH3 | 4d/Ti(IV) | −40 | 36 | 11:1 | 72 |
| 9 f | PhCH3 | 4c/Ti(IV) | −55 | 30 | 12:1 | 73 |
| 10 f | PhCH3 | 4c/Ti(IV) | −70 | 15 | 12:1 | 73 |

{kind=link}
{kind=link}
{kind=link}
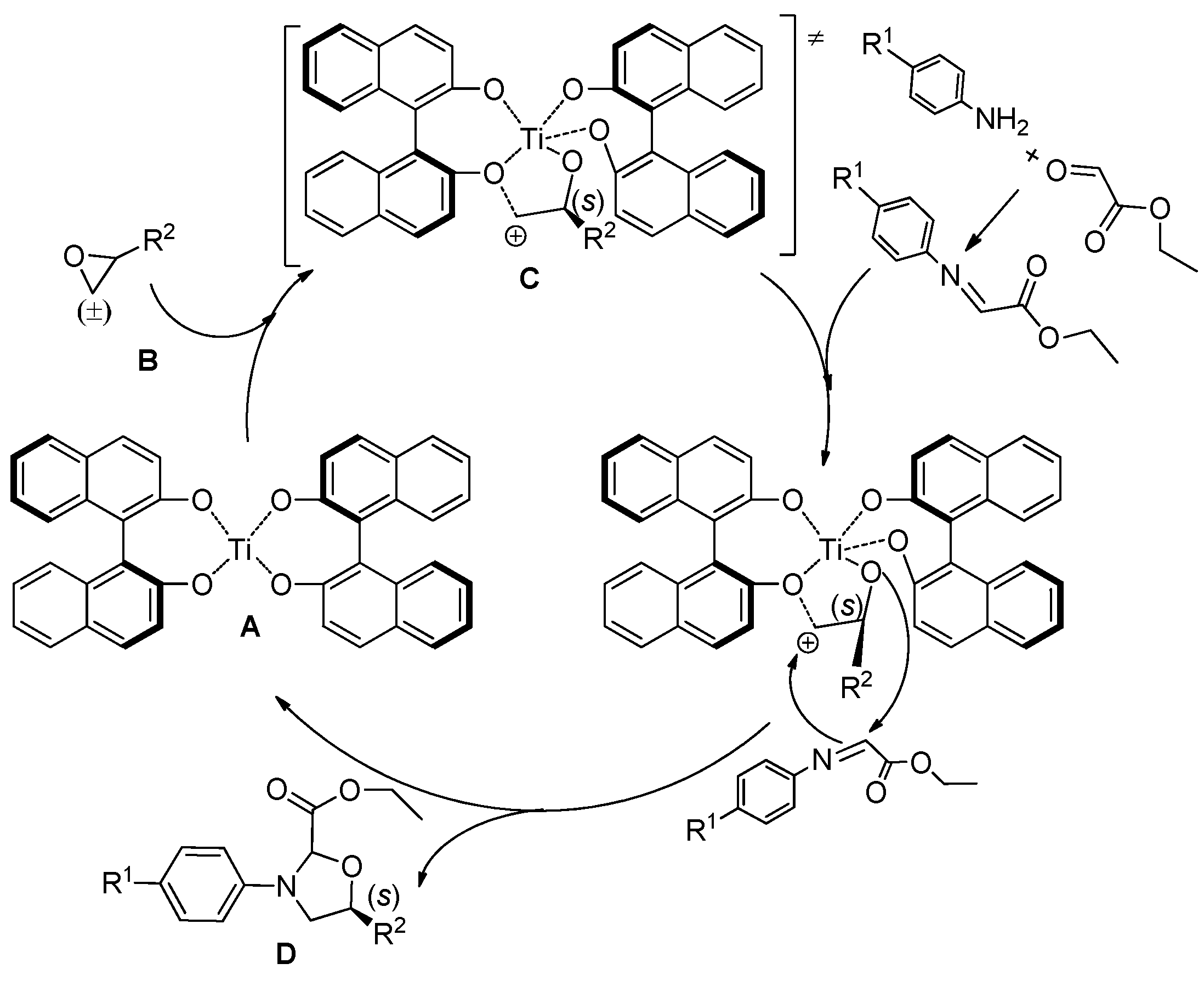{kind=link}
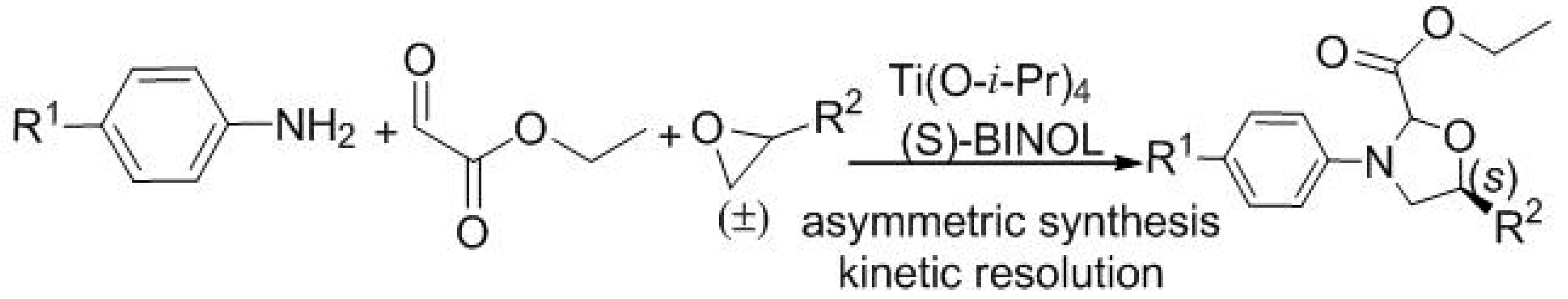{kind=link}
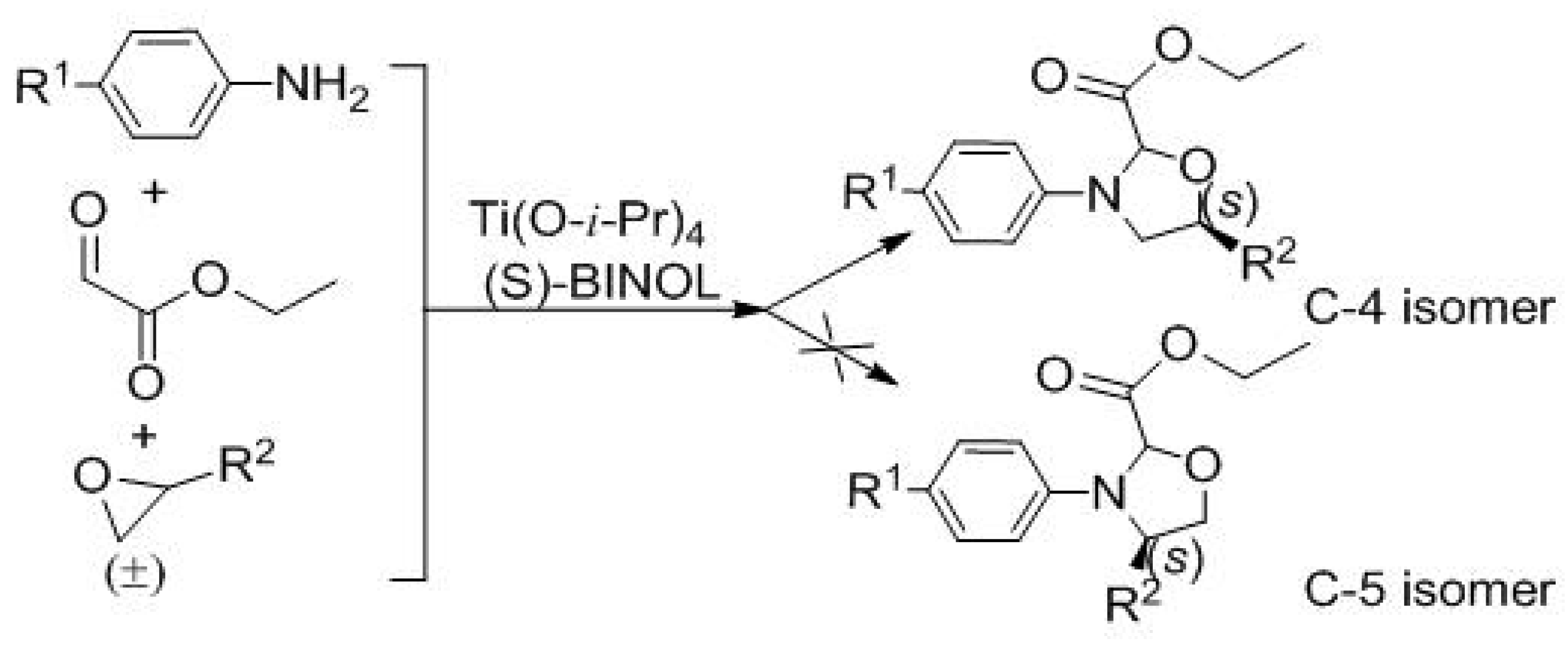
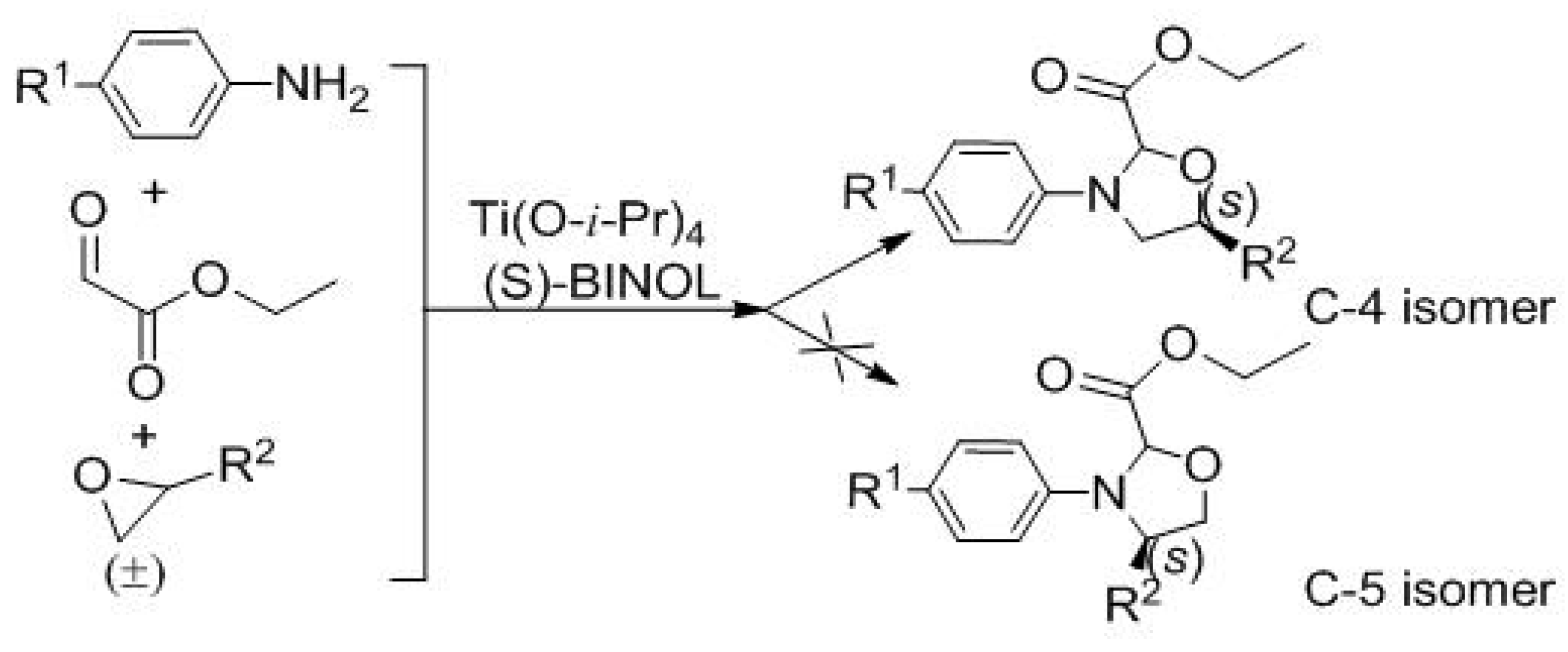
| Entry | 3 | R1 | R2 | Yield (%) b | d.r. c | ee (%) d |
|---|---|---|---|---|---|---|
| 1 | 3a | CH3O | CH2Cl | 52 | 12:1 | 43 |
| 2 | 3b | Cl | CH2Cl | 50 | 10:1 | 69 |
| 3 | 3c | CH3CH2O | CH2Cl | 47 | 10:1 | 39 |
| 4 | 3d | CH3O | CH2OCH(CH3)2 | 56 | 4:1 | 43 |
| 5 | 3e | CH3O | CH2O(CH2)3CH3 | 46 | 1.5:1 | 71 |
| 6 | 3f | CH3CH2O | CH2O(CH2)3CH3 | 53 | 3:1 | 34 |
| 7 | 3g | CH3CH2O | CH2OCH(CH3)2 | 53 | 3:1 | 72 |
| 8 | 3h | Cl | CH2O(CH2)3CH3 | 48 | 3:1 | 69 |
| 9 | 3i | Cl | CH2OCH(CH3)2 | 54 | 4:1 | 90 |
| 10 | 3j | CH3O | Ph | 42 | 1.7:1 | 84.6 |
| 11 | 3k | NO2 | CH2Cl | trace | ― | ― |
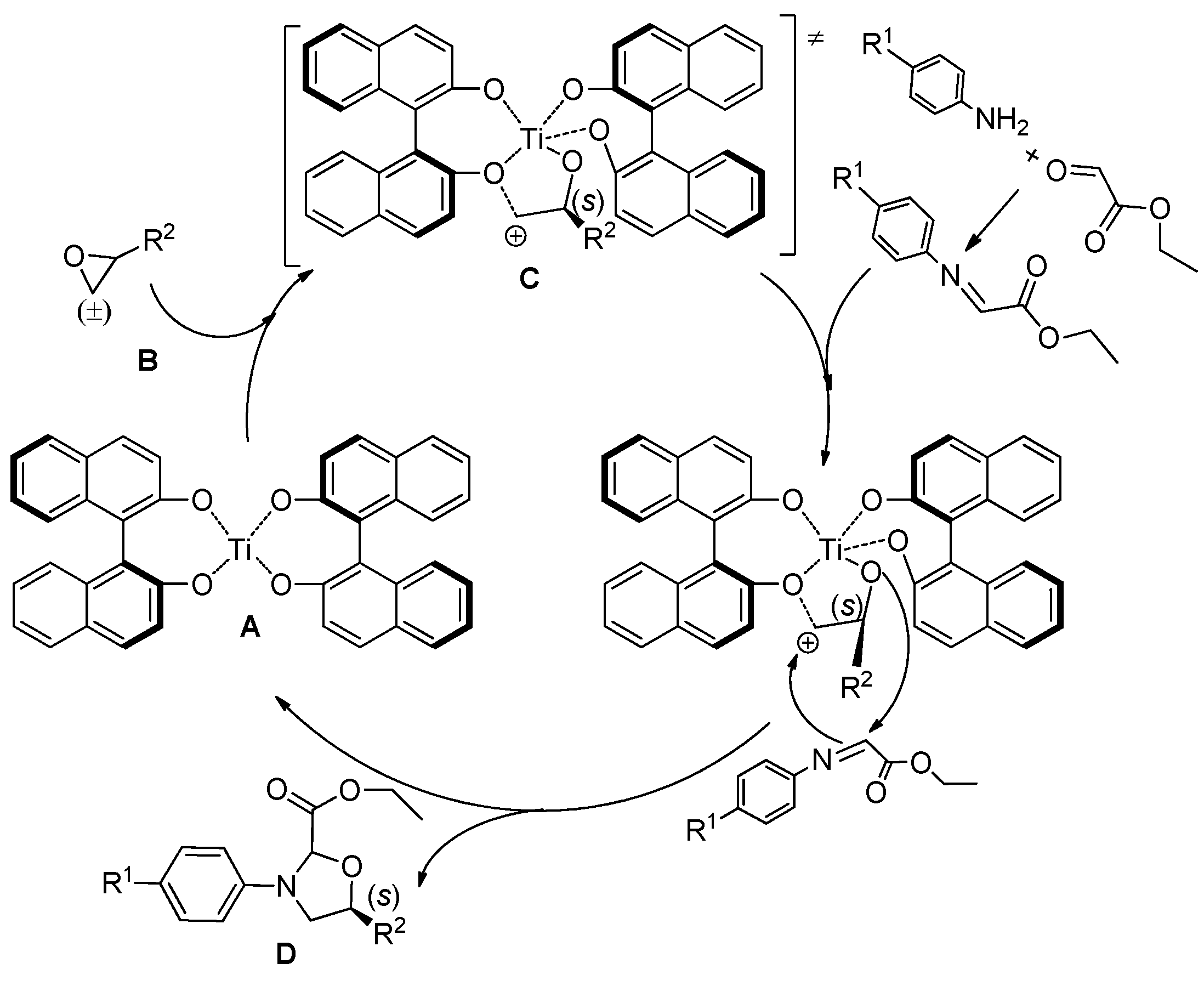
3. Experimental Section
3.1. General Procedure for the Synthesis of All 1,3-Oxazolidines
3.2. Characterization Data for All of the Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pitt, W.R.; Parry, D.M.; Perry, B.G.; GrooM, C.R. Heteroaromatic rings of the future. J. Med. Chem. 2009, 52, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.R.K.; Joshi, N.N. Chiral zinc amides as the catalysts for the enantioselective addition of diethylzinc to aldehydes. J. Org. Chem. 1997, 62, 3770–3771. [Google Scholar] [CrossRef]
- Bull, S.D.; Davies, S.G.; Nicholson, R.L.; Sanganee, H.J.; Smith, A.D. SuperQuat, (S)-4-benzyl-5,5-dimethyl-oxazolidin-2-one for the asymmetric synthesis of α-substituted-aldehydes. Tetrahedron Asymmetry 2000, 11, 3475–3479. [Google Scholar] [CrossRef]
- Davies, S.G.; Sanganee, H.J. 4-Substituted-5,5-dimethyl oxazolidin-2-ones as effective chiral auxiliaries for enolate alkylations and Michael additions. Tetrahedron Asymmetry 1995, 6, 671–674. [Google Scholar] [CrossRef]
- Hintermann, T.; Seebach, D. A useful modification of the Evans auxiliary, 4-isopropyl-5,5-diphenyloxazolidin-2-one. Helv. Chim. Acta 1998, 81, 2093–2126. [Google Scholar] [CrossRef]
- Gibson, C.; Gillon, L.; Cook, K.S. A study of 4-substituted 5,5-diaryl oxazolidin-2-ones as efficacious chiral auxiliaries. Tetrahedron Lett. 1998, 39, 6733–6736. [Google Scholar] [CrossRef]
- Ager, D.J.; Prakash, I.; Schaad, D.R. 1,2-Amino alcohols and their heterocyclic derivatives as chiral auxiliaries in asymmetric synthesis. Chem. Rev. 1996, 96, 835–875. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, G.; Amico, A.D.; Orena, M.; Sandri, S. Diastereoselective alkylation of 3-acylimidazolidin-2-ones, synthesis of (R)- and (S)-lavandulol. J. Org. Chem. 1988, 53, 2354–2356. [Google Scholar] [CrossRef]
- Yu, C.; Dai, X.; Su, W. Ytterbium(III) Triflate Catalyzed [3 + 2] cycloaddition of N-arylimines and epoxides: A novel and solvent-free synthesis of substituted 1,3-Oxazolidines. ChemInform 2007, 4, 646–648. [Google Scholar] [CrossRef]
- Anumula, R.R.; Kagga, M.; Ghanta, M.R.; Padi, P.R. Synthesis of new oxazolidinonyl/oxazolidinyl carbazole derivatives for β-blocking activity. Heterocycl. Commun. 2008, 14, 187–194. [Google Scholar] [CrossRef]
- Michaelis, D.J.; Ischay, M.A.; Yoon, T.P. Activation of N-sulfonyl oxaziridines using copper(II) catalysts: Aminohydroxylations of styrenes and 1,3-Dienes. J. Am. Chem. Soc. 2008, 130, 6610–6615. [Google Scholar] [CrossRef]
- Shaghafi, M.B.; Grote, R.E.; Jarvo, E.R. Oxazolidine synthesis by complementary stereospecific and stereoconvergent methods. Org. Lett. 2011, 13, 5188–5191. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, T.; Shiraishi, H.; Sakaguchi, S.; Ishii, Y. Synthesis of 1,3-oxazolidines from imines and epoxides catalyzed by samarium compounds. Tetrahedron Lett. 2000, 41, 3389–3393. [Google Scholar] [CrossRef]
- Poupon, E.; Francois, D.; Kunesch, N.; Husson, H.P. Reductive and oxidative transformations of the N-(cyanomethyl)oxazolidine system to expand the chiral pool of piperidines. Eur. J. Org. Chem. 2004, 2004, 4823–4829. [Google Scholar]
- Upthagrove, A.L.; Nelson, W.L. Carbinolamines, Imines, and oxazolidines from fluorinated propranolol analogs. 19F-NMR and mass spectral characterization and evidence for formation as intermediates in cytochrome p450-catalyzed N-dealkylation. Drug. Metab. Dispos. 2001, 29, 1114–1122. [Google Scholar] [PubMed]
- Anastas, P.T.; Warner, J.C. Green Chemistry, Theory and Practice; Oxford University Press: Oxford, UK, 2000; p. 135. [Google Scholar]
- Matlack, A.S. Introduction to Green Chemistry; Marcel Dekker: New York, NY, USA, 2001; p. 570. [Google Scholar]
- Trost, B.M. The atom economy—A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M. Atom economy—A challenge for organic synthesis, homogeneous catalysis leads the way. Angew. Chem. Int. Ed. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Edmonds, D.; Bulger, P.G. Cascade reactions in total synthesis. Angew. Chem. Int. Ed. 2006, 45, 7134–7186. [Google Scholar] [CrossRef] [PubMed]
- Terada, M.; Machioka, K.; Sorimachi, K. Chiral Bronsted acid-catalyzed tandem aza-ene type reaction/cyclization cascade for a one-pot entry to enantioenriched piperidines. J. Am. Chem. Soc. 2007, 129, 10336–10337. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Loh, T.P. Control of up to five stereocenters in a cascade reaction: Synthesis of highly functionalized five-membered rings. J. Am. Chem. Soc. 2008, 130, 7194–7195. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yan, S.; Hu, L.; Wang, Y.; Lin, J. Cascade reaction of isatins with heterocyclic ketene aminals: Synthesis of imidazopyrroloquinoline derivatives. Org. Lett. 2011, 13, 4782–4785. [Google Scholar] [CrossRef] [PubMed]
- Kriis, K.; Ausmees, K.; Pehk, T.; Lopp, M.; Kanger, T. A novel diastereoselective multicomponent cascade reaction. Org. Lett. 2010, 12, 2230–2233. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Guan, X.; Liu, S.; Ren, B.; Ma, X.; Guo, X.; Lv, F.; Wu, X.; Hu, W. Highly diastereoselective multicomponent cascade reactions, efficient synthesis of functionalized 1-indanols. Angew. Chem. Int. Ed. 2013, 52, 1539–1542. [Google Scholar] [CrossRef] [PubMed]
- Scroggins, S.T.; Chi, Y.; Fréchet, J.M.J. Polarity-directed one-pot asymmetric cascade reactions mediated by two catalysts in an aqueous buffer. Angew. Chem. Int. Ed. 2010, 49, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Khedkar, V.; Baskar, B.; SchÜrmann, M.; Kumar, K. Branching Cascades: A concise synthetic strategy targeting diverse and complex molecular frameworks. Angew. Chem. Int. Ed. 2011, 50, 6900–6905. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; List, B. Organocatalytic asymmetric reaction cascade to substituted cyclohexylamines. J. Am. Chem. Soc. 2007, 129, 7498–7499. [Google Scholar] [CrossRef]
- Biju, A.T.; Wurz, N.E.; Glorius, F. N-heterocyclic carbene-catalyzed cascade reaction involving the hydroacylation of unactivated alkynes. J. Am. Chem. Soc. 2010, 132, 5970–5971. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.E.; Leighton, J.L.; Carsten, D.H.; Jacobsen, E.N. Highly enantioselective ring opening of epoxides catalyzed by (salen) Cr (III) complexes. J. Am. Chem. Soc. 1995, 117, 5897–5898. [Google Scholar] [CrossRef]
- Larrow, J.F.; Schaus, S.E.; Jacobsen, E.N. Kinetic resolution of terminal epoxides via highly regioselective and enantioselective ring opening with TMSN3. An efficient, catalytic route to 1,2-amino alcohols. J. Am. Chem. Soc. 1996, 118, 7420–7421. [Google Scholar] [CrossRef]
- Tokunaga, M.; Larrow, J.F.; Kakiuchi, F.; Jacobsen, E.N. Asymmetric catalysis with water Efficient kinetic resolution of terminal epoxides by means of catalytic hydrolysis. Science 1997, 277, 936–938. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Jacobsen, E.N. Asymmetric ring opening of meso epoxides with thiols: Enantiomeric enrichment Using a bifunctional nucleophile. J. Org. Chem. 1998, 63, 5252–5254. [Google Scholar] [CrossRef]
- Ready, J.M.; Jacobsen, E.N. Asymmetric catalytic synthesis of α-aryloxy alcohols: kinetic resolution of terminal epoxides via highly enantioselective ring-opening with phenols. J. Am. Chem. Soc. 1999, 121, 6086–6087. [Google Scholar] [CrossRef]
- Jacobsen, E.N. Asymmetric catalysis of epoxide ring-opening reactions. Acc. Chem. Res. 2000, 33, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Ready, J.M.; Jacobsen, E.N. Highly active oligomeric (salen) Co catalysts for asymmetric epoxide ring-opening reactions. J. Am. Chem. Soc. 2001, 123, 2687–2688. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.M.; Kozhevnikov, I.V.; Derouane, E. Catalysts for Fine Chemical Synthesis: Hydrolysis, Oxidation and Reduction; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2002; Volume 1. [Google Scholar]
- DiMauro, E.F.; Mamai, A.; Kozlowski, M.C. Synthesis, characterization, and metal complexes of a salen ligand containing a quinoline base. Organometallics 2003, 22, 850–855. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Kakiuchi, F.; Konsler, R.G.; Larrow, J.F.; Tokunaga, M. An efficient and facile three-step synthesis of 5-amino-5-deoxy-d-pentonolactams from unprotected d-pentono-1,4-lactones. Tetrahedron Lett. 1997, 38, 773–776. [Google Scholar] [CrossRef]
- Lu, X.-B.; Liang, B.; Zhang, Y.-J.; Tian, Y.-Z.; Wang, Y.-M.; Bai, C.-X.; Wang, H.; Zhang, R. Asymmetric catalysis with CO2: Direct synthesis of optically active propylene carbonate from racemic epoxides. J. Am. Chem. Soc. 2004, 126, 3732–3733. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.-M.; Wu, G.-P.; Lin, F.; Jiang, J.-Y.; Liu, C.; Luo, Y.; Lu, X.-B. Role of the co-catalyst in the asymmetric coupling of racemic epoxides with CO2 using multichiral Co(III) complexes: Product selectivity and enantioselectivity. Chem. Sci. 2012, 3, 2094–2102. [Google Scholar] [CrossRef]
- Bhanu Prasad, B.A.; Pandey, G.; Singh, V.K. Synthesis of substituted imidazolines via [3 + 2]-cycloaddition of aziridines with nitriles. Tetrahedron Lett. 2004, 45, 1137–1141. [Google Scholar] [CrossRef]
- Gandhi, S.; Bisai, A.; Bhanu Prasad, B.A.; Singh, V.K. Studies on the reaction of aziridines with nitriles and carbonyls, synthesis of imidazolines and oxazolidines. J. Org. Chem. 2007, 72, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Benkovics, T.; Guzei, I.A.; Yoon, T.P. Oxaziridine-mediated oxyamination of indoles, an approach to 3-aminoindoles and enantiomerically enriched 3-aminopyrroloindolines. Angew. Chem. Int. Ed. 2010, 49, 9153–9157. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.K.; Sriramurthy, V. Silylmethyl-substituted aziridine and azetidine as masked 1,3- and 1,4-dipoles for formal [3 + 2] and [4 + 2] cycloaddition reactions. J. Am. Chem. Soc. 2005, 127, 16366–16367. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Williams, R.M. Concave butterfly-shaped organometallic hydrocarbons. Angew. Chem. Int. Ed. 2001, 40, 1463–1465. [Google Scholar] [CrossRef]
- Adam, W.; Peters, K.; Peters, E.M.; Schambony, S.B. Efficient control of the diastereoselectivity and regioselectivity in the singlet-oxygen ene reaction of chiral oxazolidine-substituted alkenes by a remote urea NH functionality: Comparison with dimethyldioxirane and m-chloroperbenzoic acid epoxidations. J. Am. Chem. Soc. 2001, 123, 7228–7232. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.B.; Zeitler, K.; Gschwind, R.M. Formation and stability of prolinol and prolinol ether enamines by NMR: delicate selectivity and reactivity balances and parasitic equilibria. J. Am. Chem. Soc. 2011, 133, 7065–7074. [Google Scholar] [CrossRef] [PubMed]
- Vasylyev, M.; Alper, H. Diastereoselective synthesis of hexahydropyrrolo[2,1-b]oxazoles by a rhodium-catalyzed hydroformylation/silica-promoted deformylation sequence. Angew. Chem. Int. Ed. 2009, 48, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.X.; Zhang, X.M.; Gong, L.Z. The first organocatalytic enantio- and diastereoselective 1,3-dipolar cycloaddition of azomethine ylides with nitroalkenes. ChemInform 2008, 5, 691–694. [Google Scholar] [CrossRef]
- Chen, X.H.; Zhang, W.Q.; Gong, L.Z. Asymmetric organocatalytic three-component 1,3-dipolar cycloaddition: Control of stereochemistry via a chiral brønsted acid activated dipole. J. Am. Chem. Soc. 2008, 130, 5652–5653. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Luo, Z.L.; Tao, S.W.; Tu, S.J.; Gong, L.Z. Scaffold-inspired enantioselective synthesis of biologically important spiro[pyrrolidin-3,2′-oxindoles] with structural diversity through catalytic isatin-derived 1,3-dipolar cycloadditions. Chem. Eur. J. 2012, 18, 6885–6894. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Luo, S.W.; Tao, Z.L.; He, L.; Yu, J.; Tu, S.J.; Gong, L.Z. The catalytic asymmetric 1,3-dipolar cycloaddition of ynones with azomethine ylides. Org. Lett. 2011, 13, 4680–4683. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.L.; Wang, N.X. Organocatalytic and metal-mediated asymmetric [3 + 2] cycloaddition reactions. Coord. Chem. Rev. 2012, 256, 938–952. [Google Scholar] [CrossRef]
- Wang, N.X.; Xing, Y.; Wang, Y.J. Asymmetric synthesis of chiral flavanone and chromanone derivatives. Curr. Org. Chem. 2013, 17, 1555–1562. [Google Scholar] [CrossRef]
- Povarov, L.S. α,β-Unsaturated ethers and their analogues in reactions of diene synthesis. Russ. Chem. Rev. 1967, 36, 656–670. [Google Scholar] [CrossRef]
- Jorgensen, K.A. Catalytic asymmetric hetero-diels-alder reactions of carbonyl compounds and imines. Angew. Chem. Int. Ed. 2000, 39, 3558–3588. [Google Scholar] [CrossRef]
- Lavilla, R.; Bernabeu, M.C.; Carranco, I.; Díaz, J. Dihydropyridine-based multicomponent reactions. efficient entry into new tetrahydroquinoline systems through lewis acid-catalyzed formal [4 + 2] cycloadditions. Org. Lett. 2003, 5, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Matsubara, R.; Nakamura, Y.; Kitagawa, H.; Sugiura, M. Catalytic, Asymmetric mannich-type reactions of N-acylimino esters, reactivity, diastereo- and enantioselectivity, and application to synthesis of N-acylated amino acid derivatives. J. Am. Chem. Soc. 2003, 125, 2507–2515. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, O.; de la Rosa, G.; Lavilla, R. Straightforward access to a structurally diverse set of oxacyclic scaffolds through a four-component reaction. Angew. Chem. Int. Ed. 2005, 44, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Bulatova, O.F.; Chalova, O.B.; Rakhmankulov, D.L. Structure of substituted 5-chloromethyloxazolidines. Russ. J. Org. Chem. 2001, 37, 1753–1756. [Google Scholar]
- Sample Availability: Samples of the compounds described in this paper are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, X.-W.; Zhou, Y.-Q.; Bai, C.-B.; Wang, N.-X.; Xing, Y.; Zhang, W.; Wang, Y.-J.; Lan, X.-W.; Xie, Y.; Li, Y.-H. Asymmetric Synthesis of 1,3-Oxazolidine Derivatives with Multi-Component Reaction and Research of Kinetic Resolution. Molecules 2015, 20, 17208-17220. https://doi.org/10.3390/molecules200917208
Hong X-W, Zhou Y-Q, Bai C-B, Wang N-X, Xing Y, Zhang W, Wang Y-J, Lan X-W, Xie Y, Li Y-H. Asymmetric Synthesis of 1,3-Oxazolidine Derivatives with Multi-Component Reaction and Research of Kinetic Resolution. Molecules. 2015; 20(9):17208-17220. https://doi.org/10.3390/molecules200917208
Chicago/Turabian StyleHong, Xiao-Wei, Yu-Qiang Zhou, Cui-Bing Bai, Nai-Xing Wang, Yalan Xing, Wei Zhang, Yan-Jing Wang, Xing-Wang Lan, Yu Xie, and Yi-He Li. 2015. "Asymmetric Synthesis of 1,3-Oxazolidine Derivatives with Multi-Component Reaction and Research of Kinetic Resolution" Molecules 20, no. 9: 17208-17220. https://doi.org/10.3390/molecules200917208
APA StyleHong, X.-W., Zhou, Y.-Q., Bai, C.-B., Wang, N.-X., Xing, Y., Zhang, W., Wang, Y.-J., Lan, X.-W., Xie, Y., & Li, Y.-H. (2015). Asymmetric Synthesis of 1,3-Oxazolidine Derivatives with Multi-Component Reaction and Research of Kinetic Resolution. Molecules, 20(9), 17208-17220. https://doi.org/10.3390/molecules200917208