
Design, Synthesis and Evaluation of Novel Phthalimide Derivatives as in Vitro Anti-Microbial, Anti-Oxidant and Anti-Inflammatory Agents


Abstract
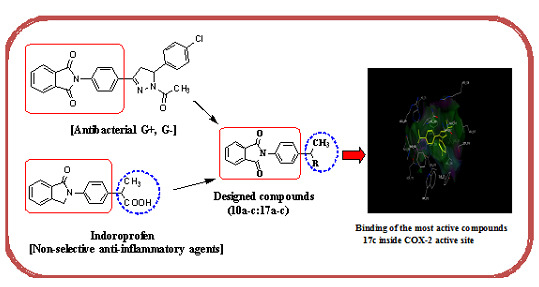
:
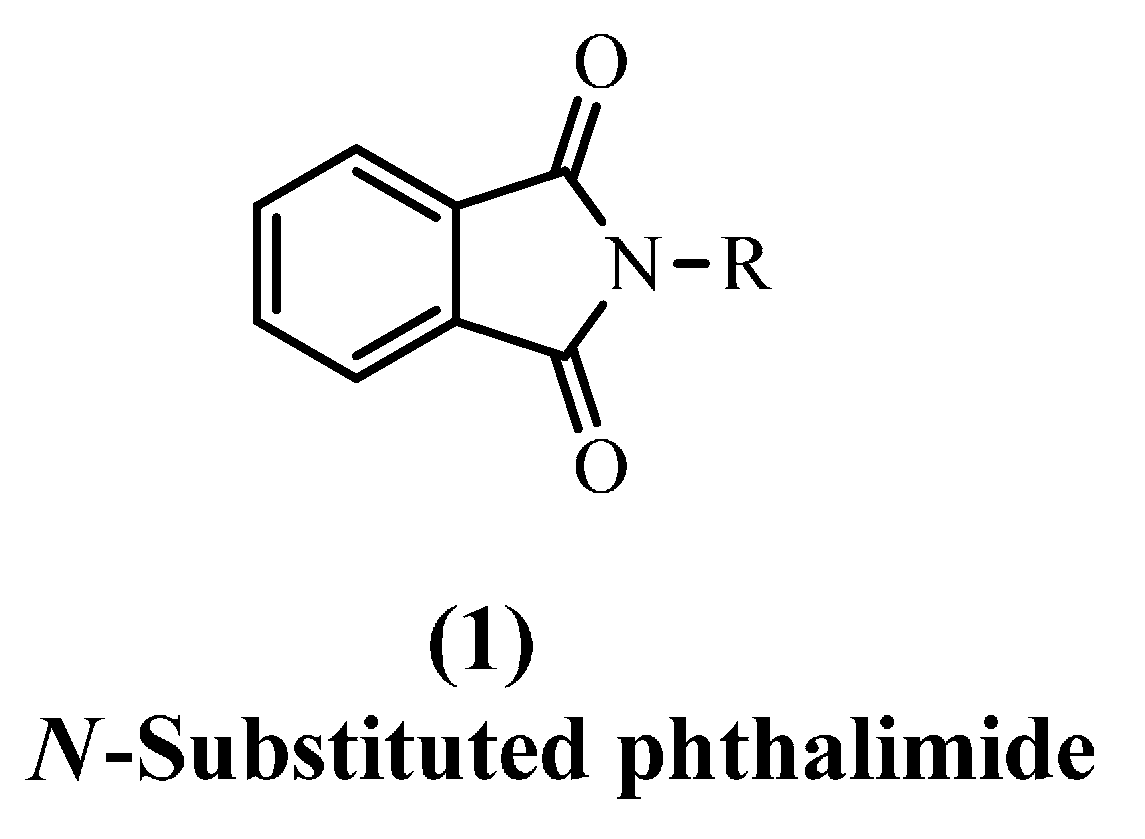
1. Introduction
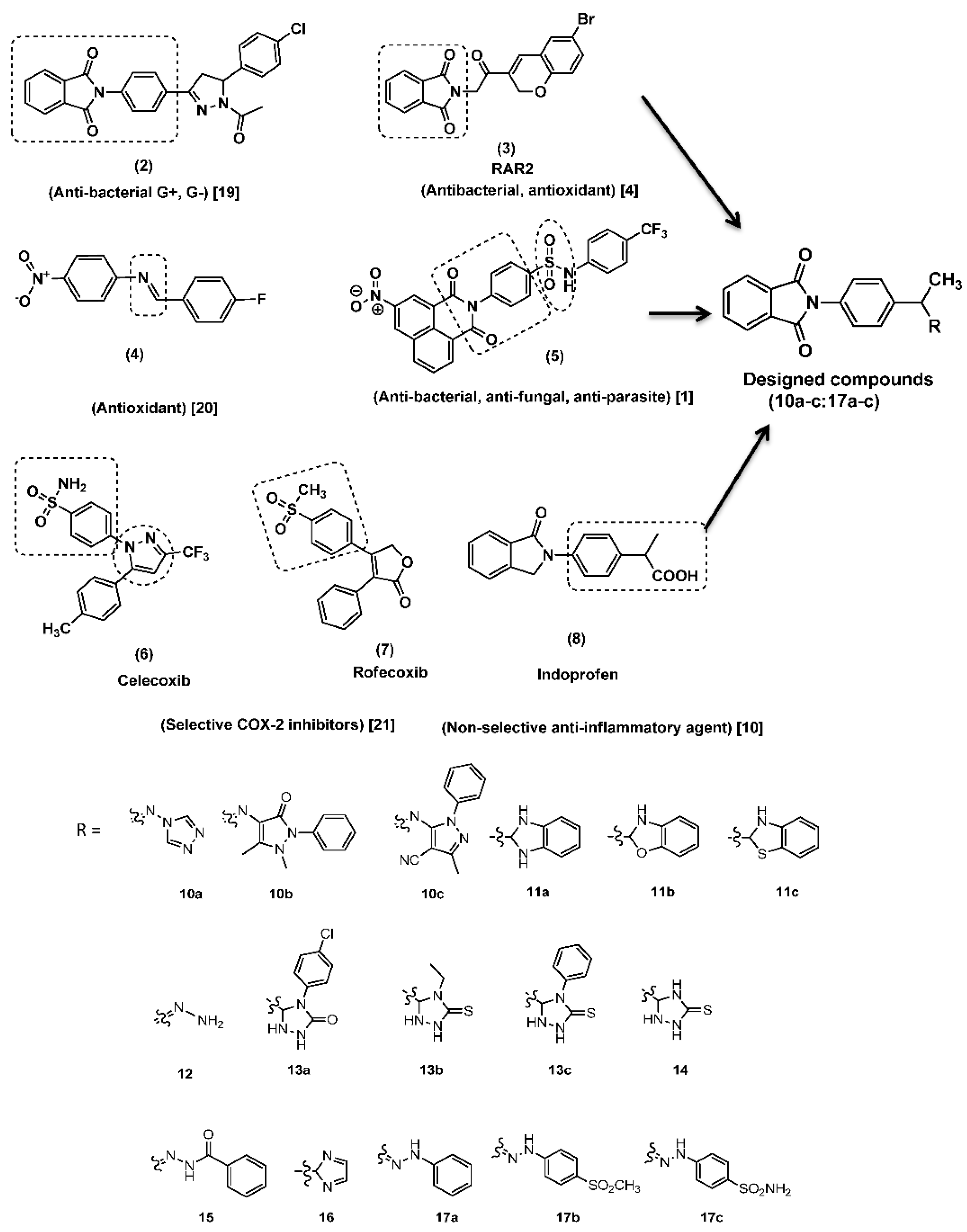
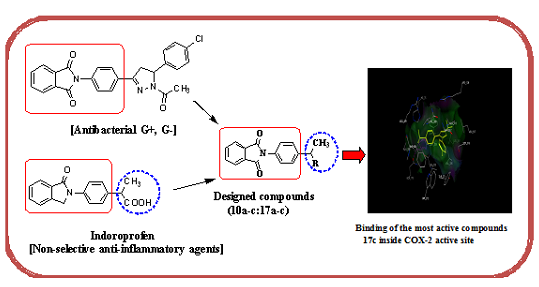
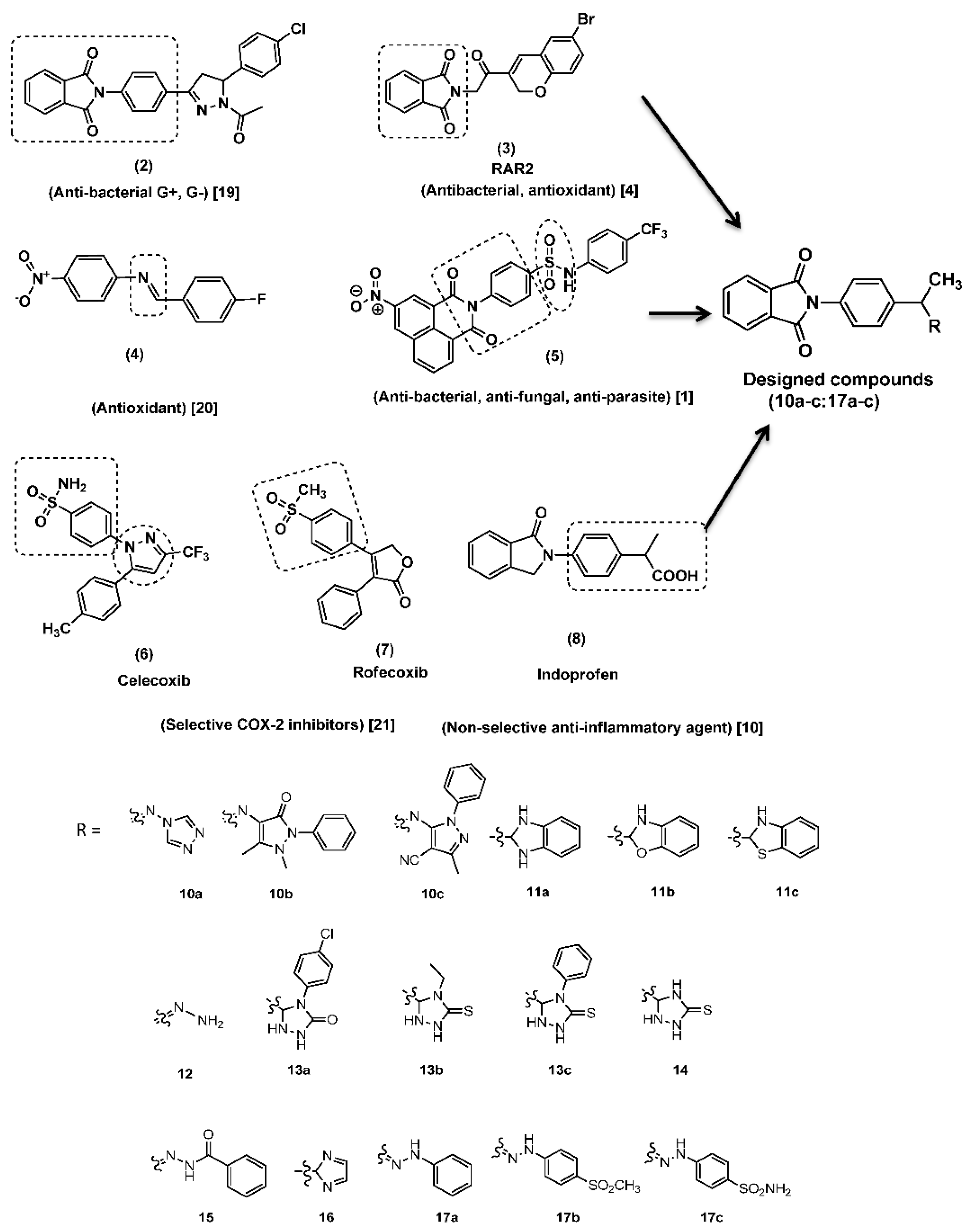
2. Results and Discussion
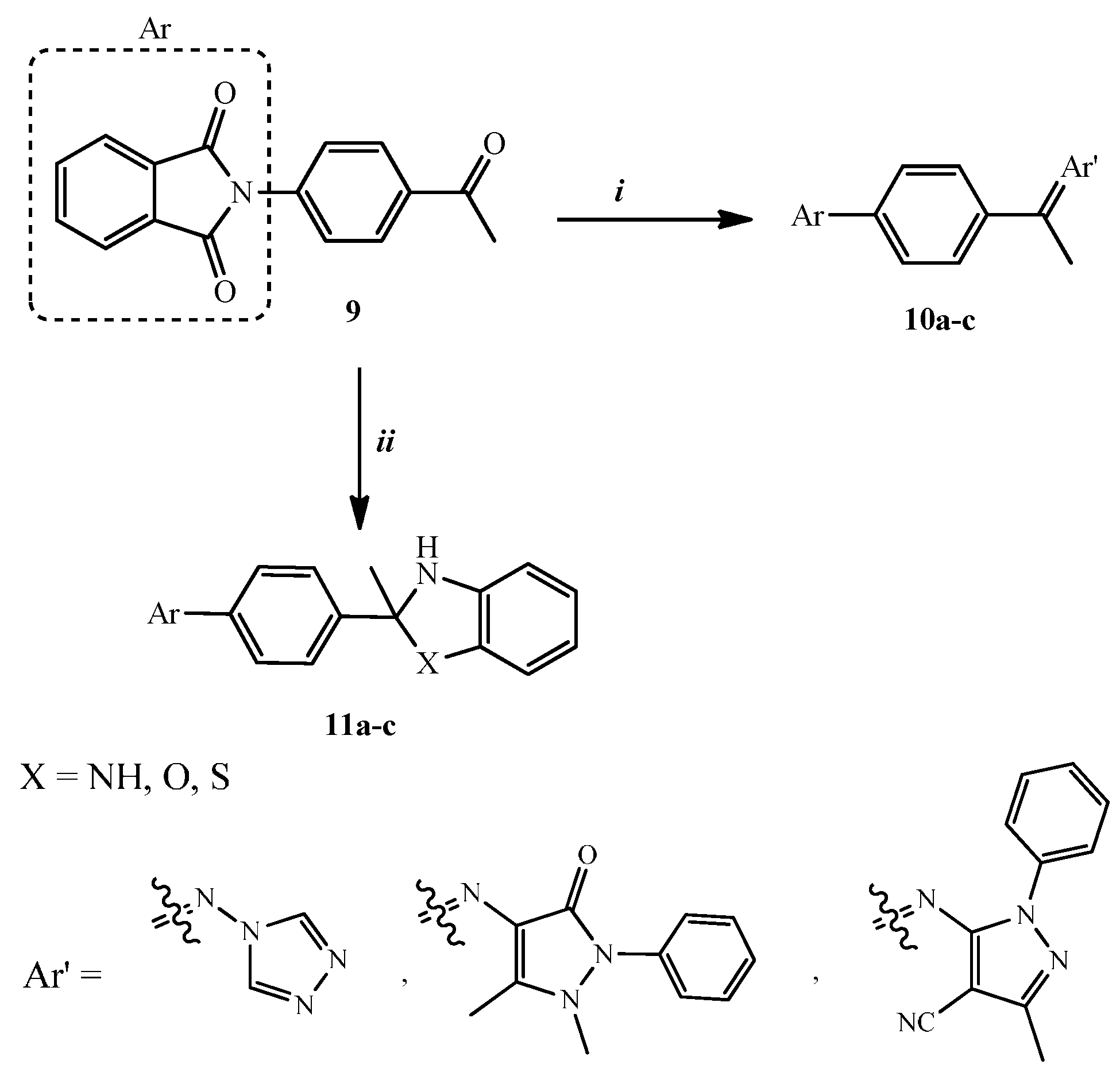
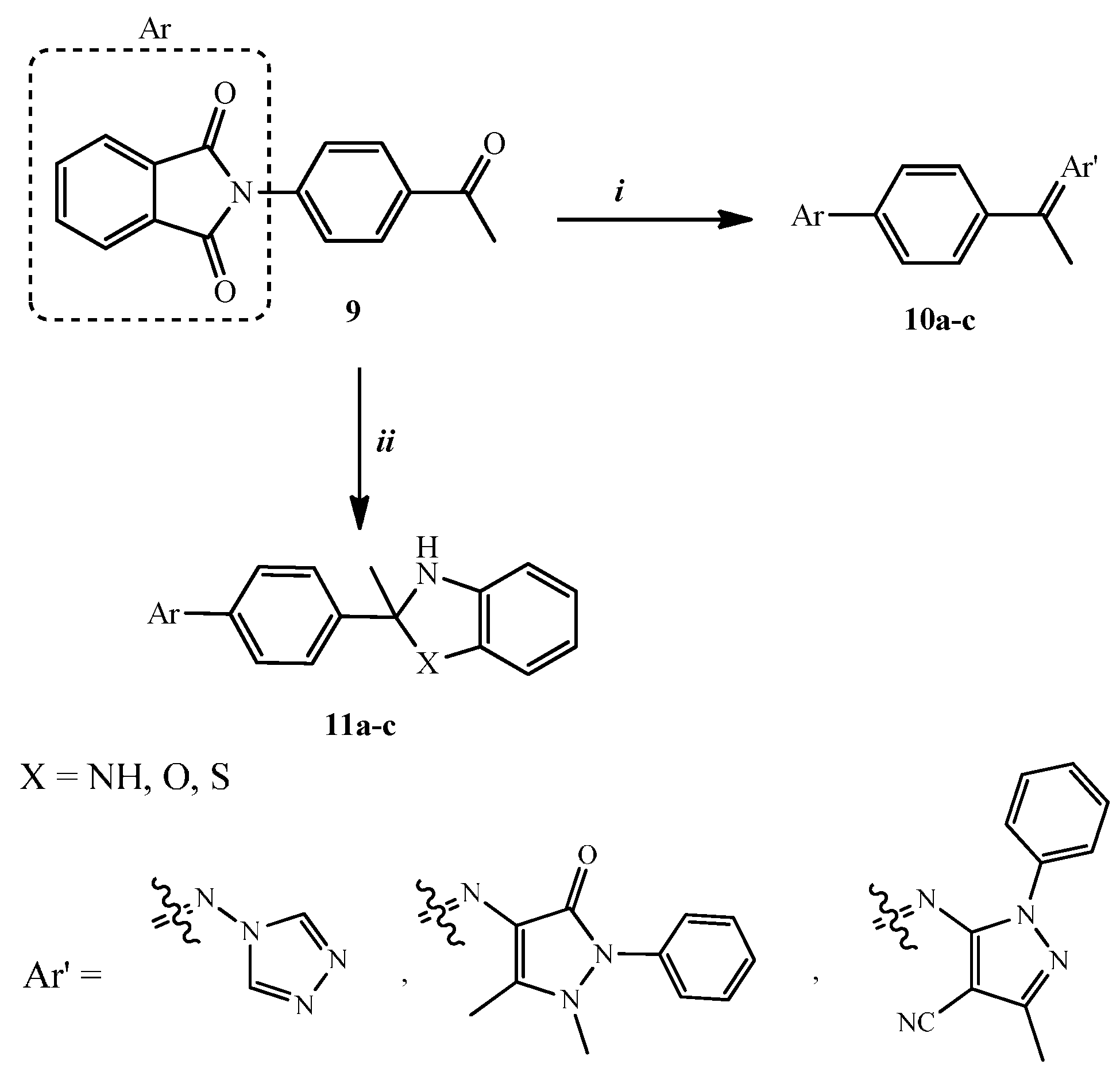
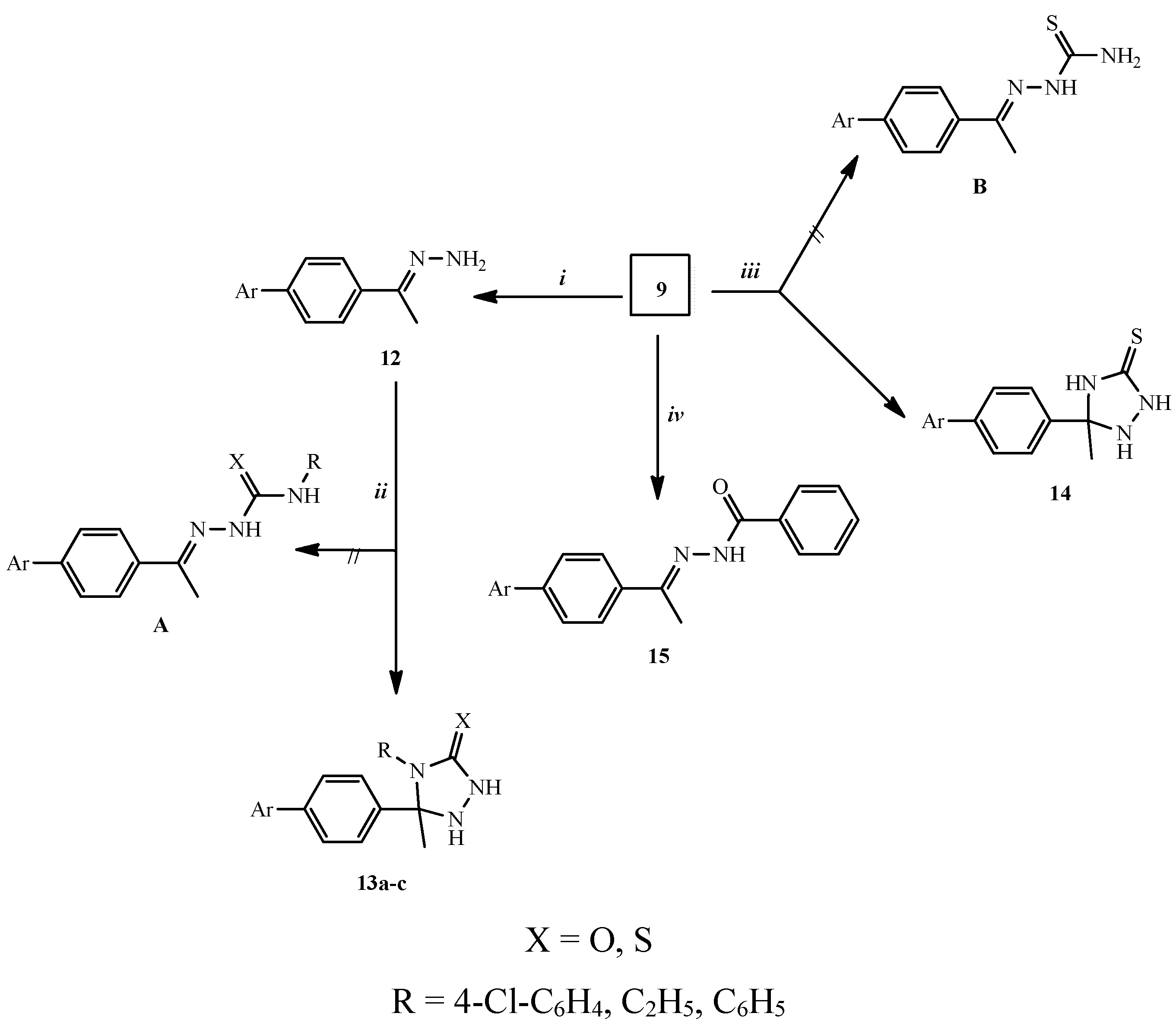
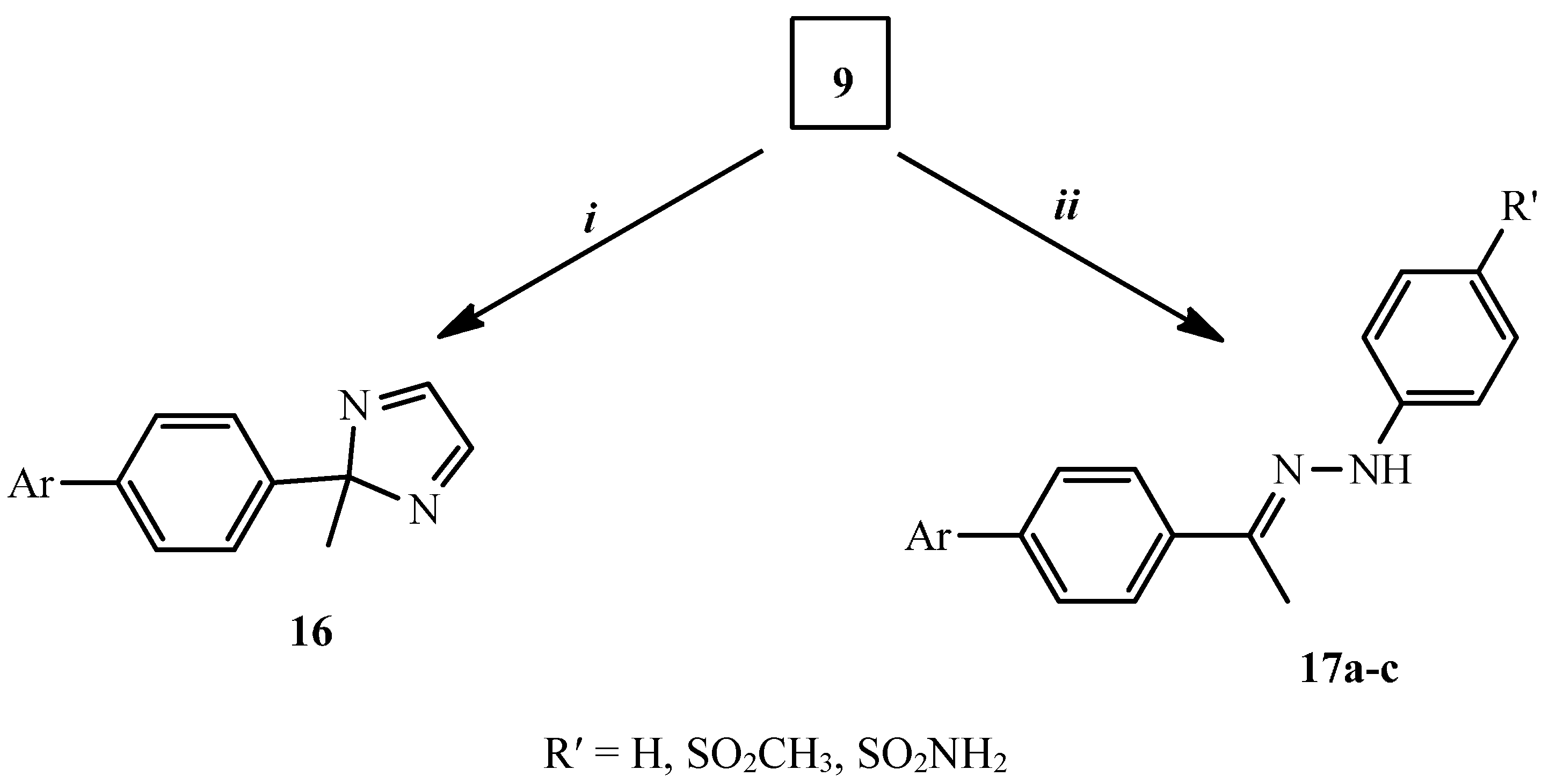
2.1. Chemical Synthesis

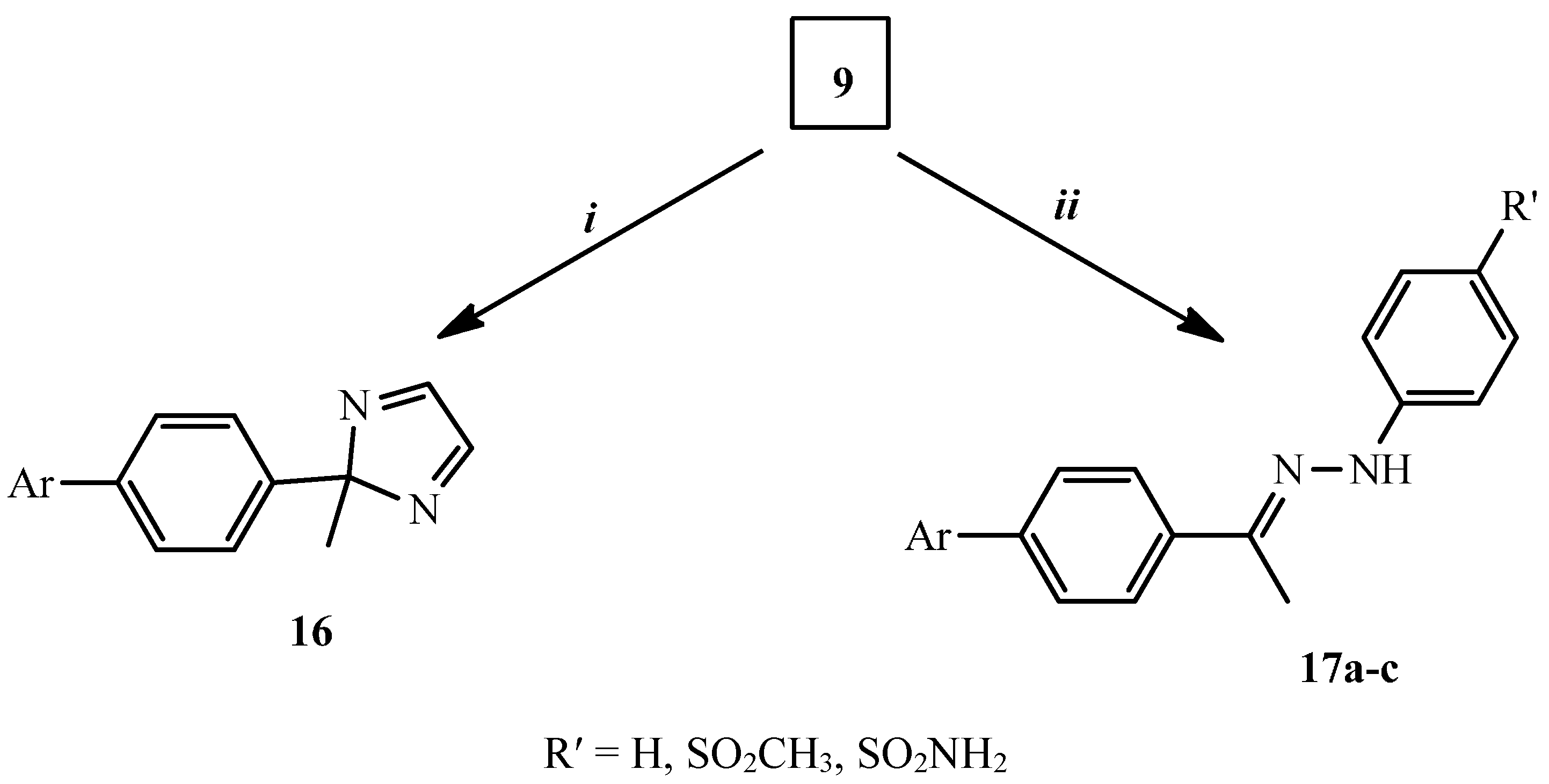
2.2. Biological Evaluation
2.2.1. Anti-Microbial Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gram Positive Rods | Gram Positive Cocci | Gram Negative Rods | Yeast | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bacillus subtilis | Listeria inocua LMG 2710 | Mycobacterium pheli | Enterococcus faecalis | Sarcina lutea | Staphylococcus aureus LMG 3242 | Escherichia coli ATCC 25922 | Escherichia coli ATCC 5087 | Psedomonas aeruginosa | Proteus vulgaris | Candida albicans | |
| 10a | 13 | - | - | - | - | 14 | - | - | - | - | 14 |
| 10b | 13 | 15 | 14.5 | 13 | - | 15 | - | - | - | - | 16 |
| 10c | - | 15 | 15 | - | - | - | - | - | 12 | - | 14 |
| 11a | 15 | 13 | 13 | 16 | - | 14.5 | 13 | - | 13 | - | 14 |
| 11b | 15 | 13 | 13 | 16.5 | - | 14 | 12 | - | 12 | - | 13 |
| 11c | - | 17 | - | - | - | 14.5 | - | - | 13 | - | 20 |
| 12 | 26 | 17 | 23 | 17.5 | 25 | 16 | 17 | 17 | 25 | - | 21 |
| 13a | 13 | 14 | - | 15.5 | - | 13 | - | - | - | - | - |
| 13b | 13 | 15.5 | 13 | 14 | 14 | 13 | - | - | - | - | 14 |
| 13c | 14 | 16 | 16 | 13.5 | 17.5 | 13 | - | - | 13 | - | 13 |
| 14 | 16 | 15 | 15.5 | 15 | 18 | - | - | - | 13 | 12 | 14 |
| 15 | 13 | - | - | 13.5 | - | - | - | - | 11 | - | 17.5 |
| 16 | 12 | 12 | - | 13 | - | - | - | - | 12 | - | 18 |
| 17a | 11 | 17 | 11 | 12 | - | - | - | - | 11 | 12 | 16.5 |
| 17b | 13 | 11 | - | 13 | - | - | - | - | 12 | - | 12 |
| 17c | - | 16 | - | 13 | - | - | - | - | 13 | - | 15 |
| AMP (a) | 22 | 30 | 42 | 30 | 54 | 31 | 38 | 44 | - | - | - |
| CTX (b) | 25 | 21 | 40 | 22 | 48 | 25 | 42 | 52 | 30 | 31 | - |
| CN (c) | 28 | 31 | 36 | 20 | 40 | 24 | 34 | 40 | 36 | 39 | - |
| FLU (d) | - | - | - | - | - | - | - | - | - | - | 40 |
| TIO (e) | - | - | - | - | - | - | - | - | - | - | 36 |
| Compound No. | Proteus vulgaris | Salmonella typhi | Pseudomonas aeruginosa 9027 | E. coli 25927 | E. coli 5087 | Sarcina lutea | Enterococcus faecalis OS4 | Mycobacterium pheli | Lactobacillus sakei LMG 2313 | Bacillus subtilis | Listeria innocua LMG 2710 | Staphylococcus aureus LMG 3242 | Staphylococcus aureus ATCC 43300 | Candida albicans ATCC 60931 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10b | na | na | na | na | na | na | na | na | na | na | na | na | na | na |
| 11a | na | na | na | na | na | na | 100 | 50 | 50 | na | 25 | 50 | na | na |
| 11b | na | na | na | na | na | na | na | na | 50 | na | na | na | na | na |
| 11c | na | na | na | na | na | na | na | na | na | na | 100 | na | na | 100 |
| 12 | na | na | 100 | na | na | 50 | 50 | 25 | 200 | 25 | 100 | na | na | 100 |
| 13c | 200 | na | 200 | 200 | 200 | na | 200 | na | 200 | 200 | 200 | 200 | 200 | na |
| 14 | na | na | na | na | na | na | na | na | na | na | na | na | na | na |
| 16 | na | na | na | na | na | na | na | na | na | na | na | na | na | na |
| 17a | na | na | na | na | na | na | na | na | na | na | na | na | na | 25 |
2.2.2. Anti-Oxidant Activity
| Compound | ORAC (Compound/Trolox) |
|---|---|
| 10a | <0.1 |
| 10b | 0.112 ± 0.007 |
| 10c | <0.1 |
| 11a | 0.751 ± 0.081 |
| 11b | 0.116 ± 0.004 |
| 11c | 0.183 ± 0.01 |
| 12 | 8.116 ± 0.343 |
| 13a | 1.169 ± 0.118 |
| 13b | 18.385 ± 0.857 |
| 13c | 13.506 ± 0.819 |
| 14 | 1.511 ± 0.045 |
| 15 | 0.192 ± 0.005 |
| 16 | <0.1 |
| 17a | 1.839 ± 0.035 |
| 17b | 0.129 ± 0.005 |
| 17c | 0.374 ± 0.007 |
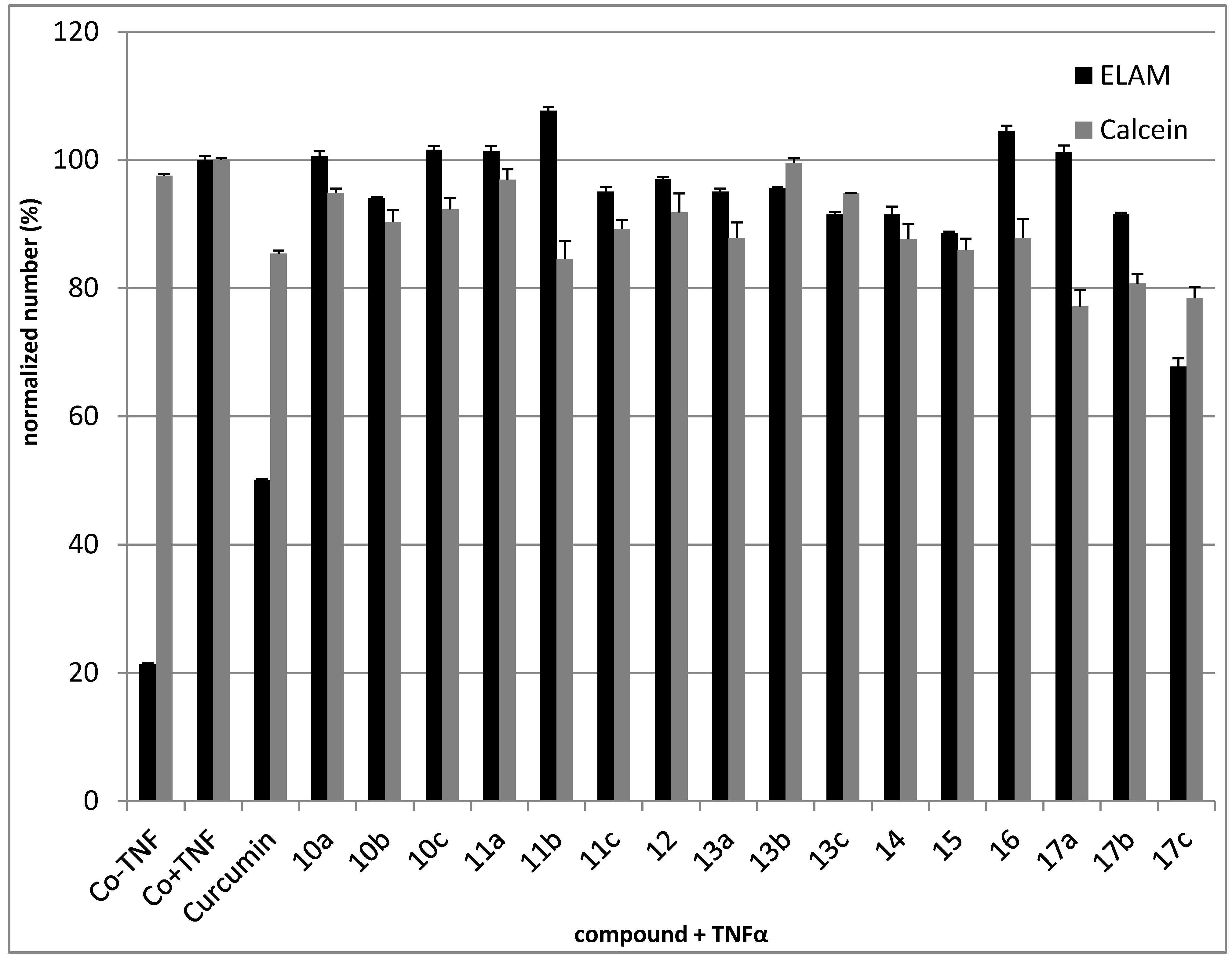
2.2.3. Anti-Inflammatory Activity
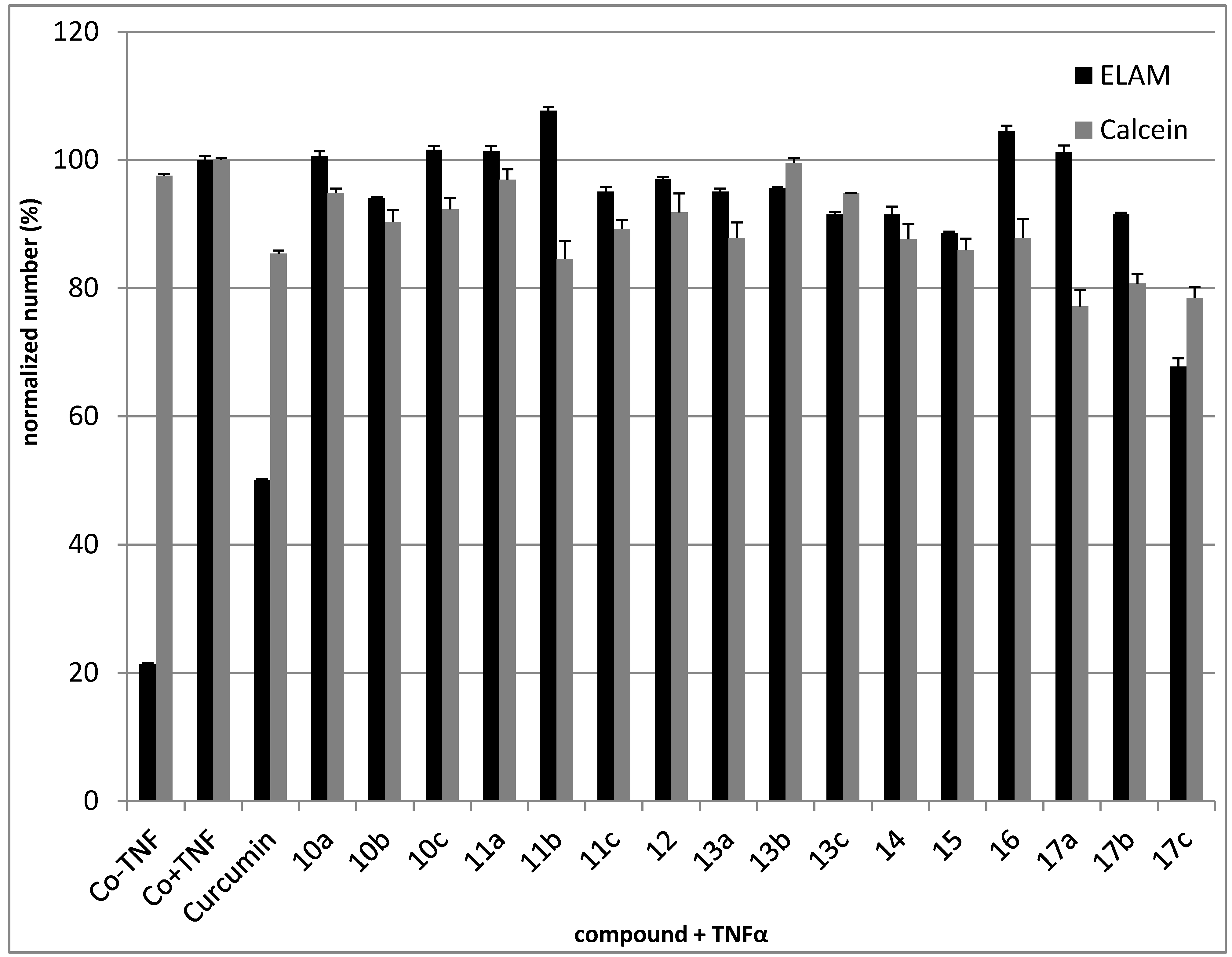
2.2.4. Cytotoxic Activity
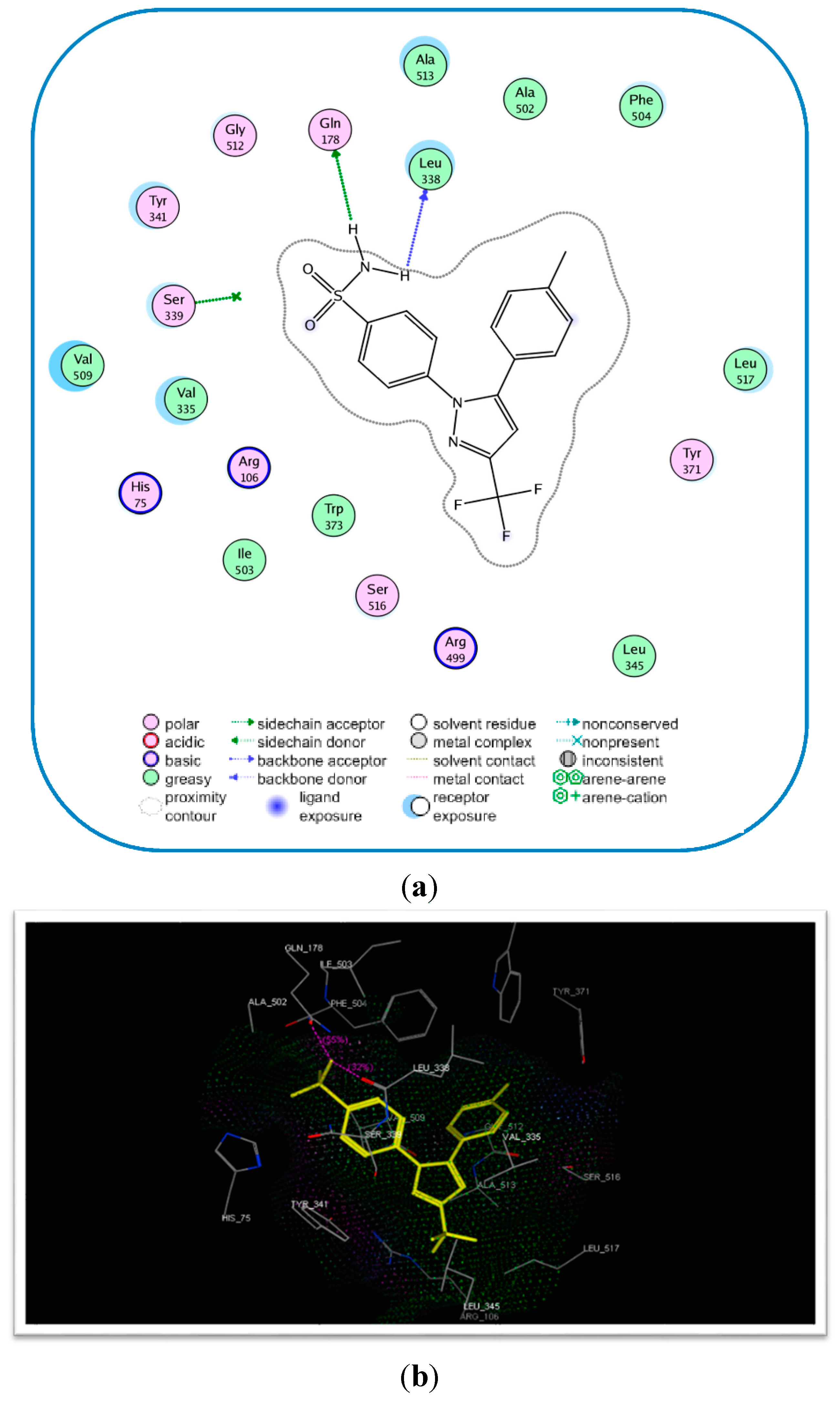
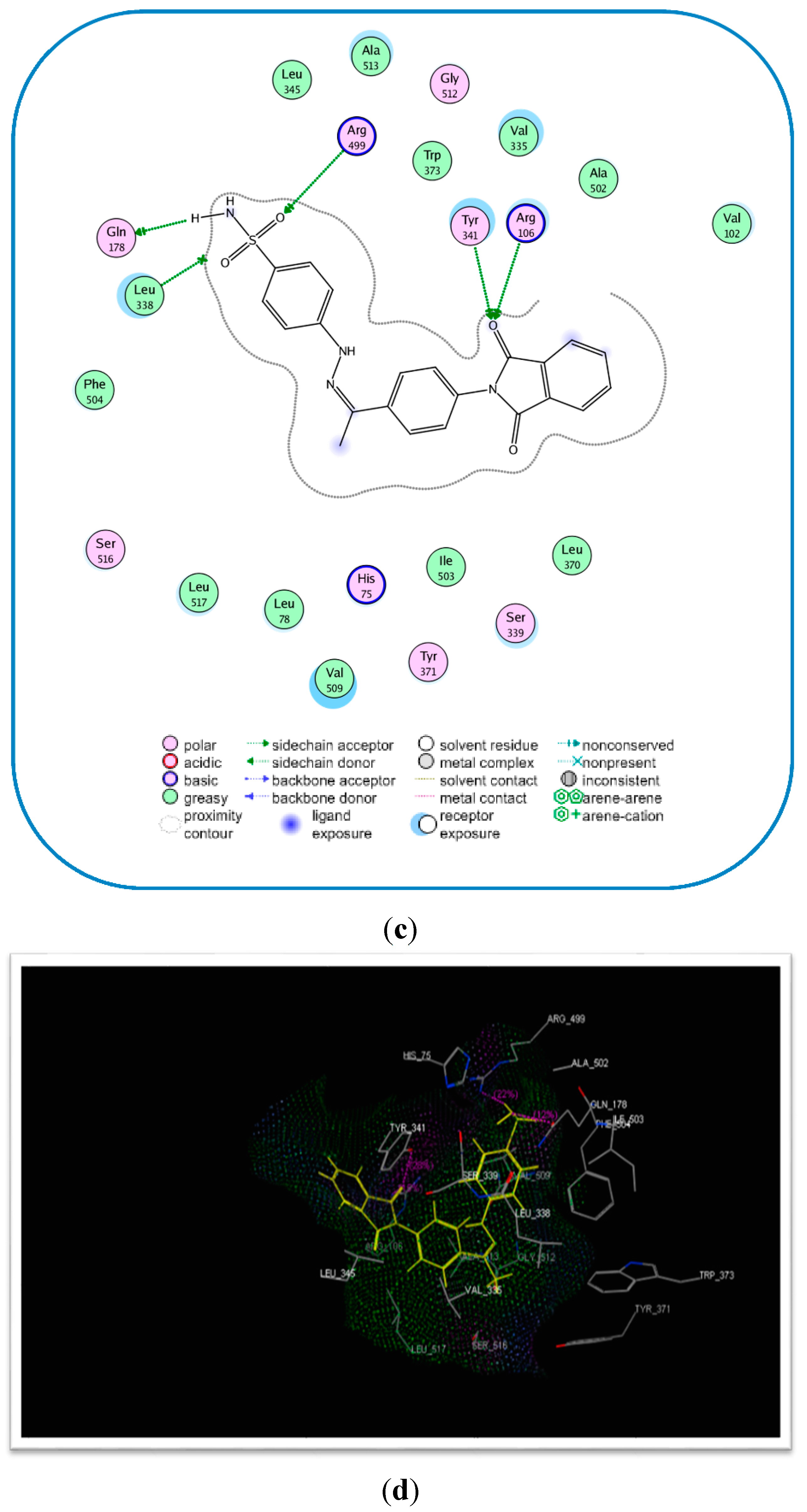
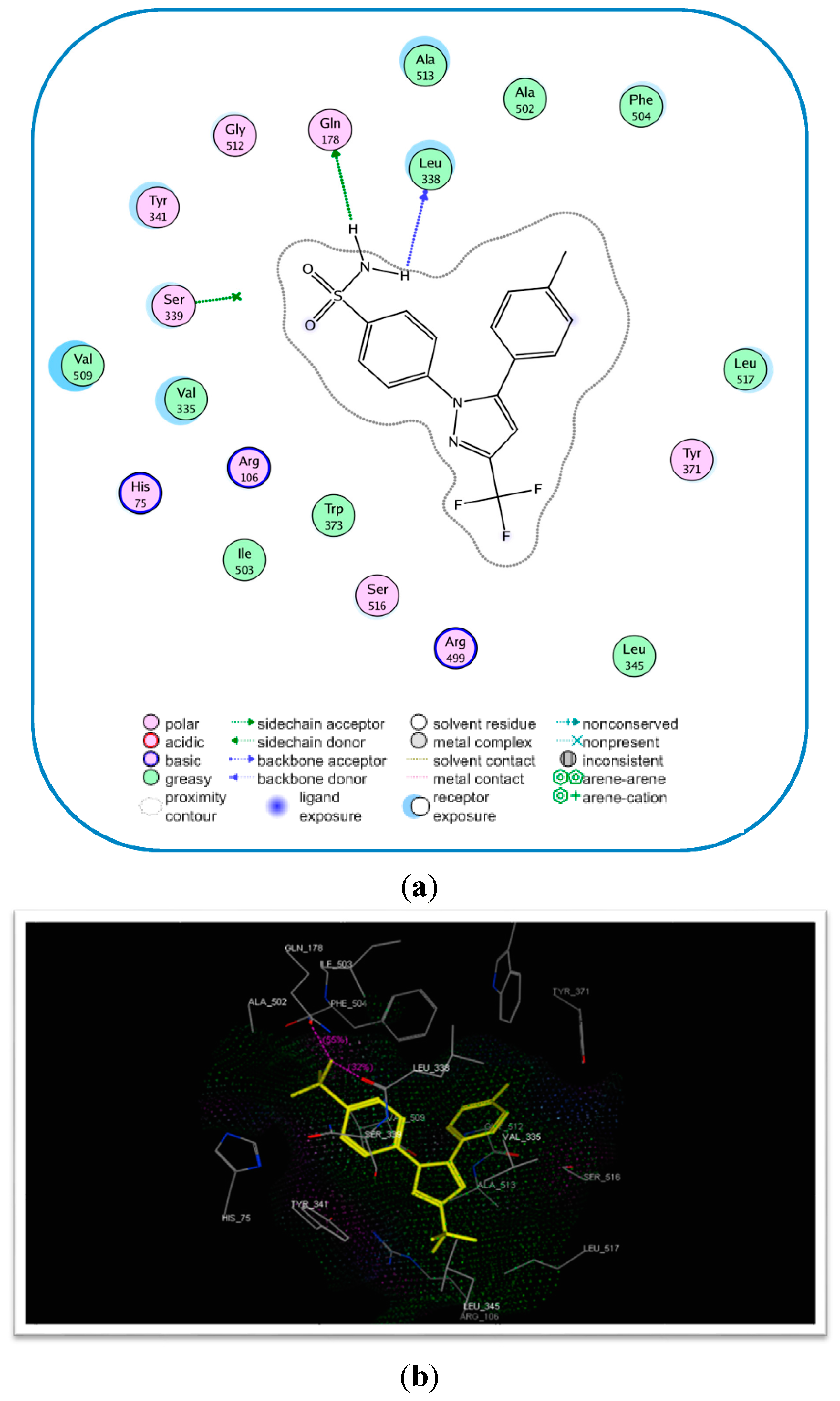
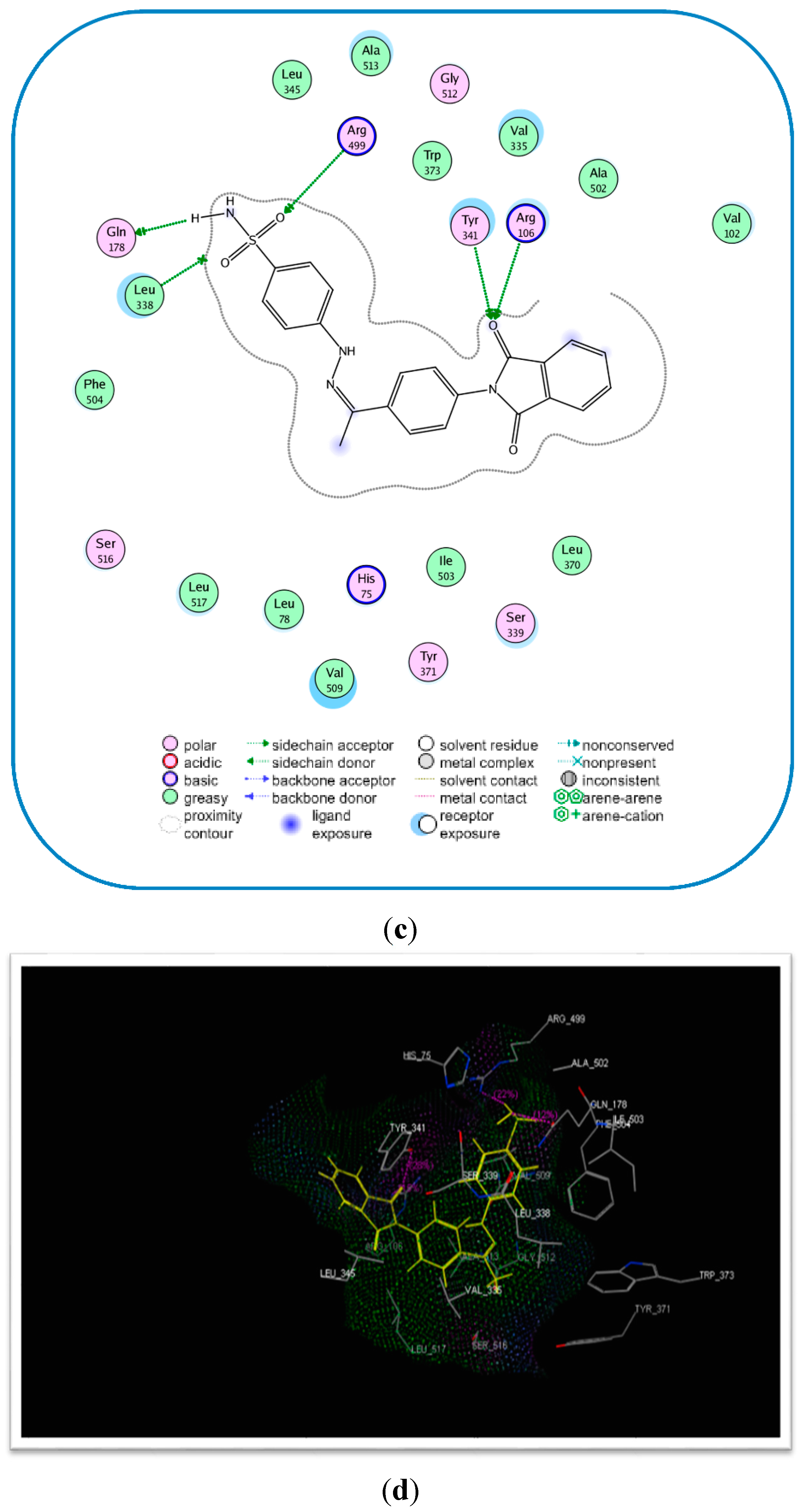
2.3. Molecular Modeling
| Compound No. | E-Refine (kcal/mol) | No. of Hydrogen Bonds | Hydrogen Bonding Residues | Distance (Å) |
|---|---|---|---|---|
| 17c | −17.89 | 4 | Tyr341 (C=O) Arg106 (C=O) Arg499 (S=O) | 2.75 2.77 2.96 |
| Celecoxib (6) | −17.27 | 2 | Gln178 (NH) Leu338 (NH) Gln178 (NH) | 2.21 2.63 2.55 |
3. Experimental Section
3.1. General Information
3.1.1. General Procedure for the Synthesis of Compounds 10a–c
3.1.2. General Procedure for the Synthesis of Compounds 11a–c
3.1.3. Synthesis of (ZE)-2-[4-(1-Hydrazonoethyl)phenyl]isoindoline-1,3-dione (12)
3.1.4. General procedure for the Synthesis of Compounds 13a–c
3.1.5. Synthesis of 2-[4-(3-Methyl-5-thioxo-1,2,4-triazolidin-3-yl)-phenyl]isoindole-1,3-dione (14)
3.1.6. Synthesis of (ZE)-Nʹ-{1-[4-(1,3-Dioxoindolin-2-yl)phenyl]ethylidene}benzohydrazide (15)
3.1.7. Synthesis of 2-[4-(2-Methyl-2H-imidazol-2-yl)phenyl]isoindole-1,3-dione (16)
3.1.8. General Procedure for the Synthesis of Compounds 17a–c
3.2. Biological Testing
3.2.1. Antimicrobial Sensitivity Test Using Agar Diffusion Method (Cup Technique)
Microorganisms
Standard Antimicrobials
Preparation of Sample Stock Solutions
Experimental Procedure
Determination of Minimum Inhibitory Concentration (MIC) Using Agar Dilution Method According to Clinical Laboratory Standards Institute (CLSI)
3.2.2. Anti-Oxidant Testing
Determination of Oxygen Radical Absorbance Capacity (ORAC)
3.2.3. Anti-Inflammatory Testing
Cell Culture
CD62E (E-Selectin, ELAM)-induction Assays
Cell-Surface ELISA ELAM
3.2.4. Cytotoxicity Testing
Molecular Modeling and Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kamal, A.; Bolla, N.R.; Srikanth, P.S.; Srivastava, A.K. Naphthalimide derivatives with therapeutic characteristics: A patent review. Expert Opin. Ther. Pat. 2013, 23, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; El-masry, A.H.; Mohamed, N.A.; Awad, G.E.A.; Habib, B.S. Synthesis, characterization and anti-microbial activity of some novel isoindole-1,3-dione derivatives. Der Pharm. Chem. 2013, 5, 97–108. [Google Scholar]
- El-Gaby, M.S.A.; Zahran, M.A.; Ismail, M.M.F.; Ammar, Y.A.A. A novel Synthesis of dibenzo[c,f]chromenes, dibenzo[c,h]chromenes and benzo[7,8]chromeno[3,4-f]isoindoles as anti-microbial agents. IlFarmaco 2000, 55, 227–232. [Google Scholar] [CrossRef]
- Rajasekaran, S.; Rao, G.K.; Pai, S.; Ranjan, A. Design, synthesis, anti-bacterial and in vitro anti-oxidant activity of substituted 2H-benzopyran-2-one derivatives. Int. J. ChemTech Res. 2011, 3, 555–559. [Google Scholar]
- Pophale, R.A.; Deodhar, M.N. Synthesis and evaluation of novel phthalimide derivatives as analgesic and anti-inflammatory agents. Der Pharm. Chem. 2010, 2, 185–193. [Google Scholar]
- Bhatnagar, A.; Sharma, P.K.; Kumar, N.; Upadhyay, A. Synthesis and in vitro evaluation of anti-oxidant and anti-inflammatory activity of 3-[4,5-dihydro-(5-substituted phenyl)-1H-pyrazol-3-yl]-2H-chromen2-one derivatives. Pharm. Chem. J. 2012, 46, 482–487. [Google Scholar] [CrossRef]
- Bansode, T.N.; Shelke, J.V.; Dongre, V.G. Synthesis and antimicrobial activity of some new N-acyl substitutedphenothiazines. Eur. J. Med. Chem. 2009, 44, 5094–5098. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.J.; Idrees, M.; Khati, N.T.; Dhond, M.G. Synthesis and anti-microbial activities of some new pyrazoles, oxadiazoles and isoxazole bearing benzofuran moiety. S. Afr. J. Chem. 2013, 66, 248–253. [Google Scholar]
- Anthony, P.; Bashir, N.; Parveen, R. Regioselectivesynthesisof 1,4-disubstituted 1,2,3-bistriazoles and their anti-fungal and anti-oxidant evaluation. Asian J. Biomed. Pharm. Sci. 2014, 4, 9–13. [Google Scholar]
- Bosquesi, P.L.; Melo, T.R.F.; Vizioli, E.O.; Santos, J.L.; Chung, M.C. Anti-inflammatory drug design using a molecular hybridization approach. Pharmaceuticals 2011, 4, 1450–1474. [Google Scholar] [CrossRef]
- Kaur, J.; Bhardwaj, A.; Huang, Z.; Knaus, E.E. N-1 and C-3 substituted indole Schiff bases as selective COX-2 inhibitors: synthesis and biological evaluation. Bioorg. Med. Chem. 2012, 22, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Kaplancikli, Z.A.; Altintop, M.D.; Ozdemir, A.; Turan, Z.G.; Khan, S.I.; Tabanca, N. Synthesis and biological evaluation of some hydrazine derivatives as anti-inflammatory agents. Lett. Drug Des. Discov. 2012, 9, 310–315. [Google Scholar] [CrossRef]
- Kushwaha, T.A. Synthesis of novel 2-benzylbenzo[d]thzole-6-sulfonamide derivatives as potential anti-inflammatory agent. J. Chem. Pharm. Sci. 2014, 7, 34–38. [Google Scholar]
- Shiradkar, M.R.; Ghodake, M.; Bothara, K.G.; Bhandari, S.V.; Nikalje, A.; Akula, K.C.; Desai, N.C.; Burange, P.J. Synthesis and anti-convulsant activity of clubbed thiazolidinone-barbituric acid and thiazolidinone–triazole derivatives. ARKIVOC 2007, XIV, 58–74. [Google Scholar]
- Moffet, R.S. Central Nervous System Depressants. VII.1 pyridylcoumarins. J. Med. Chem. 1964, 7, 446–449. [Google Scholar] [CrossRef]
- Mavrova, A.T.; Wesselinova, D.; Tsenov, Y.A.; Denkova, P. Synthesis, cytotoxicity and effects of some 1,2,4-triazole and 1,3,4-thiadiazole derivatives on immunocompetent cells. Eur. J. Med. Chem. 2009, 44, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Pertino, M.W.; Verdugo, V.; Theoduloz, C.; Hirschmann, G.S. Synthesis and anti-proliferative activity of some novel triazole derivatives from dehydroabietic acid. Molecules 2014, 19, 2523–2535. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Shrivastava, B.; Bhatia, R.; Bachwani, M.; Bachwani, M.; Khandelwal, R.; Ameta, J. Exploring potential of 1,2,4-triazole: A brief review. Pharmacol. Online 2011, 1, 1192–1222. [Google Scholar]
- Lamie, P.F. Synthesis and anti-microbial activity of some novel isoindoline-1,3-dione derivatives. J. Adv. Chem. 2014, 8, 1660–1666. [Google Scholar]
- Abdellatif, K.R.; Chowdhury, M.A.; Dong, Y.; Velázquez, C.; Das, D.; Suresh, M.R.; Knaus, E.E. Diazen-1-ium-1,2-diolated nitric oxide donor ester prodrugs of 5-(4-hydroxymethylphenyl)-1-(4-aminosulfonylphenyl)-3-trifluromethyl-1H-pyrazole and its methanesulfonyl analog: Synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. 2008, 16, 9694–9698. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 24 August 2015).
- Abdellatif, K.R.; Huang, Z.; Chowdhury, M.A.; Kaufman, S.; Knaus, E.E. A diazen-1-ium-1,2-diolated nitric oxide donor ester prodrug of 3-(4-hydroxymethylphenyl)-4-(4-methanesulfonylphenyl)-5H-furan-2-one: Synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett. 2011, 21, 3951–3956. [Google Scholar] [CrossRef] [PubMed]
- Seeley, H.W.; Van, D.P.J. A Laboratory Manual of Microbiology, 2nd ed.; Sons and Co: Bombay, India, 1975; pp. 55–80. [Google Scholar]
- Ahmed, S.H.; Amin, M.A.; Saafan, A.E.; El-Gendy, A.O.; El-Gendy, A.O.; Islam, M. Measuring susceptibility of Candida albicans biofilms towards antifungal agents. Der Pharm. Lett. 2013, 5, 376–383. [Google Scholar]
- El-Gendy, A.O.; Essam, T.M.; Amin, M.A.; Ahmed, S.H.; Nes, I.F. Clinical Screening for Bacteriocinogenic Enterococcus faecalis Isolated from Intensive Care Unit Inpatient in Egypt. J. Microb. Biochem. Technol. 2012, 4, 161–167. [Google Scholar] [CrossRef]
- Sader, H.S.; Flamm, R.K.; Jones, R.N. Antimicrobial activity of daptomycin tested against Gram-positive pathogens collected in Europe, Latin America, and selected countries in the Asia-Pacific Region. Diagn. Microbiol. Infect. Dis. 2013, 75, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Tarek, N.; Hassan, H.M.; AbdelGhani, S.M.; Radwan, H.O.; El-Gendy, A.O. Comparative chemical and anti-microbial study of nine essential oils obtained from medicinal plants growing in Egypt. Beni Suef Univ. J. Basic Appl. Sci. 2014, 3, 149–156. [Google Scholar] [CrossRef]
- Ou, B.; Hampsch-Woodill, M.; Prior, R.L. Development and validation of improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food. Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Gridling, M.; Stark, N.; Madlener, S.; Lackner, A.; Popescu, R.; Benedek, B.; Diaz, R.; Tut, F.M.; Nha Vo, T.P.; Huber, D.; et al. In vitro anti-cancer activity of two ethno-pharmacological healing plants from Guatemala Pluchea odorata and Phlebodiumdecumanum. Int. J. Oncol. 2009, 34, 1117–1128. [Google Scholar] [PubMed]
- Madlener, S.; Svacinova, J.; Kitner, M.; Kopecky, J.; Eytner, R.; Lackner, A.; Vo, T.P.; Frisch, R.; Grusch, M.; de Martin, R.; et al. In vitro anti-inflammatory and anti-cancer activities of extracts of Acalypha alopecuroidea (Euphorbiaceae). Int. J. Oncol. 2009, 35, 881–891. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds 9, 12, 14, 15, 17a–c are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamie, P.F.; Philoppes, J.N.; El-Gendy, A.O.; Rarova, L.; Gruz, J. Design, Synthesis and Evaluation of Novel Phthalimide Derivatives as in Vitro Anti-Microbial, Anti-Oxidant and Anti-Inflammatory Agents. Molecules 2015, 20, 16620-16642. https://doi.org/10.3390/molecules200916620
Lamie PF, Philoppes JN, El-Gendy AO, Rarova L, Gruz J. Design, Synthesis and Evaluation of Novel Phthalimide Derivatives as in Vitro Anti-Microbial, Anti-Oxidant and Anti-Inflammatory Agents. Molecules. 2015; 20(9):16620-16642. https://doi.org/10.3390/molecules200916620
Chicago/Turabian StyleLamie, Phoebe F., John N. Philoppes, Ahmed O. El-Gendy, Lucie Rarova, and Jiri Gruz. 2015. "Design, Synthesis and Evaluation of Novel Phthalimide Derivatives as in Vitro Anti-Microbial, Anti-Oxidant and Anti-Inflammatory Agents" Molecules 20, no. 9: 16620-16642. https://doi.org/10.3390/molecules200916620
APA StyleLamie, P. F., Philoppes, J. N., El-Gendy, A. O., Rarova, L., & Gruz, J. (2015). Design, Synthesis and Evaluation of Novel Phthalimide Derivatives as in Vitro Anti-Microbial, Anti-Oxidant and Anti-Inflammatory Agents. Molecules, 20(9), 16620-16642. https://doi.org/10.3390/molecules200916620