Abstract
A concise and expeditious approach to the total synthesis of broussonone A, a p-quinol natural compound, has been developed. The key features of the synthesis include the Grubbs II catalyst mediated cross metathesis of two aromatic subunits, and a chemoselective oxidative dearomatizationin the presence of two phenol moieties. Especially, optimization associated with the CM reaction of ortho-alkoxystyrenes was also studied, which are known to be ineffective for Ru-catalyzed metathesis reactions under conventional reaction conditions because ortho-alkoxy group could coordinate to the ruthenium center, resulting in the potential complication of catalyst inhibition.
1. Introduction
Broussonone A (1), a p-quinol natural product, was first isolated by our group from stem barks of the Korean plant Broussonetia kanzinoki Sieb (Moraceae) [1]. This plant is one of the most abundant trees in Korea, which has been extensively used as diuretic or tonic agents [1,2,3,4,5]. Broussonone A (1) exhibited inhibitory activity on pancreatic lipase with IC50 of 28.4 μM. Due to its structural features (e.g., p-quinol moiety and unrevealed stereochemistry) and interesting biological property, broussonone A lends itself as a challenging synthetic target. To date, there has been no reported total synthesis of the broussonone A.
In planning our approach, we hoped to develop a versatile and practical route that would minimize protecting group manipulations and adapt a platform that leads to a variety of analogues of 1. Herein, we report a facile synthesis of 1 in three steps from readily available starting materials, enlisting a cross metathesis (CM) of two aromatic subunits and a chemoselective oxidative dearomatization of one of two phenol groups.
2. Results and Discussion
The crucial elements of our retrosynthetic analysis of broussonone A (1) are shown in Scheme 1. We envisioned that the late-stage generation of the para-quinol B ring could be achieved through a chemoselective oxidative dearomatization in the electron-rich subunit B of 2. The requisite intermediate 2 could be obtained by a Wittig reaction of the aldehyde 3 or CM of the styrenyl ether 4 with a corresponding olefin, respectively.
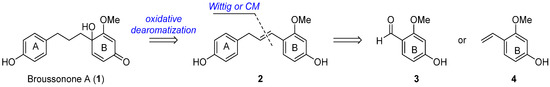
Scheme 1.
Retrosynthetic approach of broussonone A (1).
Scheme 1.
Retrosynthetic approach of broussonone A (1).
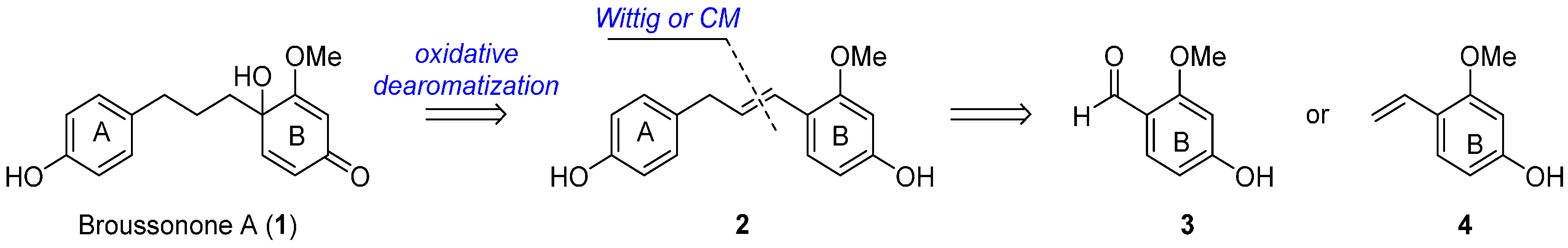
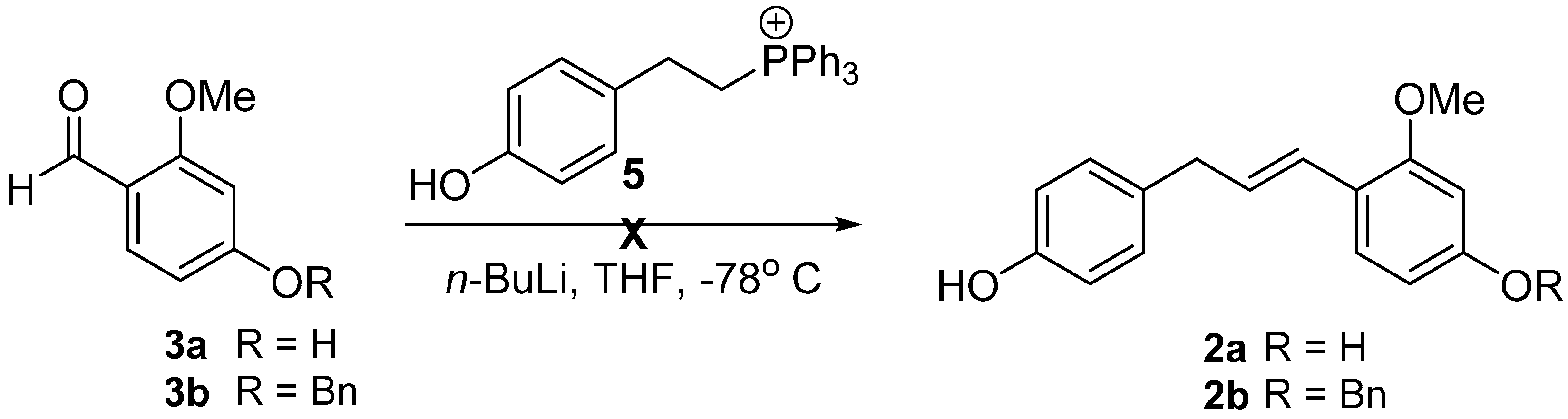
In order to synthesize the compound 2, we commenced our investigation by employing a Wittig reaction between aldehydes 3 and the corresponding phosphonium salt 5, derived from the commercially available 2-(4-hydroxyphenyl) ethanol [6,7]. However, the Wittig reaction failed to afford our desired product 2 presumably due to low reactivity of the electron-rich benzaldehydes 3 (Scheme 2) [8].
Scheme 2.
Wittig reaction to synthesize the key precursors 2.
Scheme 2.
Wittig reaction to synthesize the key precursors 2.
Having failed to access 2 through the Wittig reaction, we turned our attention to the CM strategy for the formation of 2. Grubbs catalysts have been recognized as more efficient and reliable complexes widely utilized in cross metathesis reaction for a variety of alkene chemistry [9,10,11,12]. However, ortho-alkoxystyrenes are known to be ineffective for Ru-catalyzed metathesis reactions under conventional reaction conditions because they could readily form the highly stable Ru-chelate complex 7 discouraging the catalytic cycle [13,14]. Despite the formidable challenge ahead, we were prompted to attempt the CM reaction between 4 and 6 utilizing a Grubbs protocol since it would be the most unambiguous and rapid access to the key intermediate 2 (Scheme 3).
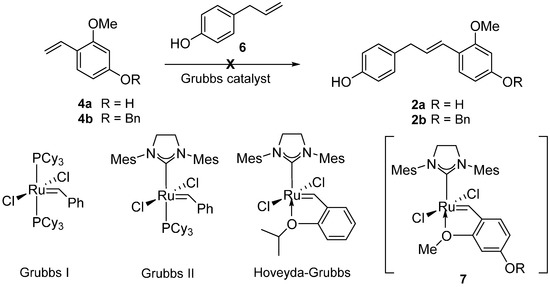
Scheme 3.
Cross metathesis (CM) reaction of 2-alkoxy styrenes 4.
Scheme 3.
Cross metathesis (CM) reaction of 2-alkoxy styrenes 4.
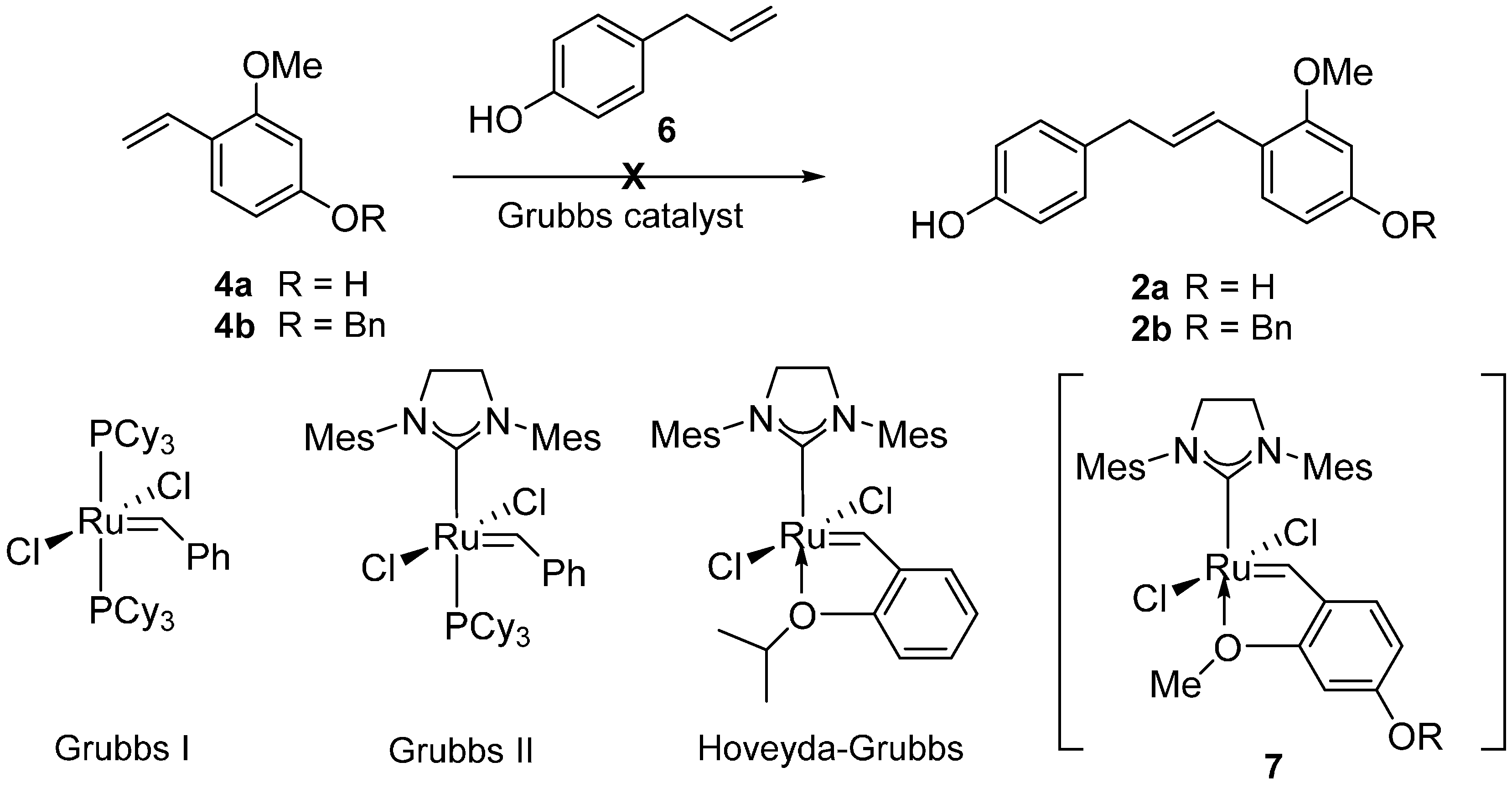
The allylphenol 6 was prepared following a literature procedure [15] and the 2-alkoxystyrene 4 was protected with a benzyl group. An initial CM attempt with the allylphenol 6 and 2-alkoxystyrenes 4 failed to provide the desired CM product and we observed the homodimer of 6 as the major product instead [16,17]. Thus, we were directed to use the homodimer 8 for the key CM reaction partner that was readily synthesized from 6 in 87% yield utilizing Grubbs I catalyst (2 mol %) [18,19].
Efforts were made to optimize the CM reaction outcome using 4b and 8 as summarized in Table 1. Several ruthenium catalysts were tested and to when the Grubbs II catalyst or the Hoveyda-Grubbs II catalyst were employed, gratifyingly we obtained desired product (entries 2–4) in low yields, whereas no product formation was observed when the Grubbs I catalyst was used (entry 1). While varying the amount of catalyst loading; we obtained the best results by using 5 mol % Grubbs II catalyst (entry 4). Further increase of catalyst loading to 10 mol % produced unfavorable results such as olefin migration in 8 (entry 5). The generally preferred solvent in CM reactions such as CH2Cl2 was not suitable for our synthesis because 8 was insoluble in CH2Cl2 (entry 6). Olefin migration of 8 rather occurred in CH2Cl2 or toluene under CM condition. Among the various solvents tested, THF was the most desirable providing the desired CM product in 41% yields. Finally, controlling the stoichiometry of the substrates 8 and 4b, we obtained the best outcome by using 2.0 equivalent of 8 and 1.0 equivalent of 4b (entries 4, 9, and 10). We were pleased to observe that slow addition of Grubbs II catalyst over 3 h delivered 2b in 61% isolated yield (entry 11) [18,19].

Table 1.
Optimization of cross metathesis reaction a. 
| Entry | 8 (equiv.) | Catalyst (mol %) | Solvent | Yield (%) b |
|---|---|---|---|---|
| 1 | 2.0 | Grubbs I (2) | THF | NR c |
| 2 | 2.0 | Grubbs II (2) | THF | 9 |
| 3 | 2.0 | Hoveyda-Grubbs II (2) | THF | 8 |
| 4 | 2.0 | Grubbs II (5) | THF | 41 |
| 5 | 2.0 | Grubbs II(10) | THF | 37 |
| 6 | 2.0 | Grubbs II (5) | CH2Cl2 | 4 |
| 7 | 2.0 | Grubbs II (5) | DCE | 12 |
| 8 | 2.0 | Grubbs II (5) | toluene | 11 |
| 9 | 1.0 | Grubbs II (5) | THF | 30 |
| 10 | 4.0 | Grubbs II (5) | THF | 36 |
| 11 | 2.0 | Grubbs II (5) | THF | 61 d |
a Unless otherwise specified, all the reaction were conducted in the presence of 8, 1.0 equiv. of 4b, and catalyst at 70 °C. b isolated yield. c No reaction. d Catalyst was slowly added over 3 h.
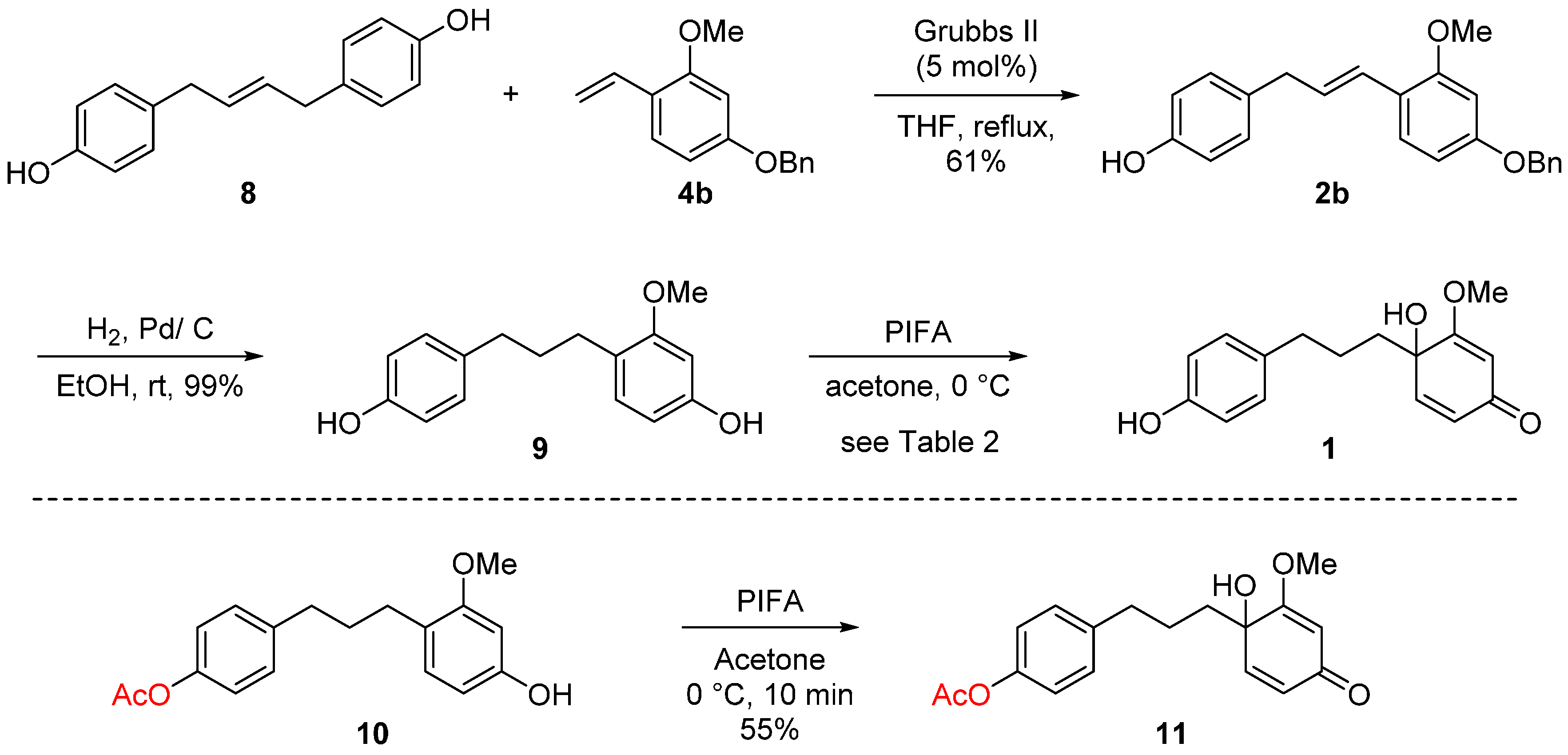
Hydrogenation of 2b with Pd/C catalyst furnished 9 [20] in 99% yield to face the final step in the total synthesis of broussonone A (Table 2). We initially anticipated the disparity in electron density of the two aromatic rings would serve as the key controlling factor for the final chemoselective dearomatization step. In recent years, hypervalent iodine reagents such as PIDA, HDIP, and PIFA have been used as oxidative reagents significantly due to their advantages including ready availability, low toxicity, ease of handling, similar activity to heavy metal catalysts, etc. [21,22,23,24]. The phenyliodine (III) diacetate (PIDA)-mediated oxidative dearomatization afforded broussonone A in low yields and mostly many unidentifiable by-products (entry 1). To our delight, replacement of PIDA with phenyliodine (III) bis(trifluoroacetate) (PIFA) accelerated the reaction and the oxidative conversion of B ring reached over 69% yield (38% yield of the desired compound 1), while at the same time the reaction accompanied the second dearmoatization to yield 12 as a major contaminant in over 31% yield (entry 3). Subsequently, the effects of various solvents were also examined. In CH3CN and THF, 12 was identified as the sole major product (entries 4–5). Using CH3CN/H2O (2/1) or acetone as solvents did not improve the ratio of 1 and 12 (entries 1–3). When the reaction was performed in the presence of 1.0 equiv. of PIFA and 1.0 equiv. of 2 in acetone at 0 °C, the best result was obtained to provide broussonone A (1) in 40% isolated yield along with 31% yield of 12 (entry 6, 71% combined yield) [25]. It should be noted that the oxidative dearomatization of the protected substrates 10 provided the Ac-protected broussonone A 11 in only 55% yield. Considering the additional steps for protection and deprotection, our strategy employing the chemoselective oxidative dearomatization in final stage could be considered competitive (Scheme 4). The spectroscopic properties (1H- and 13C-NMR, HRMS) of the synthetic broussonone A were compatible with those of the natural 1 [1].
Table 2.
Optimization of oxidative dearomatization a. 
| Entry | Hypervalent Iodine | Solvent | Yield (%) b | |
|---|---|---|---|---|
| 1 | 12 | |||
| 1 | PhI(OAc)2 | CH3CN/H2O (2/1) | 7 | 3 |
| 2 | PhI(OAc)2 | Acetone | 16 | 27 |
| 3 | PhI(OCOCF3)2 | CH3CN/H2O (2/1) | 38 | 31 |
| 4 | PhI(OCOCF3)2 | CH3CN | - | >49 |
| 5 | PhI(OCOCF3)2 | THF | - | >49 |
| 6 | PhI(OCOCF3)2 | Acetone | 40 | 31 |
| 7 | PhI(OCOCF3)2 | Acetone/H2O (2/1) | 22 | 21 |
| 8 | PhI(OCOCF3)2 | CH3OH/H2O (2/1) | - | 31 |
a 1.0 equiv. of 9 and 1.0 equiv. of oxidant was used and the reaction was stirred at 0 °C for 30 min. b isolated yield.
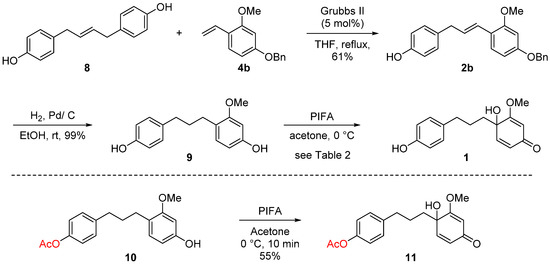
Scheme 4.
Completion of total synthesis of 1.
Scheme 4.
Completion of total synthesis of 1.
3. Experimental Section
3.1. General Information
All manipulations of compounds were performed under a nitrogen atmosphere. THF were obtained from Sigma Aldrich Co. (St. Louis, MO, USA), purified by dried over fresh Na chips. All reactant or reagent was purchased from Sigma Aldrich Co. or Tokyo Chemical Industry (Tokyo Chemical Industry, Tokyo, Japan), and used without purification. Silica gel column chromatography was performed with Silica Gel of Kieselgel 60 F254 plate (Merck Millipore Corporation, Darmstadt, Germany). All 1H-NMR and 13C-NMR spectra of organic products were recorded on Bruker DPX 400 MHz Spectrometer (Bruker Corporation, Karlsruhe, Germany) and Bruker AVANCE 500 MHz Spectrometer (Bruker Corporation, Karlsruhe, Germany). Data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant (Hz), and integration.
3.2. Synthesis
4-Allyl phenol (6). 4-Allylanisole (0.768 mL, 5.00 mmol) was dissolved in CH2Cl2 and then BBr3 (5.50 mL, 5.50 mmol) was added at 0 °C. Then the mixture was stirred at same temperature for 1 h. After the completion of the reaction, the reaction mixture was quenched with H2O and extracted with CH2Cl2. The combined organic phase were dried with Na2SO4 and concentrated in vacuo to give a crude product and it was purified by column chromatography on silica gel (EtOAc/hexanes = 1/3) to afford 4-allylphenol 6 (654 mg, 97.5%). 1H-NMR (CDCl3, 400 MHz) δ 7.06 (d, 2H, J = 8.2 Hz), 6.78 (d, 2H, J = 8.4 Hz), 5.95 (m, 1H), 5.07 (m, 2H), 3.33 (d, 2H, J = 6.7 Hz).
(E)-4,4'-(But-2-ene-1,4-diyl)diphenol (8). To a stirred solution of 4-allylphenol (6) (327 mg, 2.44 mmol) in dried CH2Cl2 was added Grubbs I catalyst (40.1 mg, 48.8 mmol) and the mixture was heated at reflux for 4.5 h. The solvent was removed under reduced pressure and purified by column chromatography on silica gel (EtOAc/hexanes = 1/4) to afforddimer 8 (256 mg, 87.3%). 1H-NMR (CD3OD, 400 MHz) δ 6.95 (d, 4H, J = 8.5 Hz), 6.68 (d, 2H, J = 8.5 Hz), 5.55 (m, 2H), 3.22 (d, 4H, J = 5.0 Hz);13C-NMR (CD3OD, 125 MHz) δ 155.1, 131.6, 130.3, 129.0, 114.7, 37.6; MS m/z (M + H)+ calculated for C16H16O2: 240.1; found 240.1.
4-(Benzyloxy)-2-methoxybenzaldehyde (3a). To a solution of 4-hydroxy-2-methoxybenzaldehyde (3) (200 mg, 1.31 mmol) in DMF was added K2CO3 (543 mg, 3.93 mmol) under N2 atmosphere and mixture was stirred at room temperature for 30 min. BnBr (0.312 mL, 2.62 mmol) was added to reaction mixture and stirred for 1.5 h. After the completion of the reaction, the reaction mixture was diluted with EtOAc and washed with H2O. The organic phase were dried with Na2SO4 and concentrated in vacuo purified by column chromatography on silica gel using (EtOAc/hexanes = 1/2) to afford aldehyde 3a (304 mg, 95.7%). 1H-NMR (CDCl3, 400 MHz) δ 7.81 (d, 1H, J = 8.6 Hz), 7.36 (m, 5H), 6.62 (dd, 1H, J = 8.7, 2.2 Hz), 6.54 (d, 1H, J = 2.2 Hz), 5.14 (s, 2H), 3.89 (s, 3H).
4-(Benzyloxy)-2-methoxy-1-vinylbenzene (4b). Under N2 atmosphere, a solution of t-BuOK (1.0 M solution in THF, 6.00 mL, 6.00 mmol) was added methyltriphenylphosphonium bromide (1.29 g, 3.60 mmol) at 0 °C and stirred at room temperature for 30 min. Then, the reaction mixture was cooled to 0 °C and a solution of 4a (291 mg, 1.20 mmol) in dry THF was added to reaction mixture and stirred for 4 h. After the completion of the reaction, the reaction mixture was quenched with saturated NH4Cl and extracted with CH2Cl2. The organic phase were dried with Na2SO4 and concentrated in vacuo and purified by column chromatography on silica gel (EtOAc/hexanes = 1/3) to afford styrene 4b (176 mg, 61.2%).1H-NMR (CD3OD, 400 MHz) δ 7.35 (m, 6H), 6.90 (dd, 1H, J = 17.8 Hz), 6.53 (m, 2H), 5.57 (dd, 1H, J = 17.8, 1.7 Hz), 5.04 (dd, 1H, J = 11.2, 1.7 Hz), 5.04 (s, 2H), 3.76 (s, 3H); 13C-NMR (CD3OD, 125 MHz) δδ 159.9, 157.9, 137.3, 131.1, 128.1, 127.5, 127.2, 126.7, 119.8, 110.7, 105.9, 98.7, 69.7, 54.6; MS m/z (M + H)+ calculated for C16H16O2: 240.1; found 240.1.
(E)-4-(3-(4-(Benzyloxy)-2-methoxyphenyl)allyl)phenol (2b). To a stirred solution of (E)-4,4'-(but-2-ene-1,4-diyl)diphenol (8) (200 mg, 0.832 mmol) and 4-(benzyloxy)-2-methoxy-1-vinylbenzene (4b) (100 mg, 0.416 mmol) in anhydrous THF was added Grubbs II catalyst (17.7 mg, 0.416 mmol) and the mixture was heated at reflux for 16 h. After the completion of the reaction, the solvent was removed under reduced pressure and the resulting residue was subjected to silica gel chromatography (EtOAc/hexanes = 1/6) to afford alkene 2b (87.8 mg, 61.0%).1H-NMR (CD3OD, 400 MHz) δ 7.43–7.27 (m, 6H), 7.01 (d, 2H, J = 8.6 Hz), 6.69 (d, 2H, J = 8.6 Hz), 6.55 (m, 3H), 6.16 (m, 1H), 5.05 (s, 2H), 3.78 (s, 3H), 3.38 (d, 2H, J = 6.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 159.2, 157.5, 153.8, 136.9, 133.0, 129.7, 128.6, 128.2, 128.0, 127.5, 127.2, 125.1, 119.9, 115.2, 105.7, 99.4, 70.2, 55.5, 39.0; MS m/z (M + H)+ calculated for C23H22O3: 346.2; found 346.2.
4-(3-(1-Hydroxy-2-methoxy-4-oxocyclohexa-2,5-dien-1-yl)propyl)phenyl acetate (11). To a solution of 4-(3-(4-hydroxy-2-methoxyphenyl)propyl)phenyl acetate (10) (30 mg, 0.10 mmol) in acetone (0.5 mL) at 0 °C was treated with PIFA (43 mg, 0.1 mmol) and stirred for 10 min at 0 °C. After the completion of the reaction, the reaction mixture was diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried with Na2SO4, concentrated in vacuo and purified by column chromatography on silica gel (EtOAc/hexanes = 1/1) to afford p-quinol 11 (17 mg, 55%). 1H-NMR (CD3OD, 400 MHz) δ 7.11 (d, 2H, J = 8.6 Hz), 6.98 (d, 2H, J = 8.5 Hz), 6.57 (d, 1H, J = 10.0 Hz), 6.16 (dd, 1H, J = 10.0, 1.6 Hz), 5.51 (d, 1H, J = 1.6 Hz), 3.75 (s, 3H), 2.55 (m, 2H), 2.28 (s, 3H), 1.95 (m, 1H), 1.80 (m, 1H), 1.37 (m, 1H), 0.84 (m, 1H); HRMS m/z (M + H)+ calculated for C18H20O5: 317.1384; found 317.1386.
4-(3-(4-Hydroxyphenyl)propyl)-3-methoxyphenol (9). To a solution of alkene 2b (101 mg, 0.292 mmol) and 10% Pd/C (12.4 mg, 0.117 mmol) in EtOH, hydrogen gas was fluxed for 10 min at room temperature. The reaction mixture was stirred under H2 atmosphere for 16 h at room temperature, then filtered over Celite, the solvent was discarded and the residue purified by column chromatography on silica gel (EtOAc/hexanes = 1/6), to afford phenol 9 (74.7 mg, 99.0%). 1H-NMR (CD3OD, 400 MHz) δ 6.95 (d, 2H, J = 8.5 Hz), 6.84 (d, 1H, J = 8.1 Hz), 6.66 (d, 2H, J = 8.5 Hz), 6.36 (d, 1H, J = 2.3 Hz), 6.27 (dd, 1H, J = 8.1, 2.3 Hz), 3.73 (s, 3H), 2.47 (m, 4H), 1.75 (m, 2H); 13C-NMR (CD3OD, 125 MHz) δ 158.3, 156.3, 154.8, 133.4, 129.7, 128.8, 121.4, 114.6, 106.1, 98.4, 54.2, 34.4, 32.2, 28.9; HRMS m/z (M + H)+ calculated for C16H18O3: 259.1334; found 259.1316.
Broussonone A (1). To a solution of 4-(3-(4-hydroxyphenyl)propyl)-3-methoxyphenol (9) (26.0 mg, 0.10 mmol) in acetone (1.0 mL) at 0 °C was treated with PIFA (42.0 mg, 0.10 mmol) and stirred for 30 min at 0 °C. The reaction mixture was diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The aqueous phase was extracted with CH2Cl2. The combined organic extracts were dried with Na2SO4, concentrated in vacuo and purified by column chromatography on silica gel (5% MeOH in CH2Cl2) to afford the synthetic broussonone A (1) (11.0 mg, 40%). 1H-NMR (CD3OD, 500 MHz) δ 6.91 (d, 2H, J = 8.5 Hz), 6.66 (d, 2H, J = 8.5 Hz), 6.61 (d, 1H, J = 10.0 Hz), 6.07 (dd, 1H, J = 10.0 Hz, 1.7 Hz), 5.52 (d, 1H, J = 1.7 Hz), 3.76 (s, 3H), 2.46 (m, 2H), 1.95 (m, 1H), 1.70 (m, 1H), 1.49 (m, 2H); 13C-NMR (CD3OD, 125 MHz) δδ 188.8, 177.1, 155.1, 148.7, 132.4, 128.9, 126.7, 114.7, 100.7, 70.8, 55.2, 37.6, 34.2, 25.3; HRMS m/z (M + H)+ calculated for C16H18O4: 274.1267; found 274.1267.
For more details of NMR spectra, please see Supplementary Materials Figures S1–S17.
4. Conclusions
In summary, we have successfully demonstrated the first total synthesis of broussonone A in three steps from the known compound 4b with a 24% overall yield. The key features of this synthetic route involve the following: (1) the Grubbs II catalyst mediated cross metathesis of two aromatic subunits; (2) the chemoselective oxidative dearomatization in the presence of two phenol moieties. These studies provide a timely contribution to the development of a practical synthetic approach to a variety of broussonone A analogues. With this practical synthesis of broussonone A now in hand, the intensive exploration of the absolute configuration of the broussonone A as well as its structural analogues will be extended.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/09/15966/s1.
Acknowledgments
This research was supported by the Medical Research Center Program (2008-0062275), the research grant of Chungbuk National University in 2012, the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2012R1A1A2038433) (to Y.-S.K.) and the Ministry of Land, Transport and Maritime Affairs, Korea (to Y.-J.L.).
Author Contributions
H.J., M.C., M.V., and Y.H.L. performed the experiments and analyzed all data. Y.-S.K. and H.L conceived and designed the experiments, K.L., N.S.C., and Y.-J.L. analyzed the HRMS data. J.T.H. and M.K.L. suggested the research work. J.-K.J. conceived and directed the investigations and composed the manuscript with revisions provided by the other authors. All authors read and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Ahn, J.H.; Liu, Q.; Lee, C.; Ahn, M.J.; Yoo, H.S.; Hwang, B.Y.; Lee, M.K. A new pancreatic lipase inhibitor from Broussonetia kanzinoki. Bioorg. Med. Chem. Lett. 2012, 22, 2760–2763. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Shin, E.; Liu, Q.; Kim, S.B.; Choi, K.M.; Yoo, H.S.; Hwang, B.Y.; Lee, M.K. Secoiridoids from the stem barks of Fraxinus rhynchophylla with pancreatic lipase inhibitory activity. Nat. Prod. Res. 2013, 27, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Andrae-Marobela, K.; Ghislain, F.W.; Okatch, H.; Majinda, R.R.T. Polyphenols: A Diverse Class of Multi-Target Anti-HIV-1 Agents. Curr. Drug Metab. 2013, 14, 392–413. [Google Scholar] [CrossRef] [PubMed]
- Kato, E.; Nakagomi, R.; Gunawan-Puteri, M.D.P.T.; Kawabata, J. Identification of hydroxychavicol and its dimers, the lipase inhibitors contained in the Indonesian spice, Eugenia polyantha. Food Chem. 2013, 136, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Kato, E.; Yama, M.; Nakagomi, R.; Shibata, T.; Hosokawa, K.; Kawabata, J. Substrate-like water soluble lipase inhibitors from Filipendula kamtschatica. Bioorg. Med. Chem. Lett. 2012, 22, 6410–6412. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Li, Z.; Tang, R.; Jia, H.; Liu, B. Novel (E)-5-styryl-2,2′-bithiophene derivatives as ligands for β-amyloid plaques. Eur. J. Med. Chem. 2011, 46, 2908–2916. [Google Scholar] [CrossRef] [PubMed]
- Grice, C.A.; Tays, K.L.; Savall, B.M.; Wei, J.; Butler, C.R.; Axe, F.U.; Bembenek, S.D.; Fourie, A.M.; Dunford, P.J.; Lundeen, K.; et al. Identification of a Potent, Selective, and Orally Active Leukotriene A4 Hydrolase Inhibitor with Anti-Inflammatory Activity. J. Med. Chem. 2008, 51, 4150–4169. [Google Scholar] [CrossRef] [PubMed]
- With electron-deficient aldehyde such as nitrobenzaldehyde, the reaction occurred efficiently with 5.
- Grubbs, R.H. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Volumes 1–3. [Google Scholar]
- Alcaide, B.; Almendros, P.; Luna, A. Grubbs’ Ruthenium-Carbenes Beyond the Metathesis Reaction: Less Conventional Non-Metathetic Utility. Chem. Rev. 2009, 109, 3817–3858. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Vila, A.M.; Monsaert, S.; Bajek, A.; Verpoort, F. Ruthenium-Based Olefin Metathesis Catalysts Derived from Alkynes. Chem. Rev. 2010, 110, 4865–4909. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J. S.; Grubbs, R.H. Alkene Chemoselectivity in Ruthenium-Catalyzed Z-Selective Olefin Metathesis. Angew. Chem. Int. Ed. 2013, 52, 9001–9004. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J.; Hoveyda, A.H. A Recyclable Ru-Based Metathesis Catalyst. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Tzeng, S.-C.; Liu, Y.-C. Peroxidase-catalyzed synthesis of neolignan and itsanti-inflammatory activity. J. Mol. Catal. B: Enzym. 2004, 32, 7–13. [Google Scholar] [CrossRef]
- Chatterjee, A.K.; Choi, T.L.; Sanders, D.P.; Grubbs, R.H. A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. [Google Scholar] [CrossRef] [PubMed]
- Gresser, M.J.; Wales, S.M.; Keller, P.A. The attempted stereoselective synthesis of chiral 2,2ʹ-biindoline. Tetrahedron 2010, 66, 6965–6976. [Google Scholar] [CrossRef]
- Previously, the ortho-alkoxy-β-methyl styrenes instead of the simple ortho-alkoxystyrenes have been used for this type CM reactions, see: Strych, S.; Trauner, D. Biomimetic Synthesis of Santalin A,B and Santarubin A,B, the Major Colorants of Red Sandalwood. Angew. Chem. Int. Ed. 2013, 52, 9509–9512. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.K.; Toste, D.; Choi, T.-L.; Grubbs, R.H. Ruthenium-Catalyzed Olefin Cross Metathesis of Styrenes as an Alternative to the Heck and Cross-Coupling Reactions. Adv. Synth. Catal. 2002, 344, 634–637. [Google Scholar] [CrossRef]
- Compound 9 is also known as broussonin B of which the only synthetic study was reported, see: de Almeida, P.A.; Fraiz, S.V.; Braz-Filho, R. Synthesis and Structural Confirmation of Natural 1,3-Diarylpropanes. J. Braz. Chem. Soc. 1999, 10, 347–353. [Google Scholar] [CrossRef]
- Rappoport, Z. The Chemistry of Phenols; Wiley: Chichester, UK, 2003; Volumes 1–2. [Google Scholar]
- Hata, K.; Hamamoto, H.; Shiozaki, Y.; Cammerer, S.B.; Kita, Y. Nucleophilic attack of intramolecular hydroxyl groups onelectron-rich aromatics using hypervalent iodine(III) oxidation. Tetrahedron 2007, 63, 4052–4060. [Google Scholar] [CrossRef]
- Plourde, G.L. Studies towards the diastereoselective spiroannulation of phenolic derivatives. Tetrahedron Lett. 2002, 43, 3597–3599. [Google Scholar] [CrossRef]
- Felpin, F.X. Oxidation of 4-arylphenol trimethylsilyl ethers to p-arylquinols using hypervalent iodine(III) reagents. Tetrahedron Lett. 2007, 48, 409–412. [Google Scholar] [CrossRef]
- The Oxone-mediated oxidative dearomitization of the corresponding ketone-contaning phenol has been reported, see: Barrada, S.; Hernandez-Torres, G.; Urbano, A.; Carreno, M.C. Total Synthesis of Natural p-Quinol. Org. Lett. 2012, 14, 5952–5955. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Sample of the compound 1 is available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).