
Mimosine Dipeptide Enantiomsers: Improved Inhibitors against Melanogenesis and Cyclooxygenase

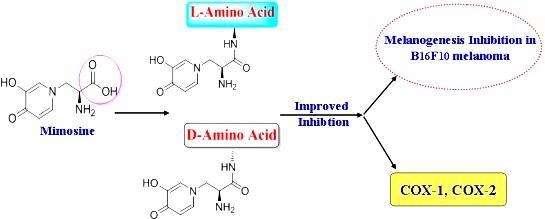
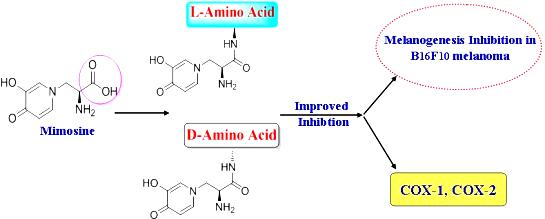
Abstract
:
1. Introduction
2. Results and Discussion
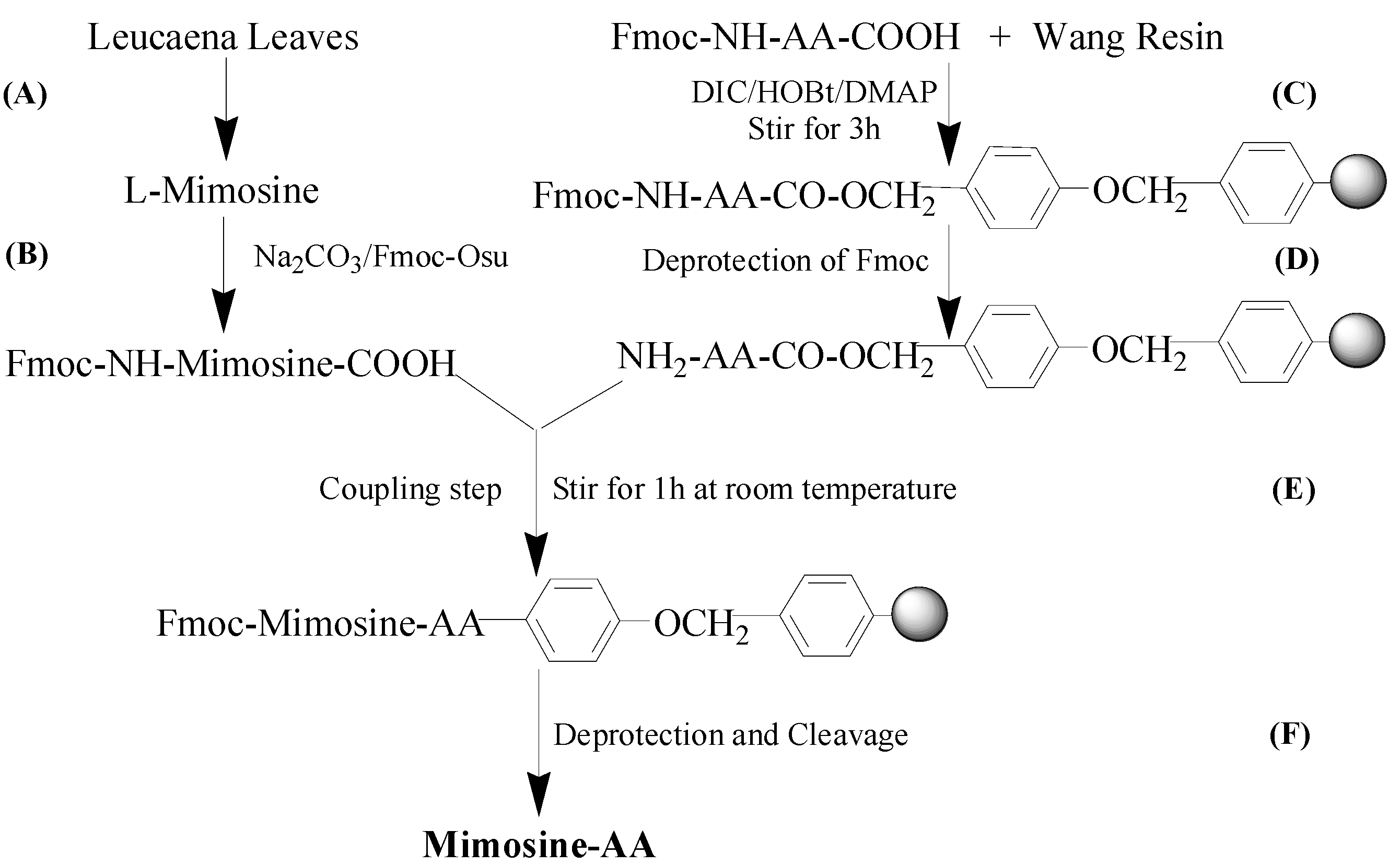
2.1. Synthesis and Tyrosinase Inhibition of Mimosine Dipeptides
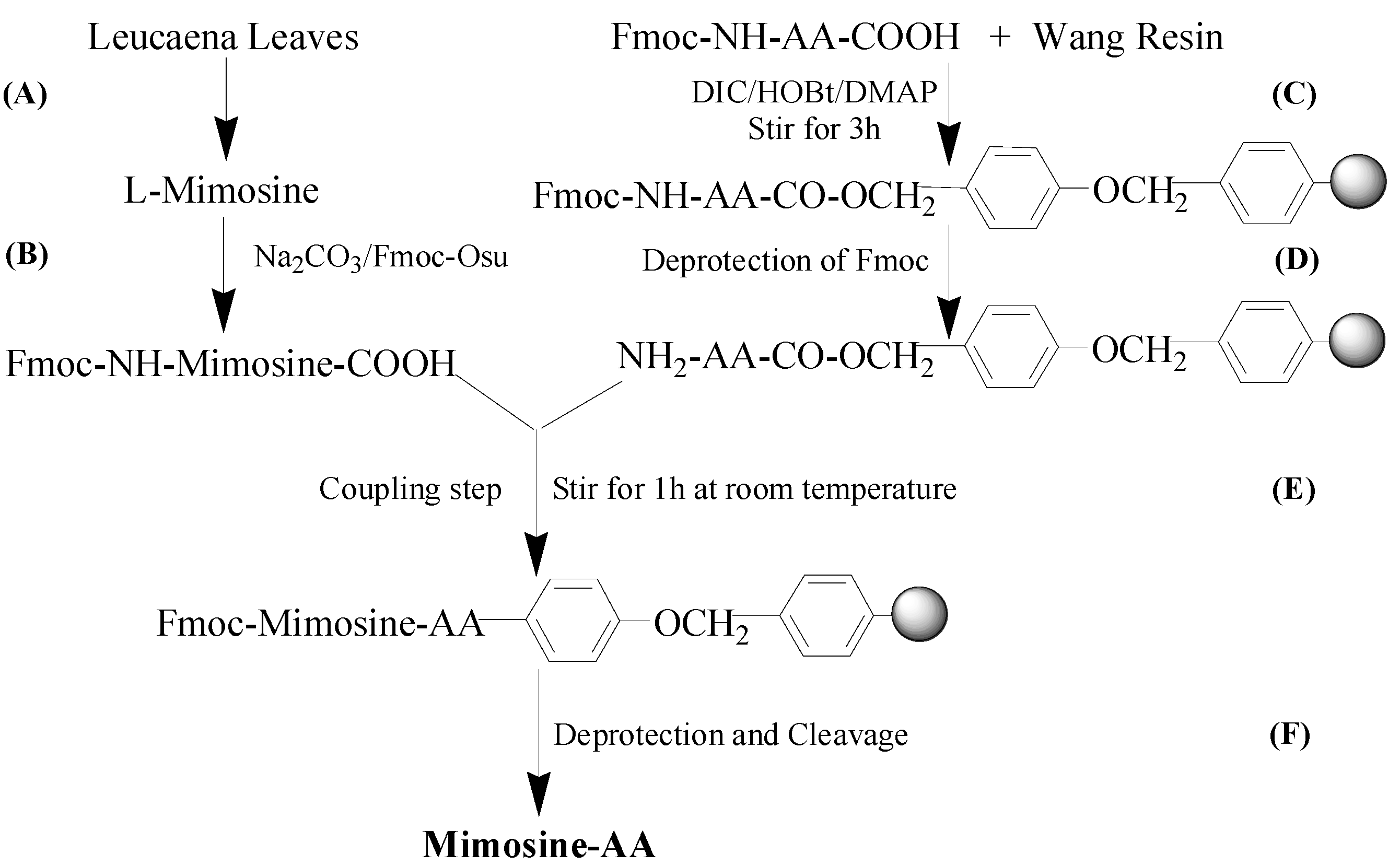


{kind=link}
{kind=link}
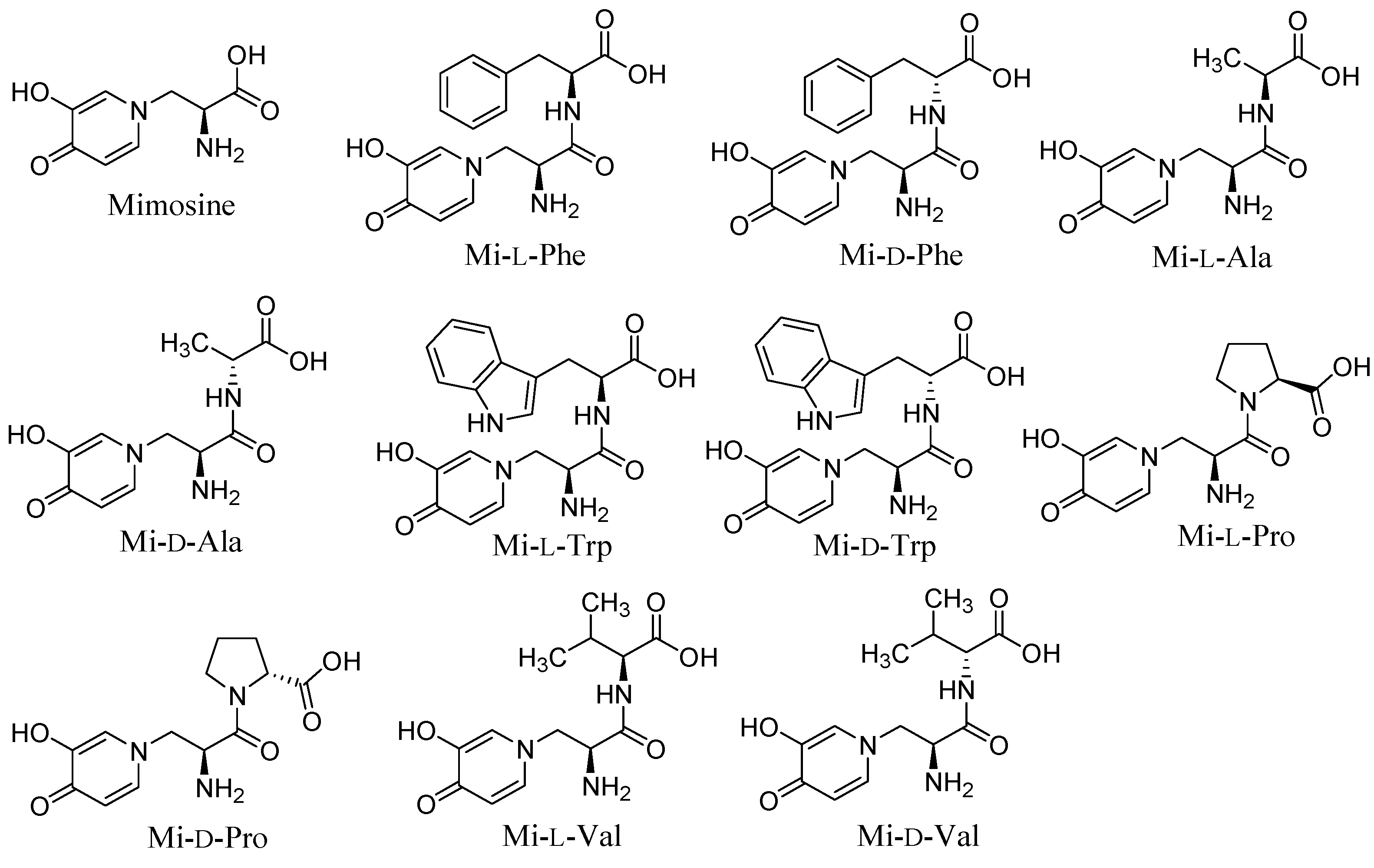{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) Mushroom Tyrosinase Inhibition |
|---|---|
| Mi-l-Phe | 24.1 ± 3.8 ab |
| Mi-d-Phe | 19.7 ± 0.8 bc |
| Mi-l-Ala | 23.1 ± 2.2 b |
| Mi-d-Ala | 17.7 ± 3.1 bc |
| Mi-l-Pro | 13.1 ± 3.3 bc |
| Mi-d-Pro | 17.2 ± 2.1 bc |
| Mi-l-Val | 11.5 ± 0.5 c |
| Mi-d-Val | 10.3 ± 0.4 c |
| Mi-l-Trp | 19.2 ± 1.5 bc |
| Mi-d-Trp | 16.9 ± 2.1 bc |
| Mimosine | 32.4 ± 1.1 ª |
| Kojic acid | 13.7 ± 1.5 bc |
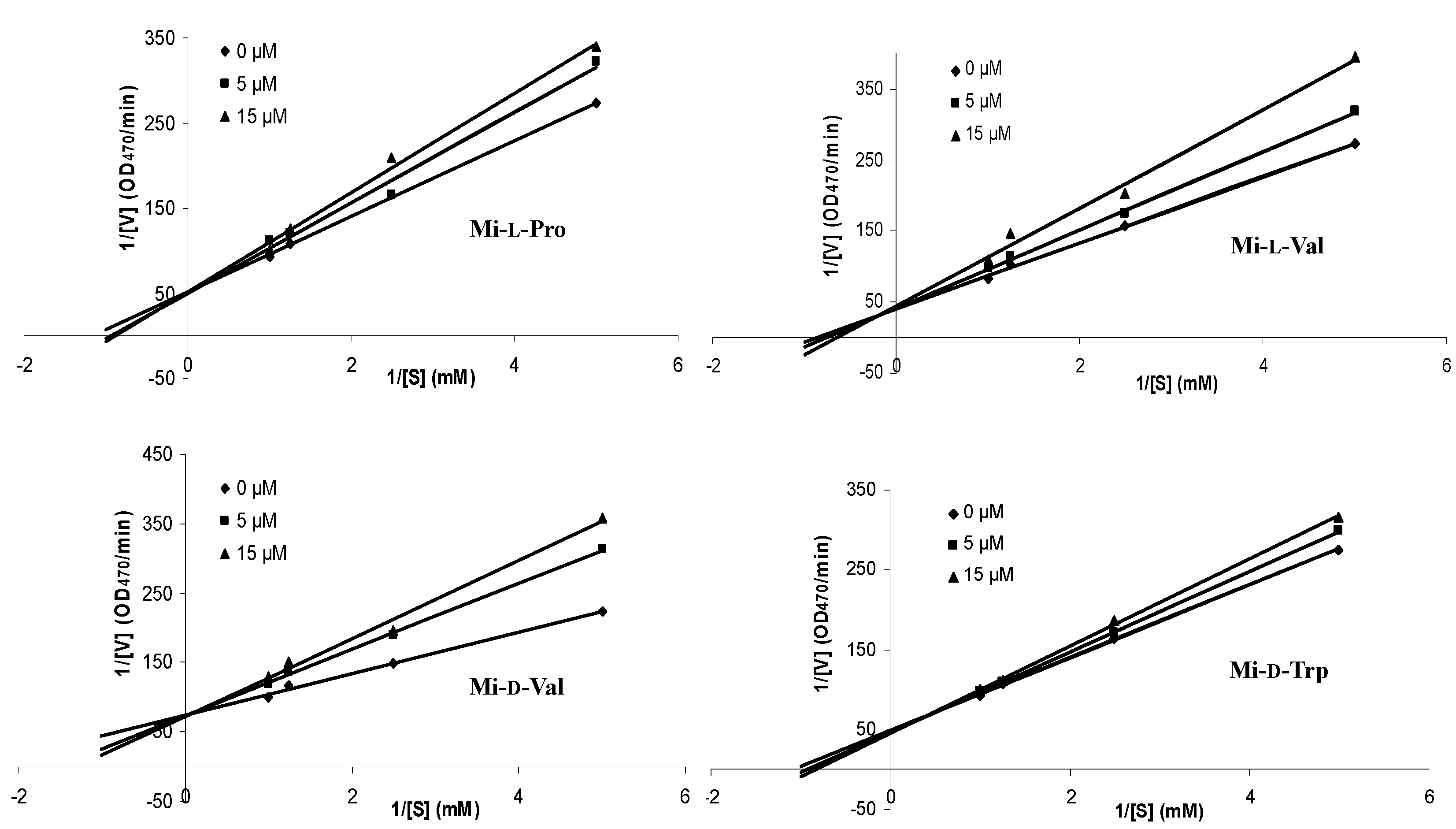
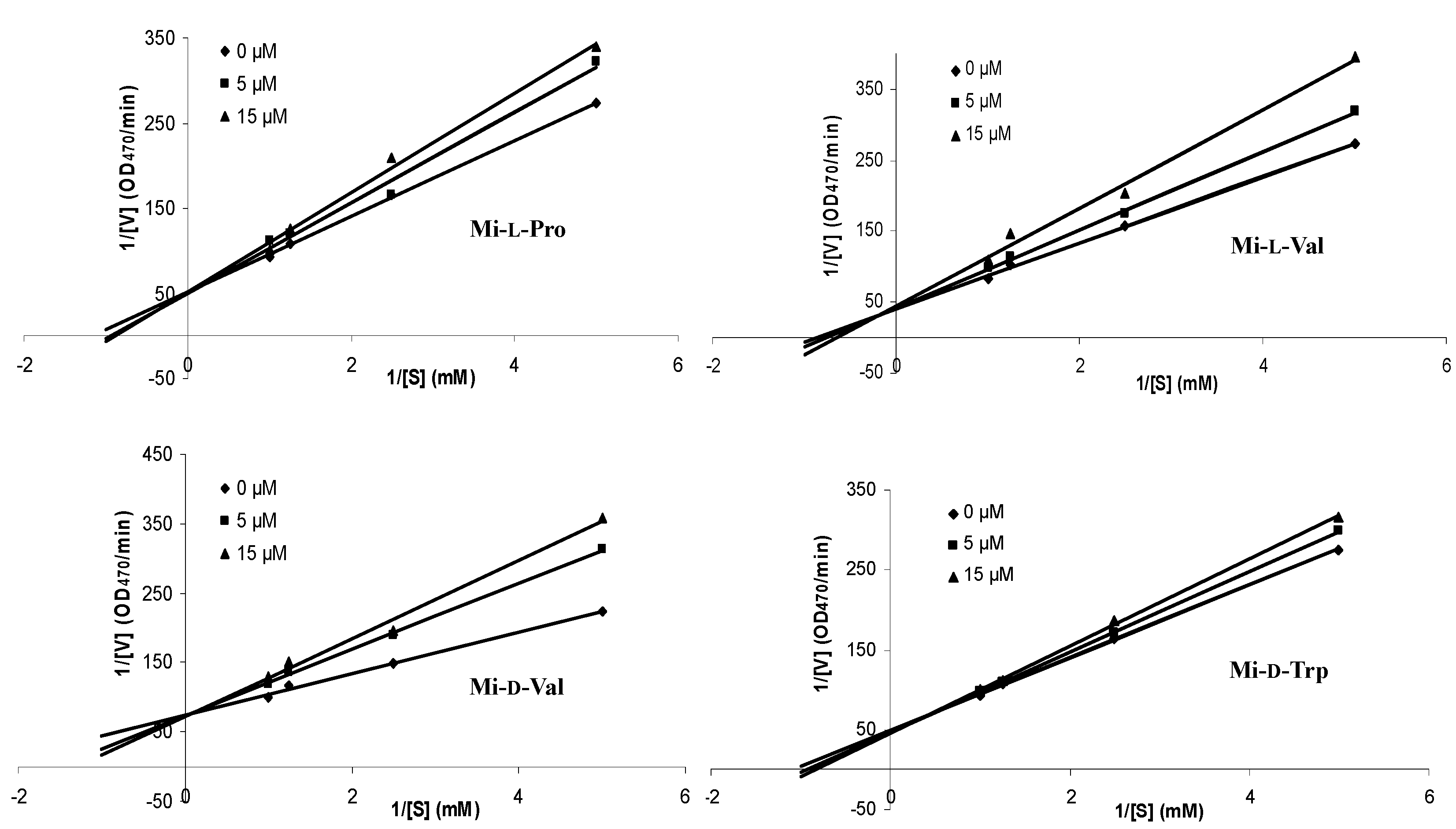
| Compound | Inhibition Type | Inhibition Constant (Ki) (mM) |
|---|---|---|
| Mi-l-Pro | competitive | 0.05 |
| Mi-l-Val | competitive | 0.03 |
| Mi-d-Val | competitive | 0.02 |
| Mi-d-Trp | competitive | 0.08 |
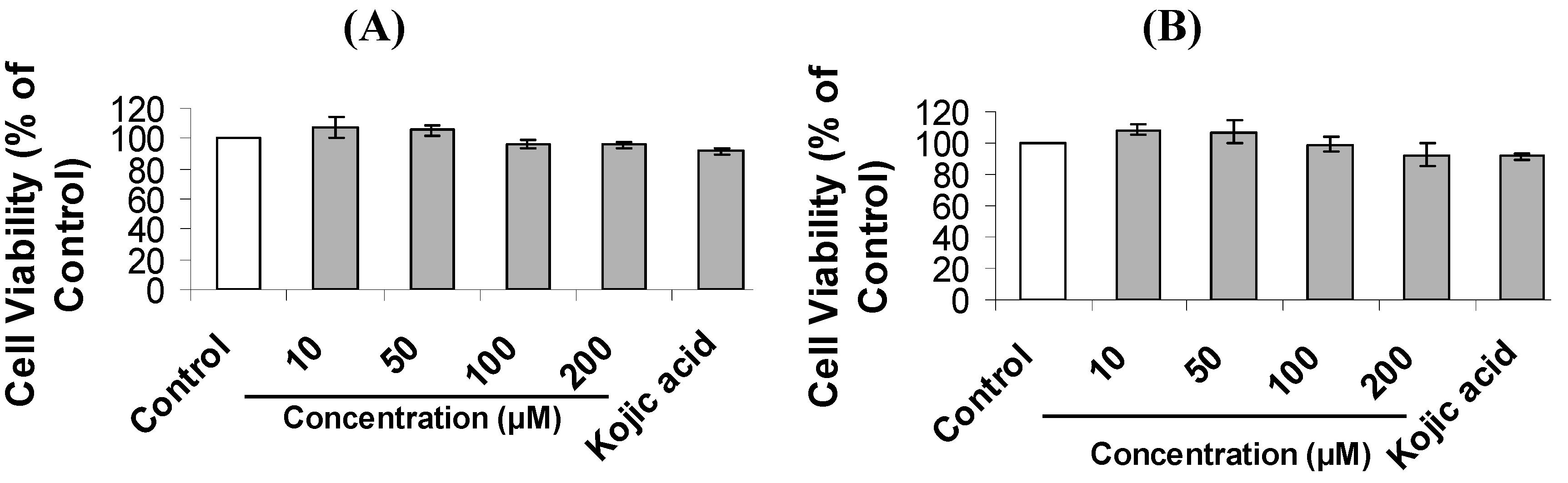
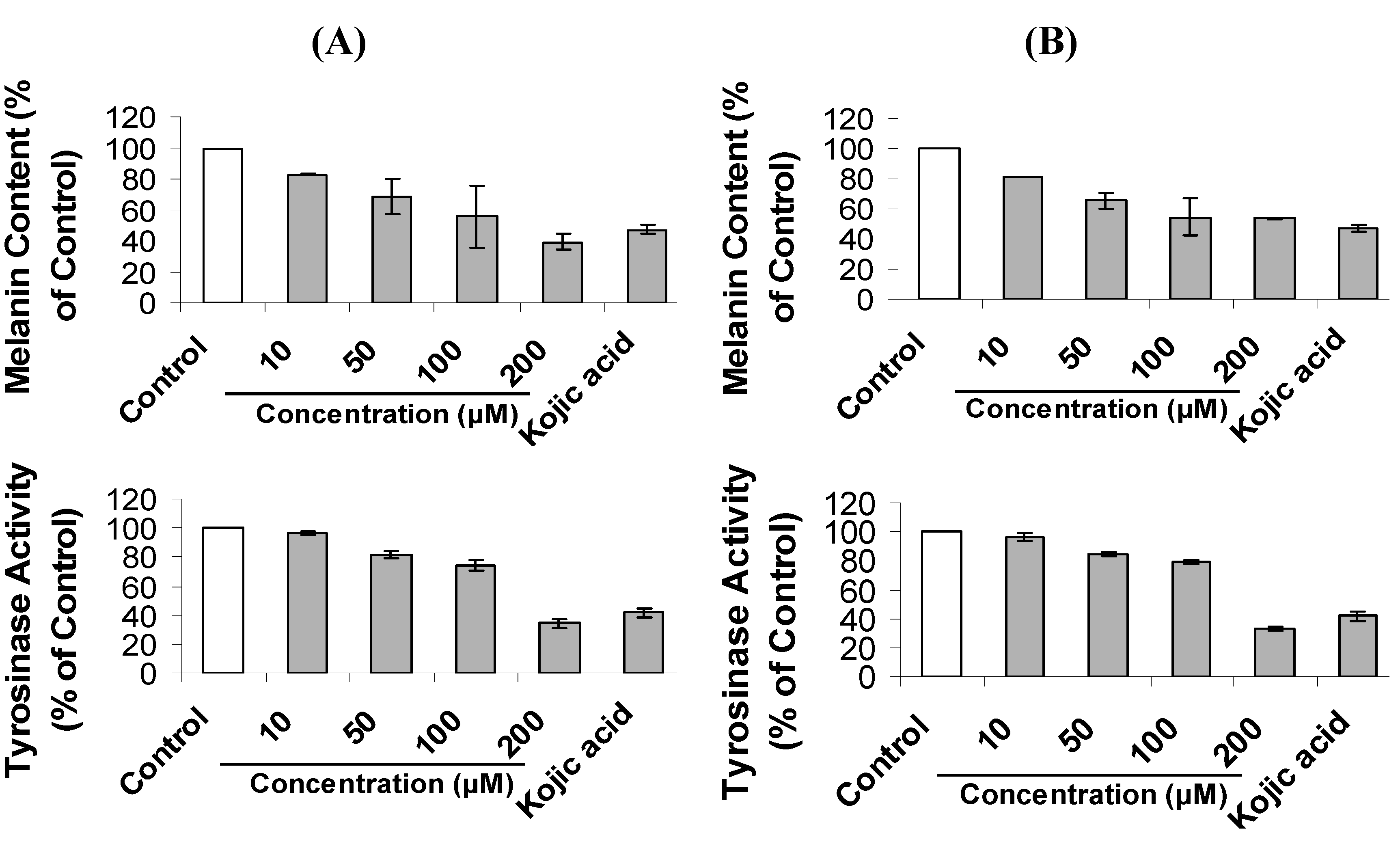
2.2. Inhibition of Melanogenesis by Mimosine Dipeptides
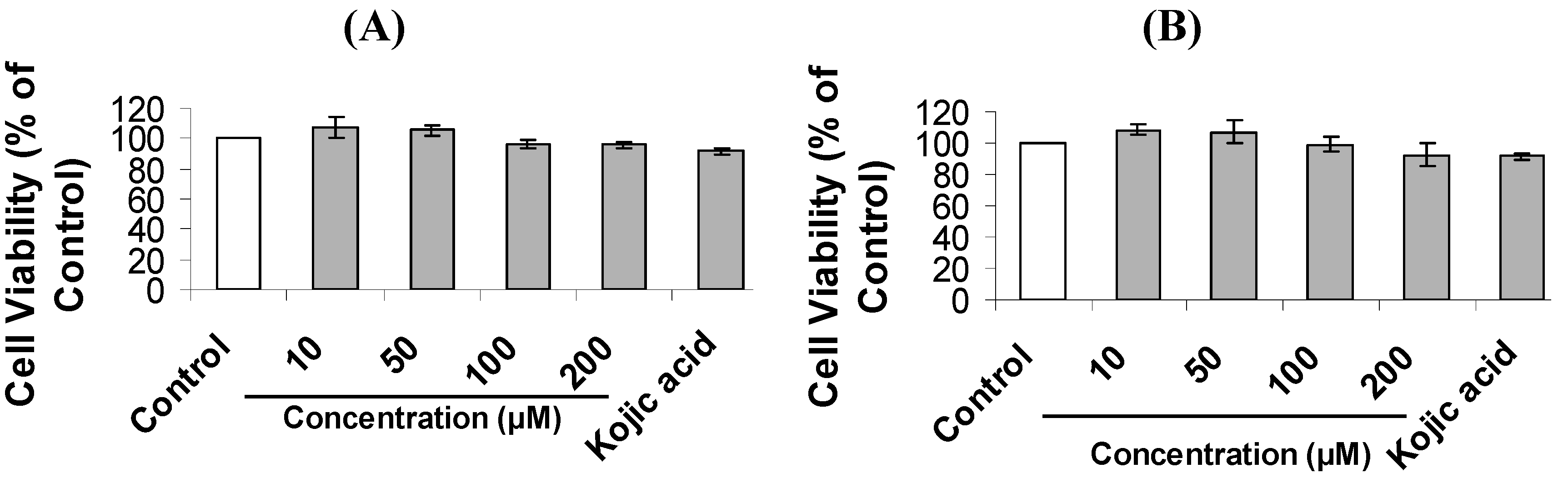
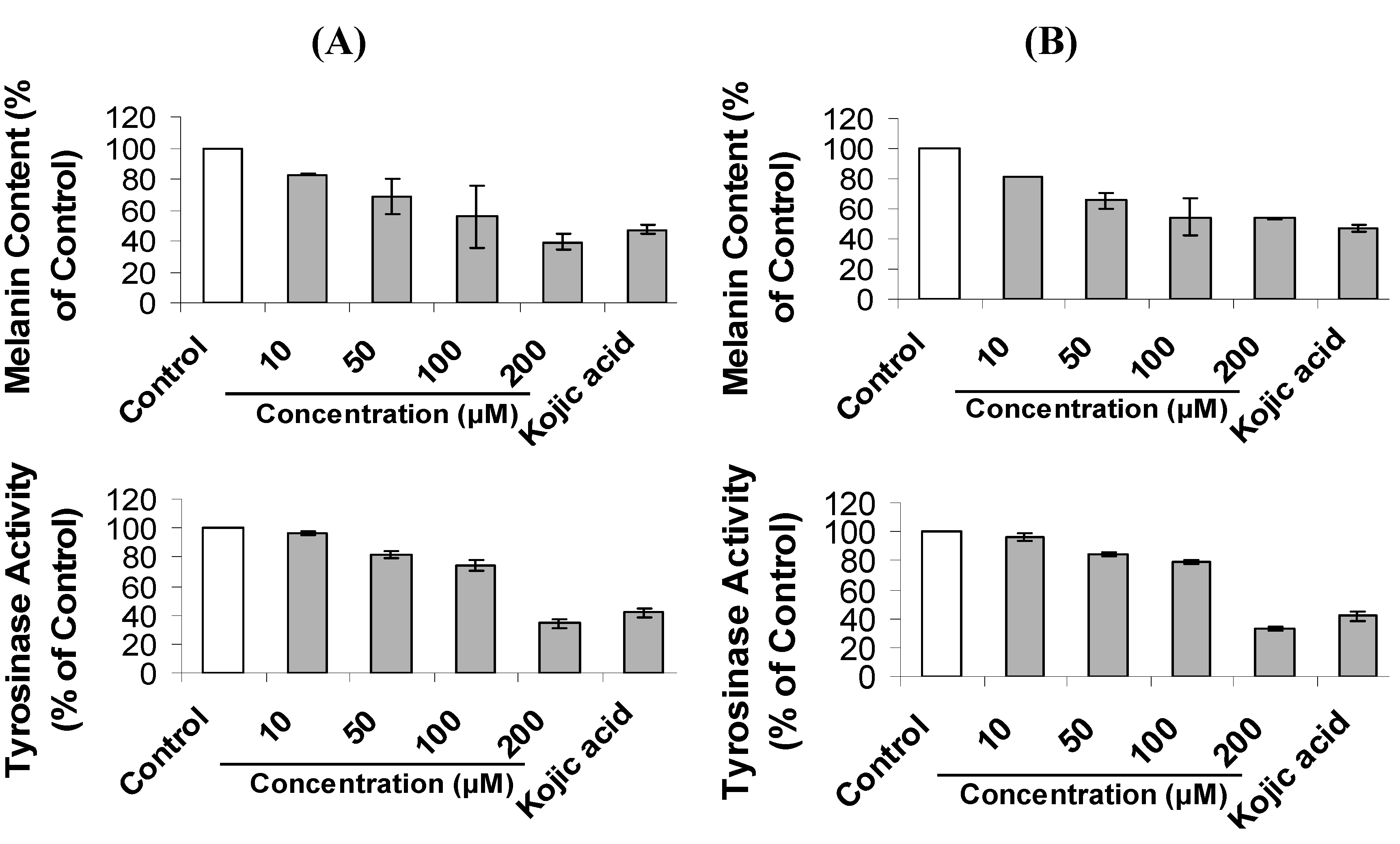
| Compound | IC50 (μM) for Cellular Tyrosinase Inhibition | IC50 (μM) for Melanin Content Inhibition |
|---|---|---|
| Mi-l-Phe | 179.2 ± 1.3 cd | 120.6 ± 8.6 de |
| Mi-d-Phe | 194.5 ± 6.2 b | 168.4 ± 1.4 c |
| Mi-l-Ala | 172.2 ± 4.6 cd | 128.0 ± 7.4 d |
| Mi-d-Ala | 180.9 ± 6.8 bc | 199.0 ± 5.2 b |
| Mi-l-Pro | 166.8 ± 7.3 def | 110.4 ± 5.3 e |
| Mi-d-Pro | 145.9 ± 5.2 h | 123.3 ± 3.0 de |
| Mi-l-Val | 172.1 ± 6.5 de | 170.5 ± 7.5 c |
| Mi-d-Val | 162.1 ± 4.1 efg | 112.2 ± 8.1 e |
| Mi-l-Trp | 156.4 ± 8.3 fg | 109.3 ± 2.7 e |
| Mi-d-Trp | 150.0 ± 2.3 gh | 116.3 ± 4.9 de |
| Mimosine | 388.4 ± 7.2 a | 225.7 ± 4.0 a |
2.3. Inhibition of Cyclooxygenase (COX) by Mimosine and Their Dipeptides
| Compound | Yield (mg) | IC50 (μM) ii | SI i COX-1/COX-2 | |
|---|---|---|---|---|
| COX-1 | COX-2 | |||
| Mi-l-Phe | 132.0 | nt | nt | nt |
| Mi-d-Phe | 48.5 | nt | nt | nt |
| Mi-l-Ala | 45.0 | nt | nt | nt |
| Mi-d-Ala | 79.0 | 19.1 ± 1.0 b | 198.4 ± 3.7 a | 0.10 |
| Mi-l-Pro | 105.5 | 17.9 ± 2.1 b | 29.3 ± 3.0 c | 0.75 |
| Mi-d-Pro | 43.0 | 21.0 ± 2.0 ab | 127.5 ± 7.7 b | 0.15 |
| Mi-l-Val | 17.0 | 24.4 ± 1.4 ab | 21.7 ± 2.0 c | 1.12 |
| Mi-d-Val | 25.0 | nt | nt | nt |
| Mi-l-Trp | 98.0 | 20.4 ± 1.6 ab | 19.3 ± 1.8 c | 0.95 |
| Mi-d-Trp | 71.5 | 26.1 ± 1.3 ab | 27.5 ± 2.2 c | 0.95 |
| Mimosine | 28.8 ± 2.3 a | 20.9 ± 2.1 c | 1.38 | |
| Indomethacin | 6.1 ± 0.9 c | 27.7 ± 1.9 c | 0.22 | |
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Mimosine Isolation from Leucaena leucocephala Leaves
3.3. Preparation of Fmoc-Mimosine
3.4. General Procedure for Synthesis of Mimosine Dipeptides
3.5. Tyrosinase Inhibition Assay
3.6. Cell Culture
3.7. Cell Viability Assay
3.8. Determination of Melanin Content in B16F10 Cells
3.9. Intracellular Tyrosinase Inhibition Assay
3.10. Cyclooxygenase (COX) Inhibition Assay
3.11. Data Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jiang, Z.; Li, S.; Liu, Y.; Deng, P.; Huang, J.; He, G. Sesamin induces melanogenesis by microphthalmia-associated transcription factor and tyrosinase up-regulation via cAMP signaling pathway. Acta Biochim. Biophys. Sin. 2011, 43, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Jang, W.H.; Park, M.; Jung, K.; Baek, H.S.; Joo, Y.H.; Park, Y.H.; Lim, K.M. A novel adamantyl benzylbenzamide derivative, AP736, suppresses melanogenesis through the inhibition of cAMP-PKA-CREB-activated microphthalmia-associated transcription factor and tyrosinase expression. Exp. Dermatol. 2013, 22, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.H.; Jung, J.H.; Lee, J.E.; Jeon, E.J.; Kim, W.; Park, C.S. Biotechnological production of arbutins (α- and β-arbutins), skin-lightening agents, and their derivatives. Appl. Microbiol. Biotechnol. 2012, 95, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Otsuka, Y.; Abe, K. Comparison of the inhibitory effects vitamin E analogues on melanogenesis in mouse B16 melanoma cells. Cytotechnology 2009, 59, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Hearing, V.J.; Tsukamoto, K. Enzymatic control of pigmentation in mammals. FASEB J. 1991, 5, 2902–2909. [Google Scholar] [PubMed]
- Gautam, R.; Jachak, S.M.; Kumar, V.; Mohan, C.G. Synthesis, biological evaluation and molecular docking studies of stellatin derivatives as cyclooxygenase (COX-1, COX-2) inhibitors and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2011, 21, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, H.; Fiebich, B.; Dannhardt, G. Cyclooxygenase-1/2 (COX-1/COX-2) and 5-lipoxygenase (5-LOX) inhibitors of the 6,7-diaryl-2,3-1H-dihydropyrrolizine type. Eur. J. Med. Chem. 2002, 37, 953–959. [Google Scholar] [CrossRef]
- Jain, H.K.; Mourya, V.K.; Agrawal, R.K. Inhibitory mode of 2-acetoxyphenyl alkyl sulfides against COX-1 and COX-2: QSAR analyses. Bioorg. Med. Chem. Lett. 2006, 16, 5280–5284. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Tacconelli, S.; Perrone, M.G.; Malerba, P.; Simone, L.; Scilimati, A.; Lavecchia, A.; Dovizio, M.; Marcantoni, E.; Bruno, A.; et al. Synthesis, pharmacological characterization, and docking analysis of a novel family of diarylisoxazoles as highly selective cyclooxygenase-1 (COX-1) inhibitors. J. Med. Chem. 2013, 56, 4277–4299. [Google Scholar] [CrossRef] [PubMed]
- Gierse, J.; Nickols, M.; Leahy, K.; Warner, J.; Zhang, Y.; Cortes-Burgos, L.; Carter, J.; Seibert, K.; Masferrer, J. Evaluation of COX-1/COX-2 selectivity and potency of a new class of COX-2 inhibitors. Eur. J. Pharmacol. 2008, 588, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Tawata, S.; Fukuta, M.; Xuan, T.D.; Deba, F. Total utilization of tropical plants Leucaena leucocephala and Alpinia zerumbet. J. Pestic. Sci. 2008, 33, 40–43. [Google Scholar]
- Upadhyay, A.; Chompoo, J.; Taira, N.; Fukuta, M.; Gima, S.; Tawata, S. Solid-phase synthesis of mimosine tetrapeptides and their inhibitory activities on neuraminidase and tyrosinase. J. Agric. Food Chem. 2011, 59, 12858–12863. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.C.Q.; Taira, N.; Tawata, S. Several herbal compounds in Okinawa plants directly inhibit the oncogenic/aging kinase PAK1. Drug Discov. Ther. 2014, 8, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.M.; Kwak, S.Y.; Seo, H.S.; Seo, J.H.; Kim, B.G.; Lee, Y.S. Kojic acid-amino acid conjugates as tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5586–5589. [Google Scholar] [CrossRef] [PubMed]
- Cabanes, J.; García-Cánovas, F.; Tudela, J.; Lozano, J.A.; García-Carmona, F. l-Mimosine, a slow-binding inhibitor of mushroom tyrosinase. Phytochemistry 1987, 26, 917–919. [Google Scholar] [CrossRef]
- Maruta, H. Herbal therapeutics that block the oncogenic kinase PAK1: A practical approach towards PAK1-dependent diseases and longevity. Phytother. Res. 2014, 28, 65–672. [Google Scholar] [CrossRef] [PubMed]
- Be Tu, P.T.; Tawata, S.; Yun, C.Y.; Kim, E.G.; Maruta, H. PAK1 is essential for the melanogenesis in skin. J. Dermatol. Sci. 2015, submitted. [Google Scholar]
- Nguyen, B.C.Q.; Taira, N.; Maruta, H.; Tawata, S. Anti-oncogenic, anti-melanogenic and anti-alopecia (hair growth-promoting) activities of several PAK1-blocking compounds from Okinawa plants. Phytother. Res. 2015, submitted. [Google Scholar]
- Zebardast, T.; Zarghi, A.; Daraie, B.; Hedayati, M.; Dadrass, O.G. Design and synthesis of 3-alkyl-2-aryl-1,3-thiazinan-4-one derivatives as selective cyclooxygenase (COX-2) inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3162–3165. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Frydas, S.; Reale, M.; Barbacane, R.C.; Gioacchino, M.D.; Felaco, M.; Trakatellis, A. Inhibition of MCP-1 and MIP-2 transcription and translation by mimosine in muscle tissue infected with the parasite Trichinella spiralis. Mol. Cell. Biochem. 2002, 229, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Tawata, S. Effective reduction and extraction of mimosine from L. leucocephala and the potential for its use as a lead compound of herbicides. In Pesticide and Alternatives; Casida, J.E., Ed.; Elsevier Science Publishers: Amsterdam, The Netherland, 1990; Vol 1, pp. 541–544. [Google Scholar]
- Tadtong, S.; Viriyaroj, A.; Vorarat, S.; Nimkulrat, S.; Suksamrarn, S. Antityrosinase and antibacterial activities of mangosteen pericarp extract. J. Health Res. 2009, 23, 99–102. [Google Scholar]
- Campos, P.M.; Horinouchi, C.D.S.; Prudente, A.S.; Cechinel-Filho, V.; Cabrini, D.A.; Otuki, M.F. Effect of a Garcinia gardneriana (Planchon and Triana) Zappi hydroalcoholic extract on melanogenesis in B16F10 melanoma cells. J. Ethnopharmacol. 2013, 148, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Nakata, A.; Yamasaki, F.; Abas, F.; Shaari, K.; Lajis, N.H.; Morita, H. Curcumin-like diarylpentanoid analogues as melanogenesis inhibitors. J. Nat. Med. 2012, 66, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of mimosine and mimosine dipeptides are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, B.C.Q.; Tawata, S. Mimosine Dipeptide Enantiomsers: Improved Inhibitors against Melanogenesis and Cyclooxygenase. Molecules 2015, 20, 14334-14347. https://doi.org/10.3390/molecules200814334
Nguyen BCQ, Tawata S. Mimosine Dipeptide Enantiomsers: Improved Inhibitors against Melanogenesis and Cyclooxygenase. Molecules. 2015; 20(8):14334-14347. https://doi.org/10.3390/molecules200814334
Chicago/Turabian StyleNguyen, Binh Cao Quan, and Shinkichi Tawata. 2015. "Mimosine Dipeptide Enantiomsers: Improved Inhibitors against Melanogenesis and Cyclooxygenase" Molecules 20, no. 8: 14334-14347. https://doi.org/10.3390/molecules200814334
APA StyleNguyen, B. C. Q., & Tawata, S. (2015). Mimosine Dipeptide Enantiomsers: Improved Inhibitors against Melanogenesis and Cyclooxygenase. Molecules, 20(8), 14334-14347. https://doi.org/10.3390/molecules200814334