
Palladium-Catalyzed One-Pot Approach to 3-(Diarylmethylene)oxindoles from Propiolamidoaryl Triflate

Abstract
:
1. Introduction
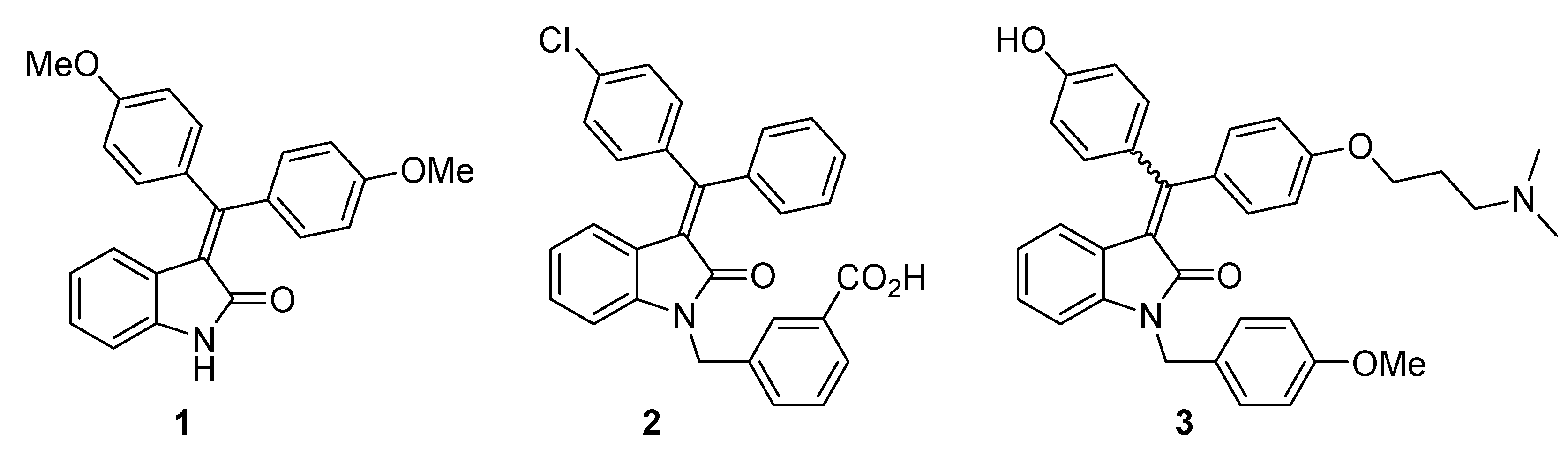
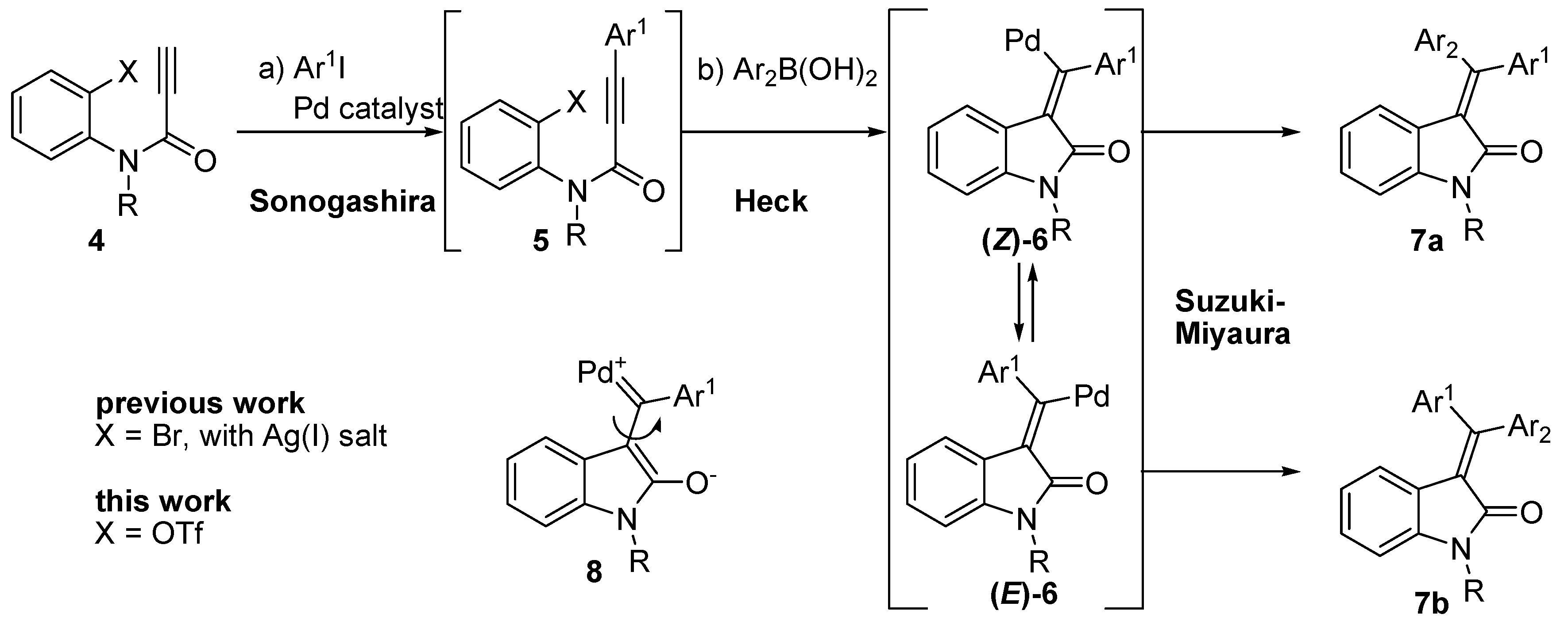
2. Results and Discussion


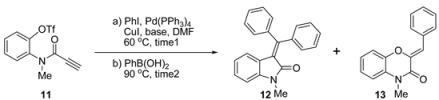
| Entry | Base | Time 1 (h) | Time 2 (h) | Yield (%) b | |
|---|---|---|---|---|---|
| 12 | 13 | ||||
| 1 | NaOAc | 1 | 12 | 64 | 7 |
| 2 | KOAc | 1 | 12 | 58 | 10 |
| 3 | K3PO4 | 1 | 12 | 68 | 20 |
| 4 | Cs2CO3 | 1 | 2 | 5 | 55 |
| 5 | K2CO3 | 1 | 4 | 64 | 29 |
| 6 | NaOAc | 3 | 12 | 61 | 7 |
| 7 | K3PO4 | 3 | 3 | 72 | 5 |
| 8 | K2CO3 | 3 | 2 | 78 | 7 |
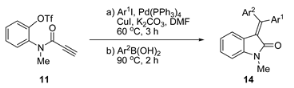
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Ar1 | Ar2 | 14 | Yield (%) b | E/Z Ratio c | Bromo Substrate (Ref. [15]) | |
|---|---|---|---|---|---|---|---|
| Yield (%) b | E/Z Ratio c | ||||||
| 1 | 4-MeOPh | 4-MeOPh | 14a | 55 | - | 85 | - |
| 2 | 4-ClPh | 4-ClPh | 14b | 54 | - | 80 | - |
| 3 | 4-NO2Ph | 4-NO2Ph | 14c | 42 | - | 67 d | - |
| 4 | Ph | 4-MeOPh | 14d | 58 | 1:1 | 52 | 1:1 |
| 5 | Ph | 4-ClPh | 14e | 65 | 5:1 | 57 | 1:1 |
| 6 | Ph | 4-NO2Ph | 14f | 59 | 3:1 | 52 | 1:1 |
| 7 | 4-MeOPh | Ph | 14d | 62 | 1.5:1 | 70 | 1.6:1 |
| 8 | 4-ClPh | Ph | 14e | 66 | 1:5 | 77 | 1:3.4 |
| 9 | 4-NO2Ph | Ph | 14f | 62 d | 1:12 | 73 d | 1:10 |
3. Experimental Section
3.1. General Information
3.2. Preparation of Propiolamidoaryl Triflate 11
3.3. General Procedure for Palladium-Catalyzed One-Pot Reaction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marti, C.; Carreira, E.M. Construction of spiro[pyrrolidine-3,3ʹ-oxindoles]—Recent Application to the synthesis of oxindole alkaloids. Eur. J. Org. Chem. 2003, 12, 2209–2219. [Google Scholar] [CrossRef]
- Kein, J.E.M.; Taylor, R.J.K. Transition-metal-mediated Routes to 3,3-disubstituted oxindoles through anilide cyclization. Eur. J. Org. Chem. 2011, 34, 6821–6841. [Google Scholar]
- Ziarani, G.M.; Gholamzadeh, P.; Lashgari, N.; Hajiabbast, P. Oxindole as starting material in organic synthesis. ARKIVOC 2013, 1, 470–535. [Google Scholar] [CrossRef]
- Sasaki, E.; Miyoshi, K.; Nozawa, Y.; Kanda, A.; Nakano, K.; Yamasaki, Y.; Miyake, H.; Mausuura, N. TAS-301, a new synthetic inhibitor of neointimal thickening after balloon injury, inhibits calcium-dependent signal transduction and cytoskeletal reorganization. Pharmacology 2001, 63, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.F.; Li, Y.Y.; Su, M.B.; Zhang, M.; Zhang, W.; Zhang, L.N.; Pang, T.; Zhang, R.T.; Liu, B.; Li, J.Y.; et al. Development of novel alkene oxindole derivatives as orally efficacious AMP-activated protein kinase activators. ACS Med. Chem. Lett. 2013, 4, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Ganguly, A.; Ghosh, A.; Yousuf, M.; Rathore, B.; Banerjee, R.; Adhikari, S. Bis-arylidene oxindoles as anti-breast-cancer agents acting via the estrogen receptor. Chem. Med. Chem. 2014, 9, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Yanada, R.; Obika, S.; Oyama, M.; Takemoto, Y. Stereoselective synthesis of 3-alkylideneoxindoles using tandem In-mediated carbometalation and Pd-catalyzed cross-Coupling reaction. Org. Lett. 2004, 6, 2825–2828. [Google Scholar] [CrossRef] [PubMed]
- Yanada, R.; Obika, S.; Inokuma, T.; Yanada, K.; Yamashita, M.; Ohta, S.; Takemoto, Y. Stereoselective synthesis of 3-alkylideneoxindoles via palladium-catalyzed domino reactions. J. Org. Chem. 2005, 70, 6972–6975. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.S.; Patch, R.J.; Player, M.R. A tandem Heck-carbocyclization/Suzuki-coupling approach to the Stereoselective synthesis of asymmetric 3,3-(diarylmethylene)indolinones. J. Org. Chem. 2005, 70, 3741–3744. [Google Scholar] [CrossRef] [PubMed]
- Shintani, R.; Yamagami, T.; Hayashi, T. Rhodium-catalyzed multicomponent-coupling reactions involving a carborhodation-cross-coupling sequence. Org. Lett. 2006, 8, 4799–4801. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Neuville, L.; Zhu, J. Palladium-catalyzed three-component synthesis of 3-(diarylmethylene)oxindoles through a domino Sonogashira/carbopalladation/C–H activation/C–C bond-forming sequence. Angew. Chem. Int. Ed. 2007, 46, 3291–3295. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Stereoselective synthesis of 3-alkylideneoxindoles by palladium-catalyzed cyclization reaction of 2-(alkynyl)aryl isocyanates with organoboron reagents. Org. Lett. 2008, 10, 4887–4889. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Peng, P.; Zhong, P.; Li, J.H. Palladium-catalyzed C–H functionalization of N-arylpropiolamides with aryliodonium salts: Selective synthesis of 3-(1-arylmethylene)oxindoles. J. Org. Chem. 2008, 73, 5476–5480. [Google Scholar] [CrossRef] [PubMed]
- Song, R.J.; Liu, Y.; Li, R.J.; Li, J.H. Selective synthesis of 3-methyleneindolin-2-ones by one-pot multicatalytic processes. Tetrahedron Lett. 2009, 50, 3912–3916. [Google Scholar] [CrossRef]
- Dong, G.R.; Park, S.; Lee, D.; Shin, K.J.; Seo, J.H. Synthesis of 3-(diarylmethylene)oxindoles via a palladium-catalyzed one-pot reaction: Sonogashira-Heck-Suzuki-Miyaura combined reaction. Synlett 2013, 24, 1993–1997. [Google Scholar] [CrossRef]
- Zargarian, D.; Alper, H. Palladium-catalyzed hydrocarboxylation of alkynes with formic acid. Organometallics 1993, 12, 712–724. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G.; Marinelli, F.; Moro, L.; Pace, P. Palladium-catalyzed hydroarylation and hydrovinylation of 3,3-dialkoxy-1-aryl-1-propynes. An approach to 3-aryl- and 3-vynylquinolines. Tetrahedron 1996, 52, 10225–10240. [Google Scholar] [CrossRef]
- Amatore, C.; Bensalem, S.; Ghalem, S.; Jutand, A. Mechanism of the carbopalladation of alkynes by aryl-palladium complexes. J. Organomet. Chem. 2004, 689, 4642–4646. [Google Scholar] [CrossRef]
- Dounay, A.B.; Overman, L.E. The asymmetric intramolecular Heck reaction in natural product total synthesis. Chem. Rev. 2003, 103, 2945–2963. [Google Scholar] [CrossRef] [PubMed]
- Sam, J.; Plampin, J.N.; Poos, G.I. Methylated 2-Amino-5-chlorobenzoxazoles. J. Org. Chem. 1958, 23, 1500–1503. [Google Scholar] [CrossRef]
- Overman, L.E.; Peterson, E.A. Enantioselective synthesis of (−)-idiospermuline. Tetrahedron 2003, 59, 6905–6919. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Chowdhury, C.; Chaudhuri, G.; Guha, S.; Mukherjee, A.K.; Kundu, N.G. Palladium-catalyzed heteroannulation leading to heterocyclic structures with two heteroatoms: A highly convenient and facile method for a totally region- and stereoselective synthesis of (Z)-2,3-dihydro-2-(ylidene)-1,4-benzo- and -naphtho[2,3-b]dioxnis. J. Org. Chem. 1998, 63, 1863–1871. [Google Scholar] [CrossRef]
- Kumar, K.S.; Rambabu, D.; Prasad, B.; Mujahid, M.; Krishna, G.R.; Rao, M.V.B.; Reddy, C.M.; Vanaja, G.R.; Kalle, A.M.; Pal, M. A new approach to construct a fused 2-ylidene chromene ring: highly regioselective synthesis of novel chromeno quinoxalines. Org. Biomol. Chem. 2012, 10, 4774–4781. [Google Scholar] [CrossRef] [PubMed]
- Loudon, J.D.; Ogg, J. 2,3-Dihydro-3-oxobenz-1,4-oxaxines. J. Chem. Soc. 1955, 739–744. [Google Scholar] [CrossRef]
- Tang, D.J.; Tang, B.X.; Li, J.H. Selective synthesis of 3-aryl quinolin-2(1H)-ones and 3-(1-arylmethylene)oxindoles involving a 2-fold arene C–H activation process. J. Org. Chem. 2009, 74, 6749–6755. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.; Park, S.; Yu, Y.; Shin, K.J.; Seo, J.H. Palladium-Catalyzed One-Pot Approach to 3-(Diarylmethylene)oxindoles from Propiolamidoaryl Triflate. Molecules 2015, 20, 14022-14032. https://doi.org/10.3390/molecules200814022
Lee D, Park S, Yu Y, Shin KJ, Seo JH. Palladium-Catalyzed One-Pot Approach to 3-(Diarylmethylene)oxindoles from Propiolamidoaryl Triflate. Molecules. 2015; 20(8):14022-14032. https://doi.org/10.3390/molecules200814022
Chicago/Turabian StyleLee, Dahye, Sunhwa Park, Yoseb Yu, Kye Jung Shin, and Jae Hong Seo. 2015. "Palladium-Catalyzed One-Pot Approach to 3-(Diarylmethylene)oxindoles from Propiolamidoaryl Triflate" Molecules 20, no. 8: 14022-14032. https://doi.org/10.3390/molecules200814022
APA StyleLee, D., Park, S., Yu, Y., Shin, K. J., & Seo, J. H. (2015). Palladium-Catalyzed One-Pot Approach to 3-(Diarylmethylene)oxindoles from Propiolamidoaryl Triflate. Molecules, 20(8), 14022-14032. https://doi.org/10.3390/molecules200814022