
Synthesis and Biological Evaluation of Lipophilic 1,4-Naphthoquinone Derivatives against Human Cancer Cell Lines

 ,
, 
Abstract
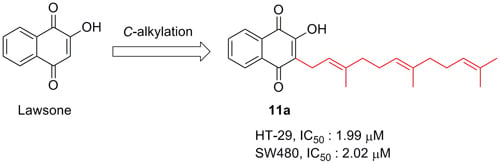
:
1. Introduction

2. Results and Discussion
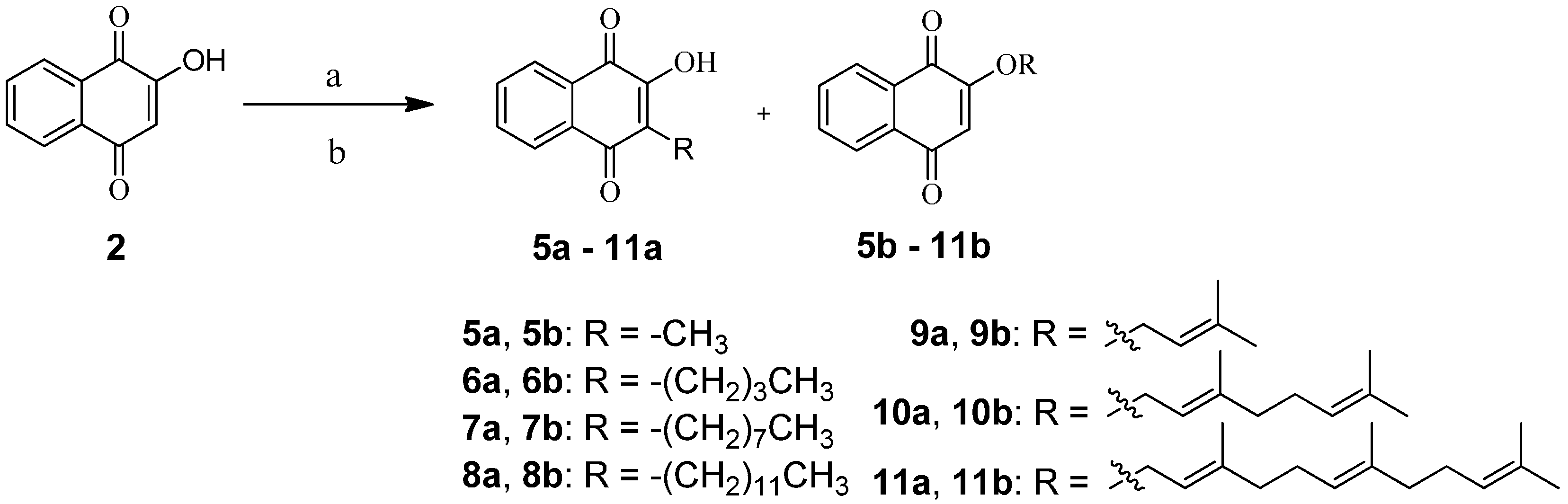
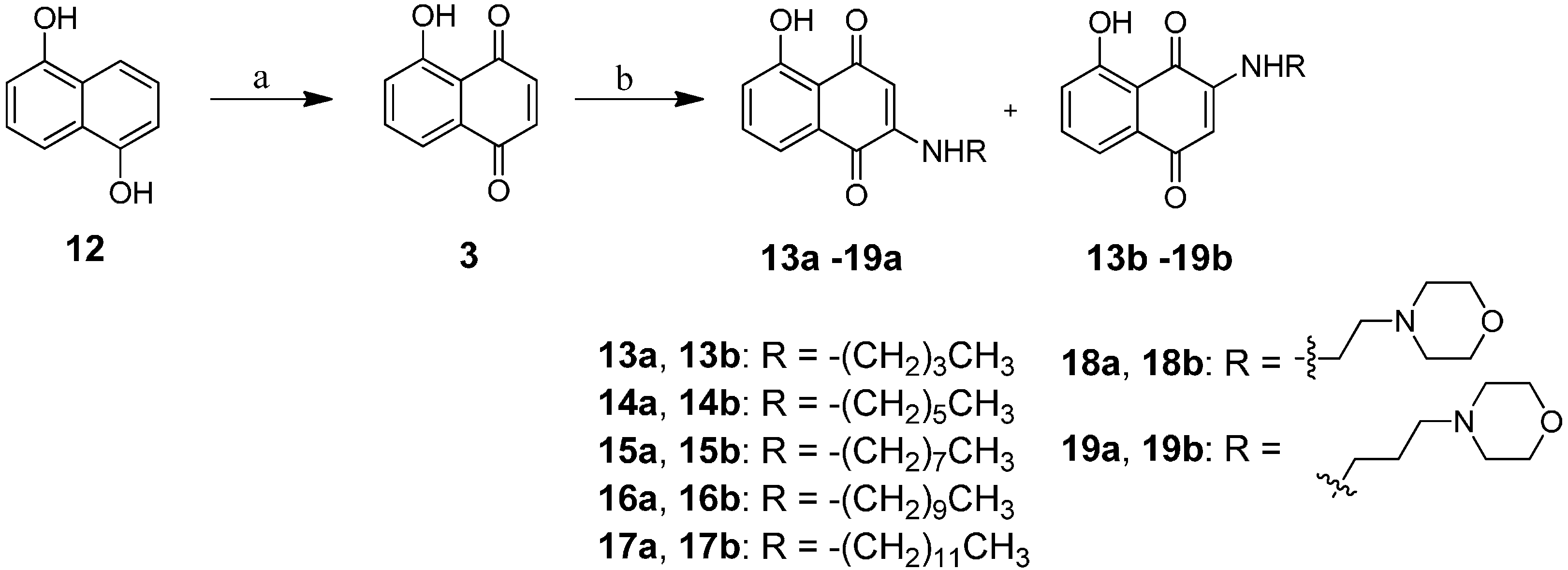
2.1. Chemistry
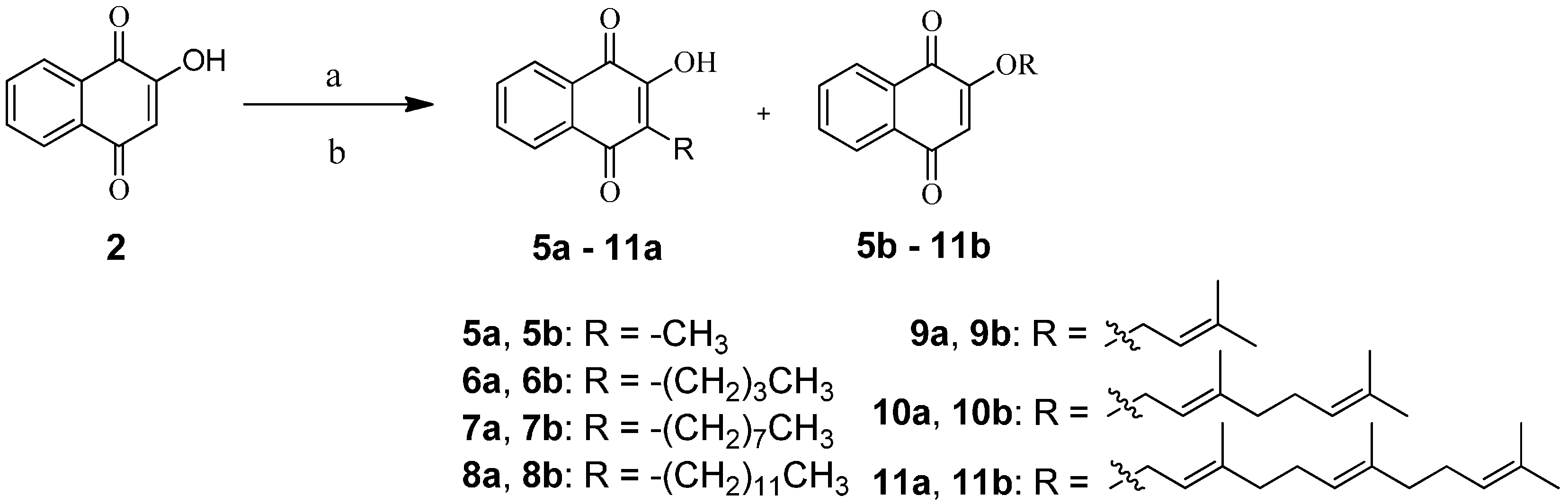

2.2. In Vitro Anti-Proliferative Activity by MTT Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | cLogP a | IC50 (μM) b (Mean ± SD) | ||||
|---|---|---|---|---|---|---|
| HT-29 Colon | SW480 Colon | HepG2 Liver | HL60 Leukemia | MCF-7 Breast | ||
| Plumbagin (1) | 2.78 | 3.67 ± 0.46 | 2.19 ± 0.65 | 2.32 ± 0.18 | 5.90 ± 1.21 | 20.89 ± 0.70 |
| Lawsone (2) | 1.68 | >100 | >100 | >100 | 45.12 ± 7.90 | >100 |
| Juglone (3) | 2.26 | >50 | >50 | >50 | 46.95 ± 0.58 | >50 |
| 5a | 2.19 | >100 | 97.09 ± 2.36 | 94.08 ± 2.63 | >100 | >100 |
| 5b | 1.78 | 8.16 ± 1.16 | 4.23 ± 0.50 | 3.32 ± 0.66 | 16.40 ± 2.84 | >25 |
| 6a | 3.79 | >100 | >100 | >100 | >100 | >100 |
| 6b | 3.22 | 14.81 ± 0.65 | 2.02 ± 0.20 | 3.67 ± 0.84 | 20.04 ± 0.79 | 31.03 ± 0.32 |
| 7a | 5.90 | 33.44 ± 1.48 | 9.73 ± 0.04 | 20.20 ± 3.52 | >100 | >50 |
| 7b | 5.34 | 28.01 ± 3.52 | 8.51 ± 0.53 | 9.66 ± 0.50 | 23.42 ± 1.24 | 20.81 ± 3.67 |
| 8a | 7.46 | >25 | >25 | >25 | >25 | >25 |
| 8b | 8.02 | >25 | >25 | >25 | >25 | >25 |
| 9a | 3.70 | >100 | >100 | 74.18 ± 2.61 | >100 | >100 |
| 9b | 3.34 | >50 | 20.72 ± 1.19 | 35.56 ± 1.13 | >50 | >50 |
| 10a | 5.73 | 12.90 ± 2.13 | 14.14 ± 0.96 | 19.10 ± 0.26 | 9.12 ± 0.69 | >100 |
| 10b | 5.37 | 42.90 ± 1.13 | 44.23 ± 1.06 | 29.10 ± 1.21 | 29.56 ± 0.88 | >100 |
| 11a | 6.82 | 1.99 ± 0.04 | 2.02 ± 0.31 | 14.01 ± 2.70 | 5.41 ± 0.88 | 47.01 ± 0.47 |
| 11b | 7.40 | 40.37 ± 0.75 | 37.16 ± 2.21 | 32.33 ± 1.46 | 35.10 ± 1.52 | >100 |
| 13a | 3.65 | 9.82 ± 1.51 | 16.17 ± 0.72 | 9.39 ± 0.12 | 45.69 ± 2.48 | >100 |
| 13b | 3.65 | 18.11 ± 1.68 | 8.28 ± 0.12 | 17.89 ± 0.14 | 6.50 ± 0.32 | 23.71 ± 0.82 |
| 14a | 4.99 | >50 | >50 | >50 | >50 | >50 |
| 14b | 4.99 | 34.53 ± 1.20 | 15.13 ± 0.49 | 17.82 ± 2.29 | >50 | >50 |
| 15a | 6.05 | >50 | >50 | >50 | >50 | >50 |
| 15b | 6.05 | >50 | 20.23 ± 2.18 | 47.06 ± 0.63 | >50 | >50 |
| 16a | 7.11 | >50 | >50 | >50 | >50 | >50 |
| 16b | 7.11 | >50 | >50 | >50 | >50 | >50 |
| 17a | 8.17 | >20 | >20 | >20 | >20 | >20 |
| 17b | 8.17 | >20 | >20 | >20 | >20 | >20 |
| 18a | 2.51 | 9.42 ± 0.45 | 8.86 ± 0.50 | 11.72 ± 0.42 | 8.42 ± 0.86 | >50 |
| 18b | 2.51 | 8.59 ± 0.67 | 5.19 ± 0.44 | 8.84 ± 0.07 | 6.49 ± 0.84 | >50 |
| 19a | 2.80 | 9.44 ± 0.32 | 4.66 ± 0.25 | 10.08 ± 0.57 | 6.35 ± 0.24 | >50 |
| 19b | 2.80 | 11.38 ± 1.51 | 3.86 ± 0.61 | 7.90 ± 0.16 | 7.76 ± 0.54 | >50 |
| Cisplatin | - | 24.07 ± 0.03 | 40.72 ± 1.18 | 36.07 ± 3.11 | >100 | >100 |
| 5-Fu | - | >100 | 32.72 ± 8.32 | 40.18 ± 7.63 | >100 | >100 |
| Doxorubicin | - | 1.70 ± 0.20 | 0.53 ± 0.07 | 0.30 ± 0.02 | 14.26 ± 0.89 | 0.39 ± 0.06 |
2.3. Cytotoxic Activity of Plumbagin (1) and Compound 11a was Exhibited in a Time-Dependent Manner
| Compound | IC50 (μM) a (Mean ± SD) | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| Plumbagin (1) | 4.72 ± 0.07 | 3.67 ± 0.46 | 2.10 ± 0.23 |
| 11a | >20 | 1.99 ± 0.04 | 0.84 ± 0.12 |
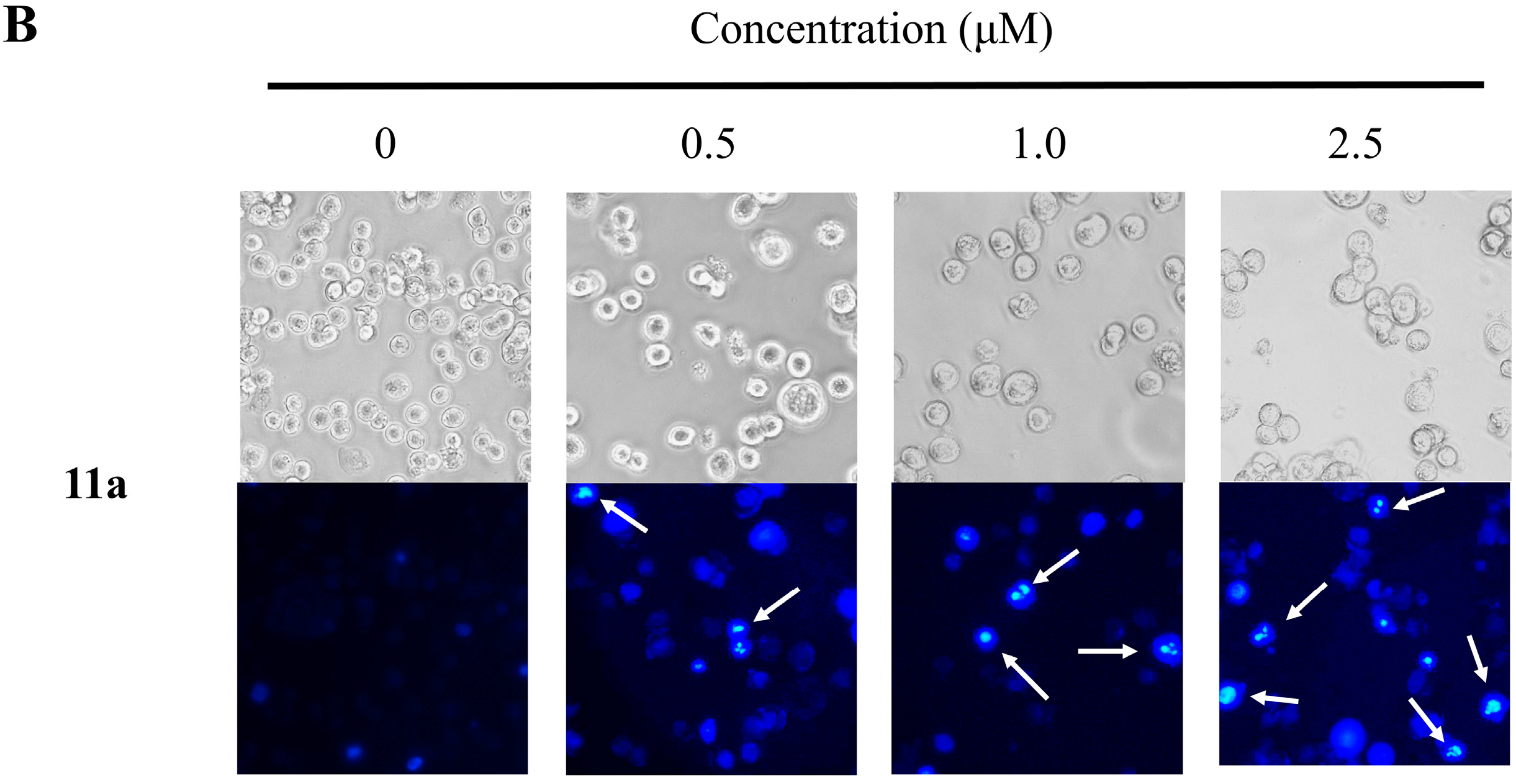
2.4. Nuclear Morphological Changes of HT-29 Cells Treated with Plumbagin (1) and Compound 11a
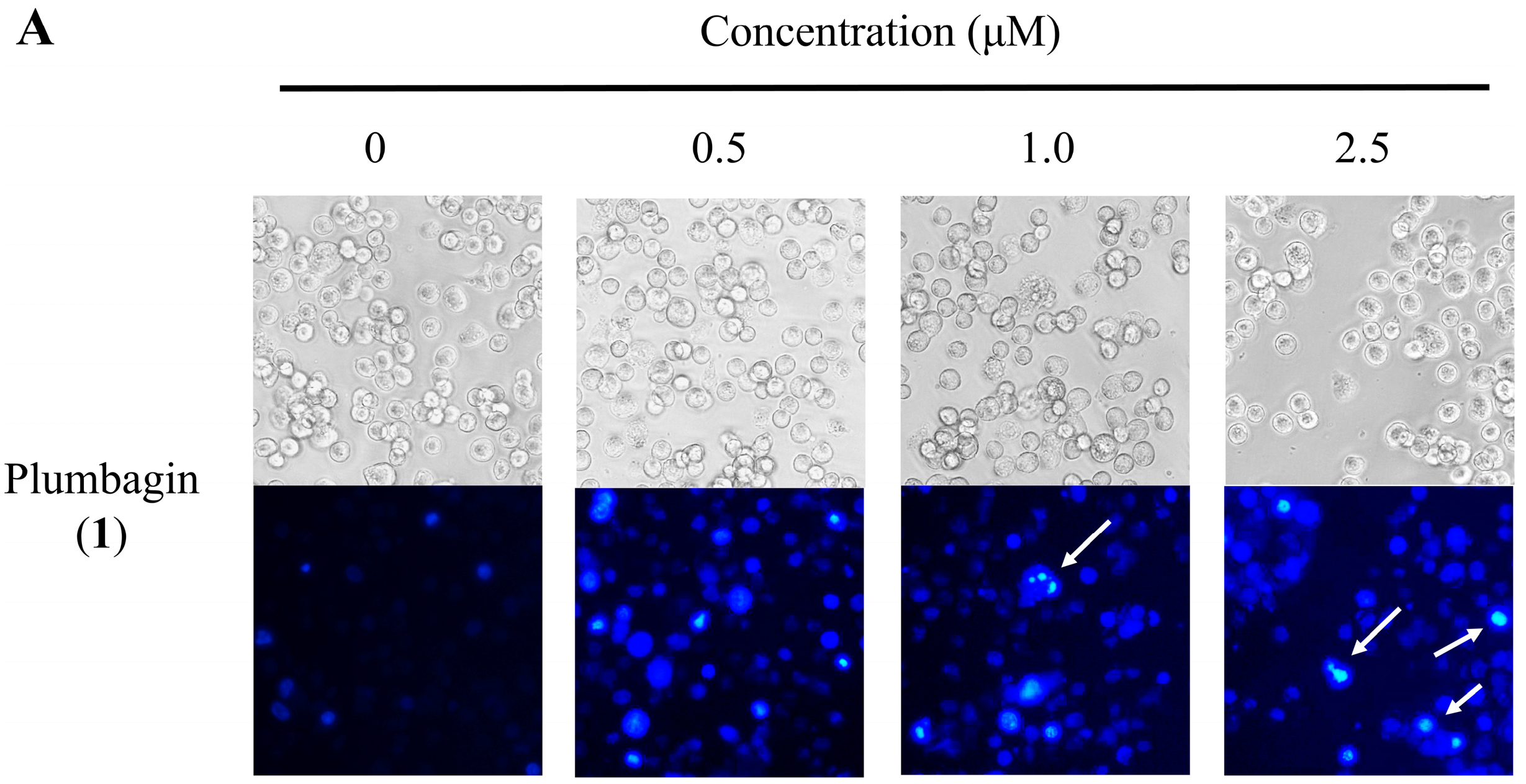
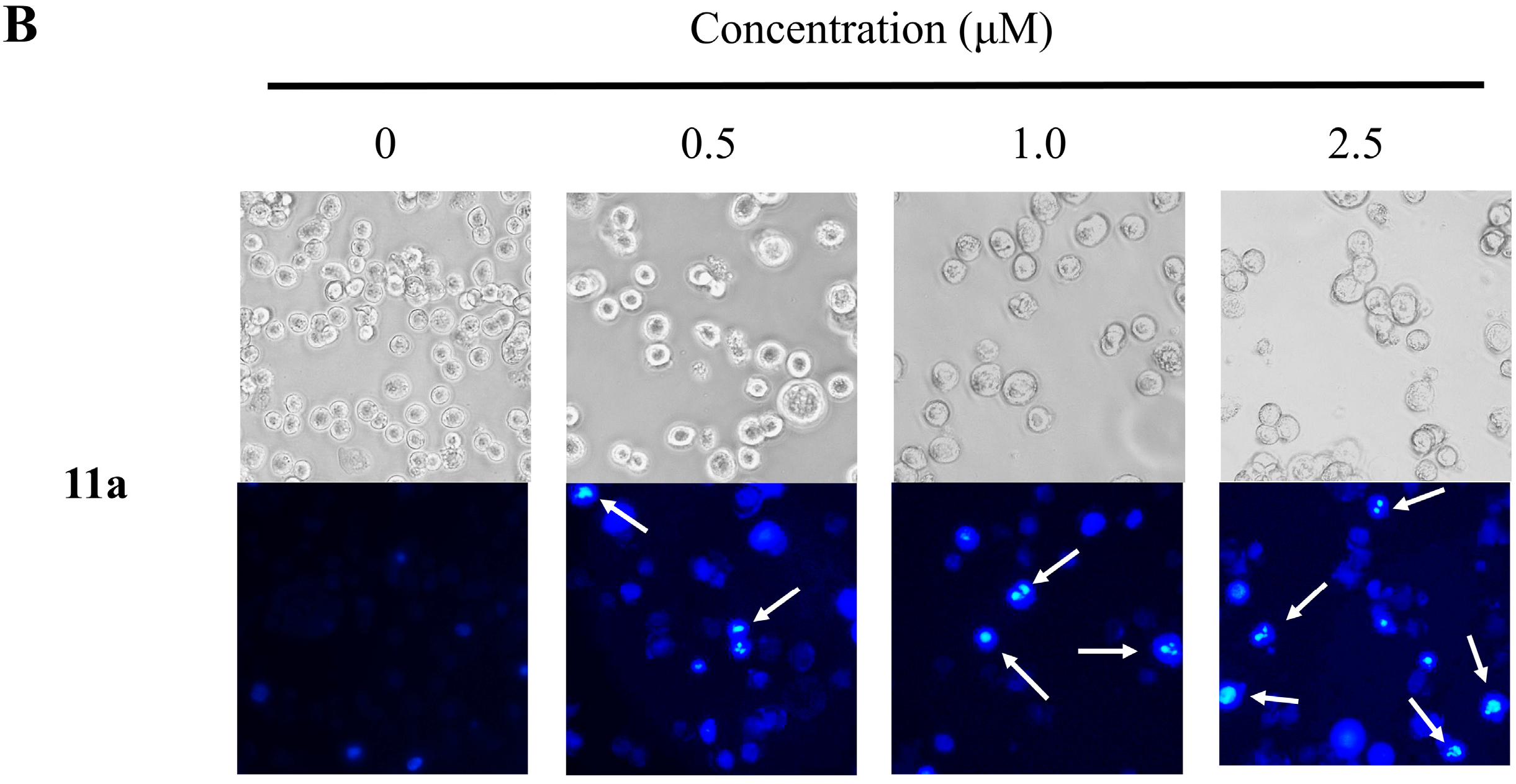
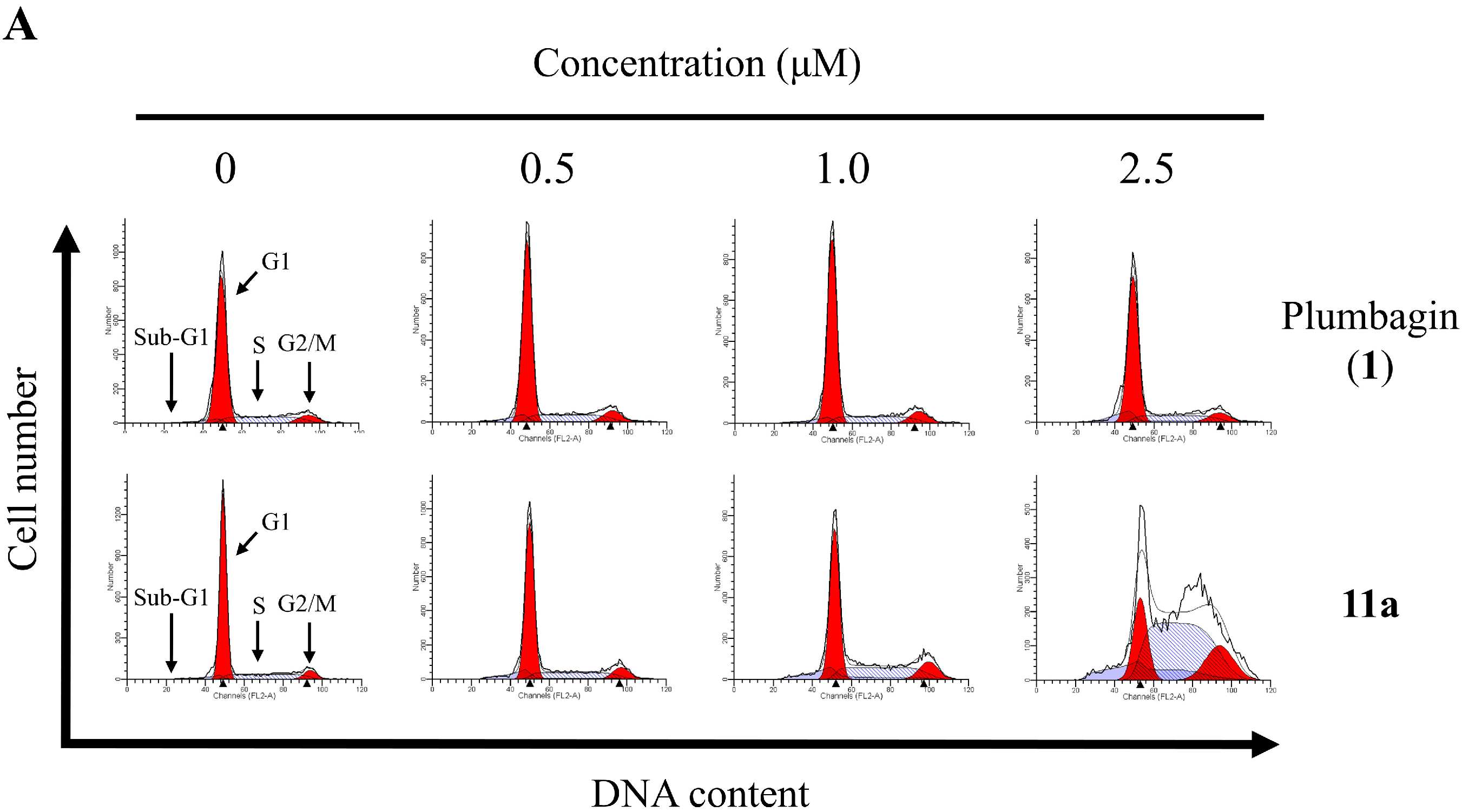
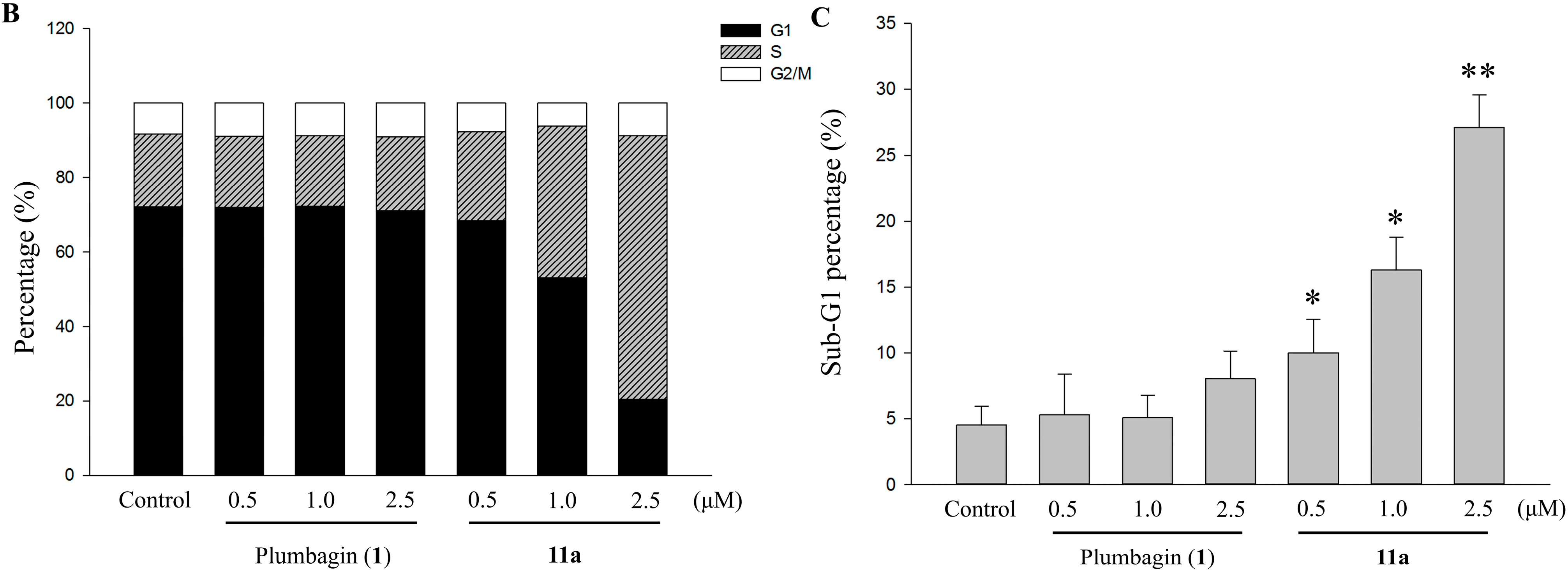
2.5. Cell Cycle Distribution Analysis Using Flow Cytometry
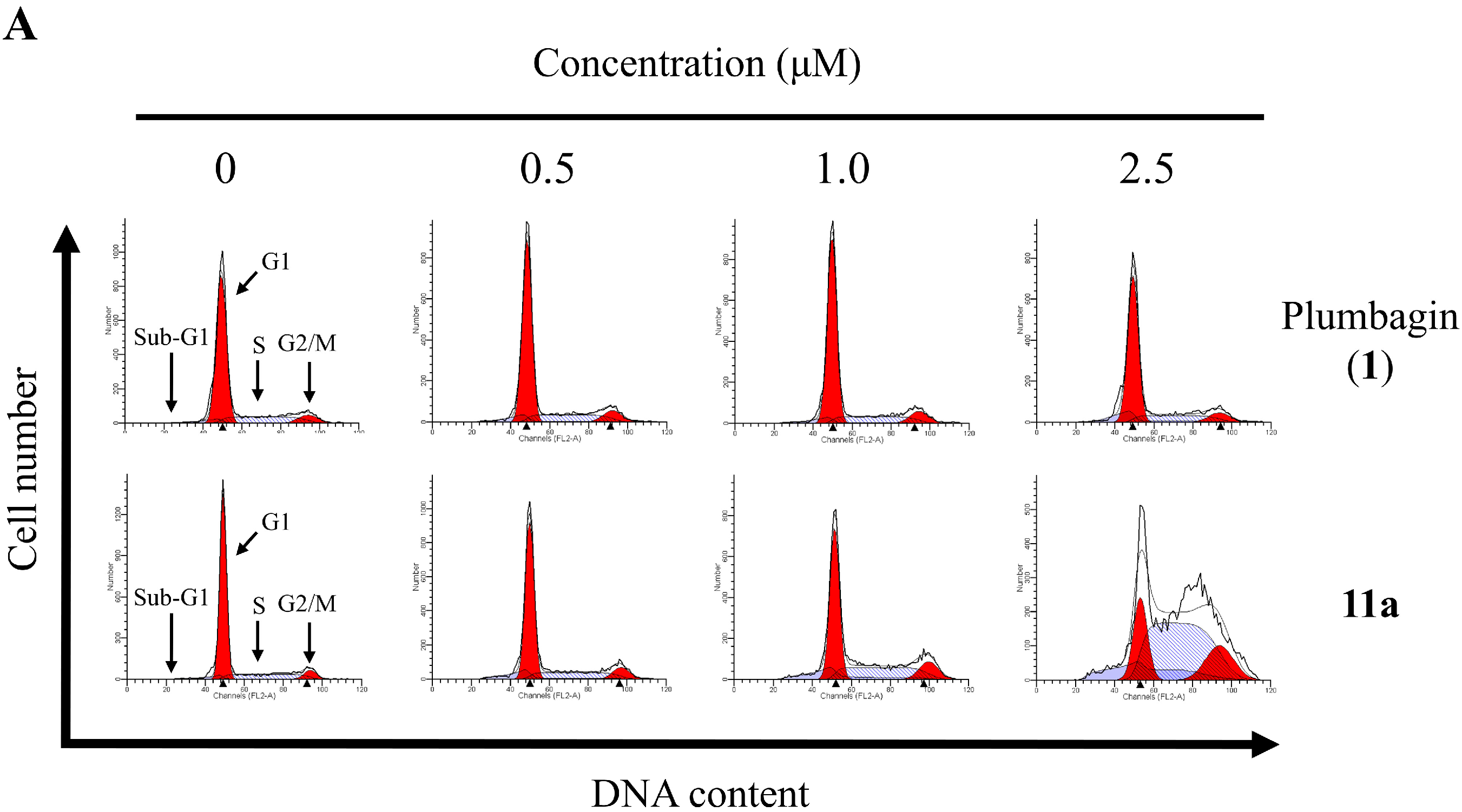
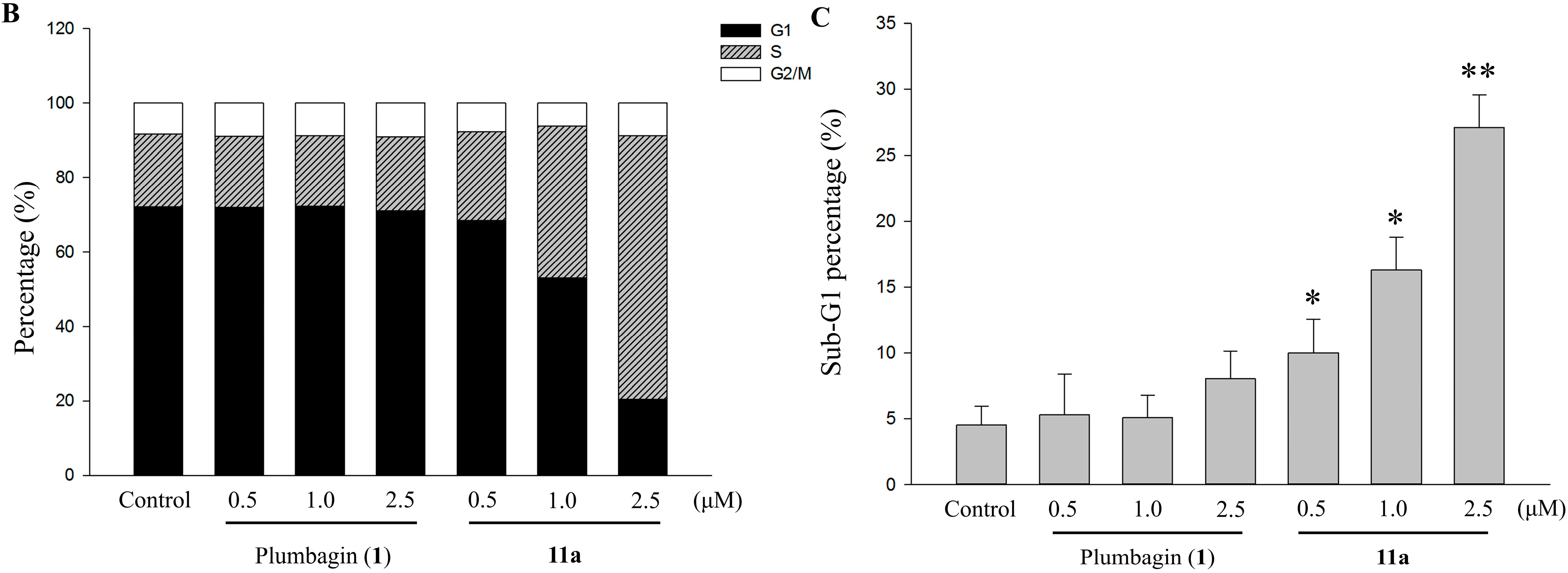
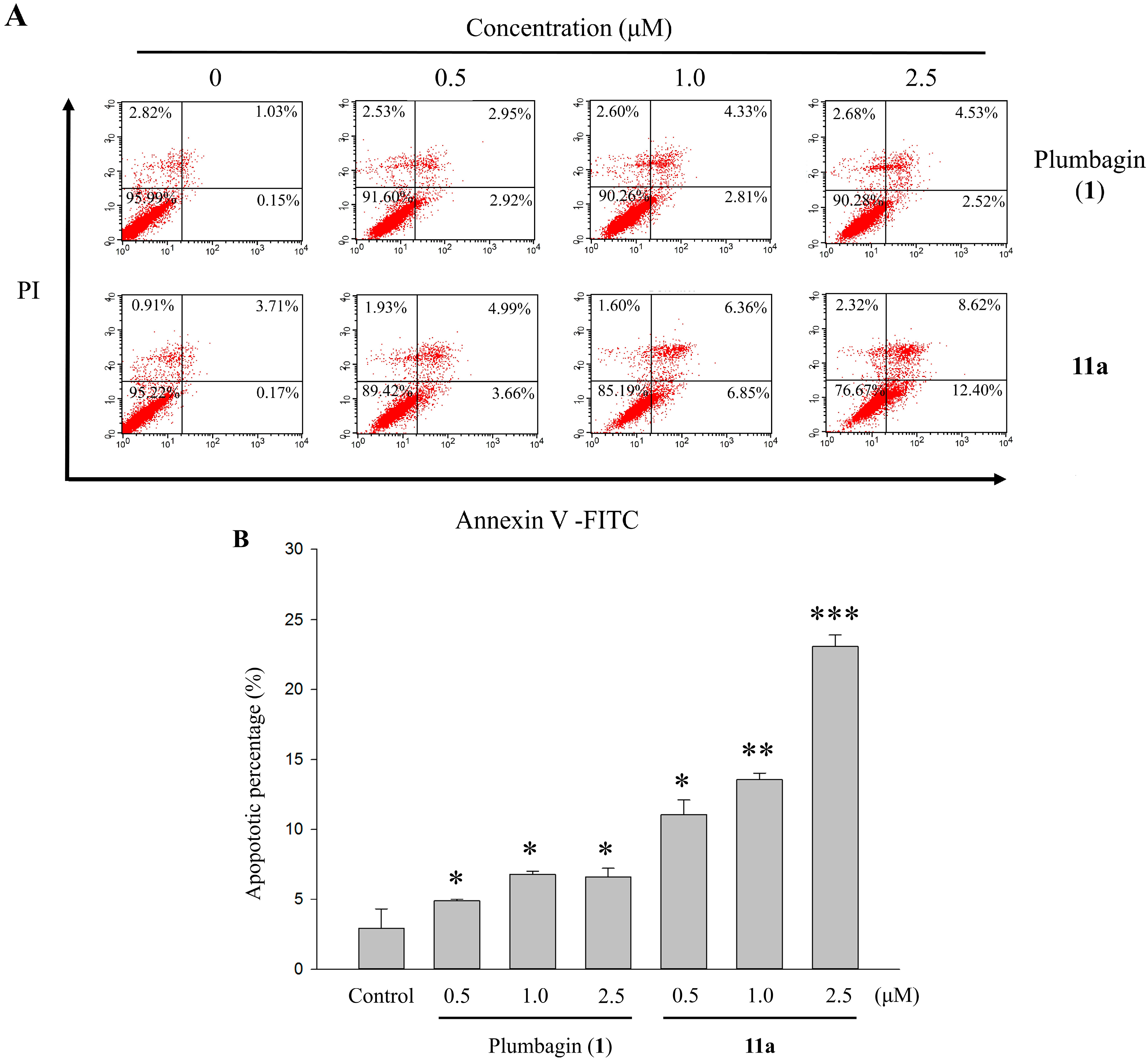
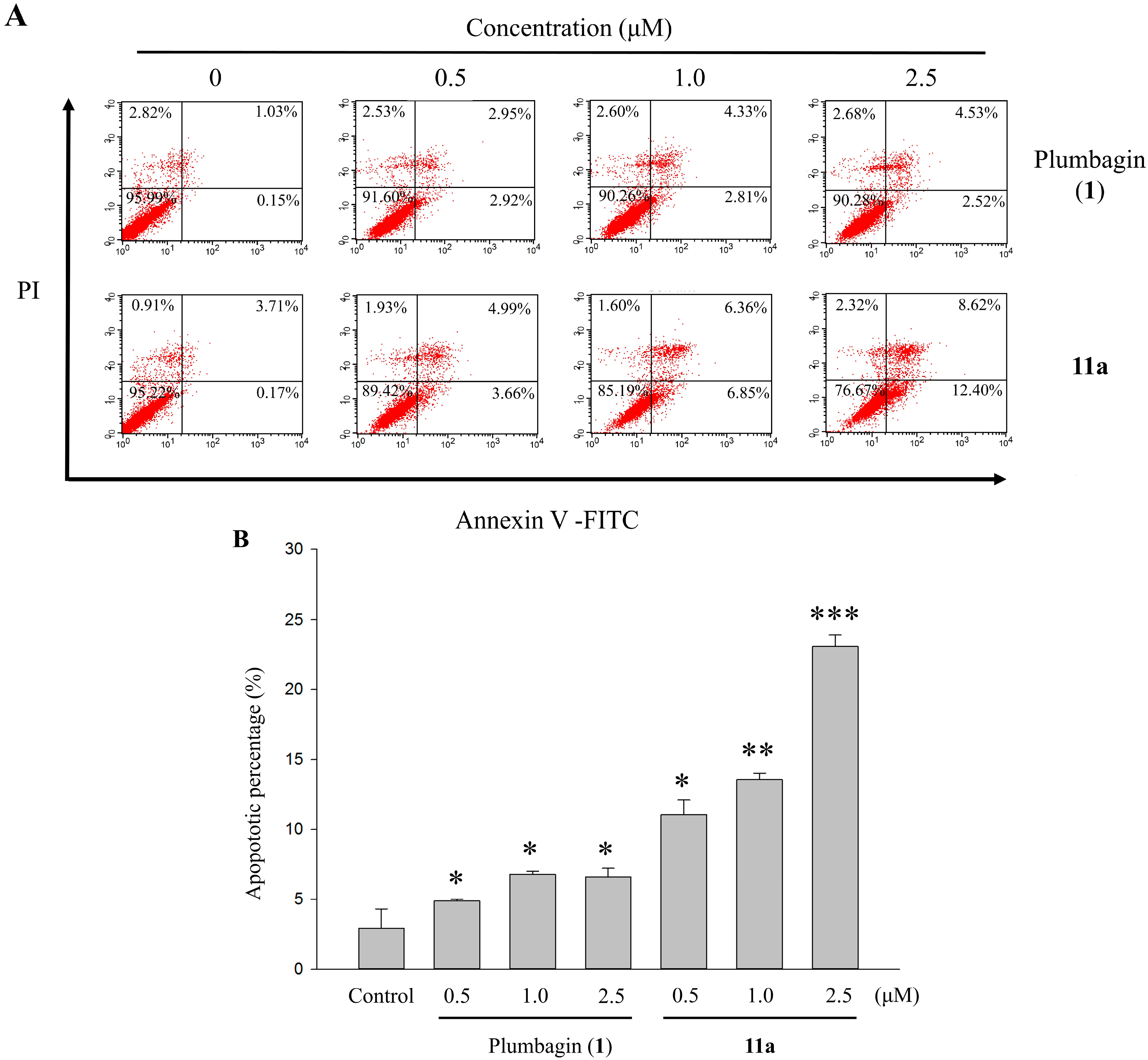
2.6. Apoptotic Analyses-Annexin V-FITC/PI Double Staining and Flow Cytometry Analyses
3. Experimental Section
3.1. General
3.2. General Synthetic Procedure for 5a,b‒11a,b
3.3. 5-Hydroxy-1,4-Naphthoquinone (Juglone, 3)
3.4. General Proceed Amination of Juglone with Alkyl Amine
3.5. Biological Activity
3.5.1. Cell Lines and Cell Culture Conditions
3.5.2. Cell Viability Assay
3.5.3. Nuclear Staining with Hoechst 33258
3.5.4. Cell Cycle Distribution Analysis
3.5.5. Assessment of Apoptotic Analysis
3.5.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- WHO Report. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/index.html (accessed on 25 June 2015).
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.R.; Babu, K.S.; Rao, R.R.; Suresh, G.; Rekha, K.; Murthy, J.M.; Rani, P.U.; Rao, J.M. Synthesis and insect antifeedant activity of plumbagin derivatives. Med. Chem. Res. 2012, 21, 578–583. [Google Scholar] [CrossRef]
- Cowan, M.M. Plant products as antimicrobial agents. Clin. Microbiol. Rev. 1999, 12, 564–582. [Google Scholar] [PubMed]
- Yang, J.T.; Li, Z.L.; Wu, J.Y.; Lu, F.J.; Chen, C.H. An oxidative stress mechanism of shikonin in human glioma cells. PLoS ONE 2014, 9, e94180. [Google Scholar] [CrossRef] [PubMed]
- McBride, T.J.; Oleson, J.J.; Woolf, D. The activity of streptonigrin against the Rauscher murine leukemia virus in vivo. Cancer Res. 1966, 26, 727–732. [Google Scholar] [PubMed]
- Keyes, S.R.; Loomis, R.; di Giovanna, M.P.; Pritsos, C.A.; Rockwell, S.; Sartorelli, A.C. Cytotoxicity and DNA crosslinks produced by mitomycin analogs in aerobic and hypoxic EMT6 cells. Cancer Commun. 1991, 3, 351–356. [Google Scholar] [PubMed]
- Reich, E.; Goldberg, I.H.; Rabinowitz, M. Structure-activity correlations of actinomycins and their derivatives. Nature 1962, 196, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Atamanyuk, D.; Zimenkovsky, B.; Atamanyuk, V.; Nektegayev, I.; Lesyk, R. Synthesis and biological activity of new thiopyrano[2,3-d]thiazoles containing a naphthoquinone moiety. Sci. Pharm. 2013, 81, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, D.; Etter, J.P.; Chettiar, S.N.; Mok, M.; Li, P.K. Antiproliferative activities and SAR studies of substituted anthraquinones and 1,4-naphthoquinones. Bioorg. Med. Chem. Lett. 2013, 23, 6864–6867. [Google Scholar]
- Didry, N.; Dubrevil, L.; Pinkas, M. Activity of anthraquinonic and naphthoquinonic compounds on oral bacteria. Pharmazie 1994, 49, 681–683. [Google Scholar] [PubMed]
- Likhitwitayawuid, K.; Kaewamatawong, R.; Ruangrungsi, N.; Krungkrai, J. Antimalarial naphthaquinones from Nepenthes thorelii. Planta Med. 1998, 64, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Mathew, N.; Paily, K.P.; Abidha; Vanamali, P.; Kalyanasundaram, M.; Balaraman, K. Macrofilaricidal activity of the plant Plumbago indica/rosea in vitro. Drug Dev. Res. 2002, 56, 33–39. [Google Scholar] [CrossRef]
- Kayser, O.; Kiderlen, A.F.; Laatsch, H.; Croft, S.L. In vitro leishmanicidal activity of monomeric and dimeric naphthoquinones. Acta Trop. 2000, 76, 131–138. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Malonne, H.; Duez, P.; Vanhaelen-Fastre, R.; Vanhaelen, M.; Fontaine, J. Cytotoxic constituents from Plumbago zeylanica. Fitoterapia 2004, 75, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Wong, Y.F.; Ge, L.; Zhang, Z.F.; Liu, Y.; Liu, L.; Zhou, H. Anti-inflammatory and analgesic effect of plumbagin through inhibition of nuclear factor-κB activation. J. Pharmacol. Exp. Ther. 2010, 335, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Thomson, R.H. Naturally Occurring Quinones, IV. Recent Advances; Blackie Academic & Professional: London, UK, 1997. [Google Scholar]
- Eldhose, B.; Gunawan, M.; Rahman, M.; Latha, M.S.; Notario, V. Plumbagin reduces human colon cancer cell survival by inducing cell cycle arrest and mitochondria-mediated apoptosis. Int. J. Oncol. 2014, 45, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Subramaniya, B.R.; Srinivasan, G.; Sadullah, S.S.; Davis, N.; Subhadara, L.B.; Halagowder, D.; Sivasitambaram, N.D. Apoptosis inducing effect of plumbagin on colonic cancer cells depends on expression of COX-2. PLoS ONE 2011, 6, e18695. [Google Scholar] [CrossRef] [PubMed]
- Pertino, M.W.; Theoduloz, C.; Palenzuela, J.A.; Afonso, M.M.; Yesilada, E.; Monsalve, F.; González, P.; Droguett, D.; Schmeda-Hirschmann, G. Synthesis and pharmacological activity of diterpenylnaphthoquinone derivatives. Molecules 2011, 16, 8614–8628. [Google Scholar] [CrossRef] [PubMed]
- Del Corral, J.M.M.; Castro, M.A.; Gordaliza, M.; Martín, M.L.; Gualberto, S.A.; Gamito, A.M.; Cuevas, C.; San Feliciano, A. Synthesis and cytotoxicity of new aminoterpenylquinones. Bioorg. Med. Chem. 2005, 13, 631–644. [Google Scholar] [CrossRef]
- Kawamura, M.; Kuriyama, I.; Maruo, S.; Kuramochi, K.; Tsubaki, K.; Yoshida, H.; Mizushina, Y. Anti-tumor effects of novel 5-O-acyl plumbagins based on the inhibition of mammalian DNA replicative polymerase activity. PLoS ONE 2014, 9, e88736. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.C.; Ibuka, T.; Huang, L.J.; Lien, J.C.; Yean, S.R.; Huang, S.C.; Lednicer, D.; Morris-Natschke, S.; Lee, K.H. Synthesis and cytotoxicity of 1,2-disubstituted naphth[2,3-d]imidazole-4,9-diones and related compounds. J. Med. Chem. 1996, 39, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, E.L.; Ríos-Luci, C.; León, L.G.; Burton, G.; Padrón, J.M.; Misico, R.I. Antiproliferative activity of synthetic naphthoquinones related to lapachol. First synthesis of 5-hydroxylapachol. Bioorg. Med. Chem. 2010, 18, 2621–2630. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, D.; Chettiar, S.N.; Etter, J.P.; Mok, M.; Li, P.K. Anticancer activity and SAR studies of substituted 1,4-naphthoquinones. Bioorg. Med. Chem. 2013, 21, 4662–4669. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lo, C.Y.; Hsu, L.C.; Chen, M.S.; Lin, Y.J.; Chen, L.G.; Kuo, C.D.; Wu, J.Y. Synthesis and anticancer activity of a novel series of 9-O-substituted berberine derivatives: A lipophilic substitute role. Bioorg. Med. Chem. Lett. 2013, 23, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Mallavadhani, U.V.; Prasad, C.V.; Shrivastava, S.; Naidu, V.G. Synthesis and anticancer activity of some novel 5,6-fused hybrids of juglone based 1,4-naphthoquinones. Eur. J. Med. Chem. 2014, 83, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Kuo, C.D.; Chu, C.Y.; Chen, M.S.; Lin, J.H.; Chen, Y.J.; Liao, H.F. Synthesis of novel lipophilic N-substituted norcantharimide derivatives and evaluation of their anticancer activities. Molecules 2014, 19, 6911–6928. [Google Scholar] [CrossRef] [PubMed]
- Hadjila, D.; Mohamed, H. Activated zeolites and heteropolyacids: An efficient catalysts for the synthesis of triacetoxyaromatic precursors of hydroxyquinones. Asian J. Chem. 2013, 25, 6112–6116. [Google Scholar]
- Ishiguro, K.; Ohira, Y.; Oku, H. Antipruritic Dinaphthofuran-7,12-dione Derivatives from the Pericarp of Impatiens balsamina. J. Nat. Prod. 1998, 61, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Bieber, L.W.; Rolim Neto, P.J.; Generino, R.M. Regioselective alkylation of substituted quinones by trialkylboranes. Tetrahedron Lett. 1999, 40, 4473–4476. [Google Scholar] [CrossRef]
- Lien, J.C.; Huang, L.J.; Teng, C.M.; Wang, J.P.; Kuo, S.C. Synthesis of 2-alkoxy 1,4-naphthoquinone derivatives as antiplatelet, antiinflammatory, and antiallergic agents. Chem. Pharm. Bull. 2002, 50, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Fieser, L.F. Naphthoquinone Antimalarials. IV-XI. Synthesis. J. Am. Chem. Soc. 1948, 70, 3174–3215. [Google Scholar] [CrossRef] [PubMed]
- Fugitt, R.B.; Schwing, G.W. Miticidal Ethers. US Patent 4,110,473, 29 August 1978. [Google Scholar]
- Kishore, N.; Mishra, B.B.; Tiwari, V.K.; Tripathi, V. Difuranonaphthoquinones from Plumbago zeylanica roots. Phytochem. Lett. 2010, 3, 62–65. [Google Scholar] [CrossRef]
- Inoue, K.; Ueda, S.; Nayeshiro, H.; Moritome, N.; Inouye, H. Biosynthesis of naphthoquinones and anthraquinones in Streptocarpus dunnii cell cultures. Phytochemistry 1984, 23, 313–318. [Google Scholar] [CrossRef]
- Fiorito, S.; Epifano, F.; Bruyere, C.; Mathieu, V.; Kiss, R.; Genovese, S. Growth inhibitory activity for cancer cell lines of lapachol and its natural and semi-synthetic derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, M. Synthesis and structure-activity relationships of novel warfarin derivatives. Bioorg. Med. Chem. 2007, 15, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Yavorskyy, A.; Shvydkiv, O.; Limburg, C.; Nolan, K.; Delauré, Y.M.C.; Oelgemöller, M. Photooxygenations in a bubble column reactor. Green Chem. 2012, 14, 888–892. [Google Scholar] [CrossRef]
- Bukhtoyarova, A.D.; Rybalova, T.V.; Ektova, L.V. Amination of 5-hydroxy-1,4-naphthoquinone in the presence of copper acetate. Russ. J. Org. Chem. 2010, 46, 855–859. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3, 5a,b‒11a,b and 13a,b‒19a,b are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.-H.; Lo, C.-Y.; Gwo, Z.-H.; Lin, H.-J.; Chen, L.-G.; Kuo, C.-D.; Wu, J.-Y. Synthesis and Biological Evaluation of Lipophilic 1,4-Naphthoquinone Derivatives against Human Cancer Cell Lines. Molecules 2015, 20, 11994-12015. https://doi.org/10.3390/molecules200711994
Wang S-H, Lo C-Y, Gwo Z-H, Lin H-J, Chen L-G, Kuo C-D, Wu J-Y. Synthesis and Biological Evaluation of Lipophilic 1,4-Naphthoquinone Derivatives against Human Cancer Cell Lines. Molecules. 2015; 20(7):11994-12015. https://doi.org/10.3390/molecules200711994
Chicago/Turabian StyleWang, Shao-Hung, Chih-Yu Lo, Zhong-Heng Gwo, Hong-Jhih Lin, Lih-Geeng Chen, Cheng-Deng Kuo, and Jin-Yi Wu. 2015. "Synthesis and Biological Evaluation of Lipophilic 1,4-Naphthoquinone Derivatives against Human Cancer Cell Lines" Molecules 20, no. 7: 11994-12015. https://doi.org/10.3390/molecules200711994
APA StyleWang, S.-H., Lo, C.-Y., Gwo, Z.-H., Lin, H.-J., Chen, L.-G., Kuo, C.-D., & Wu, J.-Y. (2015). Synthesis and Biological Evaluation of Lipophilic 1,4-Naphthoquinone Derivatives against Human Cancer Cell Lines. Molecules, 20(7), 11994-12015. https://doi.org/10.3390/molecules200711994