Abstract
In this work, the relationship between cyclophilin A (CypA) and HCV prompted us to screen a series of small molecule CypA inhibitors which were previously reported by our group. Among them, compound 1, discovered as a non-immunosuppressive anti-HCV agent with an EC50 value of 0.67 μM in a virus assay, was selected for further study. Subsequent chemical modification by O-acylation led to a novel class of molecules, among which compound 25 demonstrated the most potent anti-HCV activity in the virus assay (EC50 = 0.19 μM), but low cytotoxicity and hERG cardiac toxicity. The following studies (a solution stability assay and a simple pharmacokinetic test together with a CypA enzyme inhibition assay) preliminarily indicated that 25 was a prodrug of 1. To the best of our knowledge, 25 is probably the most potent currently reported small molecule anti-HCV agent acting via the CypA inhibitory mechanism. Consequently, our study has provided a new potential small molecule for curing HCV infection.
1. Introduction
Infection with hepatitis C virus (HCV) is well recognized as a worldwide health issue that chronically affects more than 170 million people [1,2,3]. Therefore hepatitis C has been one of the most active areas for antiviral drug development over the past two decades [4,5]. Currently, there are more than 20 directly-acting antivirals (DAAs) targeting the HCV NS3 protease, NS5B polymerase and NS5A protein in advanced clinical trials [6]. However, these DAAs have to be used in combination clinically to avoid resistance, because HCV, an RNA virus, possesses high replication and mutation rates [6]. Thus, a strategy targeting host factors essential for viral replication such as cyclophilins (Cyps) may create alternative kinds of anti-HCV agents for clinical application alone or in combination.
Cyps are a family of proteins found in vertebrates and other organisms that are proved to be identical to peptidyl-prolyl cis-trans isomerase (PPIase) [7]. Via its PPIase activity, Cyps play a crucial role in numerous cellular processes, including transcriptional regulation, immune response, protein secretion, and mitochondrial function [8,9]. Cyclophilin A (CypA), the most critical member of the Cyp family, has been extensively researched. Except for the functions mentioned above, CypA also plays a critical part in the replication of various kinds of viruses, such as human immunodeficiency virus type 1 (HIV-1), influenza virus, HCV, vesicular stomatitis virus (VSV), vaccinia virus and human papilloma virus (HPV) [10,11]. On account of the importance of CypA in the regulation of numerous biological processes, significant efforts have been made in the discovery of CypA inhibitors in the past decade [12,13,14,15,16,17].
In recent years, the relationship between HCV and CypA has been demonstrated more and more clearly [18,19,20,21]. Abundant research has shown that CypA is an important host factor for HCV proliferation as the PPIase activity of the hydrophobic gorge area in CypA plays a critical role in the HCV RNA proliferation and protein secretion [20,22]. This enzyme can enhance the rate of folding/unfolding of proteins via its PPIase activity, and thereby guarantee the correct assembly and functions of the HCV replicative complex [8]. In the latest studies HCV NS5A protein was proved to be the main ligand of CypA, which was essential for the prolyl peptide isomerization of NS5A in the process of HCV proliferation, by various methods, including nuclear magnetic resonance (NMR), isothermal titration calorimetry (ITC), and surface plasmon resonance (SPR) [23,24,25]. Thus, CypA PPIase inhibitors could hinder virus proliferation by disrupting the interaction between CypA and NS5A [23,24,25]. Currently, all reported CypA inhibitors for anti-HCV in clinical studies are cyclosporin A (CsA) or its analogues (Figure 1), such as alisporivir (DEB025), a non-immunosuppressive CsA analogue in phase III clinical trials at Novartis with an EC50 value of 0.045 μM (replicon assay), and another non-immunosuppressive CypA inhibitor SCY-635, which is in phase IIa at Scynexis with an EC50 value of 0.10 μM (replicon assay) [6,26,27]. However, these analogues might be limited in their applications when considering their synthesis difficulties and poor tolerability [28]. Furthermore, two small molecule anti-HCV CypA inhibitors, F680 and F684, are reported, but the structures of the two compounds have not been made public [28]. Therefore, a novel small molecule anti-HCV agent targeting the host factor of CypA would have extensive prospects and might provide a cure for HCV infection.
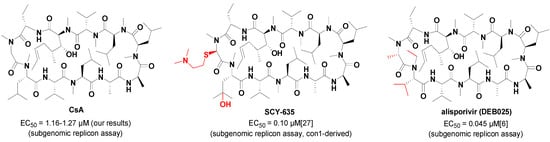
Figure 1.
Chemical structures and anti-HCV activities of CsA, SCY-635 and alisporivir.
Figure 1.
Chemical structures and anti-HCV activities of CsA, SCY-635 and alisporivir.
In consideration of the important role of CypA in numerous biological processes and related diseases, we are focused on the discovery and application of the CypA inhibitors. In a previous study, we reported a new series of small molecule CypA inhibitors with nanomolar inhibitory potencies which possess a common 1-(benzoyl)-3-(9H-fluoren-9-yl)-urea scaffold [29]. In view of the close relationship between CypA and HCV, we wondered if these compounds might possess antiviral activity as set forth. Therefore, a series of experiments reported in this article were performed aiming at discovering and developing a novel kind of anti-HCV agent.
2. Results and Discussion
2.1. Screening of Compounds for Anti-HCV Activity
Some compounds 1–13 which demonstrated potent CypA enzyme inhibitory activity in our previous work were selected for the antiviral tests. Incidentally, four newly synthesized derivatives of these compounds (compounds 14–17) were also incorporated into this assay. As this was a preliminary screen, only one kind of replicon assay was conducted with the results shown in Table 1. In the test, CsA was chosen as the control. The results were encouraging, and among compounds 1–17, compound 1 showed moderately potent anti-HCV activity with an EC50 value of 0.92 μM in the replicon assay, which was a little more potent than the control, CsA (EC50 = 1.16 μM). Furthermore, it seemed that the fluorene ring was more beneficial for the antiviral activity (compounds 1, 8 vs. 2‒4). More inspiringly, the two newly synthesized compounds 14, 15 with acetoxyl substituents demonstrated more potent activity than 1 (0.55 and 0.46 μM vs. 0.92 μM), which also suggested the chemical modification to be performed as the next step, viz. O-acylation of the hydroxyl groups on the benzene ring.

Table 1.
Chemical structures and replicon assay results of compounds 1–17 a. 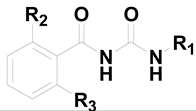
| Compound | R1 | R2 | R3 | Anti-HCV JFH1-2a Replicon Assay EC50 (μM) ± SD b |
|---|---|---|---|---|
| 1 | 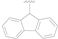 | OH | OH | 0.92 ± 0.12 |
| 2 | 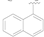 | OH | OH | >10 |
| 3 | 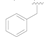 | OH | OH | >50 |
| 4 | 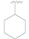 | OH | OH | >50 |
| 5 | 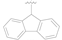 | Cl | H | >50 |
| 6 | 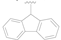 | NO2 | H | >50 |
| 7 | 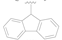 | CN | H | >50 |
| 8 | 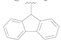 | F | H | 1.65 ± 0.32 |
| 9 | 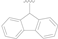 | CF3 | H | >10 |
| 10 | 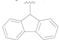 | MeS | H | >50 |
| 11 | 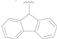 | F | F | >50 |
| 12 | 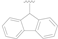 | F | MeS | >50 |
| 13 | 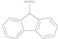 | Cl | MeO | >50 |
| 14 | 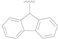 | 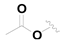 | OH | 0.55 ± 0.03 |
| 15 | 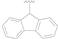 | 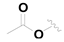 | 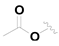 | 0.46 ± 0.05 |
| 16 | 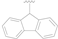 | 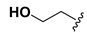 | OH | >20 |
| 17 | 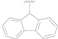 |  |  | >20 |
| CsA | 1.16 ± 0.14 |
a The synthesis and HPLC analysis data of 1–13 was according to reference [29]; the synthesis of 16–17 was described in the Supporting Information; The experimental method was described in the “Virus Assay” of the “Method”; b Each value indicates the mean ± SD of three independent experiments.
2.2. Spleen Cells Proliferation Inhibition Assay
Considering that CypA regulates numerous biological processes, selectivity should be regard as an important consideration in clinical studies for further drug development, or it may encounter clinical difficulties (e.g., alisporivir, which was put on hold by the FDA due to some side effects). Thus, before the further chemical modification of compound 1 by O-acylation, the immunosuppressive ability was evaluated firstly and CsA was selected as a control in this assay, because an anti-HCV agent via CypA inhibition should possess enough low inhibition activities against proliferation of spleen cells. The result was optimistic, as compound 1 showed quite low inhibition activities (more than 30-fold) against proliferation of spleen cells compared with its anti-HCV activity (Table 2), so it could be regarded as a a non-immunosuppressive lead compound suitable for further research.
Table 2.
Inhibition of spleen cell proliferation by compound 1 and CsA.
| Cmpd | T Spleen Cells IC50 (μM) ± SD a | B Spleen Cells IC50 (μM) ± SD a |
|---|---|---|
| 1 | 33.03 ± 5.72 | 51.93 ± 8.68 |
| CsA | 0.01 ± 0.005 | 0.04 ± 0.008 |
a Each value indicates the mean ± SD of three experiments using one mouse with triplicate sets in each assay. Half-maximal inhibitory concentrations (IC50 values) are obtained by log-probit analysis of inhibitory curves using SPSS, a windows statistical software package.
2.3. Derivatives Design and Synthesis
In light of the advance of the antiviral activities after derivatization (14, 15 vs. 1), we aimed at systematically evaluating the impact of the variation and quantity of acyloxy substituents. Thus varied kinds of substituted acyls (aroyl, straight-chain, branched-chain and cycloalkane substituted acyls) were introduced at the two hydroxyl groups of compound 1. In totally another 16 compounds 18–33 were thus designed and synthesized (Scheme 1), and their chemical structures were shown in Table 3. In general, these esters were prepared by the reaction of compound 1 and the corresponding anhydride or acyl chloride in anhydrous N,N-dimethylformamide (DMF). The differences between the two reactions are the quantity of the anhydride or acyl chloride used and the reaction temperature. When synthesizing the compounds 18–25, 0.9 equivalents of the anhydride or acyl chloride were used at a temperature below −20 °C, whereas 4 equivalents under room temperature were invariably used for the synthesis of 26–33. The synthesis of compounds 14 and 15 was also performed according to this method. Details of the synthetic procedures and structural characterizations are described in the Experimental Section. All compounds 18–33 were confirmed ≥95% purity (Supporting Information, Table S1).
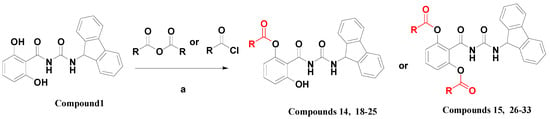
Scheme 1.
The synthesis of compounds 14–15 and 18–33.
Scheme 1.
The synthesis of compounds 14–15 and 18–33.
Reagents and conditions: (a) (i) anhydride or acyl chloride, Et3N, DMF, r.t. or −20 °C; (ii) r.t.
Table 3.
Chemical structures of compounds 18–33. 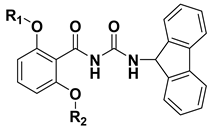
| Cmpd | R1 | R2 | Cmpd | R1 | R2 |
|---|---|---|---|---|---|
| 18 | 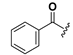 | H | 26 | 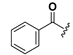 | 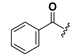 |
| 19 | 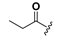 | H | 27 | 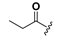 | 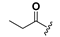 |
| 20 | 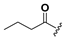 | H | 28 | 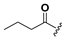 | 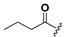 |
| 21 | 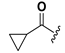 | H | 29 | 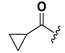 | 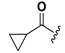 |
| 22 | 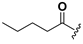 | H | 30 | 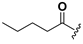 | 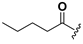 |
| 23 | 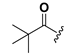 | H | 31 | 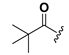 | 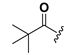 |
| 24 | 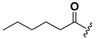 | H | 32 | 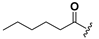 | 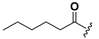 |
| 25 |  | H | 33 |  |  |
2.4. Virus Assays
Virus replicon assays (H77, JFH1 and Con1) and virus assays were performed, with CsA as a control. The results are shown in Table 4. All the ester compounds including compounds 14, 15 were tested and exhibited more potent antiviral activities than the control in three replicon assays. Furthermore, in the Con-1b replicon assay, most compounds showed similar or more potent activities compared with compound 1, except 18, 24, which were less potent than 1. The results of the H77-1a and JFH1-2a replicon assays were more exciting, as all the compounds possessed more potent antiviral activities than compound 1, except 18, 19, 24 and 33 in the JFH1-2a assay. Inspiringly, seven compounds (19, 20, 22, 23, 25, 27, 30) which were obviously more potent than compound 1 in the virus assays were identified, among which 25 exhibited the most potent anti-HCV effect with an EC50 value of 0.19 μM, that was 3.5-fold and 2.1-fold more potent than 1 and CsA, respectively. It should be noted that the antiviral activities of all compounds in the replicon assays were very close to that of the two known small molecular CypA inhibitors F680/F684. Furthermore, all the synthesized compounds showed low cytotoxicity (CC50 > 16 μM), indicating that the inhibition of HCV was not due to the death of host cells.
Further studies were conducted using one mutant type JFH1-2a virus strain (S282T, a resistance mutation in the active site that is selected by a series of nucleoside analogues including sofosbuvir, approved by the FDA, that targets HCV NS5B polymerase [30]) to demonstrate the advantage of our compounds 1 and 25 compared with sofosbuvir.
Table 4.
The anti-HCV activities of compounds 14, 15, 18–33.
| Cmpd | Anti-HCV Activity | ||||
|---|---|---|---|---|---|
| EC50 (μM) ± SD a | CC50 (μM) | ||||
| Replicon Assay | Virus Assay | ||||
| H77-1a | JFH1-2a | Con1-1b | JFH1-2a | ||
| 1 | 1.11 ± 0.33 | 0.92 ± 0.12 | 0.78 ± 0.19 | 0.67 ± 0.23 | >16 |
| 14 | 0.77 ± 0.37 | 0.55 ± 0.03 | 0.80 ± 0.23 | 0.71 ± 0.34 | >16 |
| 15 | 0.80 ± 0.13 | 0.46 ± 0.05 | 0.36 ± 0.04 | 0.74 ± 0.36 | >16 |
| 18 | 0.60 ± 0.35 | 1.05 ± 0.01 | 1.03 ± 0.02 | 0.70 ± 0.23 | >16 |
| 19 | 0.38 ± 0.22 | 0.94 ± 0.07 | 0.72 ± 0.20 | 0.47 ± 0.04 | >16 |
| 20 | 0.98 ± 0.03 | 0.70 ± 0.02 | 0.49 ± 0.02 | 0.28 ± 0.02 | >16 |
| 21 | 0.57 ± 0.20 | 0.43 ± 0.08 | 0.85 ± 0.24 | 0.78 ± 0.35 | >16 |
| 22 | 1.00 ± 0.06 | 0.47 ± 0.02 | 0.44 ± 0.10 | 0.44 ± 0.03 | >16 |
| 23 | 0.67 ± 0.05 | 0.48 ± 0.02 | 0.83 ± 0.28 | 0.53 ± 0.05 | >16 |
| 24 | 1.05 ± 0.01 | 1.08 ± 0.05 | 1.00 ± 0.01 | 0.74 ± 0.37 | >16 |
| 25 | 0.85 ± 0.21 | 0.45 ± 0.08 | 0.81 ± 0.24 | 0.19 ± 0.08 | >16 |
| 26 | 0.53 ± 0.21 | 0.36 ± 0.03 | 0.39 ± 0.05 | 1.09 ± 0.03 | >16 |
| 27 | 0.52 ± 0.27 | 0.38 ± 0.03 | 0.38 ± 0.02 | 0.42 ± 0.08 | >16 |
| 28 | 0.79 ± 0.20 | 0.84 ± 0.32 | 0.62 ± 0.15 | 0.68 ± 0.47 | >16 |
| 29 | 0.52 ± 0.14 | 0.42 ± 0.14 | 0.60 ± 0.19 | 0.74 ± 0.35 | >16 |
| 30 | 0.73 ± 0.11 | 0.62 ± 0.05 | 0.83 ± 0.26 | 0.47 ± 0.01 | >16 |
| 31 | 0.87 ± 0.27 | 0.75 ± 0.29 | 0.56 ± 0.20 | 0.84 ± 0.21 | >16 |
| 32 | 0.63 ± 0.29 | 0.38 ± 0.05 | 0.73 ± 0.34 | 0.74 ± 0.31 | >16 |
| 33 | 0.54 ± 0.15 | 0.98 ± 0.01 | 0.73 ± 0.20 | 0.95 ± 0.02 | >16 |
| CsA | 1.26 ± 0.20 | 1.16 ± 0.14 | 1.27 ± 0.32 | 0.40 ± 0.11 | >8 |
| F680, F684 b | 0.20 c | ||||
a Each value indicates the mean ± SD of three independent experiments; b Small molecular non-immunosuppressive CypA inhibitors currently in preclinical trials at INSERM for HCV therapy [4]; c Value in the genotype 1b subgenomic replicon assay [26].
As shown in Table 5, the results revealed that both 1 and 25 could maintain their anti-virus activities compared with the results in the assays using wild type JFH1-2a virus strain (0.52 μM vs. 0.56 μM and 0.27 μM vs. 0.29 μM, respectively), whereas resistance to sofosbuvir was obviously observed (0.22 μM vs. 1.14 μM). This indicated that our CypA inhibitors can be regarded as a kind of potential anti-HCV agents.
Table 5.
The anti-HCV activities of compounds 1 and 25 against mutant type virus strain.
| Cmpd | Anti-HCV Activity (Virus Assay) | ||
|---|---|---|---|
| EC50 (μM) ± SD a | CC50 (μM) | ||
| JFH1-2a (Wild Type) | JFH1-2a (S282T) | ||
| 1 | 0.52 ± 0.16 | 0.56 ± 0.13 | >20 |
| 25 | 0.27 ± 0.09 | 0.29 ± 0.07 | >20 |
| Sofosbuvir | 0.22 ± 0.07 | 1.14 ± 0.21 | >20 |
a Each value indicates the mean ± SD of three independent experiments.
2.5. Drug Combination Assay
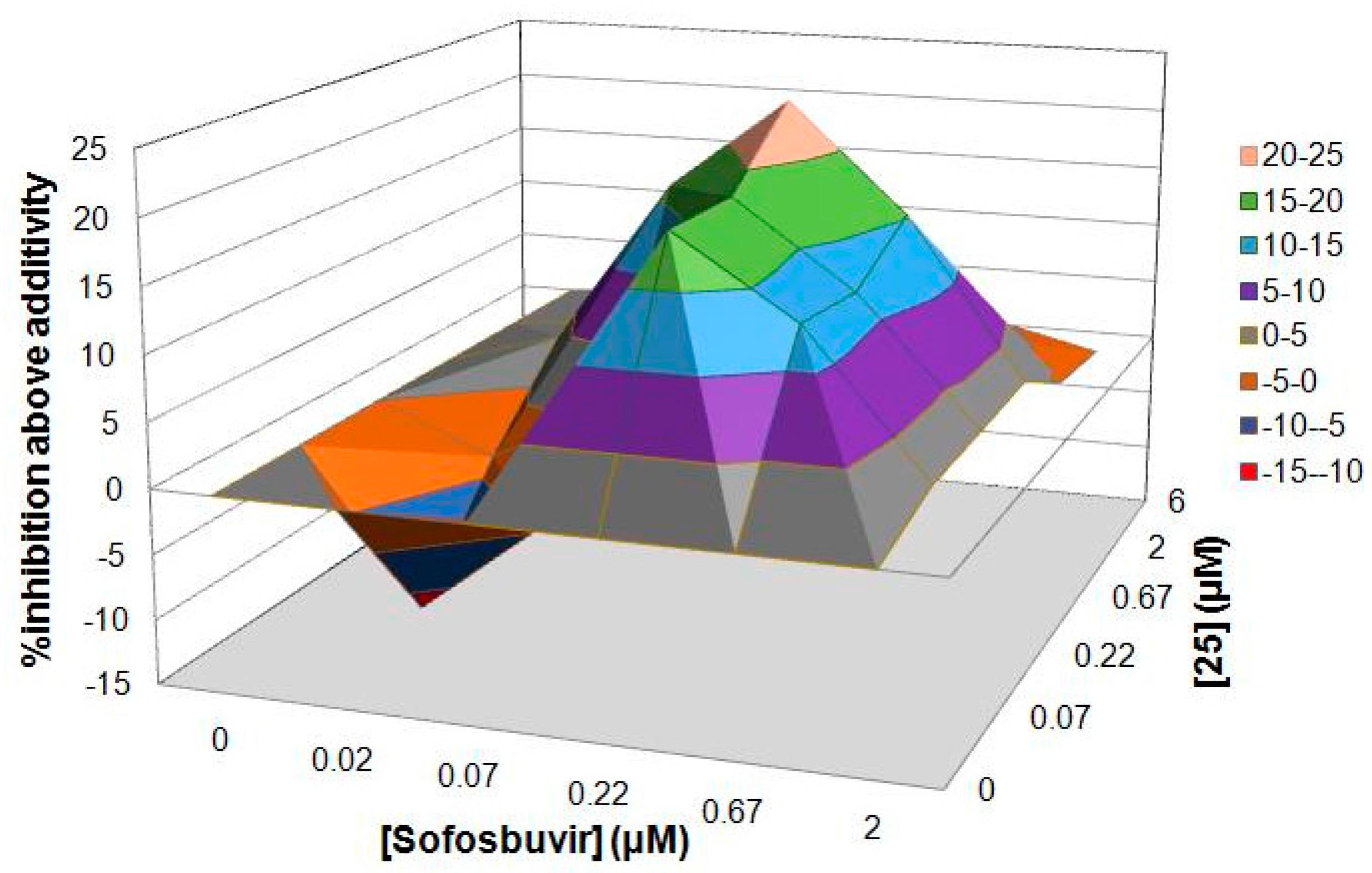
Because the inhibition of each step of the virus life cycle can affect the proliferation of a virus, we wondered whether our compounds could be combined with other anti-HCV drugs currently used in hepatitis C treatment. To investigate this hypothesis, our most potent compound 25 and sofosbuvir were employed in this study. The assay of the combination was evaluated with a HCV cell culture (JFH1-2a) system with a luciferase reporter gene. To determine whether the effects of the combination were synergistic, additive, or antagonistic, the data were analyzed using the method of Prichard and Shipman [31]. The results revealed that the 25‒sofosbuvir combination had an ntiviral effect that was more potent than the theoretical additive effects, as the maximal percent synergy extended to the range of 20% to 25%, which supported the notion that this combination was indeed synergistic (Figure 2). Nearly no evidence of significant antiviral antagonism was observed with the tested doses, as the maximal percent antagonism did not exceed almost 10%.
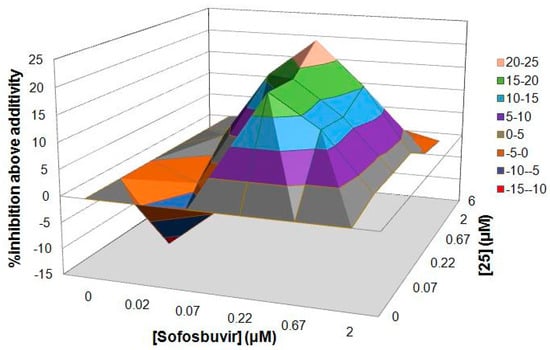
Figure 2.
Result of combination study between 25 and sofosbuvir.
Figure 2.
Result of combination study between 25 and sofosbuvir.
2.6. CypA Enzyme Inhibition Assay
The standard chymotrypsin-coupled assay results (Table 6) indicated that all the compounds remarkably reduced CypA inhibitory activities in vitro after the acylation of the hydroxyl groups. Compared with 1 (CypA enzyme inhibition: IC50 = 0.032 ± 0.002 μM), 89% of the O-acylated esters (16/18) showed found no CypA inhibition activity (IC50 > 5 μM), and only the monoacetylated compound 14 and monobenzoylated compound 18 maintained low CypA inhibitory activities (IC50 = 2.43 ± 0.50 and 1.99 ± 0.47 μM, respectively). In consideration of the potent anti-HCV activity and the ester structure features, we speculated that this kind of ester was not the active form in the antiviral process and there was probably a prodrug mechanism in these series of compounds. Therefore, several tests were conducted in the next step to make a preliminary verification of this hypothesis.
Table 6.
Enzyme inhibition activities of compounds 14, 15, 18–33.
| Cmpd | Enzyme Inhibition Assay IC50 (μM) ± SD a |
|---|---|
| 1 | 0.032 ± 0.002 |
| 14 | 2.43 ± 0.50 |
| 18 | 1.99 ± 0.47 |
| 15 and 19–33 | >5 |
a Each value indicates the mean ± SD of five independent experiments.
2.7. Preliminary Verification of the Hypothetical Prodrug Mechanism
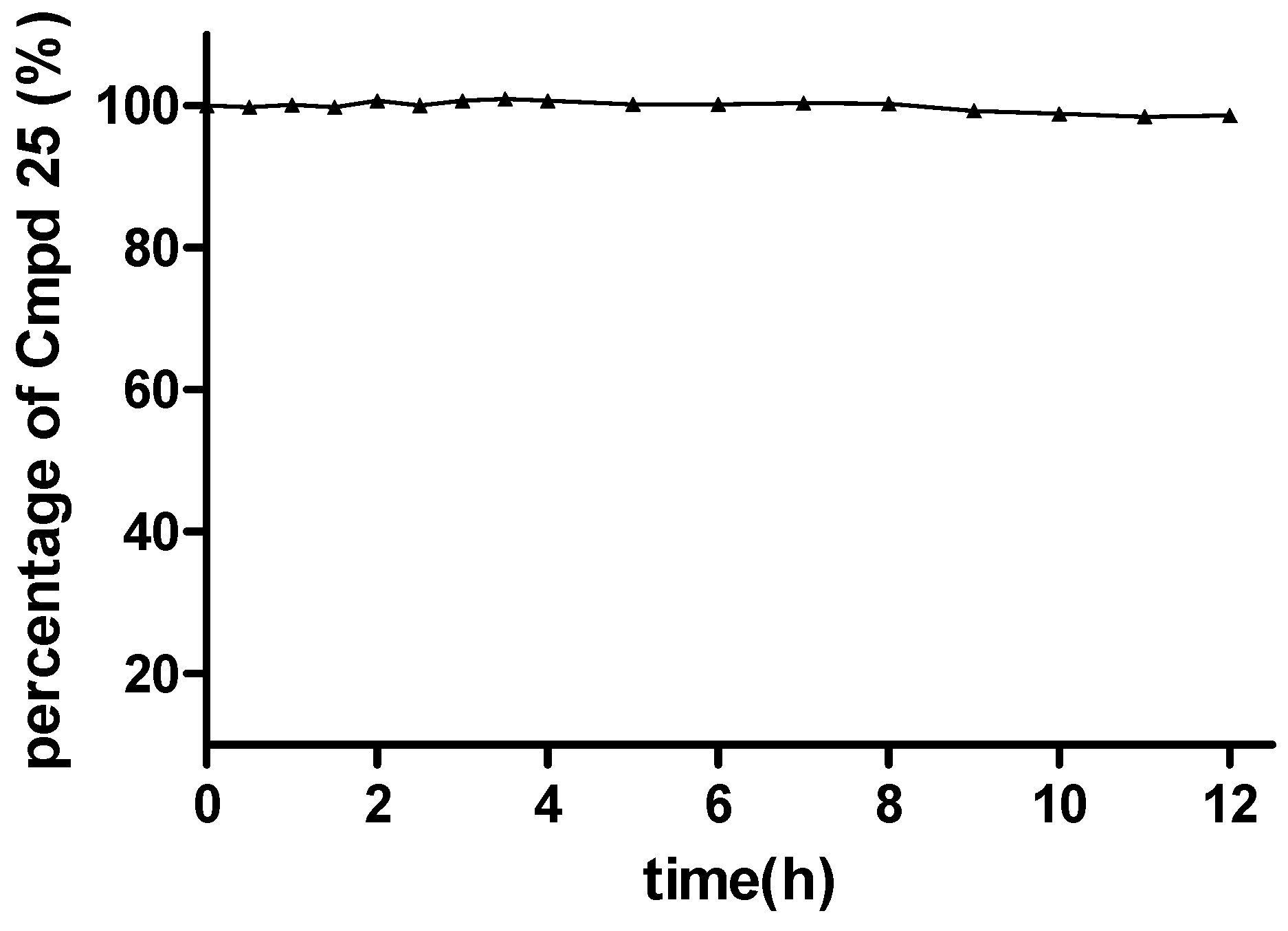
In this section, the most potent compound 25 in the virus assay was chosen for further study to make a preliminary verification of the hypothetical prodrug mechanism. Specifically, a solution stability assay and a simple pharmacokinetic test were performed, using the experimental methods described in the Supporting Information. The solution stability assay results showed that the ester moiety of 25 was quite stable in PBS buffer at pH 7.4. For example, after 12 h in the PBS buffer, 98.6% of 25 still remained in the original form (Figure 3 and Supporting Information, Table S2), indicating that this series of compounds possessed sufficient stability under in vitro physiological conditions.
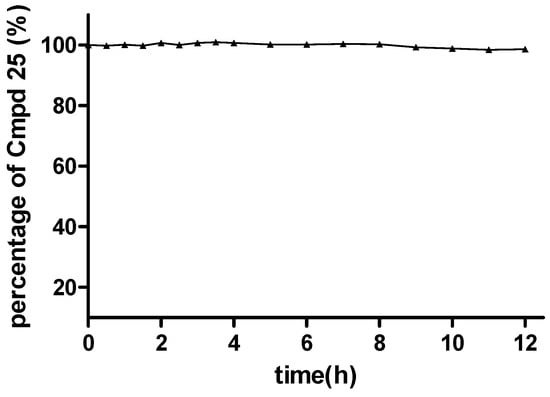
Figure 3.
Stability of 25 under pH 7.4 conditions.
Figure 3.
Stability of 25 under pH 7.4 conditions.
Moreover, the pharmacokinetic test result indicated that 25 exhibited a high release rate of 1 in vivo. After the intravenous injection of 25 (2 mg∙kg−1) into rats, both 25 and 1 could be immediately detected in the plasma. However, the plasma concentration of 1 was much higher than that of 25. After 5 min, the plasma concentration of 1 was 904.5 ± 291.9 ng∙mL−1 (mean ± SD), whereas the plasma concentration of 25 was only 20.47 ± 9.99 ng∙mL−1 (mean ± SD), indicating that 25 could be hydrolyzed into 1 rapidly (Supporting Information, Table S3). Furthermore, the AUC1/AUC25 value was about 60, which also demonstrated that the main form of 25 existing in body circulation was compound 1.
3. Experimental Section
3.1. General Information
The reagents were purchased from Adamas-beta (Shanghai, China), J & K (Beijing, China), Energy Chemical (Shanghai, China) and TCI (Shanghai, China) and used without further purification. Analytical thin-layer chromatography (TLC) was performed on HSGF 254 plates (150–200 µm thickness, Yantai Huiyou Company, Yantai, China). Yields were not optimized. Melting points were measured in capillary tubes on a SGW X-4 melting point apparatus (Shanghai Jingmiyiqi Company, Shanghai, China) without correction. Nuclear magnetic resonance (NMR) spectra were recorded on a AVANCE 400 NMR (Bruker, Karlsruhe, Germany). Chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS) used as the internal standard. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low-and high-resolution mass spectra (LRMS and HRMS) were obtained with electric ionization (EI) and electrospray ionization (ESI) on Finnigan MAT-95 (Bremen, Germany) and LCQ-DECA (Bremen, Germany) spectrometers.
1-(2-Acetoxy-6-hydroxybenzoyl)-3-(9H-fluoren-9-yl)-urea (14). To a solution of 1 (0.9 g) and triethylamine (0.35 mL) in anhydrous DMF (25 mL), was added acetic anhydride (0.21 mL) with stirring at −20 °C. The mixture was stirred at room temperature for 12 h, and then heated to 40 °C with stirring for another 24 h. The reaction mixture was poured into H2O (500 mL), extracted with dichloromethane (DCM, 200 mL) three times. The organic layers were combined, dried with anhydrous sodium sulfate, and concentrated to dryness. The residue was purified by flash column chromatography on silica gel, eluted with a mixture of THF/DCM/petroleum ether (PE) (1:5:3, v/v/v), to afford 14 (161 mg, 16%) as a white solid: mp 215–225 °C; 1H-NMR (DMSO-d6) δ: 10.84 (s, 1H), 10.45 (s, 1H), 8.69 (s, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.62 (t, J = 7.5 Hz, 2H), 7.46 (t, J = 7.4 Hz, 2H), 7.37 (t, J = 7.4 Hz, 2H), 7.27 (t, J = 8.2 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.63 (d, J = 8.0 Hz, 1H), 5.97 (d, J = 7.9 Hz, 1H), 2.13 (s, 3H); ESI-MS m/z 401.0 [M − H]−; HRMS (ESI) m/z calcd C23H18N2O5 [M − H]− 401.1137, found 401.1143.
1-(2-Benzoyloxy-6-hydroxybenzoyl)-3-(9H-fluoren-9-yl)-urea (18). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by benzoyl chloride: mp 211–214 °C; 1H-NMR (DMSO-d6) δ: 11.07 (s, 1H), 10.45 (s, 1H), 8.61 (s, 1H), 8.01 (d, J = 7.4 Hz, 2H), 7.85 (t, J = 7.7 Hz, 2H), 7.78 (t, J = 7.4 Hz, 1H), 7.58 (t, J = 7.7 Hz, 2H), 7.40 (dd, J = 14.5, 7.0 Hz, 2H), 7.38–7.30 (m, 3H), 7.26 (t, J = 7.4 Hz, 2H), 6.84 (t, J = 8.9 Hz, 2H), 5.91 (d, J = 8.2 Hz, 1H); ESI-MS m/z 463.0 [M − H]−; HRMS (ESI) m/z calcd C28H20N2O5 [M − H]− 463.1294, found 463.1299.
1-(2-Hydroxy-6-propionyloxybenzoyl)-3-(9H-fluoren-9-yl)-urea (19). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by propionic anhydride: mp 206–216 °C; 1H-NMR (acetone-d6) δ: 9.72 (s, 1H), 8.71 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.70 (d, J = 7.4 Hz, 2H), 7.46 (t, J = 7.4 Hz, 2H), 7.43–7.34 (m, 3H), 6.89 (d, J = 8.4 Hz, 1H), 6.75 (d, J = 8.1 Hz, 1H), 6.11 (d, J = 8.1 Hz, 1H), 2.60 (q, J = 7.5 Hz, 2H), 1.23–1.12 (m, 3H); ESI-MS m/z 415.0 [M − H]−; HRMS (ESI) m/z calcd C24H20N2O5 [M − H]− 415.1294, found 415.1299.
1-(2-Hydroxy-6-n-butyryloxybenzoyl)-3-(9H-fluoren-9-yl)-urea (20). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by butyric anhydride: mp 204–210 °C; 1H-NMR (acetone-d6) δ: 8.71 (s, 1H), 7.87 (d, J = 7.5 Hz, 2H), 7.71 (d, J = 7.5 Hz, 2H), 7.47 (t, J = 7.4 Hz, 2H), 7.44–7.36 (m, 3H), 6.91 (d, J = 7.7 Hz, 1H), 6.75 (d, J = 7.9 Hz, 1H), 6.12 (d, J = 8.2 Hz, 1H), 2.56 (t, J = 7.3 Hz, 2H), 1.76–1.64 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H); ESI-MS m/z 429.1 [M − H]−; HRMS (ESI) m/z calcd C25H22N2O5 [M − H]− 429.1450, found 429.1456.
1-(2-Cyclopropyl formyloxy-6-hydroxybenzoyl)-3-(9H-fluoren-9-yl)-urea (21). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by cyclopropanecarboxylic acid chloride: mp 194–199 °C; 1H-NMR (DMSO-d6) δ: 10.85 (s, 1H), 10.36 (s, 1H), 8.71 (s, 1H), 7.90 (d, J = 7.6 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.47 (t, J = 7.4 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H), 7.25 (t, J = 8.2 Hz, 1H), 6.79 (d, J = 8.2 Hz, 1H), 6.63 (d, J = 8.2 Hz, 1H), 6.00 (d, J = 8.1 Hz, 1H), 1.75 (d, J = 4.8 Hz, 1H), 0.99–0.83 (m, 4H); EI-MS m/z 428.1(M+); 165.1 (100%); HRMS (EI) m/z calcd C25H20 N2O5 (M+) 428.1372, found 428.1373.
1-(2-Hydroxy-6-valeryloxybenzoyl)-3-(9H-fluoren-9-yl)-urea (22). Prepared in the same manner as described for 14, but acetic anhydride was replaced by valeric anhydride: mp 193–199 °C; 1H-NMR (acetone-d6) δ: 8.73 (d, J = 6.2 Hz, 1H), 7.86 (d, J = 7.6 Hz, 2H), 7.70 (d, J = 7.5 Hz, 2H), 7.46 (t, J = 7.6 Hz, 2H), 7.43–7.35 (m, 3H), 6.89 (d, J = 8.3 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.11 (d, J = 8.1 Hz, 1H), 2.57 (t, J = 7.4 Hz, 2H), 1.64 (dt, J = 15.2, 7.5 Hz, 2H), 1.37 (dq, J = 14.7, 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H); ESI-MS m/z 443.0 [M − H]−; HRMS (ESI) m/z calcd C26H24N2O5 [M − H]− 443.1607, found 443.1612.
1-(2-Hydroxy-6-trimethylacetoxyloxybenzoyl)-3-(9H-fluoren-9-yl)-urea (23). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by trimethylacetic anhydride: mp 187–189 °C; 1H-NMR (acetone-d6) δ: 9.71 (s, 1H), 8.76 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.46 (t, J = 7.4 Hz, 2H), 7.37 (dd, J = 13.2, 7.3 Hz, 3H), 6.87 (d, J = 8.2 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.11 (d, J = 7.9 Hz, 1H), 1.31 (s, 9H); ESI-MS m/z 443.0 [M − H]−; HRMS (ESI) m/z calcd C26H24N2O5 [M − H]− 443.1607, found 443.1612.
1-(2-Hexanoyloxy-6-hydroxybenzoyl)-3-(9H-fluoren-9-yl)-urea (24). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by hexanoic anhydride: mp 200–222 °C; 1H-NMR (DMSO-d6) δ: 11.09 (s, 1H), 10.42 (s, 1H), 8.73 (s, 1H), 7.89 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.4 Hz, 2H), 7.46 (t, J = 7.3 Hz, 2H), 7.36 (t, J = 7.3 Hz, 2H), 7.25 (t, J = 8.2 Hz, 1H), 6.77 (d, J = 8.3 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 5.98 (d, J = 7.8 Hz, 1H), 2.39 (t, J = 7.4 Hz, 2H), 1.48 (dd, J = 14.6, 7.3 Hz, 2H), 1.22–1.03 (m, 4H), 0.72 (t, J = 7.0 Hz, 3H). ; ESI-MS m/z 457.0 [M − H]−; HRMS (ESI) m/z calcd C27H26N2O5 [M − H]− 457.1763, found 457.1769.
1-(2-Hydroxy-6-n-Octanoyloxybenzoyl)-3-(9H-fluoren-9-yl)-urea (25). Prepared in the same manner as described for 14, but the acetic anhydride was replaced by n-octanoic anhydride: mp 184–189 °C; 1H-NMR (CDCl3) δ: 11.56 (s, 1H), 9.45 (s, 1H), 8.48 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 7.6 Hz, 2H), 7.65 (d, J = 7.5 Hz, 2H), 7.43 (dd, J = 12.1, 4.8 Hz, 3H), 7.34 (t, J = 7.5 Hz, 2H), 6.89 (d, J = 8.4 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.16 (d, J = 8.4 Hz, 1H), 2.81 (t, J = 7.4 Hz, 2H), 1.85 (dt, J = 15.1, 7.4 Hz, 2H), 1.53–1.28 (m, 8H), 0.91 (t, J = 6.8 Hz, 3H); EI-MS m/z 486.2 (M+); 165.1 (100%); HRMS (EI) m/z calcd C29H30N2O5 (M+) 486.2155, found 486.2157.
1-[2,6-Bis-(acetoxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (15). To a solution of 1 (0.9 g) and triethylamine (1.38 mL) in anhydrous DMF (15 mL), was added acetic anhydride (0.93 mL) with stirring at room temperature for 12 h. The reaction mixture was poured into H2O (300 mL), and extracted with DCM (120 mL) three times. The organic layers were combined, dried with anhydrous sodium sulfate, then concentrated to dryness. The residue was purified by flash column chromatography on silica gel, eluted with a mixture of THF/DCM/PE (1:5:3, v/v/v), to afford 15 (760 mg, 69%) as a white solid: mp 191–194 °C; 1H-NMR (acetone-d6) δ: 8.64 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.69 (d, J = 7.5 Hz, 2H), 7.56 (t, J = 8.3 Hz, 1H), 7.46 (t, J = 7.4 Hz, 2H), 7.38 (t, J = 7.2 Hz, 2H), 7.18 (t, J = 8.7 Hz, 2H), 6.10 (d, J = 8.1 Hz, 1H), 2.24 (s, 6H); ESI-MS m/z 445.0 [M + H]+; HRMS (ESI) m/z calcd C25H20N2O6 [M + Na]+ 467.1219, found 467.1214.
1-[2,6-Bis-(benzoyloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (26). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by benzoyl chloride: mp 211–213 °C; 1H-NMR (acetone-d6) δ: 10.33 (s, 1H), 8.54 (d, J = 7.0 Hz, 1H), 8.18 (d, J = 8.2 Hz, 4H), 7.84–7.68 (m, 5H), 7.62 (t, J = 7.7 Hz, 4H), 7.48 (d, J = 8.3 Hz, 2H), 7.37 (t, J = 7.5 Hz, 2H), 7.31 (d, J = 7.4 Hz, 2H), 7.21 (t, J = 7.4 Hz, 2H), 5.96 (d, J = 8.3 Hz, 1H); EI-MS m/z 568.2(M+); 105.0 (100%); HRMS (ESI) m/z calcd C35H24N2O6 [M + Na]+ 591.1532, found 591.1523.
1-[2,6-Bis-(propionyloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (27). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by propionic anhydride: mp 165–167 °C; 1H-NMR (acetone-d6) δ: 9.89 (s, 1H), 8.67 (s, 1H), 7.87 (d, J = 7.6 Hz, 2H), 7.68 (d, J = 7.5 Hz, 2H), 7.58 (t, J = 8.3 Hz, 1H), 7.48 (t, J = 7.3 Hz, 2H), 7.43–7.35 (m, 2H), 7.20 (d, J = 8.3 Hz, 2H), 6.11 (d, J = 8.1 Hz, 1H), 2.59 (q, J = 7.5 Hz, 4H), 1.18 (t, J = 7.5 Hz, 6H); EI-MS m/z 472.2(M+); 165.1 (100%); HRMS (ESI) m/z calcd C27H24N2O6 [M + Na]+ 495.1532, found 495.1527.
1-[2,6-Bis-(n-butyryloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (28). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by butyric anhydride: mp 186–188 °C; 1H-NMR (ad6) δ: 9.90 (s, 1H), 8.68 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.67 (d, J = 7.5 Hz, 2H), 7.56 (t, J = 8.3 Hz, 1H), 7.47 (t, J = 7.3 Hz, 2H), 7.38 (td, J = 7.5, 1.0 Hz, 2H), 7.18 (d, J = 8.3 Hz, 2H), 6.10 (d, J = 8.1 Hz, 1H), 2.53 (t, J = 7.3 Hz, 4H), 1.78–1.62 (m, 4H), 0.96 (t, J = 7.4 Hz, 6H); EI-MS m/z 500.2 (M+); 165.1 (100%); HRMS (ESI) m/z calcd C29H28N2O6 [M + Na]+ 523.1845, found 523.1840.
1-[2,6-Bis-(cyclopropylformyloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (29). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by cyclopropanecarboxylic acid chloride: mp 183–186 °C; 1H-NMR (CDCl3) δ: 8.50 (d, J = 8.0 Hz, 1H), 7.92 (s, 1H), 7.73 (d, J = 7.5 Hz, 2H), 7.65 (d, J = 7.5 Hz, 2H), 7.49 (t, J = 8.2 Hz, 1H), 7.43 (t, J = 7.4 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.08 (d, J = 8.2 Hz, 2H), 6.17 (d, J = 8.7 Hz, 1H), 1.84 (td, J = 7.8, 4.1 Hz, 2H), 1.21–1.13 (m, 4H), 1.08–0.99 (m, 4H); EI-MS m/z 496.2(M+); 165.1 (100%); HRMS (ESI) m/z calcd C29H24N2O6 [M + Na]+519.1532, found 519.1523.
1-[2,6-Bis-(valeryloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (30). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by valeric anhydride: mp 172–174 °C; 1H-NMR (CDCl3) δ: 8.47 (d, J = 8.3 Hz, 1H), 8.01 (s, 1H), 7.72 (d, J = 7.5 Hz, 2H), 7.62 (d, J = 7.5 Hz, 2H), 7.49 (t, J = 8.2 Hz, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.07 (d, J = 8.2 Hz, 2H), 6.13 (d, J = 8.4 Hz, 1H), 2.55 (t, J = 7.5 Hz, 4H), 1.75–1.63 (m, 4H), 1.38 (tt, J = 11.3, 5.8 Hz, 4H), 0.91 (t, J = 7.3 Hz, 6H); EI-MS m/z 528.2(M+); 165.1 (100%); HRMS (ESI) m/z calcd C31H32N2O6 [M + Na]+ 551.2158, found 551.2153.
1-[2,6-Bis-(trimethylacetoxyloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (31). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by trimethylacetic anhydride: mp 202–205 °C; 1H-NMR (CDCl3) δ: 8.55 (d, J = 8.9 Hz, 1H), 7.71 (d, J = 7.5 Hz, 2H), 7.50 (t, J = 7.6 Hz, 2H), 7.49–7.37 (m, 3H), 7.28 (dd, J = 14.4, 6.9 Hz, 2H), 7.04 (d, J = 8.3 Hz, 2H), 6.13 (d, J = 8.7 Hz, 1H), 1.32 (s, 18H); EI-MS m/z 528.2(M+); 165.1 (100%); HRMS (ESI) m/z calcd C31H32N2O6 [M + Na]+ 551.2158, found 551.2153.
1-[2,6-Bis-(hexanoyloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (32). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by hexanoic anhydride: mp 162–165 °C; 1H-NMR (CDCl3) δ: 8.46 (d, J = 8.0 Hz, 1H), 7.93 (s, 1H), 7.72 (d, J = 7.6 Hz, 2H), 7.62 (d, J = 7.4 Hz, 2H), 7.50 (t, J = 8.2 Hz, 1H), 7.42 (t, J = 7.4 Hz, 2H), 7.31 (dd, J = 14.1, 6.7 Hz, 2H), 7.07 (d, J = 8.3 Hz, 2H), 6.14 (d, J = 8.2 Hz, 1H), 2.54 (t, J = 7.6 Hz, 4H), 1.69 (dd, J = 14.6, 7.2 Hz, 4H), 1.30 (dd, J = 20.4, 5.5 Hz, 8H), 0.88 (t, J = 6.8 Hz, 6H); EI-MS m/z 556.3(M+); 165.1 (100%); HRMS (ESI) m/z calcd C33H36N2O6 [M + Na]+ 579.2471, found 579.2466.
1-[2,6-Bis-(n-Octanoyloxy)benzoyl]-3-(9H-fluoren-9-yl)-urea (33). Prepared in the same manner as described for 15, but the acetic anhydride was replaced by n-octanoic anhydride: mp 152–153 °C; 1H-NMR (CDCl3) δ: 8.48 (d, J = 8.3 Hz, 1H), 8.17 (s, 1H), 7.71 (t, J = 8.3 Hz, 2H), 7.61 (d, J = 7.5 Hz, 2H), 7.51–7.45 (m, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.07 (d, J = 8.3 Hz, 2H), 6.12 (d, J = 8.4 Hz, 1H), 2.54 (t, J = 7.6 Hz, 4H), 1.77–1.63 (m, 4H), 1.34–1.17 (m, 16H), 0.87 (t, J = 6.3 Hz, 6H); HRMS (ESI) m/z calcd C37H44N2O6 [M + H]+ 613.3278, found 613.3267.
3.2. Biological Assays
3.2.1. Spleen Cells Proliferation Inhibition Assay
Preparation of Splenocyte Suspension: The spleen was aseptically taken from female ICR mice strains (6–8 weeks-old, 20 ± 2 g), crushed gently and separated into single cells by squeezing in 20 mL RPMI 1640 medium (Invitrogen, shanghai, China). The cells obtained were passed through 70 μM nylon cell strainer and centrifuged at 1000 rpm∙min−1 for 5 min at RT. Pellets were added into 10 mL erythrocytes lysis buffer (150 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA) followed by centrifugation to remove erythrocytes. After washing twice with complete medium (RPMI 1640 containing 100 U∙mL−1 of penicillin, 100 U∙mL−1 of streptomycin and 10% FBS), they were re-suspended in the complete medium and used for cell culture.
Proliferation Assay of Spleen Cells: During this assay, CsA was used as a control for evaluation of compound 1 inhibition activity. 5 × 105 spleen cells were cultured in 96-well flat-bottom microplates (Corning Incorporatrd Coring, New York, NY, USA) in RPMI 1640 complete medium containing 5 μg∙mL−1 of concanavalin A (Con A) in the presence of increasing concentration of compounds: CsA (0.01, 0.1, 0.5, 1, 2, 5, 8, 10, 100 μM); compound 1 (0.01, 0.1, 0.5, 1, 2, 5, 8, 10, 100 μM). After 72 h of incubation, the cell growth was evaluated with MTT assay. The assay plates were added 10 μL of 5 mg∙mL−1 MTT per well and incubated for 4 h. The formazan generated was solubilized with 100 μL of solution (10% SDS, 5% isobutyl alcohol, and 10 mM HCl) after incubated at 37 °C overnight. The absorption at 580 nm relative to 680 nm was measured by SpectraMax Plus 384 Reader (Molecular Devices, Shanghai, China).
3.2.2. Virus Assay
Viral Inhibition Assay: The HCV virus assay was constructed by using the method according to [32,33] with a little modification. The methods of establishing the HCV virus and subgenomic replicon cell lines were described in the Supporting Information. Huh 7.5.1 cells were seeded in 96-well plates at a density of 2 × 104 cells per well at 37 °C overnight. All of the synthetic compounds were diluted with dimethyl sulfoxide (DMSO) to 10 mM of stock solution. The initial concentration of compounds was 20 μM and then diluted at a gradient of 1:4 to the concentrations ranging from 20 μM to 0.02 μM containing 0.5% DMSO. For the HCV virus assay, serial diluted compounds were mixed with a certain titer of JFH-1 virus, and the final concentration of JFH-1 virus titer was diluted to the numbers of relative luminescence units (RLU) ranging from 20,000 to 50,000 RLU and then added to the Huh 7.5.1 cells. For the HCV replicon system, serially diluted compounds was added to the Huh 7.5.1 replicon cells and the cells were cultured for 2 days at 37 °C. Then the cells were harvested and the luminescence was detected following the manufacturer’s protocol for the Renilla-Glo™ Luciferase Assay System. EC50 was the concentration of compound at which the HCV luminescence level in the Huh7.5.1 cells is reduced by 50%. Each data point represents the average of two replicates in cell culture. The values of EC50 were plotted by the GraphPad Prism 5 software.
Cell Proliferation Assay: Huh 7.5.1 cells were seeded at a density of 2 × 104 cells per well in 96-well plates overnight. The cells were respectively incubated with serial diluted compounds for 48 h, and the viability of Huh7.5.1 cells was determined in 96-well tissue culture plates using cell proliferation reagent WST-1 (Roche, Basel, Switzerland). The absorbance (OD450 /reference OD630) was measured to detect the cytotoxicity of compounds, according to the manufacturer’s protocol of Cell Proliferation Reagent WST-1.
3.2.3. Drug Combination Assay
Huh 7.5.1 cells were seeded at 1 × 104 per well in 96-well microtiter plates. After overnight culture, Huh 7.5.1 cells were infected with JFH-1 virus with luciferase reporter gene and treated with various concentrations of 25 alone, Sofosbuvir alone, or in combinations for 24 h. Antiviral activities were determined by measuring the reduction of luciferase activities in the cells. 3D-analysis of combinations on JFH1 infection was evaluated using the method of Prichard and Shipman [31]. Combination studies for each pair of compounds were performed in triplicate. The theoretical additive surface is subtracted from the actual experimental surface, resulting in a horizontal surface that equals the zero plane when the combination is additive. A surface raising more than 25% above the zero plane indicates a synergistic effect of the combination and a surface dropping lower than 25% below the zero plane indicates antagonism.
3.2.4. CypA Enzyme Inhibition Assay
The PPIase activity of CypA was conducted in a coupled assay with chymotrypsin using the synthetic tetrapeptide N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Sigma, Shanghai, China) as described in [34] with small modifications. The assay buffer (20 mM Tris-HCl, 50 mM NaCl, pH 7.8) and CypA were precooled at 4 °C and mixed with 0.4 mg∙mL−1 chymotrypsin (20 mg∙mL−1 in 1 mM HCl). The reaction was initiated by adding peptide substrate (2 mg∙mL−1 in 500 mM LiCl in tetrahydrofuran (THF)). After a delay from the onset of mixing (usually 6 s), the cis-to-trans isomerization of the Ala-Pro peptide bond, coupled with the chymotryptic cleavage of the trans peptide, then the p-nitroanilide was quantified by the increase in absorbance at 380 nm for 60 s by a U-3010 spectrophotometer. Absorbance readings were collected every 0.1 s and the temperature was controlled at 4 °C during the assay. The progress curves were analyzed by nonlinear least-squares fit. The inhibition assays of compounds were performed in the same manner as above-mentioned. 10 µM of test compounds in DMSO, at various concentrations, was added to the CypA solution in the assay buffer. After being preincubated for 1 h at 4 °C, the assay was started by the addition of chymotrypsin and the substrate. To calculate the half-maximal inhibitory concentration (IC50), a percent of the remaining PPIase activity was plotted against the common logarithm of the compound concentration, and the data were fitted using the sigmoidal fitting model of Origin 7.0 software.
4. Conclusions
Considering the important role of CypA for HCV proliferation, we were enlightened and prompted to screen a series of compounds previously reported as CypA inhibitors and their derivatives. Fortunately, the small molecule CypA inhibitor 1 and its O-acetylated derivatives 14, 15 with potent anti-HCV activity were discovered. The subsequent assessment showed that compound 1 can be regarded as a non-immunosuppressive lead compound for further research, hence a series of acylated derivatives of the hydroxyl groups of compound 1 were prepared to give another 16 derivatives. Among these compounds, 25 exhibited the most potent antiviral activities in replicon and virus assays but low cytotoxicity (CC50 > 16 μM) and hERG (the human Ether-à-go-go-related gene) cardiac toxicity (hERG: IC50 > 15 μM, the experimental method was described in the Supporting Information). Furthermore, the solution stability assay and the simple pharmacokinetic test together with the CypA inhibition result preliminarily indicated that compound 25 was probably a prodrug of 1. The proof was set forth as follows: (1) 25 was inactive at the enzyme level (CypA enzyme inhibition: IC50 > 5 μM), whereas it was active at the cellular level (virus assay: EC50 = 0.19 μM); (2) 25 showed sufficient stability in vitro (stable in PBS buffer at pH 7.4 after 12 h), but could rapidly release 1 in vivo (AUC1/AUCA8 ≈ 60). However, further study is still necessary to explicitly demonstrate the hypothesized prodrug mechanism. To the best of our knowledge, compound 25 is probably the most potent small molecule anti-HCV agent in virus assay via CypA inhibitory mechanism. In conclusion, our present study indicated that compounds 1 and 25 could be used as a new class of potent anti-HCV agents and the further pre-clinical studies of the two compounds are currently underway in our laboratory.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/06/10342/s1.
Acknowledgments
Financial support of this research provided by the National Natural Science Foundation of China (Grants 21222211, 21372001, 91313303), the Program for New Century Excellent Talents in University (Grant NCET-12-0853), “Shu Guang” project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation (Grant 14SG28), and the Fundamental Research Funds for the Central Universities is gratefully acknowledged.
Author Contributions
W.Y. performed chemical research and wrote the main manuscript. J.Q. and H.M. conducted biological assay and wrote part of the manuscript. F.M., J.H., J.Z. and H.J. assisted with manuscript text. L.L., L.Z. and J.L. designed research, analyzed data, and wrote manuscript. All authors reviewed the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Te, H.S.; Jensen, D.M. Epidemiology of hepatitis B and C viruses: A global overview. Clin. Liver Dis. 2010, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- World Health Organization (WHO). WHO Report on the Hepatitis C, 2012; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Cui, H.; Qing, J.; Guo, Y.; Wang, Y.; Cui, L.; He, T.; Zhang, L.; Liu, L. Stapled peptide-based membrane fusion inhibitors of hepatitis C. Bioorg. Med. Chem. 2013, 21, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Qing, J.; Wang, S.; Wang, H.; Zhang, L.; Tang, Y. Design and synthesis of imidazo[1,2-α][1,8]-naphthyridine derivatives as anti-HCV agents via direct C-H arylation. Org. Biomol. Chem. 2014, 12, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Gallay, P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antiviral Res. 2013, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Peptidylprolyl cis/trans isomerases (immunophilins): Biological diversity-targets-functions. Curr. Top. Med. Chem. 2003, 3, 1315–1347. [Google Scholar] [CrossRef] [PubMed]
- Dornan, J.; Taylor, P.; Walkinshaw, M.D. Structures of immu-nophilins and their ligand complexes. Curr. Top. Med. Chem. 2003, 3, 1392–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.J.; Mei, Q.; Li, J.T.; He, H.Y. Cyclophilin A and viral infections. Biochem. Biophys. Res. Commun. 2012, 424, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Sedrani, R.; Kallen, J.; Cabrejas, L.M.M.; Papageorgiou, C.D.; Senia, F.; Rohrbach, S.; Wagner, D.; Thai, B.; Eme, A.M.J.; France, J.; et al. Sanglifehrin-cyclophilin interaction: Degradation work, synthetic macrocyclic analogues, X-ray crystal structure, and binding data. J. Am. Chem. Soc. 2003, 125, 3849–3859. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Gui, C.; Zhang, L.; Qin, Y.; Xu, Q.; Zhang, J.; Liu, H.; Shen, X.; Jiang, H. Discovering novel chemical inhibitors of human cyclophilin A: Virtual screening, synthesis, and bioassay. Bioorg. Med. Chem. 2006, 14, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Zhang, L.; Wang, F.; Gui, C.; Zhang, L.; Qin, Y.; Xu, Q.; Liu, H.; Nan, F.; et al. One novel quinoxaline derivative as a potent human cyclophilin A inhibitor shows highly inhibitory activity against mouse spleen cell proliferation. Bioorg. Med. Chem. 2006, 14, 5527–5534. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, J.; Chen, J.; Luo, X.; Zhu, W.; Shen, J.; Liu, H.; Shen, X.; Jiang, H. Strategy for discovering chemical inhibitors of human cyclophilin A: Focused library design, virtual screening, chemical synthesis and bioassay. J. Comb. Chem. 2006, 8, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Guichou, J.F.; Viaud, J.; Mettling, C.; Subra, G.; Lin, Y.L.; Chavanieu, A. Structure-based design, synthesis, and biological evaluation of novel inhibitors of human cyclophilin A. J. Med. Chem. 2006, 49, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Hijikata, M.; Hosaka, M.; Yamaji, M.; Shimotohno, K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 2003, 38, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H.L. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.L.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of Cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009, 284, 16998–17005. [Google Scholar] [CrossRef] [PubMed]
- Gaither, L.A.; Borawski, J.; Anderson, L.J.; Balabanis, K.A.; Devay, P.; Joberty, G.; Rau, C.; Schirle, M.; Bouwmeester, T.; Mickanin, C.; et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology 2010, 397, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Lopez, M.Z.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009. [Google Scholar] [CrossRef]
- Hanoulle, X.; Badillo, A.; Wieruszeski, J.M.; Verdegem, D.; Landrieu, I.; Bartenschlager, R.; Penin, F.; Lippens, G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyI cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 2009, 284, 13589–13601. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Hanoulle, X.; Chatterji, U.; Berger, C.; Snoeck, J.; Bobardt, M.; Lim, P.; Vliegen, I.; Paeshuyse, J.; Vuagniaux, G. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Robotham, J.M.; Tang, H. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J. Virol. 2012, 86, 12616–12624. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Kaul, A.; de Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporine DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; DiMassimo, B.; Rusnak, P.; Heuman, D.; Lalezari, J.; Sluder, A.; Scorneaux, B.; Mosier, S.; Kowalczyk, P.; Ribeill, Y.; et al. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J. Hepatol. 2012, 57, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Lerat, H.; Bessin, Y.; Barthe, P.; Bourget, W.; Douguet, D.; Guichou, J.F.; Pawlotsky, J.M. New cyclophilin inhibitors unrelated to cyclosporine potently inhibit HCV replication and revert HCV-induced mitochondrial dysfunction. In Hepatology; Wiley-Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Ni, S.; Yuan, Y.; Huang, J.; Mao, X.; Lv, M.; Zhu, J.; Shen, X.; Pei, J.; Lai, L.; Jiang, H.; Li, J. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach. J. Med. Chem. 2009, 52, 5295–5298. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Steuer, H.M.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Shipman, C., Jr. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 1990, 14, 181–205. [Google Scholar] [CrossRef]
- Han, Q.; Xu, C.; Wu, C.; Zhu, W.; Yang, R.; Chen, X. Compensatory mutations in NS3 and NS5A proteins enhance the virus production capability of hepatitis C reporter virus. Virus Res. 2009, 145, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liao, Q.; Yang, R.; Chen, X.; Chen, X. A novel luciferase and GFP dual reporter virus for rapid and convenient evaluation of hepatitis C virus replication. Virus Res. 2011, 155, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colon-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerasesby an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).