
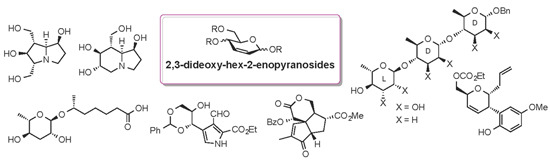
A Survey of Recent Synthetic Applications of 2,3-Dideoxy-Hex-2-enopyranosides


Abstract
:
{kind=link}
{kind=link}
{kind=link}
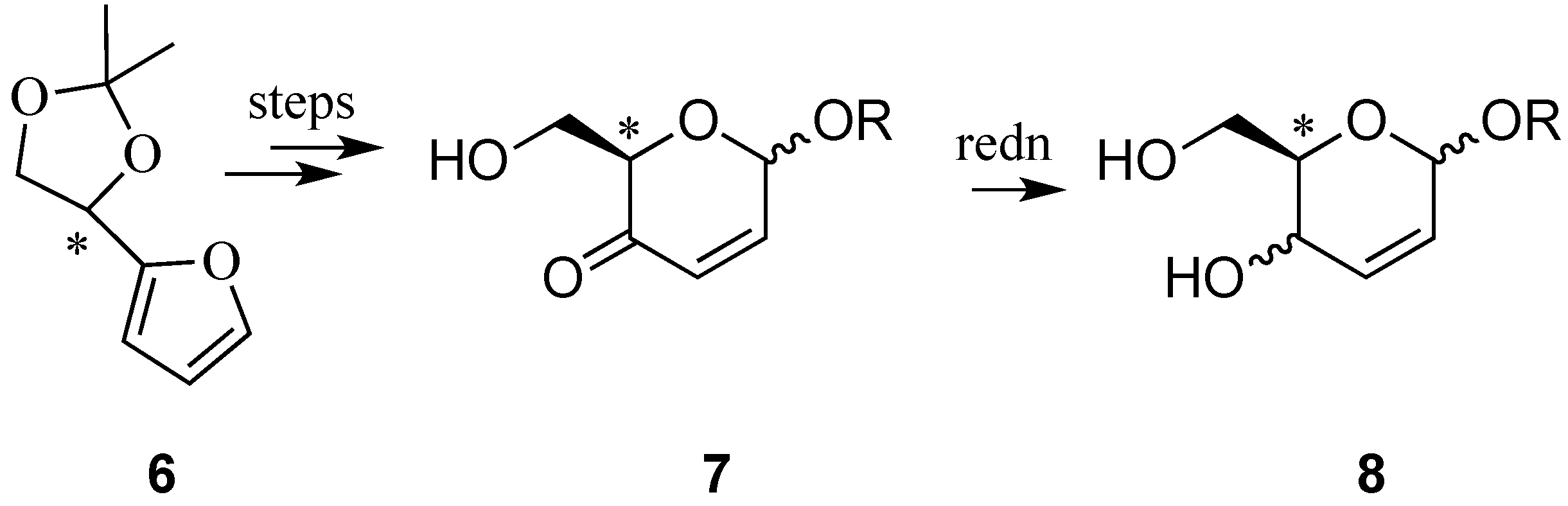{kind=link}
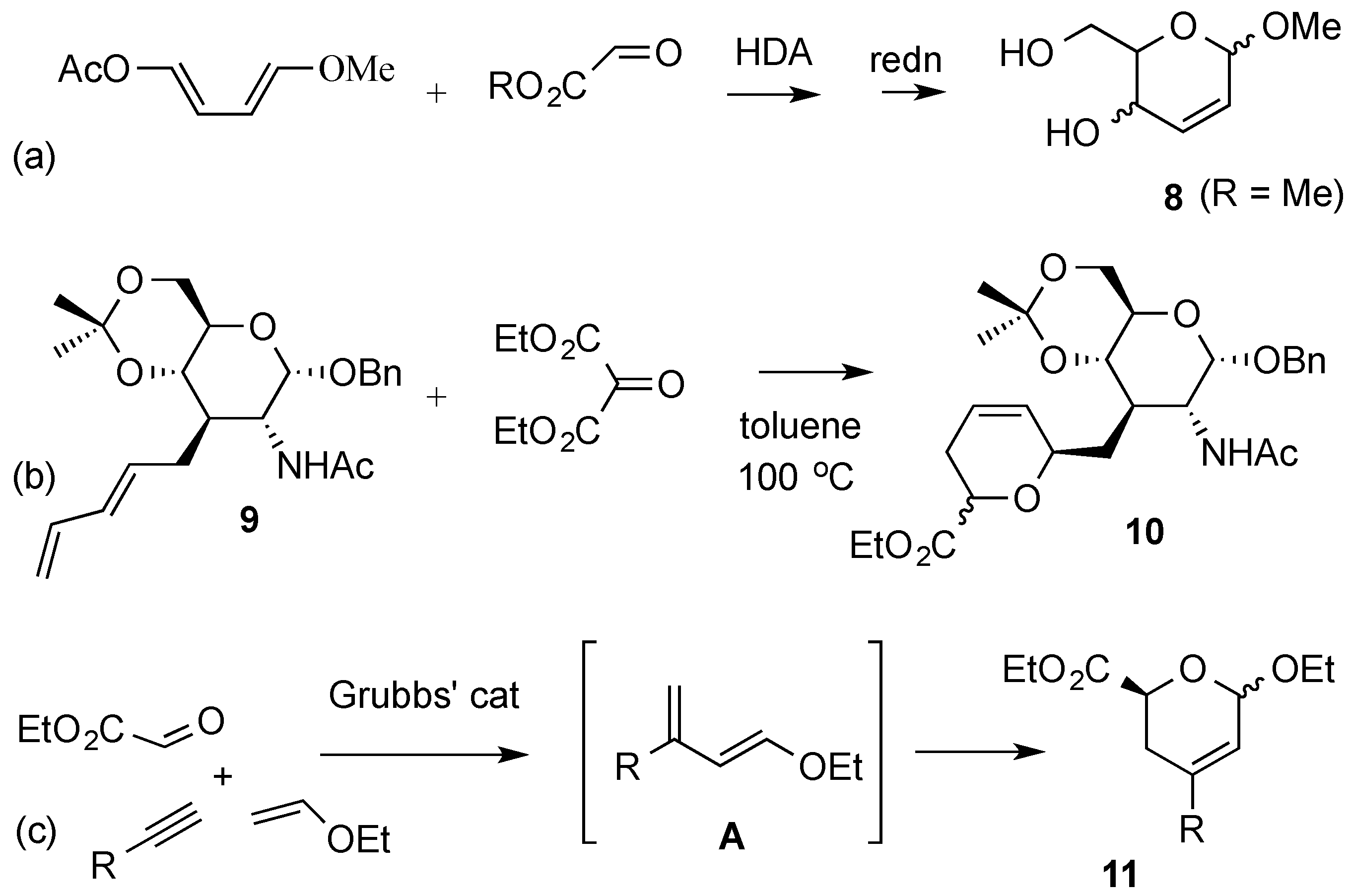{kind=link}
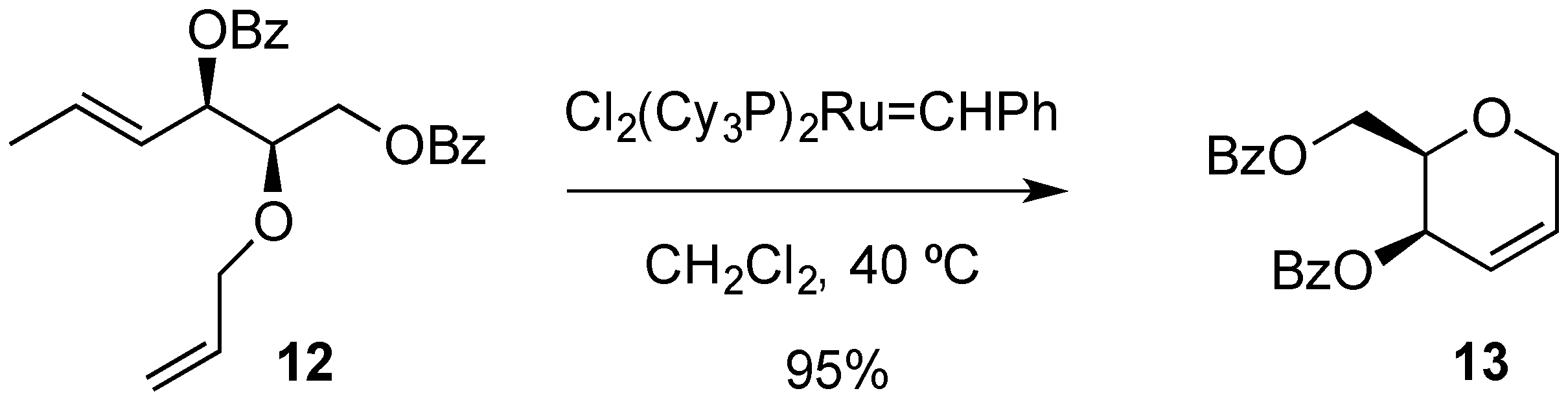{kind=link}
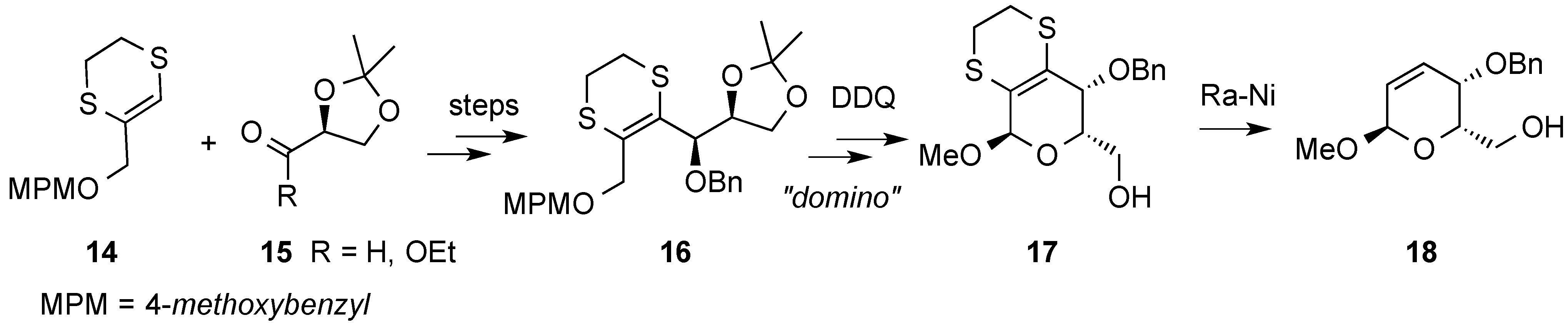{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
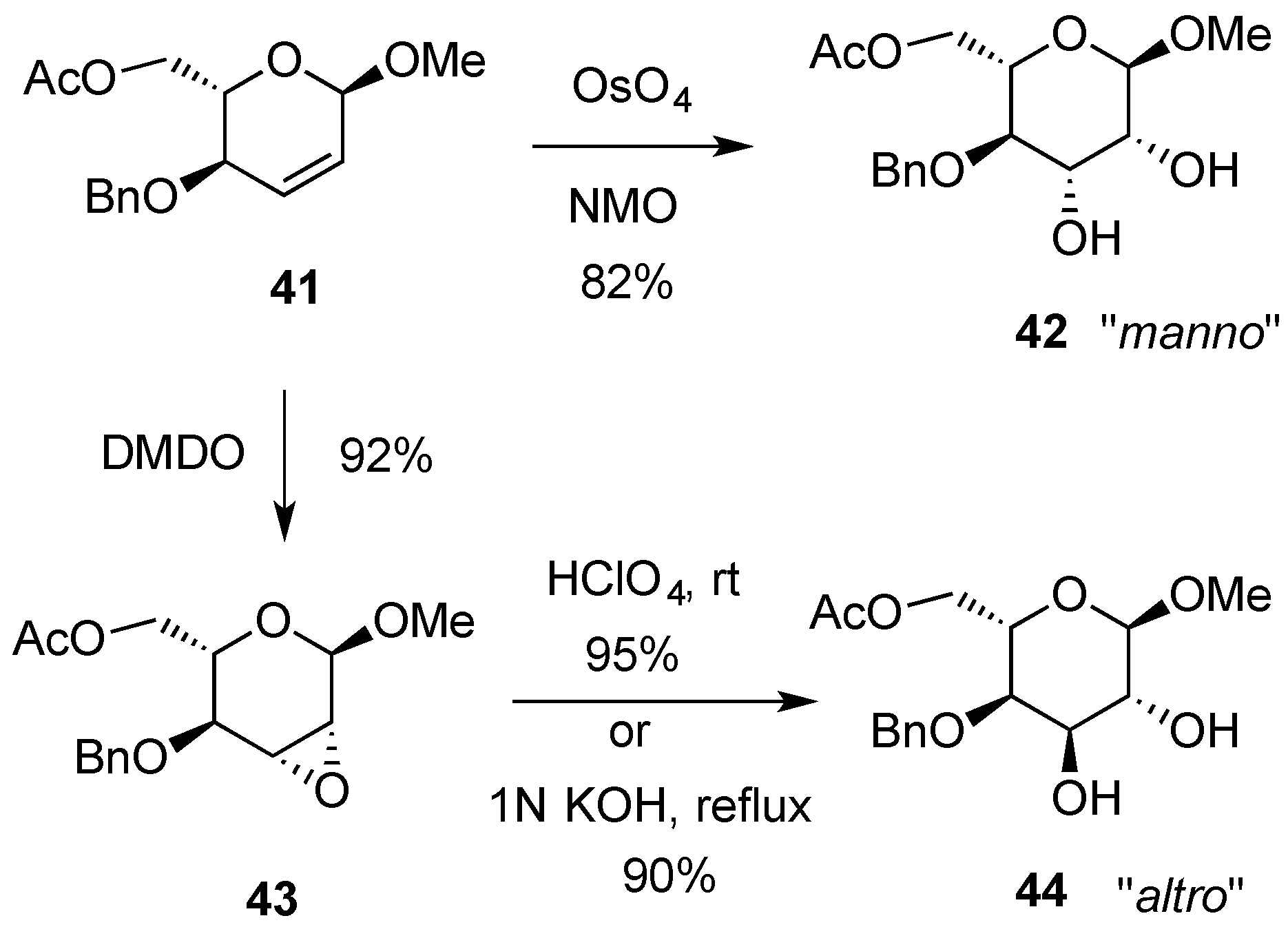{kind=link}
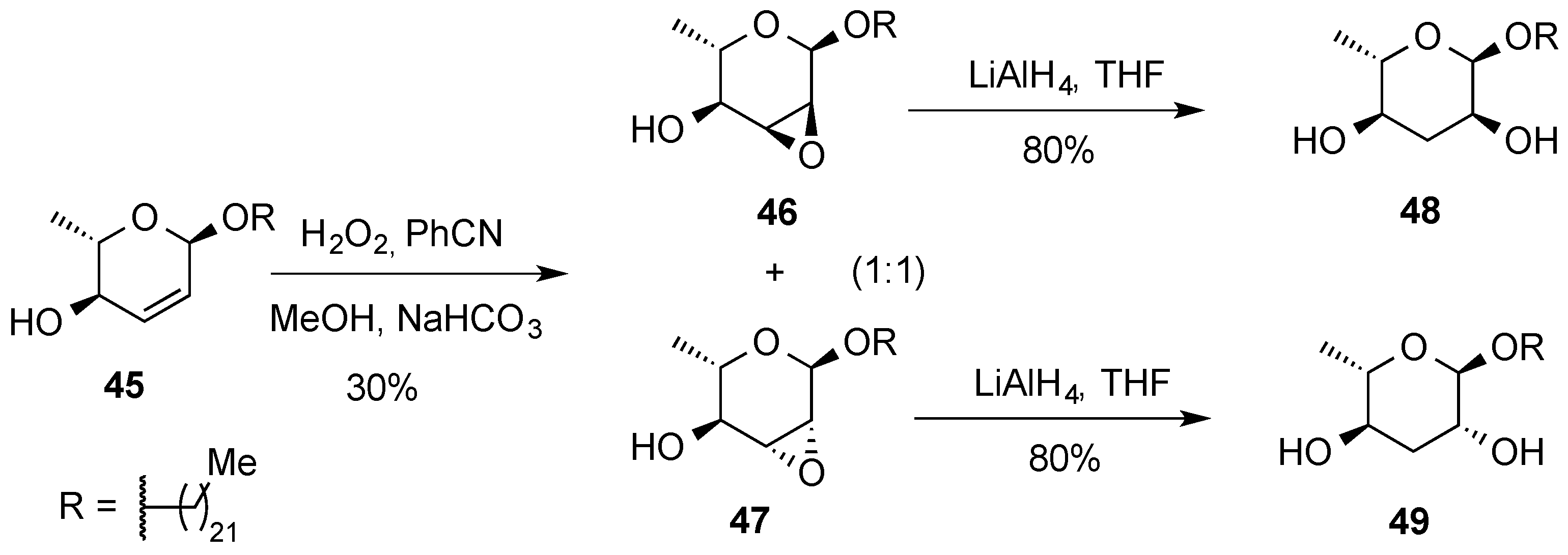{kind=link}
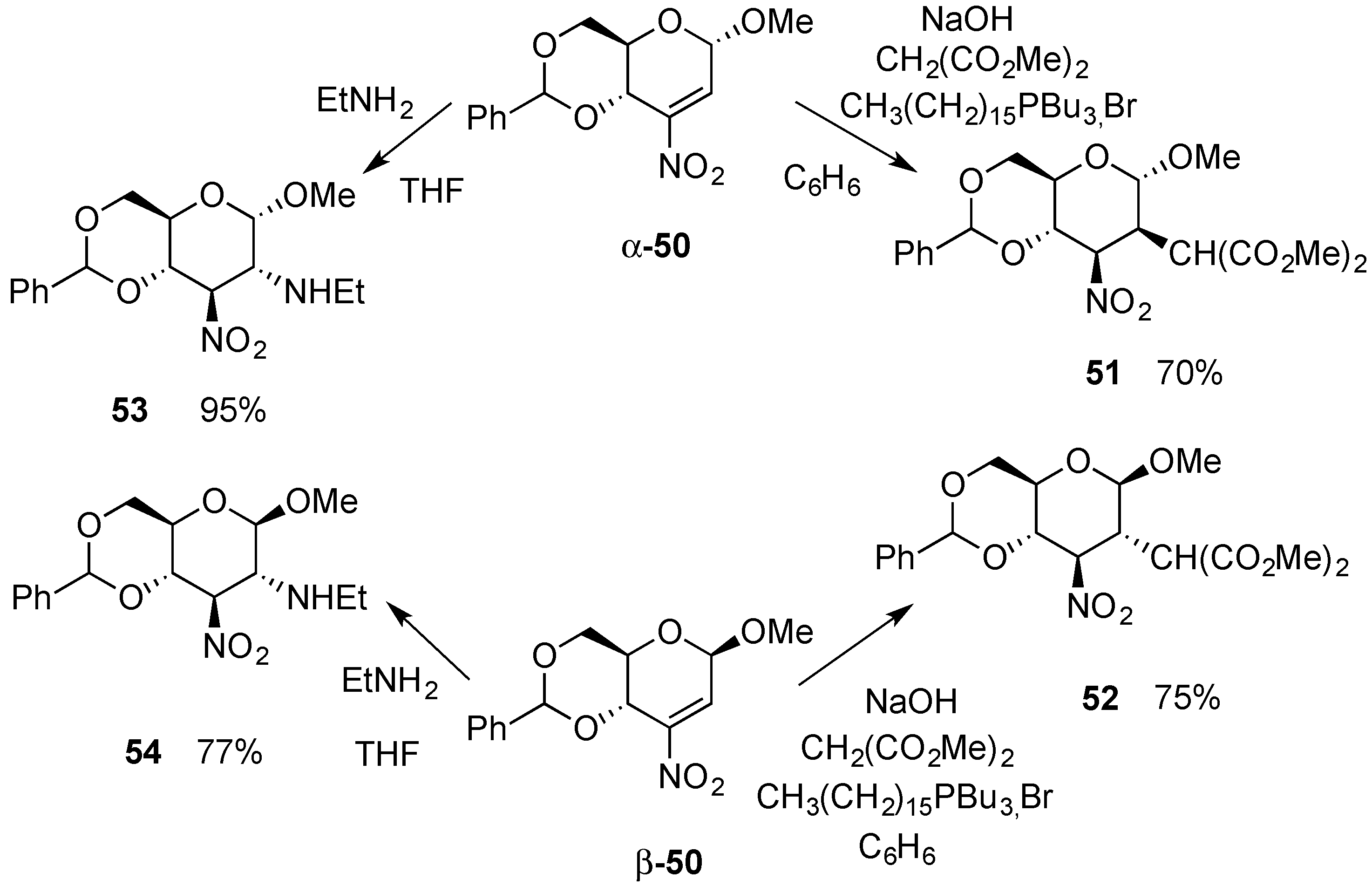{kind=link}
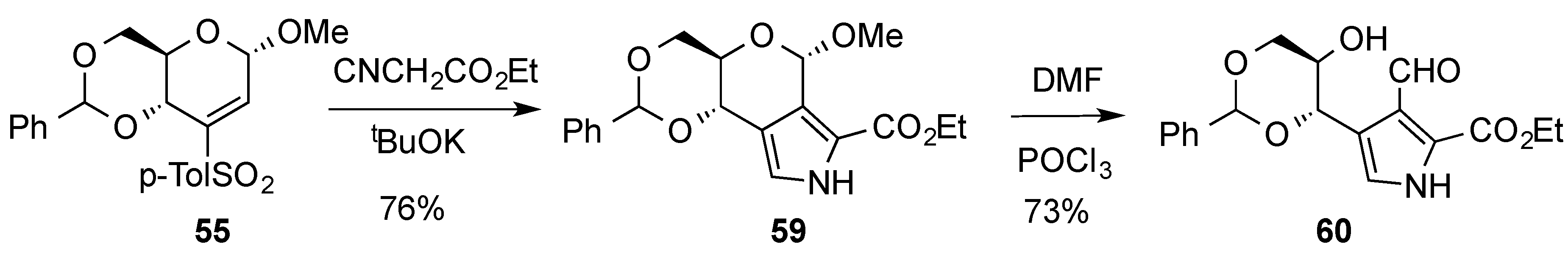{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
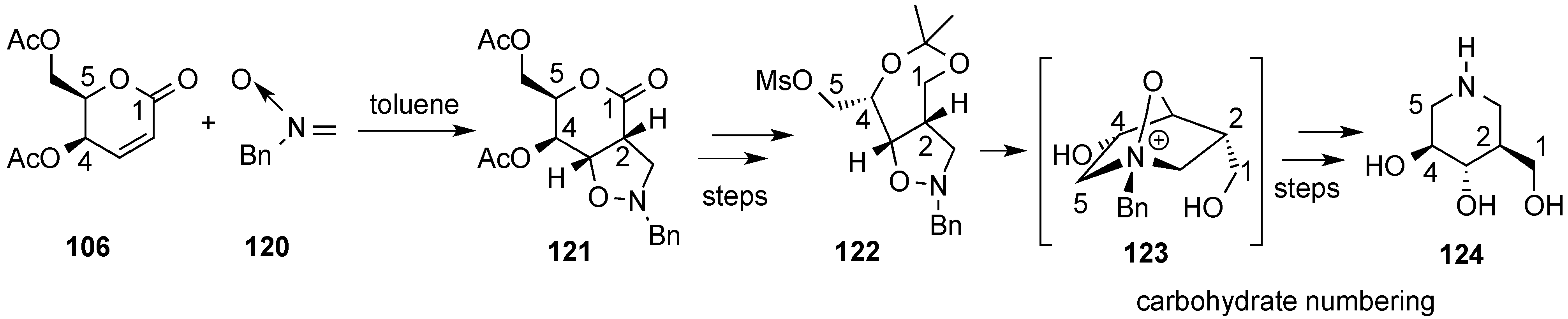{kind=link}
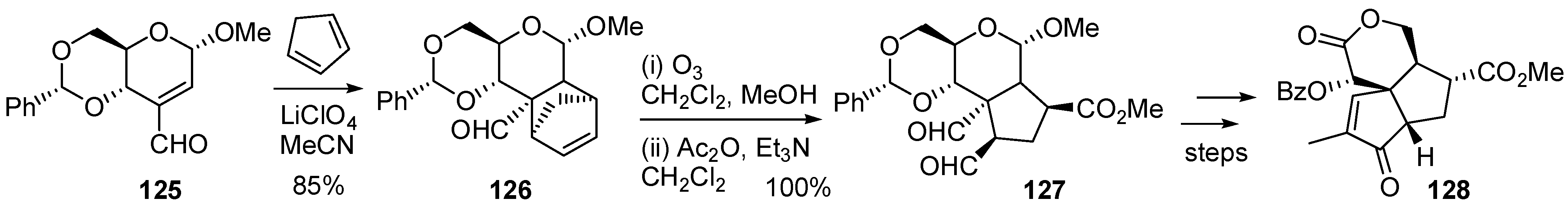{kind=link}
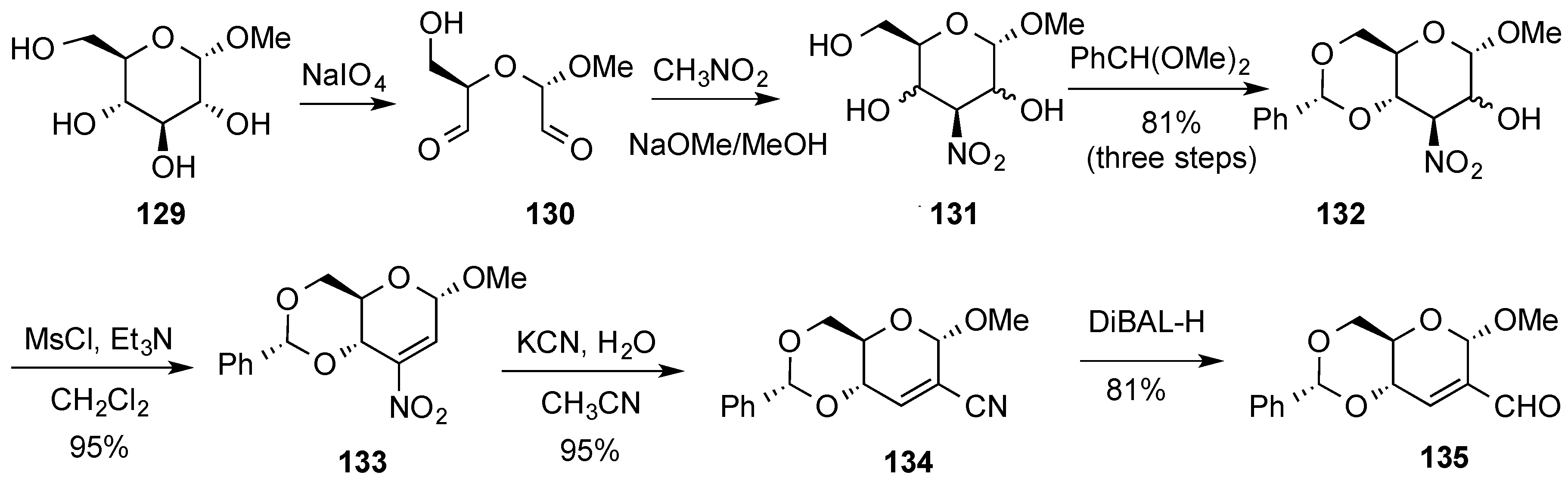{kind=link}
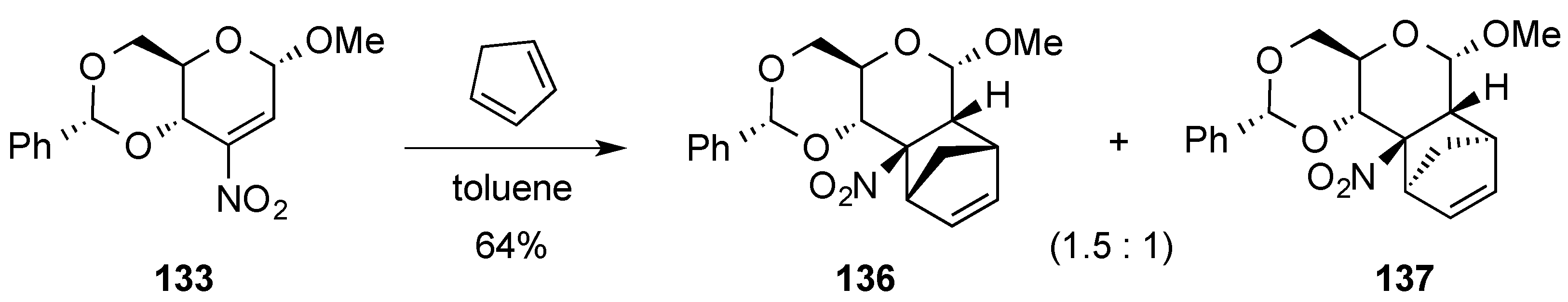{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
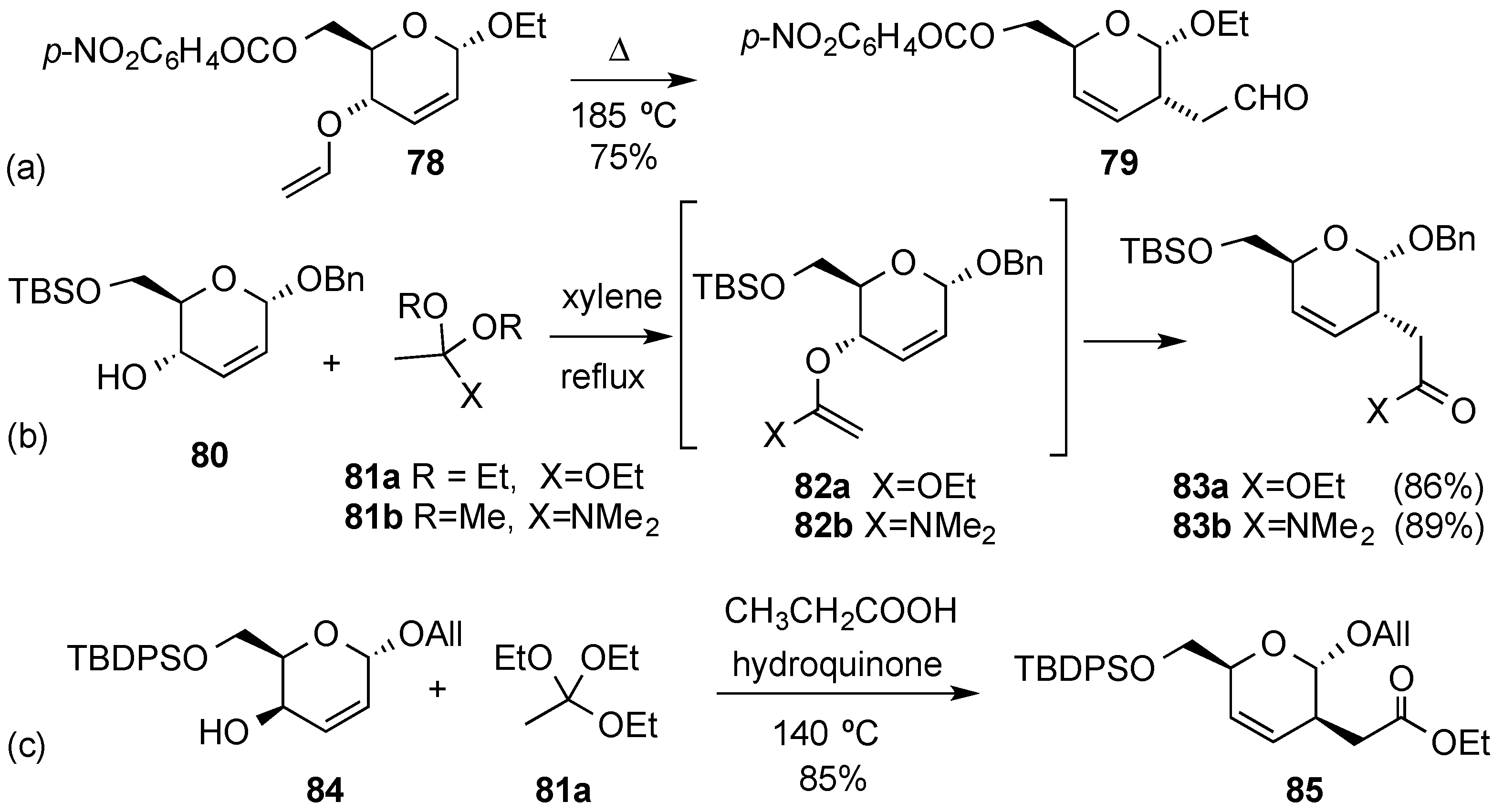
2. Synthetic Routes to Hex-2-enopyranosides
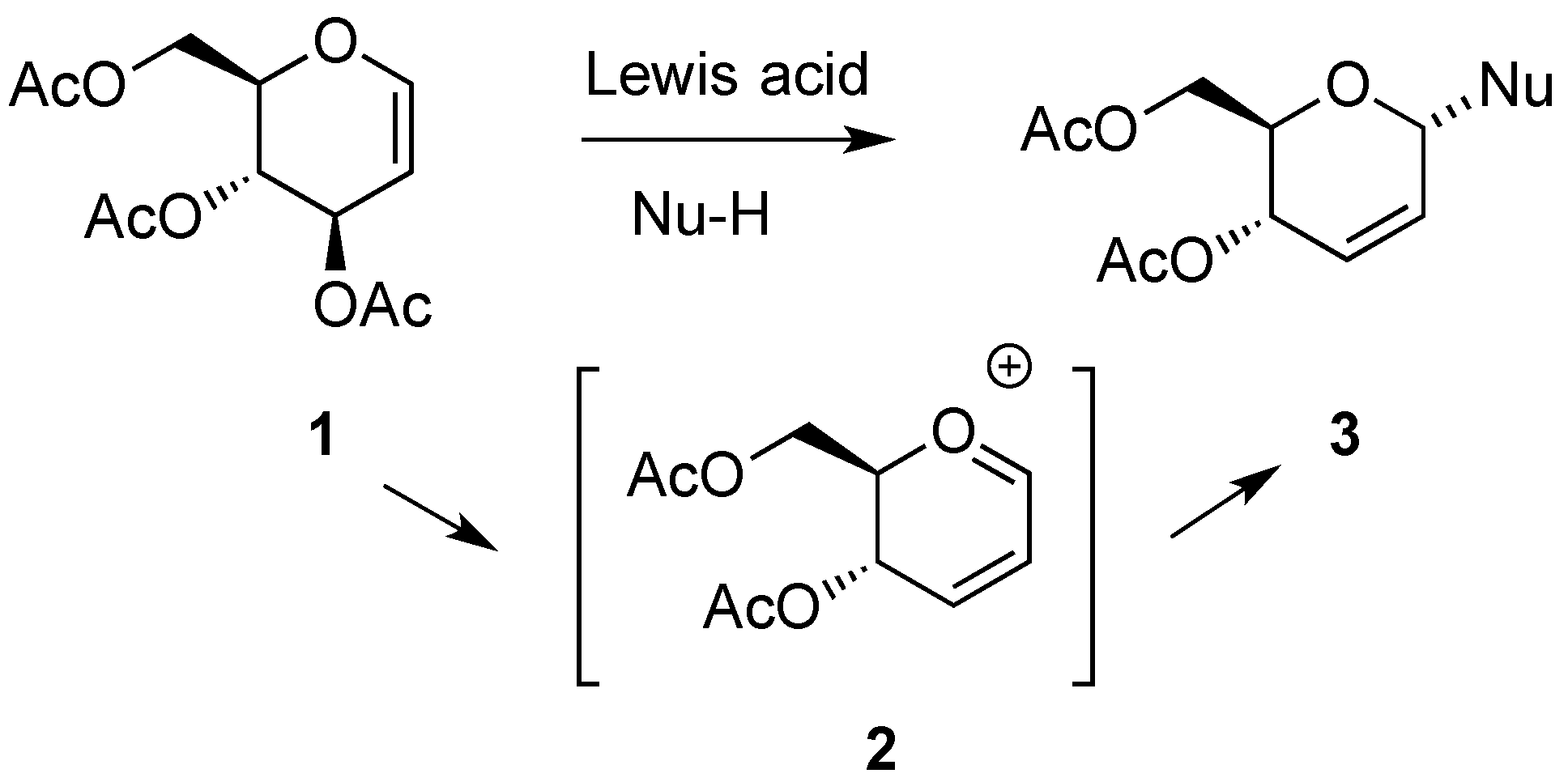
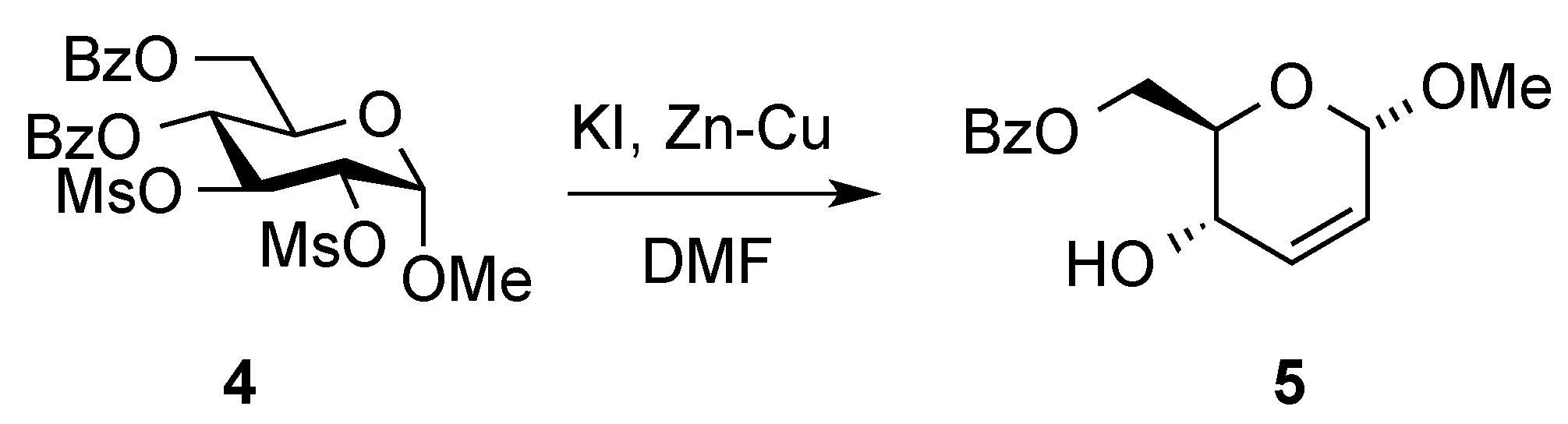
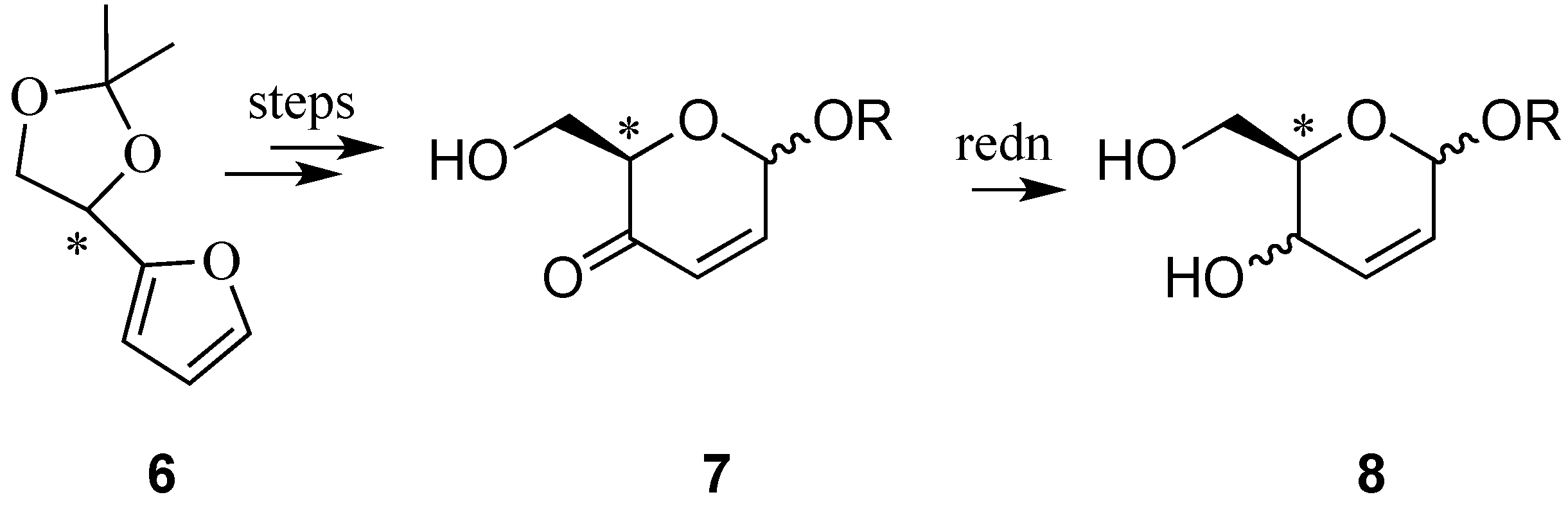
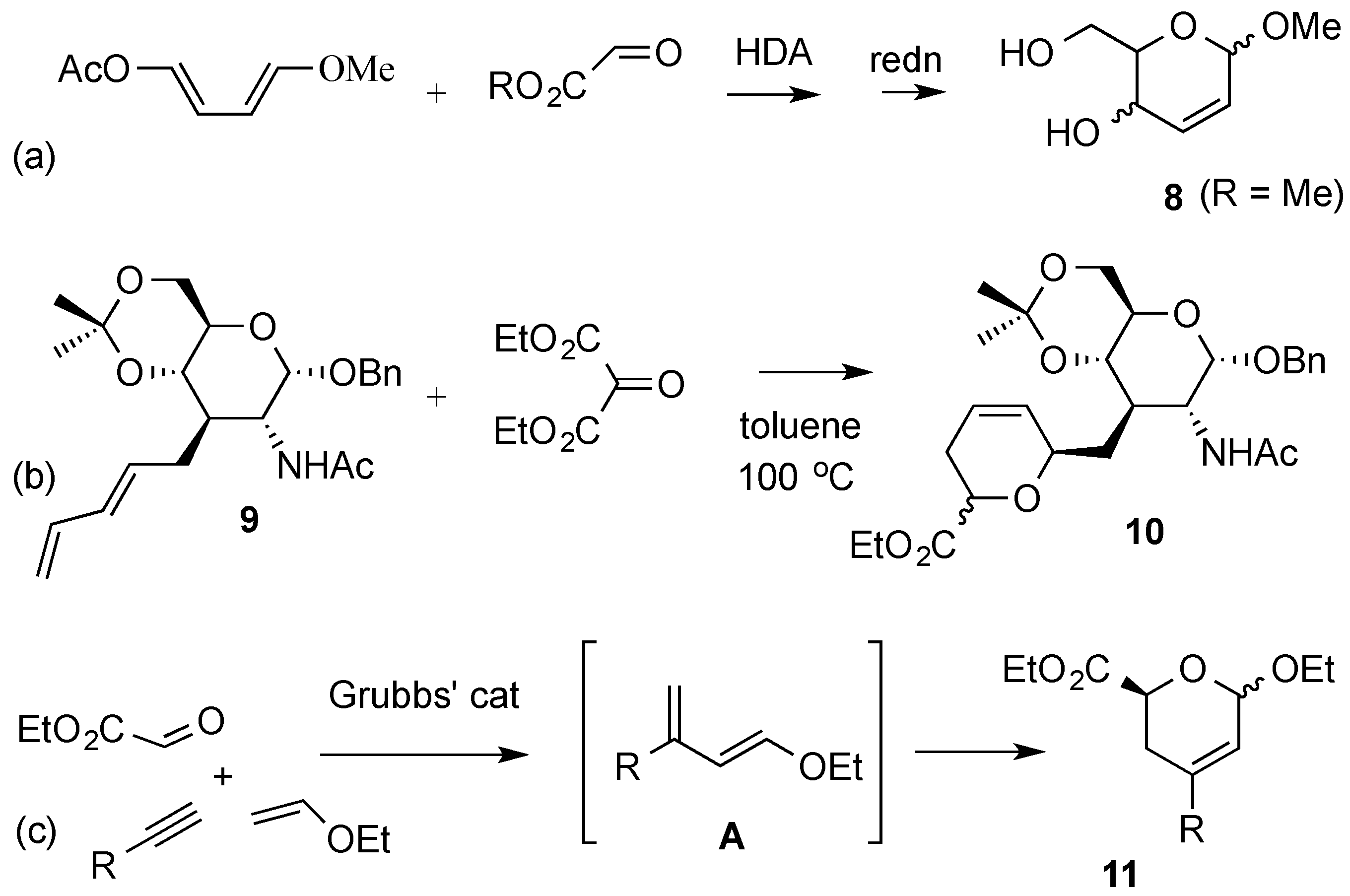
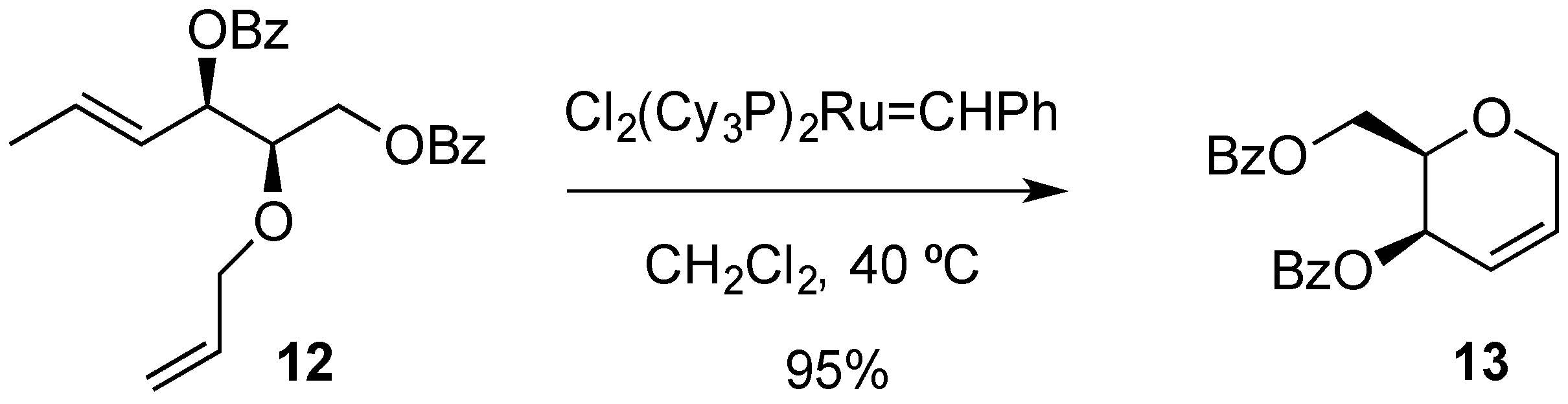
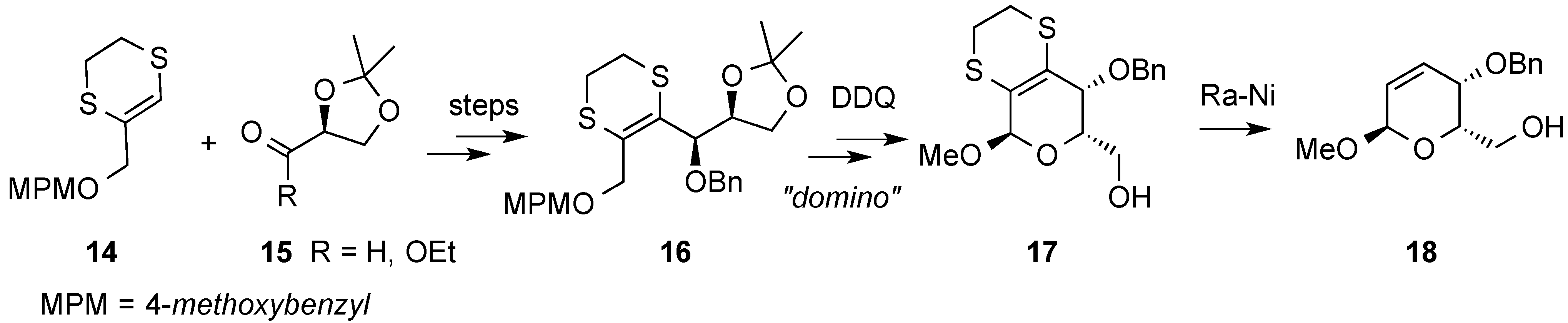
3. Reactions of Hex-2-enopyranosides
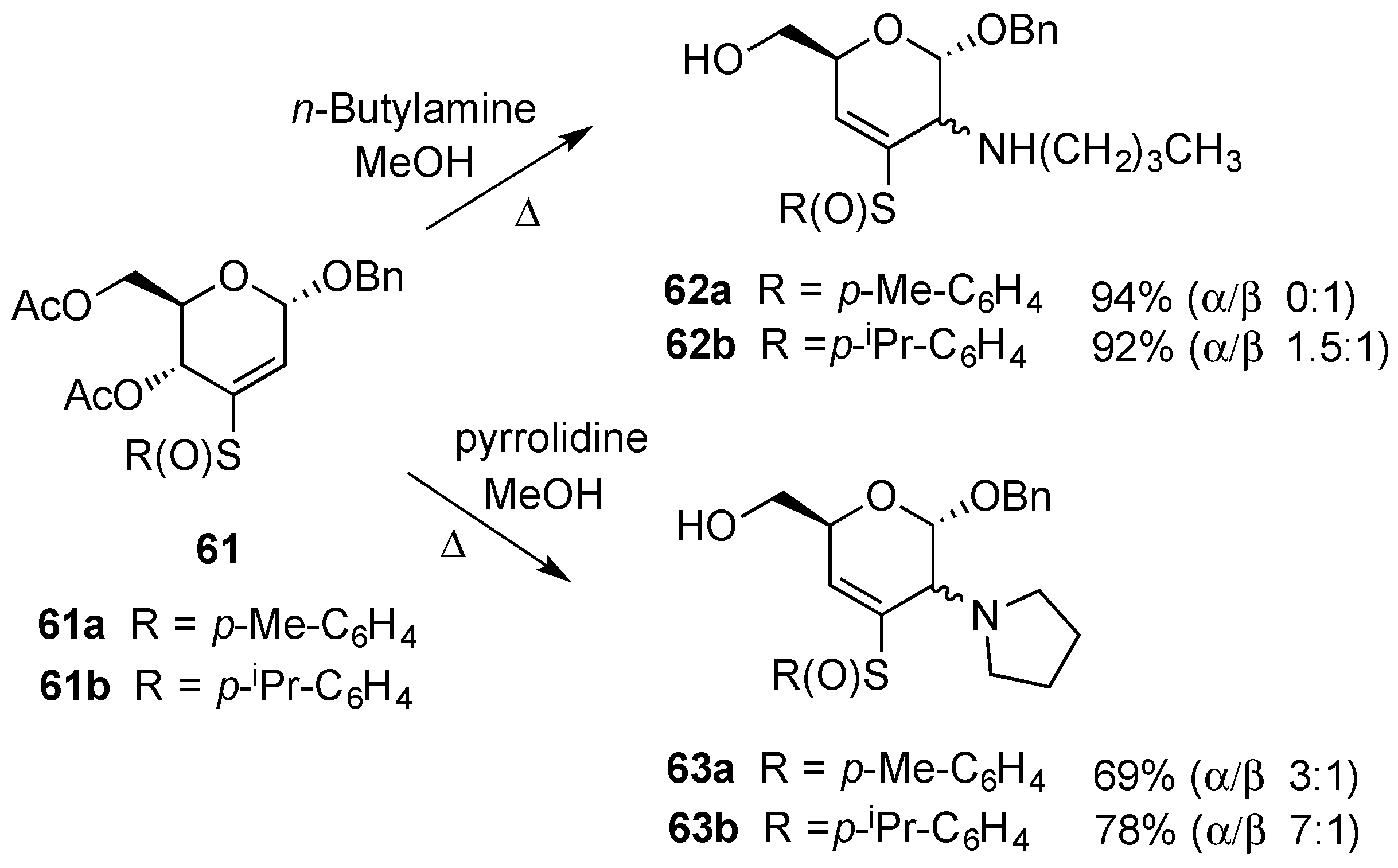
3.1. Addition Reactions
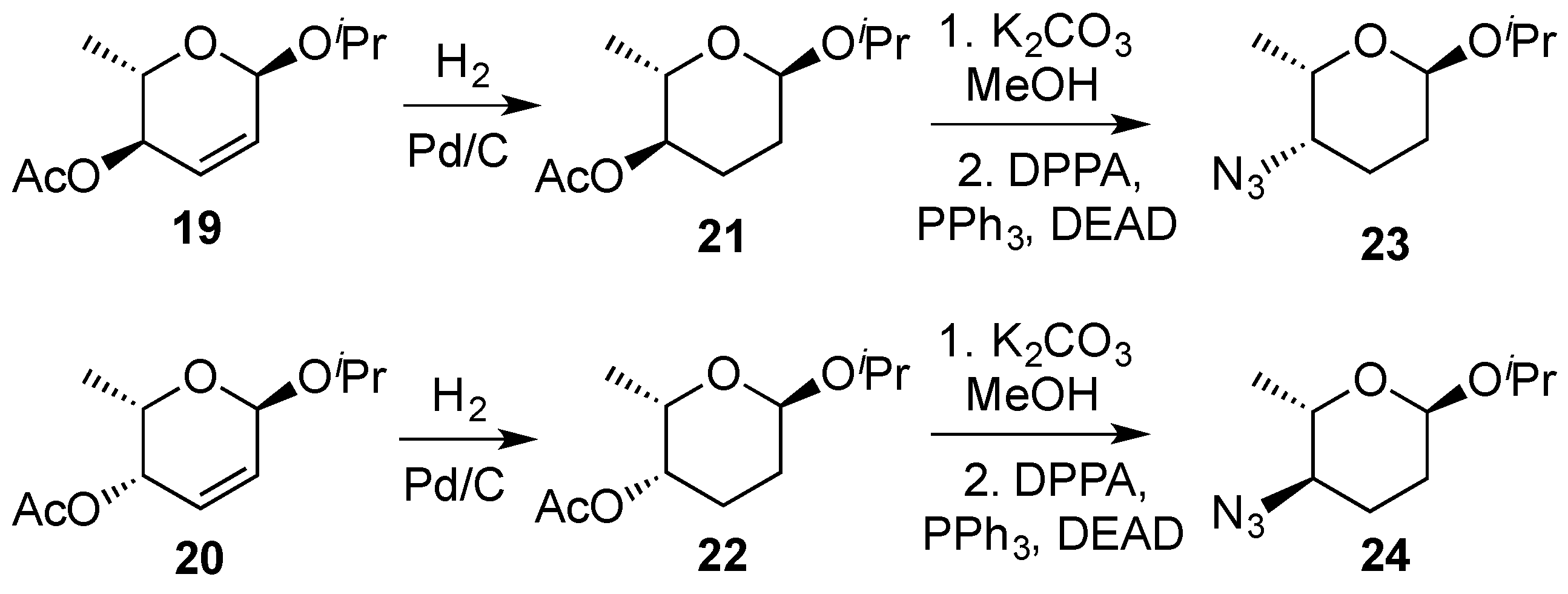
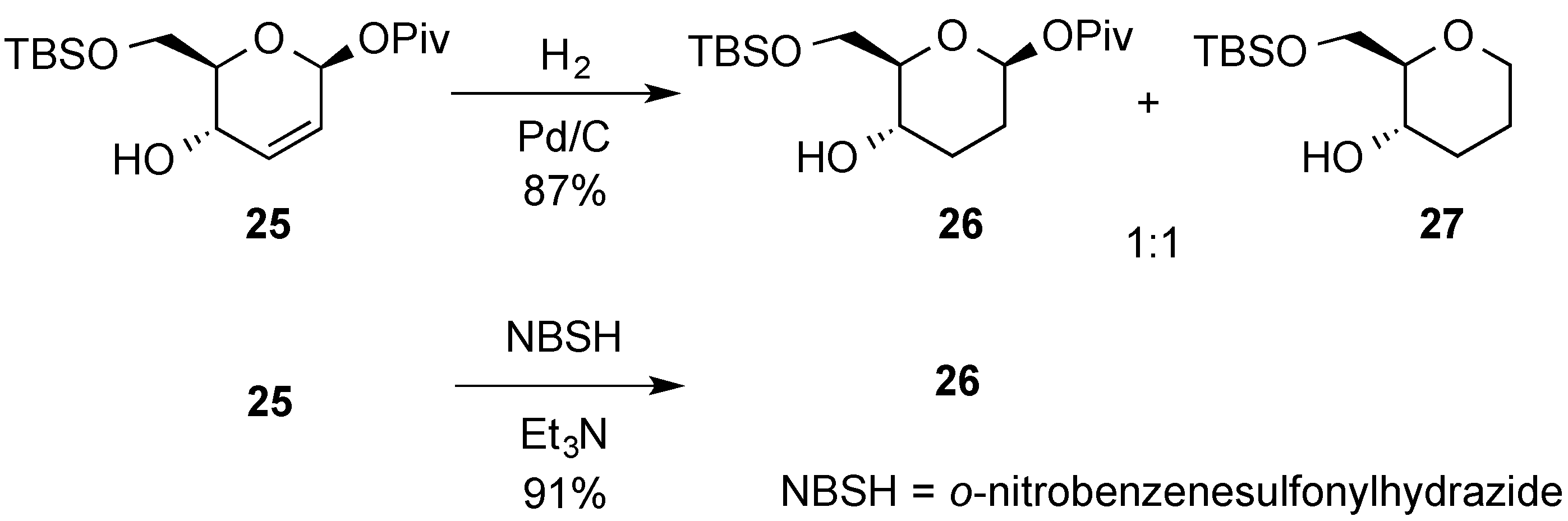
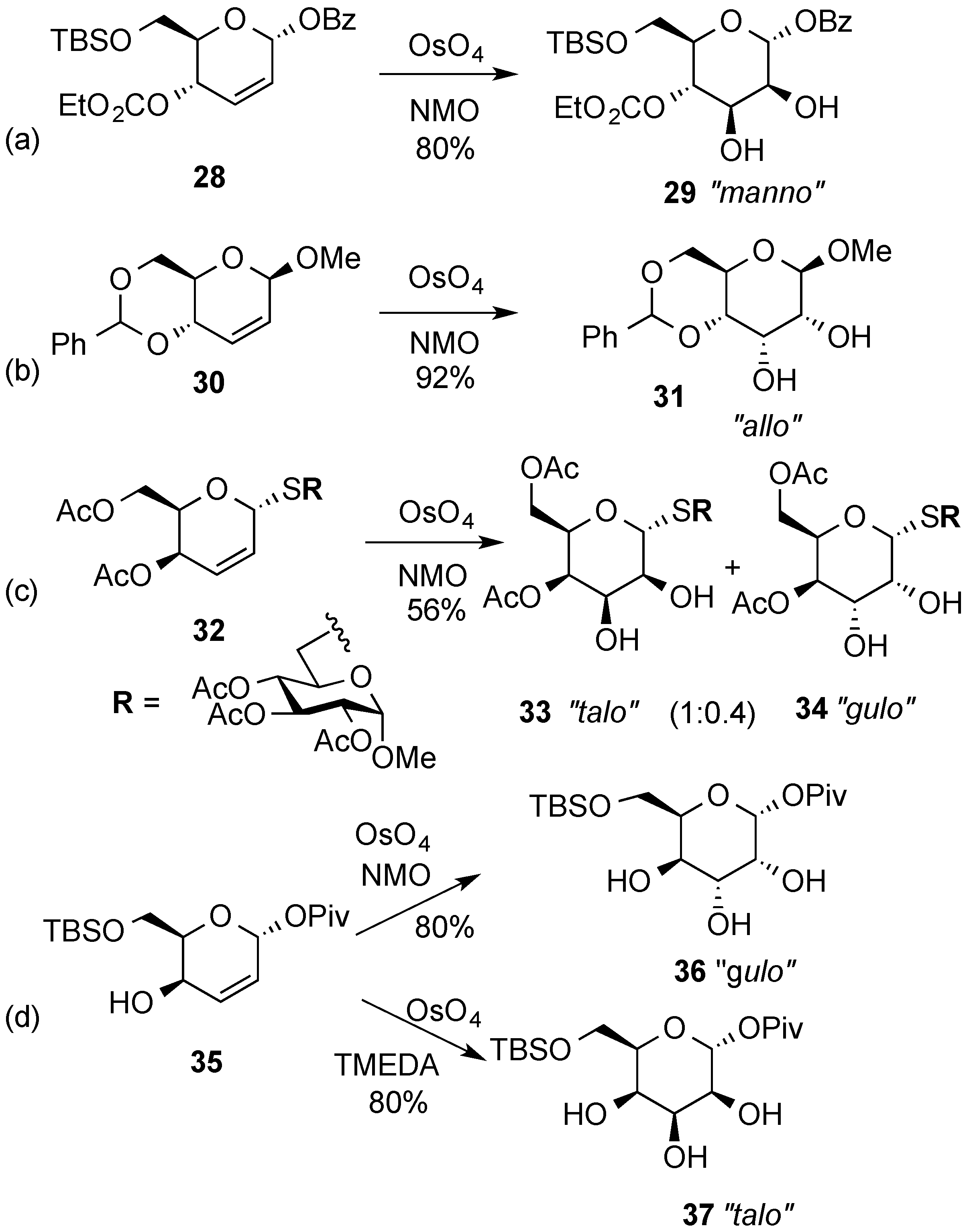
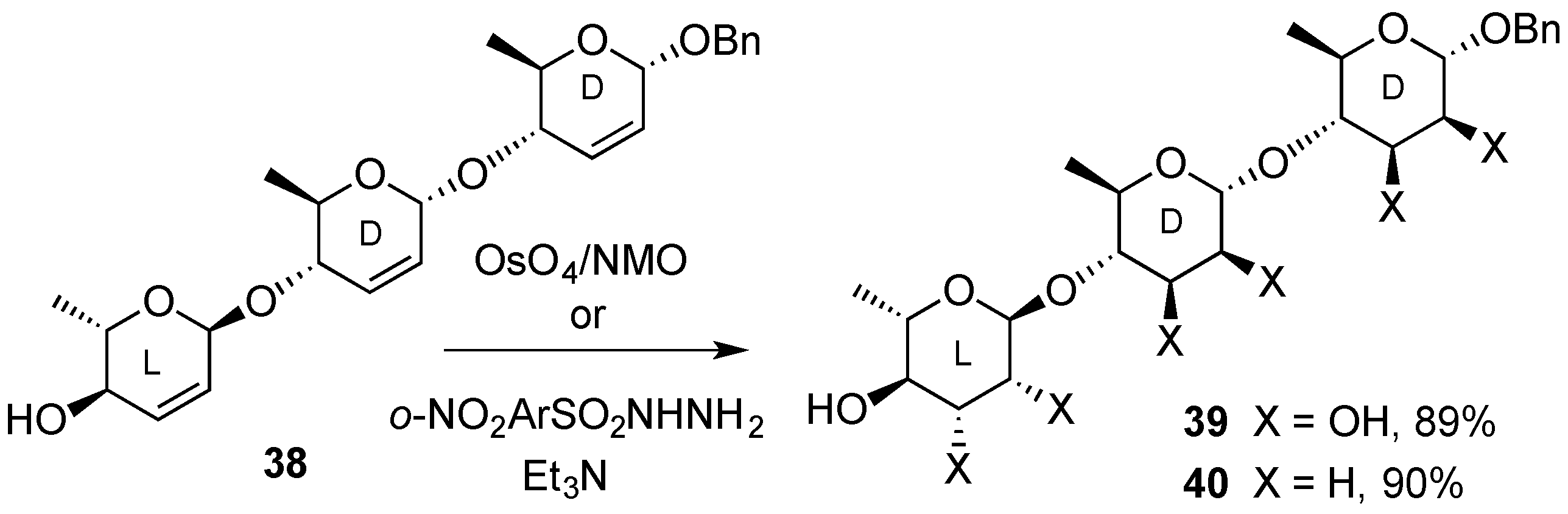
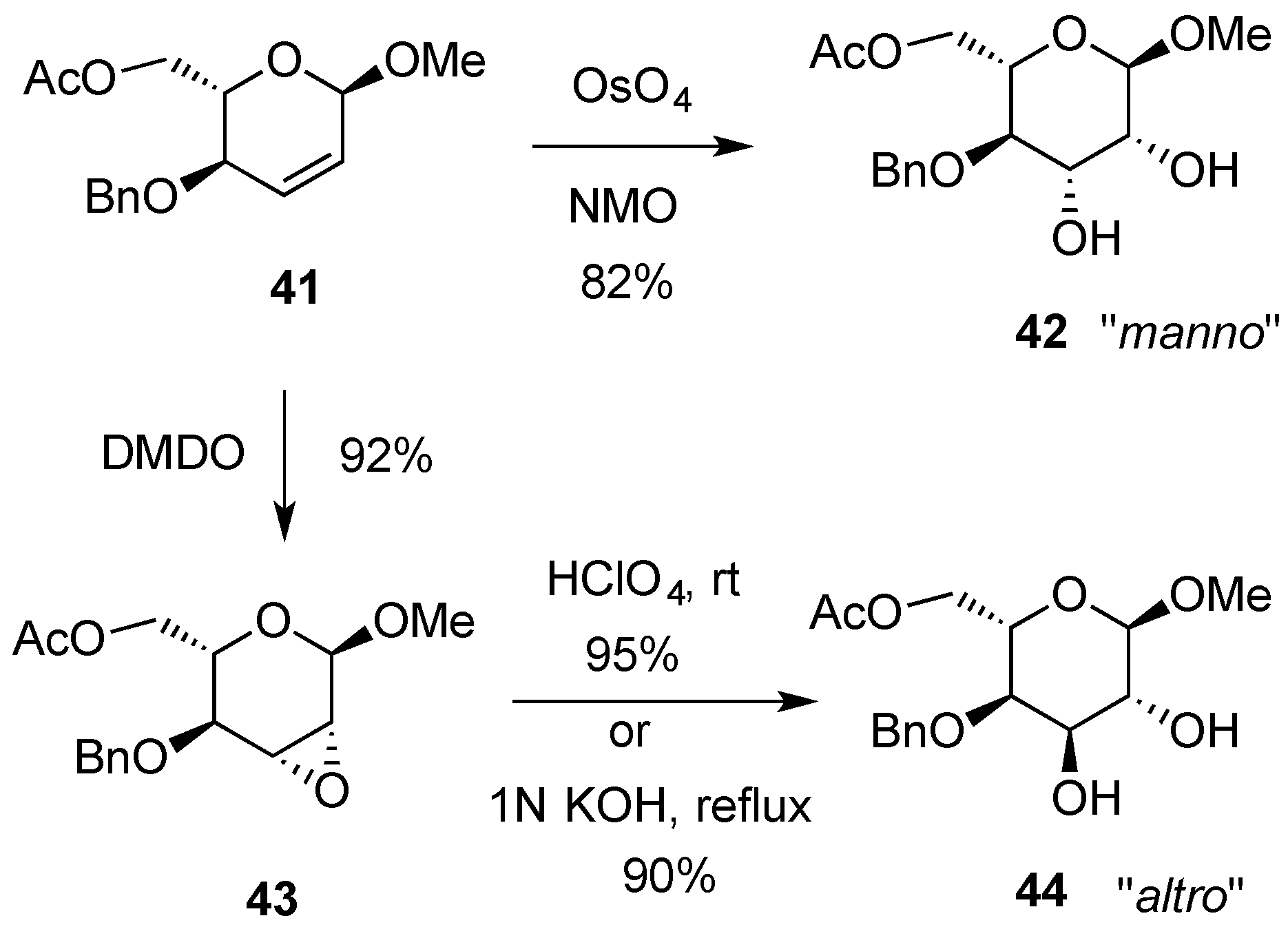
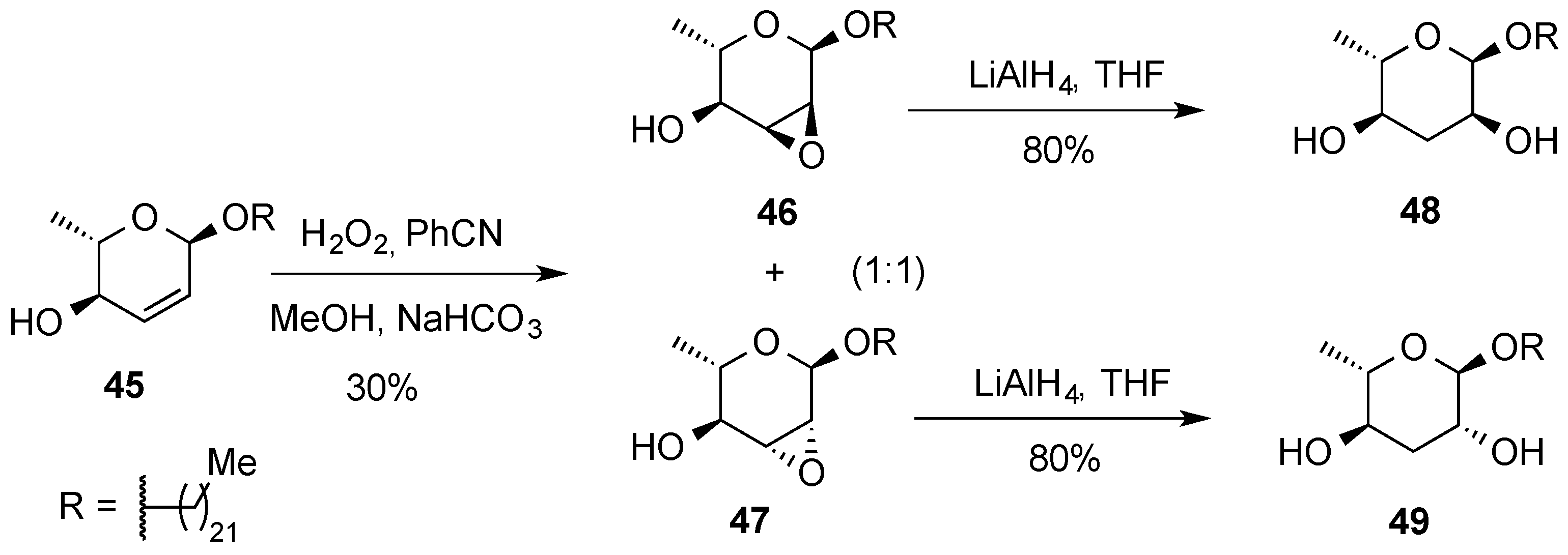
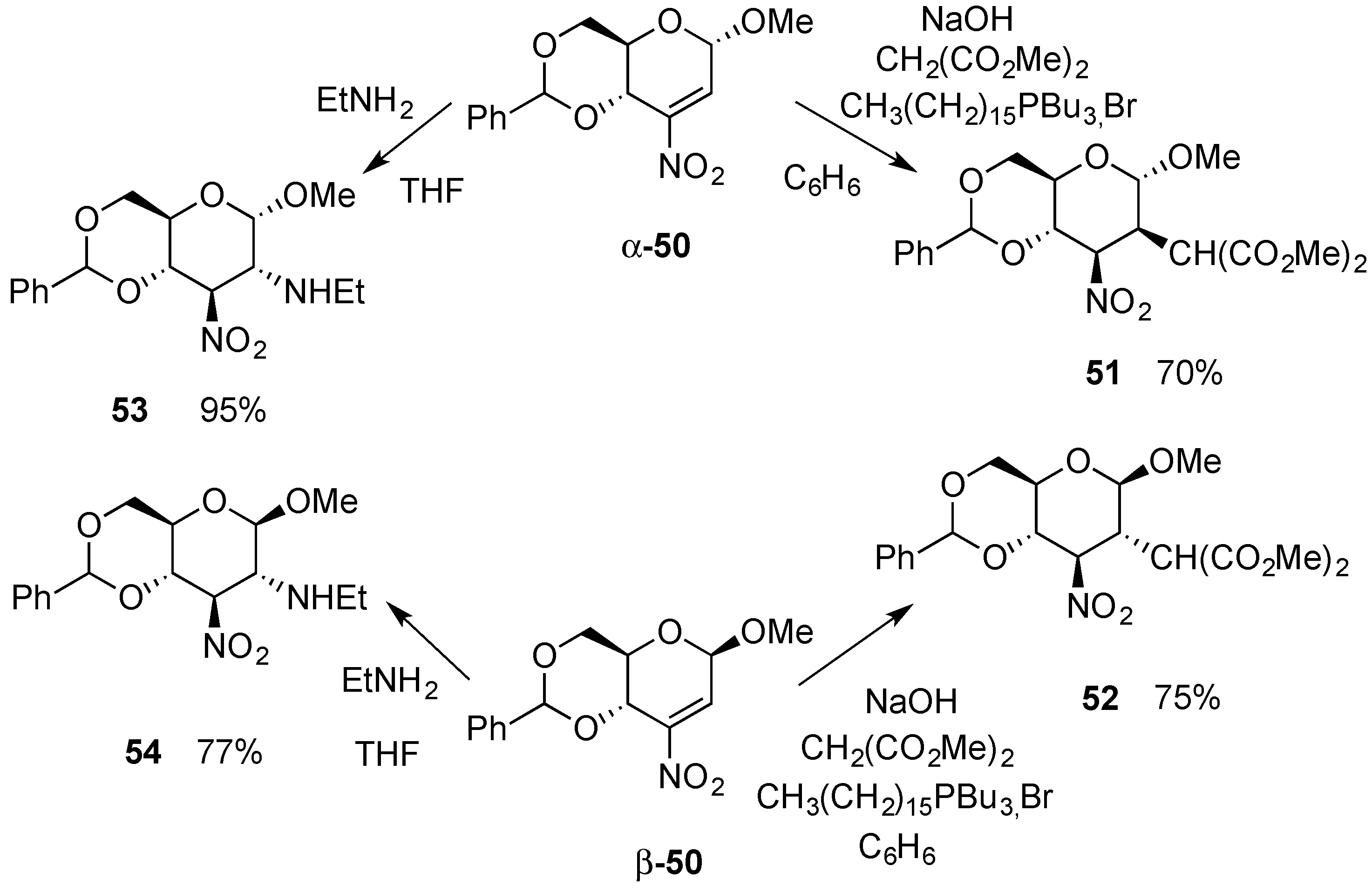
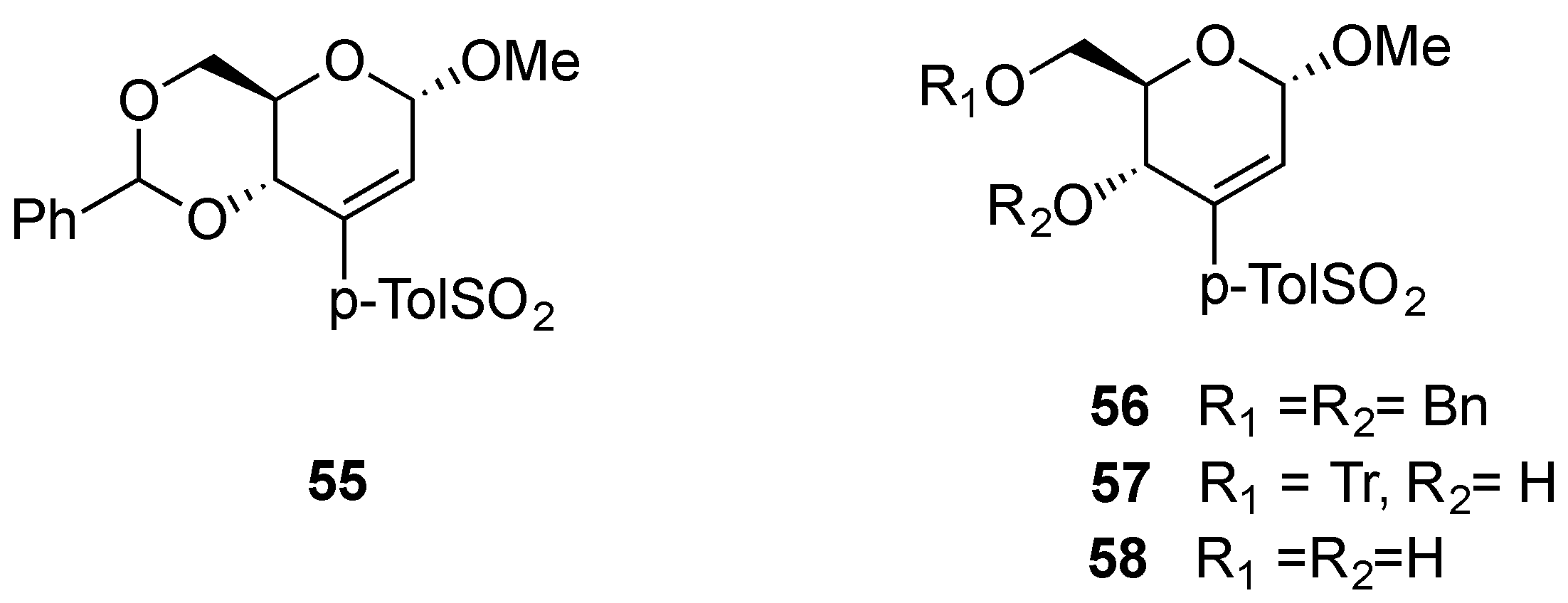
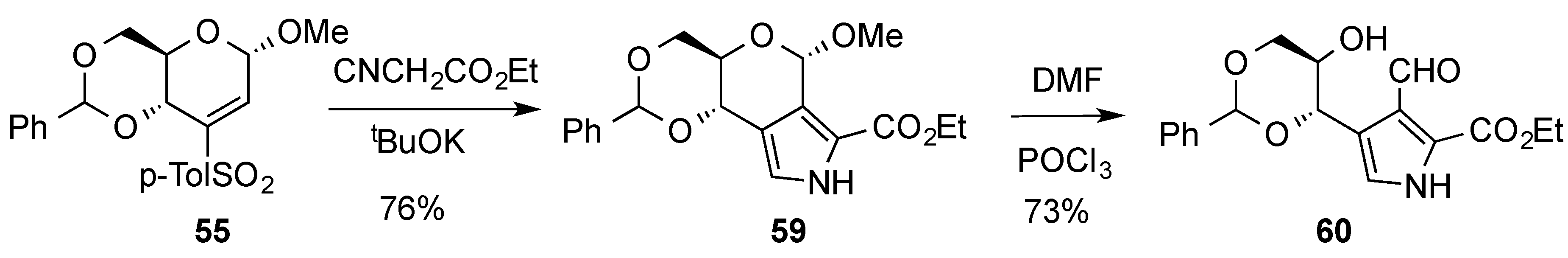
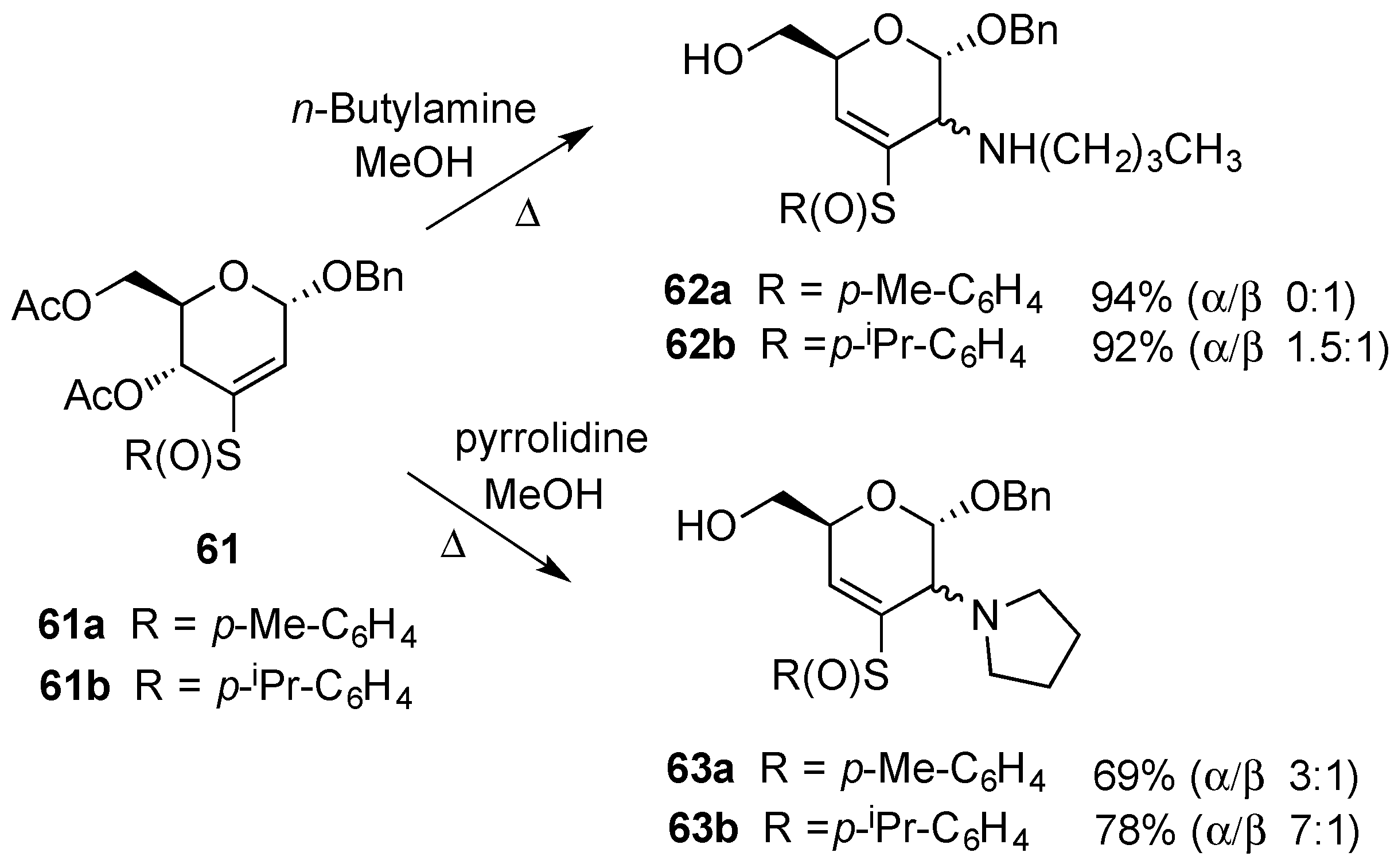
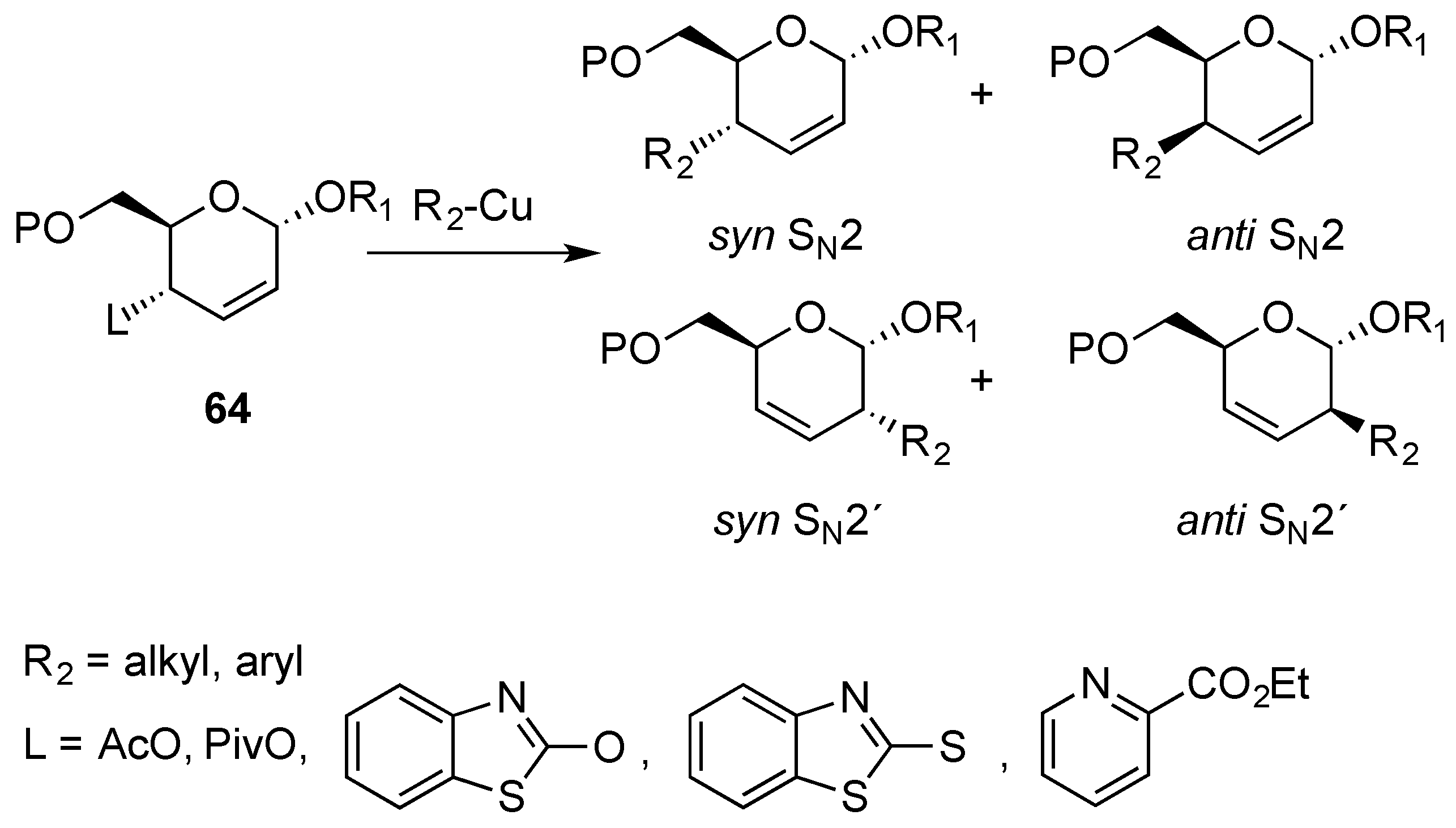
3.2. Nucleophilic Substitutions
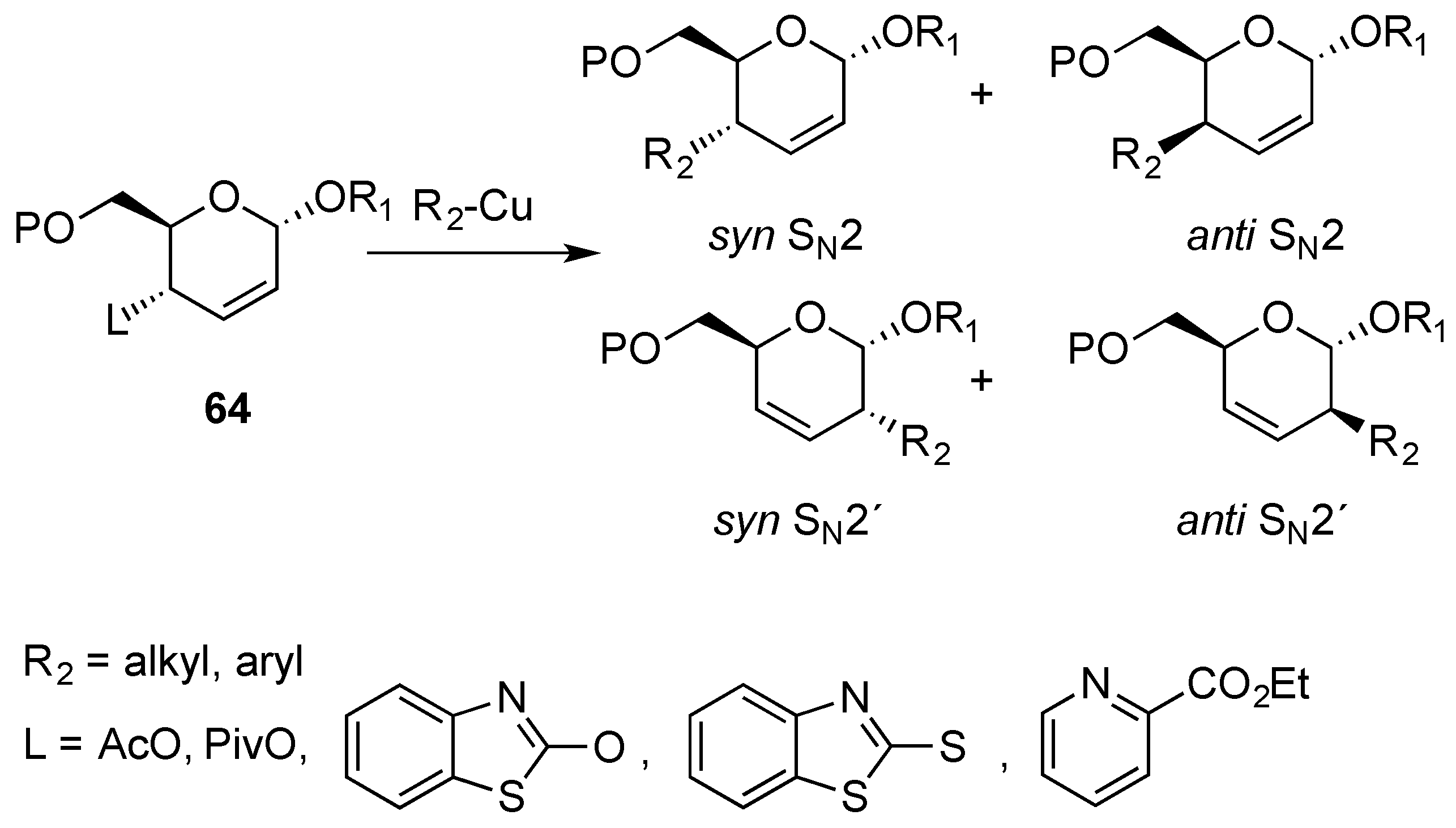
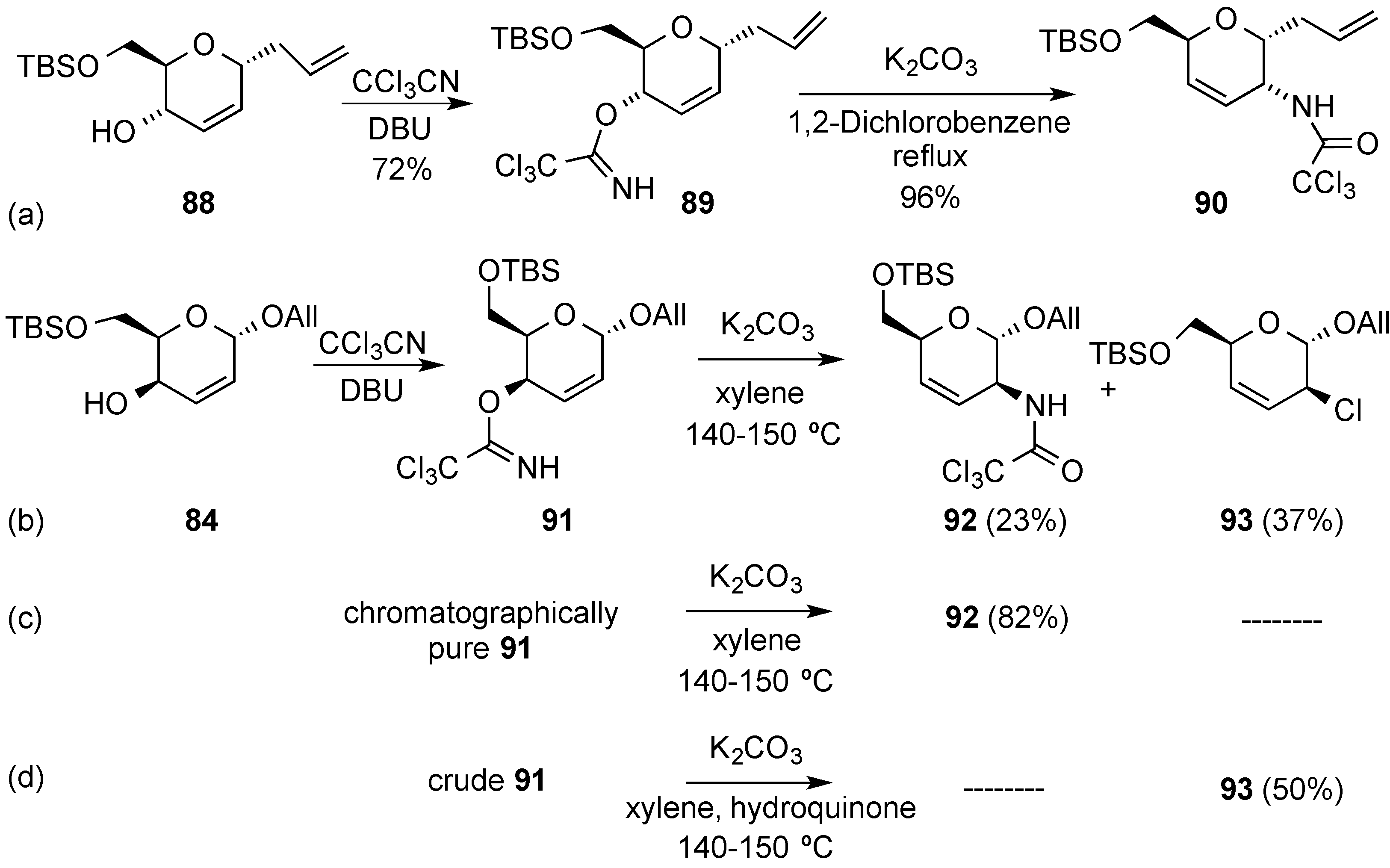
3.3. [3,3]-Sigmatropic Rearrangements
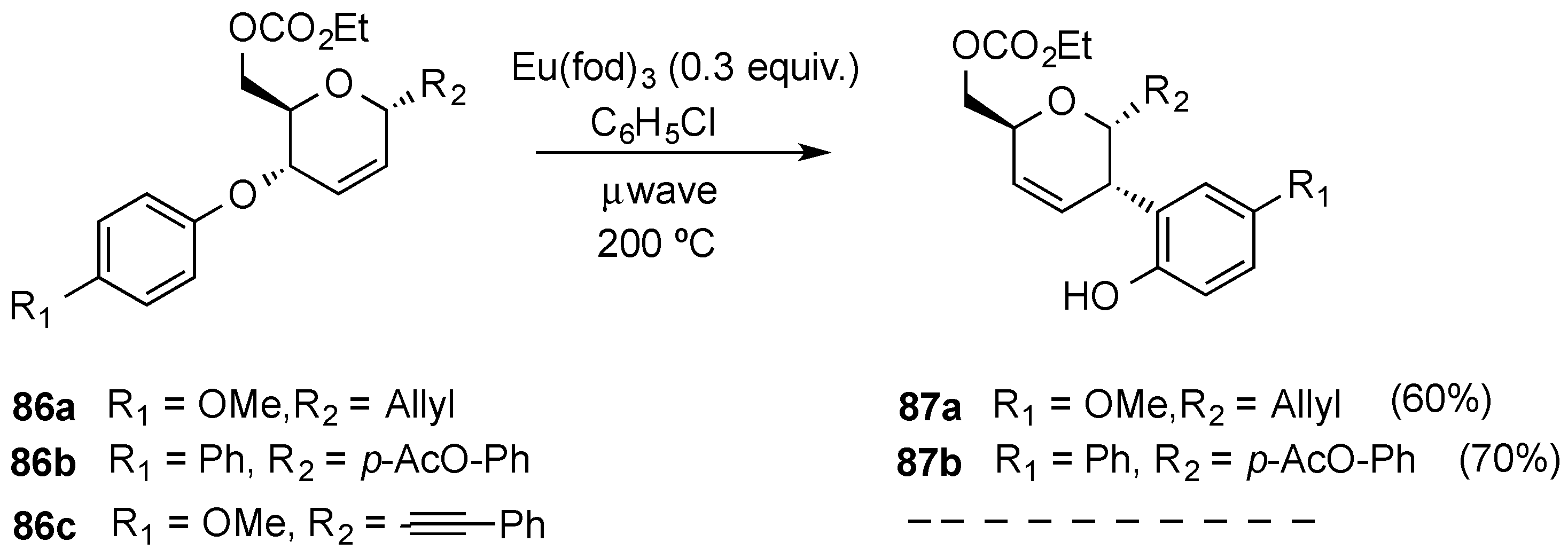
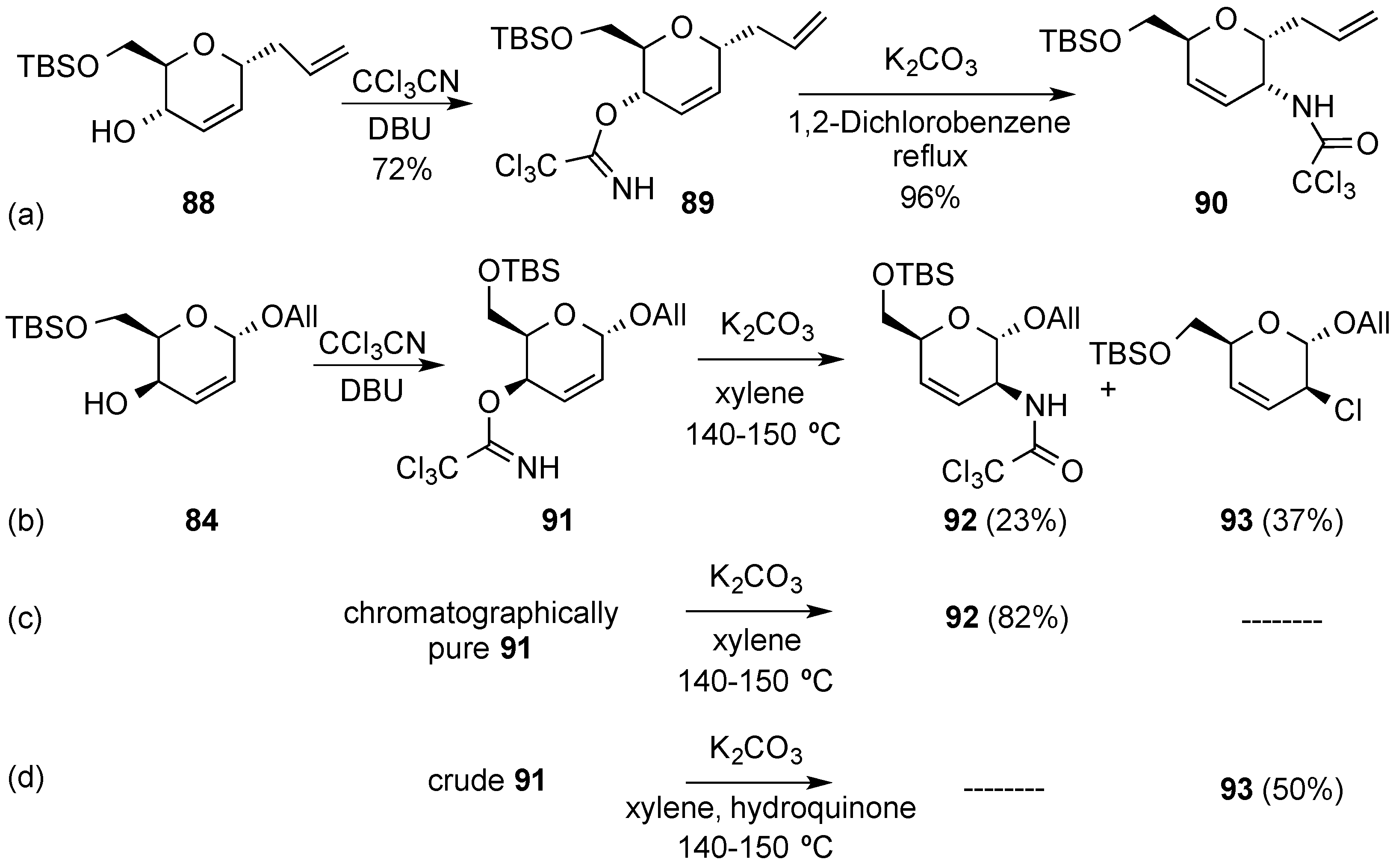
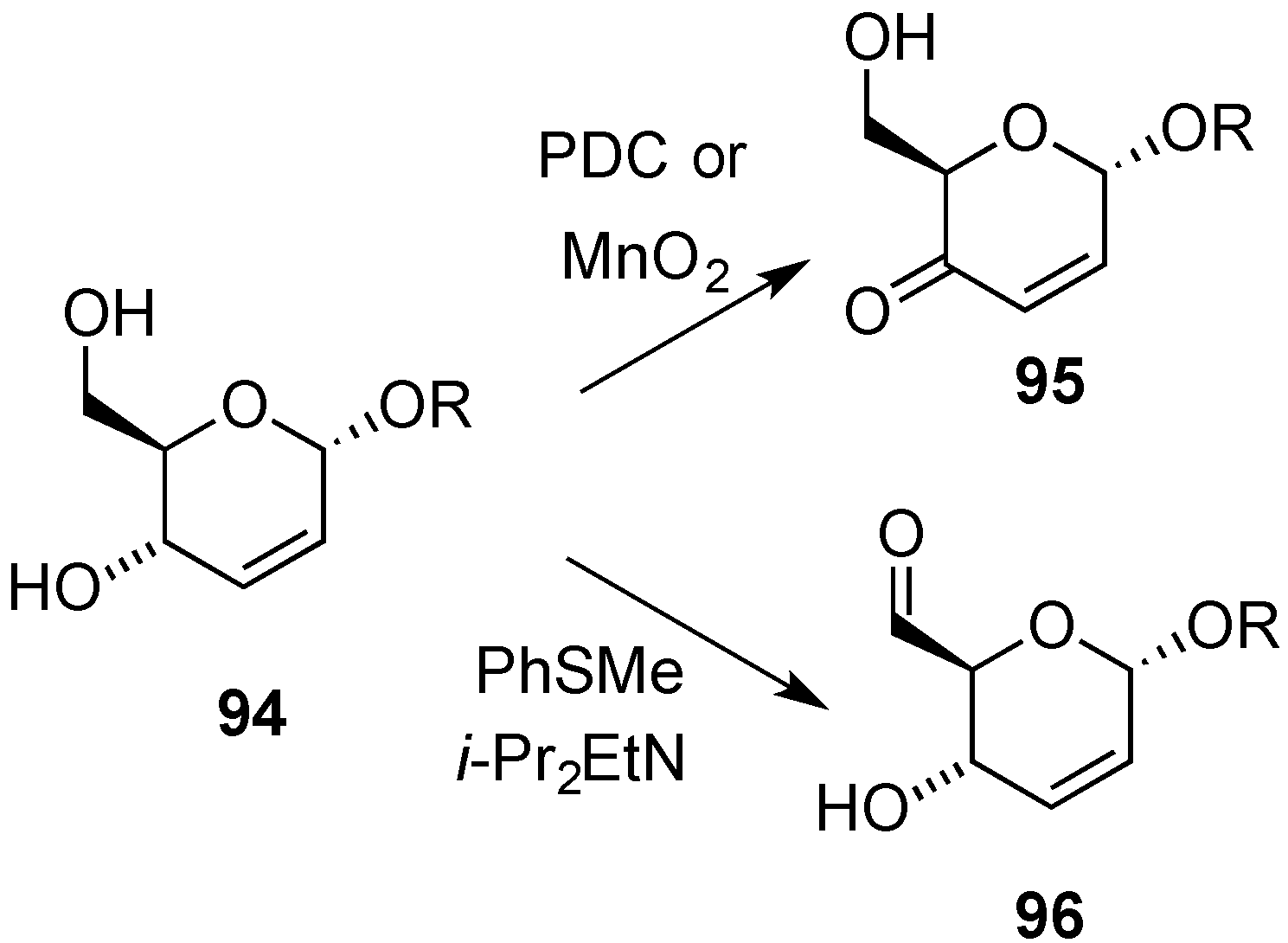
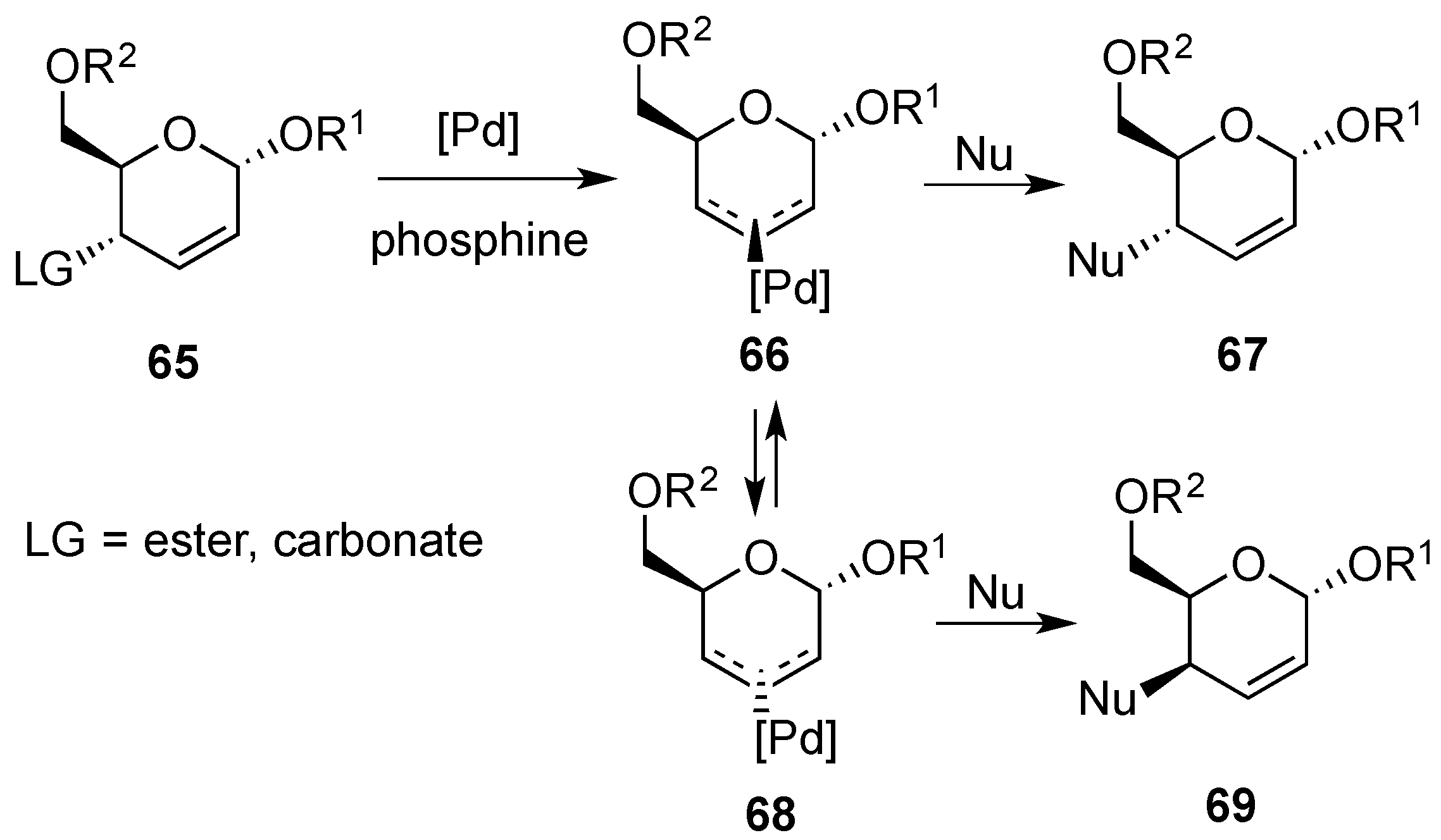
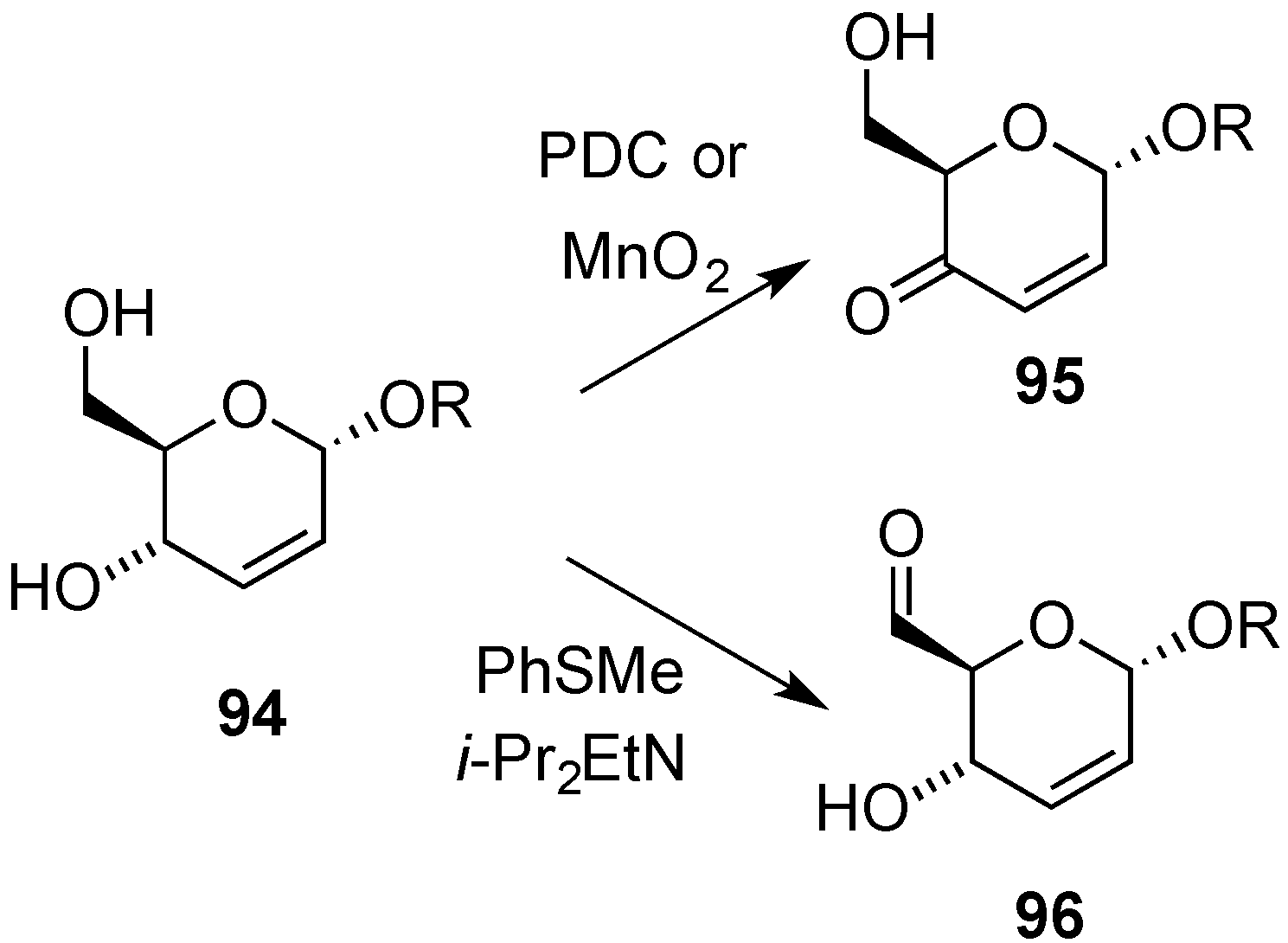
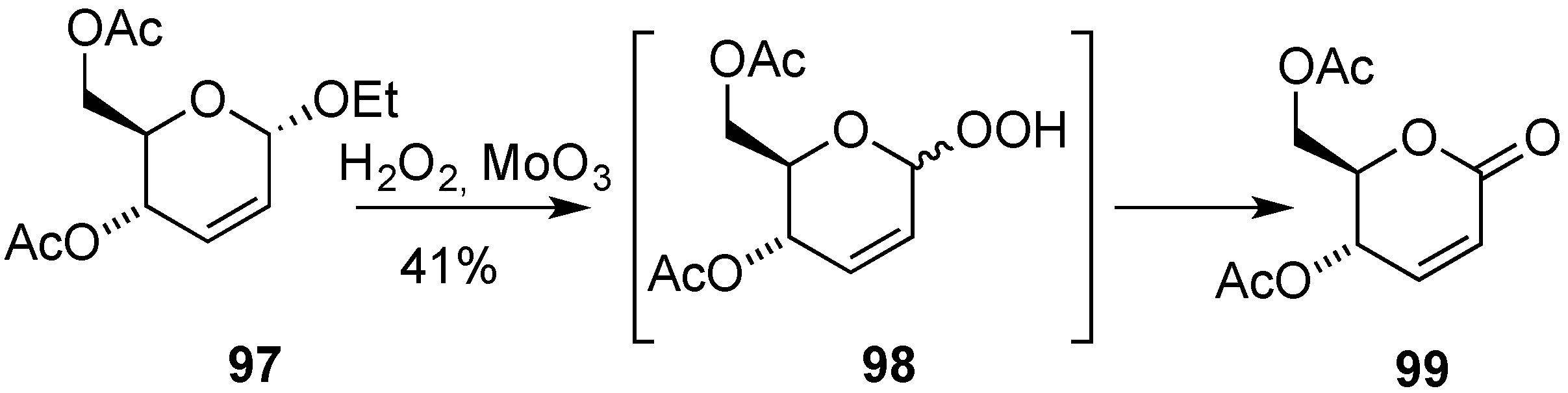
3.4. Oxidative Transformations
3.5. Cycloaddition Reactions
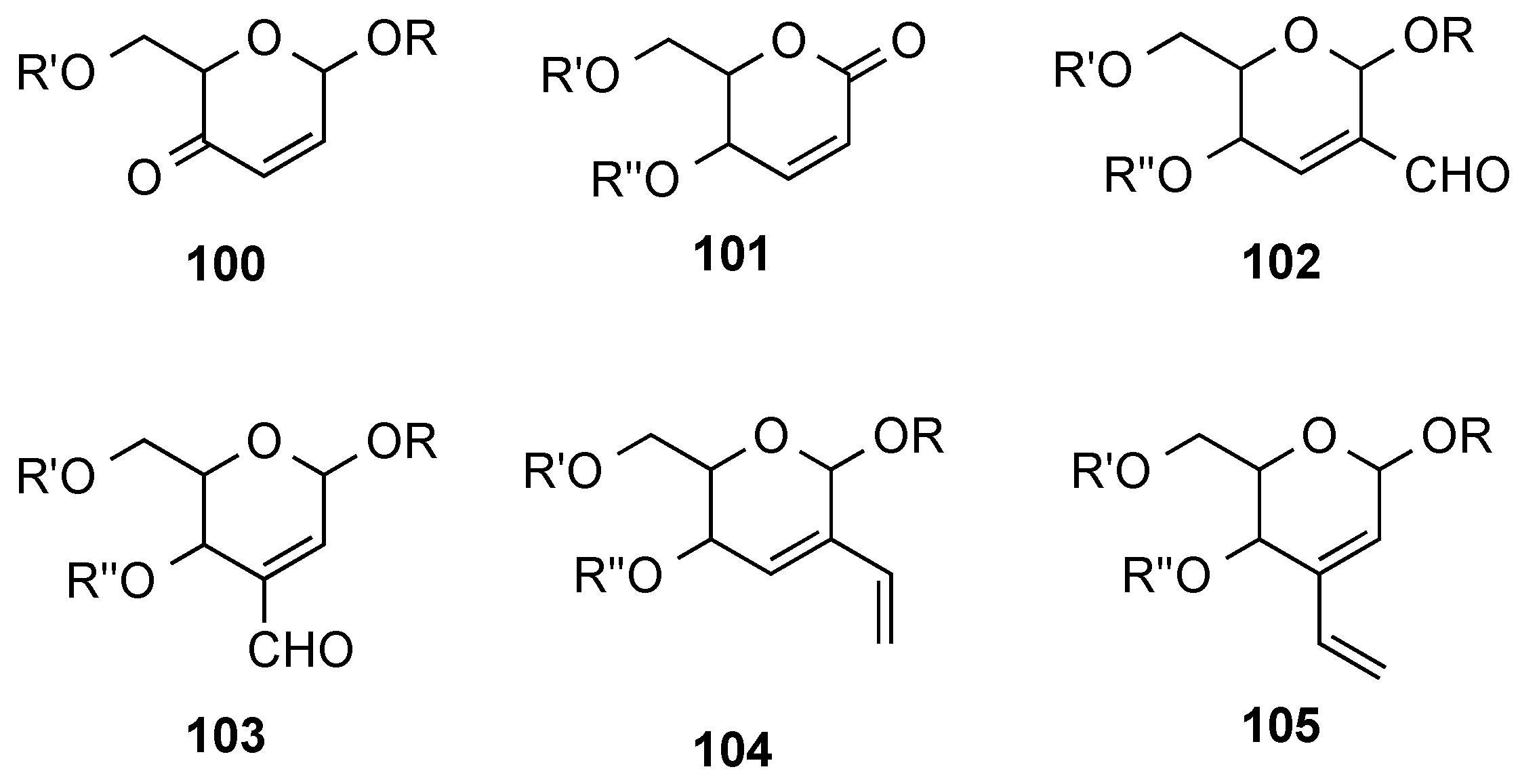
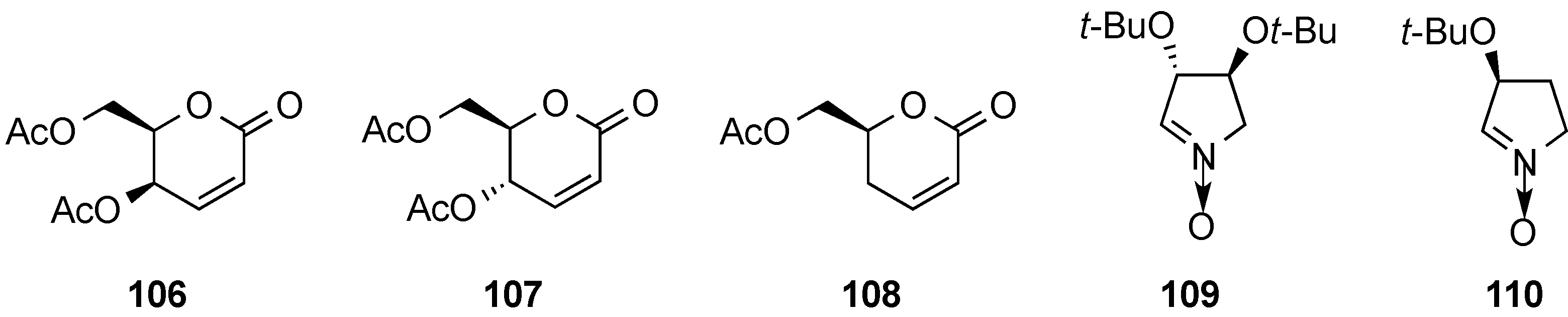
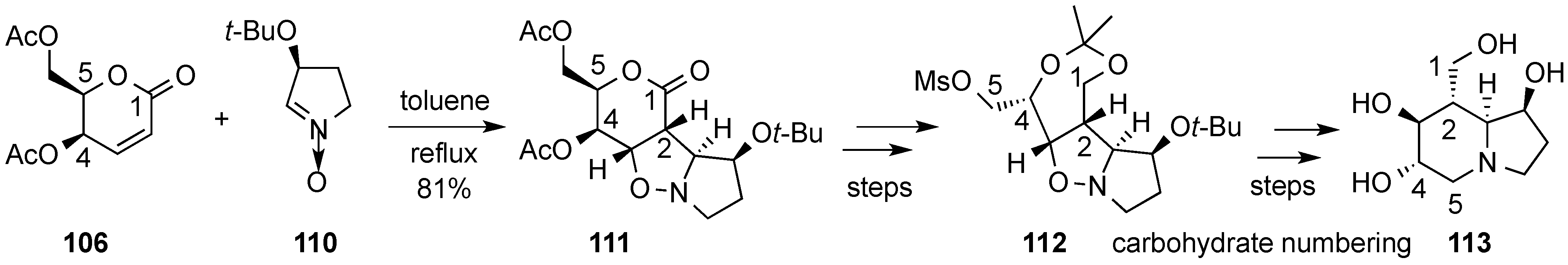
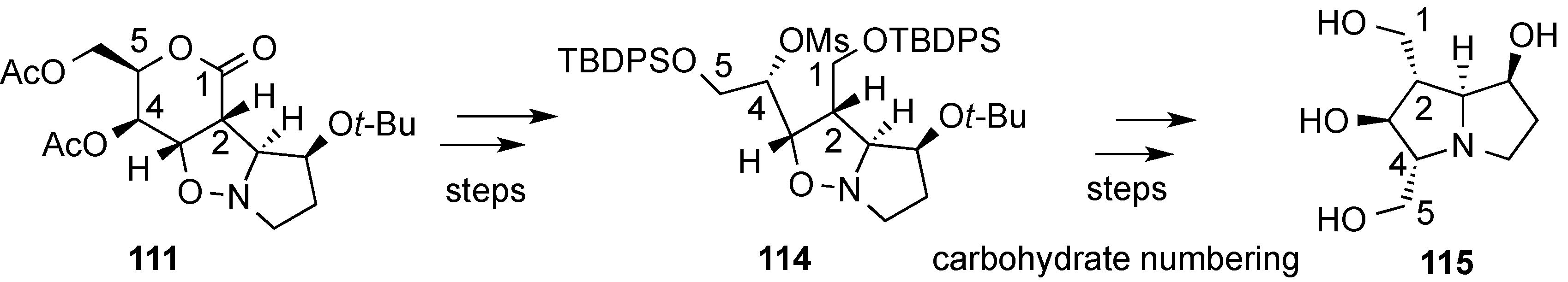

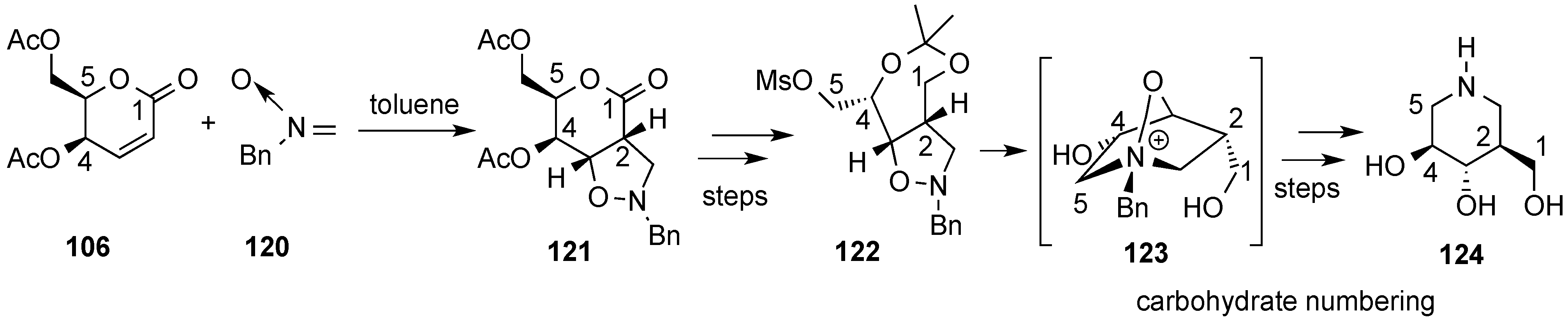
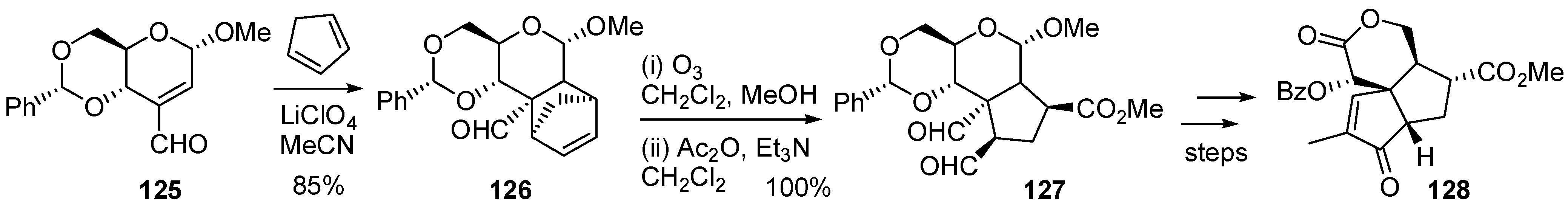
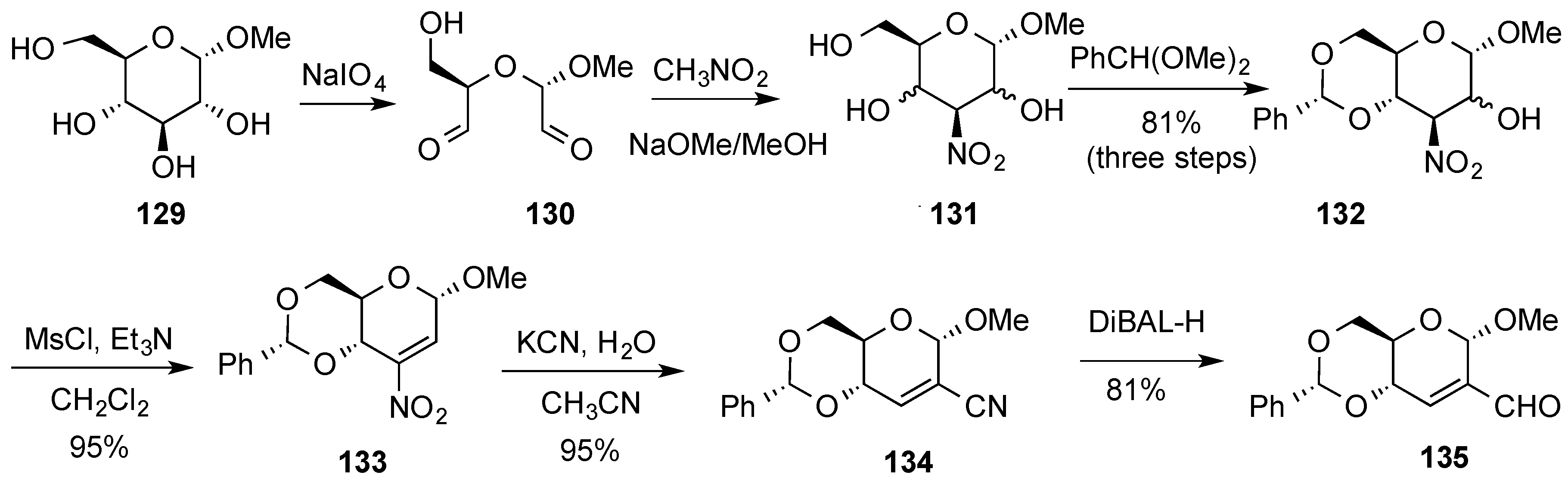
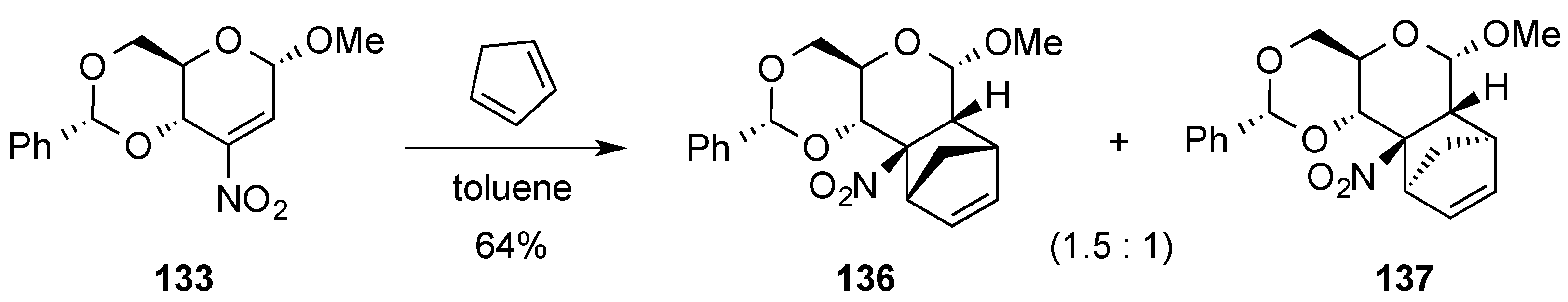
3.6. Glycosylation Reactions
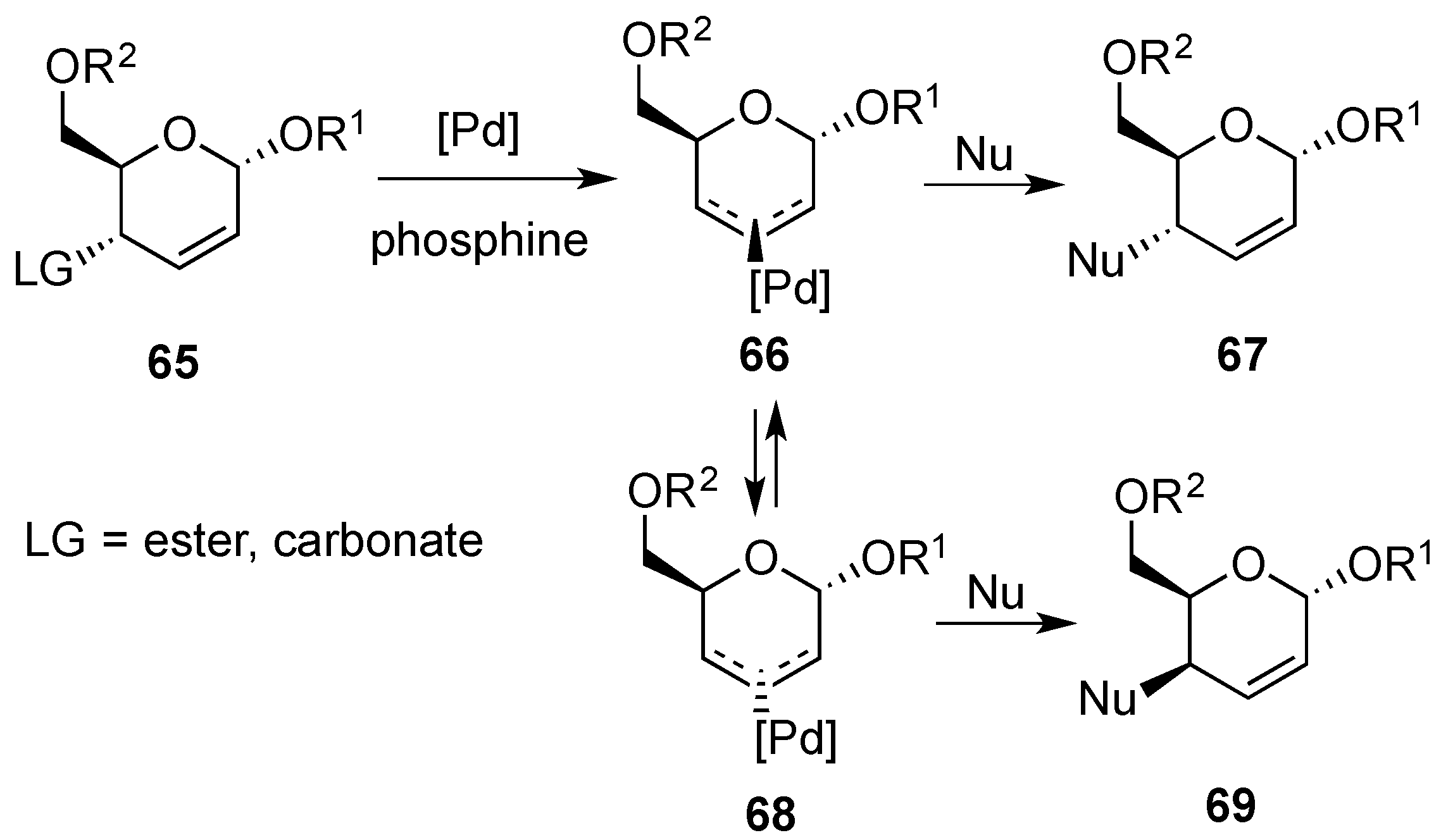
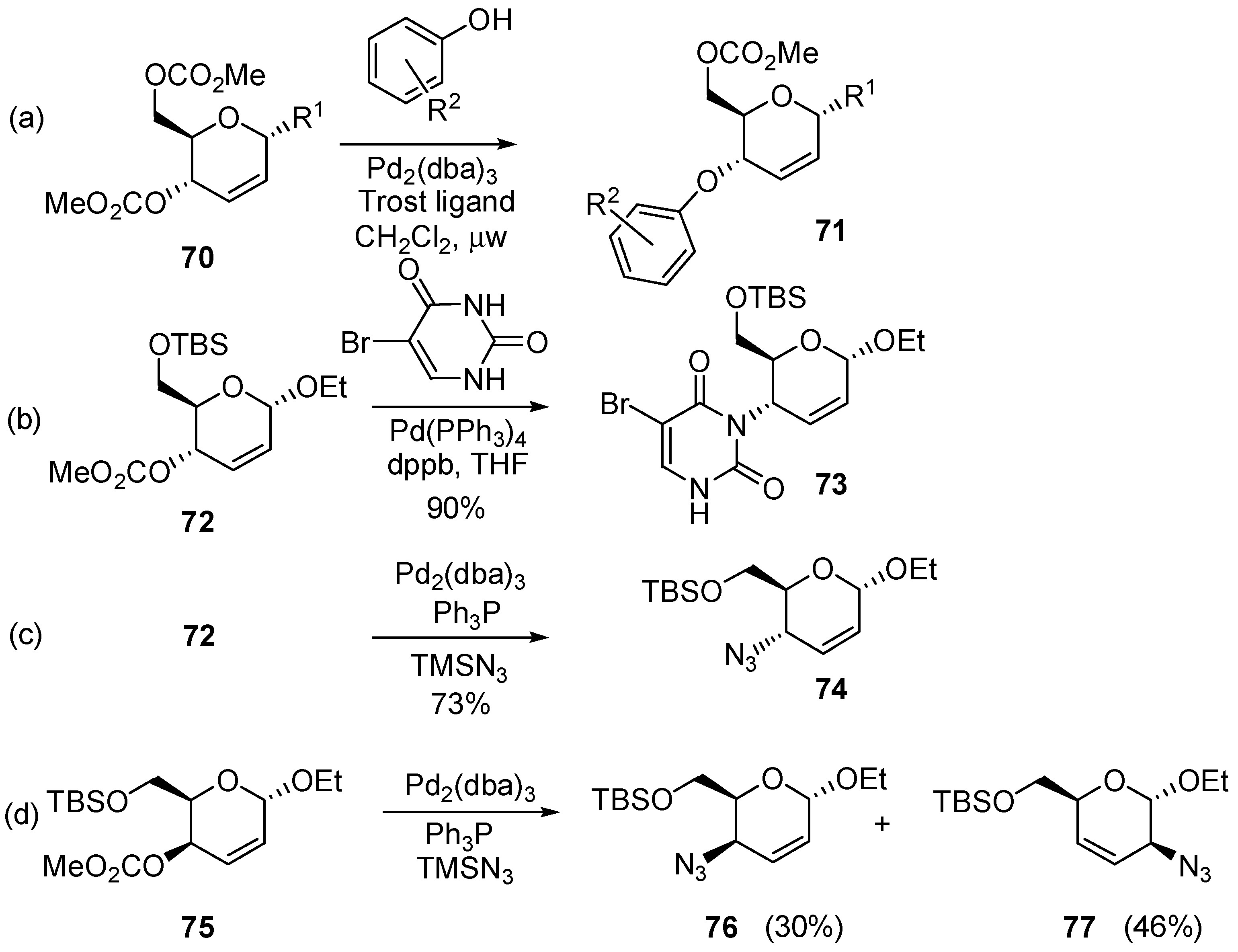
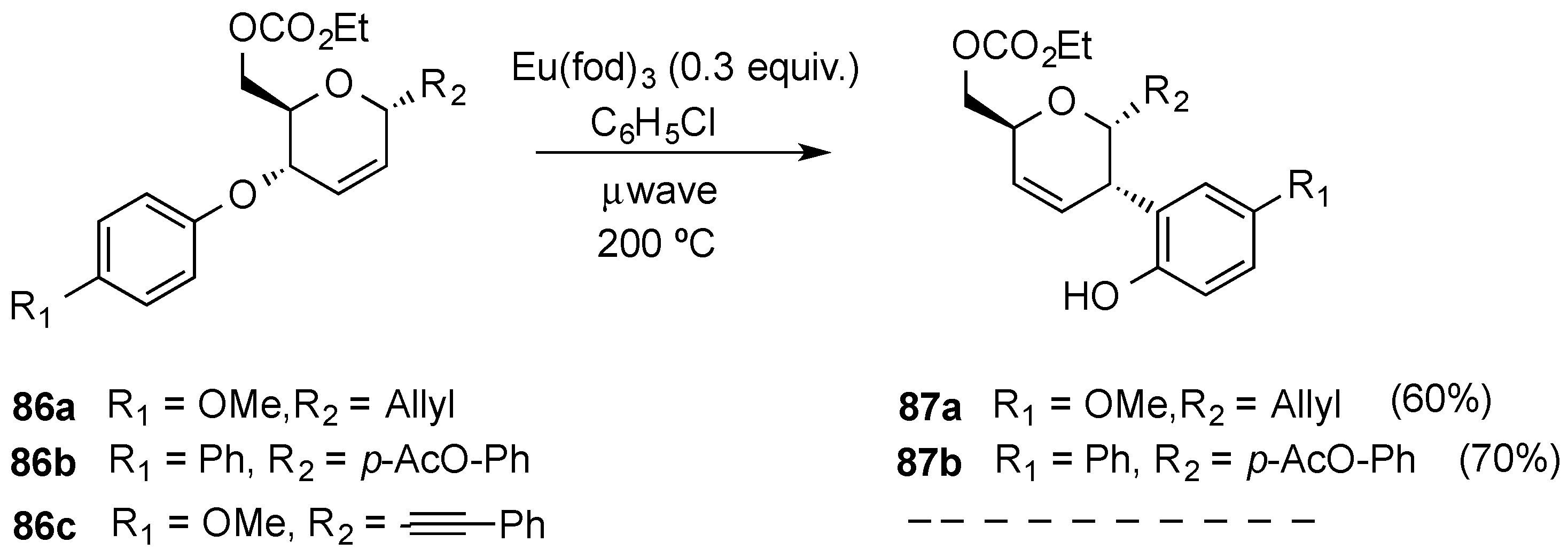
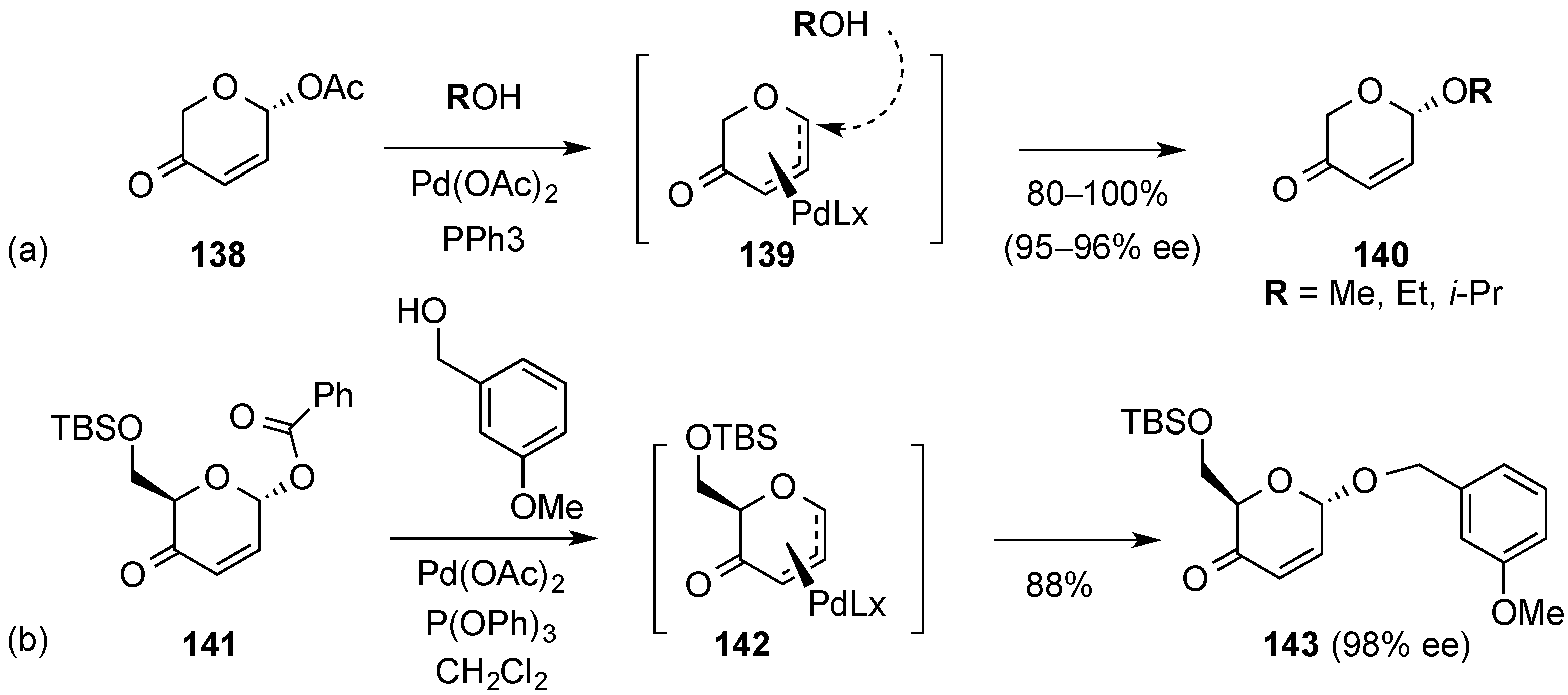
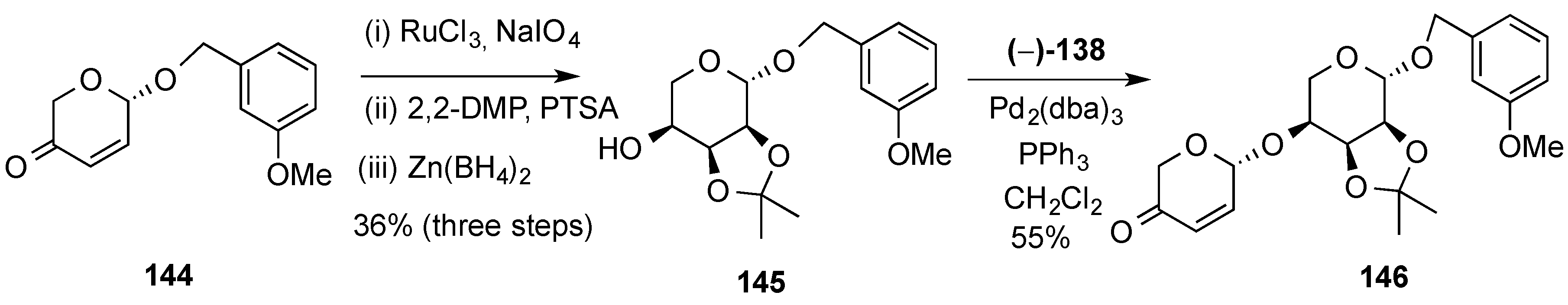
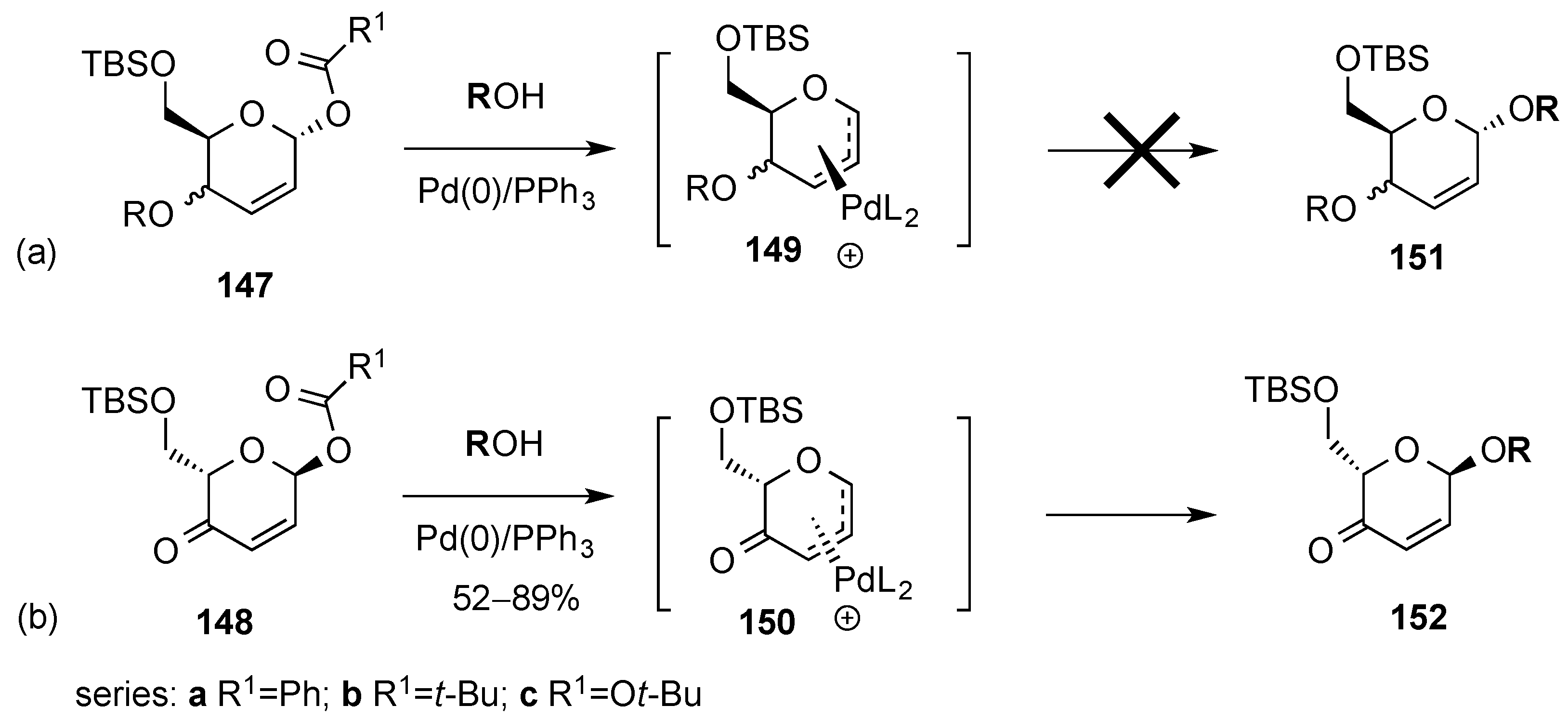
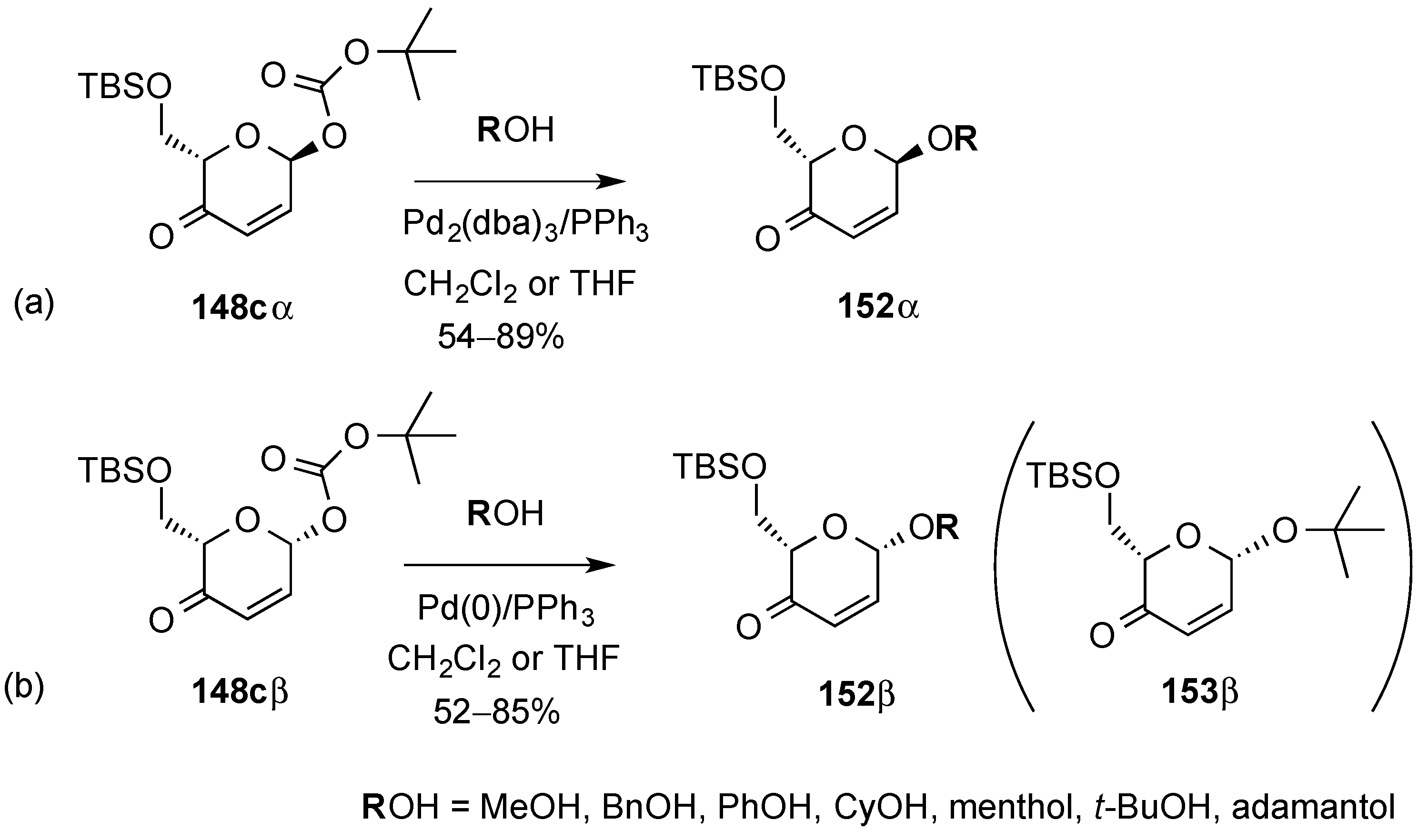
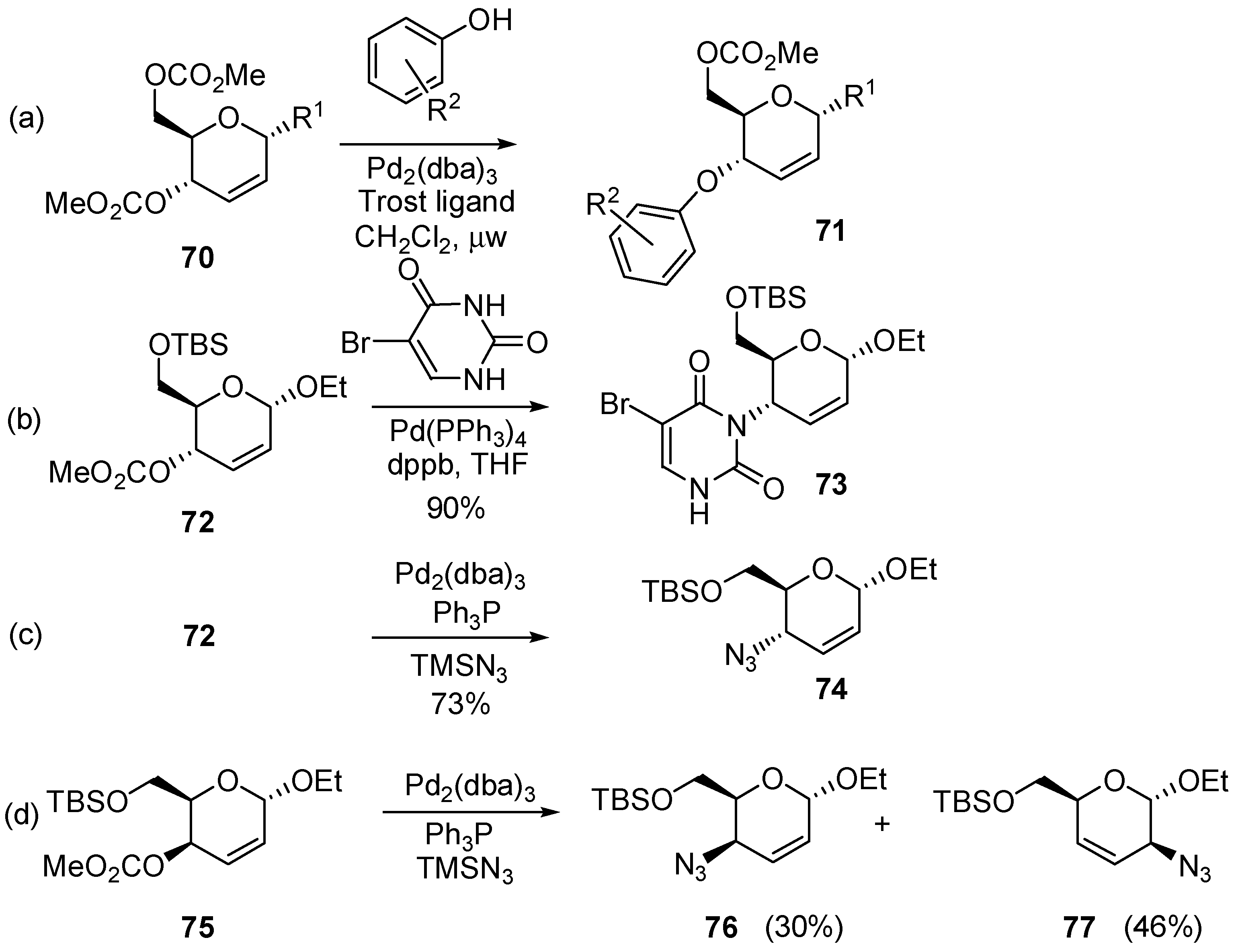
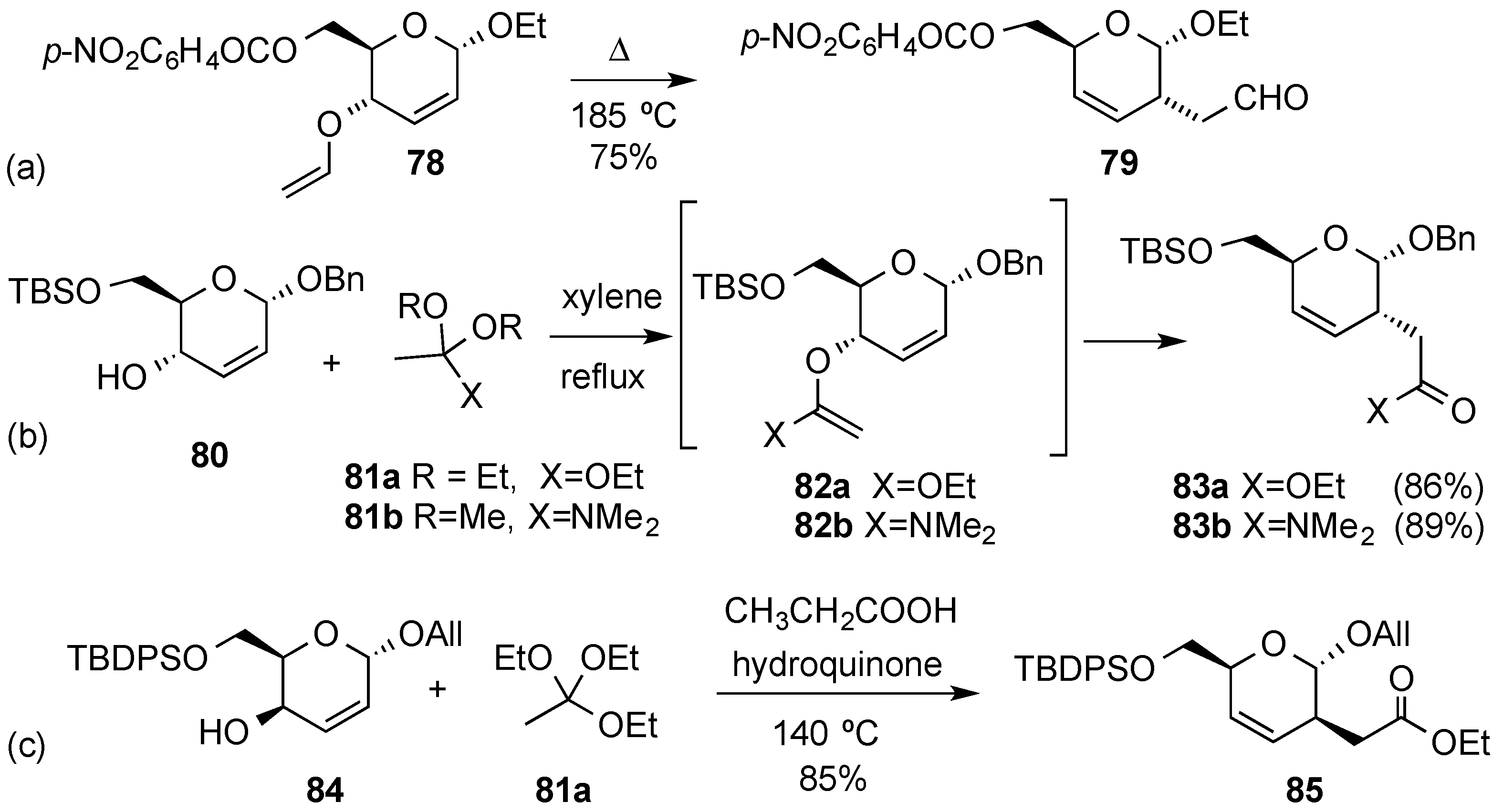
3.6.1. Palladium Mediated Glycosylation
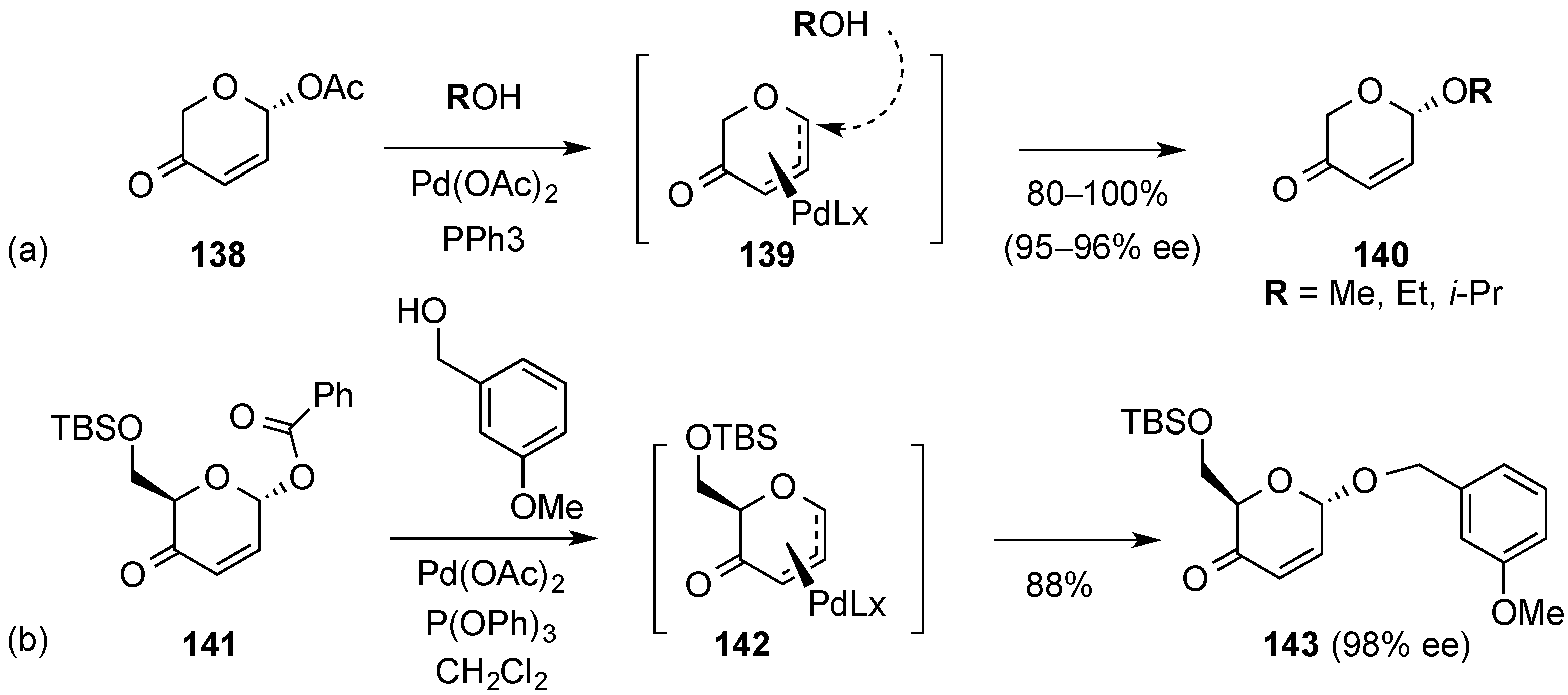
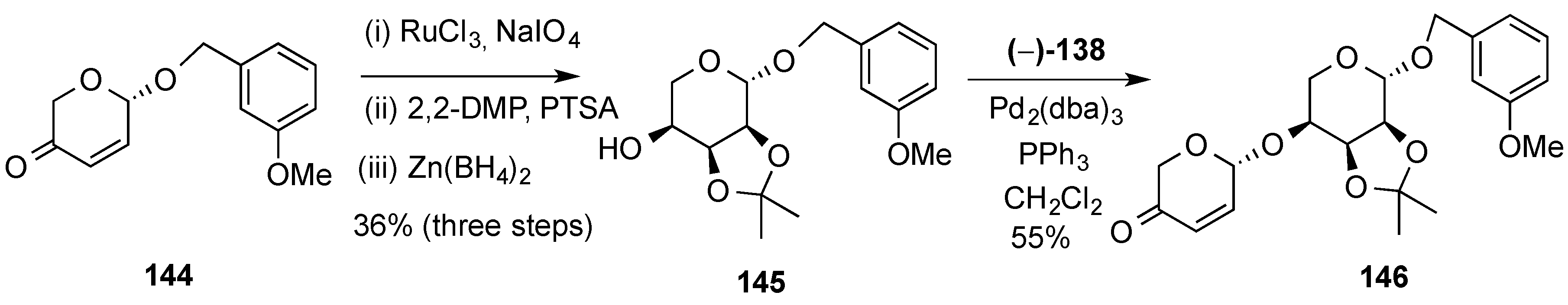
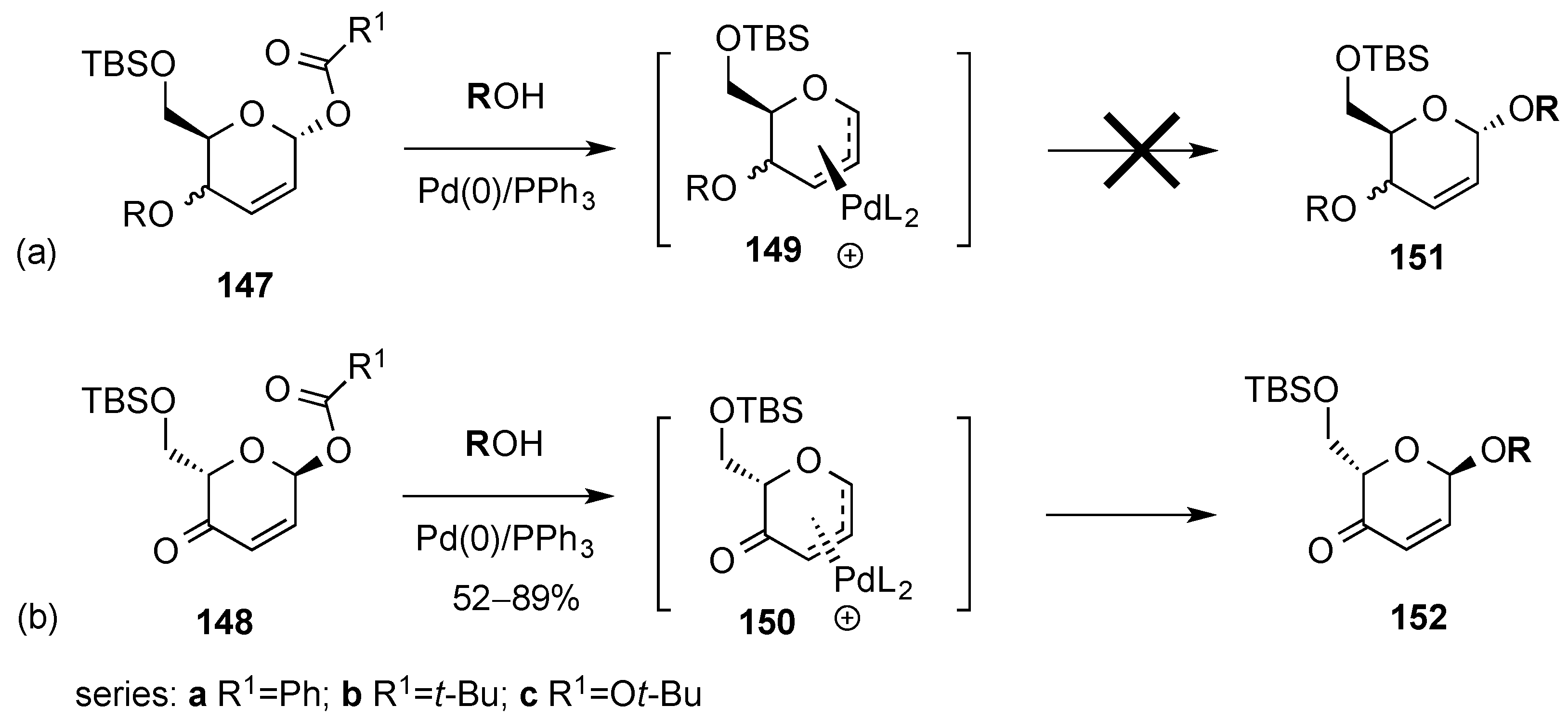
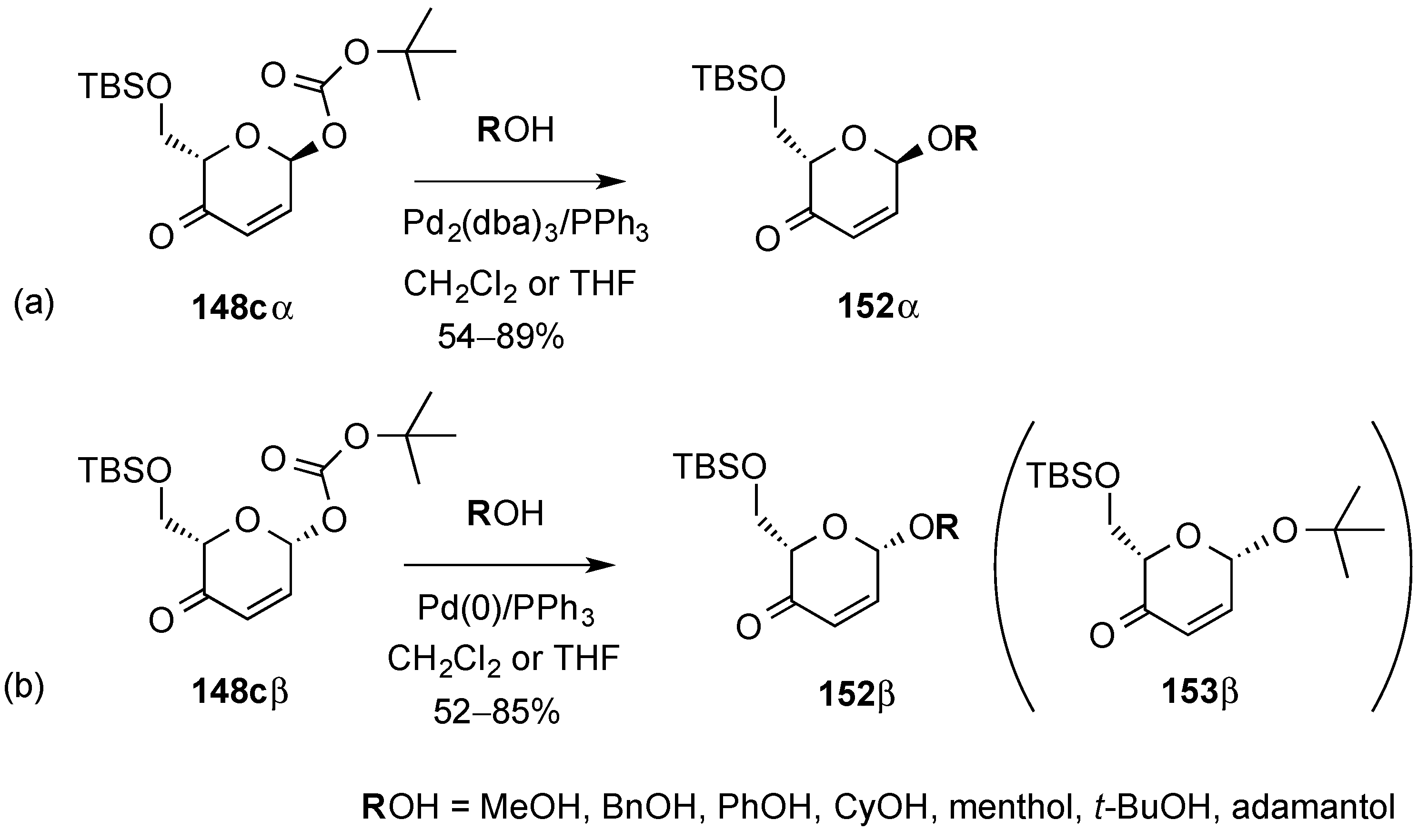
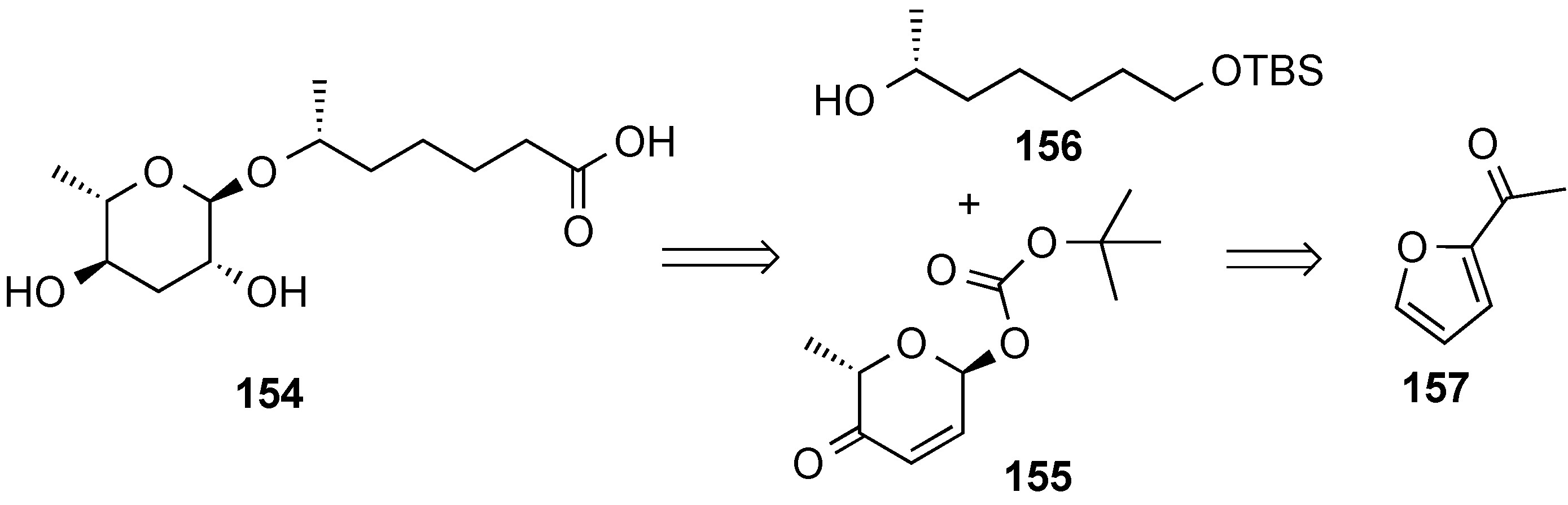
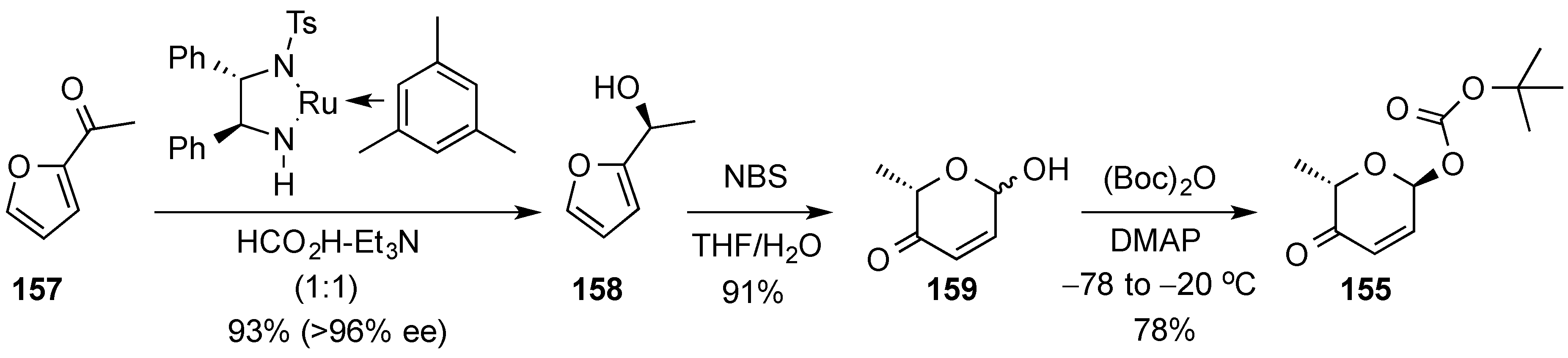
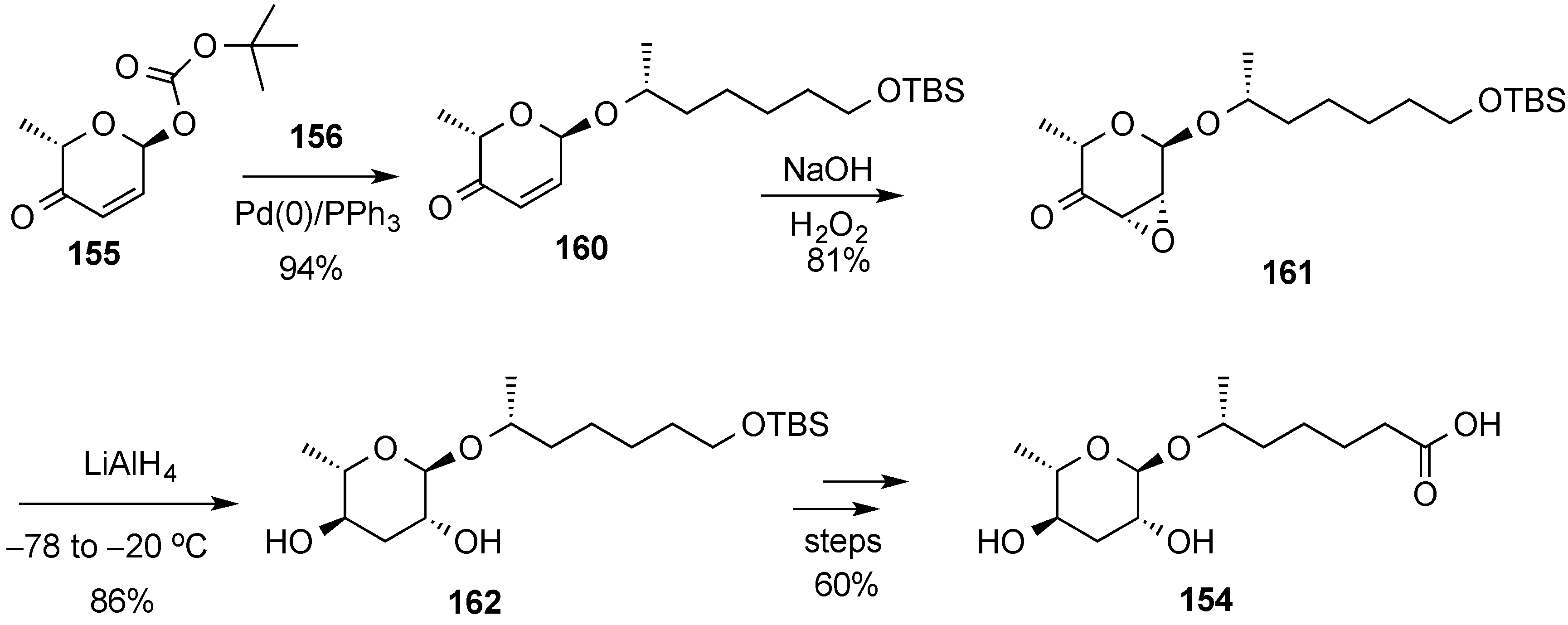
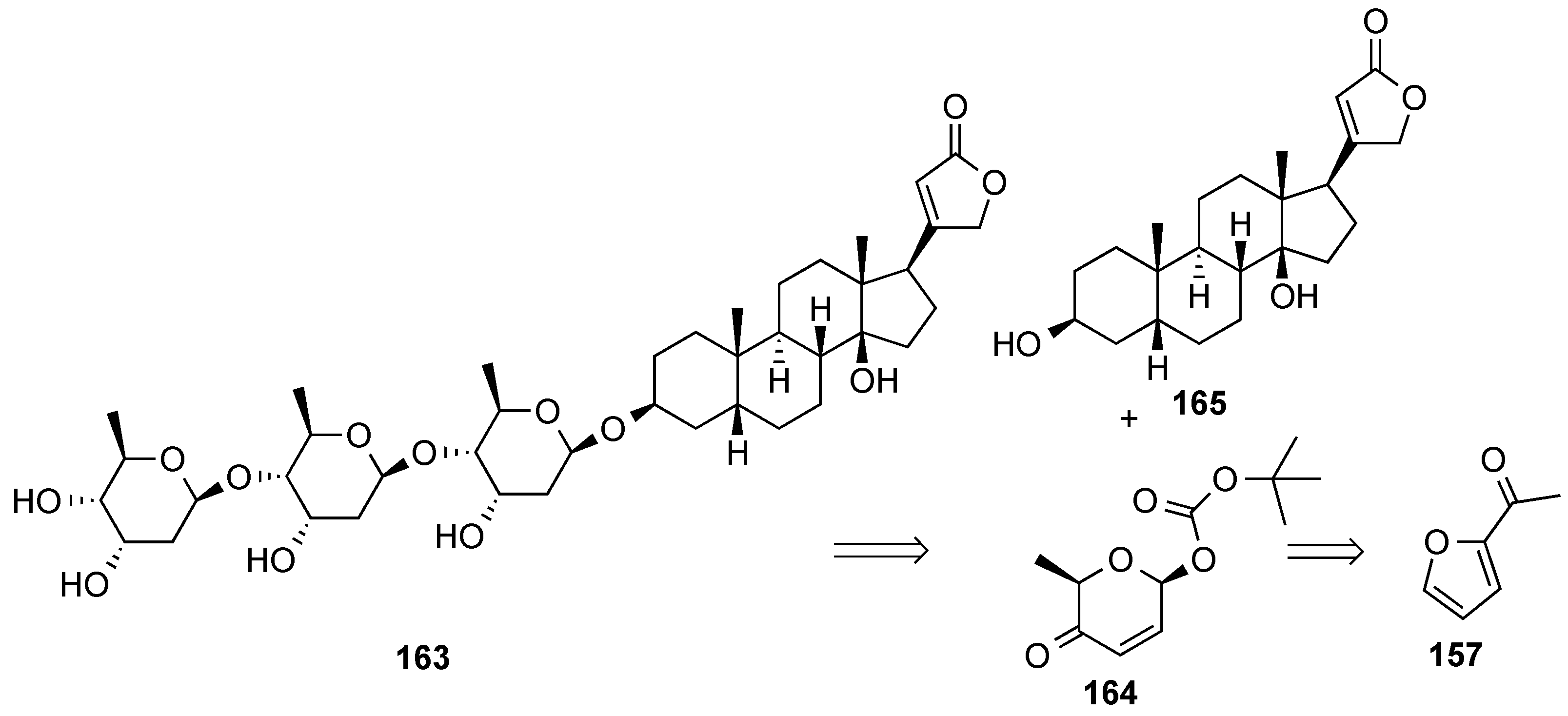
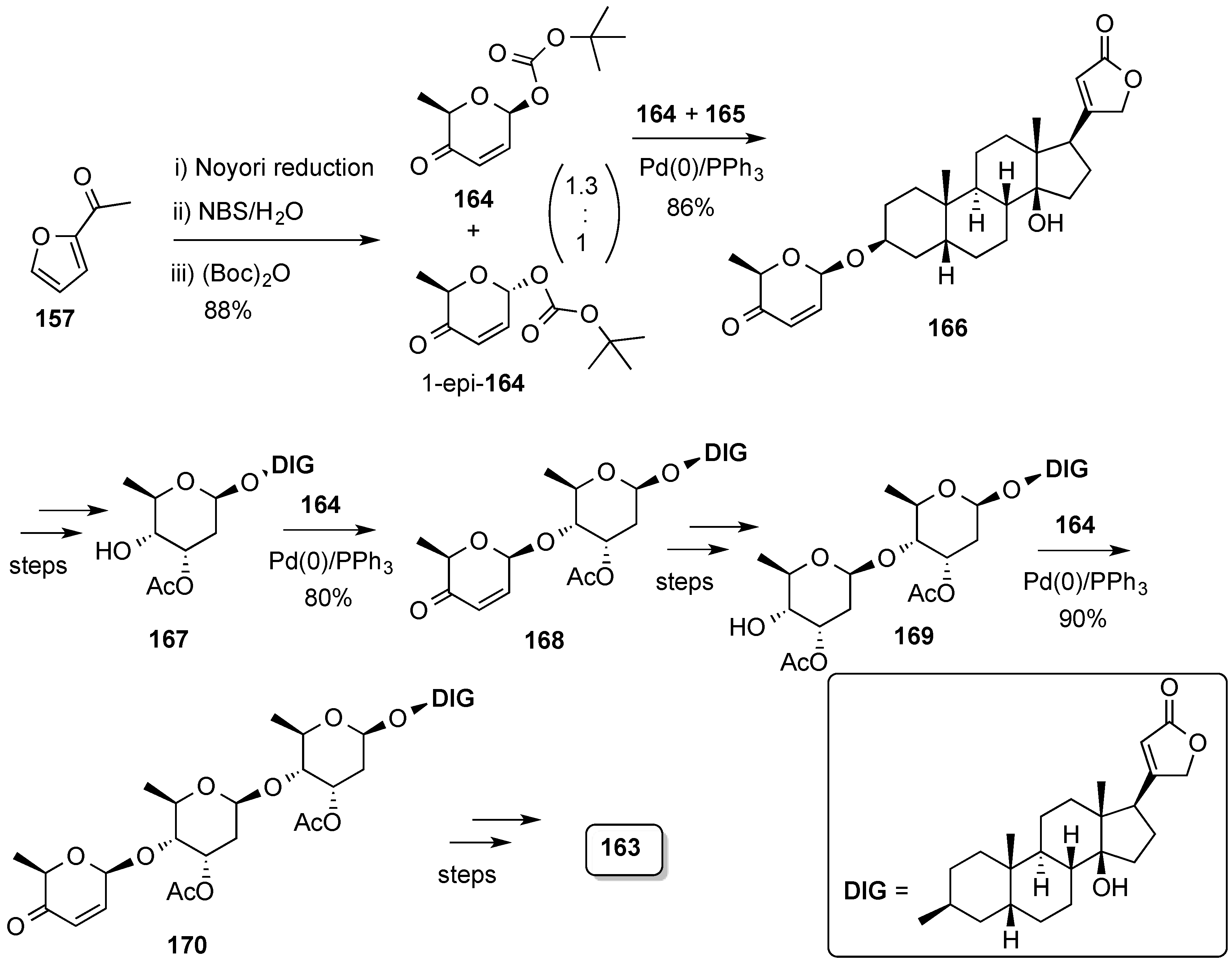
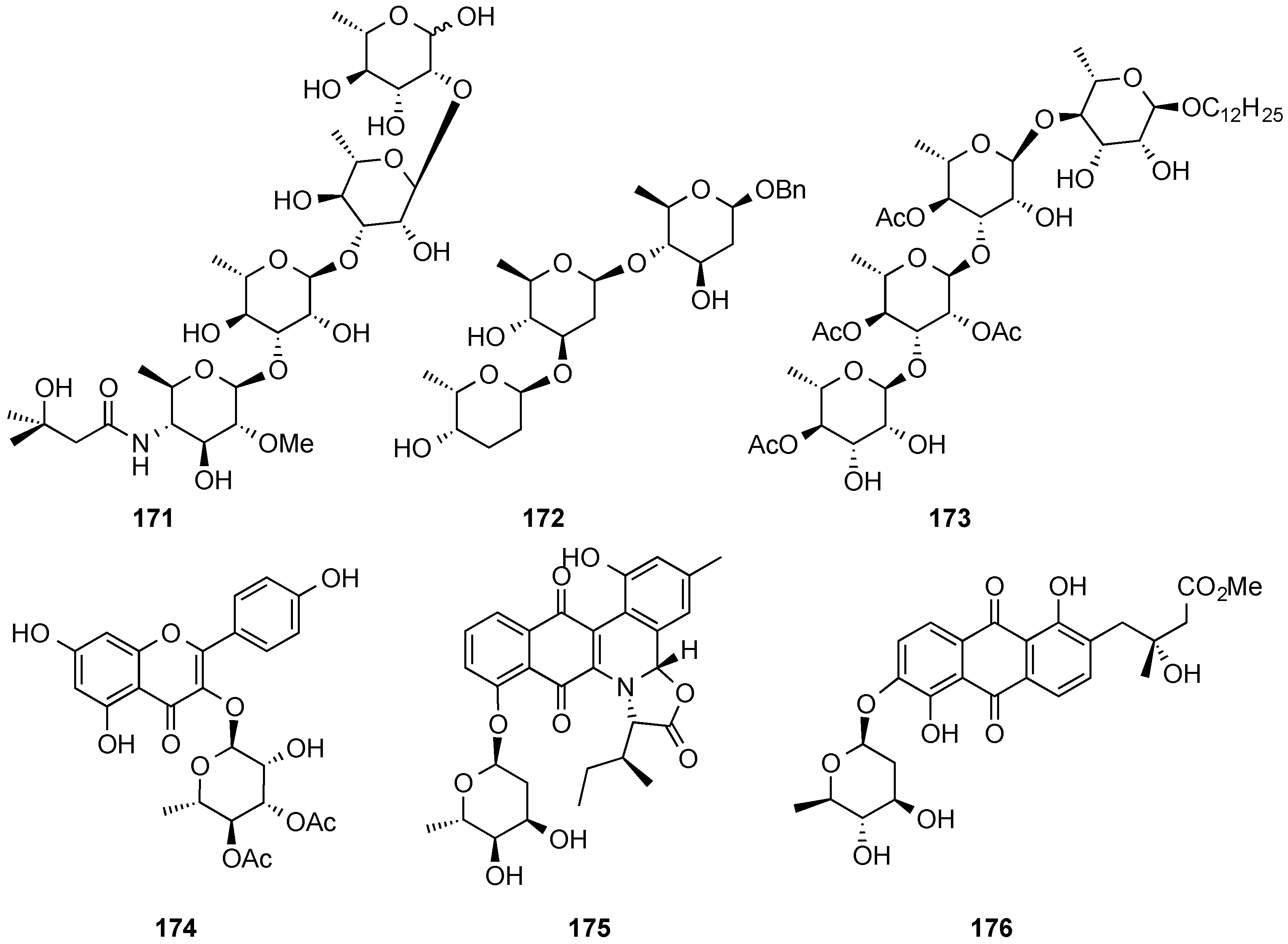
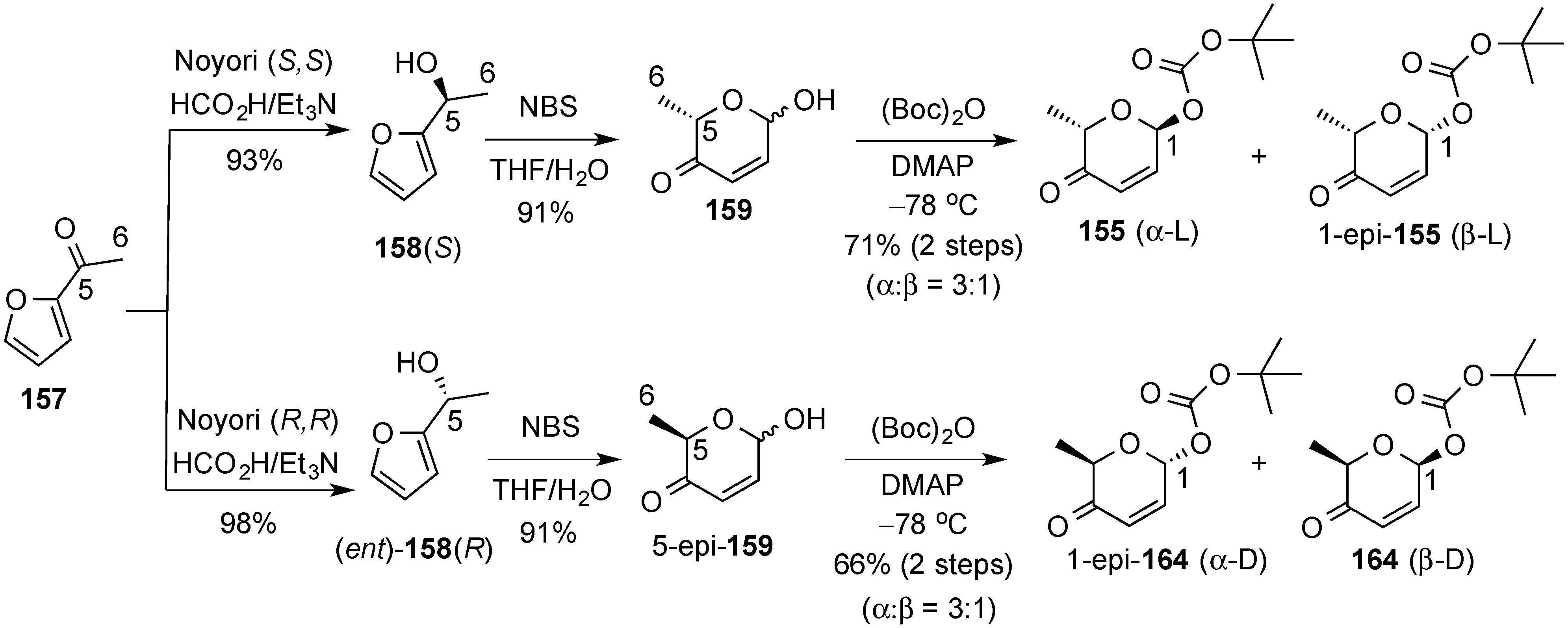
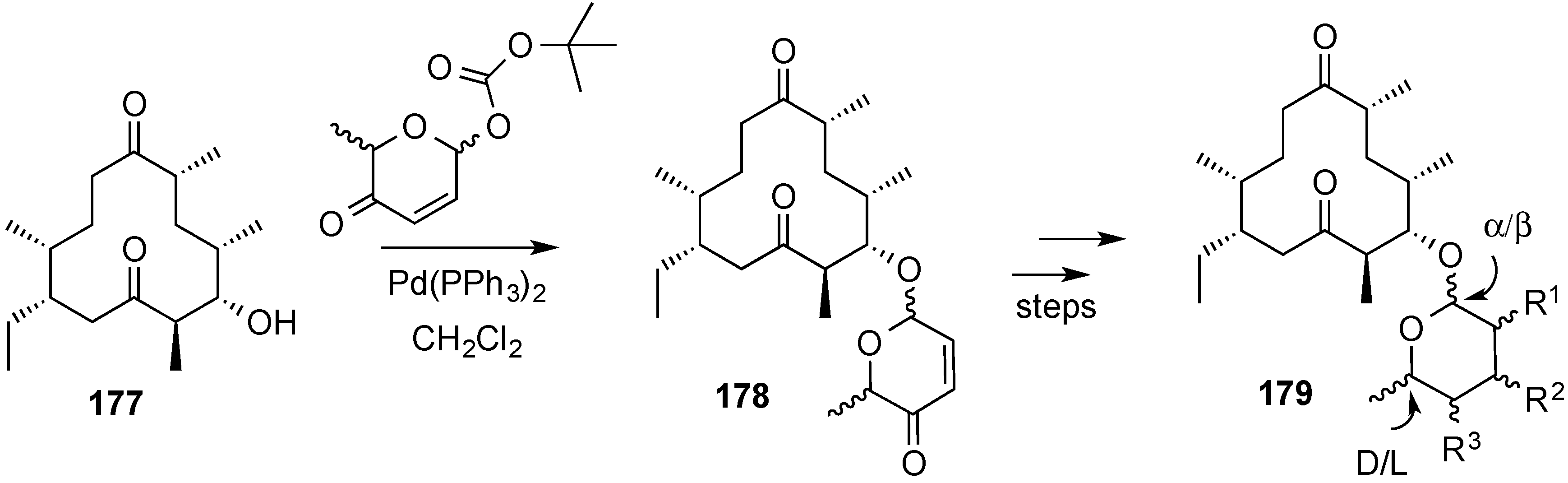
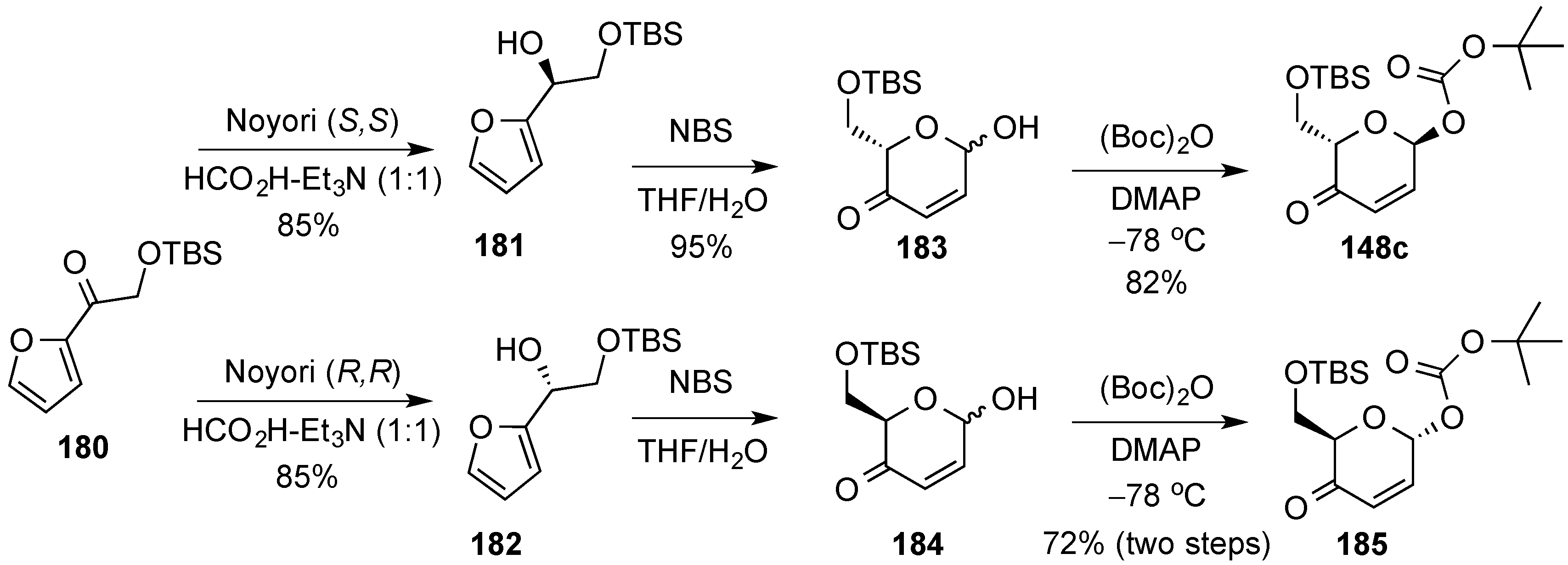
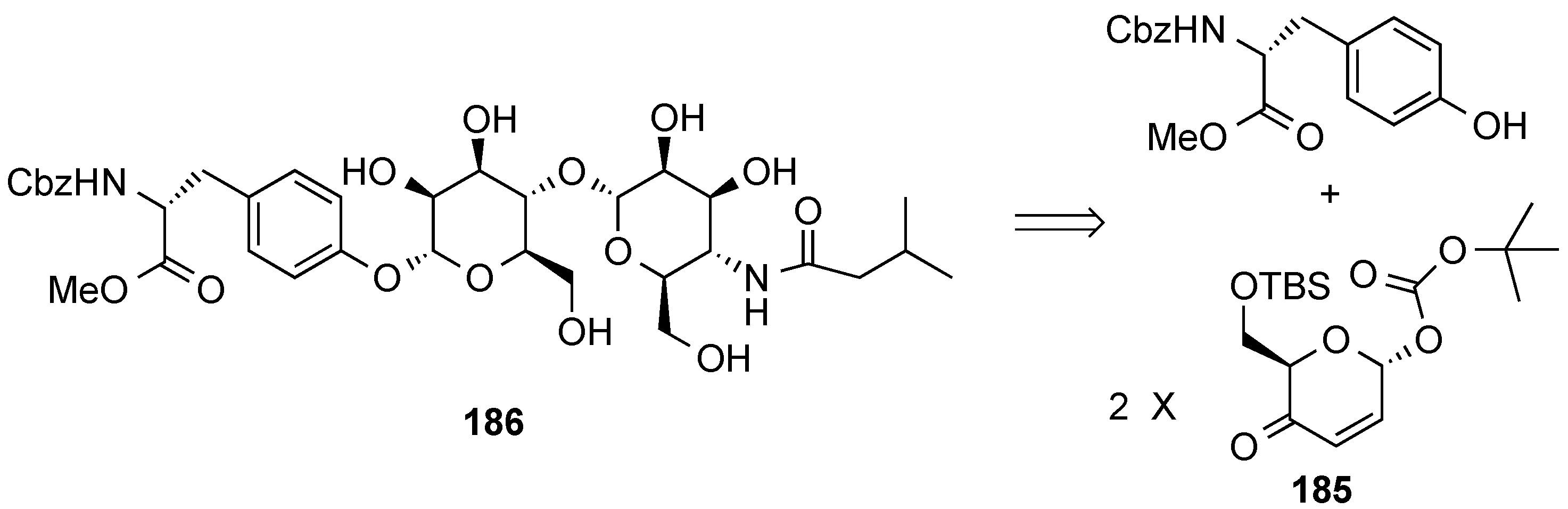
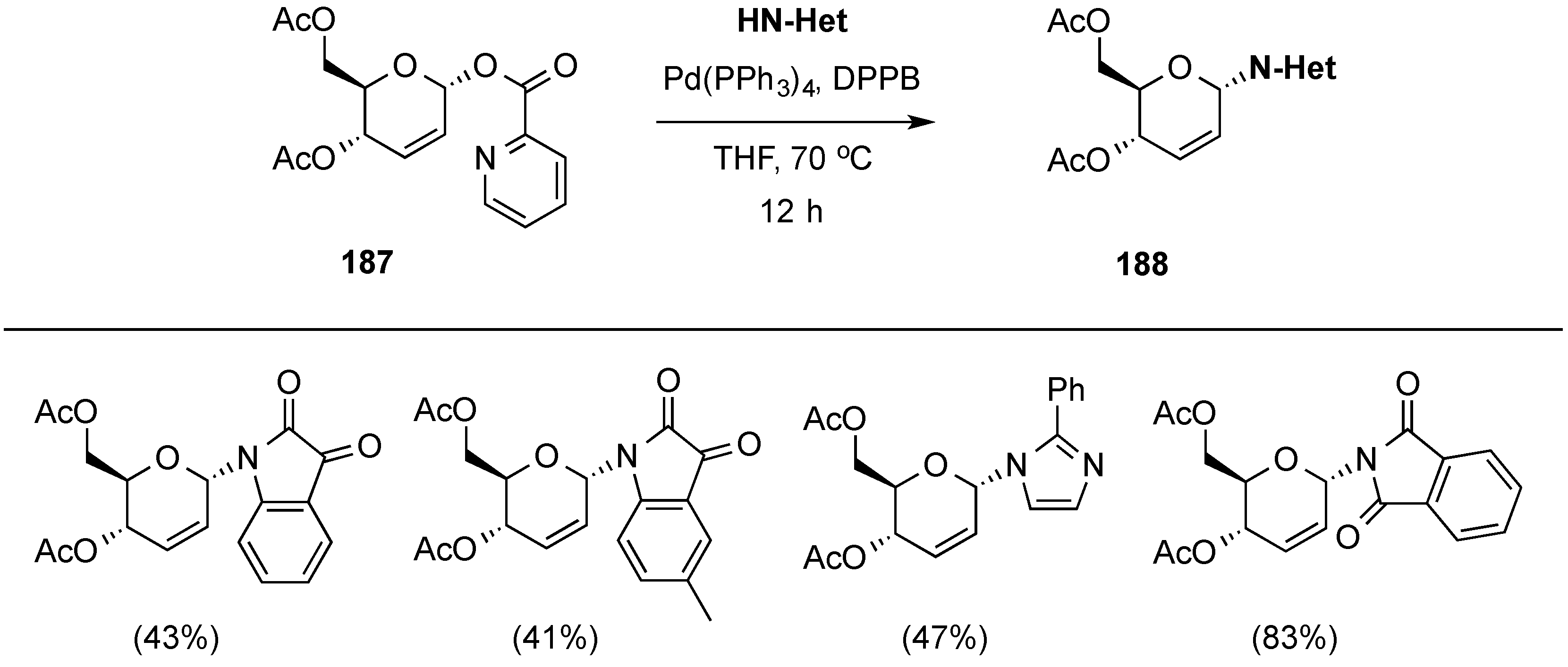
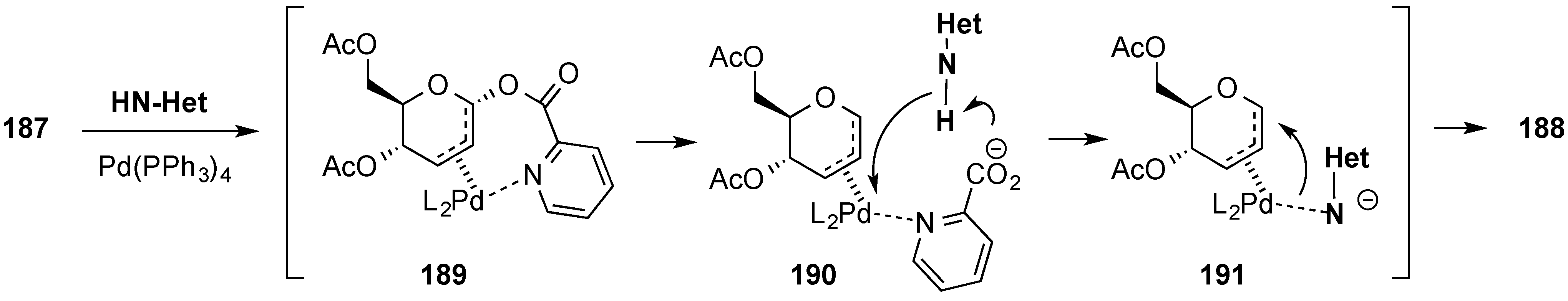
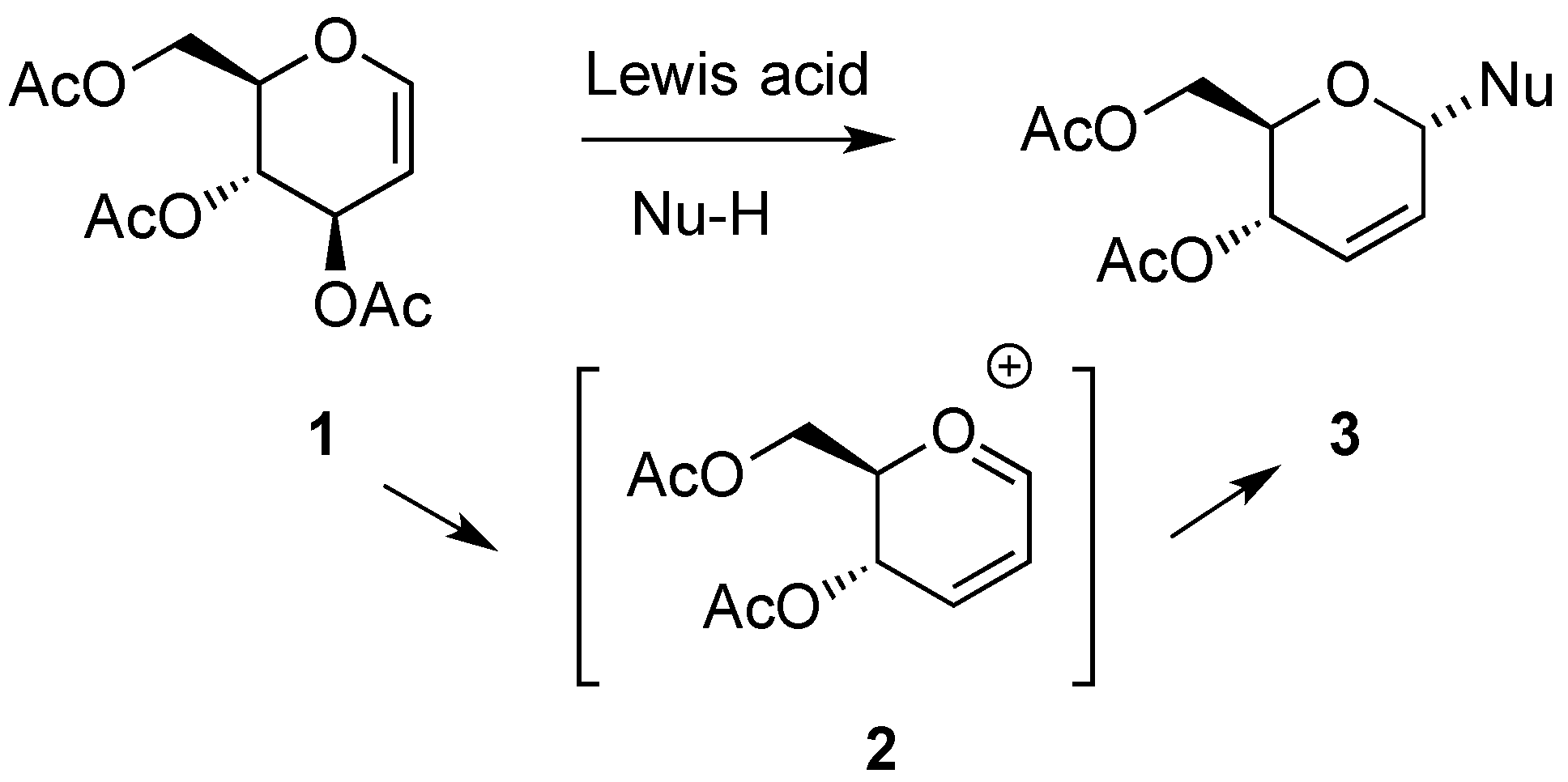
3.6.2. Lewis-Acid Mediated Glycosylation of 2,3-Unsaturated Glycosyl Acetates
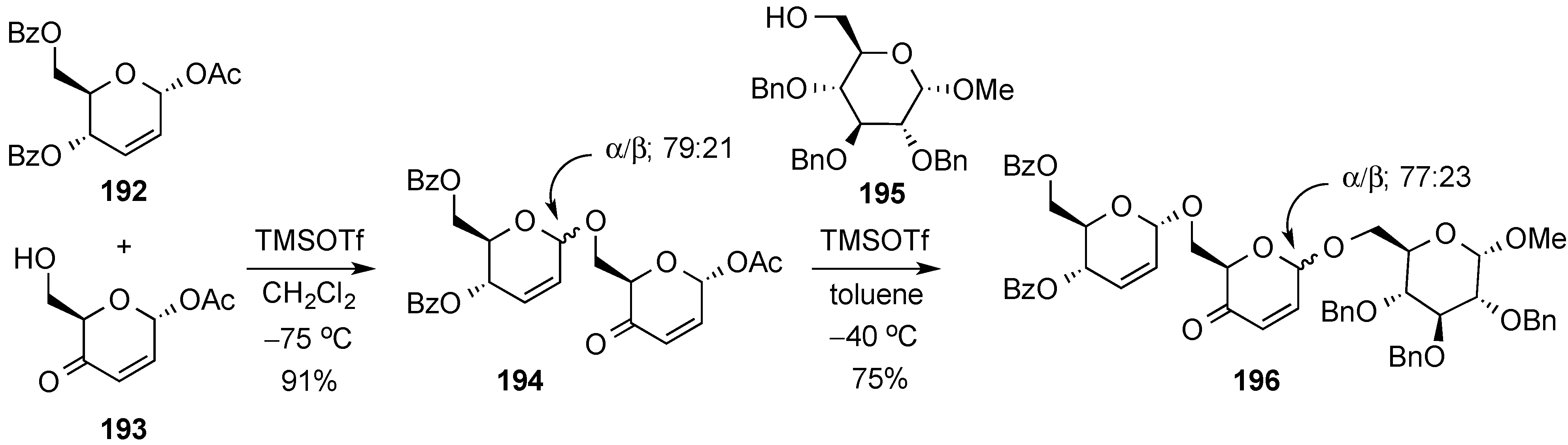
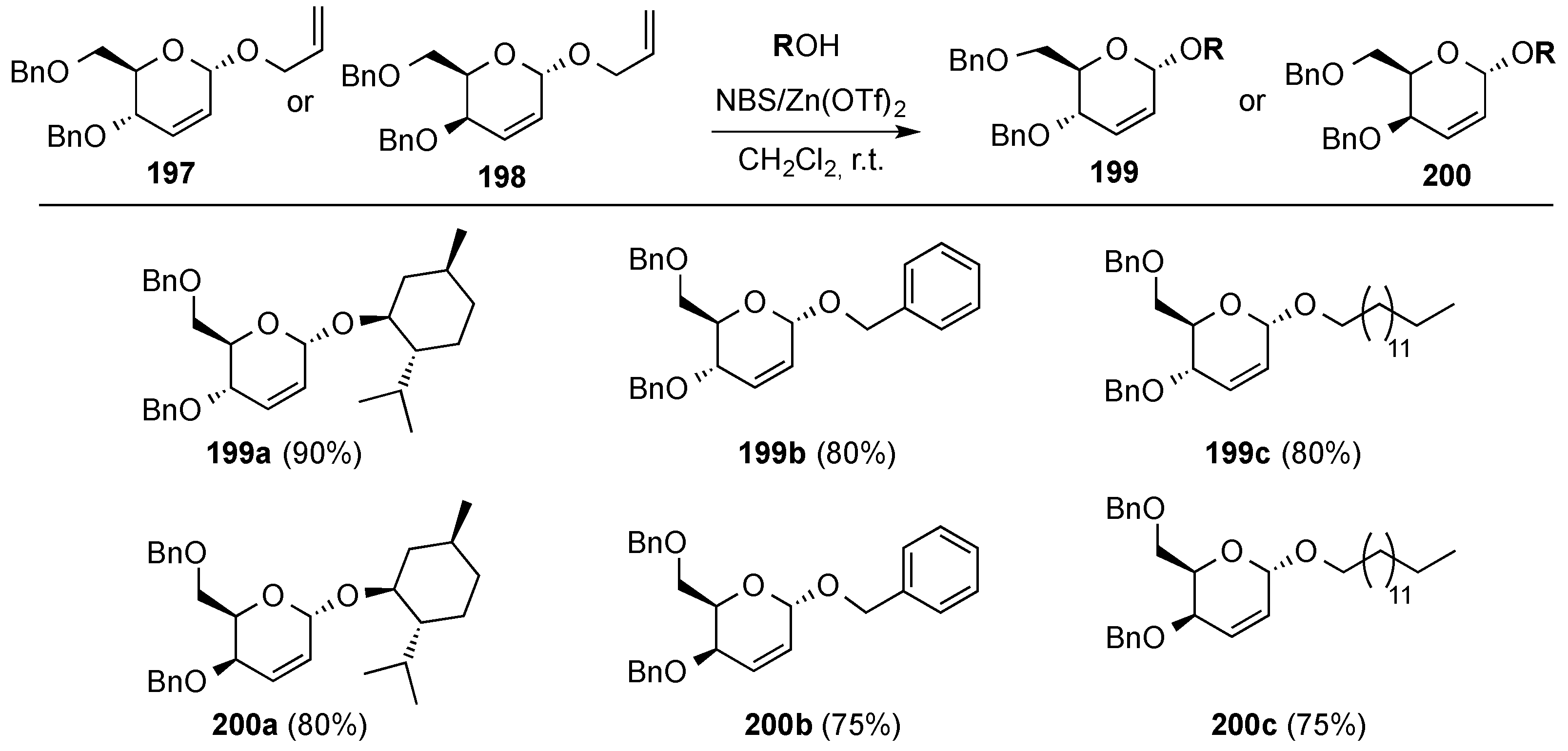
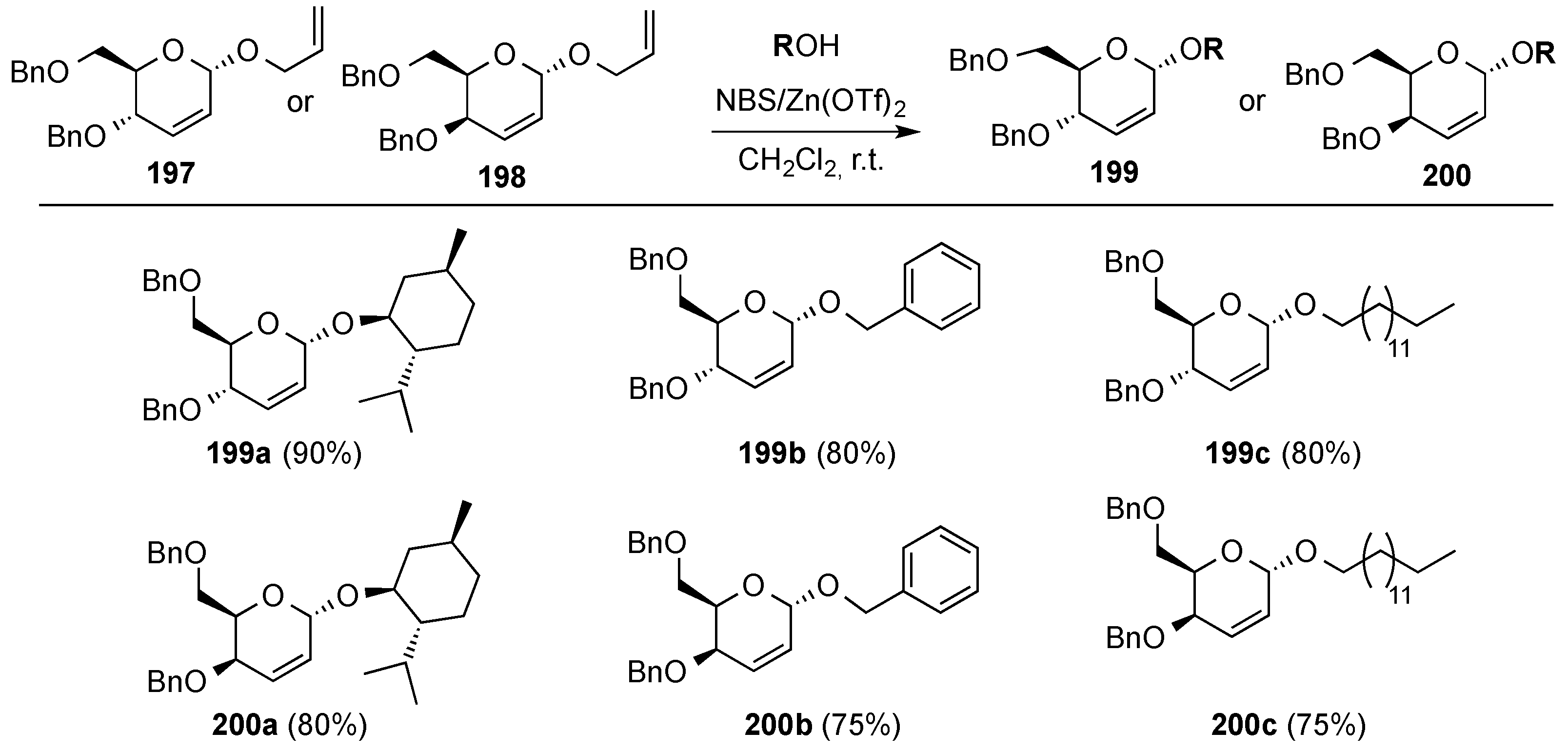
3.6.3. Halonium Ion-Mediated Glycosylation of 2,3-Unsaturated Allyl Glycosides
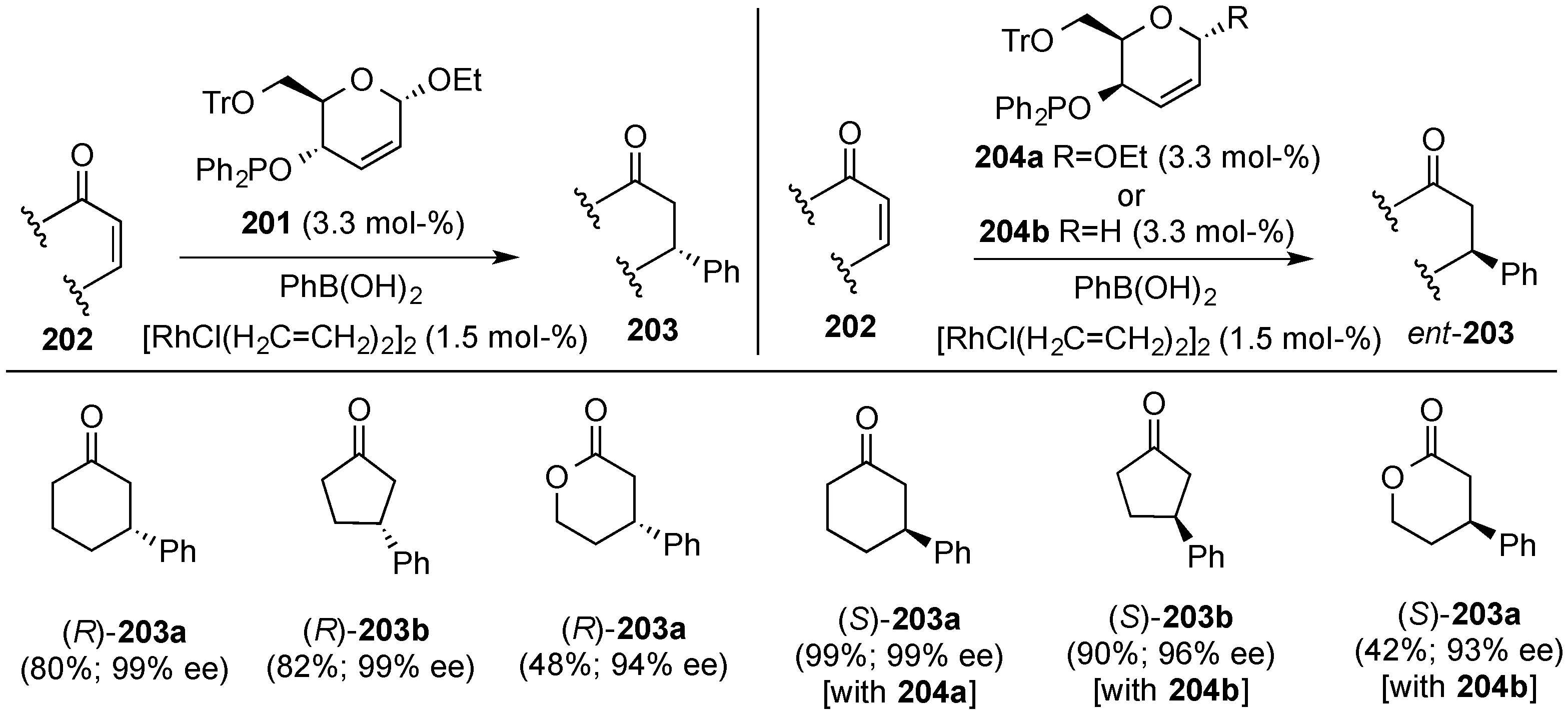
3.7. Use of 2,3-Unsaturated Hexopyranoses as Chiral Complex Ligands
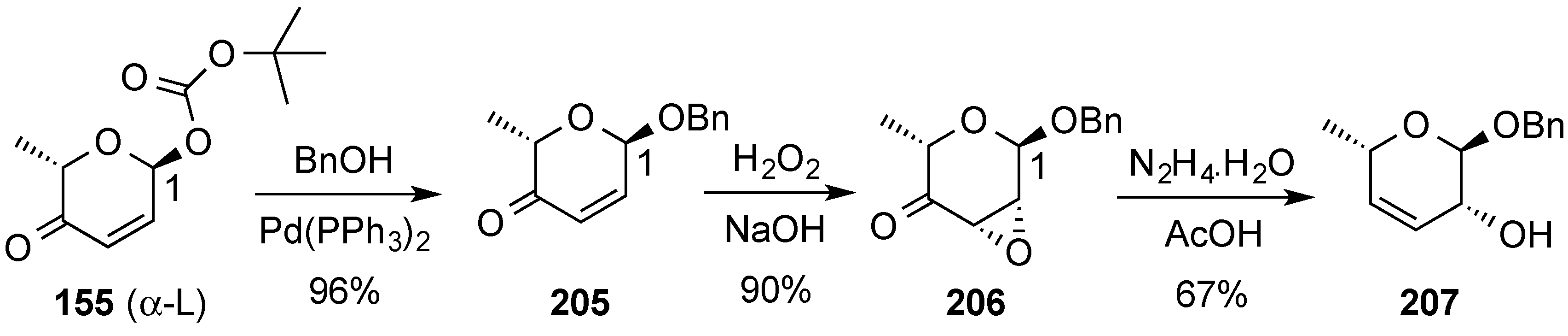
3.8. Miscellaneous
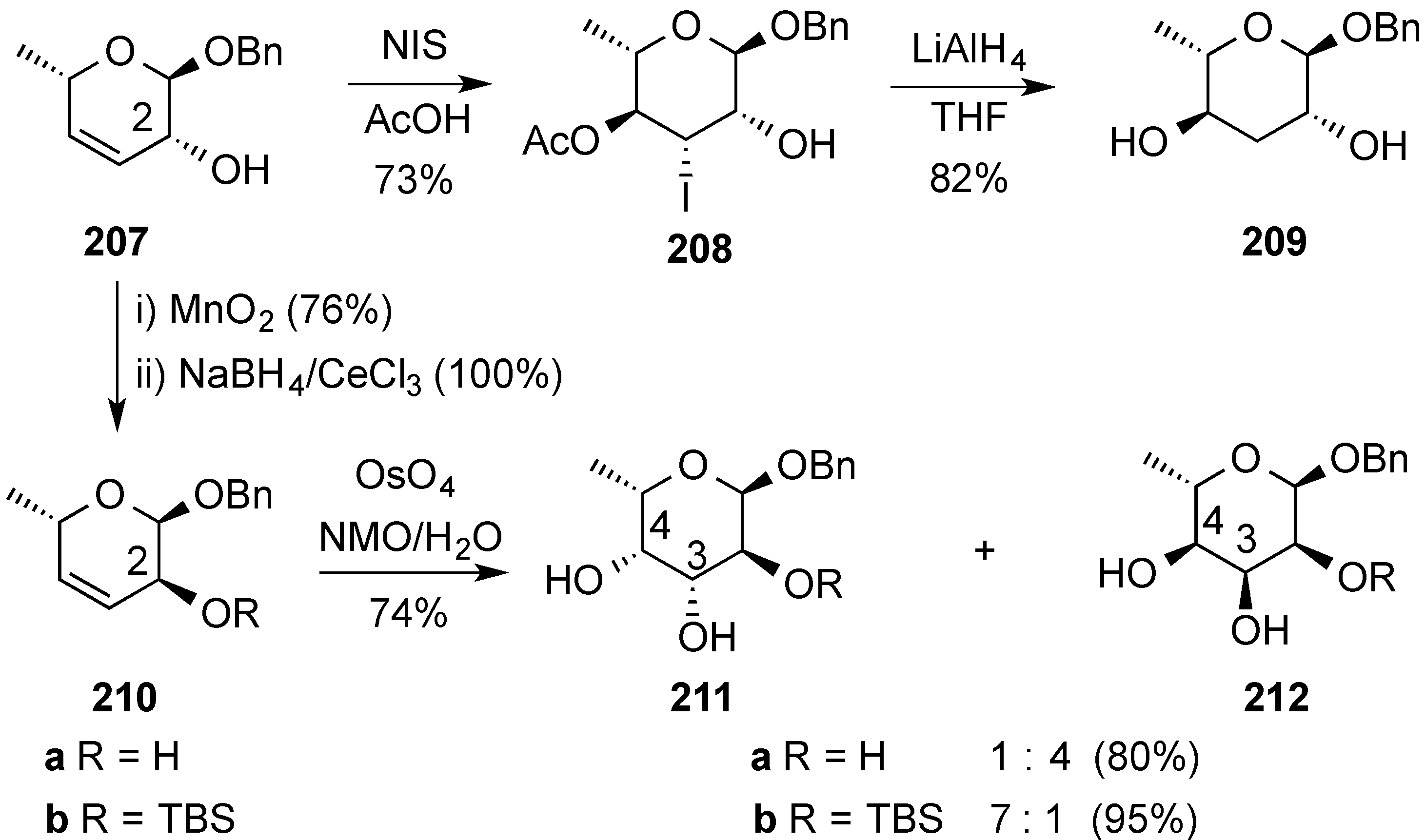
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ferrier, R.J.; Zubkov, O.A. Transformation of glycals into 2,3-unsaturated glycosyl derivatives. Org. React. 2003, 62, 569–736. [Google Scholar]
- Ferrier, R.J.; Hoberg, J.O. Synthesis and reactions of unsaturated sugars. Adv. Carbohydr. Chem. Biochem. 2003, 58, 55–119. [Google Scholar] [PubMed]
- Fischer, E. Concerning new reduction products of dextrose, glucal and hydro-glucal. Chem. Ber. 1914, 47, 196–210. [Google Scholar] [CrossRef]
- Bergmann, M. The pseudo-glucal and dihydro-pseudoglucal. Liebigs Ann. Chem. 1925, 443, 223–242. [Google Scholar] [CrossRef]
- Ferrier, R.J.; Prasad, N. Unsaturated carbohydrates. IX. Synthesis of 2,3-dideoxy-α-d-erythro-hex-2-enopyranosides from tri-O-acetyl-d-glucal. J. Chem. Soc. C 1969, 570–575. [Google Scholar] [CrossRef]
- Ferrier, R.J. Unsaturated sugars. Adv. Carbohydr. Chem. Biochem. 1969, 24, 199–266. [Google Scholar] [PubMed]
- Ferrier, R.J. Unsaturated sugars. In The Carbohydrates; Pigman, W., Horton, D., Eds.; Academic Press: New York, NY, USA, 1980; Volume 1B, p. 843. [Google Scholar]
- Fraser-Reid, B. Some progeny of 2,3-unsaturated sugars—They little resemble grandfather glucose: Twenty years later. Acc. Chem. Res. 1996, 29, 57–66. [Google Scholar] [CrossRef]
- Fraser-Reid, B. Some progeny of 2,3-unsaturated sugars—They little resemble grandfather glucose: Ten years later. Acc. Chem. Res. 1985, 18, 347–354. [Google Scholar] [CrossRef]
- Fraser-Reid, B. Some progeny of 2,3-unsaturated sugars—They little resemble grandfather glucose. Acc. Chem. Res. 1975, 8, 192–201. [Google Scholar] [CrossRef]
- The chemistry of 2,3-unsaturated derivatives on pyranoid systems had been periodically reviewed. In Carbohydrate Chemistry, Specialist Periodical Reports; Royal Society of Chemistry: Cambridge, UK, 2003; from Volume 1, 1968, to Volume 34.
- Fraser-Reid, B.; López, J.C. Unsaturated sugars: A rich platform for methodological and synthetic studies. Curr. Org. Chem. 2009, 13, 532–553. [Google Scholar] [CrossRef]
- Gómez, A.M.; Lobo, F.; Uriel, C.; López, J.C. Recent developments in the Ferrier rearrangement. Eur. J. Org. Chem. 2013, 2013, 7221–7262. [Google Scholar] [CrossRef]
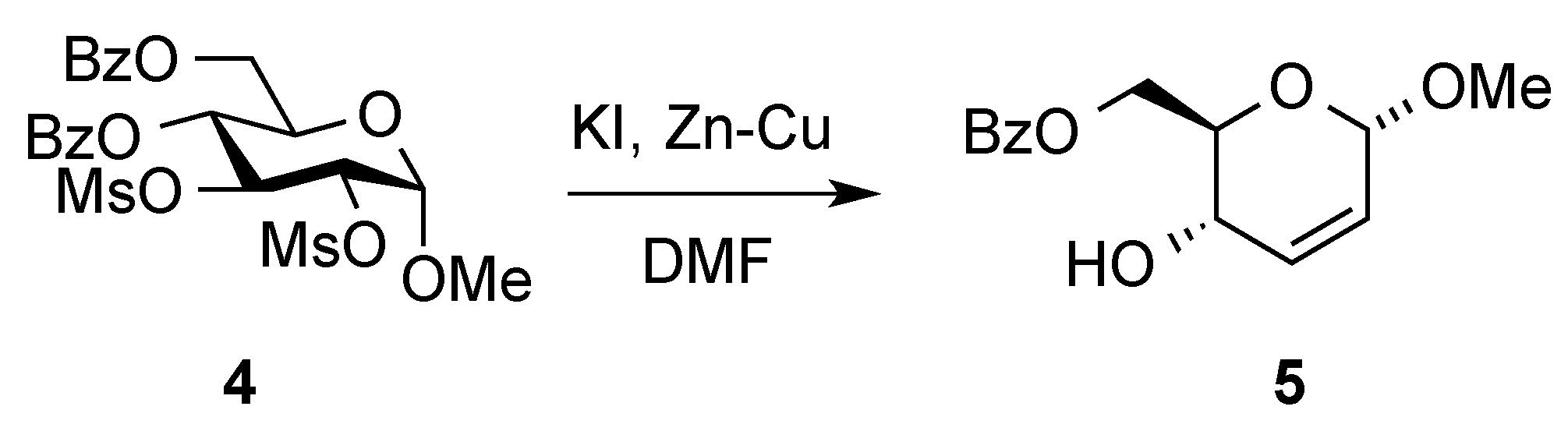
- Tipson, R.S.; Cohen, A. Action of zinc dust and sodium iodide in N,N-dimethylformamide on contiguous, secondary sulfonyloxy groups: A simple method for introducing nonterminal unsaturation. Carbohydr. Res. 1965, 1, 338–340. [Google Scholar] [CrossRef]
- Fraser-Reid, B.; Boctor, B. Some aspects of the formation of hex-2-enopyranosides from methyl 2,3-di-O-methanesu1fonyl-α-d-glucopysanosides. Can. J. Chem. 1969, 47, 393–401. [Google Scholar] [CrossRef]
- Achmatowicz, O.; Bielski, R. Stereoselective total synthesis of methyl α-D and α-l-glucopyranosides. Carbohydr. Res. 1977, 55, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Achmatowicz, O.; Bukowski, B.; Szechner, B.; Zwierzchowska, Z.; Zamojski, A. Synthesis of methyl 2,3-dideoxy-DL-alk-2-enopyranosides from furan compounds. A general approach to the total synthesis of monosaccharides. Tetrahedron 1971, 27, 1973–1996. [Google Scholar] [CrossRef]
- Shimshock, S.J.; Waltermire, R.E.; DeShong, P. A total synthesis of (±)-Tirandamycin B. J. Am. Chem. Soc. 1991, 113, 8791–8796. [Google Scholar] [CrossRef]
- Adger, B.M.; Barrett, C.; Brennan, J.; McKervey, M.A.; Murray, R.W. Oxidation of furans with dimethyldioxirane. J. Chem. Soc. Chem. Commun. 1991, 1553–1554. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of Daumone via a palladium-catalyzed glycosylation. Org. Lett. 2005, 7, 3921–3924. [Google Scholar] [CrossRef] [PubMed]
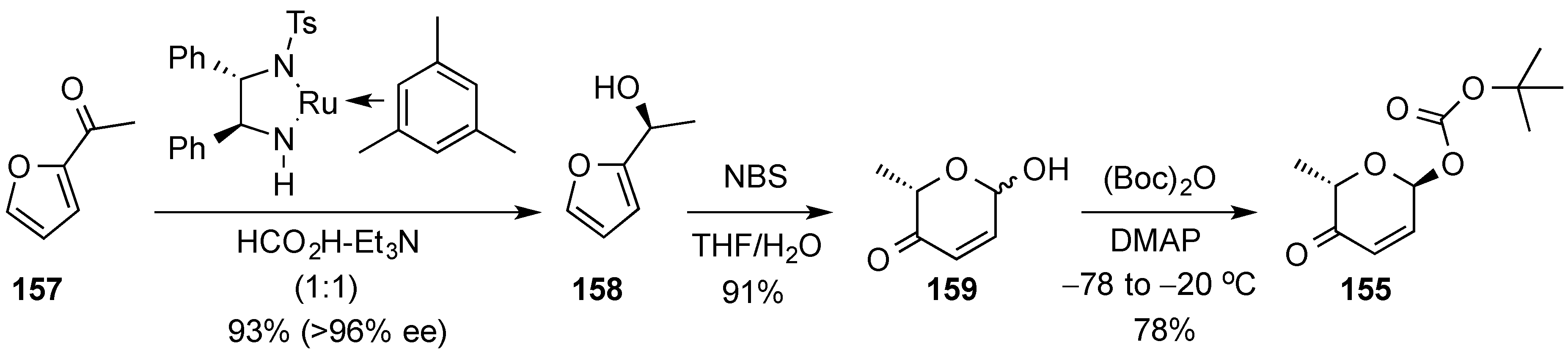
- Georgiadis, M.P.; Couladouros, E.A. Products from furans. 4. Selective oxidation of 2-furfuryl alcohol derivatives, in the presence of aryl thioethers, with N-bromosuccinimide (NBS). A new procedure for the preparation of 2H-pyran-3(6H)-ones. J. Org. Chem. 1986, 51, 2725–2727. [Google Scholar] [CrossRef]
- Kusakabe, M.; Kitano, Y.; Kobayashi, Y.; Sato, F. Preparation of optically active 2-furylcarbinols by dinetic resolution using the Sharpless reagent and their application in organic synthesis. J. Org. Chem. 1989, 54, 2085–2091. [Google Scholar] [CrossRef]
- Wahlen, J.; Moens, B.; de Vos, D.E.; Alsters, P.L.; Jacobs, P.A. Titanium silicalite 1 (TS-1) catalyzed oxidative transformations of furan derivatives with hydrogen peroxide. Adv. Synth. Catal. 2004, 346, 333–338. [Google Scholar] [CrossRef]
- Taniguchi, T.; Nakamura, K.; Ogasawara, K. Non-carbohydrate route to levoglucosenone and its enantiomer employing asymmetric dihydroxylation. Synlett 1996, 971–972. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ohnishi, H.; Ogasawara, K. An expedient preparation of chiral building blocks having levoglucosenone chromophore: A new enantiocontrolled route to (−)-β-multistriatin and (+)-exo-brevicomin. Chem. Commun. 1996, 1477–1478. [Google Scholar] [CrossRef]
- Harris, J.M.; Keranen, M.D.; O’Doherty, G.A. Syntheses of d- and l-mannose, gulose, and talose via diastereoselective and enantioselective dihydroxylation reactions. J. Org. Chem. 1999, 64, 2982–2983. [Google Scholar] [CrossRef] [PubMed]
- Deagostino, A.; Prandi, C.; Zavattaro, C.; Venturello, P. Functionalized 1-alkoxy-1,3-dienes: Their preparation and applications in synthetic organic chemistry. Eur. J. Org. Chem. 2006, 2463–2483. [Google Scholar] [CrossRef]
- Eftekhari-Sis, B.; Zirak, M. Chemistsry of α-oxoesters: A powerful tool for the synthesis of heterocycles. Chem. Rev. 2015, 115, 151–264. [Google Scholar] [CrossRef] [PubMed]
- Frihen, T.G.; Bols, M.; Pedersen, C.M. Synthesis of l-hexoses. Chem. Rev. 2015. [Google Scholar] [CrossRef]
- Bednarski, M.; Danishefsky, S. Interactivity of chiral catalysts and chiral auxiliaries in the cycloaddition of activated dienes with aldehydes: A synthesis of l-glucose. J. Am. Chem. Soc. 1986, 108, 7060–7067. [Google Scholar] [CrossRef]
- For a recent example of Danishefsky’s diene in the preparation of carbohydrate mimetics, see: Burland, P.A.; Coisson, D.; Osborn, H.M.I. Rapid synthesis of carbohydrate derivatives, including mimetics of C-linked disaccharides and C-linked aza disaccharides, using the hetero-Diels-Alder reaction. J. Org. Chem. 2010, 75, 7210–7218.
- Angerbauer, R.; Schmidt, R.R. Short synthesis of racemic uronic acids and 2,3-anhydrouronic acids. Carbohydr. Res. 1981, 89, 159–162. [Google Scholar] [CrossRef]
- Bataille, C.; Begin, G.; Guillam, A.; Lemiegre, L.; Lys, C.; Maddaluno, J.; Toupet, L. Thermal/hyperbaric heterocycloaddition of 1,4-dialkoxy-1,3-dienes: The de novo (E,Z) way to sugars. J. Org. Chem. 2002, 67, 8054–8062. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Lubineau, A.; Guillot, R.; Scherrmann, M.-C. Synthesis of C-disaccharides via a hetero-Diels-Alder reaction and further stereocontrolled transformations. Carbohydr. Res. 2008, 343, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Castagnolo, D.; Botta, L.; Botta, M. One-pot multicomponent synthesis of 2,3-dihydropyrans: New access to furanose-pyranose 1,3-C-C-linked-disaccharides. Tetrahedron Lett. 2009, 50, 1526–1528. [Google Scholar] [CrossRef]
- Lopez, J.C.; Plumet, J. Metathesis reactions of carbohydrates: Recent highlights in alkyne metathesis. Eur. J. Org. Chem. 2011, 2011, 1803–1825. [Google Scholar] [CrossRef]
- Aljarilla, A.; Lopez, J.C.; Plumet, J. Metathesis reactions of carbohydrates: Recent highlights in cross-metathesis. Eur. J. Org. Chem. 2010, 2010, 6123–6143. [Google Scholar] [CrossRef]
- Crimmins, M.T.; King, B.W.; Zuercher, W.J.; Choy, A.L. An efficient, general asymmetric synthesis of carbocyclic nucleosides: Application of an asymmetric aldol/ring-closing metathesis strategy. J. Org. Chem. 2000, 65, 8499–8509. [Google Scholar] [CrossRef] [PubMed]
- Guaragna, A.; D’Alonzo, D.; Paolella, C.; Napolitano, C.; Palumbo, G. Highly stereoselective de novo synthesis of l-hexoses. J. Org. Chem. 2010, 75, 3558–3568. [Google Scholar] [CrossRef] [PubMed]
- Guaragna, A.; Napolitano, C.; D’Alonzo, D.; Pedatella, S.; Palumbo, G. A versatile route to l-hexoses: Synthesis of l-mannose and -altrose. Org. Lett. 2006, 8, 4863–4866. [Google Scholar] [CrossRef] [PubMed]
- Guppi, S.R.; Zhou, M.; O’Doherty, G.A. De novo asymmetric synthesis of homoadenosine via a palladium-catalyzed N-glycosylation. Org. Lett. 2006, 8, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Groebke, K.; Hunziker, J.; Fraser, W.; Peng, L.; Diederichsen, U.; Zimmermann, K.; Holzner, A.; Leumann, C.; Eschenmoser, A. Why pentose- and not hexose-nucleic acids? Purine-purine pairing in homo-DNA: Guanine, isoguanine, 2,6-diaminopurine, and xanthine. Helv. Chim. Acta 1998, 81, 375–474. [Google Scholar] [CrossRef]
- Hooper, I.R. The naturally occurring aminoglycoside antibiotics. In Aminoglycoside Antibiotics; Hooper, I.R., Umezawa, H., Eds.; Springer-Verlag: New York, NY, USA, 1982. [Google Scholar]
- Zhang, G.; Shi, L.; Liu, Q.; Wang, J.; Li, L.; Liu, X. A divergent strategy for constructing a sugar library containing 2,6-dideoxy sugars and uncommon sugars with 4-substitution. Tetrahedron 2007, 63, 9705–9711. [Google Scholar] [CrossRef]
- Haukaas, M.H.; O’Doherty, G.A. Enantioselective synthesis of 2-deoxy- and 2,3-dideoxyhexoses. Org. Lett. 2002, 4, 1771–1774. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.V.; O’Brien, J.L.; Smith, A.B., III. Stereospecific synthesis of β-d-allopyranosides by dihydroxylation of β-d-erythro-2,3-dideoxyhex-2-enopyranosides. Carbohydr. Res. 2001, 334, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.; Norman, S.E.; Osborn, H.M.I. Synthesis of S-linked carbohydrate analogues via a Ferrier reaction. Tetrahedron 2008, 64, 2832–2854. [Google Scholar] [CrossRef]
- Babu, R.S.; Chen, Q.; Kang, S.-W.; Zhou, M.; O’Doherty, G.A. De novo asymmetric synthesis of all-d-, all-l-, and d-/l oligosaccharides using atom-less protecting groups. J. Am. Chem. Soc. 2012, 134, 11952–11955. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, R.J.; Prasad, N. Unsaturated carbohydrates. Part X. Epoxidations and hydroxylations of 2,3-dideoxy-α-d-hex-2-enopyranosides. The four methyl 4,6-di-O-acetyl-2,3-anhydro-α-d-hexopyranosides. J. Chem. Soc. C 1969, 575–580. [Google Scholar] [CrossRef]
- Angyal, S.J. Ring-opening of anhydrosugars of the ethylene oxide type. Chem. Ind. 1954, 1230–1231. [Google Scholar]
- Joly, J.-P.; Roze, F.; Banas, S.; Quilès, F. Synthesis and Raman spectra of 3-deoxy-α-l-rhamnosides as model sugars of the Ascaris egg shell. Tetrahedron Lett. 2010, 51, 3236–3241. [Google Scholar] [CrossRef]
- Sakakibara, T.; Yamada, M.; Sudoh, R. Phase transfer catalyzed reactions. II. Reactions of methyl 3-deoxy-3-nitro-β-d-hexopyranosides with active methylene compounds. J. Org. Chem. 1976, 41, 736–737. [Google Scholar] [CrossRef]
- Sakakibara, T.; Sudoh, R. Phase transfer catalyzed reactions. I. Highly stereoselective formation of the thermodynamically less stable manno isomers from nitro sugars with active methylene compounds. J. Org. Chem. 1975, 40, 2823–2825. [Google Scholar] [CrossRef]
- Sakakibara, T.; Tokuda, K.; Hayakawa, T.; Seta, A. Michael reactions on conformationally flexible methyl 3-C-nitro-hex-2-enopyranoside derivatives. Carbohydr. Res. 2000, 327, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Sakakibara, S. Kumazawa. Tetrahedron Lett. 1970, 11, 1645–1648. [Google Scholar] [CrossRef]
- Rajaebalee, F.J.-M. Synthèse de dérivés amines du méthyl-3-desoxy-3-nitro-α- et β-d-glucopyranoside. Carbohydr. Res. 1973, 26, 219–224. [Google Scholar] [CrossRef]
- Sakakibara, T.; Sudoh, R. Stereochemistry of nucleophilic addition reactions. 2. Kinetically controlled reaction of methyl 4,6-O-benzylidene-2,3-dideoxy-3-nitro-β-d-erythro-hex-2-enopyranoside with hydrogen cyanide. Important role of electrostatic interaction. J. Org. Chem. 1977, 42, 1746–1750. [Google Scholar] [CrossRef]
- Sakakibara, T.; Sudoh, R. Stereoselective synthesis of the thermodynamically less stable manno isomers from a nitro sugar. Carbohydr. Res. 1976, 50, 191–196. [Google Scholar] [CrossRef]
- Jegou, E.; Cleophax, J.; Leboul, J.; Gero, S.D. A facile synthesis of derivatives of lividosamine, a component of lividomycin B. Carbohydr. Res. 1975, 45, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Apostolopoulos, C.D.; Couladouros, E.A.; Georgiadis, M.P. Synthesis of sugar-amino acid conjugates via 2,3-dideoxy-hex-2-enopyranos-4-uloses. Liebigs Ann. Chem. 1994, 781–784. [Google Scholar] [CrossRef]
- Couladouros, E.A.; Constantinou- Kokotou, V.; Georgiadis, M.P.; Kokotos, G. A synthesis in one reaction vessel of 2,4-diamino sugar precursors from 2,3-dideoxy-2-enopyranos-4-uloses. Carbohydr. Res. 1994, 254, 317–324. [Google Scholar] [CrossRef]
- Sanki, A.K.; Bhattacharya, R.; Atta, A.K.; Suresh, C.G.; Pathak, T. Diastereoselective addition of planar N-heterocycles to vinyl sulfone-modified carbohydrates: A new route to isonucleosides. Tetrahedron 2008, 64, 10406–10416. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Kesarwani, M.K.; Manna, C.; Ganguly, B.; Suresh, C.G.; Pathak, T. An experimental and theoretical study on the remarkable influence of protecting groups on the selectivity of addition of amines to vinyl sulfone-modified hex-2-enopyranosides. J. Org. Chem. 2010, 75, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, B.; Sakthivel, K.; Suresh, C.G.; Pathak, T. Diastereoselective addition of amines to vinyl sulfone modified carbohydrates: A highly flexible methodology for the synthesis of new classes of deoxyaminosugars. J. Org. Chem. 2000, 65, 2637–2641. [Google Scholar] [CrossRef] [PubMed]
- Sanki, A.K.; Suresh, C.G.; Falgune, U.D.; Pathak, T. Anomeric configuration-directed diastereoselective C-C bond formation in vinyl sulfone-modified carbohydrates: A general route to branched-chain sugars. Org. Lett. 2003, 5, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.R.; Atta, A.K.; Dey, D.; Pathak, T. Densely functionalized chiral pyrroles from endocyclic, exocyclic, and acyclic vinyl sulfone-modified carbohydrates. J. Org. Chem. 2009, 74, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Jayaraman, N. Facial selectivities in the nucleophilic additions of 2,3-unsaturated 3-arylsulfinyl pyranosides. Carbohydr. Res. 2013, 380, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Cai, S.; William, R.; Liu, X.-W. Pathways leading to 3-amino- and 3-nitro-2,3-dideoxy sugars: Strategies and synthesis. RSC Adv. 2013, 3, 13594–13621. [Google Scholar] [CrossRef]
- Pauls, H.W.; Fraser-Reid, B. An efficient synthesis of ristosamine utilizing the allylic hydroxyl of an hex-2-enopyranoside. J. Org. Chem. 1983, 48, 1392–1393. [Google Scholar] [CrossRef]
- Bongini, A.; Cardillo, G.; Orena, M.; Sandri, S.; Tomasini, C. A regio- and stereoselective synthesis of methyl α-l-ristosaminide hydrochloride. Tetrahedron 1983, 39, 3801–3806. [Google Scholar] [CrossRef]
- Pauls, H.W.; Fraser-Reid, B. Stereocontrolled routes to cis-hydroxyamino sugars, part VII: Synthesis of daunosamine and ristosamine. Carbohydr. Res. 1986, 150, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Baran, P.S.; Zhong, Y.-L.; Barluenga, S.; Hunt, K.W.; Kranich, R.; Vega, J.A. Iodine(V) reagents in organic synthesis. Part 3. New routes to heterocyclic compounds via o-iodoxybenzoic acid-mediated cyclizations: Generality, scope, and mechanism. J. Am. Chem. Soc. 2002, 124, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Shibata, K.; Kinbara, S.; Takahashi, T. A divergent route to 3-amino-2,3,6-trideoxysugars including branched sugar: Synthesis of vancosamine, daunosamine, saccharosamine, and ristosamine. Chem. Lett. 2007, 36, 1372–1373. [Google Scholar] [CrossRef]
- Dorgan, B.J.; Jackson, R.F.W. Synthesis of C-linked glycosyl amino acid derivatives using organozinc reagents. Synlett 1996, 859–861. [Google Scholar] [CrossRef]
- Chapleur, Y.; Grapsas, Y. Stereospecific formation of carbon-carbon bonds in ethyl 4,6-di-O-acetyl-2,3-dideoxy-α-d-ribo-hex-3-enopyranoside. Carbohydr. Res. 1985, 141, 153–158. [Google Scholar] [CrossRef]
- Danishefsky, S.J.; Armistead, D.M.; Wincott, F.E.; Selnick, H.G.; Hungate, R. The total synthesis of Avermectin A1a. J. Am. Chem. Soc. 1989, 111, 2967–2980. [Google Scholar] [CrossRef]
- Valverde, S.; Bernabé, M.; Garcia-Ochoa, S.; Gómez, A.M. Regio- and stereochemistry of cross coupling of organocopper reagents with allyl ethers: Effect of the leaving group. J. Org. Chem. 1990, 55, 2294–2298. [Google Scholar] [CrossRef]
- Valverde, S.; Bernabe, M.; Gomez, A.M.; Puebla, P. Cross coupling reactions of 2-(allyloxy(thio))benzothiazoles with organocopper reagents in dihydropyranoid systems. Mechanistic implications of the substrate and the reagent: Regio- and stereocontrolled access to branched-chain sugars. J. Org. Chem. 1992, 57, 4546–4550. [Google Scholar] [CrossRef]
- Hyodo, T.; Katayama, Y.; Kobayashi, Y. Allylic substitution on the pyran ring. Tetrahedron Lett. 2009, 50, 3547–3549. [Google Scholar] [CrossRef]
- RajanBabu, T.V. Pd(0)-Catalyzed C-glycosylation: A facile alkylation of trifluoroacetylglucal. J. Org. Chem. 1985, 50, 3642–3644. [Google Scholar] [CrossRef]
- Dunkerton, L.V.; Euske, J.M.; Serino, A.J. Palladium(0)-assisted synthesis of C-glycopyranosyl compounds. Carbohydr. Res. 1987, 171, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; Suh, Y.-G. Selective mono-Claisen rearrangement of carbohydrate glycals. A chemical consequence of the vinylogous anomeric effect. Carbohydr. Res. 1987, 171, 161–191. [Google Scholar] [CrossRef] [PubMed]
- Brakta, M.; Lhoste, P.; Sinou, D. Palladium(0)-based approach to functionalized C-glycopyranosides. J. Org. Chem. 1989, 54, 1890–1896. [Google Scholar] [CrossRef]
- Engelbrecht, G.J.; Holzapfel, C.W. Palladium-catalyzed reactions of unsaturated carbohydrates—A route to C-glycosides. Heterocycles 1991, 32, 1267–1272. [Google Scholar] [CrossRef]
- Brescia, M.-R.; Shimshock, Y.C.; DeShong, P. Regioselectivity in the palladium-catalyzed addition of carbon nucleophiles to dihydropyran derivatives. J. Org. Chem. 1997, 62, 1257–1263. [Google Scholar] [CrossRef]
- Yeager, A.R.; Min, G.K.; Porco, J.A.; Schaus, S.E. Exploring skeletal diversity via ring contraction of glycal-derived scaffolds. Org. Lett. 2006, 8, 5065–5068. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, F.J.B.; dos Anjos, J.V.; Sinou, D.; de Melo, S.J.; Srivastava, R.M. Palladium-catalyzed alkynylation (Sonogashira coupling) at C-5 of the uracil moiety in modified unsaturated pyranosyl nucleosides. Synthesis 2007, 1890–1897. [Google Scholar]
- De Oliveira, R.N.; Cottier, L.; Sinou, D.; Srivastava, R.M. Stereocontrolled palladium(0)-catalyzed preparation of unsaturated azidosugars: An easy access to 2- and 4-aminoglycosides. Tetrahedron 2005, 61, 8271–8281. [Google Scholar] [CrossRef]
- Mrozowski, R.M.; Sansuaky, Z.M.; Vemula, R.; Wu, B.; Zhang, Q.; Lannigan, D.A.; O’Doherty, G.A. De novo synthesis and biological evaluation of C6''-substituted C4'' amide analogues of SL0101. Org. Lett. 2014, 16, 5996–5999. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, R.J.; Vethaviyasar, N. Unsaturated carbohydrates. XVII. Synthesis of branched-chain sugar derivatives by the Claisen rearrangement. J. Chem. Soc. Perkin Trans 1 1973, 1791–1793. [Google Scholar] [CrossRef]
- Krohn, K.; Flörke, U.; Gehle, D. Highly deoxygenated sugars. I. C2-Branched glucose derivatives and carbon linked deoxygenated disaccharides. J. Carbohydr. Chem. 2002, 21, 431–443. [Google Scholar] [CrossRef]
- Montero, A.; Mann, E.; Herradon, B. Preparation of sugar amino acids by Claisen-Johnson rearrangement: Synthesis and incorporation into enkephalin analogues. Eur. J. Org. Chem. 2004, 2004, 3063–3073. [Google Scholar] [CrossRef]
- Kriek, N.M.A.J.; van der Hout, E.; Kelly, P.; van Meijgaarden, K.E.; Geluk, A.; Ottenhoff, T.H.M.; van der Marel, G.A.; Overhand, M.; van Boom, J.H.; Valentijn, A.R.P.M.; et al. Synthesis of novel tetrahydropyran-based dipeptide isosters by Overman rearrangement of 2,3-didehydroglycosides. Eur. J. Org. Chem. 2003, 2003, 2418–2427. [Google Scholar] [CrossRef]
- Montero, A.; Mann, E.; Herradon, B. The Overman rearrangement in carbohydrate chemistry: Stereoselective synthesis of functionalized 3-amino-3,6-dihydro-2H-pyrans and incorporation in peptide derivatives. Tetrahedron Lett. 2005, 46, 401–405. [Google Scholar] [CrossRef]
- Montero, A.; Benito, E.; Herradon, B. Synthesis and applications of a chiral-oxygenated 3-chloro-3,6-dihydro-2H-pyran obtained under Overman rearrangement conditions. Tetrahedron Lett. 2010, 51, 277–280. [Google Scholar] [CrossRef]
- Saquib, M.; Husain, I.; Sharma, S.; Yadav, G.; Singh, V.K.; Sharma, S.K.; Shah, P.; Siddiqi, M.I.; Kumar, B.; Lal, J.; et al. 2,3-Dideoxy hex-2-enopyranosid-4-uloses as promising new anti-tubercular agents: Design, synthesis, biological evaluation and SAR studies. Eur. J. Med. Chem. 2011, 2217–2223. [Google Scholar] [CrossRef]
- Saquib, M.; Husain, I.; Kumar, B.; Shaw, A.K. Facile synthesis of enantiomerically pure 2- and 2,3-disubstituted furans catalysed by mixed Lewis acids: An easy route to 3-iodofurans and 3-(hydroxymethyl)furans. Chem. Eur. J. 2009, 15, 6041–6049. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, G.; Cano, C.; Mauzé, C. Regio- and enantioselective synthesis of novel functionalized pyrano-pyrrolidines by 1,3-dipolar cycloaddition of carbohydrates. Synlett 2005, 587–590. [Google Scholar] [CrossRef]
- Filho, J.R.F.; Srivastava, R.M.; da Silva, W.J.P.; Cottier, L.; Sinou, D. Synthesis of new branched-chain amino sugars. Carbohydr. Res. 2003, 338, 673–680. [Google Scholar] [CrossRef] [PubMed]
- By, K.; Kelly, P.A.; Kurth, M.J.; Olmstead, M.M.; Nantz, M.H. Synthesis of a C(4)-C(9) eleutheside template from d-glucal. Tetrahedron 2001, 57, 1183–1187. [Google Scholar] [CrossRef]
- Taillefumier, C.; Chapleur, Y. Enantiomerically pure decalinic structures from carbohydrates using intramolecular Diels-Alder and Ferrier carbocyclization. Can. J. Chem. 2000, 78, 708–722. [Google Scholar] [CrossRef]
- Gomez, A.M.; Lopez, J.C.; Fraser-Reid, B. Stereoselective synthesis of ethyl (Z)- and (E)-octa-2,6-dienopyranosideuronates from ethyl 2,3-dideoxy-α-d-erythro-hex-2-eno-pyranoside. Synlett 1993, 557–560. [Google Scholar] [CrossRef]
- Corey, E.J.; Kim, C.U. Improved synthetic routes to prostaglandins utilizing sulfide-mediated oxidation of primary and aecondary alcohols. J. Org. Chem. 1973, 38, 1233–1234. [Google Scholar] [CrossRef] [PubMed]
- Fraser-Reid, B.; Molino, B.F.; Magdzinski, L.; Mootoo, D.R. Tripyranoside precursors for ansamycins. Pyranosidic homologation. 6. J. Org. Chem. 1987, 52, 4505–4511. [Google Scholar] [CrossRef]
- Fraser-Reid, B.; Magdzinski, L.; Molino, B.F. New strategy for carbohydrate-based syntheses of multichiral arrays: Pyranosidic homologation. 3. J. Am. Chem. Soc. 1984, 106, 731–734. [Google Scholar] [CrossRef]
- Molino, B.F.; Magdzinski, L.; Fraser-Reid, B. Pyranosidic homologation: Part I: Extending the carbohydrate template via C6 and C4. Tetrahedron Lett. 1983, 24, 5819–5822. [Google Scholar] [CrossRef]
- Gomez, A.M.; Lopez de Uralde, B.; Valverde, S.; Lopez, J.C. A novel entry to naturally occurring 5-alkenyl α,β-unsaturated δ-lactones from d-glucose: Syntheses of (+)-acetylphomalactone and (+)-asperlin. Chem. Commun. 1997, 1647–1648. [Google Scholar] [CrossRef]
- Mieczkowski, J.; Jurczak, J.; Chmielewski, M.; Zamojski, A. The synthesis of 2,3-dideoxyhex-2-enono-1.5 lactones. Carbohydr. Res. 1977, 56, 180–182. [Google Scholar] [CrossRef]
- Bernasconi, C.; Cottier, L.; Descotes, G.; Remy, G. A related oxidation from hex-2-enopyranosides via photolysis of the corresponding 2-oxopropyl glycosides was described. Bernasconi, C.; Cottier, L.; Descotes, G.; Remy, G. Dégradations ménagées des sucres. Synthese par voie photochimique d’aldonolactones-l,5 a partir d’oxo-2 propylglycosides. Bull. Soc. Chim. Fr. 1979, 332–336. [Google Scholar]
- Jarglis, P.; Lichtenthaler, F.W. Boron trifluoride-catalyzed oxidation of glycal esters: An effective and mild method for their conversion into α,β-unsaturated lactones. Tetrahedron Lett. 1982, 23, 3781–3784. [Google Scholar] [CrossRef]
- Rollin, P.; Sinaÿ, P. A convenient, one-step oxidation of glycals to lactones using pyridinium chlorochromate. Carbohydr. Res. 1981, 98, 139–142. [Google Scholar] [CrossRef]
- Panfil, I.; Chmielewski, M. Cycloaddition of nitrones and α,β-unsaturated sugar lactones. Tetrahedron 1985, 41, 4713–4716. [Google Scholar] [CrossRef]
- Panfil, I.; Belzecki, C.; Urbanczyk-Lipkowska, Z.; Chmielewski, M. 1,3-Dipolar cycloaddition of nitrones to sugar enlactones. Tetrahedron 1991, 47, 10087–10094. [Google Scholar] [CrossRef]
- Jurczak, M.; Rabiczko, J.; Socha, D.; Chmielewski, M.; Cardona, F.; Goti, A.; Brandi, A. Diastereoselection in 1,3-dipolar cycloadditions of a chiral cyclic nitrone to α,β-unsaturated δ-lactones. Tetrahedron Asymmetry 2000, 11, 2015–2022. [Google Scholar] [CrossRef]
- Socha, D.; Jurczak, M.; Frelek, J.; Klimek, A.; Rabiczko, J.; Urbanczyk-Lipkowska, Z.; Suwinska, K.; Chmielewski, M.; Cardona, F.; Goti, A.; et al. 1,3-Dipolar cycloaddition of a nitrone derived from (S)-malic acid to α,β-unsaturated δ-lactones. Tetrahedron Asymm. 2001, 12, 3163–3172. [Google Scholar] [CrossRef]
- Lopez, J.C.; Lameignere, E.; Lukacs, G. Stereospecificity in Diels-Alder reactions of dienes and dienophiles derived from methyl 4,6-O-benzylidene-α-d-glucopyranoside. J. Chem. Soc. Chem. Commun. 1988, 706–707. [Google Scholar] [CrossRef]
- Lopez, J.C.; Lameignere, E.; Lukacs, G. Straightforward route to 2- and 3-formyl hex-1- and -2-enopyranosides and their highly stereoselective hetero Diels-Alder reaction with ethyl vinyl ether. J. Chem. Soc. Chem. Commun. 1988, 514–515. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Nguyen, S.L.; Elworthy, T.R. Preparation and Diels-Alder reactions of a pyranoid vinyl glycal: Model studies for anthraquinone aglycone and carbohydrate syntheses. Tetrahedron 1988, 44, 3355–3364. [Google Scholar] [CrossRef]
- Stecko, S.; Jurczak, M.; Panfil, I.; Furman, B.; Grzeszczyk, B.; Chmielewski, M. Synthesis of iminosugars via 1,3-dipolar cyloaddition reactions of nitrones to α,β-unsaturated sugar aldonolactones. Comptes Rendus Chim. 2011, 14, 102–125. [Google Scholar] [CrossRef]
- Pasniczek, K.; Socha, D.; Jurczak, M.; Frelek, J.; Suszczynska, A.; Urbanczyk-Lipkowska, Z.; Chmielewski, M. Double asymmetric induction in 1,3-dipolar cycloaddition of nitrones to 2,3-unsaturated sugar 1,5-lactones. J. Carbohydr. Chem. 2003, 22, 613–629. [Google Scholar] [CrossRef]
- Pasniczek, K.; Socha, D.; Jurczak, M.; Solecka, J.; Chmielewski, M. Synthesis of 8-homocastanospermine. Can. J. Chem. 2006, 84, 534–539. [Google Scholar] [CrossRef]
- Socha, D.; Pasniczek, K.; Jurczak, M.; Solecka, J.; Chmielewski, M. Synthesis of 1-homoaustraline. Carbohydr. Res. 2006, 341, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Pasniczek, K.; Jurczak, M.; Urbanczyk-Lipkowska, Z.; Chmielewski, M. Synthesis of 2,3-dihydroxy-1-epilupinine. J. Carbohydr. Chem. 2007, 26, 195–211. [Google Scholar] [CrossRef]
- Panfil, I.; Urbanczyk-Lipkowska, Z.; Chmielewski, M. Synthesis of iminosugars from α,β-unsaturated lactones and N-benzyl nitrone. Pol. J. Chem. 2005, 79, 239–249. [Google Scholar]
- Panfil, I.; Solecka, J.; Chmielewski, M. Synthesis of (−)-isofagomine. J. Carbohydr. Chem. 2006, 25, 673–684. [Google Scholar] [CrossRef]
- Stecko, S.; Pasniczek, K.; Jurczak, M.; Urbanczyk-Lipkowska, Z.; Chmielewski, M. Kinetic and thermodynamic aspects in the 1,3-dipolar cycloaddition of five-membered cyclic nitrones to α,β-unsaturated γ- and δ-lactones. Tetrahedron Asymmetry 2007, 18, 1085–1093. [Google Scholar] [CrossRef]
- Testero, S.A.; Spanevello, R.A. Enantiospecific approach toward pentalenolactone. Org. Lett. 2006, 8, 3793–3796. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.C.; Lameignere, E.; Burnouf, C.; Laborde, M.A.; Ghini, A.; Olesker, A.; Lukacs, G. Efficient routes to pyranosidic homologated conjugated enals and dienes from monosaccharides. Tetrahedron 1993, 49, 7701–7722. [Google Scholar] [CrossRef]
- Testero, S.A.; Spanevello, R.A. The first example of a highly non-symmetric ozonolysis of a sugar derived norbornene system. ARKIVOC 2003, 220–226. [Google Scholar] [CrossRef]
- Mangione, M.I.; Testero, S.A.; Suarez, A.G.; Spanevello, R.A.; Tuchages, J.-P. Synthesis and spectroscopic NMR studies of a highly stable cross-ozonide product derived from a carbohydrate system. Tetrahedron Assymetry 2006, 17, 1780–1785. [Google Scholar] [CrossRef]
- Testero, S.A.; Mangione, M.I.; Poeylaut-Palena, A.; González-Sierra, M.; Spanevello, R.A. Unsymmetrical ozonolysis of carbohydrate derived norbornene systems. Tetrahedron 2007, 63, 11410–11420. [Google Scholar] [CrossRef]
- Mangione, M.I.; Suarez, A.G.; Spanevello, R.A. Synthesis of methyl 4,6-O-benzylidene-2,3-dideoxy-2-C-formyl-α-d-erythro-hex-2-enopyranoside. Carbohydr. Res. 2005, 340, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Mangione, M.I.; Sarotti, A.M.; Suarez, A.G.; Spanevello, R.A. Experimental and theoretical study of a Diels-Alder reaction between a sugar-derived nitroalkene and cyclopentadiene. Carbohydr. Res. 2011, 346, 460–464. [Google Scholar] [CrossRef] [PubMed]
- For an excellent review on transition metal-catalyzed glycosylations, see McKay, M.J.; Nguyen, H.M. Recent advances in transition metal-catalyzed glycosylation. ACS Catal. 2012, 2, 1563–1595.
- Van der Deen, H.; van Oeveren, A.; Kellogg, R.M.; Feringa, B.L. Palladium catalyzed stereospecific allylic substitution of 5-acetoxy-2(5H)-furanone and 6-acetoxy-2H-pyran3(6H)-one by alcohols. Tetrahedron Lett. 1999, 40, 1755–1758. [Google Scholar] [CrossRef]
- Comely, A.C.; Eelkema, R.; Minnaard, A.J.; Feringa, B.L. De novo asymmetric bio- and chemocatalytic synthesis of saccharides—Stereoselective formal O-glycoside bond formation using palladium catalysis. J. Am. Chem. Soc. 2003, 125, 8714–8715. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.S.; O’Doherty, G.A. A palladium-catalyzed glycosylation reaction: The de novo synthesis of natural and unnatural glycosides. J. Am. Chem. Soc. 2003, 125, 12406–12407. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Men, H.; Lee, C. Stereoselective palladium-catalyzed O-glycosylation using glycals. J. Am. Chem. Soc. 2004, 126, 1336–337. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, C. A mild and efficient method for the stereoselective formation of C-O bonds: Palladium-catlyzed allylic etherification using zinc(II) alkoxides. Org. Lett. 2002, 4, 4369–4371. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Keranen, M.D.; Nguyen, H.; Young, V.G.; O’Doherty, G.A. Syntheses of four d- and l-hexoses via diastereoselective and enantioselective dihydroxylation reactions. Carbohydr. Res. 2000, 328, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid-triethylamine mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Babu, R.S.; Zhou, M.; O’Doherty, G.O. De novo synthesis of oligosaccharides using a palladium-catalyzed glycosylation reaction. J. Am. Chem. Soc. 2004, 126, 3428–3429. [Google Scholar] [CrossRef] [PubMed]
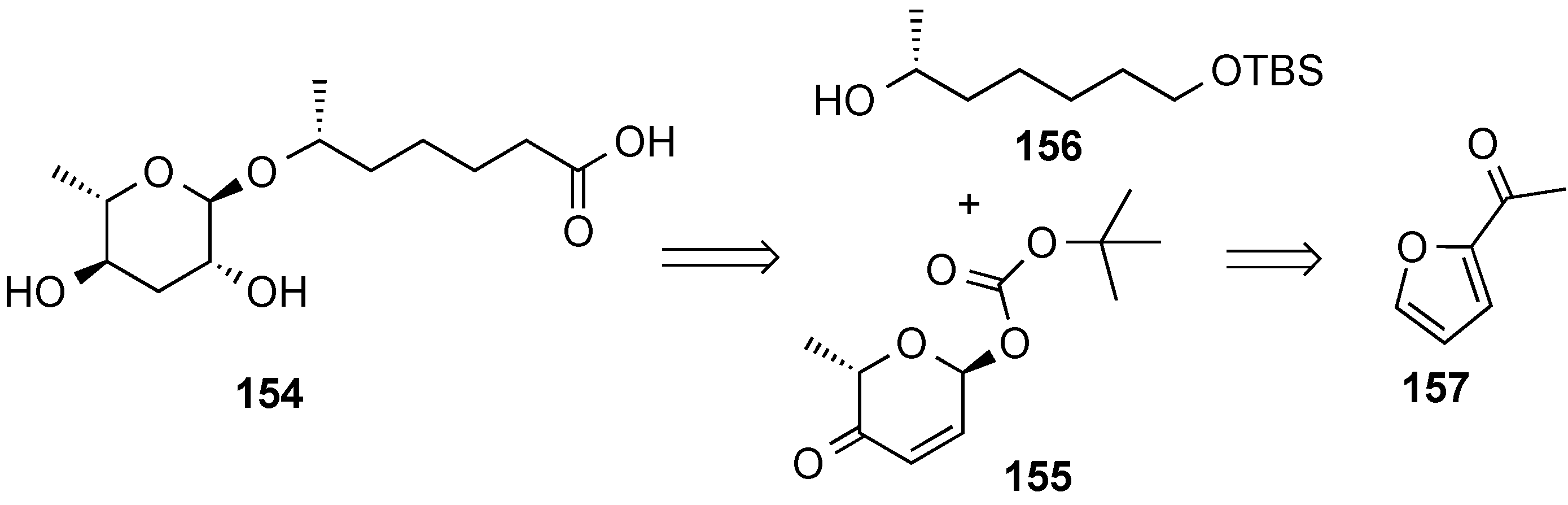
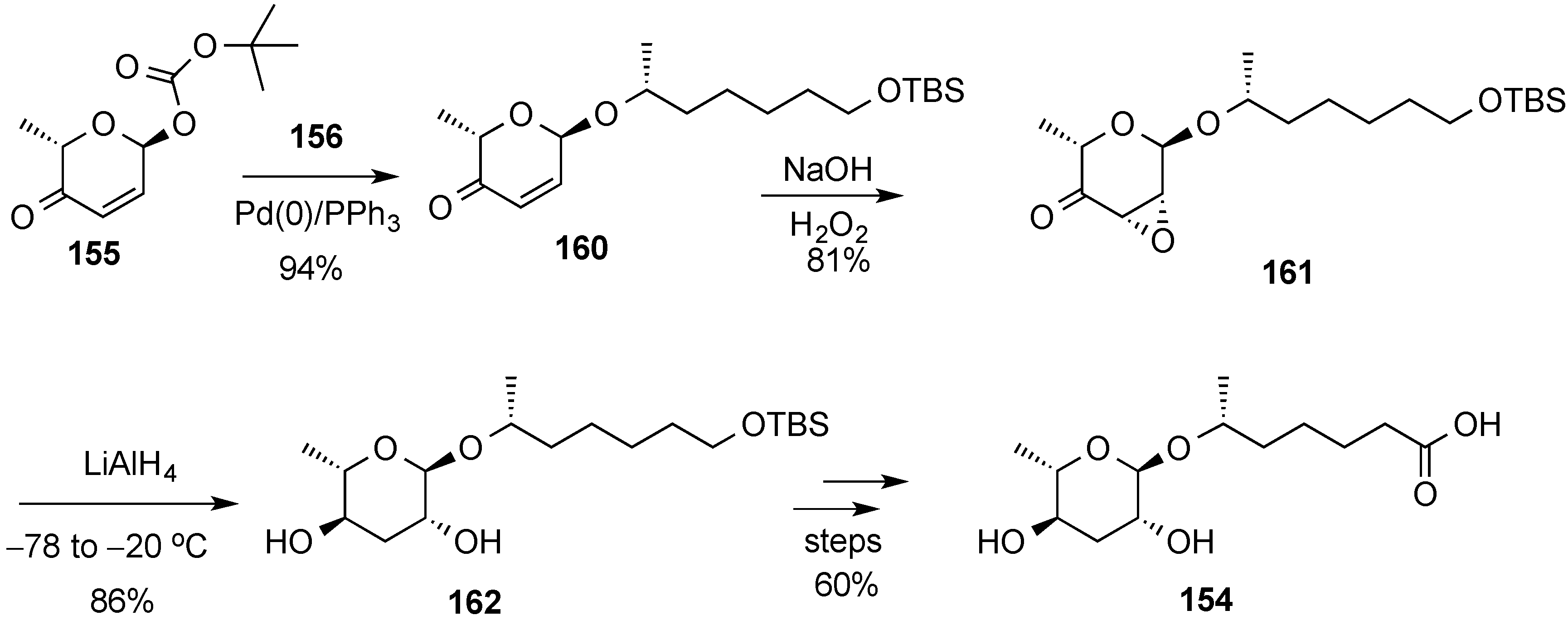
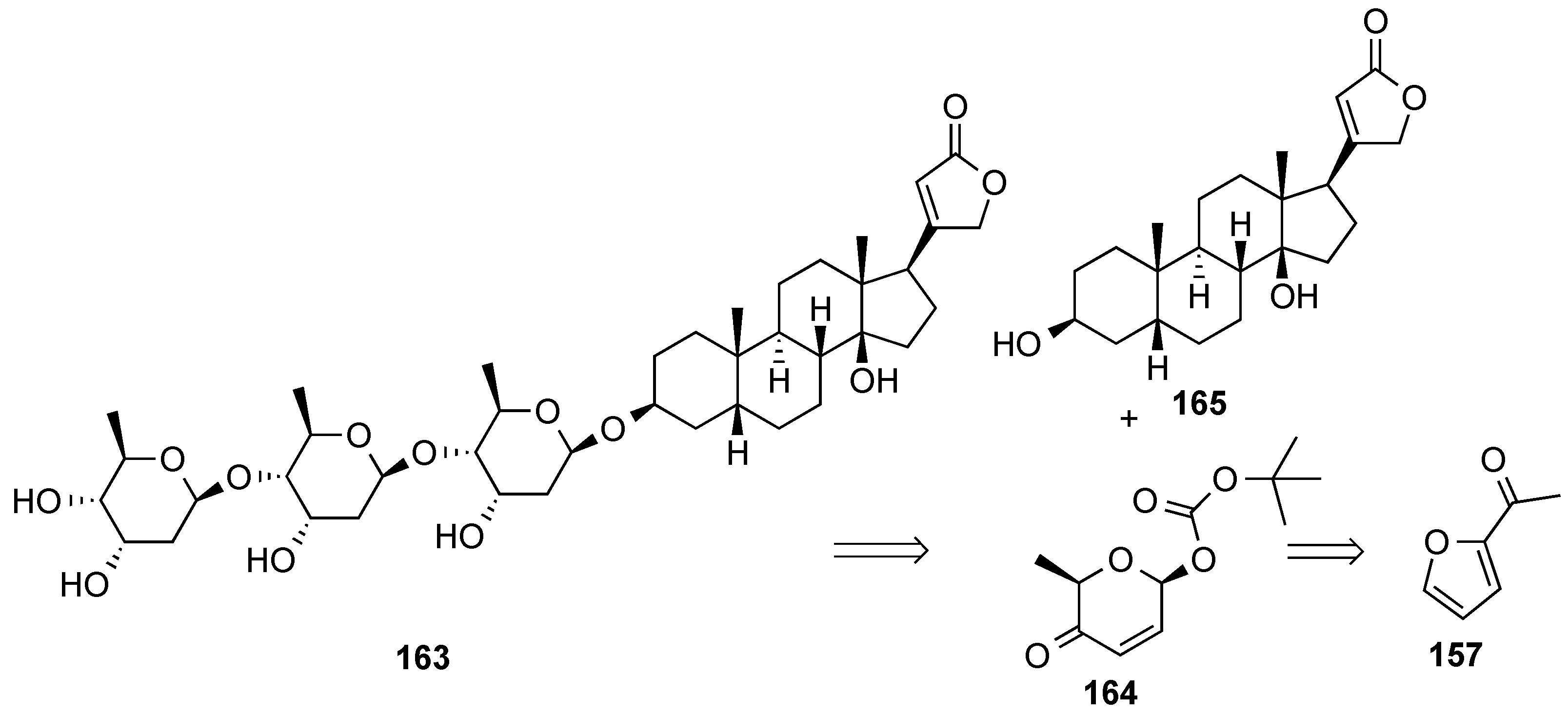
- Zhou, M.; O’Doherty, G.A. A stereoselective synthesis of digitoxin and digitoxigen mono- and bisdigitoxoside from digitoxigenin via a palladium-catalyzed glycosylation. Org. Lett. 2006, 8, 4339–4342. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; O’Doherty, G.A. De Novo Approach to 2-Deoxy-β-glycosides: Asymmetric Syntheses of Digoxose and Digitoxin. J. Org. Chem. 2007, 72, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of anthrax tetrasaccharide by a palladium-catlyzed glycosylation reaction. Angew. Chem. Int. Ed. Engl. 2007, 46, 5206–5208. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of anthrax tetrasaccharide and related tetrasaccharide. J. Org. Chem. 2008, 73, 5211–5220. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.L.; Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of rhamno di- and tri-saccharides related to the anthrax tetrasaccharide. Tetrahedron 2013, 69, 3432–3436. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; O’Doherty, G.A. De novo synthesis of the trisaccharide subunit of landomycins A and E. Org. Lett. 2008, 10, 2283–2286. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Li, M.; O’Doherty, G.A. Synthesis of several cleistrioside and cleistetroside natural products via a divergent de novo asymmetric approach. Org. Lett. 2010, 12, 5466–5469. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; O’Doherty, G.A. De novo asymmetric syntheses of SL0101 and its analogues via a palladium-catalyzed glycosylation. Org. Lett. 2006, 8, 5149–5152. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Sharif, E.U.; O’Doherty, G.A. Total synthesis of Jadomycin A and a carbasugar analogue of jadomycin B. Angew. Chem. Int. Ed. Engl. 2010, 49, 9492–9495. [Google Scholar] [CrossRef] [PubMed]
- Sharif, E.U.; O’Doherty, G.A. Biosynthesis and total synthesis studies on the jadomycin family of natural products. Eur. J. Org. Chem. 2012, 2095–2108. [Google Scholar] [CrossRef]
- Chen, Q.; Zhong, Y.; O’Doherty, G.A. Convergent de novo synthesis of vineomycinone B2 methyl ester. Chem. Commun. 2013, 49, 6806–6808. [Google Scholar] [CrossRef]
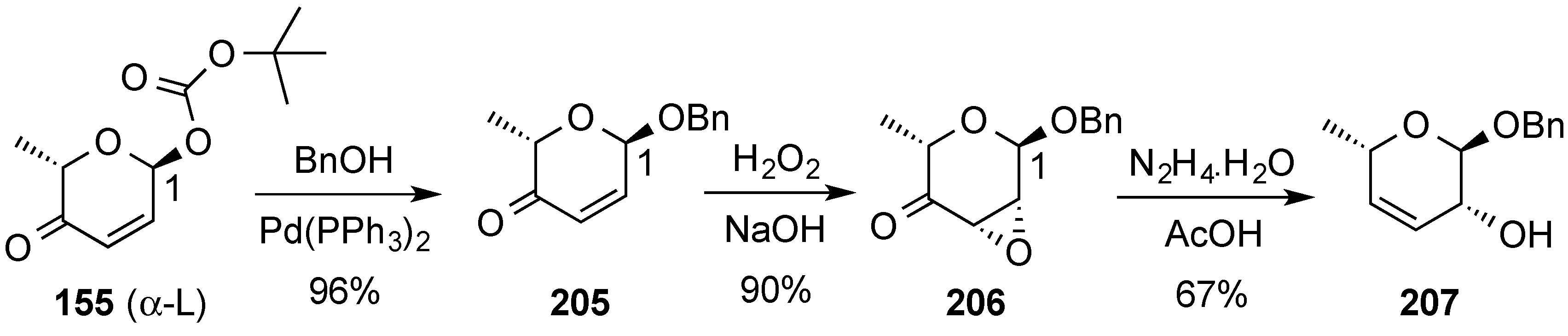
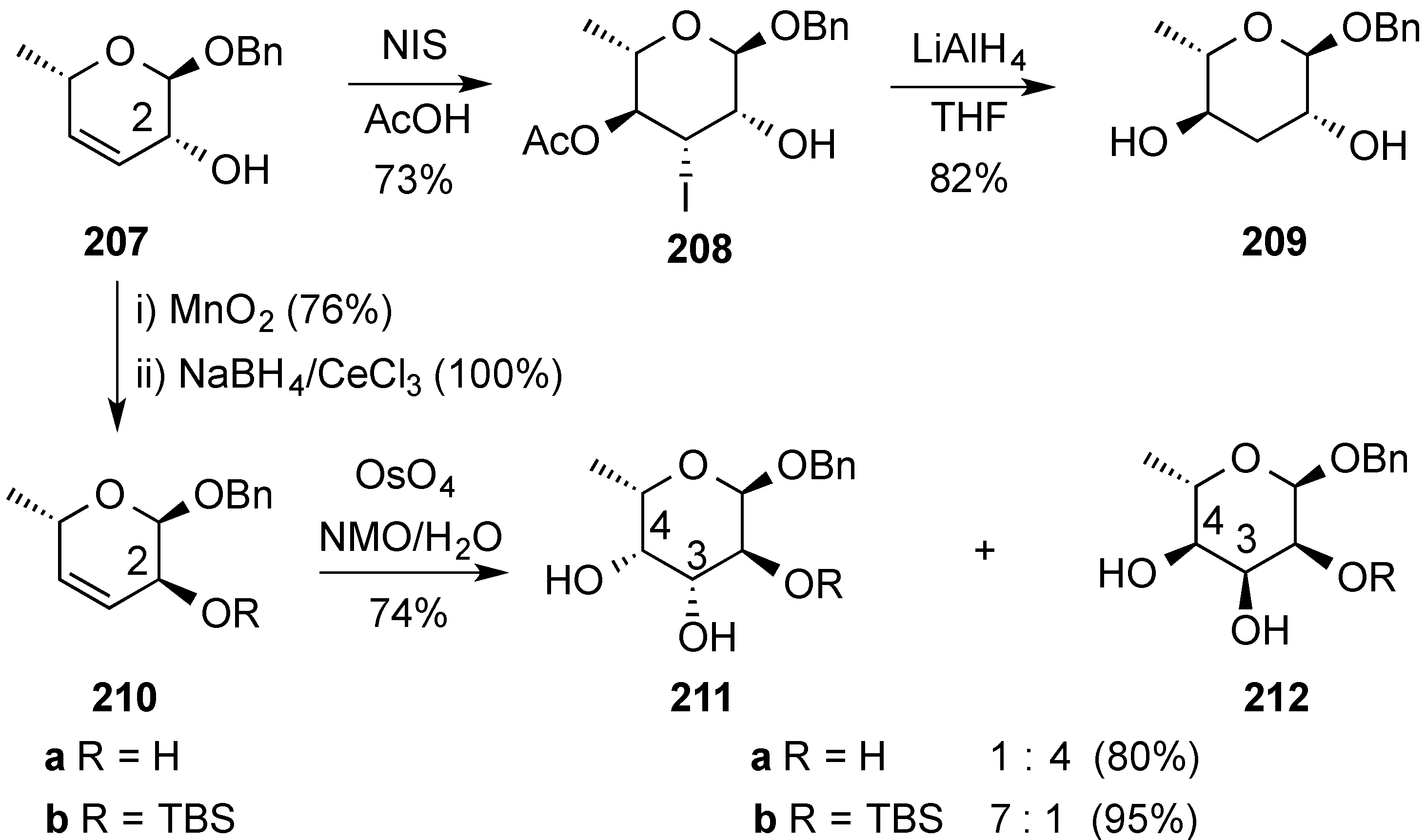
- Aljahdali, A.Z.; Shi, P.; Zhong, Y.; O’Doherty, G.A. De novo asymmetric synthesis of the pyranoses: From monosaccharides to oligosaccharides. Adv. Carbohydr. Chem. Biochem. 2013, 69, 55–123. [Google Scholar] [PubMed]
- Borisova, S.A.; Guppi, S.R.; Kim, H.J.; Wu, B.; Penn, J.H.; Liu, H.-W.; O’Doherty, G.A. A de novo approach to the synthesis of glycosylated methymycin analogues with structural and stereochemical diversity. Org. Lett. 2010, 12, 5150–5153. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; O’Doherty, G.A. De novo asymmetric synthesis and biological evaluation of the trisaccharide portion of PI-080 and vineomycin B2. Org. Lett. 2008, 10, 4529–4532. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Scott, J.; O’Doherty, G.A. Synthesis of 7-oxa-phomopsolide E and its C-4 epimer. Tetrahedron Lett. 2004, 45, 1005–1009. [Google Scholar] [CrossRef]
- Guppi, S.R.; O’Doherty, G.A. Synthesis of aza-analogues of the glycosylated tyrosine portion of mannopeptimycin-E. J. Org. Chem. 2007, 72, 4966–4969. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.S.; Guppi, S.R.; O’Doherty, G.A. Synthetic studies toward mannopectimycin-E: Synthesis of the O-linked tyrosine 1,4-α,α-manno,manno-pyranosyl pyranoside. Org. Lett. 2006, 8, 1605–1608. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Xiang, S.-H.; Leng, W.-L.; Hoang, K.L. M.; Liu, X.-W. Palladium-catalyzed glycosylation: Novel synthetic approach to diverse N-heterocyclic glycosides. Org. Lett. 2015, 17, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, S.; Sasaki, K.; Wang, S.; Watanabe, T.; Takahashi, D.; Toshima, K. Effective and chemoselective glycosylations using 2,3-unsaturated sugars. Org. Biomol. Chem. 2010, 8, 3164–3178. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, S.; Wang, S.; Watanabe, T.; Sasaki, K.; Takahashi, D.; Toshima, K. Chemoselective glycosylations using 2,3-unsaturated-4-keto glycosyl donors. Org. Biomol. Chem. 2010, 8, 988–990. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Aga, M.A.; Rouf, A.; Shah, B.A.; Taneja, S.C. 2,3-Unsaturated allyl glycosides as glycosyl donors for selective α-glycosylation. J. Org. Chem. 2011, 76, 3506–3510. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, T.; Ozuduru, G.; Grugel, H.; Albrecht, F.; Telligmann, S.M.; Boysen, M.M.K. More than just sweet—Sugar-derived stereodifferentiating agents for asymmetric synthesis. Synthesis 2011, 17, 2685–2708. [Google Scholar]
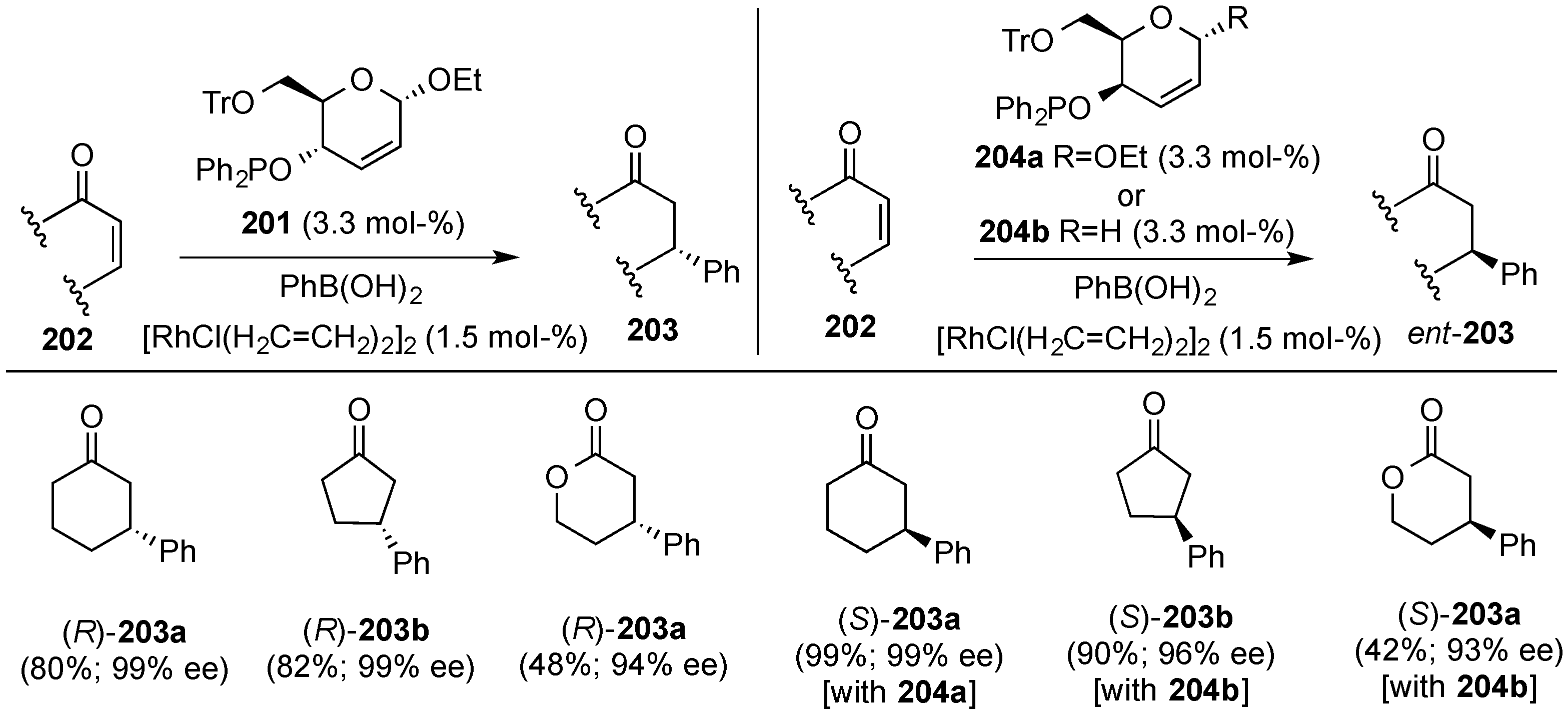
- Minuth, T.; Boysen, M.M.K. Novel, efficient alkene-phosphinite hybrid ligand based on d-glucose. Org. Lett. 2009, 11, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- Grugel, H.; Albrecht, F.; Minuth, T.; Boysen, M.M.K. Efficient pseudo-enantiomeric carbohydrate olefin ligands. Org. Lett. 2012, 14, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Cuccarese, M.F.; Wang, H.-Y.L.; O’Doherty, G.A. Application of the Wharton rearrangement for the de novo synthesis of pyranosides with ido, manno, and colito stereochemistry. Eur. J. Org. Chem. 2013, 3067–3075. [Google Scholar] [CrossRef]
- Wharton, P.; Bohlen, D. Hydrazine reduction of α,β-epoxy ketones to allylic alcohols. J. Org. Chem. 2011, 76, 3506–3510. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.L.; O’Doherty, G.A. De novo synthesis of deoxy sugar via a Wharton rearrangement. Chem. Commun. 2011, 47, 10251–10253. [Google Scholar] [CrossRef]
- An alternative approach to α-ascarilose from keto-epoxide 201 had previously been described: Shan, M.; Xing, Y.; O’Doherty, G.A. De novo asymmetric synthesis of an a-6-deoxyaltropyranoside as well as its 2-/3-deoxy and 2,3-dideoxy congeners. J. Org. Chem. 2009, 74, 5961–5966.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez, A.M.; Lobo, F.; Miranda, S.; Lopez, J.C. A Survey of Recent Synthetic Applications of 2,3-Dideoxy-Hex-2-enopyranosides. Molecules 2015, 20, 8357-8394. https://doi.org/10.3390/molecules20058357
Gomez AM, Lobo F, Miranda S, Lopez JC. A Survey of Recent Synthetic Applications of 2,3-Dideoxy-Hex-2-enopyranosides. Molecules. 2015; 20(5):8357-8394. https://doi.org/10.3390/molecules20058357
Chicago/Turabian StyleGomez, Ana M., Fernando Lobo, Silvia Miranda, and J. Cristobal Lopez. 2015. "A Survey of Recent Synthetic Applications of 2,3-Dideoxy-Hex-2-enopyranosides" Molecules 20, no. 5: 8357-8394. https://doi.org/10.3390/molecules20058357
APA StyleGomez, A. M., Lobo, F., Miranda, S., & Lopez, J. C. (2015). A Survey of Recent Synthetic Applications of 2,3-Dideoxy-Hex-2-enopyranosides. Molecules, 20(5), 8357-8394. https://doi.org/10.3390/molecules20058357