
Structure Based Modeling of Small Molecules Binding to the TLR7 by Atomistic Level Simulations

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Homology Modeling
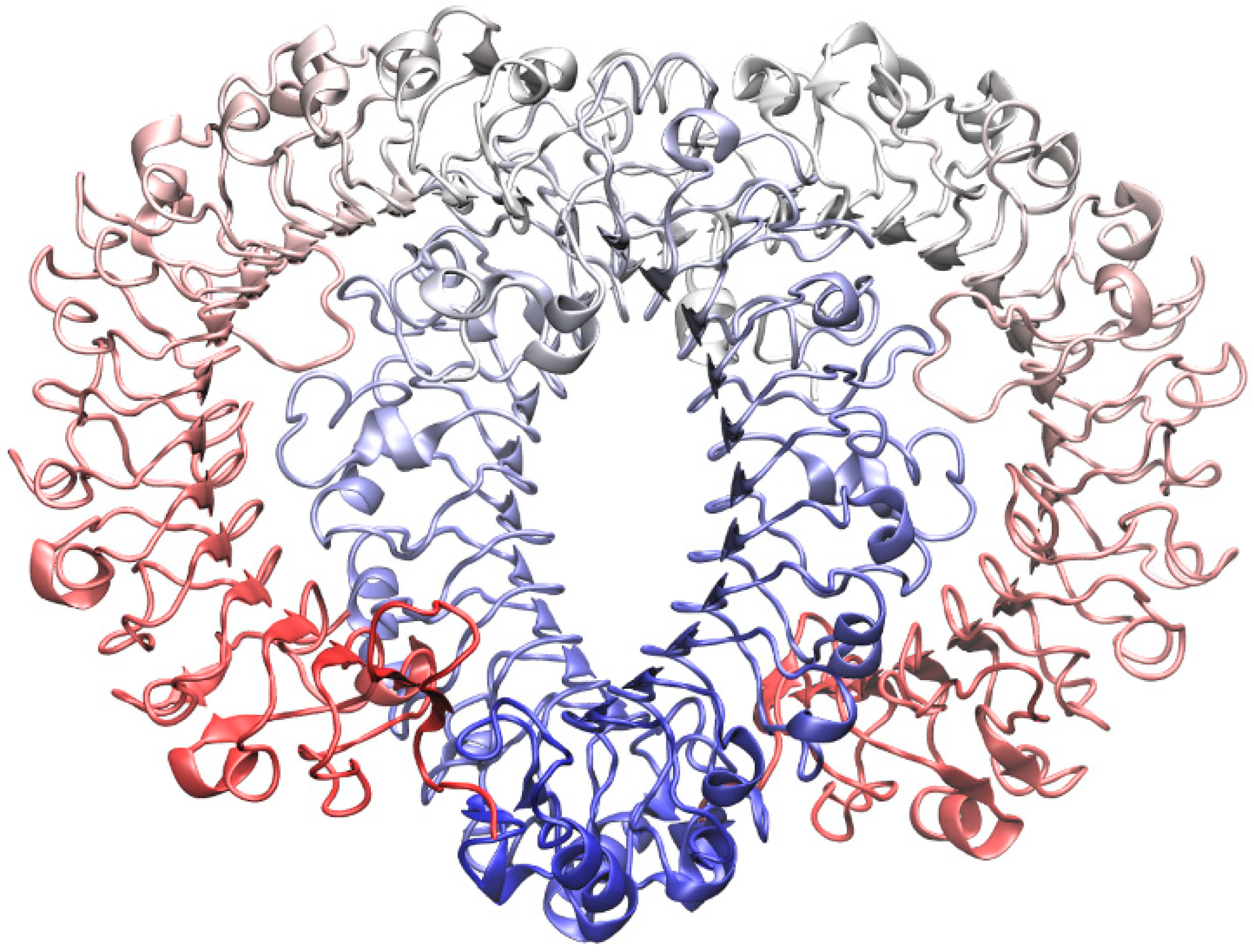

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | ERRAT Quality Factor | Procheck | |||
|---|---|---|---|---|---|
| Core | Additional | Generously | Disallowed | ||
| TLR8 | 84.364 | 79.0 | 20.1 | 0.5 | 0.4 |
| TLR7 | 73.861 | 69.0 | 28.1 | 1.9 | 1.0 |
| Minimized TLR7 1 | 76.506 | 70.9 | 26.1 | 2.0 | 1.0 |
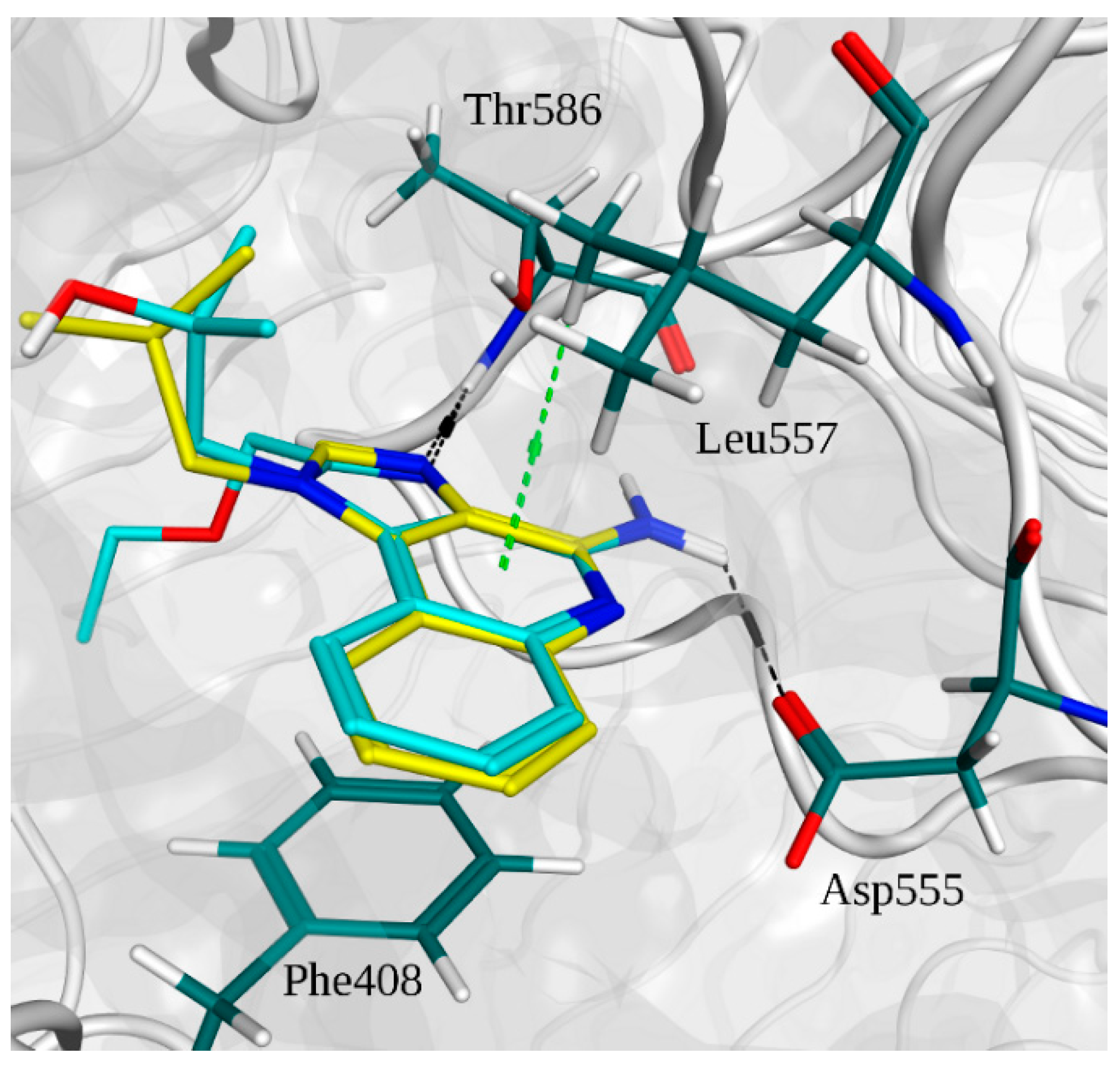
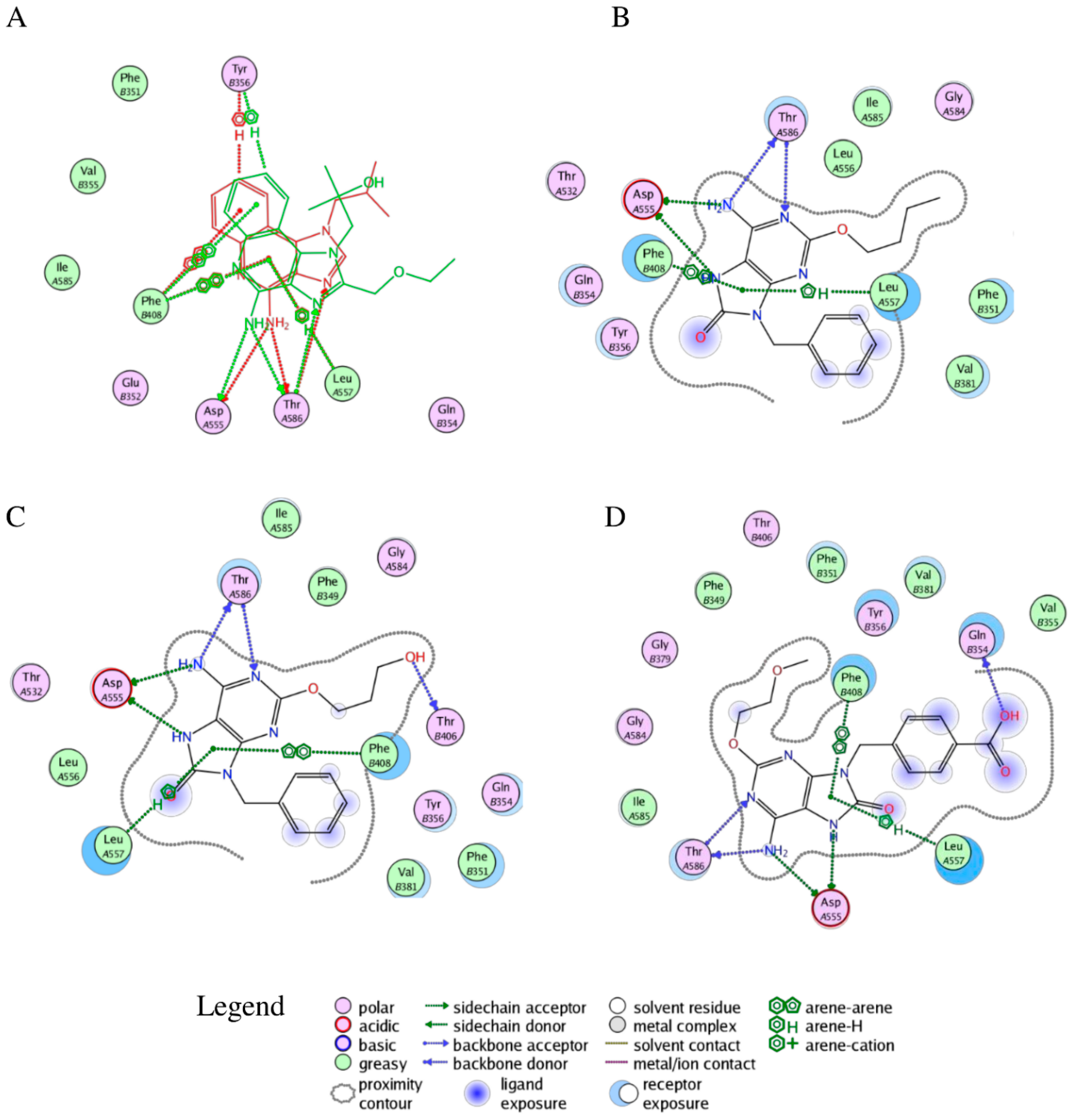
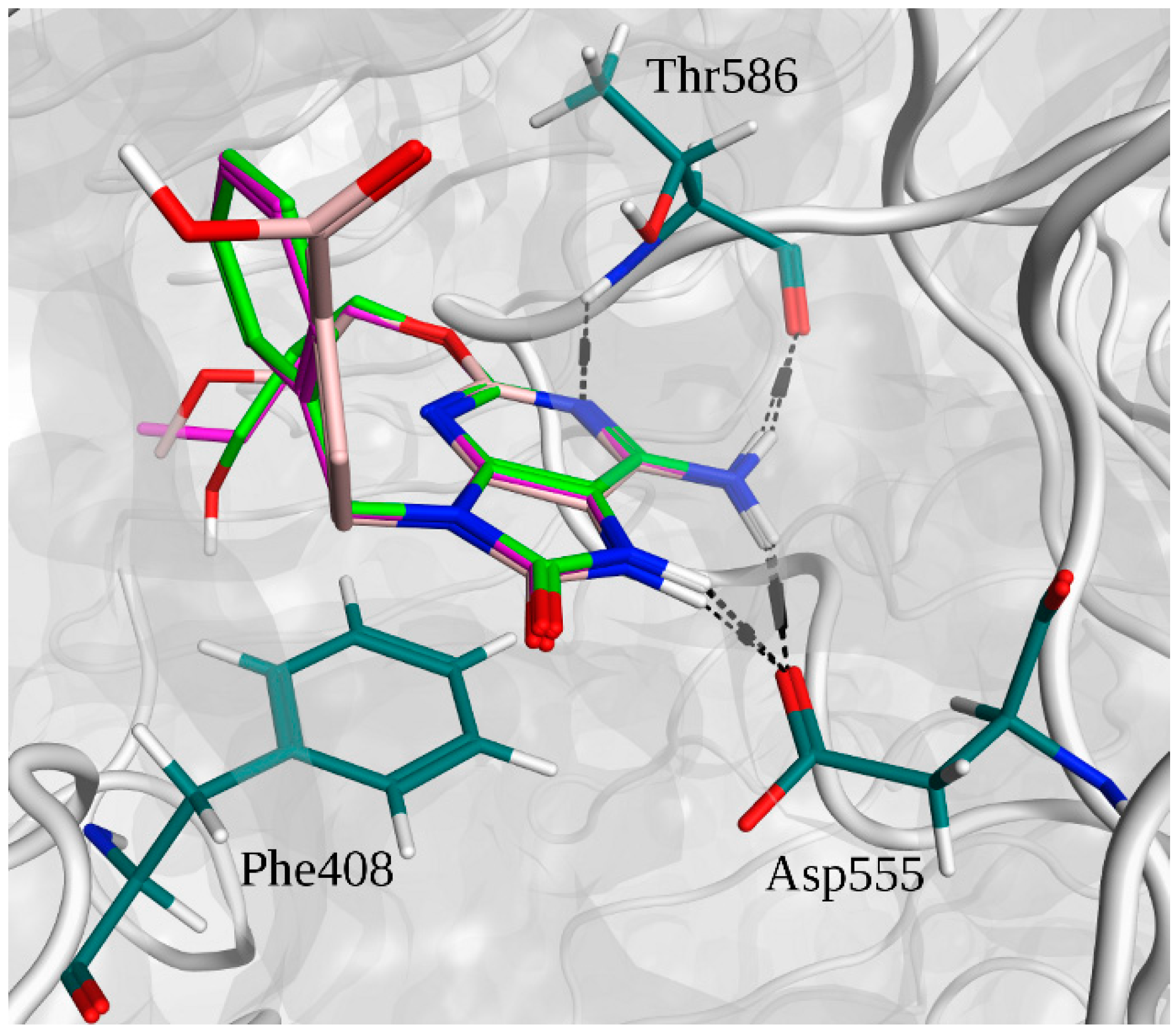
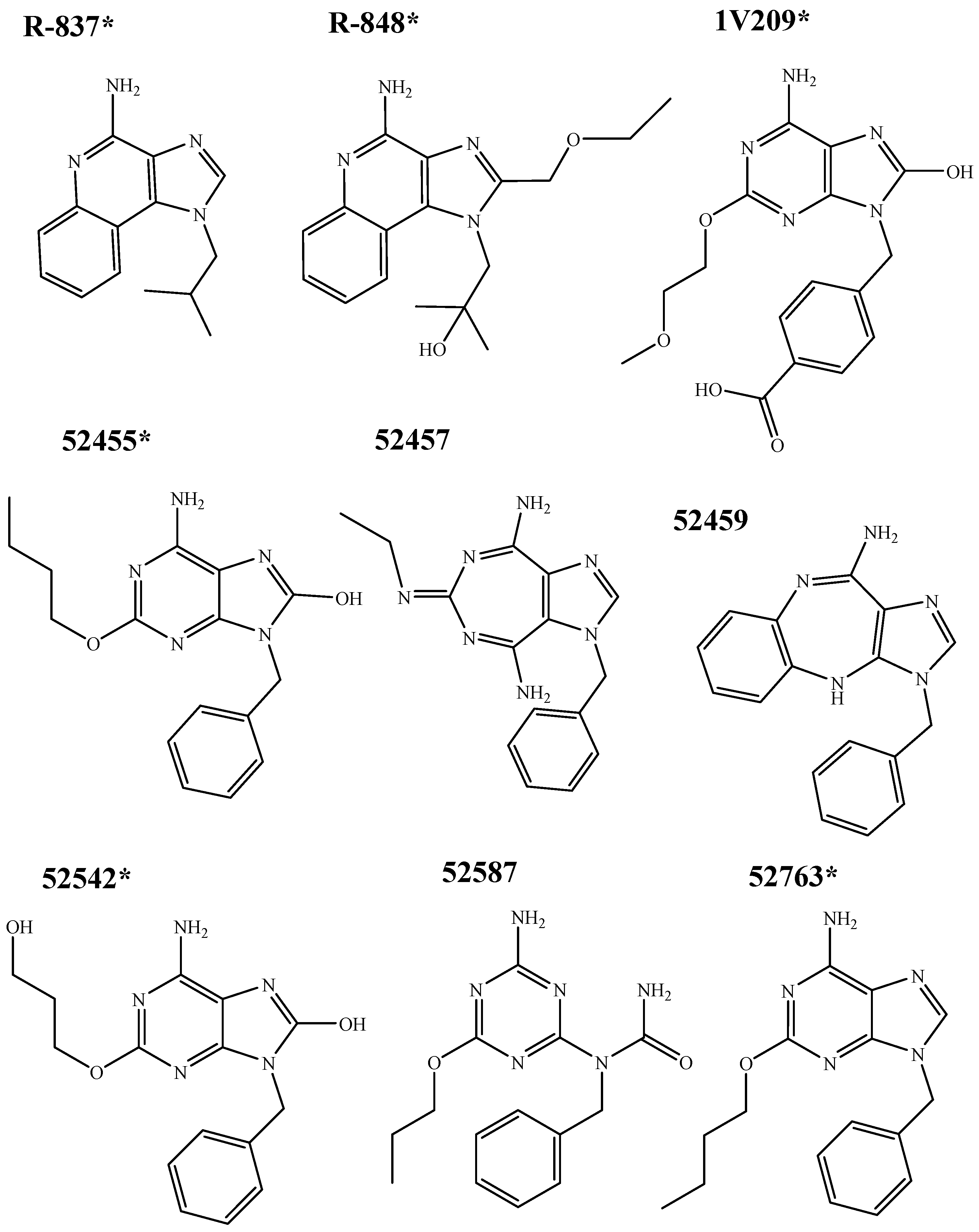
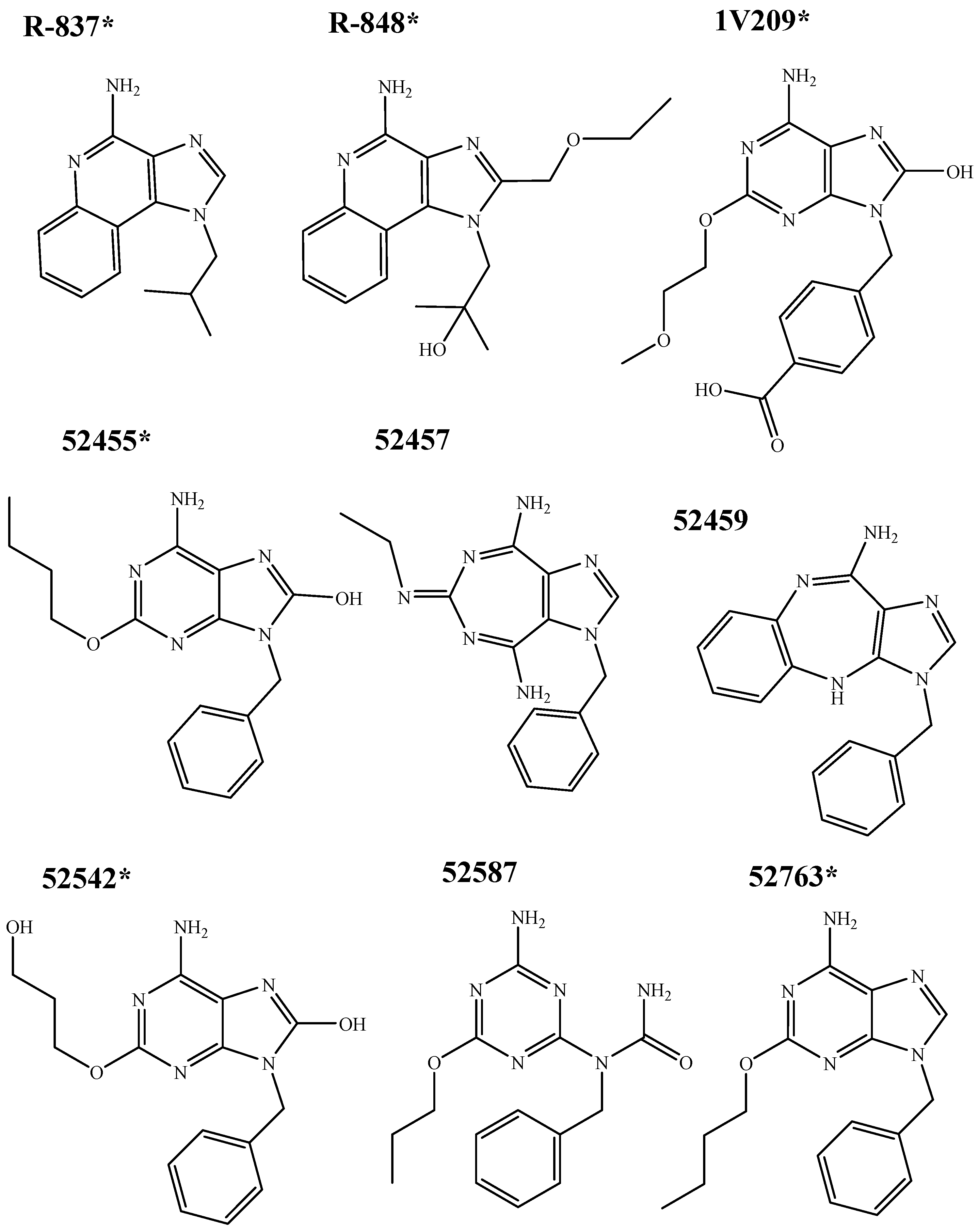
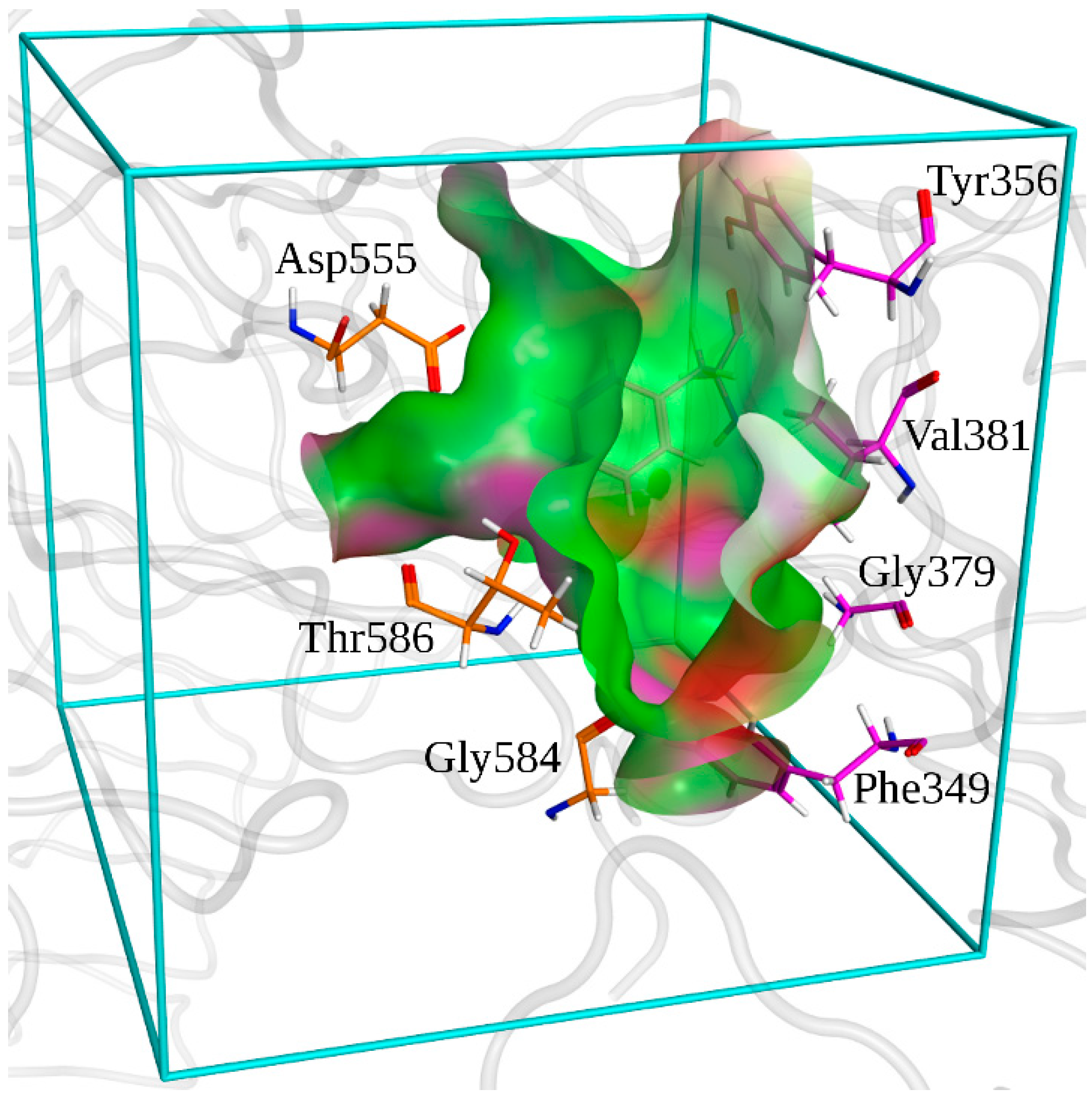
2.2. Molecular Docking
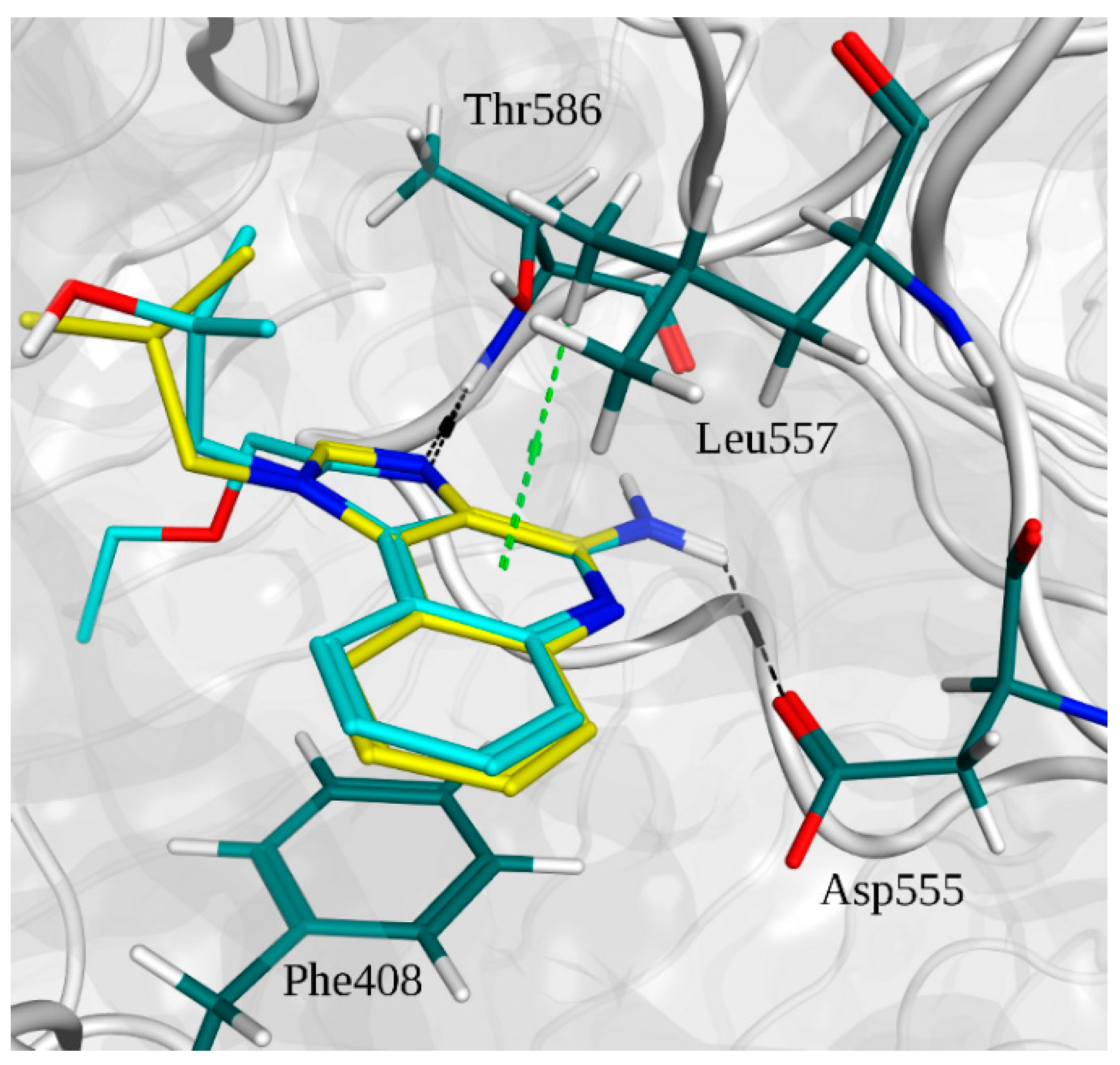
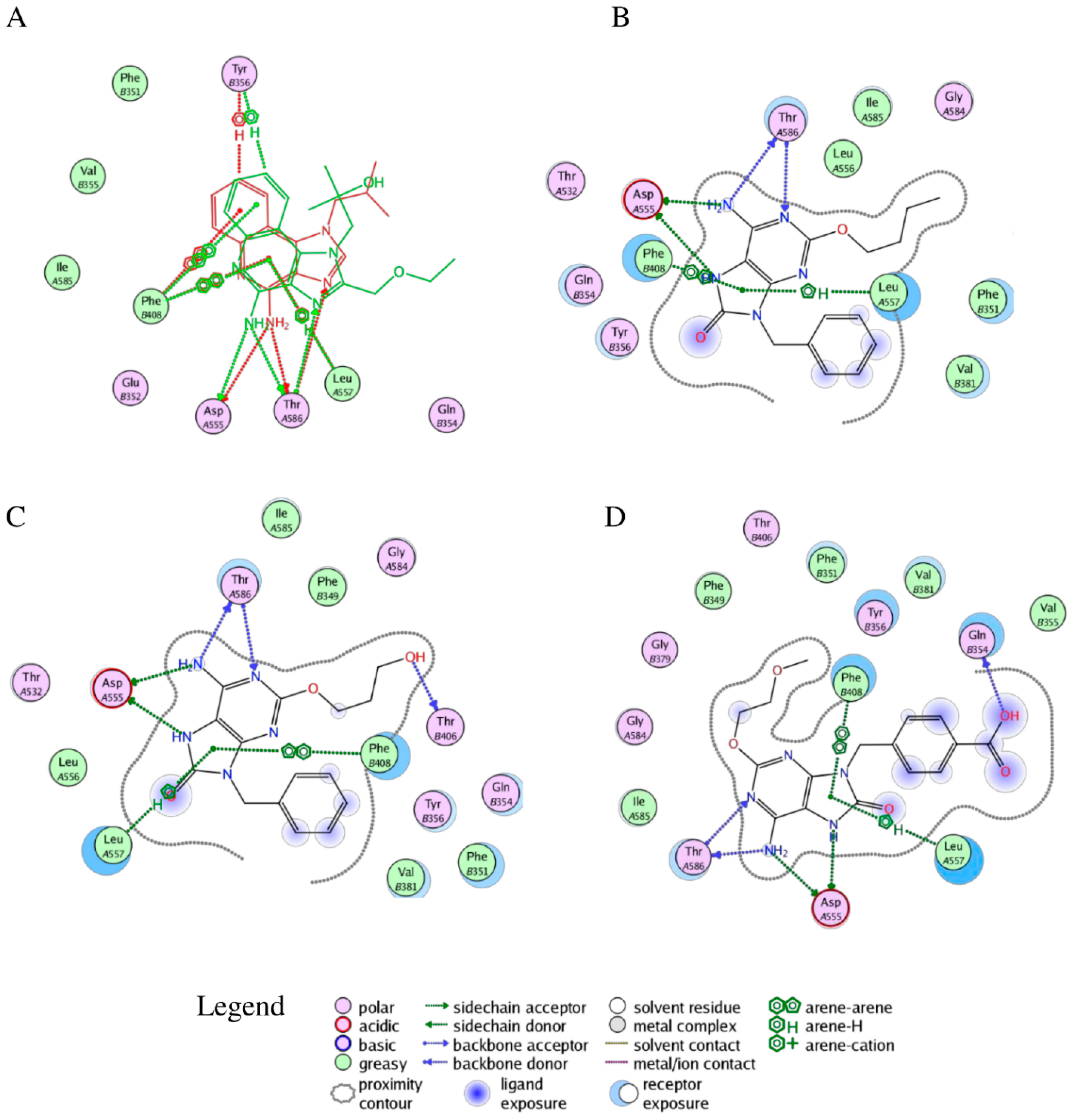
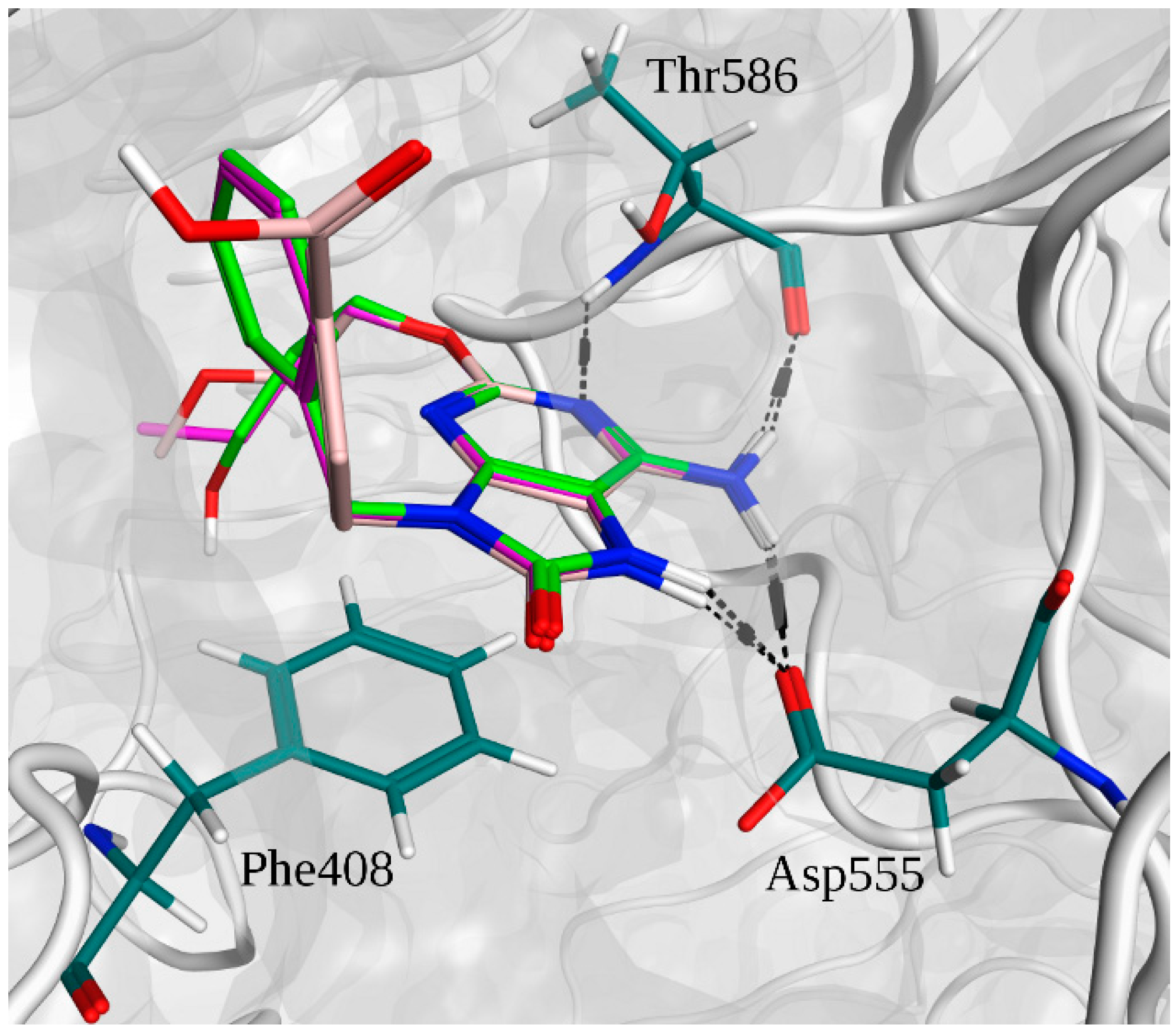
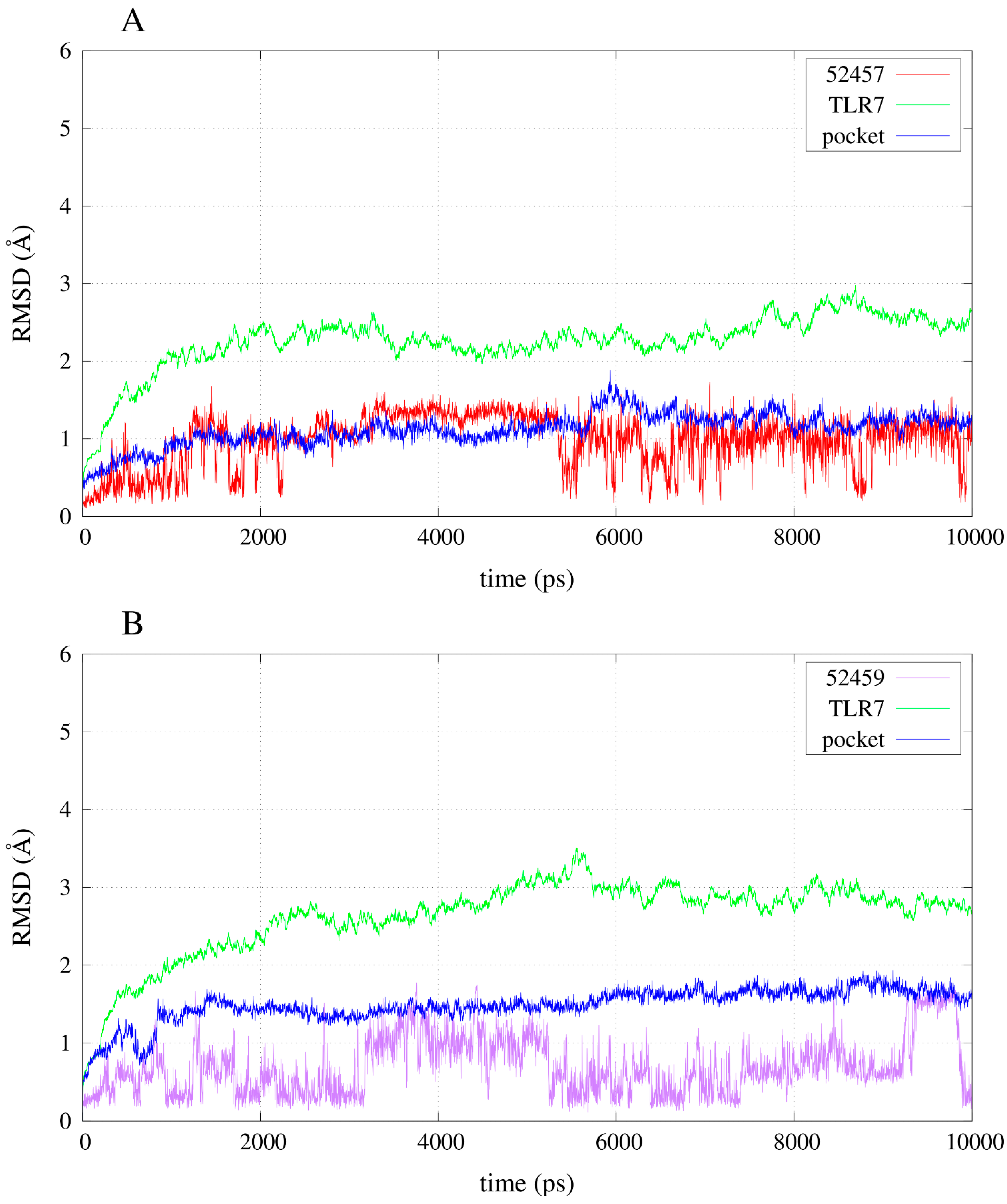
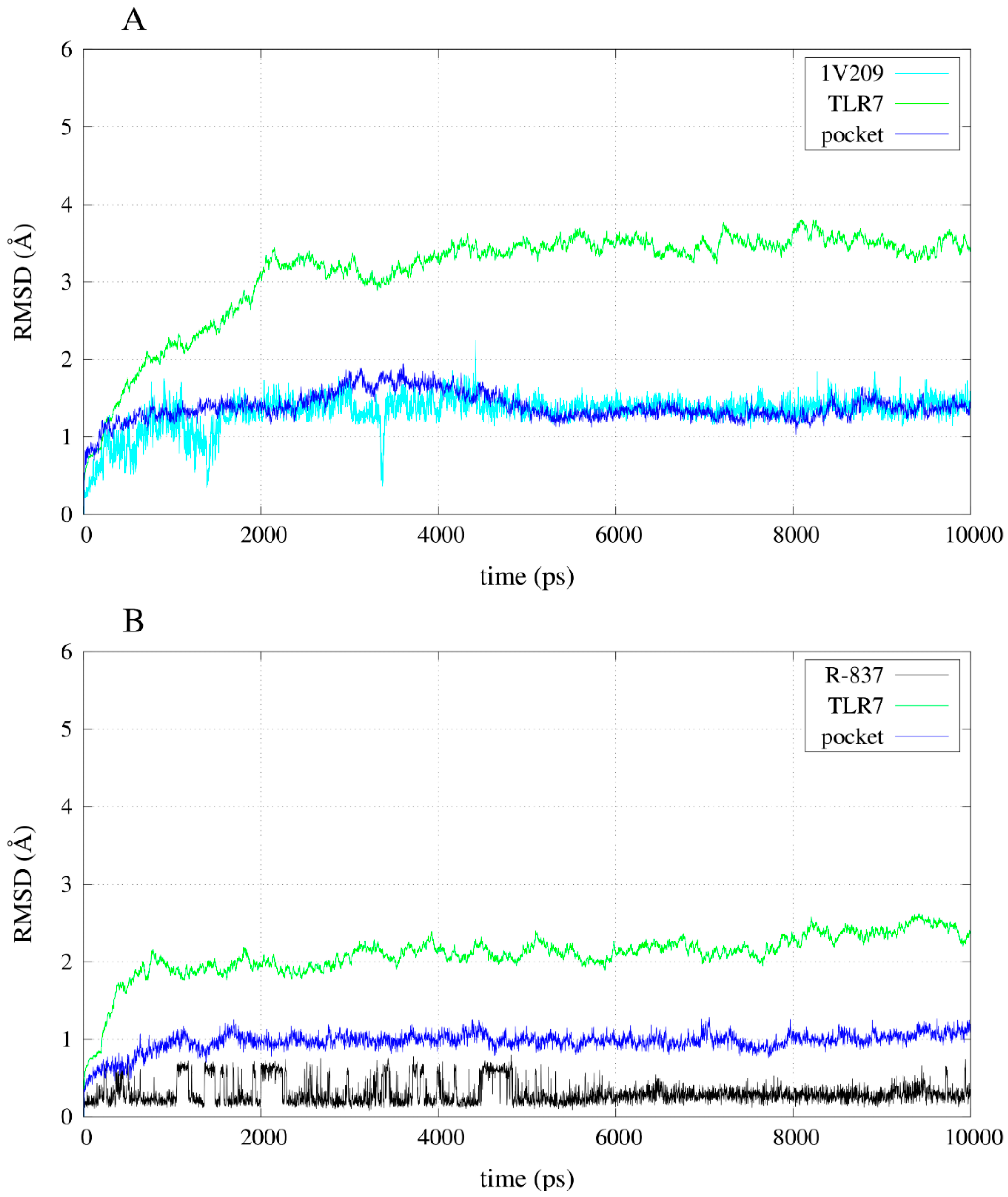
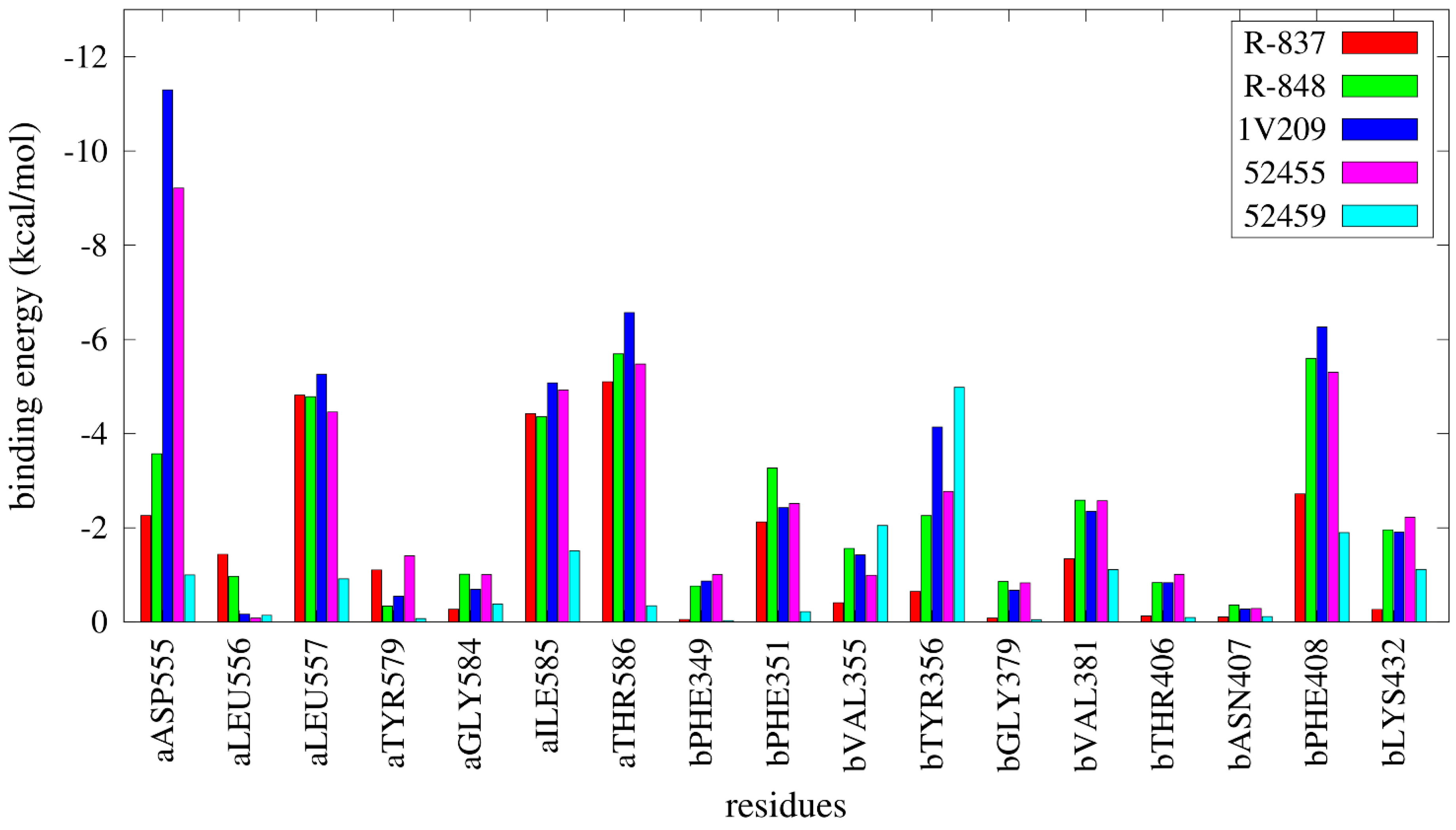
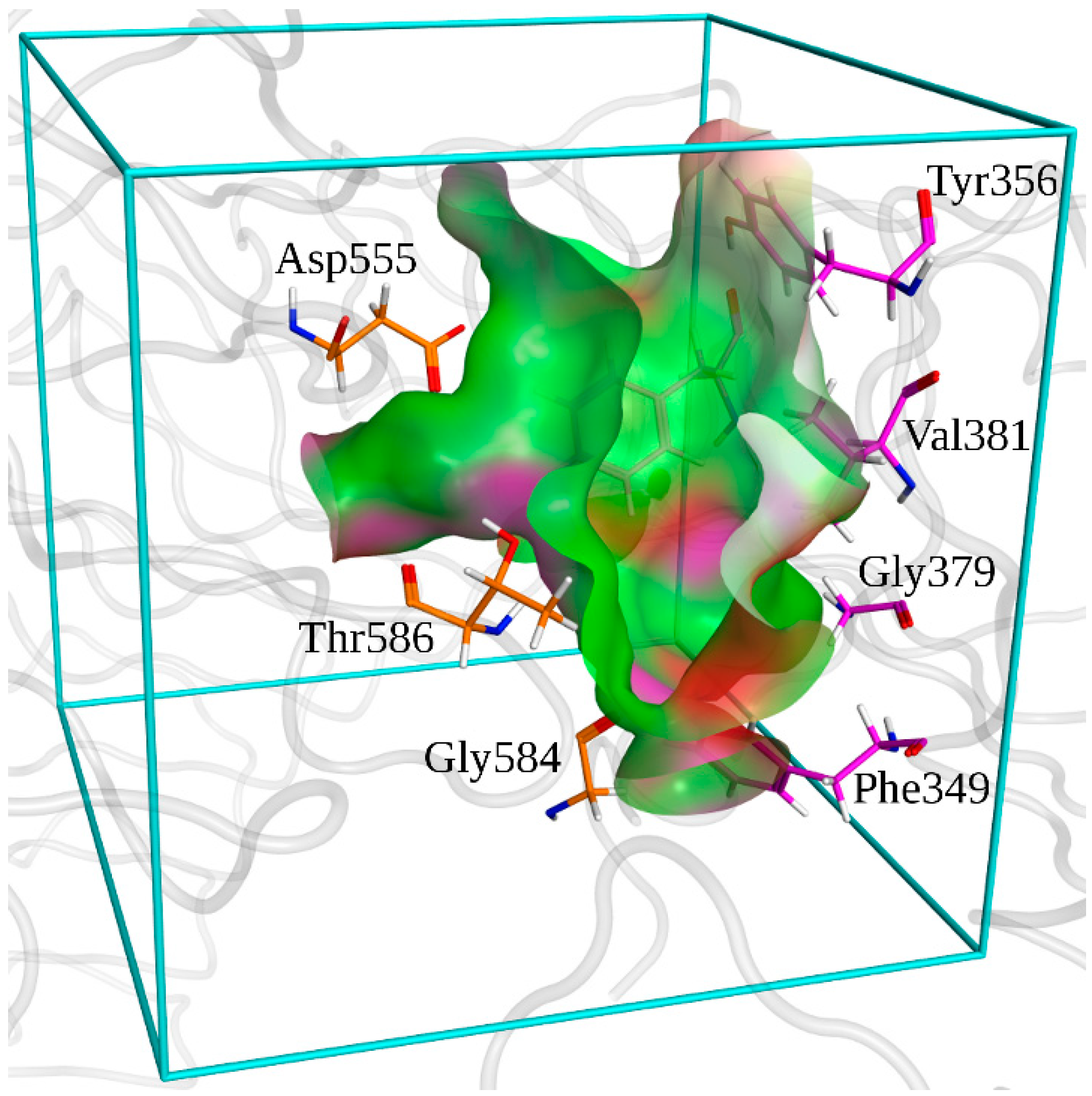
2.3. Molecular Dynamics Simulations
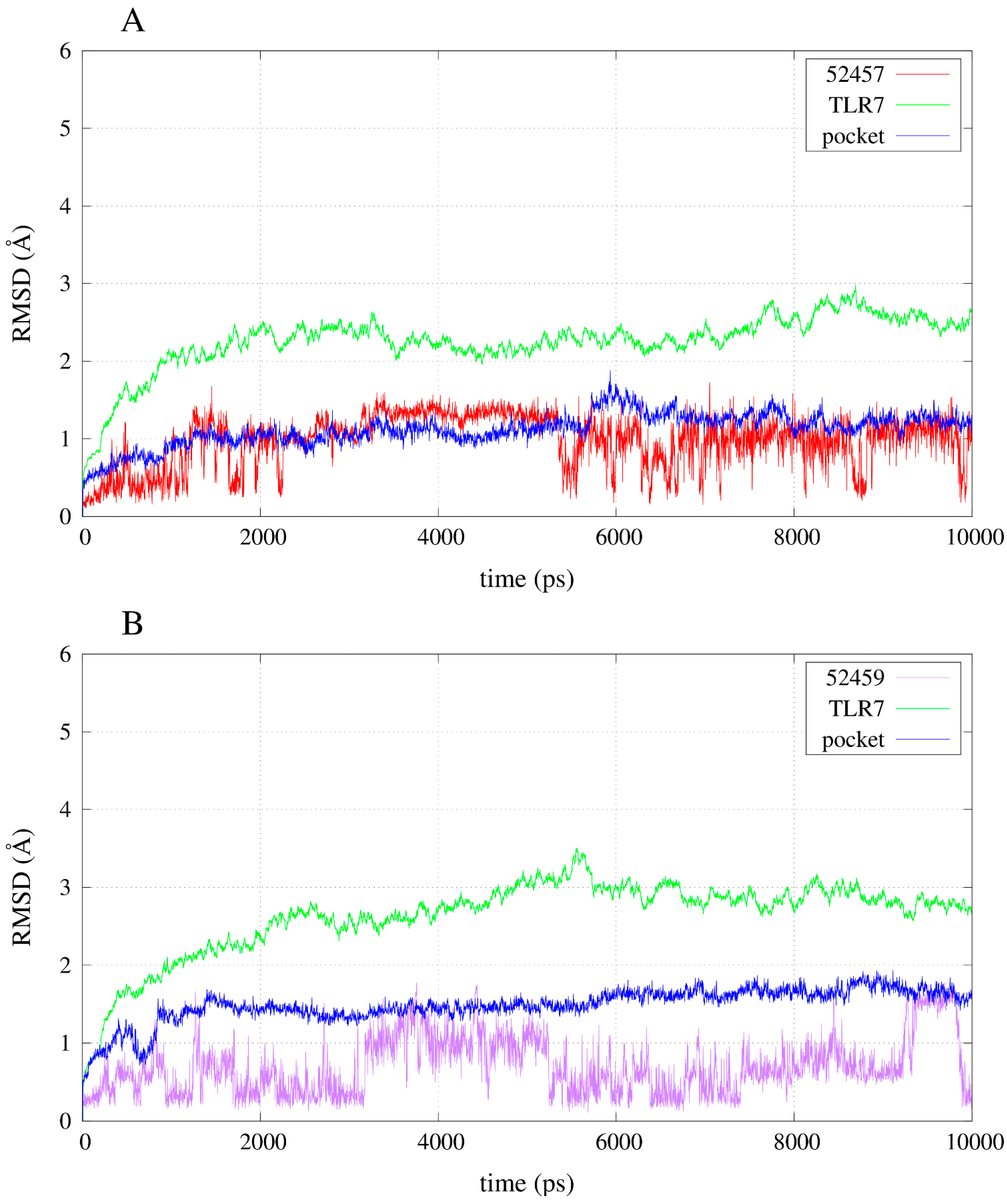
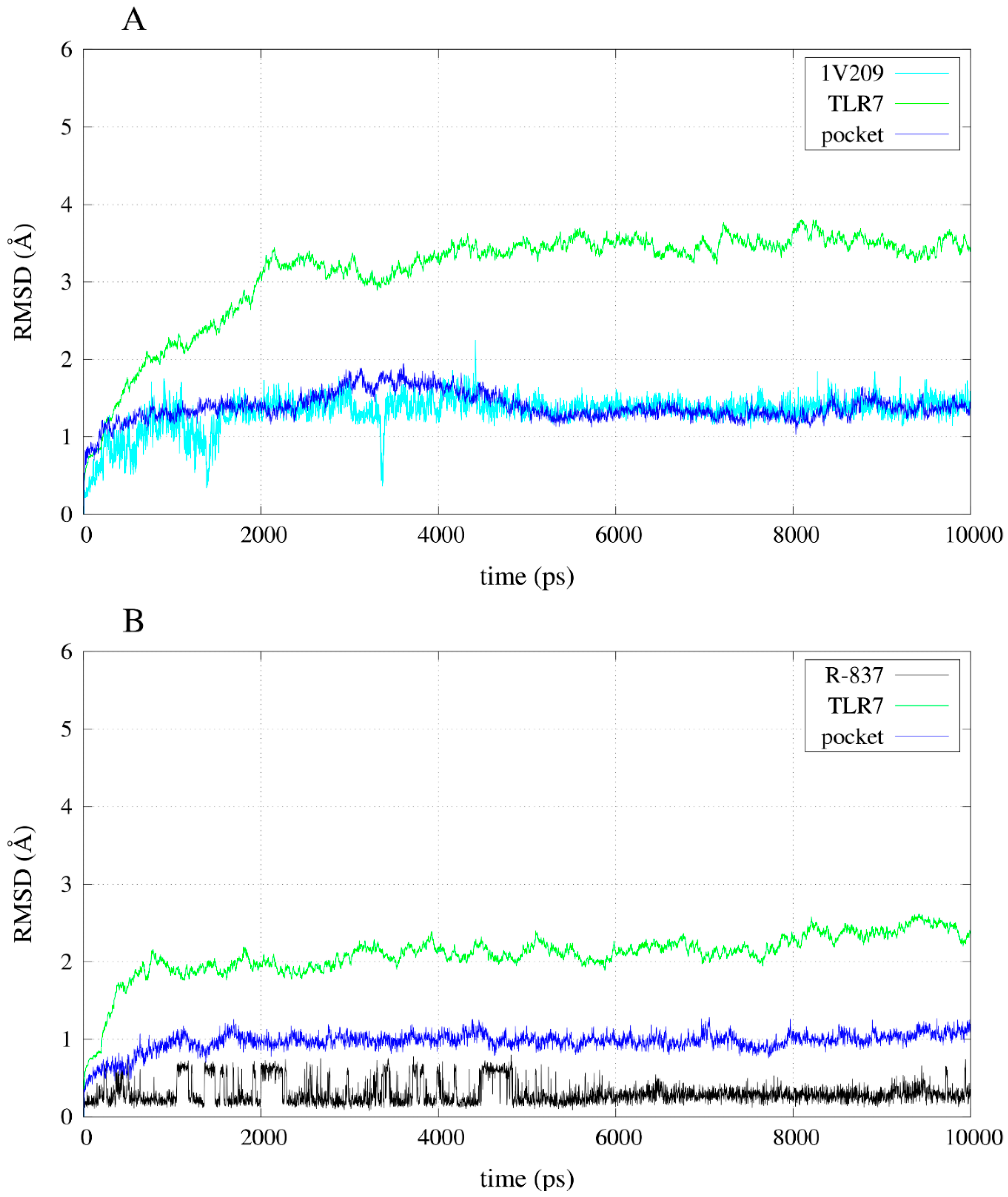
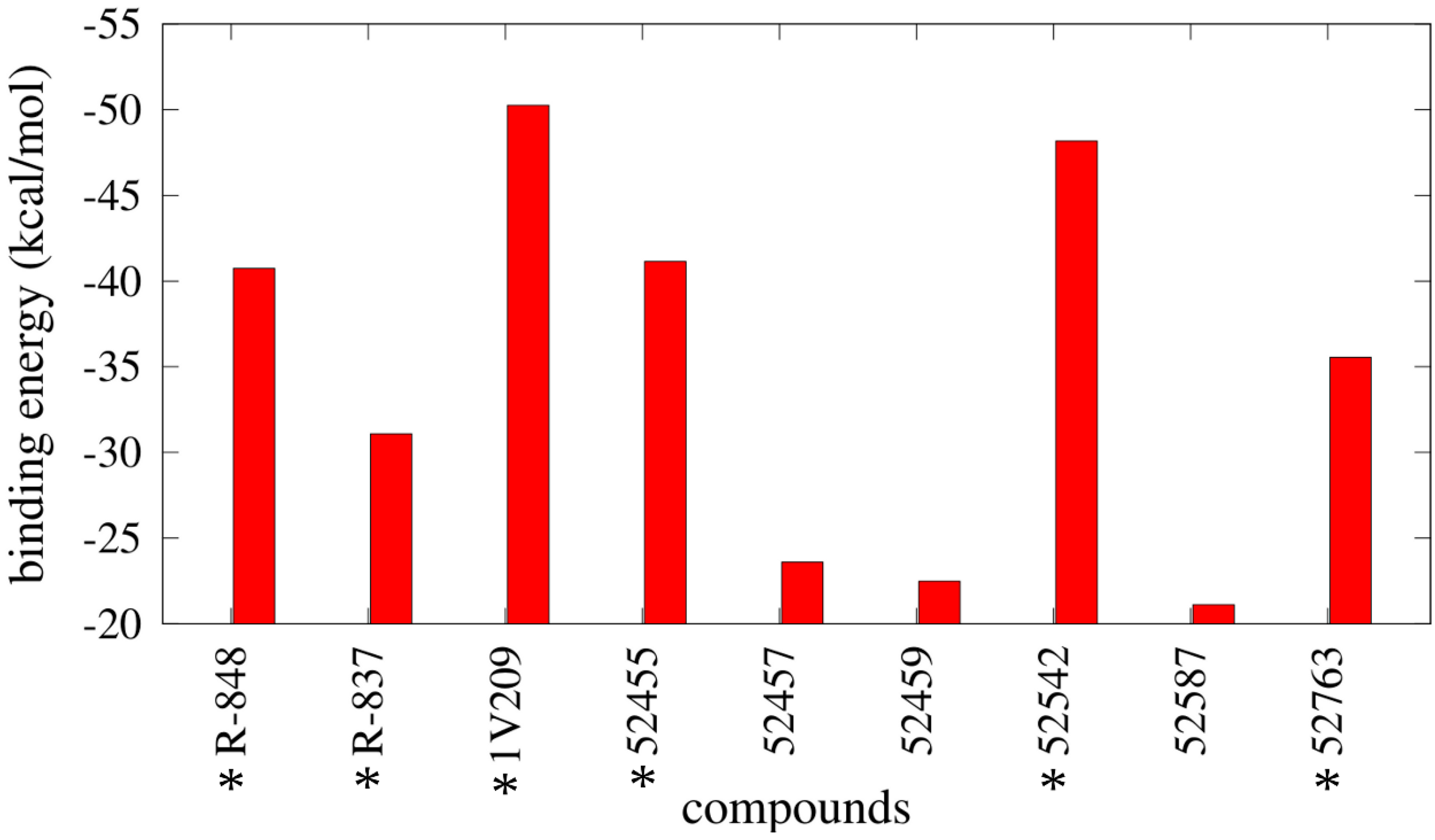
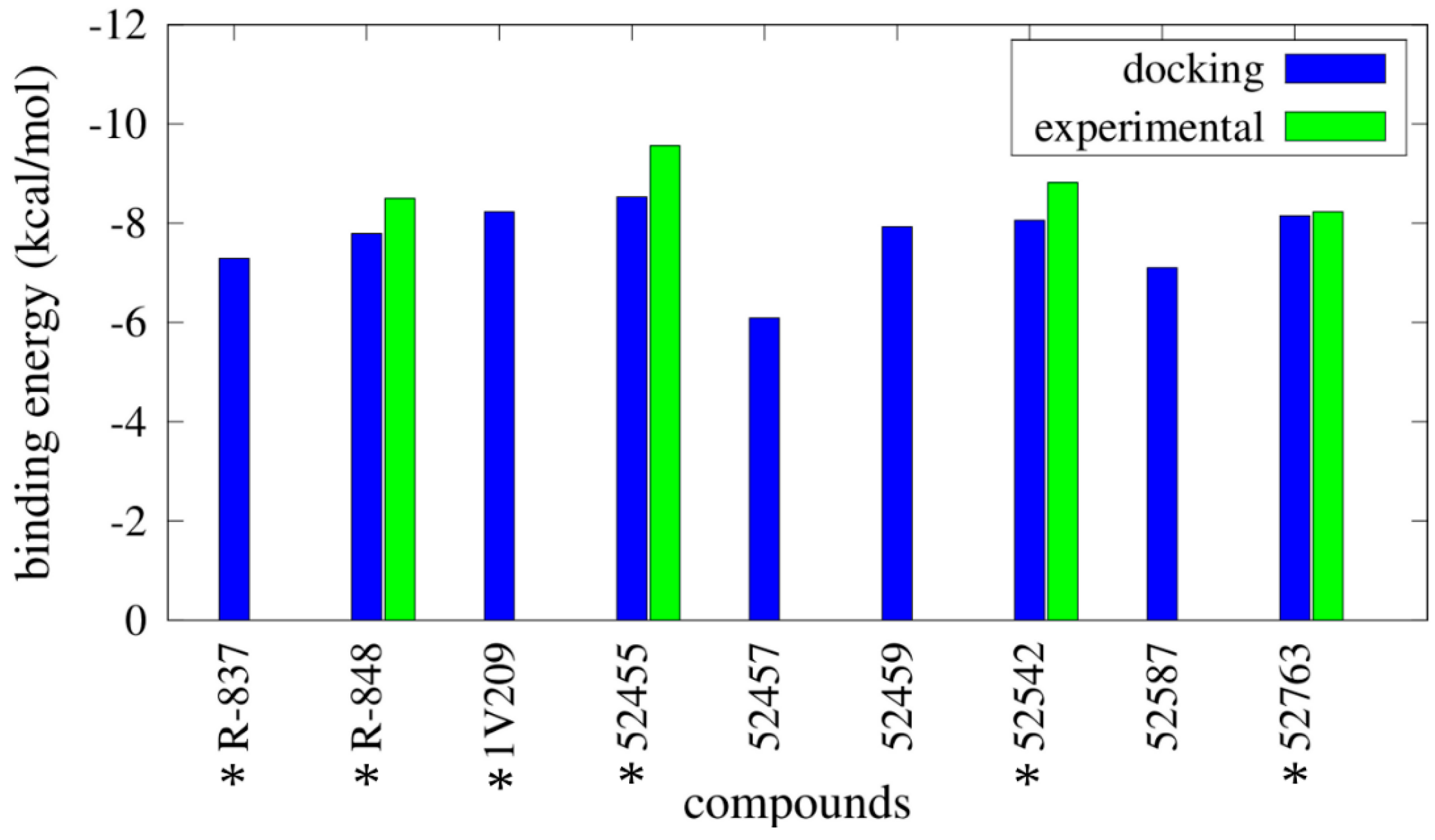
2.4. Free Energy Estimates
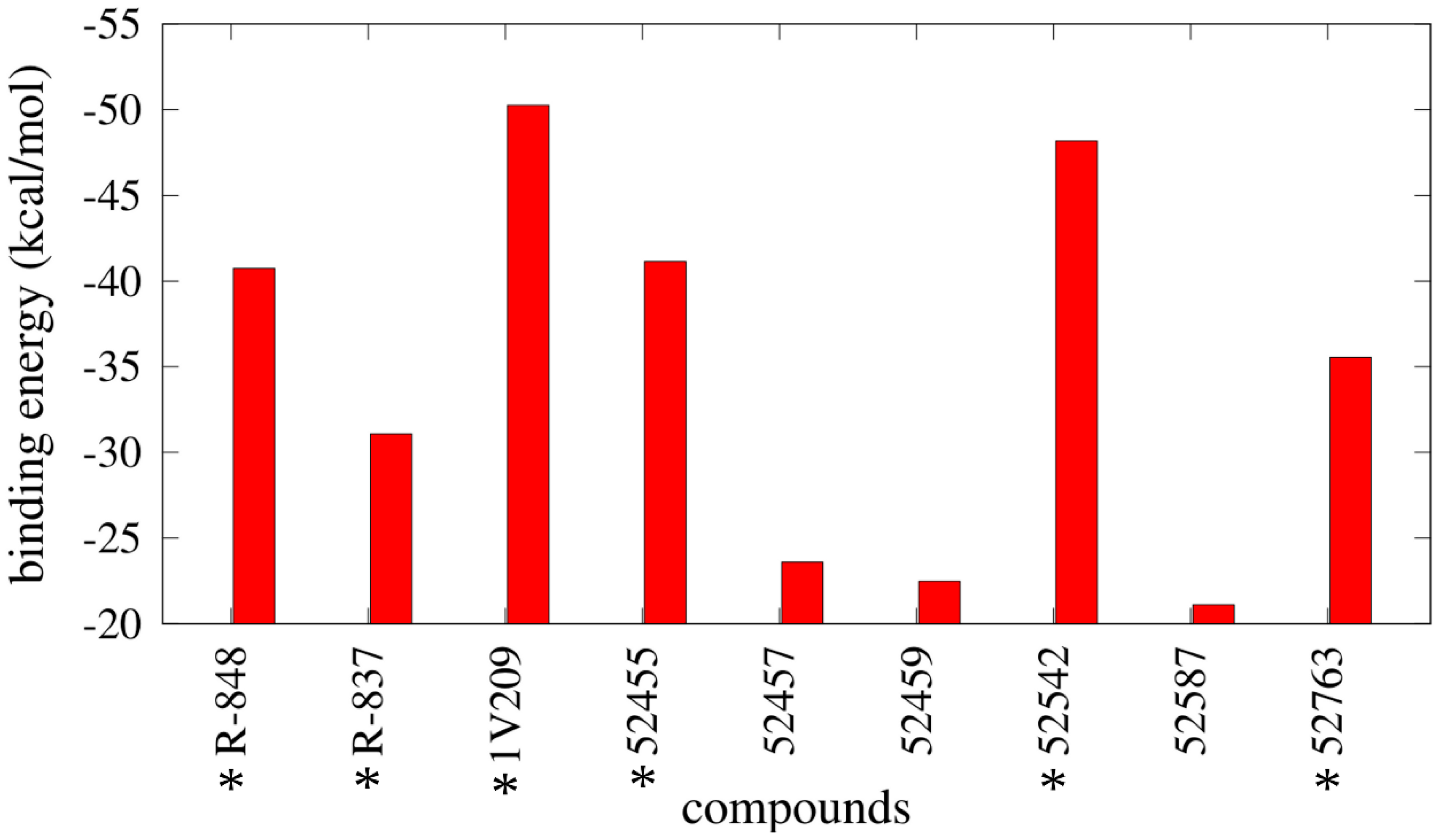
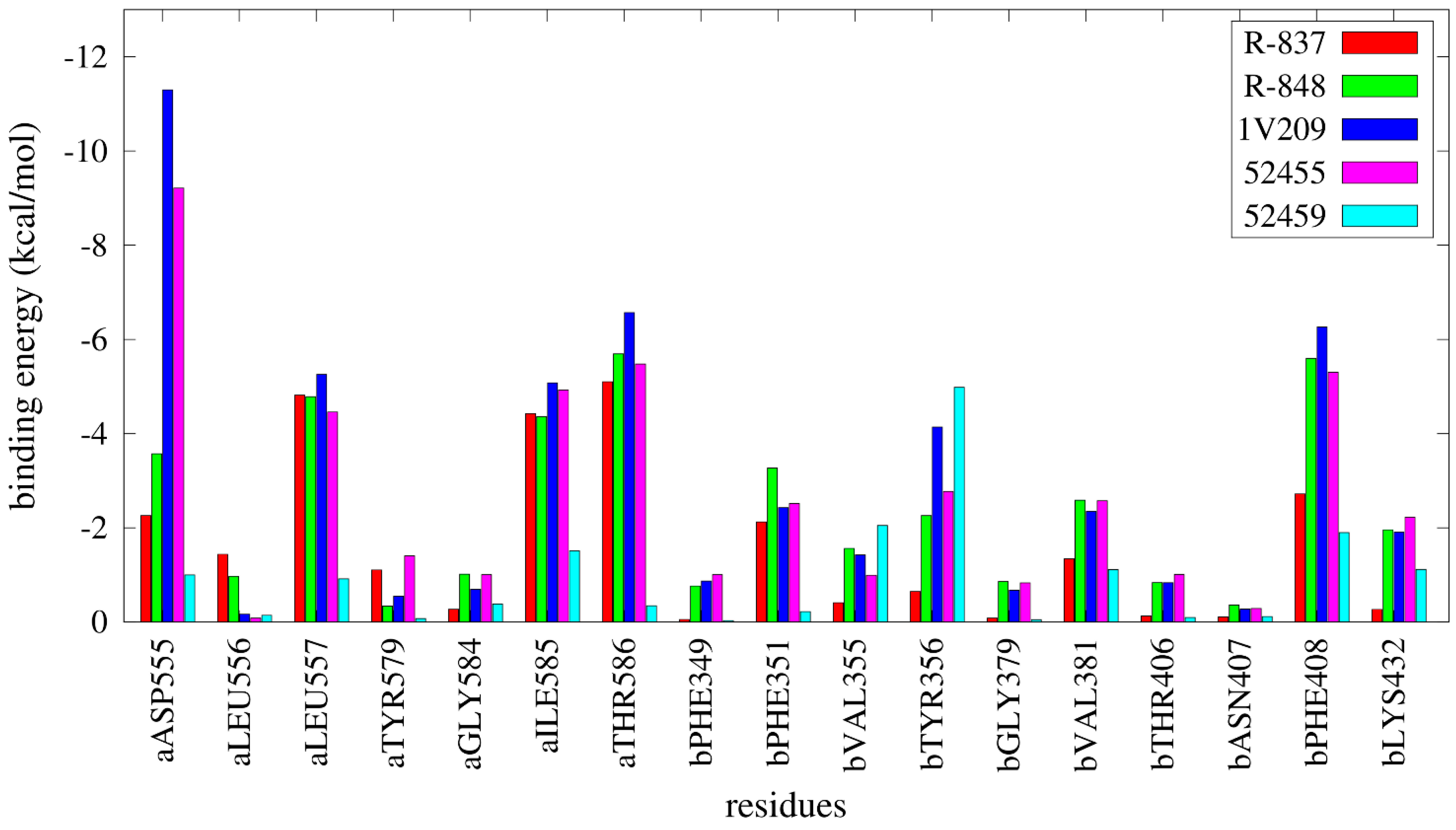
3. Discussion
4. Experimental Section
4.1. Homology Modeling
4.2. Molecular Docking
- -
- 1000 initial poses of the small molecules were generated in the binding pocket;
- -
- -
4.3. Molecular Dynamics
4.4. Free Energy Estimates
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.J.M.; Ponsaerts, P.; Berneman, Z.N.; van Tendeloo, V.F.I. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist 2008, 13, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Kodys, K.; Szabo, G. Impaired expression and function of toll-like receptor 7 in hepatitis C virus infection in human hepatoma cells. Hepatology 2010, 51, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Innate sensing of self and non-self RNAs by Toll-like receptors. Trends Mol. Med. 2006, 12, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.P.; Schön, M. TLR7 and TLR8 as targets in cancer therapy. Oncogene 2008, 27, 190–199. [Google Scholar] [CrossRef] [PubMed]
- So, E.Y.; Ouchi, T. The application of Toll like receptors for cancer therapy. Int. J. Biol. Sci. 2010, 6, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Koski, G.K.; Czerniecki, B.J. Combining Innate Immunity with Radiation Therapy for Cancer Treatment. Clin. Cancer Res. 2005, 11, 7–11. [Google Scholar] [PubMed]
- Ochi, A.; Graffeo, C.S.; Zambirinis, C.P.; Rehman, A.; Hackman, M.; Fallon, N.; Barilla, R.M.; Henning, J.R.; Jamal, M.; Rao, R.; et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J. Clin. Investig. 2012, 122, 4118–4129. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M.; Consortium, U. UniProt Knowledgebase: A hub of integrated protein data. Database (Oxford) 2011, 2011, bar009. [Google Scholar] [CrossRef]
- Maschalidi, S.; Hässler, S.; Blanc, F.; Sepulveda, F.E.; Tohme, M.; Chignard, M.; van Endert, P.; Si-Tahar, M.; Descamps, D.; Manoury, B. Asparagine endopeptidase controls anti-influenza virus immune responses through TLR7 activation. PLoS Pathog. 2012, 8, e1002841. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.M.; Shepherd, D.; Gileadi, U.; Aichinger, M.C.; Kessler, B.M.; Edelmann, M.J.; Essalmani, R.; Seidah, N.G.; Reis E Sousa, C.; Cerundolo, V. Processing of Human Toll-like Receptor 7 by Furin-like Proprotein Convertases Is Required for Its Accumulation and Activity in Endosomes. Immunity 2013, 39, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, C.; Ramsauer, K.; Kubarenko, A.V.; Debasitis, J.C.; Leykin, I.; Weber, A.N.R.; Siggs, O.M.; Beutler, B.; Zhang, P.; Otten, G.; et al. A point mutation in the amino terminus of TLR7 abolishes signaling without affecting ligand binding. J. Immunol. 2011, 186, 4213–4222. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Petricevic, B.; Wessner, B.; Sachet, M.; Vrbanec, D.; Spittler, A.; Bergmann, M. CL097, a TLR7/8 ligand, inhibits TLR-4—Dependent activation of IRAK-M and BCL-3 expression. Shock 2009, 32, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Forsbach, A.; Samulowitz, U.; Völp, K.; Hofmann, H.-P.; Noll, B.; Tluk, S.; Schmitz, C.; Wader, T.; Müller, C.; Podszuweit, A.; et al. Dual or triple activation of TLR7, TLR8, and/or TLR9 by single-stranded oligoribonucleotides. Nucleic Acid Ther. 2011, 21, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Jähnisch, H.; Wehner, R.; Tunger, A.; Kunze, A.; Oehrl, S.; Schäkel, K.; Rohayem, J.; Bornhäuser, M.; Tonn, T.; Bachmann, M.; et al. TLR7/8 agonists trigger immunostimulatory properties of human 6-sulfo LacNAc dendritic cells. Cancer Lett. 2013, 335, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.J.; O’Neill, L.A.J. New developments in Toll-like receptor targeted therapeutics. Curr. Opin. Pharmacol. 2012, 12, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Meng, T.-C.; Tomai, M.A. The antiviral activity of Toll-like receptor 7 and 7/8 agonists. Drug News Perspect. 2008, 21, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, E.C.; Liu, H.; Schwartz, M.J.; Scherr, D.S. Toll-like receptor 7 agonist therapy with imidazoquinoline enhances cancer cell death and increases lymphocytic infiltration and proinflammatory cytokine production in established tumors of a renal cell carcinoma mouse model. J. Oncol. 2012, 2012, 103298. [Google Scholar] [CrossRef] [PubMed]
- Imiquimod—National Cancer Institute. Available online: http://www.cancer.gov/cancertopics/druginfo/imiquimod (accessed on 19 June 2014).
- Chang, Y.C.; Madkan, V.; Cook-Norris, R.; Sra, K.; Tyring, S. Current and potential uses of imiquimod. South. Med. J. 2005, 98, 914–920. [Google Scholar] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 2002, 3, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Bilu, D.; Sauder, D.N. Imiquimod: Modes of action. Br. J. Dermatol. 2003, 149 (Suppl.), 5–8. [Google Scholar] [PubMed]
- Henriques, L.; Palumbo, M.; Guay, M.-P.; Bahoric, B.; Basik, M.; Kavan, P.; Batist, G. Imiquimod in the treatment of breast cancer skin metastasis. J. Clin. Oncol. 2014, 32, e22–e25. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Chuang, T.-H.; Redecke, V.; She, L.; Pitha, P.M.; Carson, D.A.; Raz, E.; Cottam, H.B. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: Activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2003, 100, 6646–6651. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Surber, C.; French, L.E.; Stockfleth, E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin. Investig. Drugs 2013, 22, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Forsbach, A.; Müller, C.; Montino, C.; Kritzler, A.; Nguyen, T.; Weeratna, R.; Jurk, M.; Vollmer, J. Negative regulation of the type I interferon signaling pathway by synthetic Toll-like receptor 7 ligands. J. Interferon Cytokine Res. 2012, 32, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Vultaggio, A.; Nencini, F.; Pratesi, S.; Maggi, L.; Guarna, A.; Annunziato, F.; Romagnani, S.; Parronchi, P.; Maggi, E. The TLR7 ligand 9-benzyl-2-butoxy-8-hydroxy adenine inhibits IL-17 response by eliciting IL-10 and IL-10-inducing cytokines. J. Immunol. 2011, 186, 4707–4715. [Google Scholar] [CrossRef]
- Weterings, J. 7-Hydro-8-oxo-adenine Derivatives as Potent TLR7 Ligands. Available online: https://openaccess.leidenuniv.nl/bitstream/handle/1887/13284/Chapter.2.pdf?sequence=10 (accessed on 19 June 2014).
- Heil, F.; Ahmad-Nejad, P.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Gellert, T.; Dietrich, H.; Lipford, G.; Takeda, K.; Akira, S.; et al. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur. J. Immunol. 2003, 33, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Chan, M.; Norton, J.T.; Wu, C.C.N.; Yao, S.; Cottam, H.B.; Tawatao, R.I.; Corr, M.; Carson, D.A.; Daniels, G.A. Additive melanoma suppression with intralesional phospholipid-conjugated TLR7 agonists and systemic IL-2. Melanoma Res. 2010, 21, 66–75. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z. Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Gong, J.; Jamitzky, F.; Heckl, W.M.; Stark, R.W.; Rössle, S.C. Homology modeling of human Toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci. 2009, 18, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Gong, J.; Rössle, S.C.; Jamitzky, F.; Heckl, W.M.; Stark, R.W. A leucine-rich repeat assembly approach for homology modeling of the human TLR5–10 and mouse TLR11–13 ectodomains. J. Mol. Model. 2011, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jin, H.; Sun, L.; Zhang, L.; Sun, G.; Wang, Z.; Yu, Y. Toll-like receptor 7 agonists: Chemical feature based pharmacophore identification and molecular docking studies. PLoS ONE 2013, 8, e56514. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-Y.; Gajewski, M.; Danani, A.; Tuszynski, J.A. Homology and Molecular Dynamics Models of Toll-Like Receptor 7 Protein and Its Dimerization. Chem. Biol. Drug Des. 2014, 83, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Amber Home Page. Available online: http://ambermd.org/ (accessed on 12 March 2015).
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2013.08. Available online: http://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 2 February 2014).
- Kelly, K.; Labute, P. The A* Search and Applications to Sequence Alignment. Available online: http://www.chemcomp.com/journal/astar.htm (accessed on 10 June 2014).
- Kelly, K. Multiple Sequence and Structure Alignment in MOE. Available online: http://www.chemcomp.com/journal/align.htm (accessed on 10 June 2014).
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- AmberTools 12 Reference Manual. Available online: http://ambermd.org/doc12/AmberTools12.pdf (accessed on 10 March 2015).
- Hoffmann, R. An Extended Hückel Theory. I. Hydrocarbons. J. Chem. Phys. 1963, 39, 1397. [Google Scholar] [CrossRef]
- Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry. Available online: http://www.ccl.net/cca/documents/proton/ (accessed on 31 March 2014).
- Wang, J.; Wang, W.; Kollmann, P.; Case, D. Antechamber, An Accessory Software Package for Molecular Mechanical Calculation. J. Comput. Chem. 2005, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Goodsell, D. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Schrödinger Release 2013–3: LigPrep, version 2.8, Schrödinger, LLC, New York, NY. Available online: http://www.schrodinger.com/productpage/14/10/ (accessed on 24 January 2014).
- Nussinov, R.; Wolfson, H.J. Efficient detection of three-dimensional structural motifs in biological macromolecules by computer vision techniques. Proc. Natl. Acad. Sci. USA 1991, 88, 10495–10499. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, H.J.; Rigoutsos, I. Geometric hashing: An overview. IEEE Comput. Sci. Eng. 1997, 4, 10–21. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Forli, S. Raccoon|AutoDock VS: An Automated Tool for Preparing AutoDock Virtual Screenings. Available online: http://autodock.scripps.edu/resources/raccoon (accessed on 7 March 2014).
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M. Ligand Interaction Diagrams. Available online: http://www.chemcomp.com/journal/ligintdia.htm (accessed on 27 November 2014).
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A. The Generalized Born Model: Its Foundation, Applications, and Limitations. Available online: http://people.cs.vt.edu/~onufriev/PUBLICATIONS/gbreview.pdf (accessed on 9 June 2014).
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Genheden, S.; Kuhn, O.; Mikulskis, P.; Hoffmann, D.; Ryde, U. The normal-mode entropy in the MM/GBSA method: Effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012, 52, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Introduction to Computational Chemistry; John Wiley & Sons: Chicester, UK, 2013; p. 624. [Google Scholar]
- Karplus, M.; Kushick, J.N. Method for estimating the configurational entropy of macromolecules. Macromolecules 1981, 14, 325–332. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Jayaram, B.; Liu, Y.; Beveridge, D.L. A modification of the generalized Born theory for improved estimates of solvation energies and pK shifts. J. Chem. Phys. 1998, 109, 1465. [Google Scholar] [CrossRef]
- Onufriev, A.; Case, D.A.; Bashford, D. Effective Born radii in the generalized Born approximation: The importance of being perfect. J. Comput. Chem. 2002, 23, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A. Chapter 7 Implicit Solvent Models in Molecular Dynamics Simulations: A Brief Overview. Annu. Rep. Comput. Chem. 2008, 4, 125–137. [Google Scholar]
- Zhu, J.; Shi, Y.; Liu, H. Parametrization of a Generalized Born/Solvent-Accessible Surface Area Model and Applications to the Simulation of Protein Dynamics. J. Phys. Chem. B 2002, 106, 4844–4853. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kommission für Technologie und Innovation KTI. Available online: http://www.kti.admin.ch/?lang=en (accessed on 15 February 2015).
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, F.; Deriu, M.A.; Licandro, G.; Prunotto, A.; Danani, A.; Tuszynski, J.A. Structure Based Modeling of Small Molecules Binding to the TLR7 by Atomistic Level Simulations. Molecules 2015, 20, 8316-8340. https://doi.org/10.3390/molecules20058316
Gentile F, Deriu MA, Licandro G, Prunotto A, Danani A, Tuszynski JA. Structure Based Modeling of Small Molecules Binding to the TLR7 by Atomistic Level Simulations. Molecules. 2015; 20(5):8316-8340. https://doi.org/10.3390/molecules20058316
Chicago/Turabian StyleGentile, Francesco, Marco A. Deriu, Ginevra Licandro, Alessio Prunotto, Andrea Danani, and Jack A. Tuszynski. 2015. "Structure Based Modeling of Small Molecules Binding to the TLR7 by Atomistic Level Simulations" Molecules 20, no. 5: 8316-8340. https://doi.org/10.3390/molecules20058316
APA StyleGentile, F., Deriu, M. A., Licandro, G., Prunotto, A., Danani, A., & Tuszynski, J. A. (2015). Structure Based Modeling of Small Molecules Binding to the TLR7 by Atomistic Level Simulations. Molecules, 20(5), 8316-8340. https://doi.org/10.3390/molecules20058316