
Copper(I) Complexes of Mesoionic Carbene: Structural Characterization and Catalytic Hydrosilylation Reactions

Abstract
:
1. Introduction
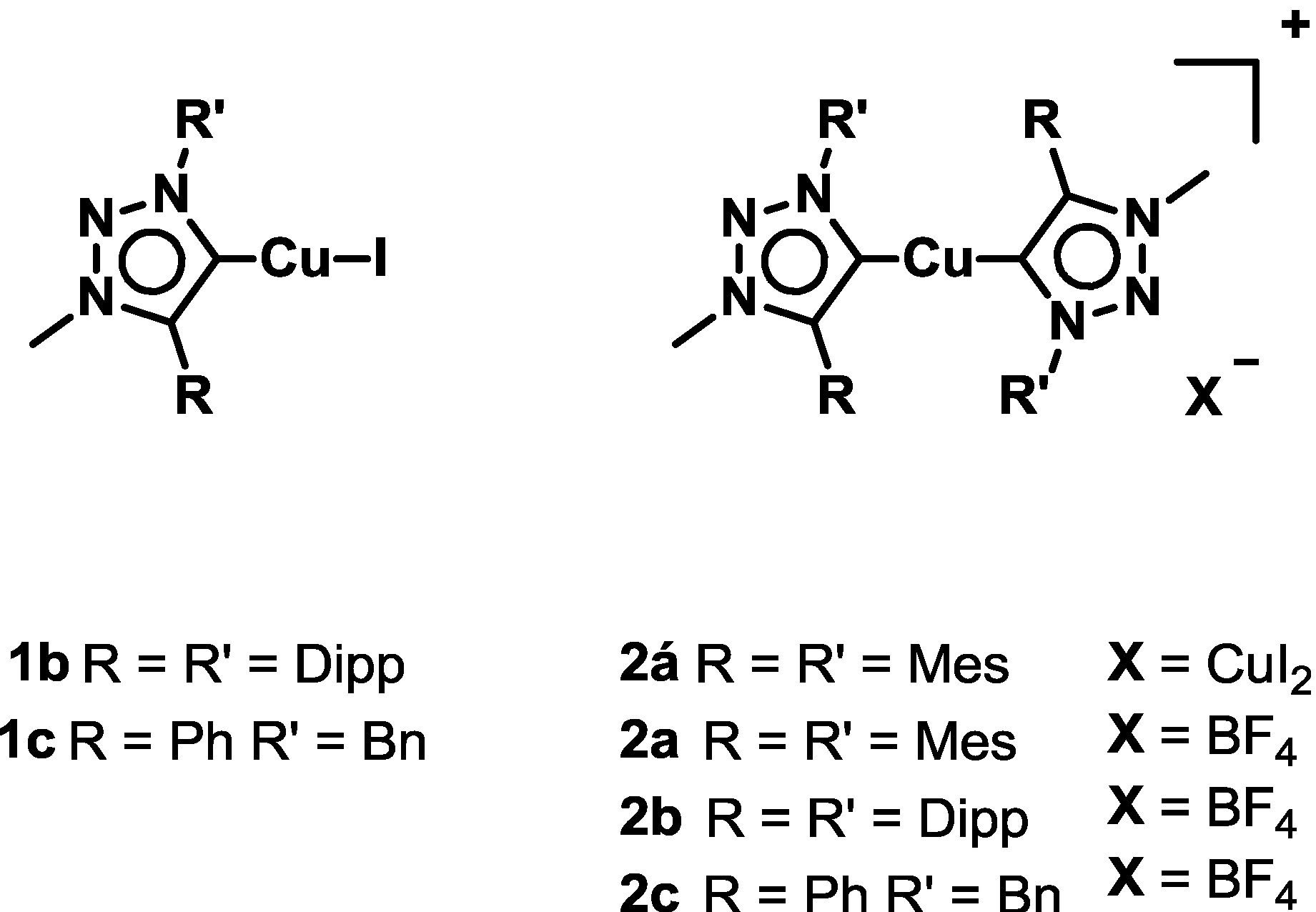
2. Results and Discussion
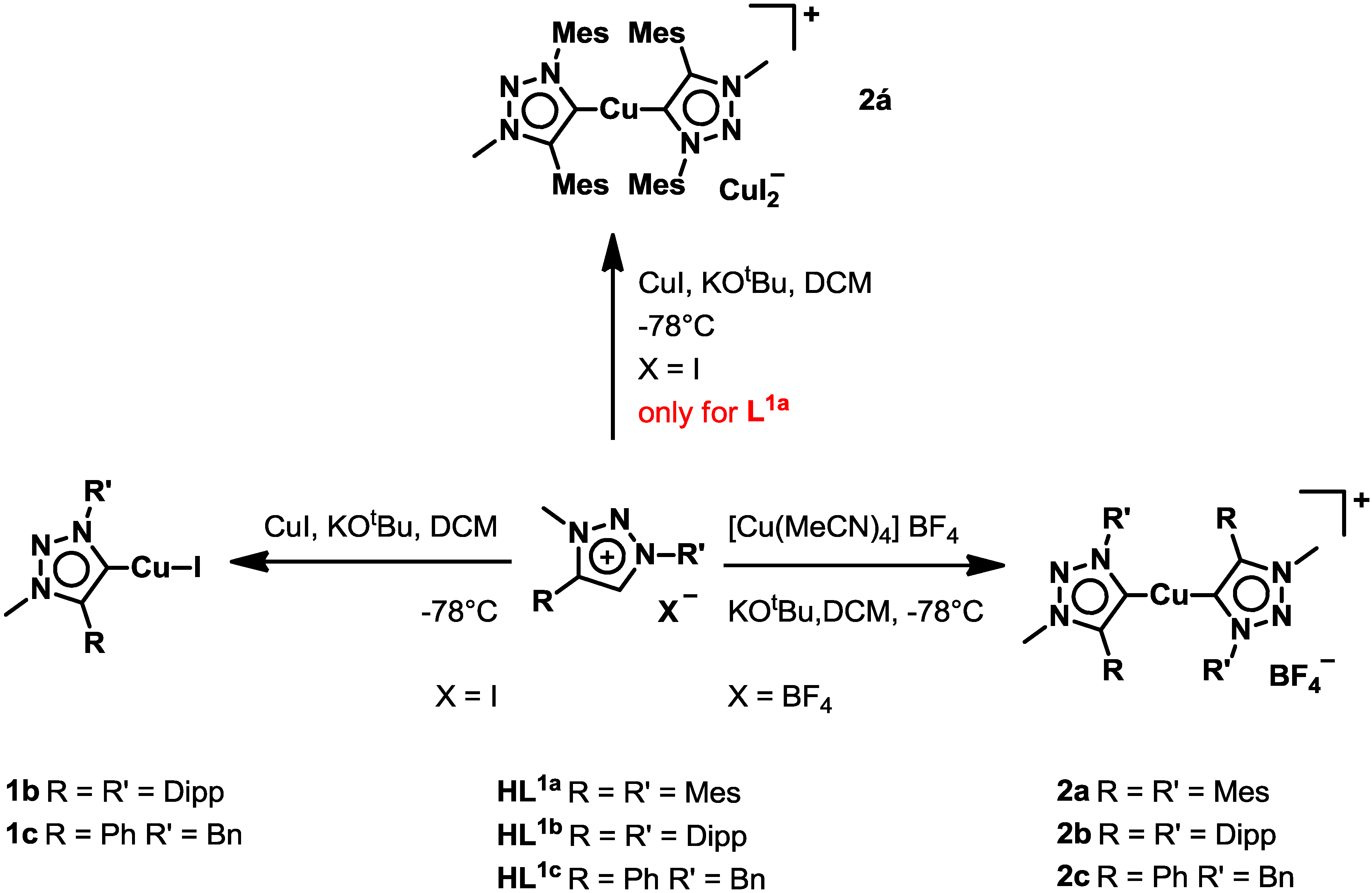
2.1. Synthesis and Characterization of Ligands and Complexes
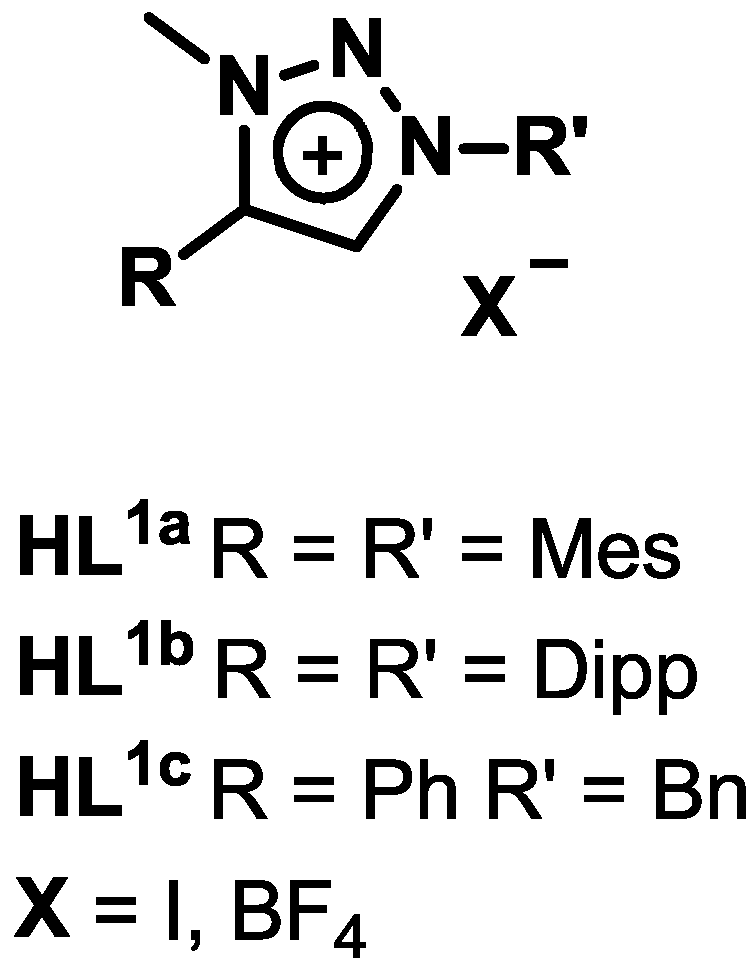
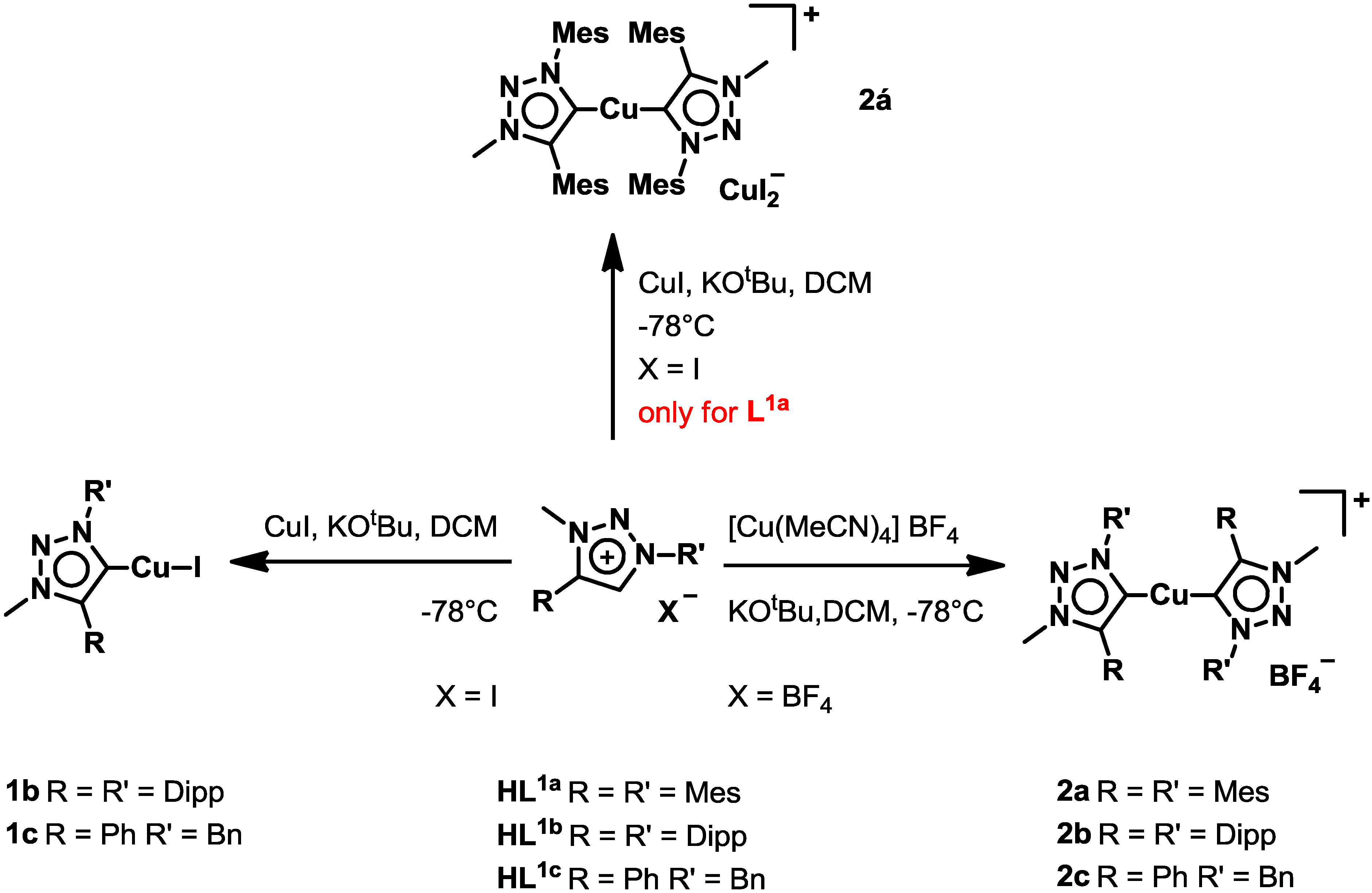
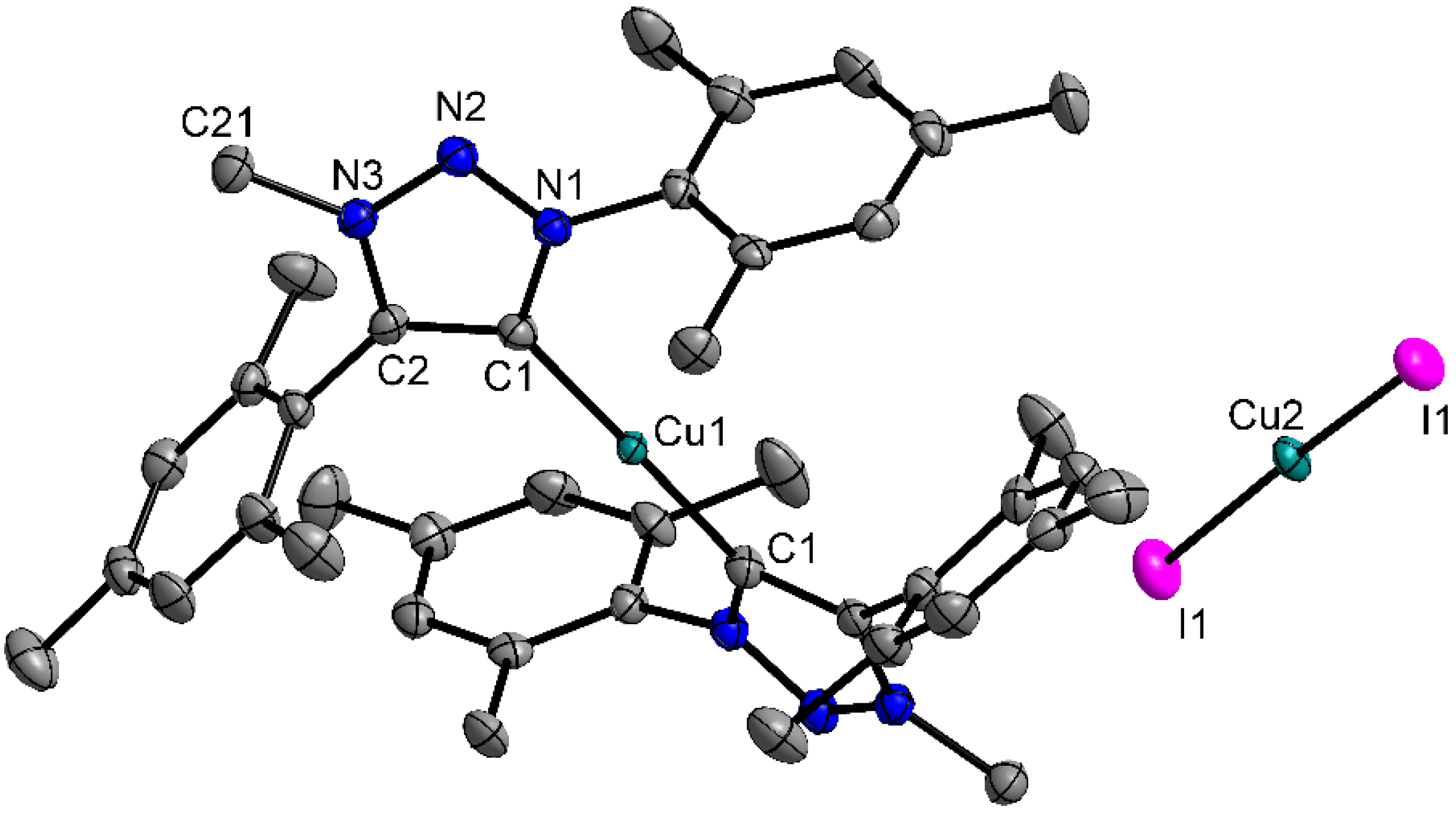
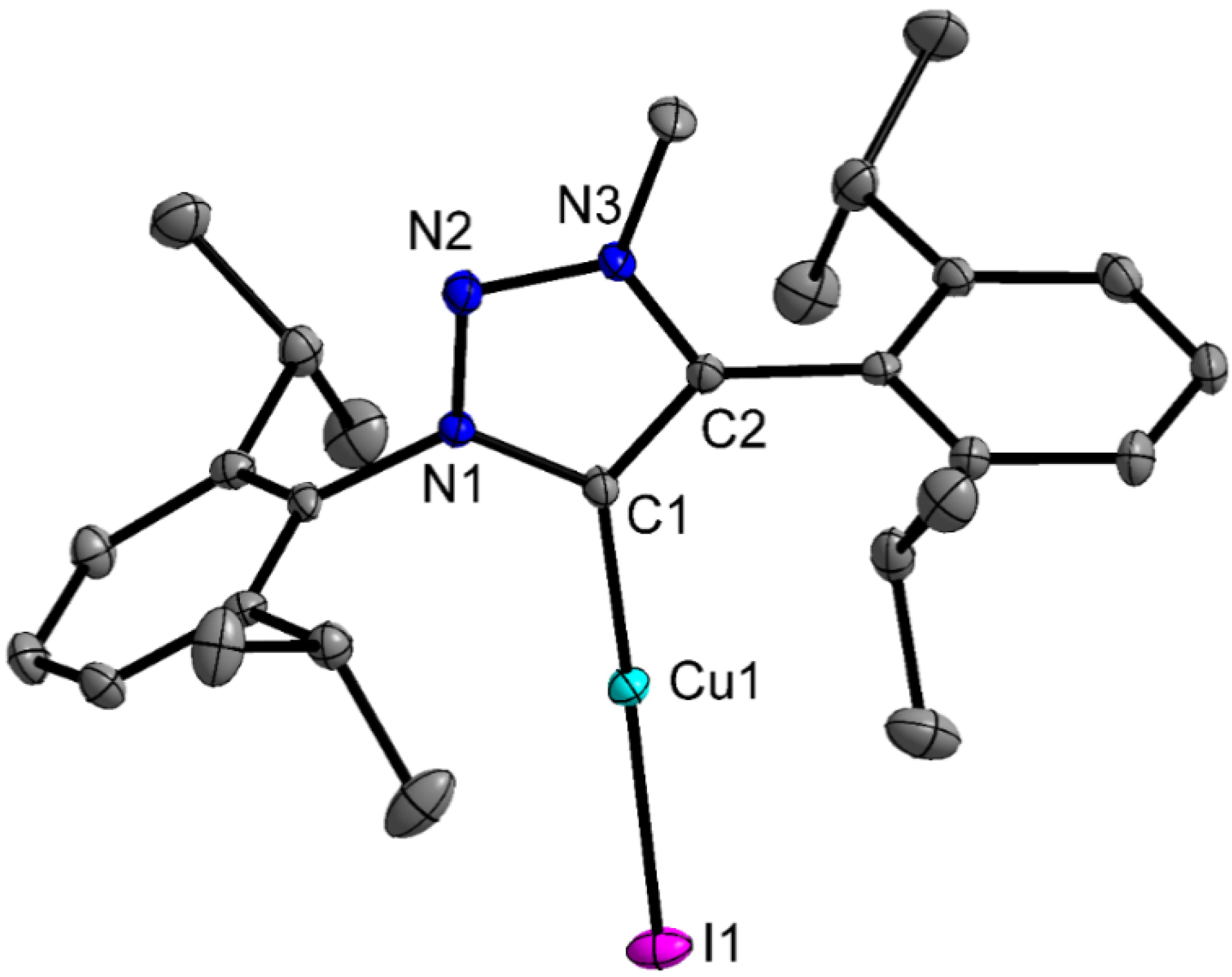
2.2. Crystal Structures

{kind=link}
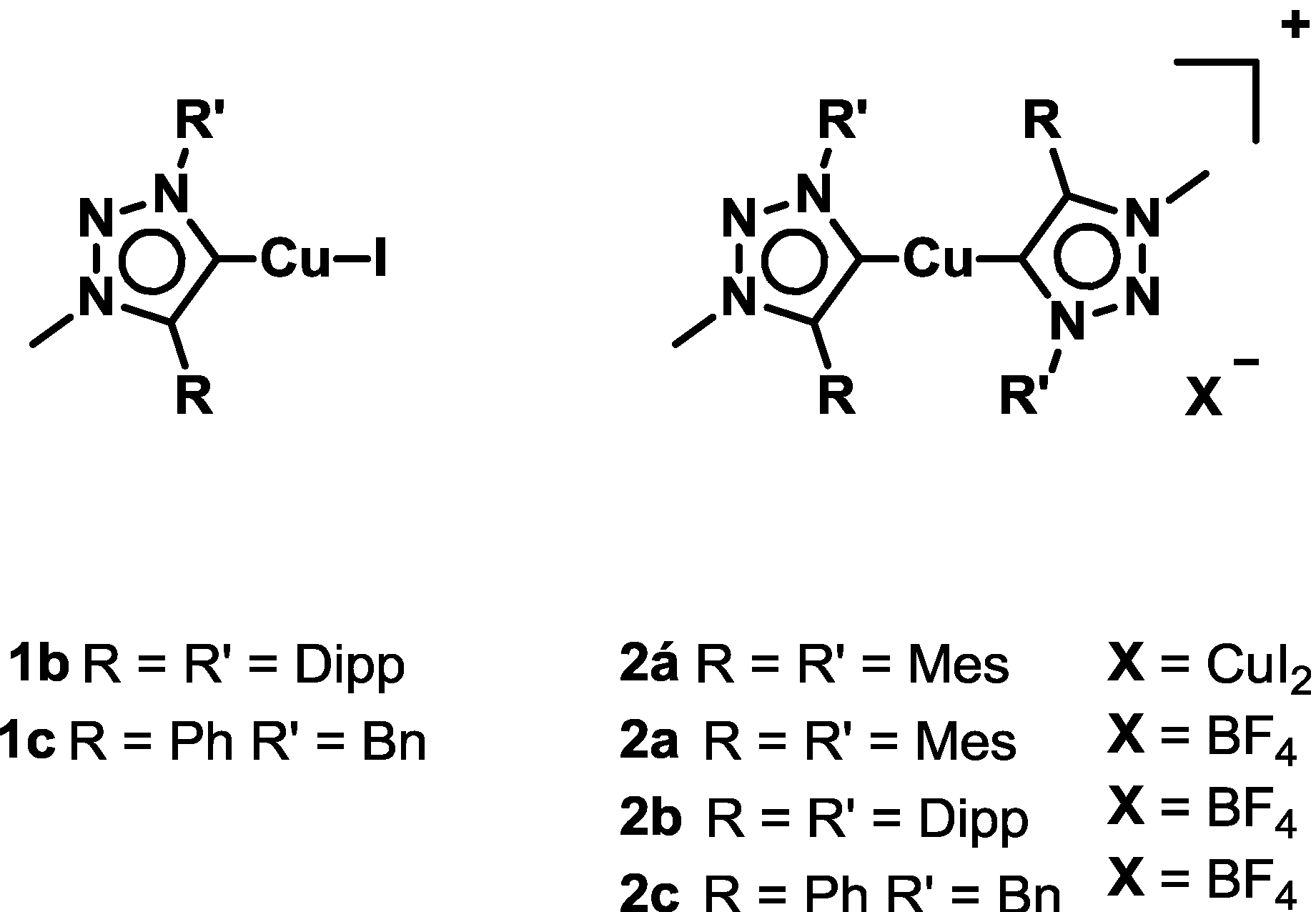{kind=link}
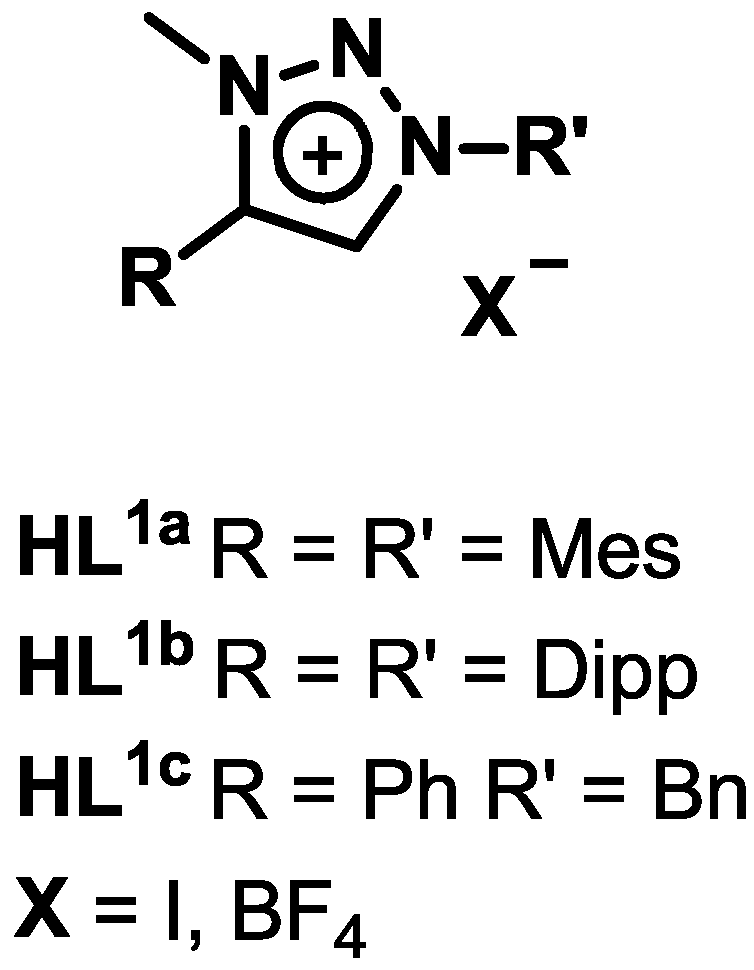{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
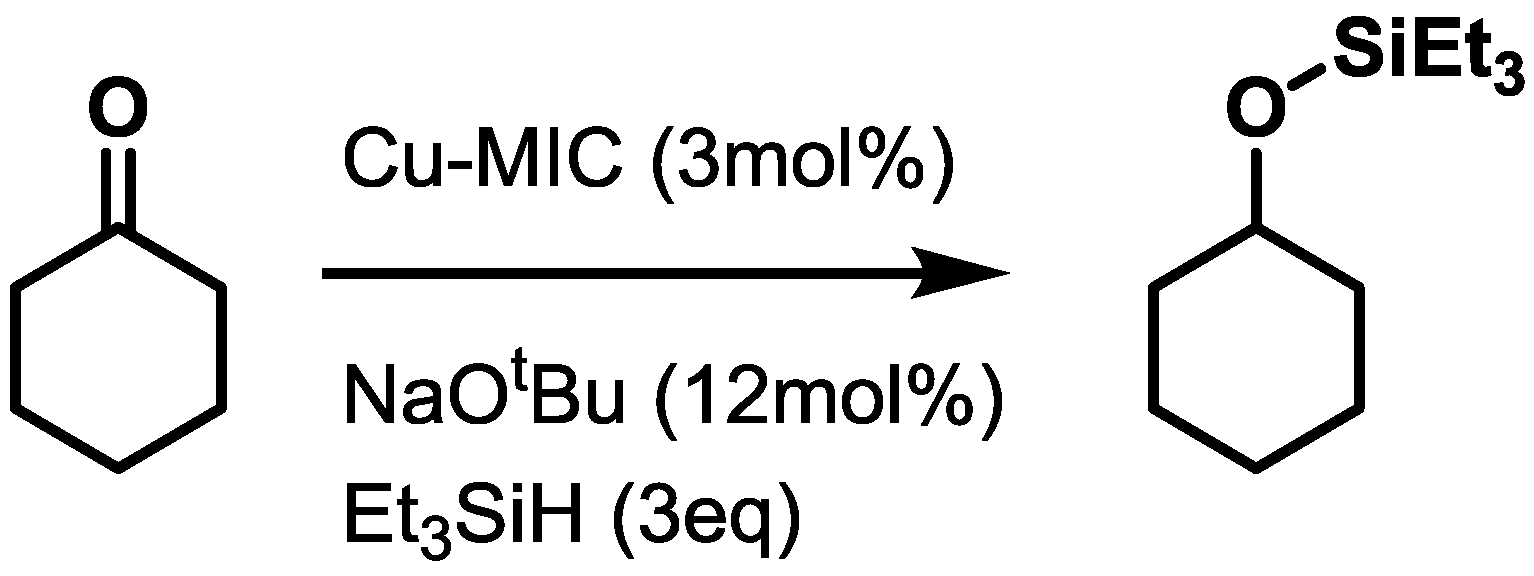{kind=link}
{kind=link}
| 2á | 1b | |
|---|---|---|
| Chemical formula | C42H50N6Cu1 Cu1I2 | C27H37N3Cu1I1 |
| Mr | 1019.76 | 594.04 |
| Crystal system, Space group | Monoclinic P2/c | orthorhombic Pbca |
| a (Å) | 12.969(4) | 16.3003(7) |
| b (Å) | 8.262(2) | 18.3924(8) |
| c (Å) | 20.397(6) | 18.5609(7) |
| α(°) | 90 | 90 |
| β (°) | 94.489(7) | 90 |
| γ (°) | 90 | 90 |
| V (Å3) | 2179(1) | 5564.6(4) |
| Z | 2 | 8 |
| Densitiy (g cm−3) | 1.554 | 1.418 |
| F(000) | 1016 | 2416 |
| Radiation Type | MoKα | MoKα |
| μ (mm−1) | 2.428 | 1.912 |
| Crystal size | 0.42 × 0.37 × 0.09 | 0.5 × 0.4 × 0.3 |
| Meas. Refl. | 22,753 | 71,218 |
| Indep. Refl. | 3869 | 7418 |
| Obsvd. [ I > 2σ(I)] refl. | 3000 | 6334 |
| Rint | 0.0419 | 0.1543 |
| R [F2 > 2σ(F2)], wR(F2), S | 0.0376, 0.1086, 1.048 | 0.0448, 0.1276, 1.106 |
| Δρmax, Δρmin (e Å−3) | 1.497, −0.607 | 1.970, −1.977 |
| CCDC | 965,501 |
| Atoms | 2á | 1b |
|---|---|---|
| Cu1-C1 | 1.877(5) | 1.893(3) |
| Cu1-I1 | - | 2.394(1) |
| Cu2-I1 | 2.403(1) | - |
| C1-C2 | 1.379(7) | 1.388(4) |
| C2-N3 | 1.367(6) | 1.365(4) |
| N3-N2 | 1.325(6) | 1.325(3) |
| N2-N1 | 1.334(5) | 1.329(3) |
| N1-C1 | 1.371(6) | 1.371(3) |
| C1-Cu1-C1 | 178.3(3) | - |
| C1-Cu1-I1 | - | 175.0(1) |
| I1-Cu2-I1 | 173.8(1) | - |
| N1-C1-C2 | 102.7(4) | 102.0(2) |
| trz-trz | 38.8(2) | - |
| Ntrz-R | 78.7(2) | 82.2(1) |
| Ctrz-R | 84.4(2) | 86.0(1) |
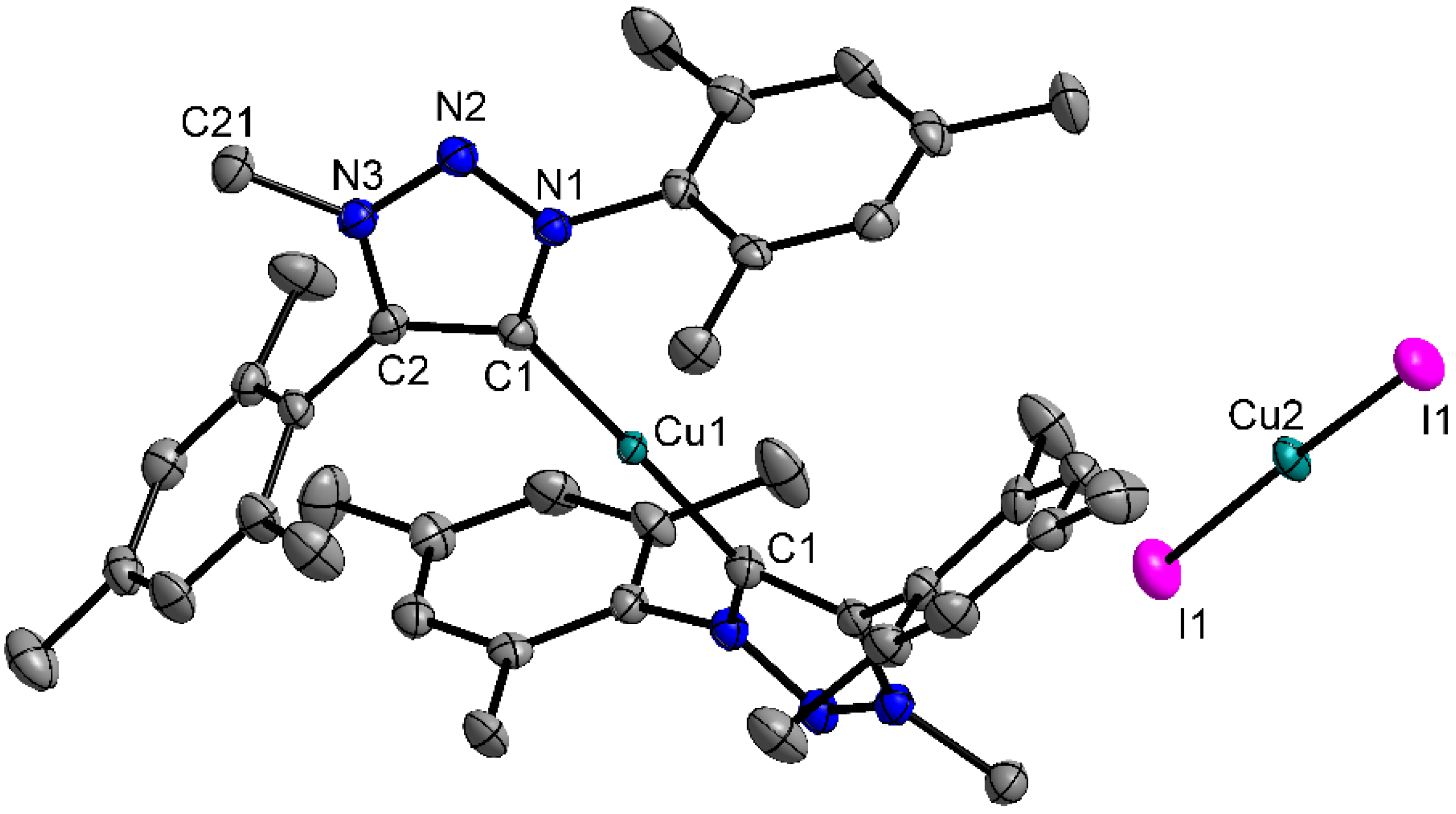
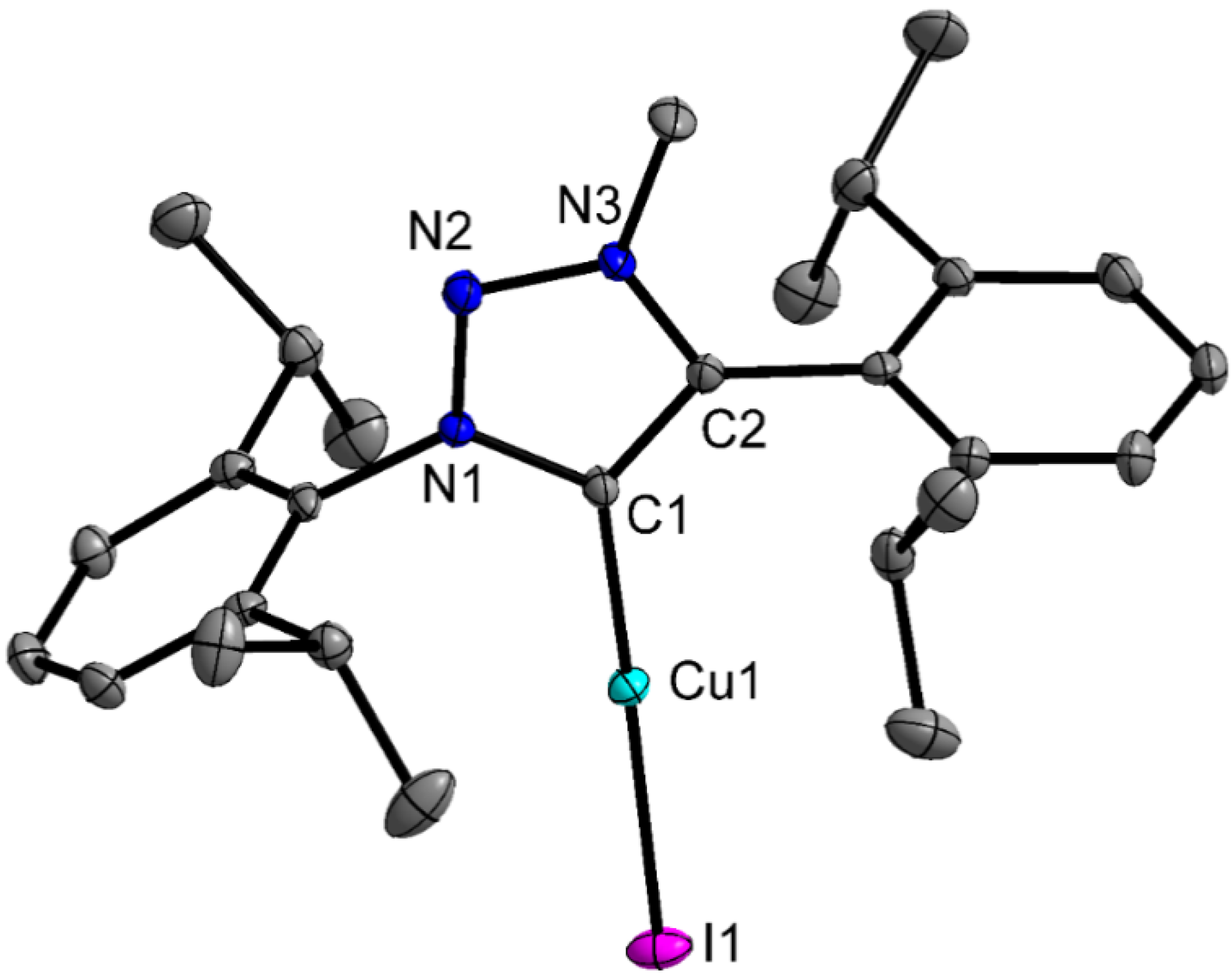
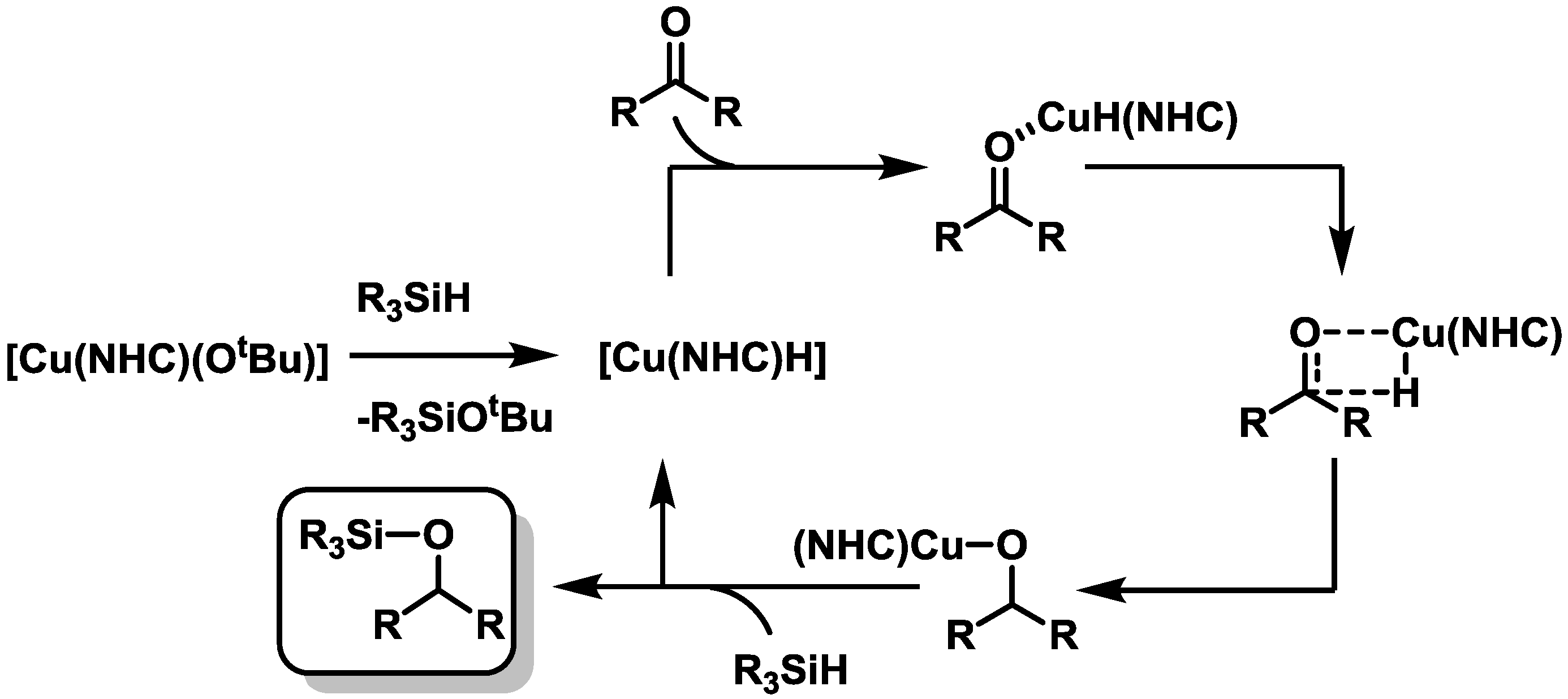
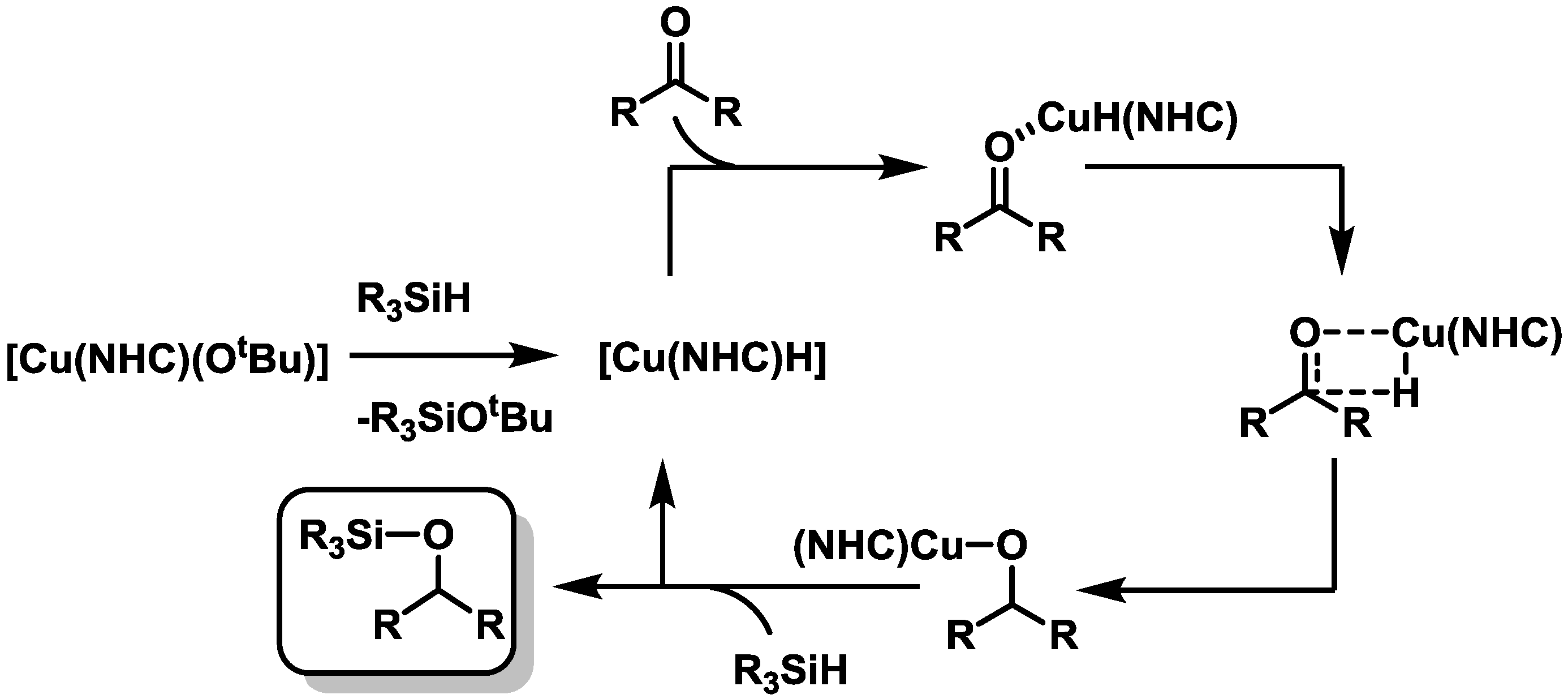
2.3. Catalysis
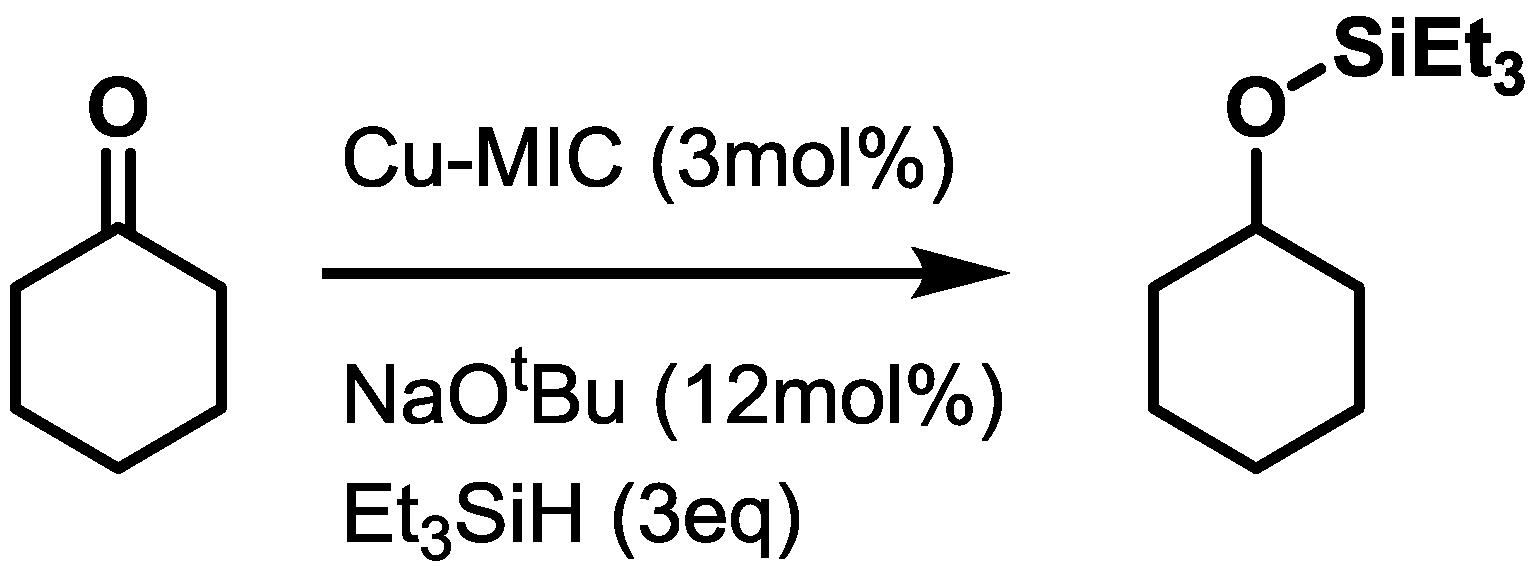
| Catalyst | Conversion [%] |
|---|---|
| 2á | >99 |
| 1b | 91 |
| 1c | 97 |
| 2a | >99 |
| 2b | 99 |
| 2c | >99 |
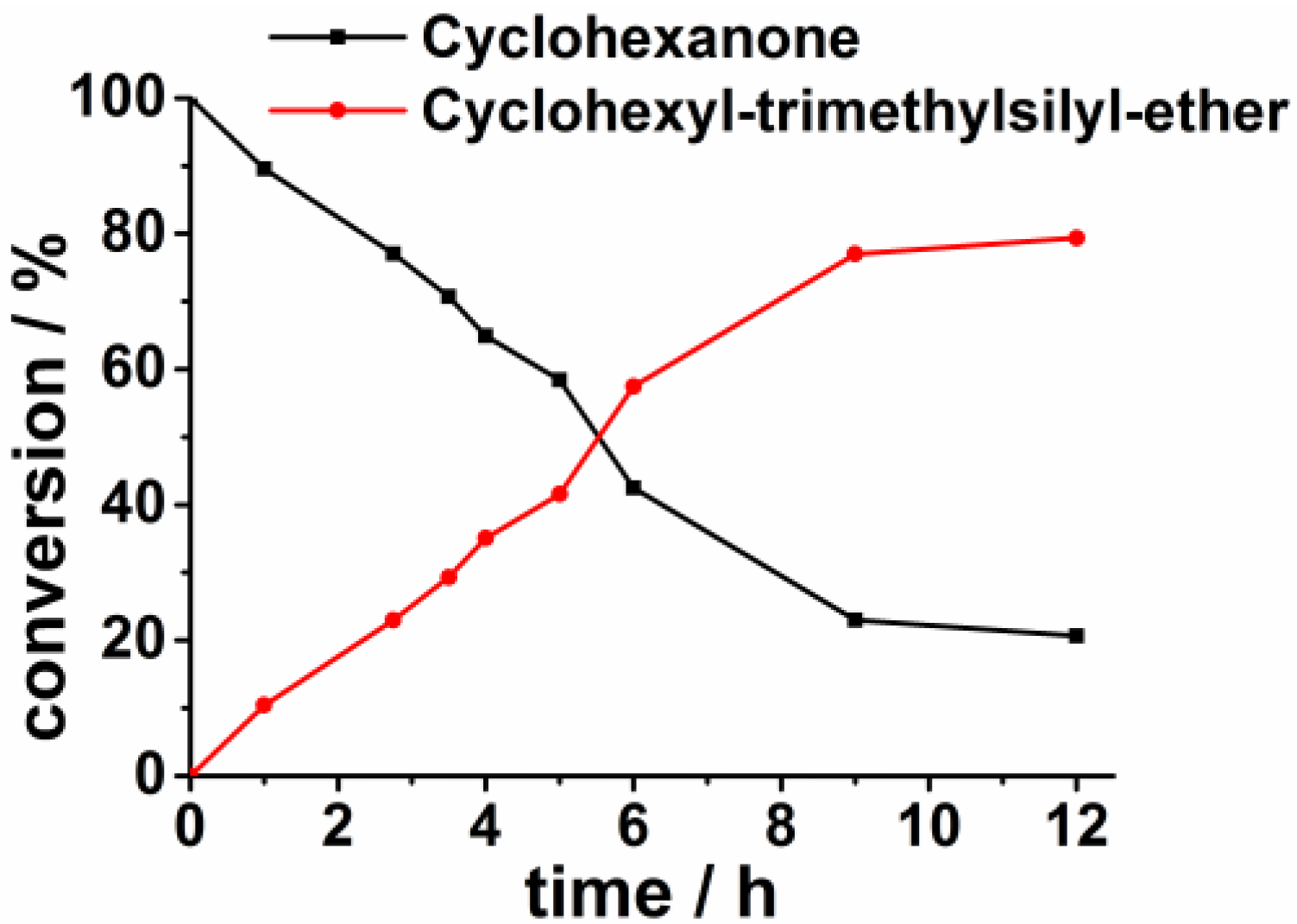
3. Experimental Section
3.1. General Information
3.2. X-ray Crystallography
3.3. Synthesis of Triazolium Salts
3.3.1. Synthesis of Copper(I)-Carbene Complexes
3.3.2. Hydrosilylations of Cyclohexanone
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry; Wiley-Interscience: New York, NY, USA, 2001. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry; Kluwer Academic/Plenum: New York, NY, USA, 2001. [Google Scholar]
- Caycho, J.R.; Tellado, F.G.; Aramas, P.; Juan, J.; Tellado, M. Sodium borohydride-amberlyst-15 (H+): An effective reductor for hindered and unreactive ketones in aprotic solvent. Tetrahedron. Lett. 1997, 38, 277–280. [Google Scholar] [CrossRef]
- Yakabe, S.; Hirano, M.; Morimoto, T. Alumina-assisted reduction of carbonyl compounds with sodium borohydride in hexane. Can. J. Chem. 1998, 76, 1916–1921. [Google Scholar]
- Yun, J.; Buchwald, S.L. Titanium-catalyzed asymmetric ketone hydrosilylation: The effect of catalyst activation protocol and additives on the rate and enantioselectivity. J. Am. Chem. Soc. 1999, 121, 5640–5644. [Google Scholar] [CrossRef]
- Tao, B.; Fu, G.C. Application of a new family of P,N ligands to the highly enantioselective hydrosilylation of aryl alkyl and dialkyl ketones. Angew. Chem. Int. Ed. 2002, 41, 3892–3894. [Google Scholar] [CrossRef]
- Reyes, C.; Prock, A.; Giering, W.P. Kinetic study of the hydrosilylation of acetophenone by [Rh(COD)Cl]2/(R)-BINAP. Organometallics 2002, 21, 546–554. [Google Scholar] [CrossRef]
- Hashimoto, H.; Aratani, I.; Kabuto, C.; Kira, M. Stoichiometric hydrosilylation of nitriles and catalytic hydrosilylation of imines and ketones using a μ-silane diruthenium complex. Organometallics 2003, 22, 2199–2201. [Google Scholar] [CrossRef]
- Saito, M.; Nishibayashi, Y.; Uemura, S. Synthesis of dinuclear complexes bearing metalloporphyrin-phosphine hybrid ligands and their catalytic activity toward hydrolilylation of ketones. Organometallics 2004, 23, 4012–4017. [Google Scholar] [CrossRef]
- Ojima, I. The Hydrosilylation Reaction: The Chemistry of Organosilicon Compounds; Patai, S., Rapaport, Z., Eds.; Wiley: New York, NY, USA, 1989; p. 1479. [Google Scholar]
- Ojima, I.; Li, Z.; Zhu, J. Recent Advances in the Hydrosilylation Reaction: Chemistry of Organic Silicon Compounds; Rappaport, Z., Apeloig, Y., Eds.; Wiley: New York, NY, USA, 1998; Volume 2, p. 1687. [Google Scholar]
- Jurkauskas, V.; Sadighi, J.P.; Buchwald, S.L. Conjugate reduction of α,β-unsaturated carbonyl compounds catalyzed by a copper carbene complex. Org. Lett. 2003, 5, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Kauer Zinn, F.; Stevens, E.D.; Nolan, S.P. (NHC)CuI (NHC = N-heterocyclic carbene) complexes as efficient catalysts for the reduction of carbonyl compounds. Organometallics 2004, 23, 1157–1160. [Google Scholar] [CrossRef]
- Díez-González, S.; Kaur, H.; Kauer Zinn, F.; Stevens, E.D.; Nolan, S.P. A simple and efficient copper-catalyzed procedure for the hydrosilylation of hindered and functionalized ketones. J. Org. Chem. 2005, 70, 4784–4796. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Scott, N.M.; Nolan, S.P. Cationic copper(I) complexes as efficient precatalysts for the hydrosilylation of carbonyl compounds. Organometallics 2006, 25, 2355–2358. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. Copper, silver and gold complexes in hydrosilylation reactions. Acc. Chem. Res. 2008, 41, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Stevens, E.D.; Scott, N.M.; Petersen, J.L.; Nolan, S.P. Synthesis and characterization of [Cu(NHC)2]X complexes: Catalytic and mechanistic studies of hydrosilylation reactions. Chem. Eur. J. 2008, 14, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Escudero-Adán, E.C.; Benet-Buchholz, J.; Stevens, E.D.; Slawin, A.M.Z.; Nolan, S.P. [(NHC)CuX] complexes: Synthesis, characterization and catalytic activities in reduction reactions and Click chemistry. On the advantage of using well-defined catalytic systems. Dalton Trans. 2010, 39, 7595–7606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, J.; Hou, Z. Highly efficient catalytic hydrosilylation of carbon dioxide by an N-heterocyclic carbene copper catalyst. Chem. Commun. 2013, 49, 4782–4784. [Google Scholar] [CrossRef]
- John, F.; McGettigan, C.; Stryker, J.M. Selective reduction of alkynes to cis-alkenes by hydrometallation using [PPh3)CuH]6. Tetrahedron. Lett. 1990, 31, 2397–2400. [Google Scholar] [CrossRef]
- Brestensky, D.M.; Stryker, J.M. Regioselective conjugate reduction and reductive silylation of α,β-unsaturated aldehydes using [PPh3)CuH]6. Tetrahedron. Lett. 1989, 30, 5677–5680. [Google Scholar] [CrossRef]
- Mahoney, W.S.; Brestensky, D.M.; Stryker, J.M. Selective hydride-mediated conjugate reduction of α,β-unsaturated carbonyl compounds using [PPh3)CuH]6. J. Am. Chem. Soc. 1988, 110, 291–293. [Google Scholar] [CrossRef]
- Brestensky, D.M.; Huseland, D.E.; McGettigan, C.; Stryker, J.M. Simplified, one-pot procedure for the synthesis of [PPh3)CuH]6, a stable copper hydride for conjugate reductions. Tetrahedron. Lett. 1988, 29, 3749–3752. [Google Scholar] [CrossRef]
- Chiu, P.; Li, Z.; Fung, K.C.M. An expedient preparation of Stryker’s reagent. Tetrahedron Lett. 2003, 44, 455–457. [Google Scholar] [CrossRef]
- Deutsch, C.; Krause, N.; Lipshutz, B.H. CuH-catalyzed reactions. Chem. Rev. 2008, 108, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Vergote, T.; Nahra, F.; Merschaert, A.; Riant, O.; Peeters, D.; Leyssens, T. Mechanistic insight into the(NHC)copper(I)-catalyzed hydrosilylation of ketones. Organometallics 2014, 33, 1953–1963. [Google Scholar] [CrossRef]
- Gruendemann, S.; Kovacevic, A.; Albrecht, M.; Faller, J.W.; Crabtree, R.H. Abnormal ligand binding and reversible ring hydrogenation in the reaction of imidazolium salts with IrH5(PPh3)2. J. Chem. Am. Soc. 2002, 124, 10473–10481. [Google Scholar] [CrossRef]
- Han, Y.; Huynh, H.V.; Tan, G.L. Palladium(II) pyrazolin-4-ylidenes: Remote N-heterocyclic carbene complexes and their catalytic application in aqueous Suzuki-Miyaura coupling. Organometallics 2007, 26, 6581–6585. [Google Scholar] [CrossRef]
- Schuster, O.; Raubenheimer, H.G.; Albrecht, M. Beyond conventional N-heterocyclic carbenes: Abnormal, remote and other classes of NHC ligands with reduced heteroatom stabilization. Chem. Rev. 2009, 109, 3445–3478. [Google Scholar] [CrossRef] [PubMed]
- Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Crystalline 1H-1,2,3-triazole-5-ylidenes: New stable mesoionic carbenes (MICs). Angew. Chem. Int. Ed. 2010, 49, 4759–4762. [Google Scholar] [CrossRef]
- Crabtree, R.H. Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord. Chem. Rev. 2013, 257, 755–766. [Google Scholar] [CrossRef]
- Donnelly, K.F.; Petronilho, A.; Albrecht, M. Application of 1,2,3-triazolylidenes as versatile NHC-type ligands: Synthesis, properties and application in catalysis and beyond. Chem. Commun. 2013, 49, 1145–1159. [Google Scholar] [CrossRef]
- Crowley, J.D.; Lee, A.; Kilpin, K.J. 1,3,4-Trisubstituted-1,2,3-triazole-5-ylidene ‘Click’ carbene ligands: Synthesis, catalysis and self-assembly. Aust. J. Chem. 2011, 64, 1118–1132. [Google Scholar] [CrossRef]
- Arnold, P.L.; Pearson, S. Abnormal N-heterocyclic carbenes. Coord. Chem. Rev. 2007, 251, 596–609. [Google Scholar] [CrossRef]
- Schweinfurth, D.; Deibel, N.; Weisser, F.; Sarkar, B. With Click towards new ligands. Nachr. Chem. 2011, 59, 937–941. [Google Scholar] [CrossRef]
- Mathew, P.; Neels, A.; Albrecht, M. 1,2,3-Triazolylidenes as versatile abnormal carbene ligands for late transition metals. J. Am. Chem. Soc. 2008, 130, 13534–13535. [Google Scholar] [CrossRef] [PubMed]
- Lalrempuia, R.; McDaniel, N.D.; Müller-Bunz, H.; Bernhard, S.; Albrecht, M. Water oxidation catalyzed by strong carbene-type donor-ligand complexes of iridium. Angew. Chem. Int. Ed. 2010, 49, 9765–9768. [Google Scholar] [CrossRef]
- Karthikeyan, T.; Sankararaman, S. Palladium complexes with abnormal N-heterocyclic carbene ligands derived from 1,2,3-triazolium ions and their application in Suzuki coupling. Tetrahedron Lett. 2009, 50, 5834–5837. [Google Scholar] [CrossRef]
- Inomata, S.; Hiroki, H.; Terashima, T.; Ogata, K.; Fukuzawa, S. 1,2,3-Triazol-5-ylidene-palladium complex catalyzed Mizoroki-Heck and Sonogashira coupling reactions. Tetrahedron 2011, 67, 7263–7267. [Google Scholar] [CrossRef]
- Terashima, T.; Inomata, S.; Ogata, K.; Fukuzawa, S. Synthetic, structural and catalytic studies of well-defined allyl 1,2,3-triazol-5-ylidene (tzNHC) palladium complexes. Eur. J. Inorg. Chem. 2012, 1387–1393. [Google Scholar] [CrossRef]
- Hohloch, S.; Hettmanczyk, L.; Sarkar, B. Introducing potential hemilability into “Click” triazoles and triazolylidenes: Synthesis and characterization of d6-metal complexes and oxidation catalysis. Eur. J. Inorg. Chem. 2014, 3164–3171. [Google Scholar] [CrossRef]
- Bolje, A.; Hohloch, S.; Urankar, D.; Pevec, A.; Gazvoda, M.; Sarkar, B.; Kosmrlj, J. Exploring the scope of pyridyl- and picolyl-functionalized 1,2,3-triazol-5-ylidenes in bidentate coordination to ruthenium(II) cymene chloride complexes. Organometallics 2014, 33, 2588–2598. [Google Scholar] [CrossRef]
- Bolje, A.; Kosmrlj, J. A selective approach to pyridine appended 1,2,3-triazolium salts. Org. Lett. 2013, 15, 5084–5087. [Google Scholar] [CrossRef] [PubMed]
- Hohloch, S.; Suntrup, L.; Sarkar, B. Arene-ruthenium(II) and -iridium(III) complexes with “Click”-based pyridyl-triazoles, bis-triazoles, and chelating abnormal carbenes: Applications in catalytic transfer hydrogenation of nitrobenzene. Organometallics 2013, 32, 7376–7385. [Google Scholar] [CrossRef]
- Hohloch, S.; Frey, W.; Su, C.-Y.; Sarkar, B. Abnormal carbenes derived from the 1,5-cycloaddition product between azides and alkynes: Structural characterization of Pd(II) complexes and their catalytic properties. Dalton Trans. 2013, 42, 11355–11358. [Google Scholar] [CrossRef] [PubMed]
- Keske, E.C.; Zenkina, O.V.; Wang, R.; Crudden, C.M. Synthesis and structure of silver and rhodium 1,2,3-triazol-5-ylidene mesoionic carbene complexes. Organometallics 2012, 31, 456–461. [Google Scholar] [CrossRef]
- Hohloch, S.; Kaiser, S.; Duecker, F.L.; Bolje, A.; Maity, R.; Kosmrlj, J.; Sarkar, B. Catalytic oxygenation of sp3 “C-H” bonds with Ir(III) complexes of chelating triazoles and mesoionic carbenes. Dalton Trans. 2015, 44, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Maity, R.; Hohloch, S.; Su, C.-Y.; van der Meer, M.; Sarkar, B. Cyclometalled mono- and dinuclear IrIII complexes with “Click”-derived triazoles and mesoionic carbenes. Chem. Eur. J. 2014, 20, 9952–9961. [Google Scholar] [CrossRef] [PubMed]
- Maity, R.; van der Meer, M.; Sarkar, B. Redox-active multinuclear Pd(II) complexes with bis- and tris-mesoionic carbenes. Dalton Trans. 2015, 44, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.R.; Young, P.C.; Lucas, N.T.; Lee, A.-L.; Crowley, J.D. Gold(I) and palladium(II) complexes of 1,2,4-trisubstituted 1,2,3-triazol-5-ylidene “Click” carbenes: Systematic study of the electronic and steric influence on catalytic activity. Organometallics 2013, 32, 7065–7076. [Google Scholar] [CrossRef] [PubMed]
- Rostostev, V.V.; Greem, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I) catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes and azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Hohloch, S.; Su, C.-Y.; Sarkar, B. Copper(I) complexes of normal and abnormal carbenes and their use as catalysts for the Huisgen [3+2] cycloaddition between azides and alkynes. Eur. J. Inorg. Chem. 2011, 3067–3075. [Google Scholar] [CrossRef]
- Hohloch, S.; Scheiffele, D.; Sarkar, B. Activating azides and alkynes for the Click reaction with [Cu(aNHC)2I] or [Cu(aNHC)2]+ (aNHC = triazole-derived abnormal carbenes): Structural characterization and catalytic properties. Eur. J. Inorg. Chem. 2013, 3956–3965. [Google Scholar] [CrossRef]
- Hohloch, S.; Sarkar, B.; Nauton, L.; Cisnetti, F.; Gautier, A. Are Cu(I) mesoionic NHC carbenes associated with nitrogen additives the best Cu-carbene catalysts for the azide-alkyne click reaction in solution? A case study. Tetrahedron Lett. 2013, 54, 1808–1812. [Google Scholar] [CrossRef]
- Nakamura, T.; Ogata, K.; Fukuzawa, S. Synthesis of dichlorobis(1,4-dimesityl-1H-1,2,3-triazol-5-ylidene) palladium [PdCl2(TMes)2] and its application to Suzuki-Miyaura coupling reaction. Chem. Lett. 2010, 39, 920–922. [Google Scholar] [CrossRef]
- Nakamura, T.; Terashima, T.; Ogata, K.; Fukuzawa, S.-I. Copper(I) 1,2,3-triazol-5-ylidene complexes as efficient catalysts for the click reactions of azides and alkynes. Org. Lett. 2011, 13, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Inomata, H.; Ogata, K.; Fukuzawa, S.; Hou, Z. Direct C-H carboxylation with carbon dioxide using 1,2,3-triazol-5-ylidene copper(I) complexes. Org. Lett. 2012, 14, 3986–3989. [Google Scholar] [CrossRef] [PubMed]
- Kilpin, K.J.; Paul, U.S.D.; Lee, A.-L.; Crowley, J.D.; Nolan, J.D. Gold(I) “click” 1,2,3-triazolylidenes: synthesis, self-assembly and catalysis. Chem. Commun. 2011, 47, 1, 328–330. [Google Scholar]
- Díez-González, S.; Correao, A.; Cavallo, L.; Nolan, S.P. (NHC)Copper(I)-catalyzed [3+2] cycloaddition of azides and mono- or disubstituted alkynes. Chem. Eur. J. 2006, 12, 7558–7564. [Google Scholar] [CrossRef] [PubMed]
- Díez-González, S.; Stevens, E.D.; Nolan, S.P. A [(NHC)CuCl] as a latent Click catalyst. Chem. Commun. 2008, 4747–4749. [Google Scholar] [CrossRef]
- Díez-González, S.; Nolan, S.P. [(NHC)2Cu]X complexes as efficient catalysts for the azide-alkyne click chemistry at low catalyst loading. Angew. Chem. Int. Ed. 2008, 47, 8881–8884. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELX-97; Program for Crystal Structure Refinement: Göttingen, Germany, 1997. [Google Scholar]
- Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 22 April 2015).
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hohloch, S.; Duecker, F.L.; Van der Meer, M.; Sarkar, B. Copper(I) Complexes of Mesoionic Carbene: Structural Characterization and Catalytic Hydrosilylation Reactions. Molecules 2015, 20, 7379-7395. https://doi.org/10.3390/molecules20047379
Hohloch S, Duecker FL, Van der Meer M, Sarkar B. Copper(I) Complexes of Mesoionic Carbene: Structural Characterization and Catalytic Hydrosilylation Reactions. Molecules. 2015; 20(4):7379-7395. https://doi.org/10.3390/molecules20047379
Chicago/Turabian StyleHohloch, Stephan, Fenja Leena Duecker, Margarethe Van der Meer, and Biprajit Sarkar. 2015. "Copper(I) Complexes of Mesoionic Carbene: Structural Characterization and Catalytic Hydrosilylation Reactions" Molecules 20, no. 4: 7379-7395. https://doi.org/10.3390/molecules20047379
APA StyleHohloch, S., Duecker, F. L., Van der Meer, M., & Sarkar, B. (2015). Copper(I) Complexes of Mesoionic Carbene: Structural Characterization and Catalytic Hydrosilylation Reactions. Molecules, 20(4), 7379-7395. https://doi.org/10.3390/molecules20047379