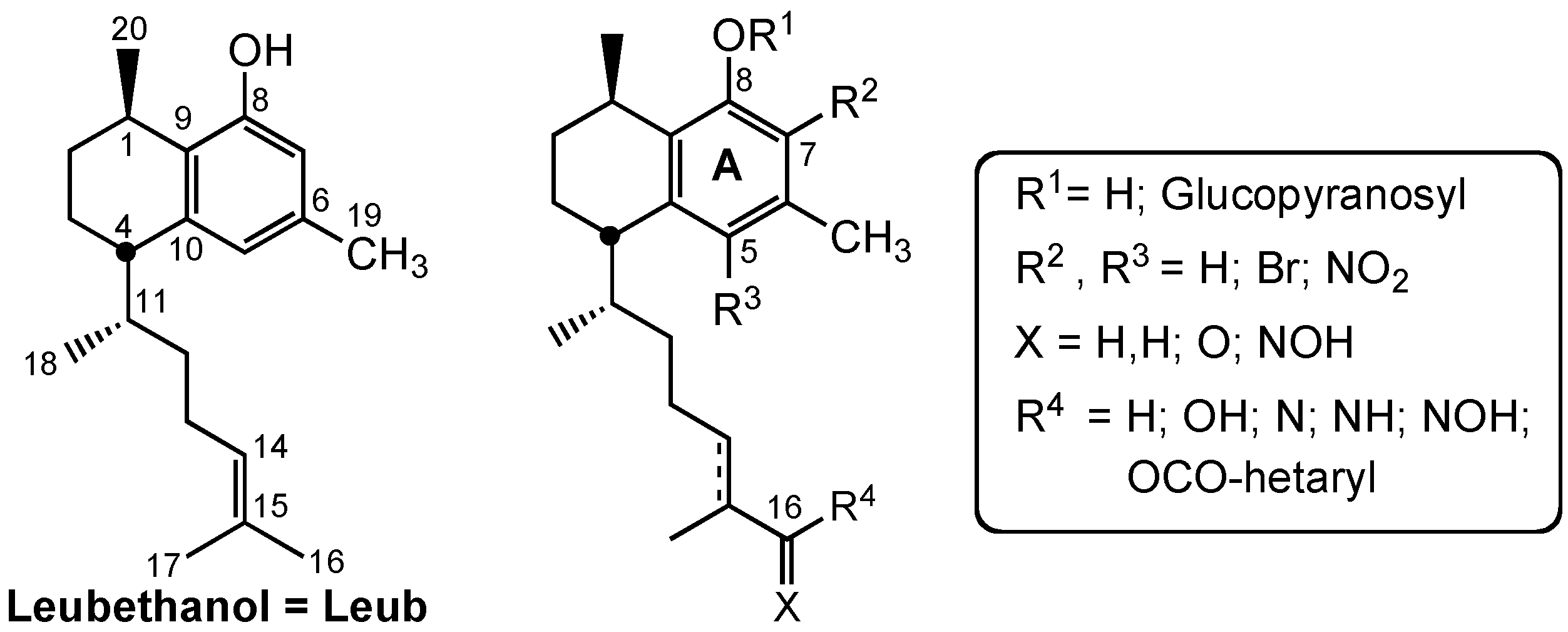
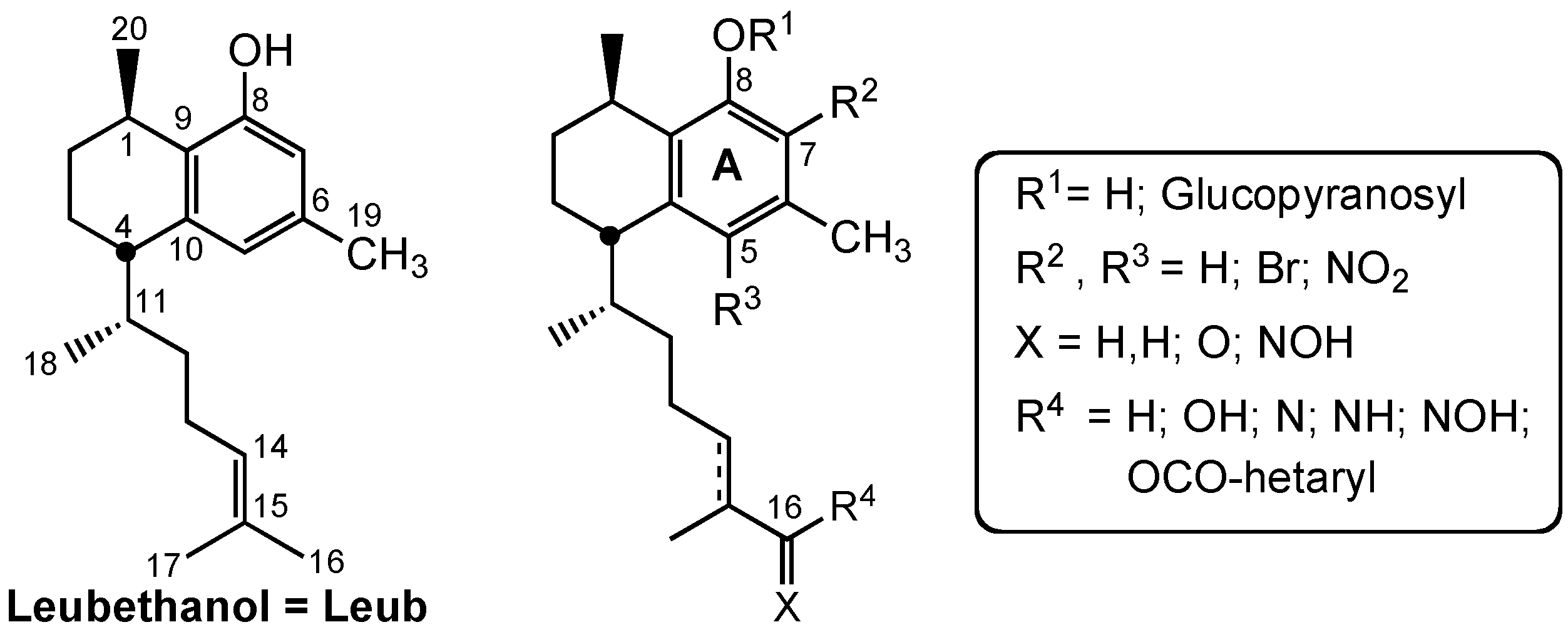
3.2. Chemistry
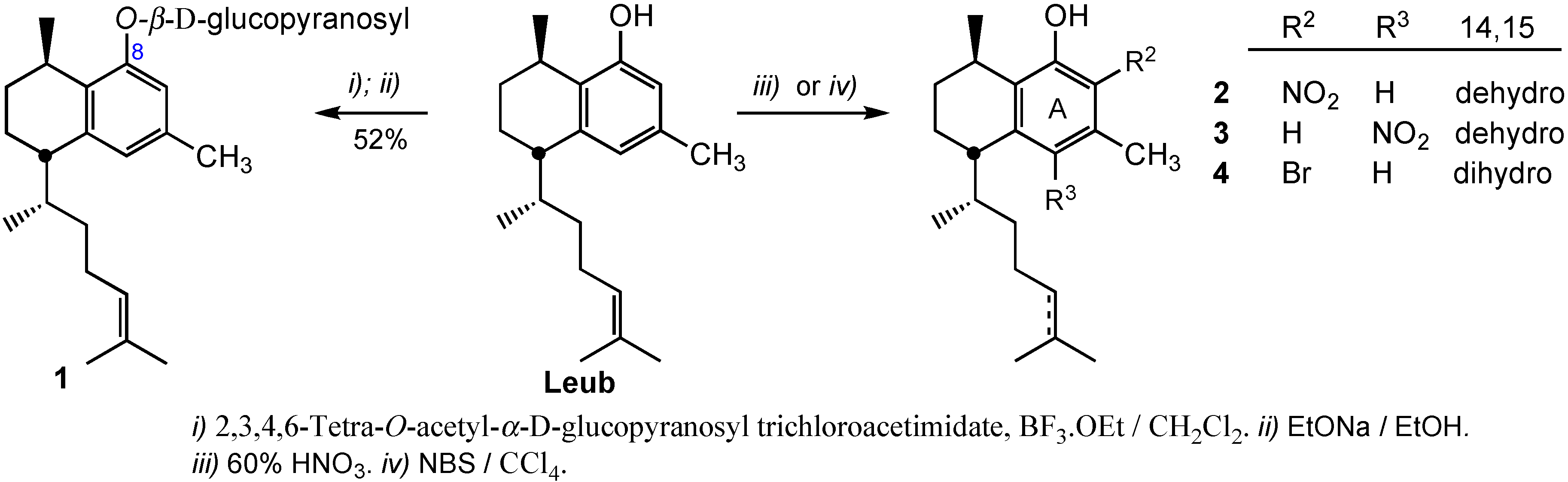
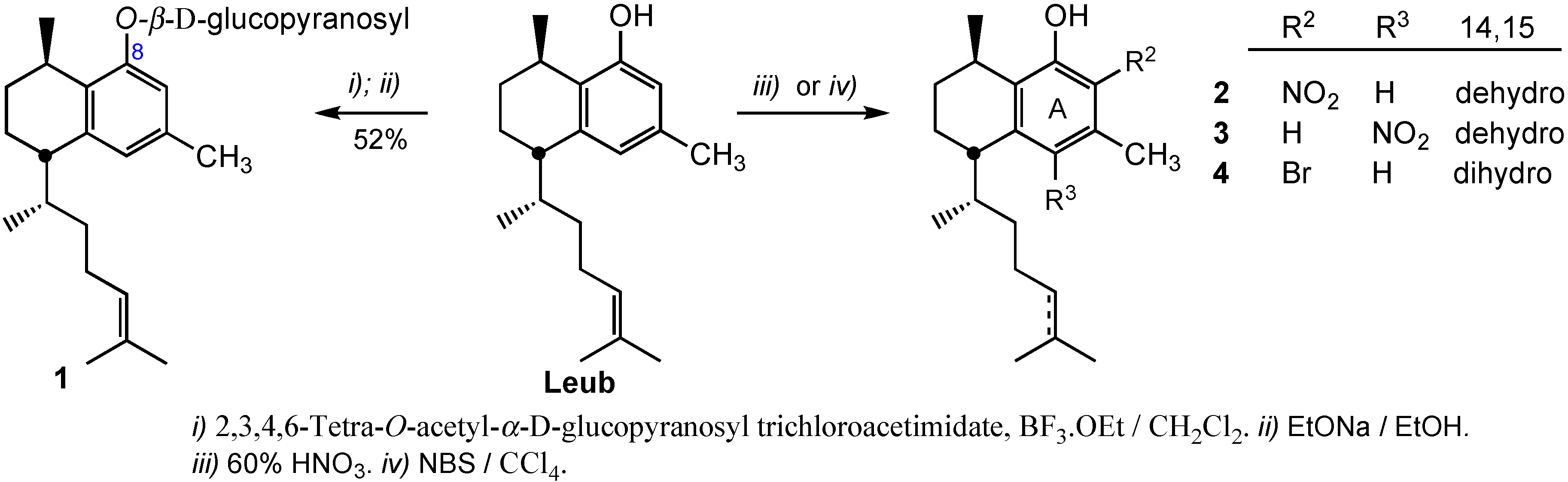
3.2.1. Preparation of 8-O-(2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl)leubethanol
To a stirred solution of leubethanol (112 mg, 0.40 mmol) in dry CH2Cl2 (5 mL) at −20 °C, BF3·Et2O (75 μL, 0.60 mmol) with a syringe was added. Immediately, the suspension turned into a light-yellow solution. The mixture was stirred for 45 min and then, a solution of 2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl trichloroacetimidate (292 mg, 0.59 mmol), recently obtained, in CH2Cl2 (2 mL) was added. The reaction was maintained for 4 h at room temperature. After that, the solution was diluted with CH2Cl2 and was washed with 10% NaHCO3, brine, water and dried over sodium sulphate. The organic layer was concentrated to give an oil, that was purified by column chromatography with n-hexane/ethyl acetate (95:5) as eluent to give 207 mg (80%) of 8-O-(2,3,4,6-tetra-O-acetyl-β-d-glucopyranosyl) leubethanol. IR: 2956, 2928, 2870, 1755, 1696, 1612, 1606, 1575, 1516, 1450, 1378, 1226, 1108, 1043, 832, 750 cm−1; 1H-NMR (CDCl3): δ 0.95 (3H, d, J = 6.8 Hz), 1.11 (3H, d, J = 5.7 Hz), 1.1–1.3 (2H, m), 1.54 (3H, s), 1.65, (3H, s), 1.16–2.0 (7H, m), 2.03 (3H, s), 2.04 (3H, s), 2.05 (3H, s), 2.09 (3H, s), 2.26 (3H, s), 2.53 (1H, m), 2.88 (1H, m), 3.89 (1H, m), 4.21 (2H, m), 5.00 (1H, d, J = 6.8), 5.15 (1H, t, J = 8.8 Hz), 5.33 (3H, m), 6.71 (1H, s), 6.72 (1H, s); 13C-NMR (CDCl3): δ 17.7, 18.8, 19.3, 20.7, 21.6, 22.7 (4C), 25.8, 26.2, 26.8, 27.5, 33.2, 42.4, 37.7, 62.4, 68.6, 71.2, 71.9, 72.9, 99.3, 113.8, 124.9 (2C), 129.9, 131.1, 134.8, 140.9, 154.6; 169.3, 169.6, 170.4, 170.7; HRMS (ESI+) for C34H48O10Na [M+Na]+ calcd. 639.3145; found 639.3110.
3.2.2. Preparation of 8-O-(β-d-Glucopyranosyl)leubethanol (1)
2,3,4,6-Tetra-O-acetyl-β-d-glucopyranosyl leubethanol (160 mg, 0.25 mmol) was dissolved in dry EtOH (5 mL) and a solution of sodium ethoxide (28 mg Na, 1.25 mmol) in EtOH was added. After stirring the mixture at room temperature for 2 h the reaction was completed. The solution was taken to dryness to obtain a residue that was purified by column chromatography with CH2Cl2/MeOH (8:2) as eluent to give 58 mg (52%) of compound 1. IR: 3412, 2923, 2862, 1652, 1612, 1575, 1458, 1376, 1268, 1075, 841 cm−1; 1H-NMR (400 MHz, CDCl3): δ 0.95 (3H, d, J = 6.7 Hz), 0.96–1.30 (4H, m), 1.30 (3H, d, J = 6.8 Hz), 1.51 (3H, s), 1.60–2.0 (8H,m), 1.62 (3H, s), 2.24 (3H, s), 2.57 (1H, m), 3.28 (1H, m), 3.32 (1H, m), 3.41 (1H, m), 3.49 (1H, m), 3.70 (1H, dd, J = 12.1, 5.8), 3.89 (1H, dd, J = 12.1, 2.0), 4.87 (1H, d, J = 7.3 Hz), 4.93 (1H, t, J = 6.8 Hz), 6.63 (1H, s), 6.79 (1H, s); 13C-NMR (50.3 MHz, CDCl3): δ 17.7, 19.1, 20.3, 21.6, 22.2, 25.9, 27.4, 27.5, 28.6, 34.6, 39.5, 43.7, 62.7, 71.6, 78.1, 78.4, 75.2, 102.2, 113.7, 124.6, 125.9, 132.5, 132.0, 136.0, 141.2, 156.6; HRMS (ESI+) for C26H41O6 [M+H]+ calcd. 449.2903; found 449.2937.
3.2.3. Preparation of 7-Nitroleubethanol (2) and 5-Nitroleubethanol (3)
To a solution of leubethanol (56 mg, 0.19 mmol) in n-hexane (2 mL) 65% HNO3 (16 μL, 0.23 mmol) was added and the mixture stirred for 8 h. Then, ethyl acetate (30 mL) was added and the organic layer was washed with 5% NaHCO3, water and dried over Na2SO4. The solvent was removed under vacuum to provide a crude (60 mg), that was purified by silica flash chromatography using n-hexane/ethyl acetate (95:5) as eluent, to give 23 mg (37%) of 7-nitroleubethanol (2) and 16 mg (26%) of 5-nitroleubethanol (3).
Compound 2. IR: 3450, 2959, 2927, 2870, 1606, 1575, 1516, 1453, 1352, 1239, 1181, 1128, 1052 cm−1; 1H-NMR (CDCl3): δ 0.83 (3H, d, J = 6.8 Hz), 1.18 (3H, d, J = 6.8 Hz), 1.3–1.1 (2H, m), 1.52 (3H, s), 1.65 (3H, s), 2.0–1.6 (7H, m), 2.21 (3H, s), 3.12 (2H, m), 4.93 (1H, t, J = 6.8 Hz), 5.37 (1H, br s), 6.50 (1H, s); 13C-NMR (CDCl3): δ 17.6, 18.2, 18.5, 19.0, 21.5, 25.4, 25.7, 26.6, 26.7, 34.7, 37.7, 38.0, 114.7, 124.5, 128.4, 129.0, 131.6, 134.0, 146.3, 154.1; HRMS (ESI+) for C20H29NO3Na [M+Na]+ calcd. 354.2045; found 354.2042.
Compound 3. IR: 2959, 2928, 2871, 1601, 1577, 1540, 1454, 1406, 1339, 1272, 1201, 1109, 1056 cm−1. 1H-NMR (CDCl3) δ 0.99 (3H, d, J = 6.8 Hz), 1.19 (3H, d, J = 6.8 Hz), 1.3–1.1 (2H, m), 1.54 (3H, s), 1.65 (3H, s), 2.0–1.6 (7H, m), 2.57 (3H, s), 3.24 (1H, m), 4.95 (1H, t, J = 6.8 Hz), 6.63 (1H, s), 11.27 (1H, br s); 13C-NMR (CDCl3): δ 17.7, 18.8, 18.8, 22.9, 22.9, 25.8, 26.2, 26.9, 27.0, 33.4, 37.9, 42.9, 124.5, 124.7, 131.5, 131.6, 132.2, 132.6, 140.9, 154.1; HRMS (ESI+) for C20H29NO3Na [M+Na]+ calcd. 354.2045; found 354.2033.
3.2.4. Preparation of 7-Bromo-14,15-dihydroleubethanol (4)
A solution of leubethanol (306 mg, 1.1 mmol) in ethanol (10 mL) was treated with H
2 under 10% Pd-C catalysis to provide 14,15-dihydroleubethanol in 95% yield [
12]. Then, 14,15-dihydroleubethanol (54 mg, 0.18 mmol) was dissolved in carbon tetrachloride (3 mL) and NBS (35 mg, 0.20 mmol) was added. The mixture was refluxed for 4 h, followed to 1 h at 0 °C. The solid residue was filtered off and the solvent was evaporated under reduced pressure to give a crude oil that was purified by silica flash chromatography using
n-hexane/ethyl acetate (98:2) as eluent giving 44 mg (67%) of compound
4. IR: 3511, 2952, 2927, 2869, 1599, 1559, 1459, 1399, 1379, 1312, 1235, 1207, 1171, 1040, 980 cm
−1.
1H-NMR (CDCl
3): δ 0.83 (6H, d,
J = 6.8 Hz), 0.94 (3H, d,
J = 6.8 Hz), 1.10–1.30 (2H, m), 1.19 (3H, d,
J = 7.2 Hz), 1.50–2.0 (8H, m), 2.34 (3H, s), 2.54 (1H, m), 3.18 (1H, m), 5.65 (1H, br s), 6.66 (1H, s);
13C-NMR (CDCl
3): δ (ppm) 18.9, 19.4, 21.0, 22.6, 22.8, 23.0, 25.6, 27.2, 27.5, 28.0, 33.6, 38.5, 39.2, 42.2, 110.4, 123.3, 128.2, 133.8, 139.9, 149.3; HRMS (ESI
+) for C
20H
32BrO [M+H]
+: calcd 367.1637; found 367.1646.
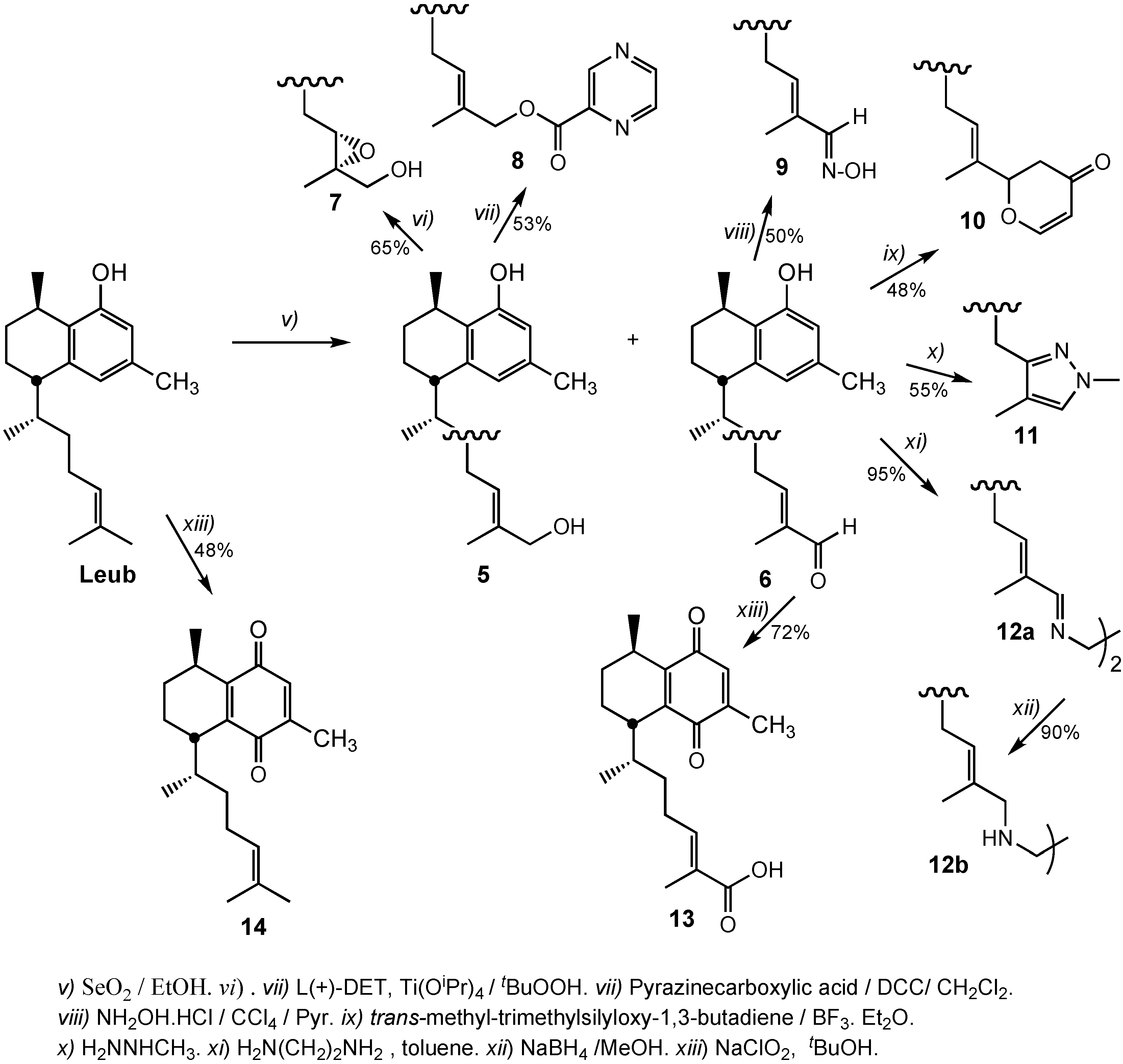
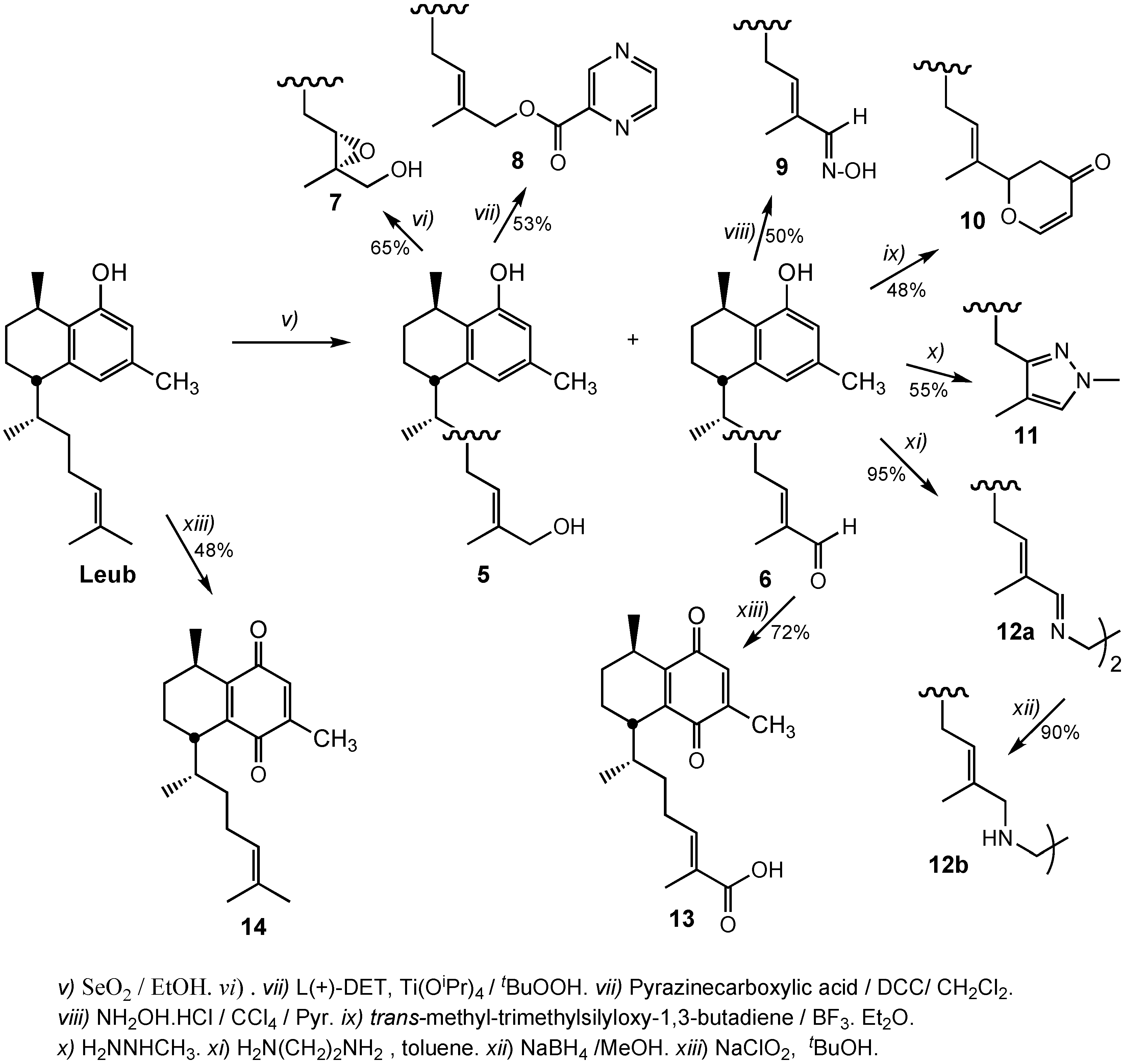
3.2.5. Preparation of 14,15-α-Epoxy-16-hydroxyleubethanol (7)
l-(+)DET (60 μL, 0.34 mmol) was dissolved in CH
2Cl
2 (3.5 mL) under nitrogen and the solution cooled to −23 °C, then Ti(
i-PrO)
4 (300 μL, 6M) was added and the mixture stirred for 15 min. After that, a solution of compound
5 (25 mg, 0.08 mmol) in CH
2Cl
2 (1 mL) [
12], was added and the mixture stirred for 5 min. Then, a solution of 3M of
tert-butyl hydroperoxide (TBHP) in toluene (0.2 mL, 0.6 mmol) was added and the mixture stirred for 12 h. The reaction was quenched by the addition of a 10% aq. solution of tartaric acid (1 mL) and was stirred for 30 min at −23 °C, followed of 1 h at room temperature. The solution was filtered off through Celite, and the residue washed with diethyl ether. The organic layer was washed with water and reduced to a volume of 50 mL, treated with 1 M solution of NaOH (3 mL) at 0 °C for 45 min. The organic phase was washed with brine. The aqueous layer was extracted with Et
2O and washed with brine. The combined organic layers were dried over sodium sulphate and concentrated to give an oil, which was purified by column chromatography with
n-hexane/ethyl acetate (7:3) as eluent to give 13.6 mg (65%) of 14,15-α-epoxy-16-hydroxy-leubethanol (
7) in ≈91% ee. IR: 3408, 2951, 2927, 2869, 1616, 1580, 1457, 1421, 1377, 1332, 1285, 1254, 1035, 842 cm
−1;
1H-NMR (CDCl
3): δ 0.98 (3H, d,
J = 6.8 Hz), 1.20 (3H, d,
J = 6.8 Hz), 1.21 (3H, s), 2.24 (3H, s), 2.60 (1H, m), 2.93 (1H, m), 3.07 (1H, m), 3.63 (2H, m), 4.77 (1H, br s), 6.43 (1H, s), 6.57 (1H, s);
13C-NMR (CDCl
3): δ 14.2, 18.8, 21.1 (2C), 26.6, 26.6, 27.4, 30.0, 38.5, 39.2, 42.3, 60.4 (2C), 65.3, 114.4, 122.2, 126.3, 135.3, 140.7, 153.1; HRMS (ESI) for C
20H
31O
2 [M+H]
+: calcd 303.2324; found 303.2369.
3.2.6. Preparation of (E)-6R-((1S,4R)-5-Hydroxy-4,7-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)-2-methylhept-2-enyl pyrazine-2-carboxylate (8)
A solution of pyrazinecarboxylic acid (13.6 mg, 0.11mmol), allylic alcohol 5 (31.5 mg. 0.1 mmol), and N,N-dicyclohexylcarbodiimide (22.7 mg, 0.11 mmol) in 5 mL of dichloromethane was stirred for 24 h at room temperature. The mixture was filtered off and the filtrate washed with water, 5% HCl, and with water again, dried over sodium sulphate and the solvent removed under vacuum to give a crude oil that was purified by silica flash chromatography using n-hexane/ethyl acetate (9:1) as eluent to give 10 mg (33%) of starting material and 22 mg (53%) of pyrazine ester derivative 8. IR: 3452, 2949, 2919, 1702, 1616, 1580, 1453, 1332, 1247, 1001, 890 cm−1; 1H-NMR (CDCl3); δ 0.96 (3H, d, J = 6.8 Hz), 1.1–1.6 (6H, m), 1.18 (3H, d, J = 7.2 Hz), 1.68 (3H, s), 1.9–2.1 (6H, m), 2.21 (3H, s), 2.56 (1H, m), 3.05 (1H, m), 4.78 (2H, s), 5.47 (1H, t, J = 7.2 Hz), 6.43 (1H, s), 6.55 (1H, s), 8.73 (1H, dd, J = 1.6, 2,4 Hz); 8.75 (1H, d, J = 2,4 Hz), 9.29 (1H, d, J = 1.6 Hz); 13C-NMR (CDCl3): δ 14.1, 18.7, 19.3, 21,1, 21.1, 26.2, 26.6, 27.4, 32.8, 38.2, 42.3, 72.2, 113.3, 122.3, 126.3.128.9, 131.8, 135.1, 140.7, 143.6, 144.5, 146.2, 147.5, 153.2, 163.8. HRMS (ESI+) for C25H32N2O3Na [M+Na]+: calcd 431.2311; found 431.2304.
3.2.7. Preparation of 16-Hydroximinoleubethanol (9)
The 16-oxoleubethanol 6 (43 mg, 0.14 mmol) dissolved in methanol (5 mL) was treated with hydroxylamine hydrochloride (97 mg, 1.4 mmol) and 5 drops of pyridine and the mixture was refluxed for 14 h. The solvent was removed under vacuum and the crude dissolved in dichloromethane, the organic layer was washed with water and dried over Na2SO4. Removal of the solvent gave 40 mg of a crude, that was purified by silica flash chromatography using n-hexane/ethyl acetate (9:1) as eluent, to provide 20 mg (50%) of 9. IR: 3367, 2922, 2862, 1616, 1580, 1454, 1372, 1321, 1285, 1253, 1171, 1054, 995 cm−1; 1H-NMR (CDCl3): δ 0.99 (3H, d, J = 6.8 Hz), 1.3–1.6 (4H, m), 1.20 (3H, d, J = 6.8 Hz), 1.77 (3H, s), 1.8–2.2(6H, m), 2.24 (3H, s), 2.60 (1H, m), 3.06 (1H, m), 5.59 (1H, t, J = 7.6 Hz), 6.43 (1H, s), 6.56 (1H, s), 7.66 (1H, s), 8.10 (1H, br s); 13C-NMR (CDCl3): δ 11.9, 18.6, 19.2, 21.0, 21.4, 26.5, 26.5, 27.5, 32.5, 38.2, 42.2, 113.3, 122.3, 126.2, 130.2, 135.2, 139.9, 140.6, 153.0, 154.9; HRMS (ESI+) for C20H30NO2 [M+H]+ calcd. 316. 2277; found 316.2271.
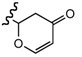
3.2.8. Preparation of 2-((E)-6R-((1S,4R)-5-Hydroxy-4,7-dimethyl-1,2,3,4-tetrahydronaphthalen-1-yl)hept-2-en-2-yl)-2H-pyran-4(3H)-one (10)
To a solution of trans-methoxytrimethylsilyloxy-1,3-butadiene (40 mg, 0.23 mmol) in dry diethyl ether (5 mL) under nitrogen atmosphere at −30 °C aldehyde 6 (75 mg, 0.23 mmol) dissolved in diethyl ether (2 mL) and boron trifluoride diethyl etherate (45 μL, 0.23 mmol) were added. The mixture was maintained for 4 hours under stirring and the reaction was quenched by the addition of Et3N (96 μL, 0.69 mmol) and water (2 mL). The mixture was treated with diethyl ether, washed with brine and the organic layer dried with Na2SO4. The crude was dissolved in dichloromethane (5 mL) and 4 drops of trifluoroacetic acid was added. One hour later, the solvent was evaporated under reduced pressure to give a crude that was purified by silica flash chromatography using n-hexane/ethyl acetate (9:1) as eluent giving 20 mg (26%) of starting material and 38 mg (48%) of dihydropyranone derivative 10. IR: 3402, 2953, 2923, 2869, 1661, 1588, 1502, 1454, 1413, 1278, 1226, 1174, 1050, 984 cm−1; 1H-NMR (CDCl3): δ 0.99 (3H, d, J = 6.8 Hz), 1.19 (3H, d, J = 6.9 Hz), 2.23 (3H, s), 2.35 (3H, s), 2.59 (1H, m), 2.73 (1H, dd, J = 16.7, 14.5 Hz), 3.08 (1H, m). 4.69 (1H, dd, J = 14.5, 2.4 Hz), 5.42 (2H, m), 6.44 (1H, s), 6.56 (1H, s), 7.39 (1H, dt, J = 6.1, 1.0 Hz); 13C-NMR (CDCl3): δ 11.9, 18.7, 19.2, 21.1, 21.1, 26.0, 26.5, 27.5, 32.7, 38.2, 40.6, 42.2, 84.5, 106.7, 113.3, 122.2, 126.3, 130.8, 131.2, 135.0, 140.6, 153.2, 163.6, 193.2; HRMS (ESI+) for C24H32O3Na [M+Na]+ calcd. 391.2249; found 391.2239.
3.2.9. Preparation of (5S,8R)-5-((2R)-4-(1,4-Dimethyl-1H-pyrazol-3-yl)butan-2-yl)-3,8-dimethyl-5,6,7,8-tetrahydronaphthalen-1-ol (11)
To a stirring solution of 16-oxoleubethanol 6 (19 mg, 0.06 mmol) in 2-propanol (2 mL), methylhydrazine (26 μL, 0.5 mmol) was added and the mixture refluxed for 5 hours. The solvent was removed under reduced pressure to give a crude oil that was purified by silica flash chromatography using n-hexane/ethyl acetate (9:1) as eluent to give 11 mg (55%) of the pyrazole 11. IR: 3209, 2929, 2864, 1613, 1578, 1451, 1419, 1371, 1321, 1285, 1260, 1172, 1059 cm−1. 1H-NMR (CDCl3): δ 1.02 (3H, d, J = 6.8 Hz), 1.10–1.30 (2H, m), 1.18 (3H, d, J = 7.2 Hz), 1.50–203 (8H, m), 1.85 (3H, s), 2.20 (3H, s), 2.59 (1H, m), 3.02 (1H, m), 3.75 (3H, s), 5.31 (1H, br s), 6.40 (1H, s), 6.50 (1H, br s), 7.00 (1H, br s); 13C-NMR (CDCl3): δ 8.2, 18.7, 19.6, 21.1, 21.2, 25.0, 26.7, 27.5, 33.0, 38.2, 38.3, 42.4, 113.2, 113.5, 122.2, 126.3, 129.5, 135.0, 140.8, 151.5, 153.3; HRMS (ESI+) for C21H31N2O [M+H]+: calcd 327.2436; found 367.2439.
3.2.10. Preparation of (5S,5'S,8R,8'R)-5,5'-((2R,2'R,5E,5'E,7Z,7'Z)-7,7'-(Ethane-1,2-diylbis(azan-1-yl-1-ylidene))bis(6-methylhept-5-ene-2-yl-7-ylidene))bis(3,8-dimethyl-5,6,7,8-tetrahydronaphthalen-1-ol) (12a)
To a solution of compound 6 (91 mg, 0.3 mmol) in dry toluene (1.5 mL), ethyendiamine (10 mL, 0.15 mmol) and anhydrous Na2SO4 (900 mg) were added. The mixture was maintained at room temperature for 19 h. The solid was filtered, and the solvent was removed under reduced pressured to isolated 92 mg (95%) of the diimine derivative 12a. IR: 3447, 2925, 1654, 1624, 1458, 1119 cm−1. 1H-NMR (CDCl3): δ 0.96 (3H, d, J = 7.6 Hz), 1.18 (3H, d, J = 6.8 Hz), 1.20–1.60 (4H, m), 1.76 (3H, s), 1.8–2.10 (6H, m), 2.19 (3H, s), 2.56 (1H, m), 3.07 (1H, m), 3.70 (2H, s), 5.69 (1H, t, J = 6.8 Hz), 6.38 (1H, s), 6.51 (1H, s), 7.69 (1H, s); 13C-NMR (CDCl3) δ 12.4, 19.6, 20.0 (2C), 22.0, 27.5, 27.7, 28.4, 33.3, 39.2, 43.1, 62.1, 114.1, 122.7, 127.5, 135.9, 136.3, 141.5, 144.1, 154.5, 168.7. HRMS (ESI+) for C42H61N2O2 [M+H]+ calcd. 625.4732; found 625.4728.
3.2.11. Preparation of (5S,5'S,8R,8'R)-5,5'-((2R,2'R,5E,5'E)-7,7'-(Ethane-1,2-diylbis(azanediyl))-bis(6-methylhept-5-ene-7,2-diyl))bis(3,8-dimethyl-5,6,7,8-tetrahydronaphthalen-1-ol) (12b)
Diimine 12a (83 mg, 0.13 mmol) was dissolved in ethanol (2 mL), and a solution of NaBH4 (10 mg, 0.26 mmol) in anhydrous ethanol (1 mL) was added. The mixture was maintained for 3 h at 40 °C, cooled, diluted with water (5 mL), treated with a 5% NaOH, and extracted with dichloromethane. The organic layer was washed with brine, dried over K2CO3, and the solvent was removed under reduced pressure to isolated 79 mg (90%) of ethylendiamine derivative 12b. IR: 3450, 2925, 1654, 1617, 1578, 1458, 840 cm−1.1H-NMR (CDCl3): δ 0.89 (3H, d, J = 6.8 Hz), 1.00–1.25 (4H, m), 1.11 (3H, d, J = 6.0 Hz), 1.50–2.0 (8H, m), 1.75 (3H, s), 2.15 (3H, s), 2.50 (1H, m), 2.52 (1H, m), 3.01 (2H, m), 5.09 (1H, br. s), 6.33 (1H, s), 6.48 (1H, s); 13C-NMR (CDCl3): δ 14.7, 18.8, 19.4, 21.2 (2C), 26.1, 26.6, 26.5, 33.2, 38.2, 42.4, 47.7, 57.3, 113.3, 121.7, 126.8, 127.5, 132.4, 134.8, 140.7, 153.9; HRMS (ESI+) for C42H65N2O2 [M+H]+ calcd. 629.5042; found 629.5038.
3.2.12. Preparation of (R,E)-6-((1S,4R)-4,7-Dimethyl-5,8-dioxo-1,2,3,4,5,8-hexahydronaphthalen-1-yl)-2-methylhept-2-enoic acid (13)
The aldehyde 6 (60 mg, 0.17 mmol) was dissolved in tert-butyl alcohol (4 mL) and 2-methyl-2-butene (1 mL). Then, a solution of sodium chlorite (141 mg, 1.56 mmol) and sodium hydrogen phosphate (126 mg, 1.05 mmol) in water (2 mL) was added dropwise over a 10 minute period. The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressured, and the residue dissolved in water (10 mL) and extracted with hexane (2 × 8 mL). The combined organic layers were dried with Na2SO4, filtered and evaporated under reduced pressure to give 40 mg (72%) of the quinone 13. IR: 3520, 2930, 2870, 1710, 1690, 1660, 1606, 1516, 1453, 1281, 1128, 890 cm−1; 1H-NMR (CDCl3): δ 0.85 (3H, d, J = 6.8 Hz), 1.06 (3H, d, J = 6.8 Hz), 1.10–1.30 (2H, m), 1.79 (3H, s), 1.60–2.20 (7H, m), 2.00 (3H, s), 2.78 (1H, m), 2.93 (1H, m), 6.52 (1H, s), 6.80 (1H, t, J = 6.8 Hz), 8.20 (1H, br s); 13C-NMR (CDCl3): δ 11.9, 15.8, 18.5, 19.3, 21.0, 25.2, 26.6, 26.8, 33.4, 35.8, 36.7, 127.1, 133.4, 144.9, 145.0, 145.4, 147.4, 173.2, 187.1, 188.4; HRMS (ESI+) for C20H26O4Na [M+Na]+ calcd. 353.1729; found 353.1714.
3.2.13. Preparation of (5R,8S)-2,5-Dimethyl-8-((E,R)-6-methylhept-5-en-2-yl)-5,6,7,8-tetrahydronaphthalene-1,4-dione (14)
Leubethanol (74 mg, 0.26 mmol) was dissolved in in tert-butyl alcohol (9 mL) and 2-methyl-2-butene (2 mL). Then, a solution of sodium chlorite (216 mg, 3.4 mmol) and sodium hydrogen phosphate (210 mg, 1.8 mmol) in water (2 mL) was added dropwise over a 10 minute period. The reaction mixture was stirred at room temperature overnight. The solvent was removed under reduced pressured, the residue dissolved in 10 mL of water and extracted with hexane (2 × 10 mL). The combined organic layers were dried with Na2SO4, filtered and evaporated under reduced pressure to give 30 mg of starting material and 35 mg (48%) of the quinone 14. IR: 2957, 2926, 1718, 1676, 1601, 1540, 1410, 1272, 1201, 1109, 890 cm−1; 1H-NMR (CDCl3): δ 0.78 (3H, d, J = 6.8 Hz), 1.01 (3H, d, J = 6.8 Hz), 1.10–1.40 (2H, m), 1.49 (3H, s),1.58 (3H, s), 1.60–2.20 (7H, m), 1.95 (3H, s), 2.70 (1H, m), 2.87 (1H, m), 4.93 (1H, t, J = 7.2 Hz); 6.44 (1H, s); 13C-NMR (CDCl3): δ 15.7, 17.6, 18.7, 19.0, 21.1, 25.6, 25.7, 26.3, 26.5, 34.8, 36.6, 37.6, 124.1, 131.5, 133.4, 143.2, 145.5, 147.0, 187.3, 188.4; HRMS (ESI+) for C20H29O2 [M+H]+ calcd. 301.2168; found 301.2164.
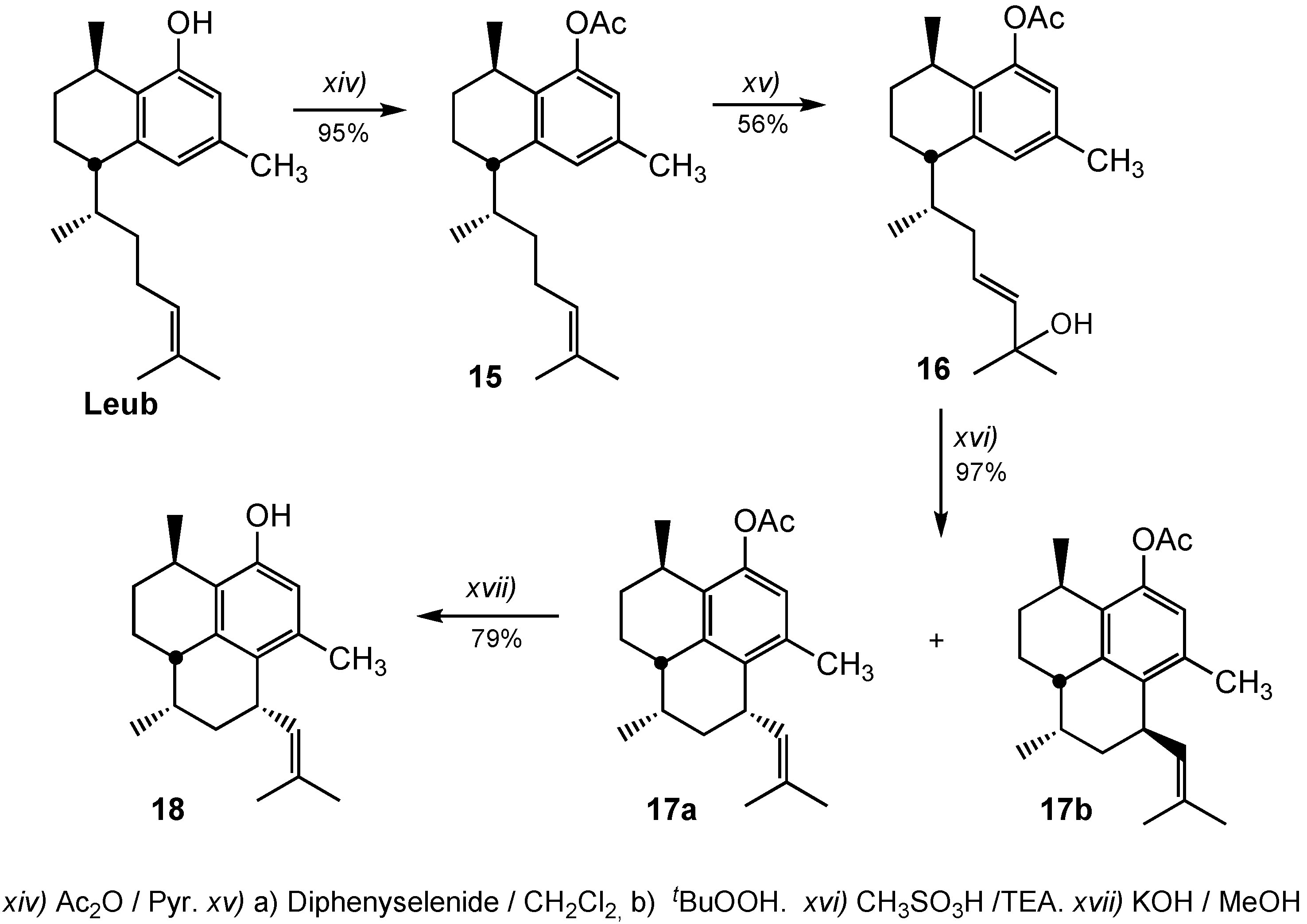
3.2.14. Preparation of 8-Acetoxyleubethanol (15)
Distilled Ac2O (0.5 mL) was added to a solution of leubethanol (97 mg, 0.33 mmol) in pyridine (2 mL). After 12 hours the reaction was quenched with ice, one hour later the mixture was extracted with EtOAc and the organic layer was washing with 10% HCl, 5% NaHCO3, brine and finally dried over Na2SO4.The solvent was evaporated under reduced pressure to give a crude oil that was purified by chromatography on silica gel n-hexane/EtOAc (95:5) to give 104 mg of acetylated Leub, compound 15 (95%). IR: 2954, 2926, 2870, 1765, 1619, 1571, 1451, 1370, 1207, 1048 cm−1. 1H-NMR (CDCl3): δ 0.99 (3H, d, J = 7.1 Hz, 3H), 1.15 (3H, d, J = 6.8 Hz), 1.59 (3H, s), 1.68 (3H, s), 2.31 (6H, s), 2.61 (1H, m), 2.95 (1H, m), 5.01 (1H, t, J = 7.1 Hz), 6.70 (1H, s), 6.88 (1H, s); 13C-NMR (CDCl3): δ 17.7, 18.8, 19.3, 21.2 (2C), 21.8, 25.8, 26.3, 27.2, 27.4, 33.2, 38.0, 42.3, 120.3, 124.9, 127.6, 131.2, 131.6, 135.1, 141.2, 148.7, 169.7; HRMS (ESI+) for C22H33O2 [M+H]+: calcd. 329.2481; found 329.2502.
3.2.15. Preparation of (5S,8R)-5-((E,R)-6-Hydroxy-6-methylhept-4-en-2-yl)-3,8-dimethyl-5,6,7,8-tetrahydronaphthalen-1-yl acetate (16)
To a magnetic stirred, ice-cooled solution of diphenyldiselenide (590 mg, 1.89 mmol) in dry dichloromethane (7 mL) was slowly added chilled 33% of hydrogen peroxide (0.21 mL, 64.2 mg, 1.89 mmol). After stirring vigorously for 30 min (white crystals deposit within 10 min), powered anhydrous magnesium sulfate (300 mg) was added and the mixture was stirred for an additional 30 min in the ice-bath. The ice bath was removed, compound 15 (408 mg, 0.86 mmol) was added and the mixture stirred vigorously for 6 h at 25 °C. Chilled 3M tert-butyl hydroperoxide (1.4 mL, 4.3 mmol) was added to the reaction mixture which had been immersed in an ice bath; then, after removing the ice bath, the mixture was stirred for 20 h at 25 °C to give a pale orange solution with a lot of white precipitated. The white precipitate (PhSeO2H and hydrated MgSO4), was filter off and washed with ethyl acetate. The filtrate was concentrated to give an oil. The oil was dissolved in ethyl acetate (100 mL) and washed with 5% Na2CO3 water, 10% FeSO4, water, (sat)NaHCO3, water and brine, successively and then, it was dried over Na2SO4. The solvent was evaporated under reduced pressure to give a crude oil that was purified by chromatography on silica gel n-hexane/ethyl acetate (98:2) to give 152 mg of starting material and 245 mg (56%) of allyl alcohol 16. IR: 3454, 2950, 2922, 2872, 1763, 1618, 1571, 1453, 1370, 1207, 1140, 971, 881 cm−1. 1H-NMR (CDCl3): δ 0.95 (3H, d, J = 6.8 Hz), 1.12 (3H, d, J = 6.8 Hz), 1.22 (3H, s), 1.23 (3H, s), 2.28 (3H, s), 2.29 (3H, s), 2.62 (1H, m), 2.91 (1H, m), 5.3–5.6 (2H, m), 6.67 (1H, s), 6.87 (1H, s); 13C-NMR (CDCl3): δ 18.8, 19.3, 21.1, 21.1, 21.6, 27.2, 27.2, 29.7, 29.8, 36.3, 39.3, 41.6, 70.6, 120.4, 126.3, 127.8, 131.6, 135.3, 139.0, 141.1, 148.7, 169.8; HRMS (ESI+) for C22H32O3Na [M+Na]+: calcd. 367.2249; found 367.2295.
3.2.16. Preparation of (3R,7S,9R,9aS)-3,6,9-Trimethyl-7-(2-methylprop-1-enyl)-2,3,7,8,9,9a-hexahydro-1H-phenalen-4-yl acetate (17a) and (3R,7R,9S,9aS)-3,6,9-Trimethyl-7-(2-methylprop-1-enyl)-2,3,7,8,9,9a-hexahydro-1H-phenalen-4-yl acetate (17b)
A solution of allylic alcohol 16 (70 mg, 0.20 mmol) was cooled to −78 °C and treated dropwise with methanesulfonic acid (65 μL, 1.0 mmol). The solution was warmed to −40 °C and stirred for 12 h, and then triethylamine (0.50 mL) was added. The mixture was warmed to 20 °C, filtered off through a small plug of silica gel with hexane, and concentrate in vacuo to afford 63 mg (97%) of a mixture of compounds 17a + 17b (2:1).
Compound 17a. IR: 2925, 2863, 1762, 1598, 1452, 1370, 1209, 1036 cm−1. 1H-NMR (CDCl3): δ 0.86 (3H, d, J = 7.1 Hz), 1.19 (3H, d, J = 6.9 Hz), 1.67 (3H, s), 1.72 (3H, s), 2.12 (3H, s), 2.29 (3H, s), 2.70 (1H, m), 3.02 (1H, m), 3.65 (1H, m), 5.16 (1H, m), 6.68 (1H, s); 13C-NMR (CDCl3): δ 16.0, 17.6, 19.9, 21.1, 22.7, 25.3, 27.1, 28.6, 30.8, 31.5, 34.7, 37.3, 39.9, 121.7, 128.8, 131.4, 132.6, 135.0, 135.5, 137.2, 146.9, 169.7; HRMS (ESI+) for C22H30O2Na [M+Na]+: calcd. 349.2143; found 349.2154.
Compound 17b. IR: 2925, 2863, 1762, 1598, 1452, 1370, 1209, 1036 cm−1. 1H-NMR (CDCl3) δ 0.72 (3H, d, J = 7.1 Hz), 1.20 (3H, d, J = 6.9 Hz), 1.68 (3H, s), 1.72 (3H, s), 2.13 (3H, s), 2.29 (3H, s), 2.70 (1H, m), 3.02 (1H, m), 3.63 (1H, m), 3.82 (1H, ddd, J = 8.7, 8.7, 8.7 Hz); 5.01 (1H, br d, J = 8.7 Hz,), 6.66 (1H, br s); 13C-NMR (CDCl3): δ 13.6, 17.4, 20.0, 21.1, 23.0, 25.4, 27.2, 28.7, 31.2, 31.4, 33.6, 37.5, 40.3, 121.6, 129.0, 129.8, 131.3, 135.1, 135.2, 138.1, 149.6, 169.7.
3.2.17. Preparation of (3R,7S,9R,9aS)-3,6,9-Trimethyl-7-(2-methylprop-1-enyl)-2,3,7,8,9,9a-hexahydro-1H-phenalen-4-ol (18)
Acetyl compound 17a (53 mg, 0.16 mmol) was dissolved in KOH/MeOH (5 mL, 2M) and stirred for 12 h. After that time, the solvent was evaporated, the residue was dissolved in water and extracted with ethyl acetate. The organic layer was dried over Na2SO4, and the solvent was evaporated under reduced pressure to give a crude oil that was purified by chromatography on silica gel n-hexane/ ethyl acetate (98:2) to give 35 mg 63%) of alcohol compound 18. IR: 3516, 2958, 2921, 2866, 1589, 1450, 1412, 1377, 1237, 1177, 1031, 908, 732 cm−1. 1H-NMR (CDCl3): δ 0.70 (3H, d, J = 7.0 Hz); 1.28 (3H, d, J = 6.8 Hz); 1.67 (3H, s); 1.71 (3H, s); 2.10 (3H, s); 2.68 (1H, m); 3.11 (1H, m); 3.61 (1H, q, J = 9.0 Hz); 4.98 (1H, dt, J = 9.3, 1.3 Hz); 6.42 (1H, s); 13C-NMR (CDCl3): δ 13.3, 17.4, 19.9, 22.8, 27.8, 28.5, 29.4, 31.4, 32.0, 33.2, 37.7, 40.8, 115.4, 125.7, 128.4, 129.6, 130.6, 135.1, 138.8, 151.4. HRMS (ESI+) for C20H29O [M+H]+: calcd. 385.2218; found 385.2255.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}