
Epigallocatechin-3-Gallate Protects HUVECs from PM2.5-Induced Oxidative Stress Injury by Activating Critical Antioxidant Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
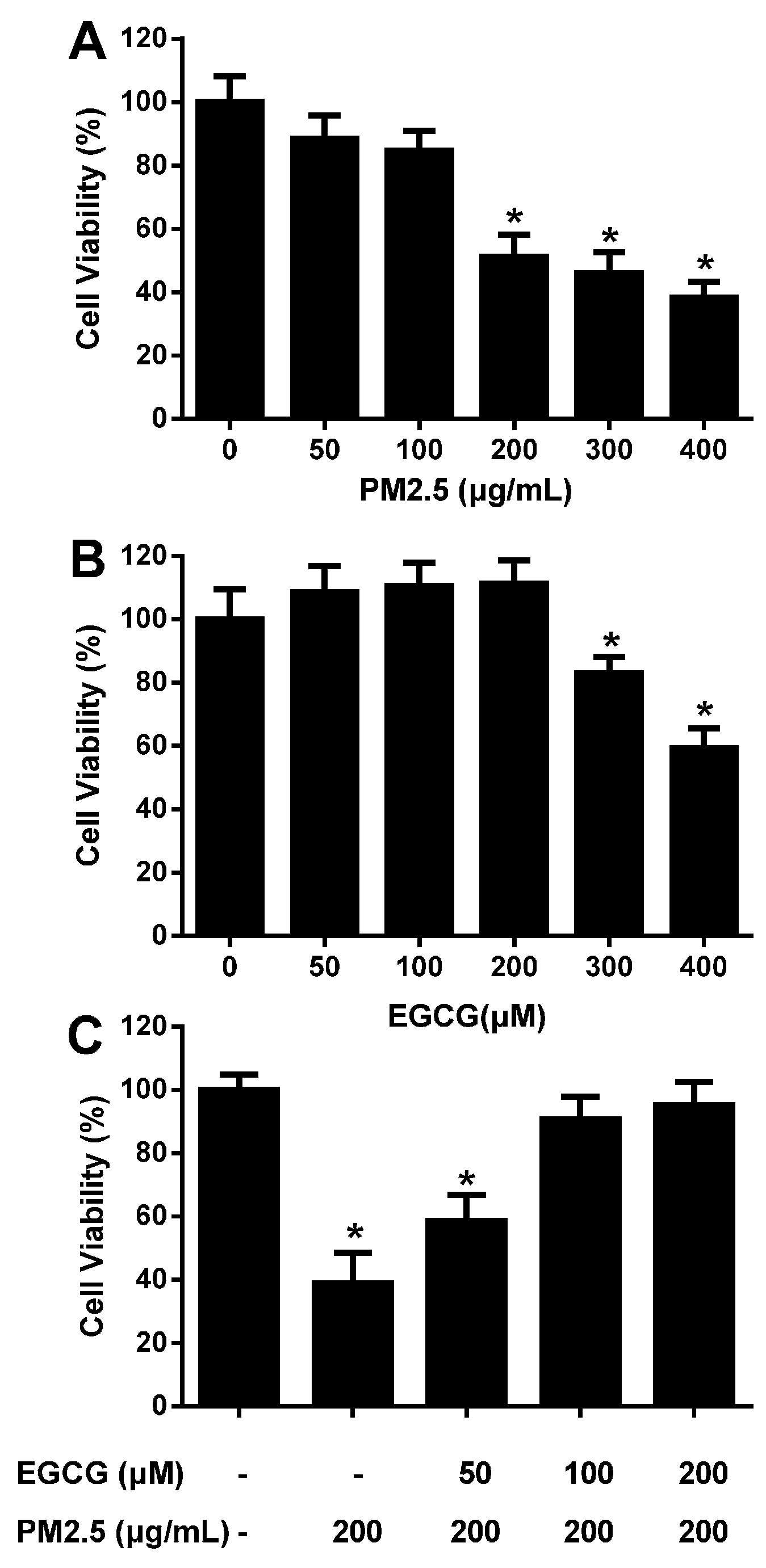
2.1. EGCG Prevented PM2.5-Induced Reduction in HUVEC Viability
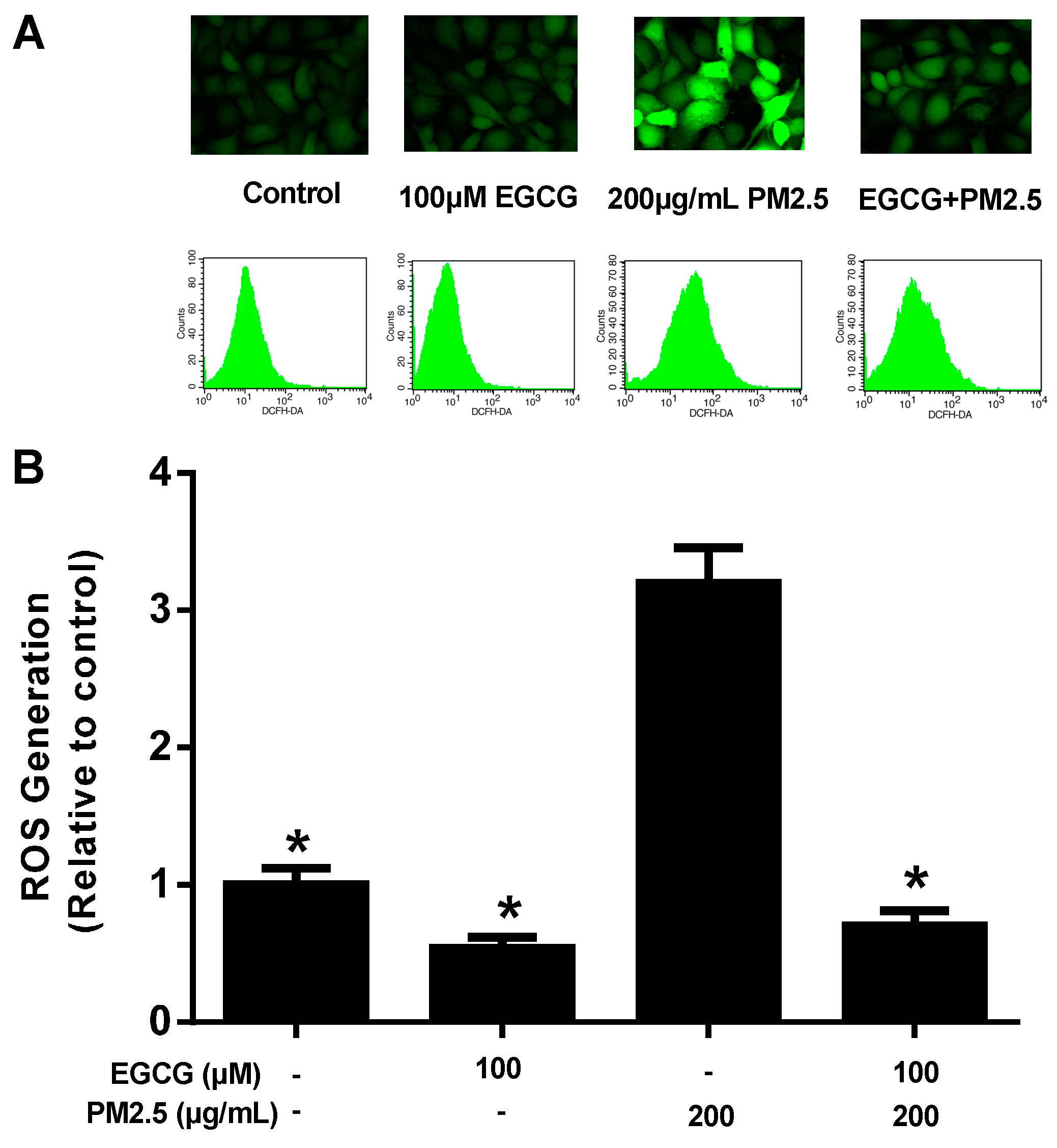
2.2. EGCG Reduced PM2.5-Induced ROS Production in HUVECs
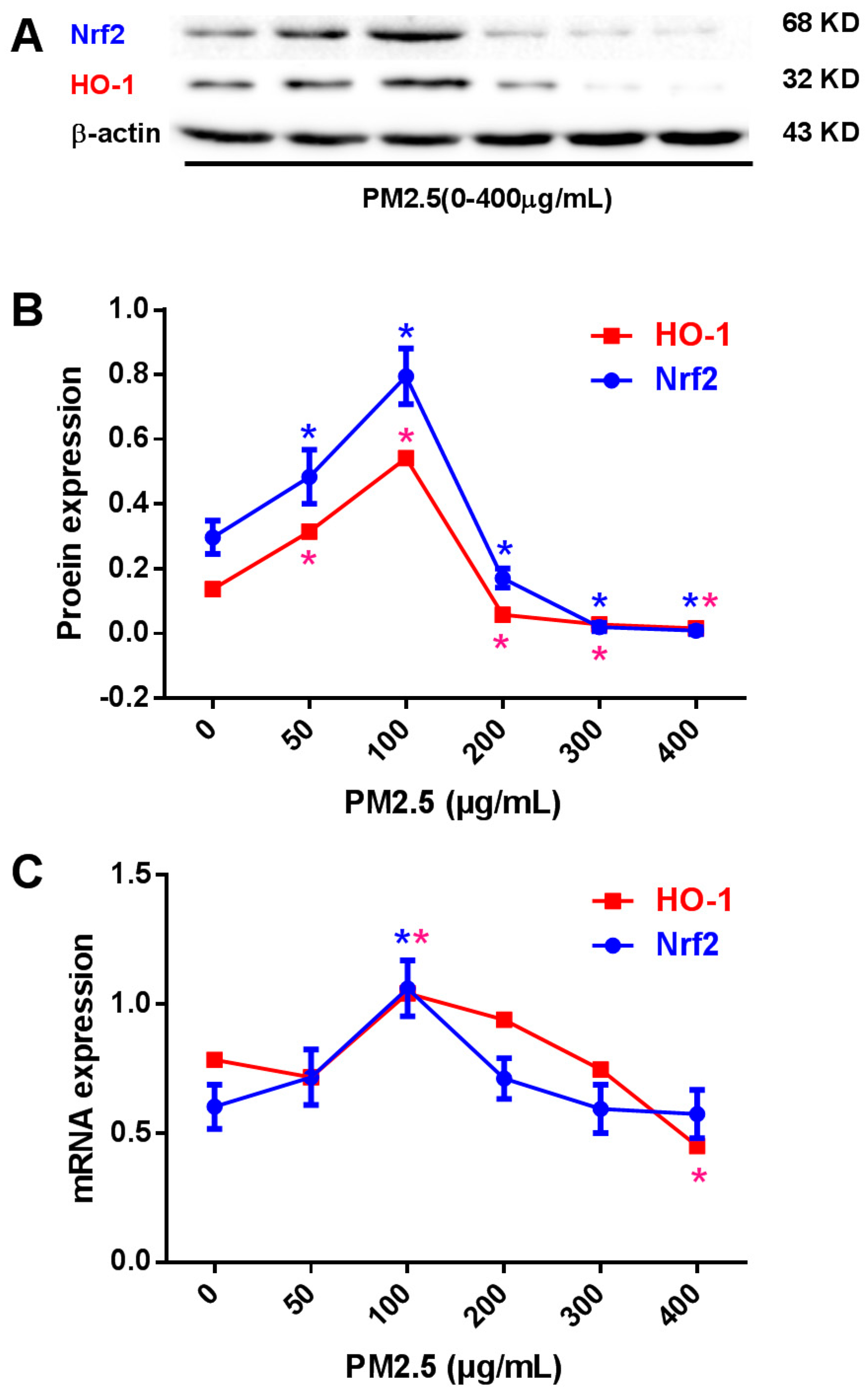
2.3. Effects of PM2.5 on Nrf2 and HO-1 Expression in HUVECs
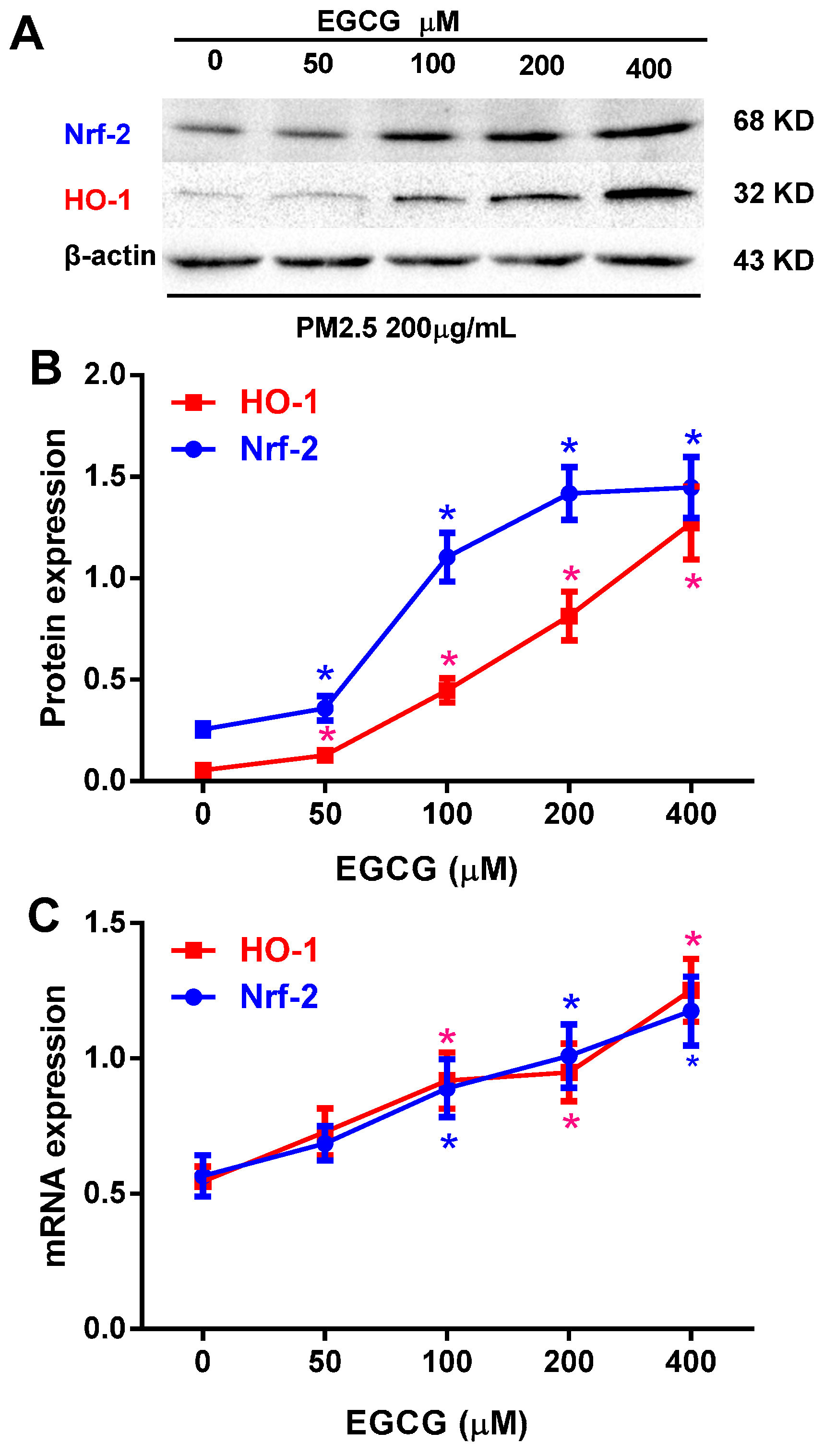
2.4. EGCG Upregulated Nrf2 and HO-1 Expression in PM2.5-Treated HUVECs
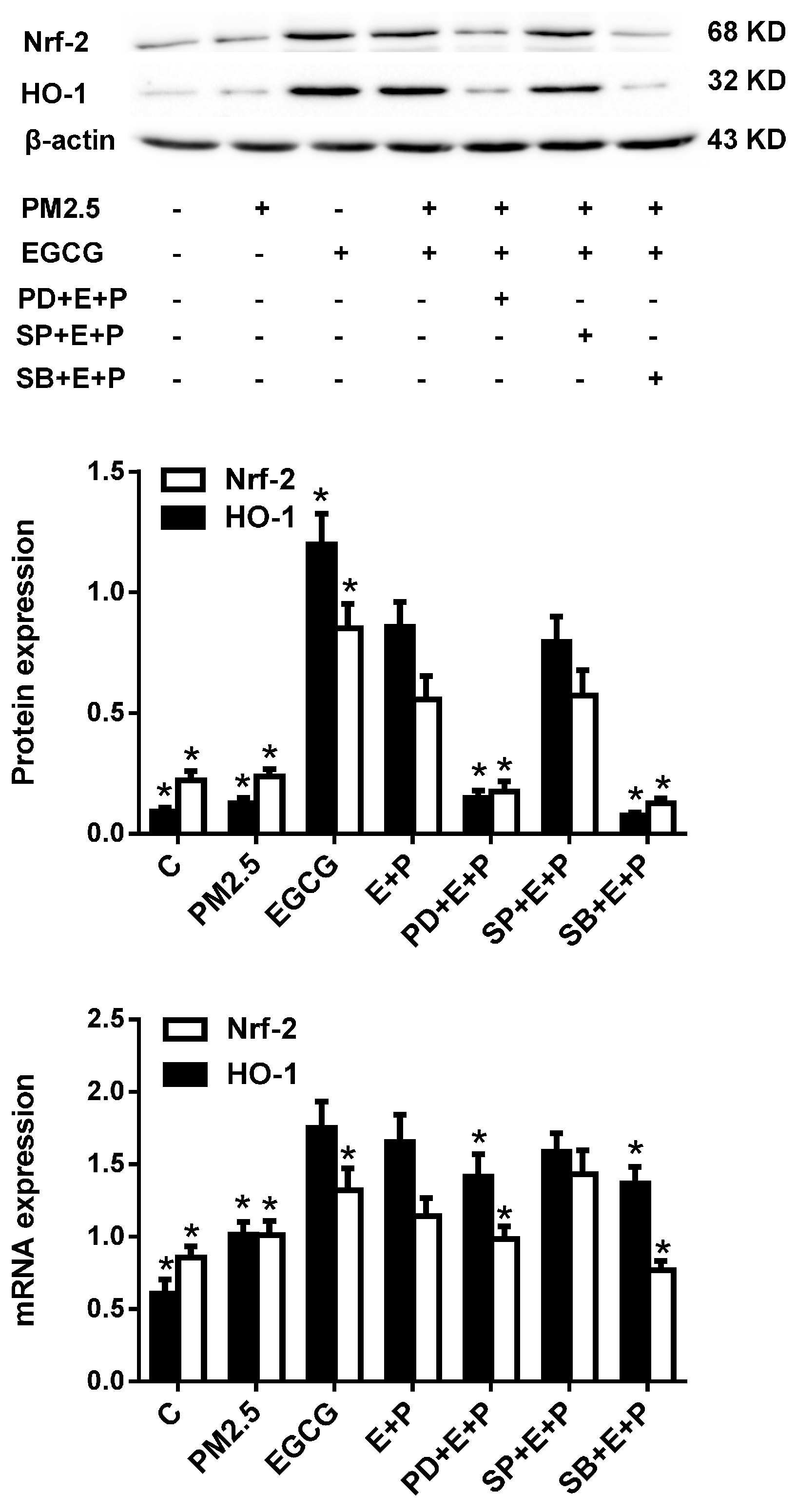
2.5. Inhibiting either ERK1/2 or P38 MAPK Abrogated the EGCG-Induced Upregulation of Nrf2 and HO-1 in HUVECs Exposed to PM2.5
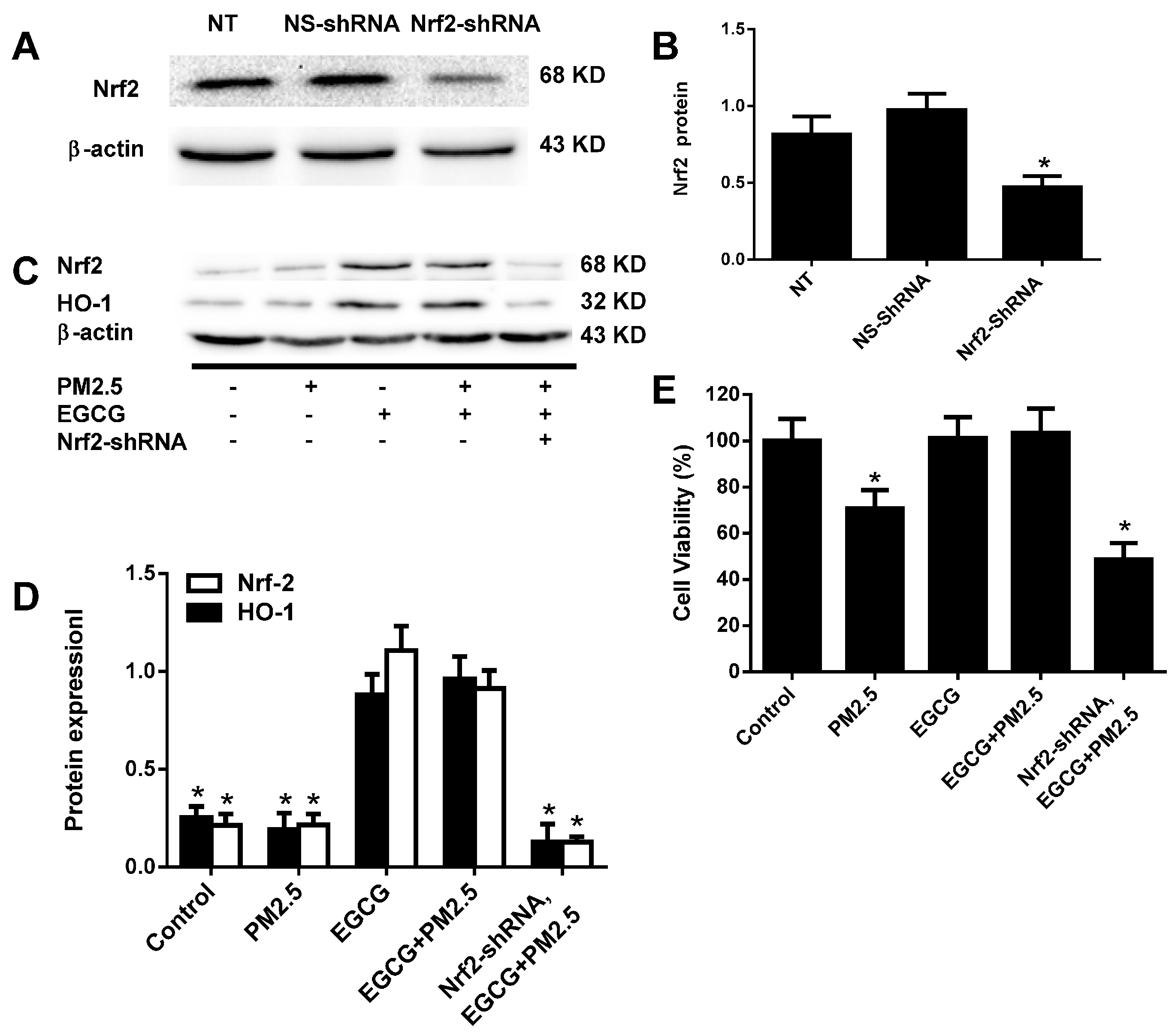
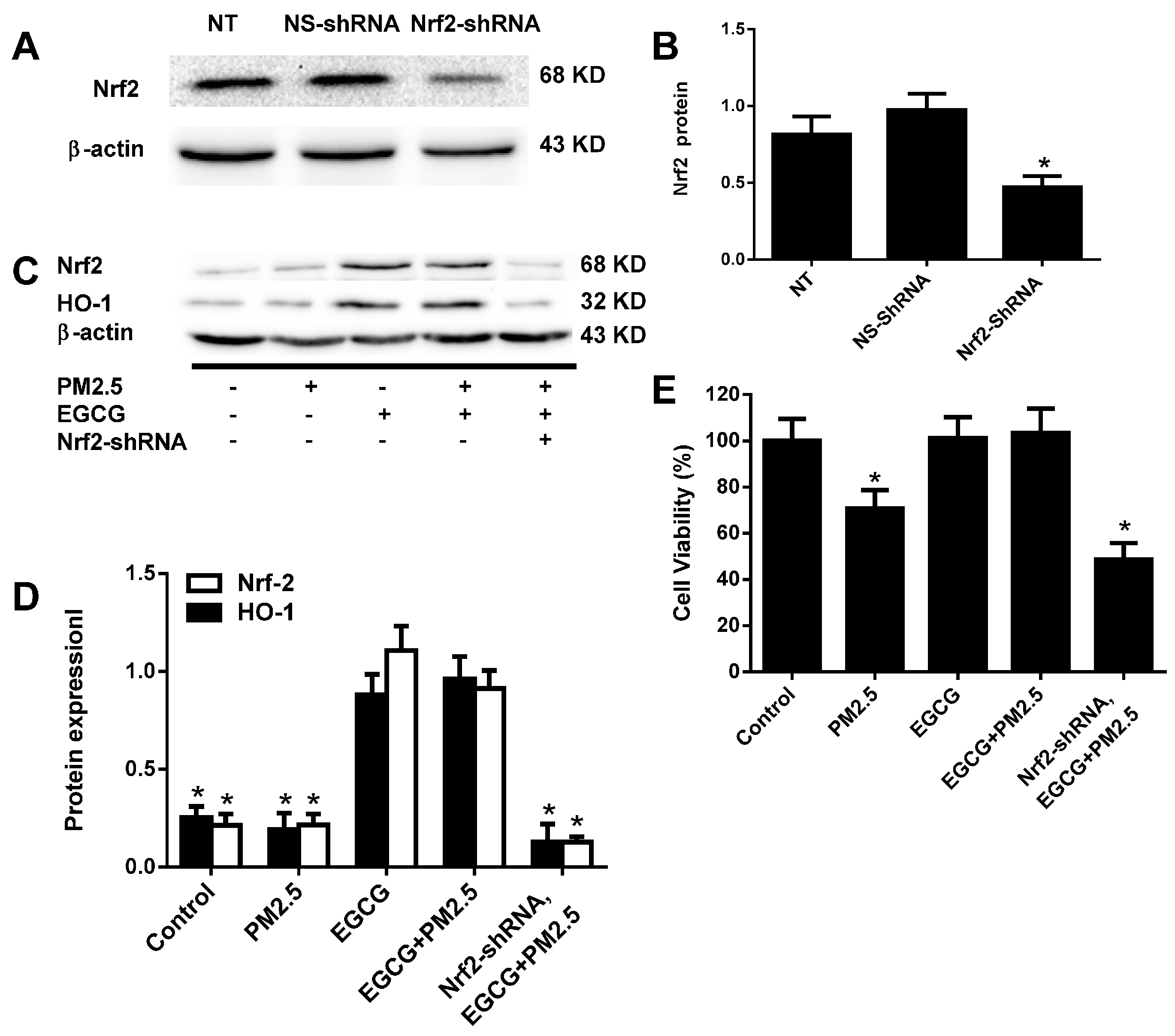
2.6. Nrf2 Silencing Abolished the EGCG-Induced Upregulation of Nrf2 and HO-1 in HUVECs Exposed to PM2.5
3. Discussion
4. Experimental Section
4.1. Reagents and Cell Culture
4.2. Ambient PM2.5 Water-Soluble Extracts
4.3. Cytotoxicity Assays
4.4. Determining Intracellular ROS Production
4.5. Western Blotting
4.6. Real-Time PCR
4.7. Nrf2-shRNA Transient Transfection
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beelen, R.; Hoek, G.; van Den Brandt, P.A.; Goldbohm, R.A.; Fischer, P.; Schouten, L.J.; Jerrett, M.; Hughes, E.; Armstrong, B.; Brunekreef, B. Long-term effects of traffic-related air pollution on mortality in a Dutch cohort (NLCS-AIR study). Environ. Health Perspect. 2008, 116, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Puett, R.C.; Yanosky, J.D.; Hart, J.E.; Paciorek, C.J.; Schwartz, J.D.; Suh MacIntosh, H.H.; Speizer, F.E.; Laden, F. Chronic fine and coarse particulate exposure, mortality, and coronary heart disease in the Nurses’ Health Study. Environ. Health Perspect. 2009, 117, 1702–1706. [Google Scholar] [PubMed]
- Puett, R.C.; Hart, J.E.; Suh, H.; Mittleman, M.; Laden, F. Particulate matter exposures, mortality, and cardiovascular disease in the health professionals follow-up study. Environ. Health Perspect. 2011, 119, 1130. [Google Scholar] [CrossRef]
- Cesaroni, G.; Badaloni, C.; Gariazzo, C.; Stafoggia, M.; Sozzi, R.; Davoli, M.; Forastiere, F. Long-term exposure to urban air pollution and mortality in a cohort of more than a million adults in Rome. Environ. Health Perspect. 2013, 121, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Hoek, G.; Krishnan, R.M.; Beelen, R.; Peters, A.; Ostro, B.; Brunekreef, B.; Kaufman, J.D. Long-term air pollution exposure and cardio-respiratory mortality: A review. Environ. Health. 2013, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Knutsen, S.F.; Shavlik, D.; Beeson, W.L.; Petersen, F.; Ghamsary, M.; Abbey, D. The association between fatal coronary heart disease and ambient particulate air pollution: Are females at greater risk? Environ. Health Perspect. 2005, 113, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Crouse, D.L.; Peters, P.A.; van Donkelaar, A.; Goldberg, M.S.; Villeneuve, P.J.; Brion, O.; Khan, S.; Atari, D.O.; Jerrett, M.; Pope, C.A., III; et al. Risk of nonaccidental and cardiovascular mortality in relation to long-term exposure to low concentrations of fine particulate matter: A Canadian national-level cohort study. Environ. Health Perspect. 2012, 120, 708–714. [Google Scholar]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Ross, A.H.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar]
- Suwa, T.; Hogg, J.C.; Quinlan, K.B.; Ohgami, A.; Vincent, R.; van Eeden, S.F. Particulate air pollution induces progression of atherosclerosis. J. Am. Coll. Cardiol. 2002, 39, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Dockery, D.W.; Muller, J.E.; Mittleman, M.A. Increased particulate air pollution and the triggering of myocardial infarction. Circulation 2001, 103, 2810–2815. [Google Scholar] [CrossRef] [PubMed]
- Glantz, S.A. Air pollution as a cause of heart disease: Time for action. J. Am. Coll. Cardiol. 2002, 39, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, P.; Golino, P. Free radicals and antioxidants in cardiovascular diseases. Expert. Rev. Cardiovasc. Ther. 2005, 3, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Fiotakis, K.; Vlachogianni, T. Airborne particulate matter and human health: Toxicological assessment and importance of size and composition of particles for oxidative damage and carcinogenic mechanisms. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2008, 26, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Trovato-Salinaro, A.; Cambria, M.T.; Locascio, M.S.; Rienzo, L.D.; Condorelli, D.F.; Mancuso, C.; De Lorenzo, A.; Calabrese, E.J. The hormetic role of dietary antioxidants in free radical-related diseases. Curr. Pharm. Des. 2010, 16, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ferrandiz, M.; Devesa, I. Inducers of heme oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Deliv. 2010, 11, 1504. [Google Scholar]
- Wang, G.; Hamid, T.; Keith, R.J.; Zhou, G.; Partridge, C.R.; Xiang, X.; Kingery, J.R.; Lewis, R.K.; Li, Q.; Rokosh, D.G.; et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 2010, 121, 1912–1925. [Google Scholar]
- Kroon, P.A.; Iyer, A.; Chunduri, P.; Chan, V.; Brown, L. The cardiovascular nutrapharmacology of resveratrol: Pharmacokinetics, molecular mechanisms and therapeutic potential. Curr. Med. Chem. 2010, 17, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Tkachev, V.O.; Menshchikova, E.B.; Zenkov, N.K. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Moscow) 2011, 76, 407–422. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001, 174, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Li, C.P.; Qin, G.; Shi, R.Z.; Zhang, M.S.; Lv, J.Y. Ginsenoside Rg1 reduces toxicity of PM 2.5 on human umbilical vein endothelial cells by upregulating intracellular antioxidative state. Environ. Toxicol. Pharmacol. 2013, 35, 21–29. [Google Scholar]
- Tang, Y.; Jacobi, A.; Vater, C.; Zou, X.; Stiehler, M. Salvianolic acid B protects human endothelial progenitor cells against oxidative stress-mediated dysfunction by modulating Akt/mTOR/4EBP1, p38 MAPK/ATF2, and ERK1/2 signaling pathways. Biochem. Pharmacol. 2014, 90, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Miller, W.H.; Al-Benna, S.; Brosnan, M.J.; Drummond, R.D.; McBride, M.W.; Dominiczak, A.F. Strategies to reduce oxidative stress in cardiovascular disease. Clin. Sci. (Lond.) 2004, 106, 219–234. [Google Scholar] [CrossRef]
- Knaapen, A.M.; Borm, P.J.; Albrecht, C.; Schins, R.P. Inhaled particles and lung cancer. Part A: Mechanisms. Int. J. Cancer. 2004, 109, 799–809. [Google Scholar] [CrossRef]
- Pullikotil, P.; Chen, H.; Muniyappa, R.; Greenberg, C.C.; Yang, S.; Reiter, C.E.; Lee, J.W.; Chung, J.H.; Quon, M.J. Epigallocatechin gallate induces expression of heme oxygenase-1 in endothelial cells via p38 MAPK and Nrf-2 that suppresses proinflammatory actions of TNF-α. J. Nutr. Biochem. 2012, 23, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hsieh, C.W.; Lai, P.H.; Lin, J.B.; Liu, Y.C.; Wung, B.S. Upregulation of endothelial heme oxygenase-1 expression through the activation of the JNK pathway by sublethal concentrations of acrolein. Toxicol. Appl. Pharmacol. 2006, 214, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Wung, B.S.; Hsu, M.C.; Wu, C.C.; Hsieh, C.W. Piceatannol upregulates endothelial heme oxygenase-1 expression via novel protein kinase C and tyrosine kinase pathways. Pharmacol. Res. 2006, 53, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Burnett, R.T.; Thurston, G.D.; Thun, M.J.; Calle, E.E.; Krewski, D.; Godleski, J.J. Cardiovascular mortality and long-term exposure to particulate air pollution epidemiological evidence of general pathophysiological pathways of disease. Circulation 2004, 109, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Alfranca, A.; Iñiguez, M.A.; Fresno, M.; Redondo, J.M. Prostanoid signal transduction and gene expression in the endothelium: Role in cardiovascular diseases. Cardiovasc. Res. 2006, 70, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Mattei, E.; Rivera, E.; Gioda, A.; Sanchez-Rivera, D.; Roman-Velazquez, F.R.; Jimenez-Velez, B.D. Use of human bronchial epithelial cells (BEAS-2B) to study immunological markers resulting from exposure to PM 2.5 organic extract from Puerto Rico. Toxicol. Appl. Pharmacol. 2010, 243, 381–389. [Google Scholar]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.C.; Song, T.Y.; Yeh, Y.C.; Huang, C.Y.; Yang, S.F.; Chiu, T.H.; Tsai, K.H.; Chen, K.L.; Wu, Y.J.; Tsai, C.S.; et al. EGCG protects against oxidized LDL-induced endothelial dysfunction by inhibiting LOX-1-mediated signaling. J. Appl. Physiol. 2010, 108, 1745–1756. [Google Scholar]
- Lee, J.M.; Johnson, J.A. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J. Biochem. Mol. Biol. 2004, 37, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.-Z.; Wang, Z.-J.; Bai, F.; Qin, X.-J.; Cao, J.; Lv, J.-Y.; Zhang, M.-S. Epigallocatechin-3-Gallate Protects HUVECs from PM2.5-Induced Oxidative Stress Injury by Activating Critical Antioxidant Pathways. Molecules 2015, 20, 6626-6639. https://doi.org/10.3390/molecules20046626
Yang G-Z, Wang Z-J, Bai F, Qin X-J, Cao J, Lv J-Y, Zhang M-S. Epigallocatechin-3-Gallate Protects HUVECs from PM2.5-Induced Oxidative Stress Injury by Activating Critical Antioxidant Pathways. Molecules. 2015; 20(4):6626-6639. https://doi.org/10.3390/molecules20046626
Chicago/Turabian StyleYang, Guang-Zhao, Zhao-Jun Wang, Feng Bai, Xiao-Jiang Qin, Jing Cao, Ji-Yuan Lv, and Ming-Sheng Zhang. 2015. "Epigallocatechin-3-Gallate Protects HUVECs from PM2.5-Induced Oxidative Stress Injury by Activating Critical Antioxidant Pathways" Molecules 20, no. 4: 6626-6639. https://doi.org/10.3390/molecules20046626
APA StyleYang, G.-Z., Wang, Z.-J., Bai, F., Qin, X.-J., Cao, J., Lv, J.-Y., & Zhang, M.-S. (2015). Epigallocatechin-3-Gallate Protects HUVECs from PM2.5-Induced Oxidative Stress Injury by Activating Critical Antioxidant Pathways. Molecules, 20(4), 6626-6639. https://doi.org/10.3390/molecules20046626