
Bioavailability Enhancement of Paclitaxel via a Novel Oral Drug Delivery System: Paclitaxel-Loaded Glycyrrhizic Acid Micelles

Abstract
:1. Introduction
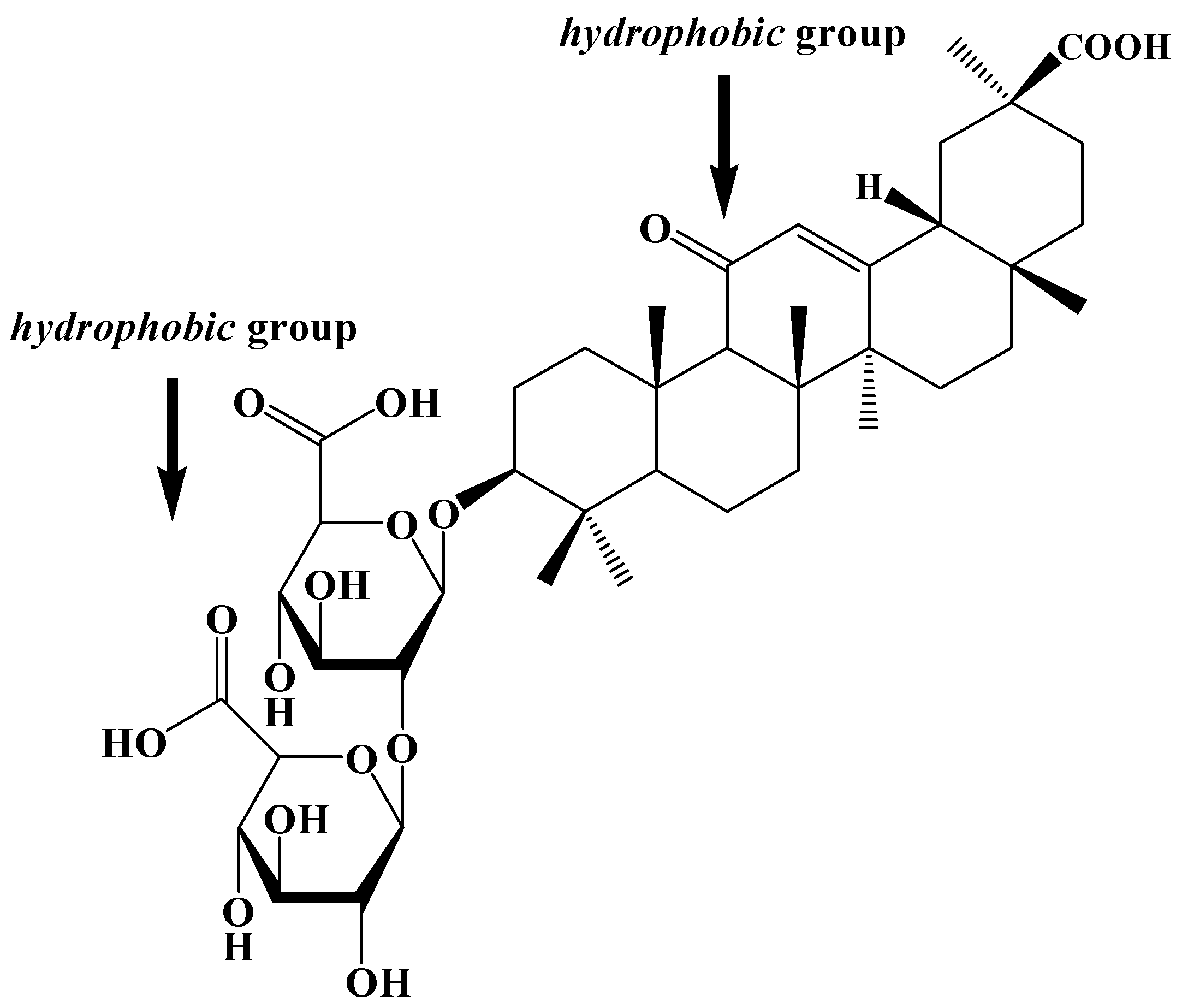
2. Results and Discussions
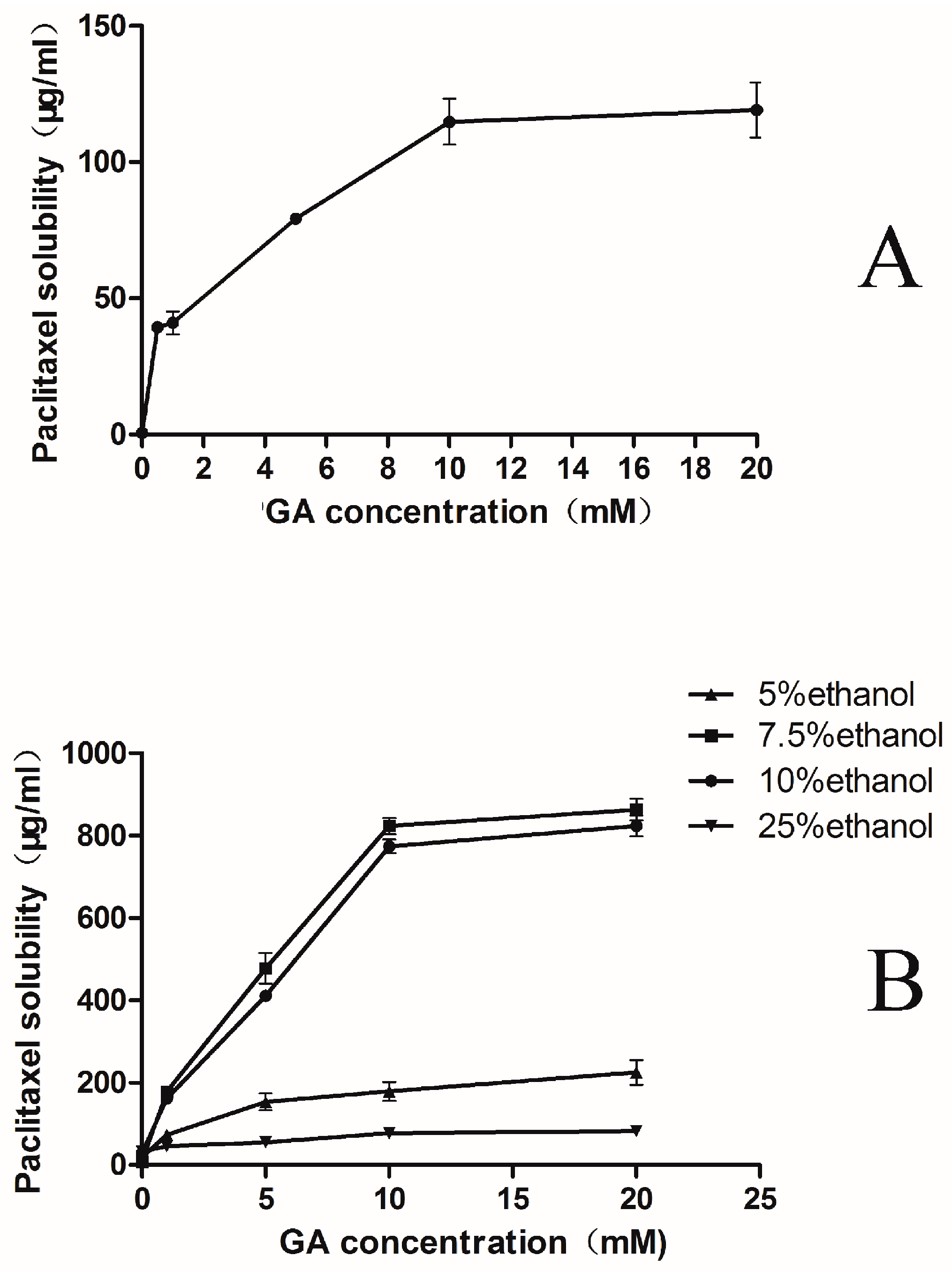
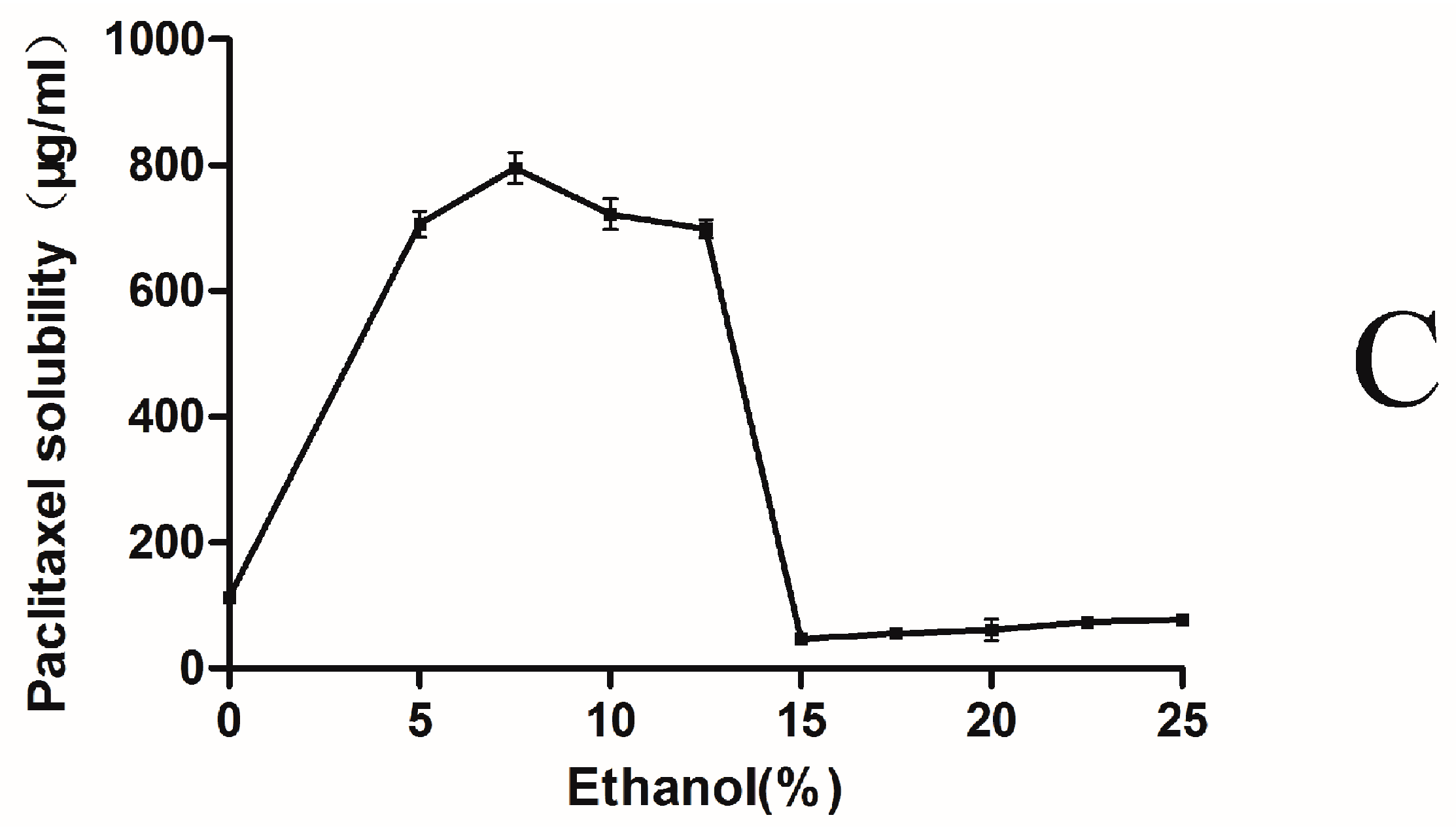
2.1. Phase-Solubility Test
2.2. Characterization of the PTX-Loaded GA Micelles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Size (nm) | Zeta Potential (mV) | Polydispersity Index |
|---|---|---|---|
| Taxol® | 15.1 ± 0.4 | 3.0 ± 0.2 | 0.333 ± 0.03 |
| Bare GA micelle | 82.3 ± 6.4 | −10.0 ± 1.1 | 0.243 ± 0.03 |
| PTX-loaded GA micelle | 245.4 ± 5.6 | −45.7 ± 1.8 | 0.192 ± 0.02 |
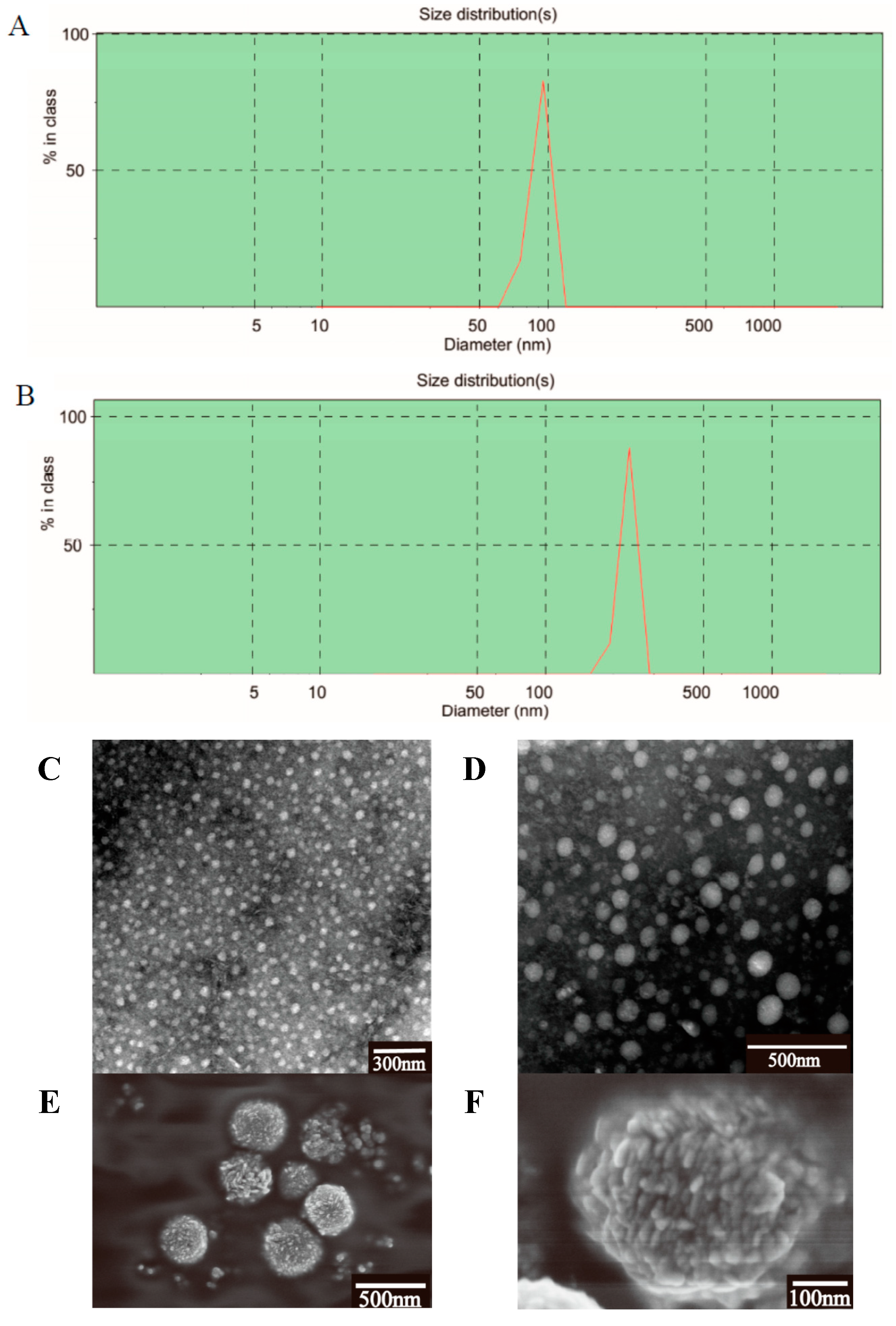
2.3. Micelles Stability
| Time | Size (nm) | Polydispersity Index | Zeta Potential (mV) | Encapsulation Efficiency (%) |
|---|---|---|---|---|
| 0 month | 249.4 ± 4.5 | 0.181 ± 0.02 | −45.7 ± 1.8 | 89.97 ± 0.82 |
| 1 month | 247.7 ± 1.2 | 0.183 ± 0.02 | −47.6 ± 2.7 | 87.64 ± 1.27 |
| 2 month | 242.3 ± 9.0 | 0.187 ± 0.05 | −48.9 ± 2.1 | 84.07 ± 3.24 |
| 3 month | 227.8 ± 9.0 | 0.192 ± 0.03 | −51.3 ± 3.2 | 82.57 ± 4.36 |
2.4. In Vitro Release
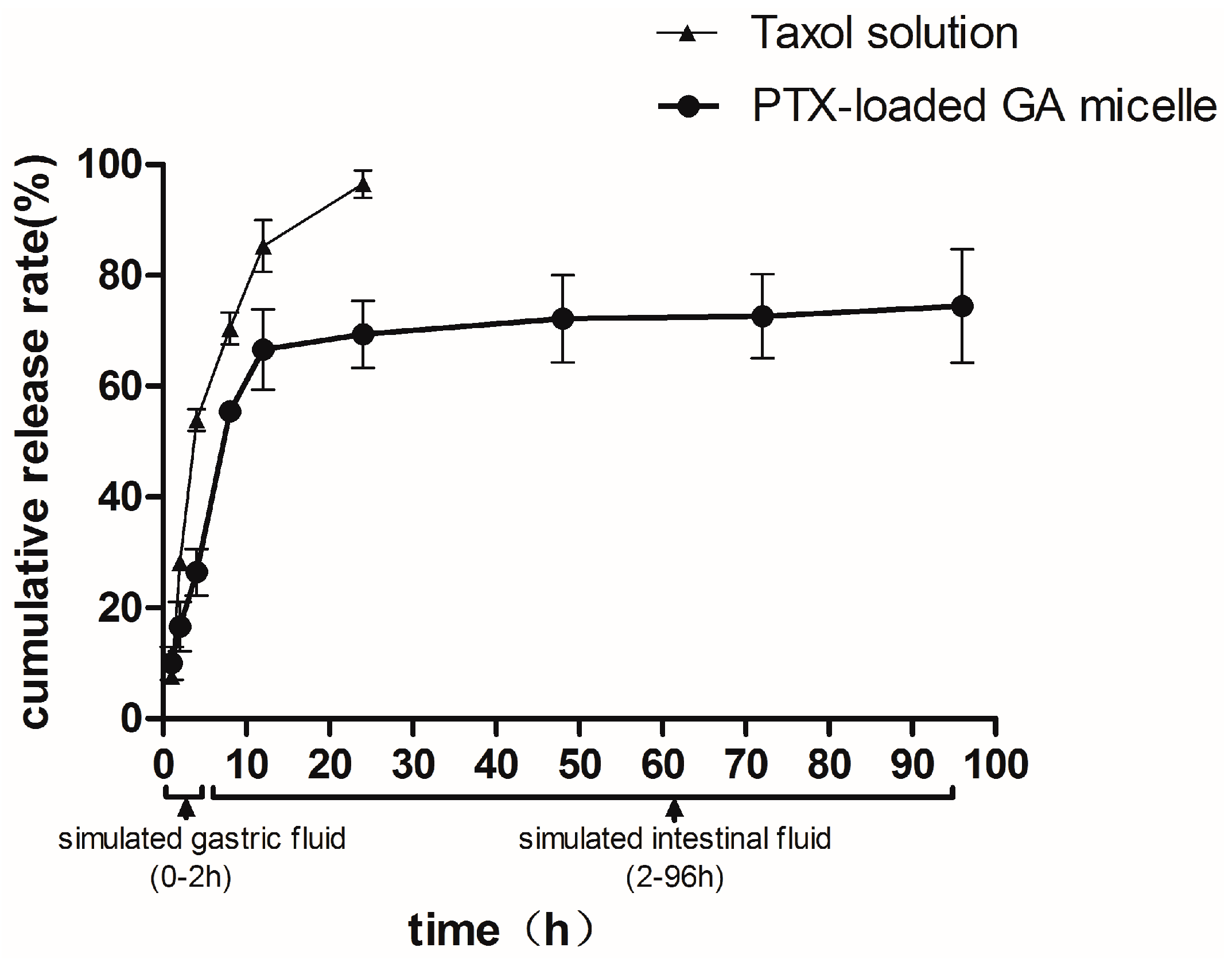
2.5. DSC Analysis
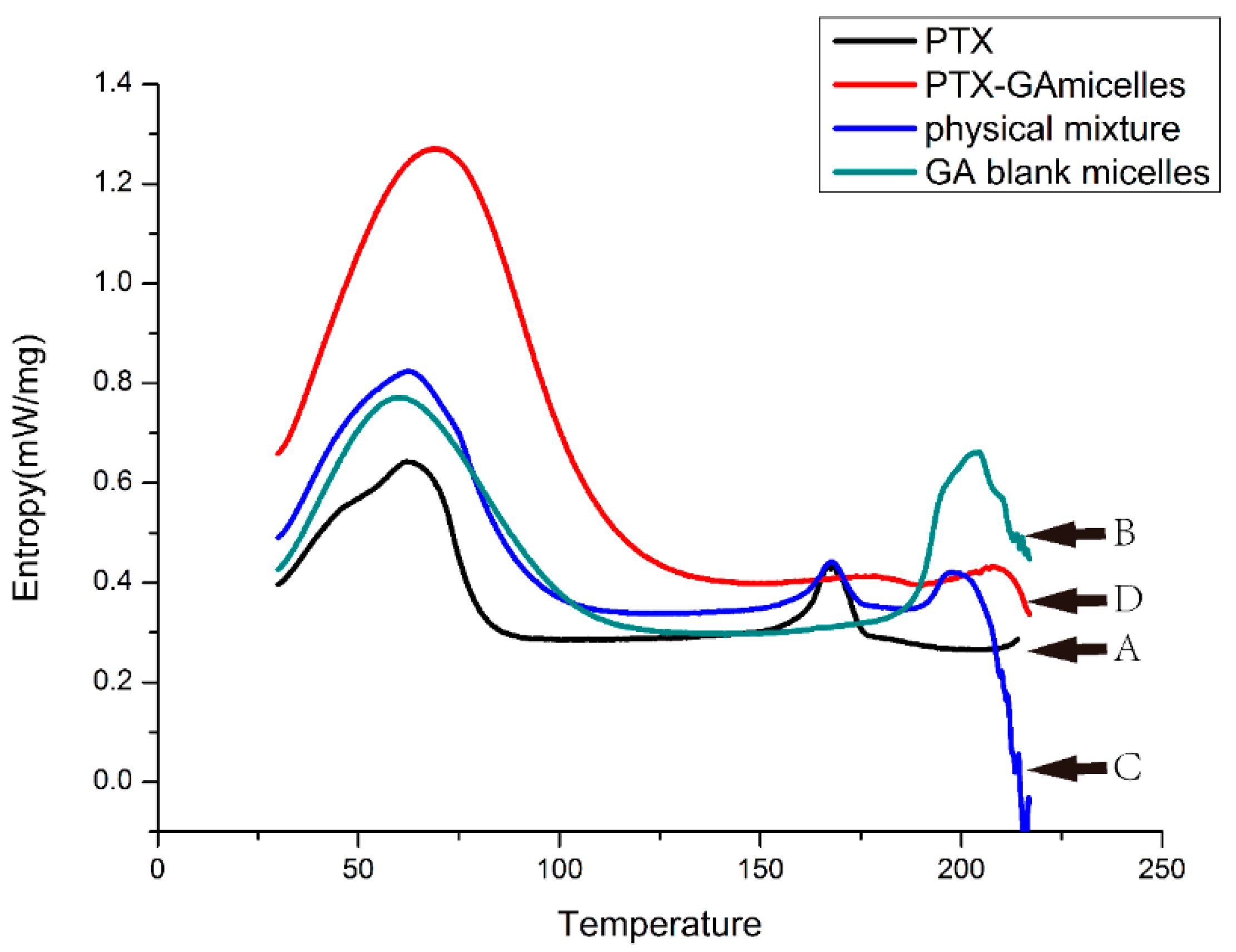
2.6. Pharmacokinetic Studies in Rats
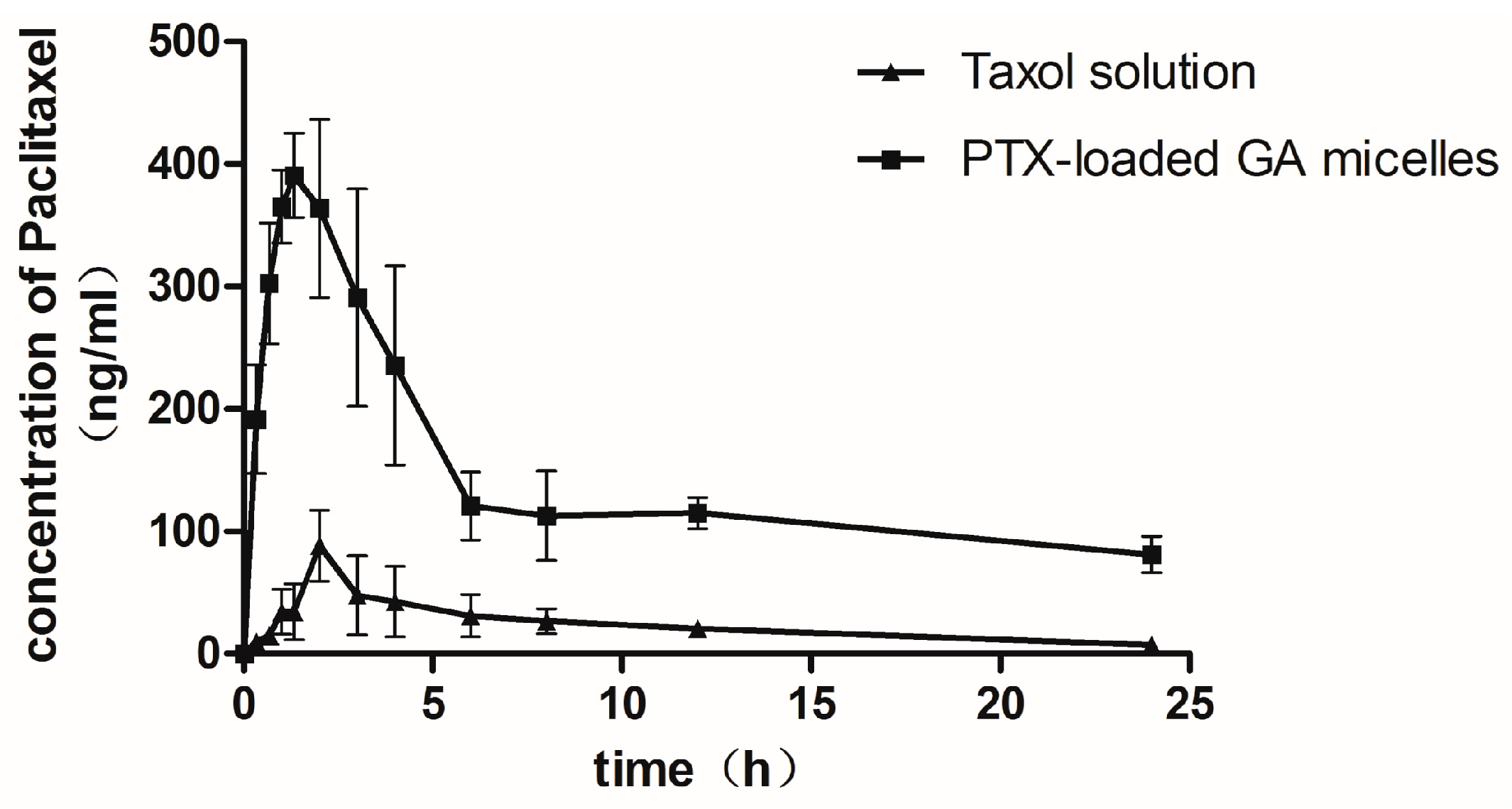
| Parameters | Taxol (Oral) | PTX-Loaded-GA Micelle (Oral) | Taxol (I.V.) |
|---|---|---|---|
| Cmax (μg/mL) | 0.10 ± 0.01 | 0.46 ± 0.10 * | 20.034 ± 5.701 |
| tmax (h) | 2.60 ± 0.89 | 1.63 ± 0.92 | —— |
| AUC0–24 h (μg·h/mL) | 0.57 ± 0.12 | 3.42 ± 1.02 * | 10.26 ± 1.42 |
| t1/2 (h) | 9.49 ± 3.52 | 11.76 ± 5.76 | 12.33 ± 7.91 |
| MRT(h) | 8.18 ± 1.12 | 9.08 ± 1.34 | 3.56 ± 1.58 |
| CL(L/h) | 7.47 ± 0.98 | 1.09 ± 0.23 * | 0.46 ± 0.10 |
| Absolute bioavailability(%) | 1.68 | 10.0 * | 100 |
| Relative bioavailability(%) | 100 | 596.38 | —— |
2.7. Intestinal Absorption of PTX by in Situ Closed Loop Method
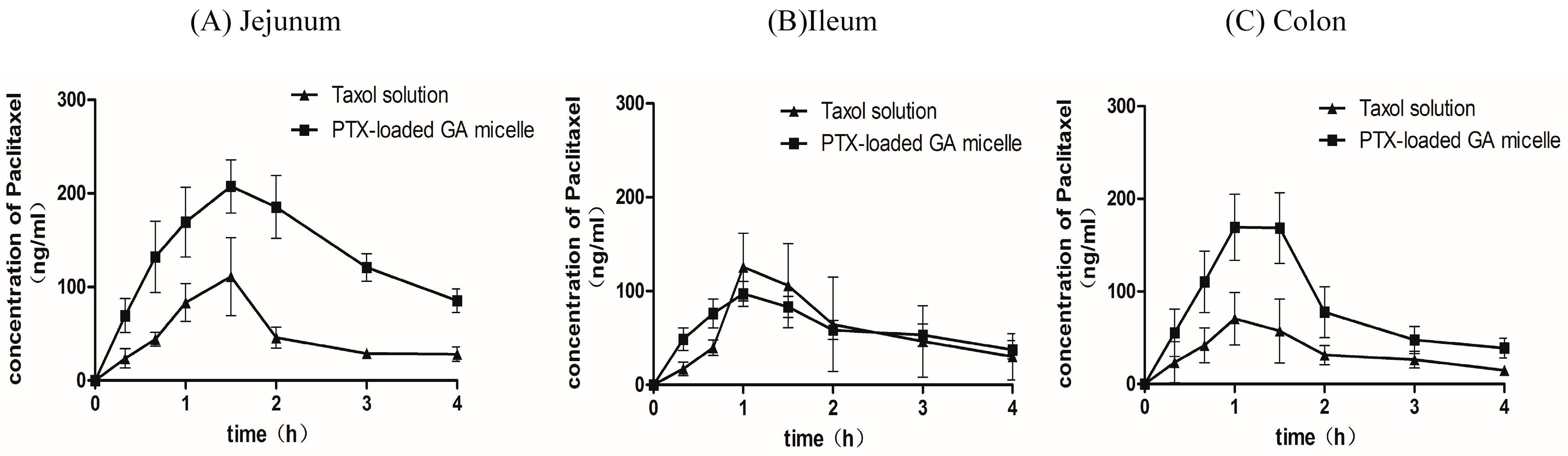
| Parameters | Cmax (μg/mL) | tmax (h) | AUC0–4 h(μg·h/mL) | t1/2 (h) |
|---|---|---|---|---|
| Jejunum | ||||
| Taxol® | 0.12 ± 0.08 | 1.30 ± 0.27 | 0.19 ± 0.10 | 1.65 ± 0.48 |
| PTX-loaded GA micelle | 0.21 ± 0.06 | 1.60 ± 0.22 | 0.54 ± 0.20* | 2.32 ± 1.33 |
| Ileum | ||||
| Taxol® | 0.23 ± 0.12 | 1.20 ± 0.27 | 0.23 ± 0.12 | 1.34 ± 0.57 |
| PTX-loaded GA micelle | 0.24 ± 0.10 | 1.10 ± 0.22 | 0.24 ± 0.10 | 2.48 ± 1.28 |
| Colon | ||||
| Taxol® | 0.08 ± 0.03 | 1.07 ± 0.48 | 0.14 ± 0.04 | 1.92 ± 0.79 |
| PTX-loaded GA micelle | 0.18 ± 0.03 * | 1.20 ± 0.27 | 0.31 ± 0.13 * | 1.73 ± 0.73 |
3. Experimental Section
3.1. Chemicals
3.2. Animals
3.3. Methods
3.3.1. Solubility Test
3.3.2. Preparation of PTX-Loaded GA Micelle
3.3.3. Particle Size, Size Distribution and Zeta Potential
3.3.4. Transmission Electron Microscopy (TEM)
3.3.5. Scanning Electron Microscopy (SEM)
3.3.6. Determination of the Drug Encapsulation Efficiency and Drug Loading Rate
3.3.7. The Storage Stability of PTX-Loaded GA Micelle
3.3.8. In Vitro Release of Drug-Loaded Micelles Solution
3.3.9. Differential Scanning Calorimetry (DSC) Analysis
3.3.10. Pharmacokinetic Studies in Rats
3.3.11. Intestinal Absorption of PTX
3.3.12. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singla, A.K.; Garg, A.; Aggarwal, D. Paclitaxel and its formulations. Int. J. Pharm. 2002, 235, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Beijnen, J.H.; Huizing, M.T.; Ten, B.H.W.; Veenhof, C.H.; Vermorken, J.B.; Giaccone, G.; Pinedo, H.M. Bioanalysis, pharmacokinetics, and pharmacodynamics of the novel anticancer drug paclitaxel (Taxol). Semin Oncol 1994, 21 (Suppl. S8), 53–62. [Google Scholar] [PubMed]
- Sparreboom, A.; van Asperen, J.; Mayer, U.; Schinkel, A.H.; Smit, J.W.; Meijer, D.K.; Borst, P.; Nooijen, W.J.; Beijnen, J.H.; van Tellingen, O. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 1997, 94, 2031–2035. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sarti, F.; Perera, G.; Bernkop-Schnurch, A. Development and in vivo evaluation of an oral drug delivery system for paclitaxel. Biomaterials 2011, 32, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.D.; Ren, F.; Li, G.F.; Liu, S.J. Preparation of paclitaxel-loaded polybutylcyanoacrylate nanoparticles. Nan Fang Yi Ke Da Xue Xue Bao 2010, 30, 763–766. [Google Scholar] [PubMed]
- Campos, F.C.; Victorino, V.J.; Martins-Pinge, M.C.; Cecchini, A.L.; Panis, C.; Cecchini, R. Systemic toxicity induced by paclitaxel in vivo is associated with the solvent cremophor EL through oxidative stress-driven mechanisms. Food Chem. Toxicol. 2014, 68, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Russell-Jones, G.J. Use of targeting agents to increase uptake and localization of drugs to the intestinal epithelium. J. Drug Target 2004, 12, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Nornoo, A.O.; Zheng, H.; Lopes, L.B.; Johnson-Restrepo, B.; Kannan, K.; Reed, R. Oral microemulsions of paclitaxel: In situ and pharmacokinetic studies. Eur. J. Pharm. Biopharm. 2009, 71, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, Y.M. Taxol-loaded block copolymer nanospheres composed of methoxy poly(ethylene glycol) and poly(epsilon-caprolactone) as novel anticancer drug carriers. Biomaterials 2001, 22, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.; Feng, S.S. Preparation and characterization of poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials 2003, 24, 5037–5044. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, R.; Caputo, O.; Gasco, M.R. Preparation and characterization of solid lipid nanospheres containing paclitaxel. Eur. J. Pharm. Sci. 2000, 10, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Yoncheva, K.; Calleja, P.; Agueros, M.; Petrov, P.; Miladinova, I.; Tsvetanov, C.; Irache, J.M. Stabilized micelles as delivery vehicles for paclitaxel. Int. J. Pharm. 2012, 436, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, F.Z.; Yang, H.; Zhou, J.; Yao, J.; Zhang, T.; Zhang, Q. Enhanced oral bioavailability of paclitaxel in pluronic/LHR mixed polymeric micelles: Preparation, in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2012, 47, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Torne, S.J.; Ansari, K.A.; Vavia, P.R.; Trotta, F.; Cavalli, R. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded nanosponges. Drug Deliv. 2010, 17, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Khandavilli, S.; Panchagnula, R. Nanoemulsions as versatile formulations for paclitaxel delivery: Peroral and dermal delivery studies in rats. J. Investig. Dermatol. 2007, 127, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cabrer, P.; Campos, F. Liposomes and nanotechnology in drug development: Focus on neurological targets. Int. J. Nanomedicine 2013, 8, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Hamman, J.H. Chitosan based polyelectrolyte complexes as potential carrier materials in drug delivery systems. Mar. Drugs 2010, 8, 1305–1322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, N.; Wang, K.; Shi, C.; Zhang, L.; Luan, Y. A review of polypeptide-based polymersomes. Biomaterials 2014, 35, 1284–1301. [Google Scholar] [CrossRef] [PubMed]
- Fiore, C.; Eisenhut, M.; Ragazzi, E.; Zanchin, G.; Armanini, D. A history of the therapeutic use of liquorice in Europe. J. Ethnopharmacol. 2005, 99, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S. A drug over the millennia: Pharmacognosy, chemistry, and pharmacology of licorice. Yakugaku Zasshi 2000, 120, 849–862. [Google Scholar] [PubMed]
- Selyutina, O.Y.; Polyakov, N.E.; Korneev, D.V.; Zaitsev, B.N. Influence of glycyrrhizin on permeability and elasticity of cell membrane: Perspectives for drugs delivery. Drug Deliv. 2014, 1–8. [Google Scholar] [CrossRef]
- Zhao, M.X.; Ji, L.N.; Mao, Z.W. beta-Cyclodextrin/glycyrrhizic acid functionalised quantum dots selectively enter hepatic cells and induce apoptosis. Chemistry 2012, 18, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Davey, A.K.; Chen, Y.X.; Wang, J.P.; Liu, X.Q. Effects of diammonium glycyrrhizinate on the pharmacokinetics of aconitine in rats and the potential mechanism. Xenobiotica 2009, 39, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Radwant, M.A.; Aboul-Enein, H.Y. The effect of oral absorption enhancers on the in vivo performance of insulin-loaded poly(ethylcyanoacrylate) nanospheres in diabetic rats. J. Microencapsul. 2002, 19, 225–235. [Google Scholar] [CrossRef] [PubMed]
- James, K.C.; Stanford, J.B. The solubilising properties of liquorice. J. Pharm. Pharmacol. 1962, 14, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Kornievskaya, V.S.; Kruppa, A.I.; Polyakov, N.E.; Leshina, T.V. Effect of glycyrrhizic acid on lappaconitine phototransformation. J. Phys. Chem. B 2007, 111, 11447–11452. [Google Scholar] [CrossRef] [PubMed]
- Vervaet, C.; Byron, P.R. Drug-surfactant-propellant interactions in HFA-formulations. Int. J. Pharm. 1999, 186, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Koizumi, M.; Adachi, I.; Kawakami, J. Inhibition of P-glycoprotein-mediated transport by terpenoids contained in herbal medicines and natural products. Food Chem. Toxicol. 2006, 44, 2033–2039. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Peng, R.X.; Kong, R.; Yu, J.P. Effects of 18 alpha-glycyrrhizic acid on rat liver cytochrome P450 isoenzymes and phase II transferase. Yao Xue Xue Bao 2001, 36, 321–324. [Google Scholar] [PubMed]
- Tolstikova, T.G.; Khvostov, M.V.; Bryzgalov, A.O. The complexes of drugs with carbohydrate-containing plant metabolites as pharmacologically promising agents. Mini Rev. Med. Chem. 2009, 9, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Nafisi, S.; Manouchehri, F.; Bonsaii, M. Study on the interaction of glycyrrhizin and glycyrrhetinic acid with RNA. J. Photochem. Photobiol. B 2012, 111, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; McShane, M.J. Loading of hydrophobic materials into polymer particles: Implications for fluorescent nanosensors and drug delivery. J. Am. Chem. Soc. 2005, 127, 13448–13449. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, S.; Papp, M.; Park, K.; Pinal, R. Hydrotropic solubilization of poorly water-soluble drugs. J. Pharm. Sci. 2010, 99, 3953–3965. [Google Scholar] [CrossRef] [PubMed]
- Konnova, T.A.; Faizullin, D.A.; Haertle, T.; Zuev, Y.F. beta-casein micelle formation in water-ethanol solutions. Dokl. Biochem. Biophys. 2013, 448, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Paik, S.Y.; Ryu, J.; Choi, K.O.; Kang, T.S.; Lee, J.K.; Song, C.W.; Ko, S. Dynamic light scattering-based method to determine primary particle size of iron oxide nanoparticles in simulated gastrointestinal fluid. Food Chem. 2014, 161, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.Q.; Wu, X.L.; Du, Y.Z.; You, J.; Yuan, H. Cellular uptake and cytotoxicity of shell crosslinked stearic acid-grafted chitosan oligosaccharide micelles encapsulating doxorubicin. Eur. J. Pharm. Biopharm. 2008, 69, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Mignet, N.; Seguin, J.; Ramos, R.M.; Brulle, L.; Touil, Y.S.; Scherman, D.; Bessodes, M.; Chabot, G.G. Development of a liposomal formulation of the natural flavonoid fisetin. Int. J. Pharm. 2012, 423, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Sun, S.; Li, X.; Piao, H.; Piao, H.; Cui, F.; Fang, L. alpha-Tocopherol succinate-modified chitosan as a micellar delivery system for paclitaxel: Preparation, characterization and in vitro/in vivo evaluations. Int. J. Pharm. 2012, 423, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Agueros, M.; Ruiz-Gaton, L.; Vauthier, C.; Bouchemal, K.; Espuelas, S.; Ponchel, G.; Irache, J.M. Combined hydroxypropyl-beta-cyclodextrin and poly(anhydride) nanoparticles improve the oral permeability of paclitaxel. Eur. J. Pharm. Sci. 2009, 38, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, G.; Satturwar, P.; Jones, M.C.; Furtos, A.; Leroux, J.C. Polymeric micelles for oral drug delivery. Eur. J. Pharm. Biopharm. 2010, 76, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liang, S.; John, J.; Hsiao, C.H.; Wei, X.; Liang, D.; Xie, H. Development of PLGA-based itraconazole injectable nanospheres for sustained release. Int. J. Nanomedicine 2013, 8, 4521–4531. [Google Scholar] [PubMed]
- Kulkarni, S.A.; Feng, S.S. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm. Res. 2013, 30, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Hom, F.S. Phase solubility of solid species formed by magnesium aluminate from aqueous solutions containing sulfate ions. J. Pharm. Sci. 1963, 52, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Wang, Y.M.; Adachi, I.; Horikoshi, I. Pharmacokinetic study of taxol-loaded poly(lactic-co-glycolic acid) microspheres containing isopropyl myristate after targeted delivery to the lung in mice. Biol. Pharm. Bull. 1996, 19, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, Y.; Chen, Y.; Ye, J.; Sha, X.; Fang, X. Multifunctional Pluronic P123/F127 mixed polymeric micelles loaded with paclitaxel for the treatment of multidrug resistant tumors. Biomaterials 2011, 32, 2894–2906. [Google Scholar] [CrossRef] [PubMed]
- Fetih, G.; Habib, F.; Okada, N.; Fujita, T.; Attia, M.; Yamamoto, A. Nitric oxide donors can enhance the intestinal transport and absorption of insulin and [Asu(1,7)]-eel calcitonin in rats. J. Control Release 2005, 106, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds of PTX and GA are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.-H.; Zhang, Q.; Liang, Q.-Y.; Wang, S.-Q.; Zhao, B.-X.; Wang, Y.-T.; Cai, Y.; Li, G.-F. Bioavailability Enhancement of Paclitaxel via a Novel Oral Drug Delivery System: Paclitaxel-Loaded Glycyrrhizic Acid Micelles. Molecules 2015, 20, 4337-4356. https://doi.org/10.3390/molecules20034337
Yang F-H, Zhang Q, Liang Q-Y, Wang S-Q, Zhao B-X, Wang Y-T, Cai Y, Li G-F. Bioavailability Enhancement of Paclitaxel via a Novel Oral Drug Delivery System: Paclitaxel-Loaded Glycyrrhizic Acid Micelles. Molecules. 2015; 20(3):4337-4356. https://doi.org/10.3390/molecules20034337
Chicago/Turabian StyleYang, Fu-Heng, Qing Zhang, Qian-Ying Liang, Sheng-Qi Wang, Bo-Xin Zhao, Ya-Tian Wang, Yun Cai, and Guo-Feng Li. 2015. "Bioavailability Enhancement of Paclitaxel via a Novel Oral Drug Delivery System: Paclitaxel-Loaded Glycyrrhizic Acid Micelles" Molecules 20, no. 3: 4337-4356. https://doi.org/10.3390/molecules20034337
APA StyleYang, F.-H., Zhang, Q., Liang, Q.-Y., Wang, S.-Q., Zhao, B.-X., Wang, Y.-T., Cai, Y., & Li, G.-F. (2015). Bioavailability Enhancement of Paclitaxel via a Novel Oral Drug Delivery System: Paclitaxel-Loaded Glycyrrhizic Acid Micelles. Molecules, 20(3), 4337-4356. https://doi.org/10.3390/molecules20034337