
Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents


Abstract
:1. Background
2. Classification of HDAC Family
3. Histone Deacetylases and Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal Dependent | |||||
|---|---|---|---|---|---|
| Class | Members | Size (aa) | Cellular Localization | Physiological Function | X-ray Crystal |
| I | HDAC1 | 483 | Nucleus | Cell survival and proliferation | Yes |
| HDAC2 | 488 | Nucleus | Cell proliferation, Insulin resistance | Yes (core domain) | |
| HDAC3 | 428 | Nucleus | Cell survival and proliferation | Yes | |
| HDAC8 | 377 | Nucleus | Cell proliferation | Yes | |
| IIA | HDAC4 | 1084 | Nucleus/Cytoplasm | Regulation of skeletogenesis and gluconeogenesis | Yes (catalytic & glutamine rich domains) |
| HDAC5 | 1122 | Nucleus/Cytoplasm | Cardiovascular growth and function, gluconeogenesis, cardiac myocytes and endothelial cell function | No | |
| HDAC7 | 912 | Nucleus/Cytoplasm | Thymocyte differentiation, endothelial function, glucogenesis | Yes (catalytic domain) | |
| HDAC9 | 1069 | Nucleus/Cytoplasm | Homologous recombination, thymocyte differentiation, cardiovascular growth and function | No (structure is known for aa 138–158) | |
| IIB | HDAC6 | 1215 | Cytoplasm | Cell motility, control of cytoskeletal dynamics | Yes (zinc finger and ubiquitin binding domains) |
| HDAC10 | 669 | Cytoplasm | Homologous recombination, Autophagy mediated cell- survival | No | |
| IV | HDA11 | 347 | Nucleus | Immunomodulators-DNA replication | No |
| NAD+ Dependent | |||||
| III | SIRT 1 | 747 | Nucleus, Cytoplasm | Aging, redox regulation, cell survival, autoimmune system regulation | Yes (catalytic domain) |
| SIRT 2 | 389 | Nucleus | Cell survival-cell migration and invasion | Yes | |
| SIRT 3 | 399 | Mitochondria | Urea Cycle, Redox balance, ATP regulation, metabolism, apoptosis and cell signaling | Yes | |
| SIRT 4 | 314 | Mitochondria | Energy metabolism, ATP regulation, metabolism, apoptosis and cell signaling | No | |
| SIRT 5 | 310 | Mitochondria | Urea cycle, Energy metabolism, ATP regulation, metabolism, apoptosis and cell signaling | Yes | |
| SIRT 6 | 355 | Nucleus | Metabolic regulation | Yes | |
| SIRT 7 | 400 | Nucleus | Apoptosis | No | |
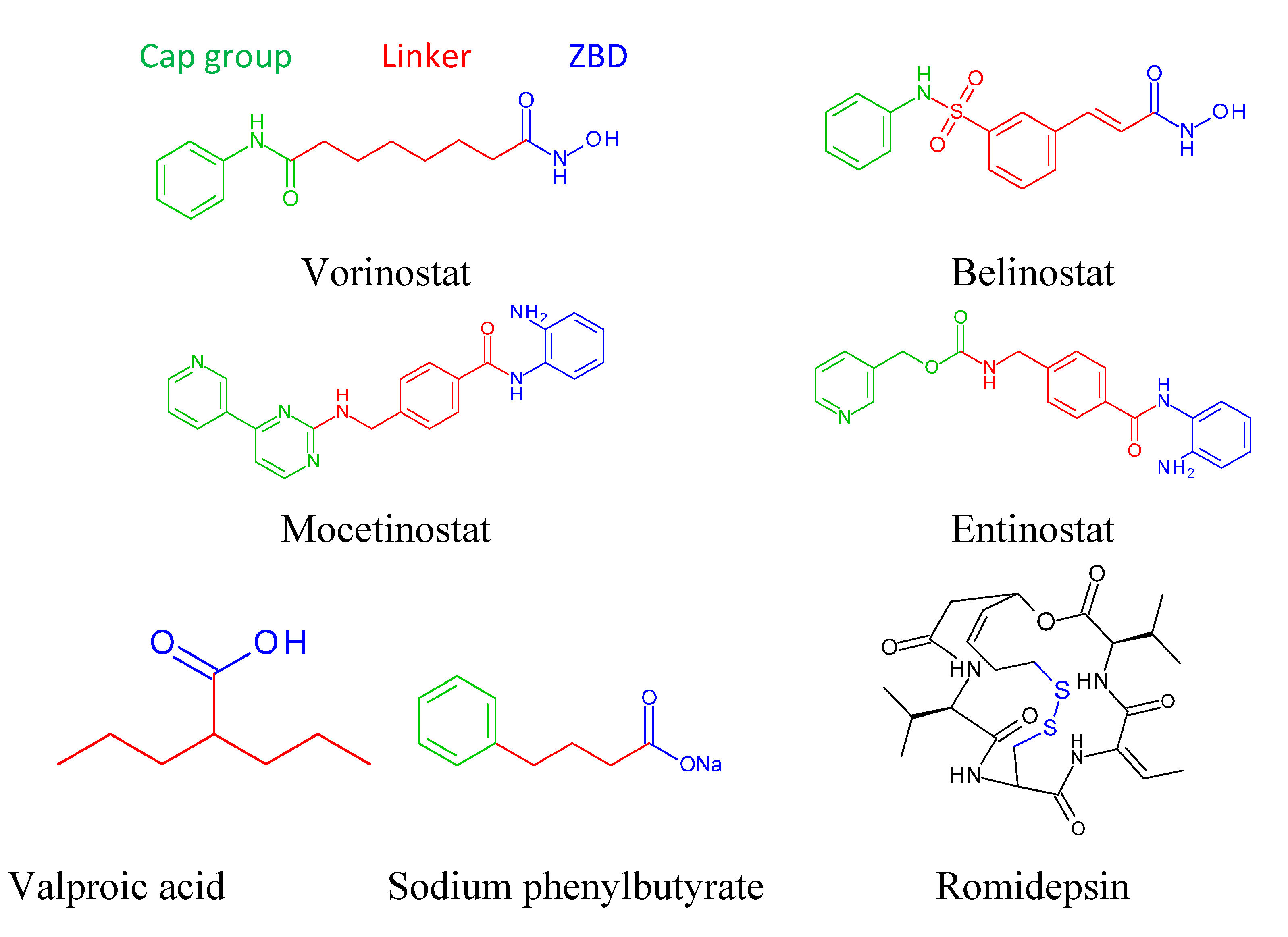
4. FDA Approved Drugs
4.1. Vorinostat
4.2. Romidepsin (Depsipeptide, ISTODAX)
4.3. Belinostat (Beleodaq)
5. Different Classes of HDAC Inhibitors
5.1. Hydroxamic Acid Derivatives
5.1.1. Abexinostat (PCI-24781)
5.1.2. Pracinostat (SB939)
5.1.3. Resminostat
5.1.4. Givinostat
5.1.5. Panobinostat (LB589)
5.1.6 CUDC-101
5.2. Benzamide Derivatives
| Hydroxamic Acid Based HDAC Inhibitors (HDACi) | HDAC Specificity (Class) | In Vitro Potency | Combination | Cancer Types | Reference |
|---|---|---|---|---|---|
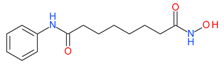 Vorinostat (SAHA) | I and II | nM | Temozolomide + radiation | Glioblastoma Multiforme (GBM) | [95] |
| CHOP | Peripheral T-cell lymphoma (PTCL) | [43] | |||
| - | Gastrointestinal(GI) | [44] | |||
| Whole brain radiation | Brain metastasis | [37] | |||
| 5-fluorouracil/leucovorin(5FU/LV) | Refractory colorectal and solid tumors | [96,97] | |||
| Hydroxychloroquine | Advanced solid tumors | [98] | |||
| Marizomib | Melanoma, Pancreatic and Lung cancer | [99] | |||
| Bortezomib | Multiple myeloma | [100] | |||
| 5-fluorouracil | Metastatic colorectal | [101] | |||
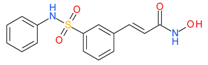 Belinostat (Beleodaq) | I and II | μM | - | Malignant pleural mesothelioma | [55] |
| - | Epithelial & microcapillary ovarian cancers | [56] | |||
| - | Thymic epithelial tumor(TETs) | [57] | |||
| - | Myelodysplastic syndrom (MDS) | [58] | |||
| Carboplatin | Platinum resistant ovarian cancer | [59] | |||
| Carboplatin + Paclitaxel | Ovarian cancer | [60] | |||
| - | Acute myeloid leukemia (AML) | [61] | |||
| Cisplatin + doxorubicin + cyclophosphamide | Thymic epithelial tumors | [62] | |||
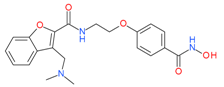 Abexinostat(PCI-24781) | I and II | nM | - | Advanced solid tumors | [70] |
| Pazopanib | Metastatic solid tumor | [95] | |||
| Cisplatin+radiation | Nasopharyngeal carcinoma (NPC) | [102] | |||
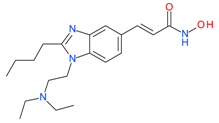 Pracinostat (SB939) | I, II and IV | μM | - | Myelofibrosis(MF) | [72] |
| - | Advanced solid tumors | [73] | |||
| - | Refractory solid tumors | [74] | |||
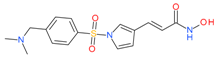 Resminostat | I and II | μM | - | Advanced solid tumors | [75] |
| - | Relapsed/refractory Hogdkin Lymphoma (HL) | [77,78] | |||
| or Sorafenib | Advanced hepatocellular carcinoma (HCC) | [79] | |||
| - | Colorectal carcinoma | [95] | |||
 Givinostat (ITF-2357) | I and II | nM | - | Myeloproliferative neoplasms(MPN) | [80] |
| Hydroxycarbamide | Polycythemia vera | [83] | |||
 Panobinostat | I and II | μM | - | Small cell lung cancer (SCLC) | [84] |
| - | Myelofibrosis(MF) | [85] | |||
| - | Advanced solid tumors | [86] | |||
| - | Cutaneous T-cell lymphoma | [87] | |||
| - | Relapsed/refractory hogdkins lymphoma | [88] | |||
| - | Myelodysplastic syndrome (MDS) | [89] | |||
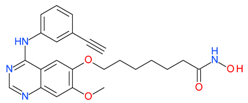 CUDC-101 | I and II | nM | - | Advanced solid tumors | [92–94] |
5.2.1. Mocetinostat (MGCD0103)
5.2.2. Entinostat
5.3. Short Chain Fatty Acids
Valproic Acid
| HDACi | HDAC Specificity (Class) | In Vitro Potency | Combination | Cancer Types | Reference |
|---|---|---|---|---|---|
| Benzamide Based HDAC Inhibitors (HDACi) | |||||
 Mocetinostat (MGCD0103) | I and IV | μM | - | Leukemia | [104] |
| - | Myelodysplastic syndrome (MDS) | [104] | |||
| - | Chronic lymphocytic leukemia (CLL) | [106] | |||
| - | Advanced solid tumors | [107] | |||
| - | Relapsed Hodgkin’s lymphoma | [108] | |||
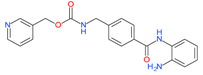 Entinostat (MS-275) | I | μM | 13-cis retinoic acid(CRA) | Advanced solid tumors | [109] |
| Erlotinib | NSCLC | [110] | |||
| Exemestane | Breast cancer | [111] | |||
| - | Refractory solid tumors and lymphoma | [112] | |||
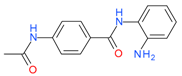 Tacedinaline (CI994) | I | μM | - | Advanced solid tumor | [119] |
| Short Chain Fatty Acid Based HDAC Inhibitors (HDACi) | |||||
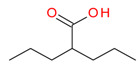 Valproic acid | I | mM | - | Refractory solid or central nervous system (CNS) tumors | [113] |
| - | Neuroendocrine tumors(NET) | [114] | |||
| Bevacizumab | Colorectal, Prostate, Breast, melanoma | [115] | |||
| Decitabine | NSCLC | [116] | |||
| S-1 | Pancreatobiliary | [117] | |||
| Hydralazine | Solid cancer | [118] | |||
 Phenylbutyrate | I and II | mM | - | Refractory solid tumor or lymphoma | [95] |
| - | Recurrent brain tumor | [95] | |||
| Azacitidine | Acute myeloid leukemia or MDS | [95] | |||
| Azacitidine | Prostate cancer | [95] | |||
| Azacitidine | NSCLC | [95] | |||
| Cyclic Peptide Based HDAC Inhibitors (HDACi) | |||||
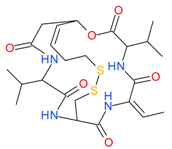 Romidepsin (Depsipeptide) | I | nM | - | Relapsed or refractory PTCL | [48,49] |
| Bortezomib | NSCLC | [50] | |||
| Abraxane | Inflammatory breast cancer | [95] | |||
| Gemcitabine | Pancreatic, Breast, NSCLC, Ovarian | [52] | |||
| - | Thyroid cancer | [53] | |||
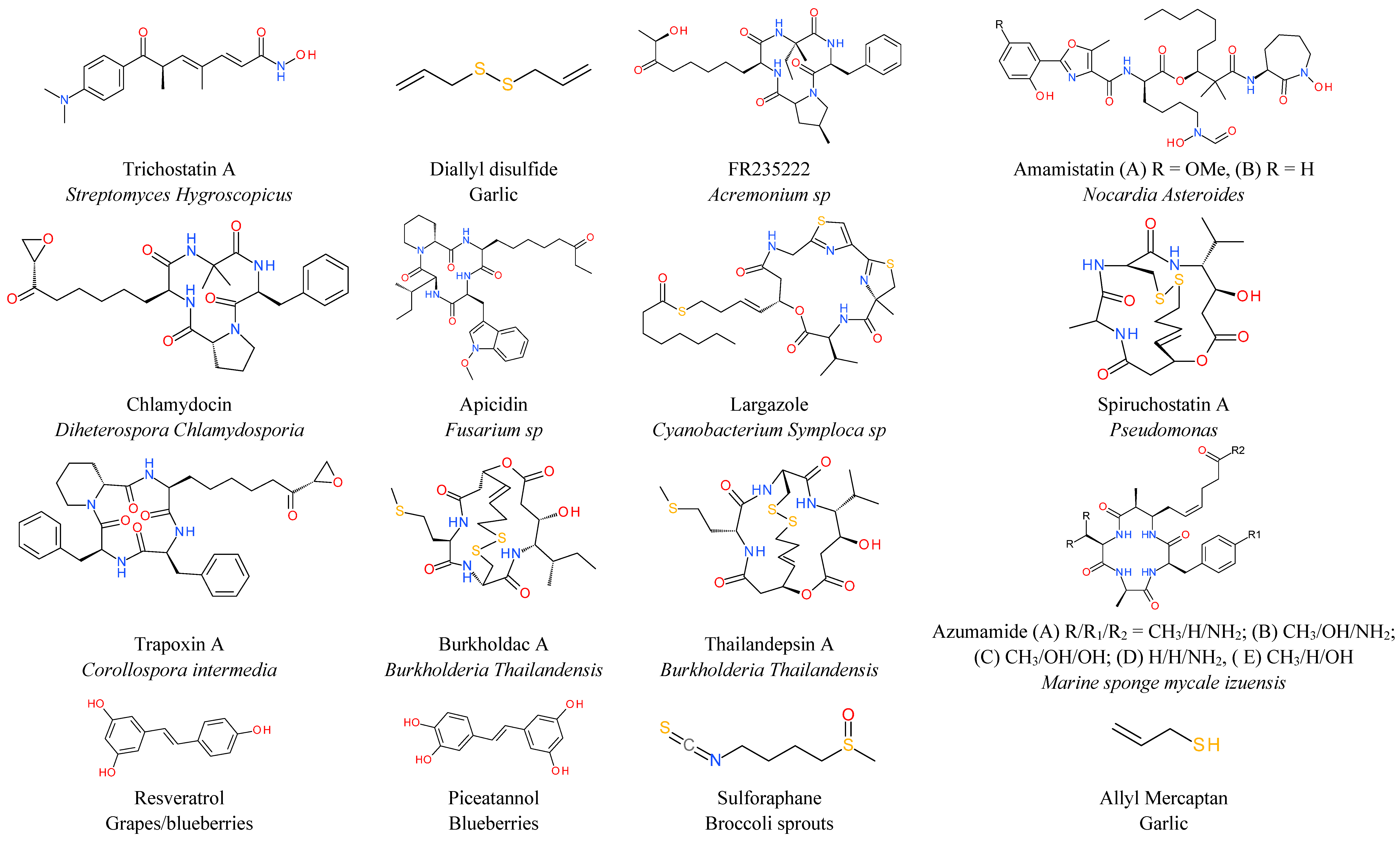
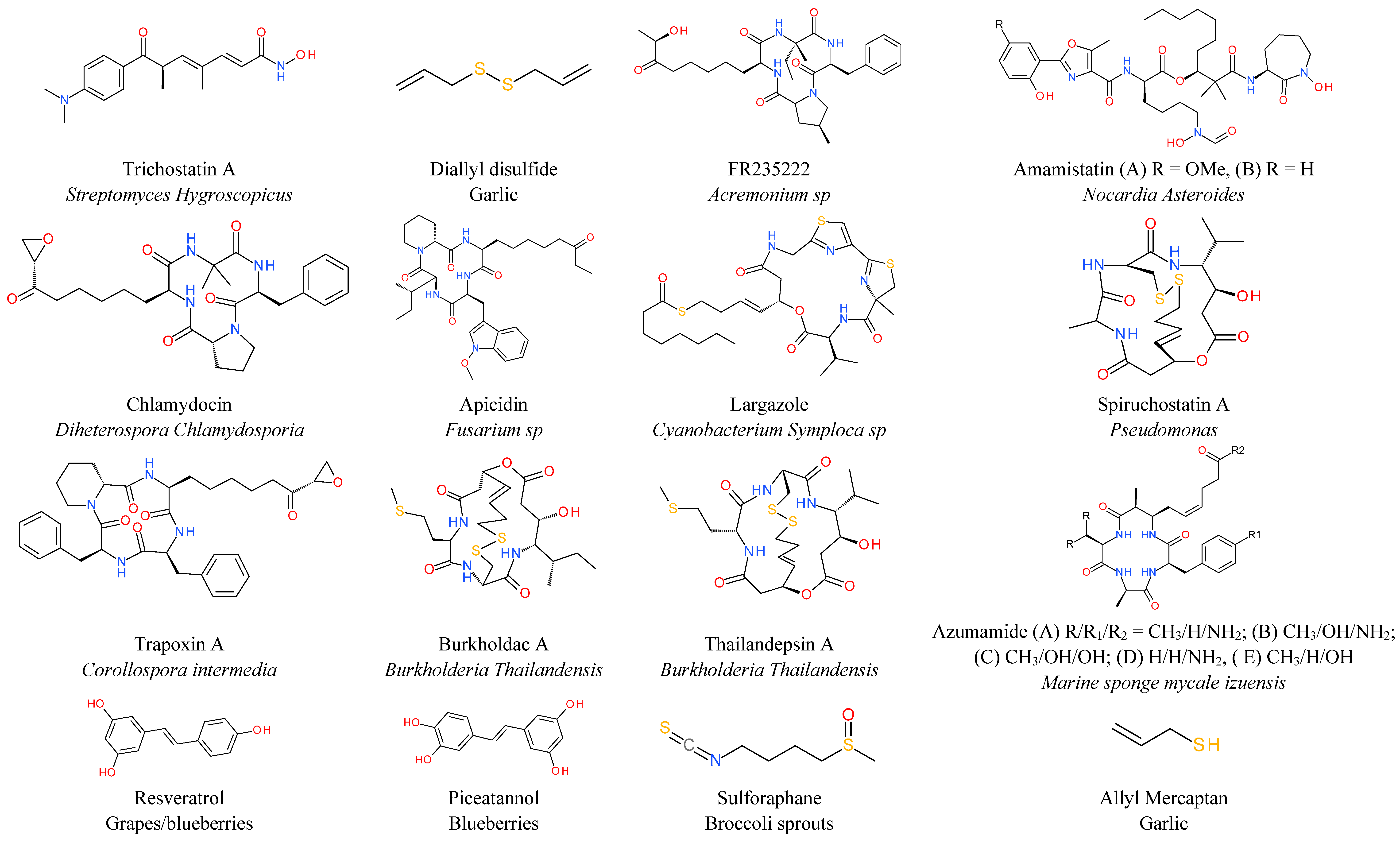
6. Natural HDAC Inhibitors
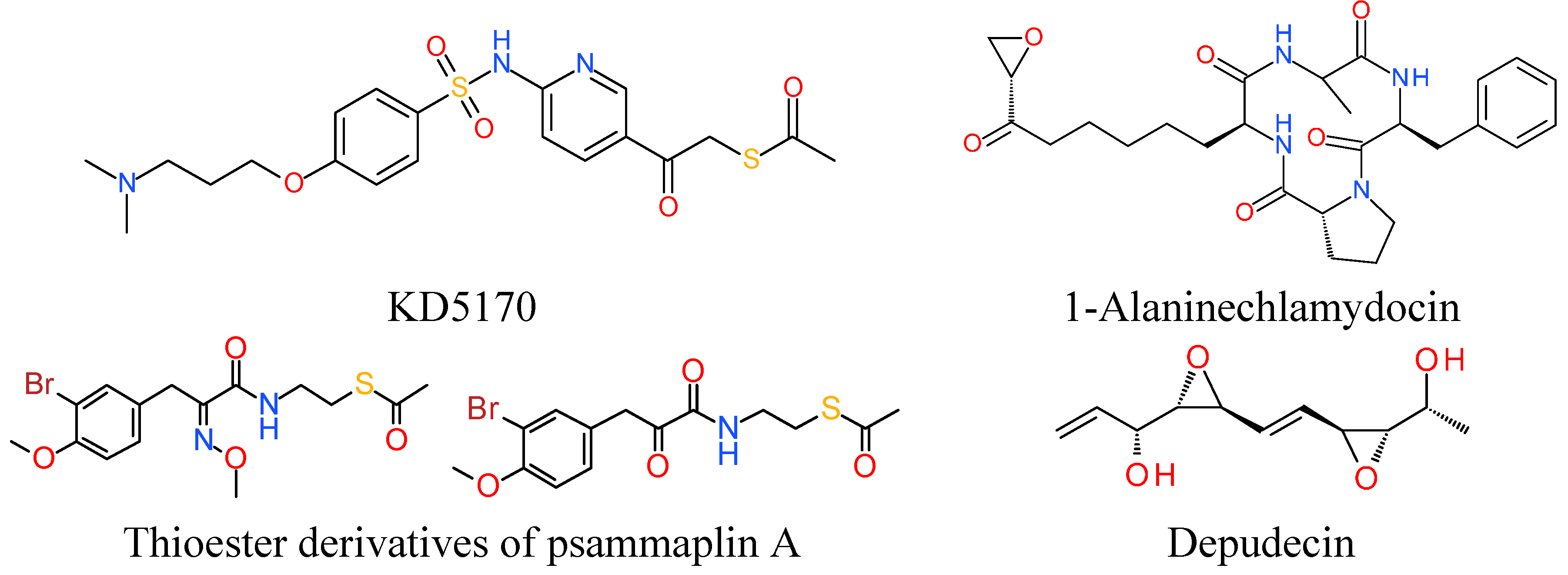
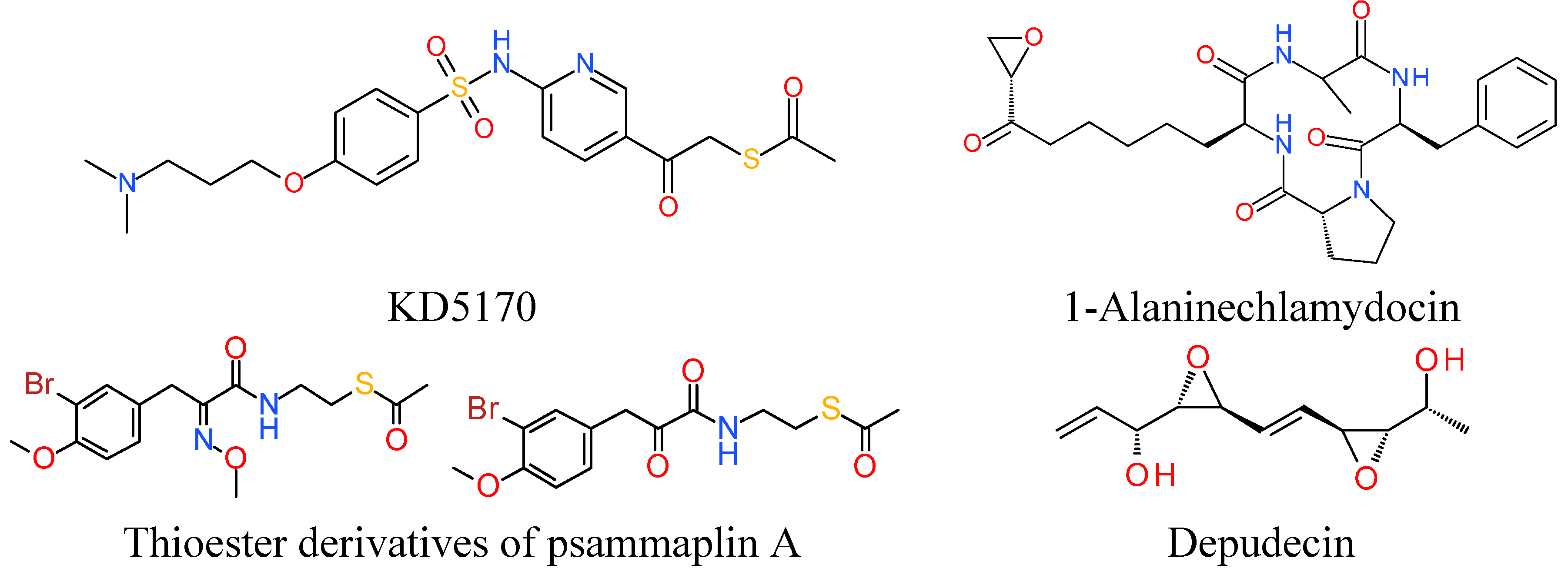
7. Miscellaneous
7.1. Thioester Based HDACi
7.2. Epoxide Based HDACi
7.3. Electrophilic Ketone Based HDACi
8. Toxicity in Clinical Trials
9. Basic Structure of Zinc Binding HDAC Inhibitors
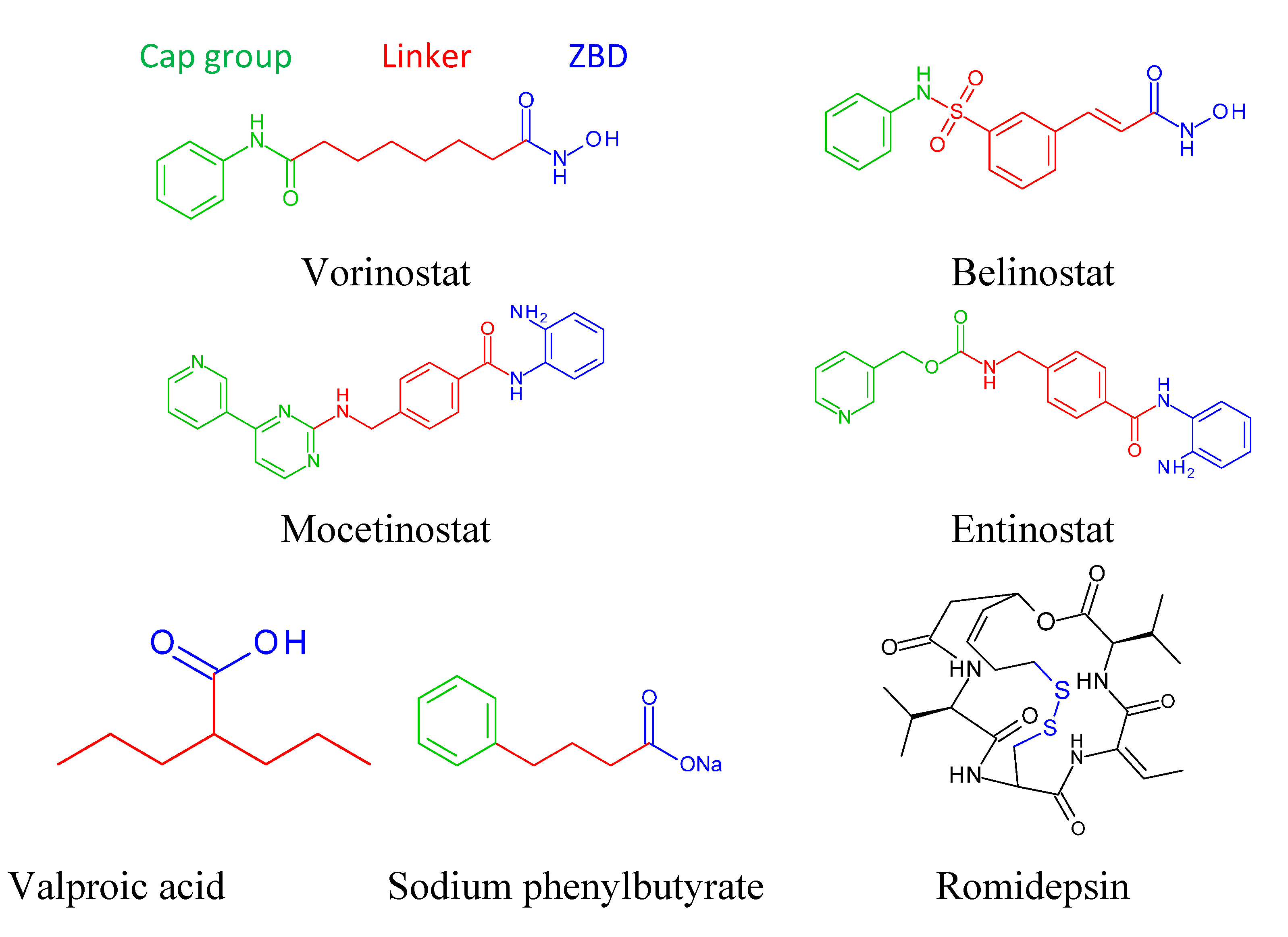
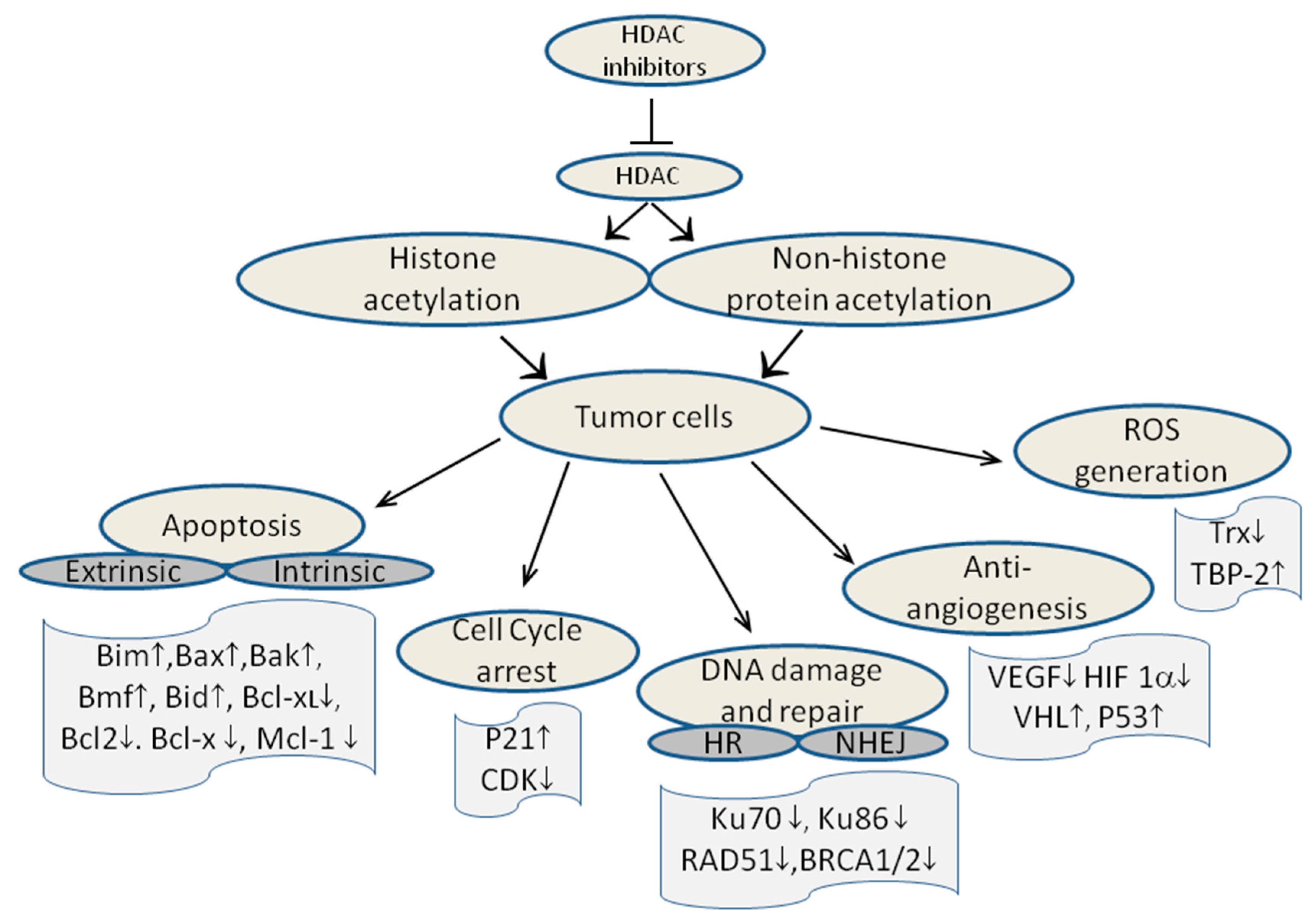
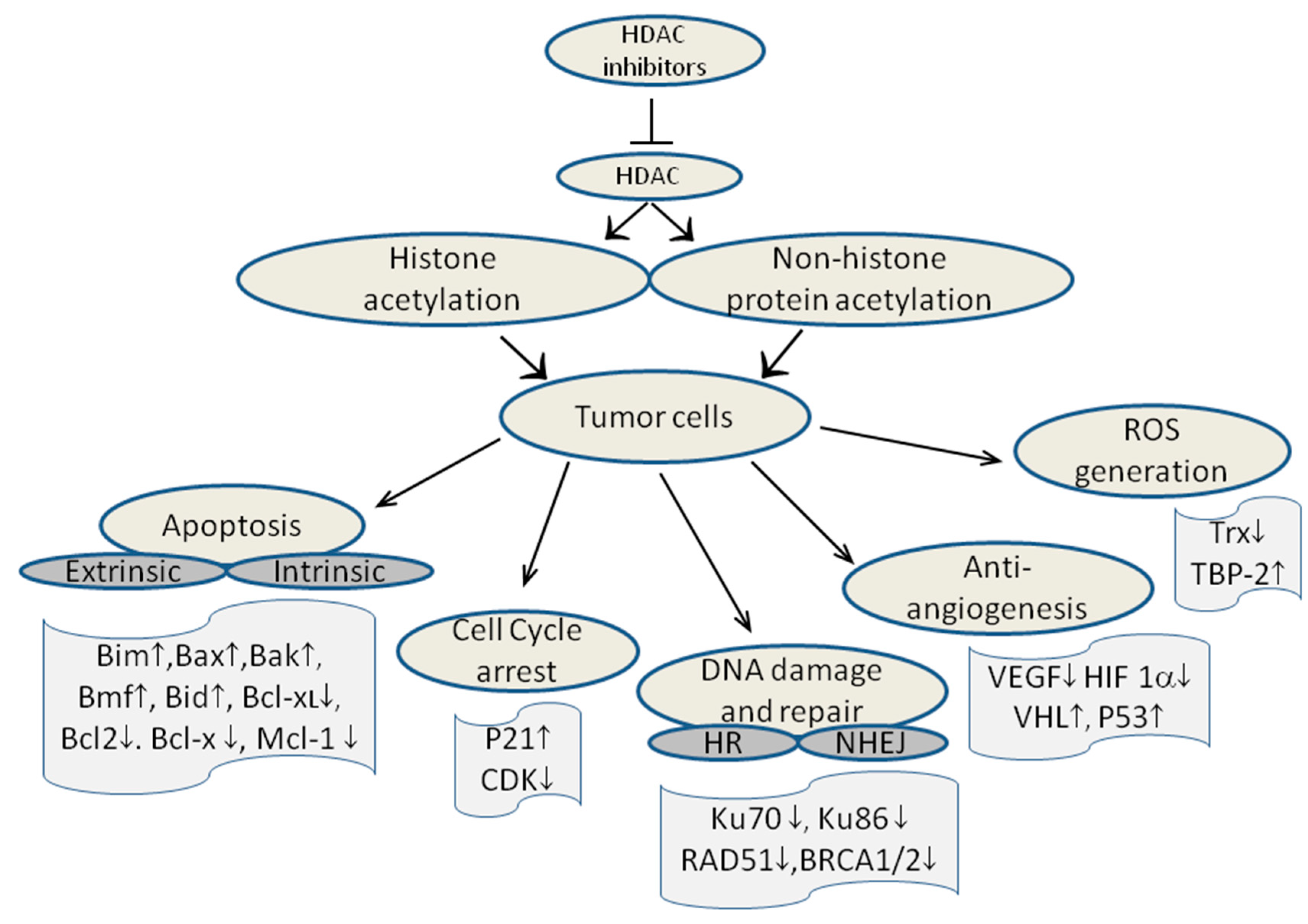
10. Mechanism of action of HDAC inhibitors
11. How to Obtain Novel HDAC Inhibitors?
12. Molecular Modeling Based Studies
13. Quantitative Structure Activity Relationship of HDAC Inhibitors
14. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S. The nucleosome: From genomic organization to genomic regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Brosch, G.; Loidl, P.; Graessle, S. Histone modifications and chromatin dynamics: A focus on filamentous fungi. FEMS Microbiol. Rev. 2008, 32, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Gregoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Kiermer, V.; Dequiedt, F.; Verdin, E. The emerging role of class II histone deacetylases. Biochem. Cell Biol. 2001, 79, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.R.; Xu, Y.F.; Wang, X.B.; Gong, J.P.; Liu, Z.J. Suppression of histone deacetylase 11 promotes expression of il-10 in kupffer cells and induces tolerance following orthotopic liver transplantation in rats. J. Surg. Res. 2012, 174, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Cheng, F.; Wang, H.W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Buglio, D.; Khaskhely, N.M.; Voo, K.S.; Martinez-Valdez, H.; Liu, Y.J.; Younes, A. HDAC11 plays an essential role in regulating ox40 ligand expression in hodgkin lymphoma. Blood 2011, 117, 2910–2917. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Seto, E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor cdt1. J. Biol. Chem. 2009, 284, 11446–11453. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Lee, H.G.; Camins, A.; Pallas, M.; Casadesus, G.; Smith, M.A.; Zhu, X. The sirtuin pathway in ageing and alzheimer disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2011, 10, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [PubMed]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef] [PubMed]
- Borradaile, N.M.; Pickering, J.G. Nad+, sirtuins, and cardiovascular disease. Curr. Pharm. Des. 2009, 15, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Suzuki, T.; Yoshimitsu, M.; Arima, N.; Honda, S.I.; Soeda, S. Anticancer agents targeted to sirtuins. Molecules 2014, 19, 20295–20313. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at lys16 and trimethylation at lys20 of histone h4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Seto, E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharmacol. 2011, 206, 39–56. [Google Scholar] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Trans. Res. 2011, 3, 166–179. [Google Scholar]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. Fda approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase ii multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Pohlman, B.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Ben-Yehuda, D.; Hillen, U. Final Results of a Phase II Trial of Belinostat (PXD101) in Patients with Recurrent or Refractory Peripheral or Cutaneous T-Cell Lymphoma. In Proceedings of the 51st ASH Annual Meeting and Exposition, New Orleans, LA, USA, 5–8 December 2009; p. 920.
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. Fr901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. (Tokyo) 1994, 47, 301–310. [Google Scholar] [CrossRef]
- FDA approves Beleodaq to treat rare, aggressive form of non-Hodgkin lympoma. Available online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm403929.htm (accessed on 10 September 2014).
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 2014, 21, 2642–2664. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the tsa and saha inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Lawrence, Y.R.; Choy, H.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Judy, K.D.; Farrell, C.J.; Moshel, Y.; Berger, A.C.; et al. Vorinostat as a radiosensitizer for brain metastasis: A phase i clinical trial. J. Neurooncol. 2014, 118, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Bruchim, I.; Fishman, A.; Werner, H. The mechanism of action of the histone deacetylase inhibitor vorinostat involves interaction with the insulin-like growth factor signaling pathway. PLoS One 2011, 6, e24468. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lax, S.F. Molecular genetic pathways in various types of endometrial carcinoma: From a phenotypical to a molecular-based classification. Virchows Arch. 2004, 444, 213–223. [Google Scholar] [CrossRef]
- Ma, T.; Galimberti, F.; Erkmen, C.P.; Memoli, V.; Chinyengetere, F.; Sempere, L.; Beumer, J.H.; Anyang, B.N.; Nugent, W.; Johnstone, D.; et al. Comparing histone deacetylase inhibitor responses in genetically engineered mouse lung cancer models and a window of opportunity trial in patients with lung cancer. Mol. Cancer Ther. 2013, 12, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Saelen, M.G.; Ree, A.H.; Kristian, A.; Fleten, K.G.; Furre, T.; Hektoen, H.H.; Flatmark, K. Radiosensitization by the histone deacetylase inhibitor vorinostat under hypoxia and with capecitabine in experimental colorectal carcinoma. Radiat. Oncol. 2012, 7. [Google Scholar] [CrossRef]
- Oki, Y.; Younes, A.; Copeland, A.; Hagemeister, F.; Fayad, L.E.; McLaughlin, P.; Shah, J.; Fowler, N.; Romaguera, J.; Kwak, L.W.; et al. Phase I study of vorinostat in combination with standard chop in patients with newly diagnosed peripheral T-cell lymphoma. Br. J. Haematol. 2013, 162, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Hamaguchi, T.; Shirao, K.; Chin, K.; Hatake, K.; Noguchi, K.; Otsuki, T.; Mehta, A.; Ohtsu, A. Evaluation of safety, pharmacokinetics, and efficacy of vorinostat, a histone deacetylase inhibitor, in the treatment of gastrointestinal (gi) cancer in a phase i clinical trial. Int. J. Clin. Oncol. 2013, 18, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. Discovery and development of saha as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Prince, H.M.; Kirschbaum, M.H.; Zain, J.; Allen, S.L.; Jaffe, E.S.; Ling, A.; Turner, M.; Peer, C.J.; et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 2011, 117, 5827–5834. [Google Scholar] [CrossRef] [PubMed]
- Karthik, S.; Sankar, R.; Varunkumar, K.; Ravikumar, V. Romidepsin induces cell cycle arrest, apoptosis, histone hyperacetylation and reduces matrix metalloproteinases 2 and 9 expression in bortezomib sensitized non-small cell lung cancer cells. Biomed. Pharmacother. 2014, 68, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.M.; Chu, K.; Boley, K.M.; Ye, Z.; Liu, H.; Wright, M.C.; Moraes, R.; Zhang, X.; Green, T.L.; Barsky, S.H.; et al. The class I HDAC inhibitor romidepsin targets inflammatory breast cancer tumor emboli and synergizes with paclitaxel to inhibit metastasis. J. Exp. Ther. Oncol. 2013, 10, 219–233. [Google Scholar] [PubMed]
- Jones, S.F.; Infante, J.R.; Spigel, D.R.; Peacock, N.W.; Thompson, D.S.; Greco, F.A.; McCulloch, W.; Burris, H.A., 3rd. Phase 1 results from a study of romidepsin in combination with gemcitabine in patients with advanced solid tumors. Cancer Investig. 2012, 30, 481–486. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Luchenko, V.; Peer, C.J.; Ghafourian, K.; Reynolds, J.; Draper, D.; Frye, R.; Woo, S.; Venzon, D.; Wright, J.; et al. Phase i trial of a new schedule of romidepsin in patients with advanced cancers. Clin. Cancer Res. 2013, 19, 4499–4507. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase ii study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.A.; Tsao, M.S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Rajan, A.; Berman, A.; Kelly, R.J.; Szabo, E.; Lopez-Chavez, A.; Trepel, J.; Lee, M.J.; Cao, L.; Espinoza-Delgado, I.; et al. Phase ii study of belinostat in patients with recurrent or refractory advanced thymic epithelial tumors. J. Clin. Oncol. 2011, 29, 2052–2059. [Google Scholar] [CrossRef] [PubMed]
- Cashen, A.; Juckett, M.; Jumonville, A.; Litzow, M.; Flynn, P.J.; Eckardt, J.; LaPlant, B.; Laumann, K.; Erlichman, C.; DiPersio, J. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann. Hematol. 2012, 91, 33–38. [Google Scholar] [CrossRef]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; Disilvestro, P.A.; Fader, A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: A gynecologic oncology group study. Gynecol. Oncol. 2012, 125, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Dizon, D.S.; Damstrup, L.; Finkler, N.J.; Lassen, U.; Celano, P.; Glasspool, R.; Crowley, E.; Lichenstein, H.S.; Knoblach, P.; Penson, R.T. Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.H.; Foon, K.A.; Frankel, P.; Ruel, C.; Pulone, B.; Tuscano, J.M.; Newman, E.M. A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: A california cancer consortium study. Leuk. Lymphoma 2014, 55, 2301–2304. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Rajan, A.; Szabo, E.; Tomita, Y.; Carter, C.A.; Scepura, B.; Lopez-Chavez, A.; Lee, M.J.; Redon, C.E.; Frosch, A.; et al. A phase I/II trial of belinostat in combination with cisplatin, doxorubicin and cyclophosphamide in thymic epithelial tumors: A clinical and translational study. Clin. Cancer Res. 2014, 20, 5392–5402. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.; Sung, F.; Tao, Q.; Poon, F.F.; Lui, V.W.; Yeo, W.; Chan, S.L.; Chan, A.T. The preclinical activity of the histone deacetylase inhibitor PXD101 (belinostat) in hepatocellular carcinoma cell lines. Investig. New Drugs 2010, 28, 107–114. [Google Scholar] [CrossRef]
- Savickiene, J.; Treigyte, G.; Valiuliene, G.; Stirblyte, I.; Navakauskiene, R. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anticancer Drugs 2014, 25, 938–949. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J. Clin. Oncol. 2006, 24, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Richon, V.M.; O’Connor, O.; Curley, T.; MacGregor-Curtelli, B.; Tong, W.; Klang, M.; Schwartz, L.; Richardson, S.; Rosa, E.; et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003, 9, 3578–3588. [Google Scholar] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, saha) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Richon, V. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer 2006, 95, S2–S6. [Google Scholar] [CrossRef]
- Sholler, G.S.; Currier, E.A.; Dutta, A.; Slavik, M.A.; Illenye, S.A.; Mendonca, M.C.F.; Dragon, J.; Roberts, S.S.; Bond, J.P. PCI-24781 (abexinostat), a novel histone deacetylase inhibitor, induces reactive oxygen species-dependent apoptosis and is synergistic with bortezomib in neuroblastoma. J. Cancer Ther. Res. 2013, 2. [Google Scholar] [CrossRef]
- Fouliard, S.; Robert, R.; Jacquet-Bescond, A.; du Rieu, Q.C.; Balasubramanian, S.; Loury, D.; Loriot, Y.; Hollebecque, A.; Kloos, I.; Soria, J.C.; et al. Pharmacokinetic/pharmacodynamic modelling-based optimisation of administration schedule for the histone deacetylase inhibitor abexinostat (S78454/PCI-24781) in phase I. Eur. J. Cancer 2013, 49, 2791–2797. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Liu, J.; Ren, W.; Wei, W.; Wang, S.; Lahat, G.; Zhu, Q.S.; Bornmann, W.G.; McConkey, D.J.; Pollock, R.E.; et al. Combining pci-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin. Cancer Res. 2009, 15, 3472–3483. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Cardama, A.; Kantarjian, H.; Estrov, Z.; Borthakur, G.; Cortes, J.; Verstovsek, S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk. Res. 2012, 36, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Razak, A.R.; Hotte, S.J.; Siu, L.L.; Chen, E.X.; Hirte, H.W.; Powers, J.; Walsh, W.; Stayner, L.A.; Laughlin, A.; Novotny-Diermayr, V.; et al. Phase I clinical, pharmacokinetic and pharmacodynamic study of sb939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2011, 104, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.P.; Bernstein, M.; Samson, Y.; Wall, D.A.; Desai, S.; Nicksy, D.; Wainman, N.; Eisenhauer, E.; Baruchel, S. A phase i study of histone deacetylase inhibitor, pracinostat (sb939), in pediatric patients with refractory solid tumors: Ind203 a trial of the ncic ind program/c17 pediatric phase i consortium. Pediatr. Blood Cancer 2013, 60, 1868–1874. [Google Scholar] [CrossRef] [PubMed]
- Brunetto, A.T.; Ang, J.E.; Lal, R.; Olmos, D.; Molife, L.R.; Kristeleit, R.; Parker, A.; Casamayor, I.; Olaleye, M.; Mais, A.; et al. First-in-human, pharmacokinetic and pharmacodynamic phase i study of resminostat, an oral histone deacetylase inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 5494–5504. [Google Scholar] [CrossRef] [PubMed]
- Mandl-Weber, S.; Meinel, F.G.; Jankowsky, R.; Oduncu, F.; Schmidmaier, R.; Baumann, P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (mm) cells. Br. J. Haematol. 2010, 149, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Walewski, J.; Paszkiewicz-Kozik, E.; Borsaru, G.; Moicean, A.; Warszewska, A.; Strobel, K.; Biggi, A.; Hauns, B.; Mais, A.; Henning, S.W. Resminostat in Relapsed or Refractory Hodgkin Lymphoma: Initial Results of the Saphire phase II Trial with a Novel Oral Histone Deacetylase (HDAC) Inhibitor. In Proceedings of the 52nd ASH Annual Meeting and Exposition, Orlando, FL, USA, 4–7 December 2010.
- Walewski, J.; Paszkiewicz-Kozik, E.; Warszewska, A.; Borsaru, G.; Moicean, A.; Hellmann, A.; Mayer, J.; Hauns, B.; Mais, A.; Henning, S.W. Final Results of the Phase II Saphire Trial of Resminostat (4sc-201) in Patients with Relapsed/Refractory Hodgkin Lymphoma. In Proceedings of the 53rd ASH Annual Meeting and Exposition, San Diego, CA, USA, 10–13 December 2011.
- Bitzer, M.; Ganten, T.M.; Woerns, M.A.; Siveke, J.T.; Dollinger, M.M.; Scheulen, M.E.; Wege, H.; Giannini, E.G.; Cillo, U.; Trevisani, F. Resminostat in advanced hepatocellular carcinoma (HCC): Overall survival subgroup analysis of prognostic factors in the shelter trial. J. Clin. Oncol. 2013, 31, e15088. [Google Scholar]
- Rambaldi, A.; Dellacasa, C.M.; Finazzi, G.; Carobbio, A.; Ferrari, M.L.; Guglielmelli, P.; Gattoni, E.; Salmoiraghi, S.; Finazzi, M.C.; di Tollo, S.; et al. A pilot study of the histone-deacetylase inhibitor givinostat in patients with jak2v617f positive chronic myeloproliferative neoplasms. Br. J. Haematol. 2010, 150, 446–455. [Google Scholar] [PubMed]
- Amaru Calzada, A.; Pedrini, O.; Finazzi, G.; Leoni, F.; Mascagni, P.; Introna, M.; Rambaldi, A.; Golay, J. Givinostat and hydroxyurea synergize in vitro to induce apoptosis of cells from JAK2(V617F) myeloproliferative neoplasm patients. Exp. Hematol. 2013, 41, 253–260. [Google Scholar]
- Furlan, A.; Monzani, V.; Reznikov, L.L.; Leoni, F.; Fossati, G.; Modena, D.; Mascagni, P.; Dinarello, C.A. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol. Med. 2011, 17, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Vannucchi, A.M.; Martinelli, V.; Ruggeri, M.; Nobile, F.; Specchia, G.; Pogliani, E.M.; Olimpieri, O.M.; Fioritoni, G.; Musolino, C.; et al. A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br. J. Haematol. 2013, 161, 688–694. [Google Scholar] [CrossRef] [PubMed]
- De Marinis, F.; Atmaca, A.; Tiseo, M.; Giuffreda, L.; Rossi, A.; Gebbia, V.; D’Antonio, C.; dal Zotto, L.; Al-Batran, S.E.; Marsoni, S.; et al. A phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in pretreated patients with small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 1091–1094. [Google Scholar]
- Mascarenhas, J.; Lu, M.; Li, T.; Petersen, B.; Hochman, T.; Najfeld, V.; Goldberg, J.D.; Hoffman, R. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br. J. Haematol. 2013, 161, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Oizumi, S.; Minami, H.; Kitagawa, K.; Komatsu, Y.; Fujiwara, Y.; Inada, M.; Yuki, S.; Kiyota, N.; Mitsuma, A.; et al. Phase I dose-escalating study of panobinostat (LBH589) administered intravenously to japanese patients with advanced solid tumors. Investig. New Drugs 2012, 30, 1950–1957. [Google Scholar] [CrossRef]
- Fukutomi, A.; Hatake, K.; Matsui, K.; Sakajiri, S.; Hirashima, T.; Tanii, H.; Kobayashi, K.; Yamamoto, N. A phase I study of oral panobinostat (LBH589) in japanese patients with advanced solid tumors. Investig. New Drugs 2012, 30, 1096–1106. [Google Scholar] [CrossRef]
- Younes, A.; Sureda, A.; Ben-Yehuda, D.; Zinzani, P.L.; Ong, T.C.; Prince, H.M.; Harrison, S.J.; Kirschbaum, M.; Johnston, P.; Gallagher, J.; et al. Panobinostat in patients with relapsed/refractory hodgkin’s lymphoma after autologous stem-cell transplantation: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Al-Ali, H.K.; Gattermann, N.; Haase, D.; Janzen, V.; Krauter, J.; Gotze, K.; Schlenk, R.; Nolte, F.; Letsch, A.; et al. Phase 2 study of oral panobinostat (LBH589) with or without erythropoietin in heavily transfusion-dependent ipss low or int-1 mds patients. Leukemia 2014, 28, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Dimicoli, S.; Jabbour, E.; Borthakur, G.; Kadia, T.; Estrov, Z.; Yang, H.; Kelly, M.; Pierce, S.; Kantarjian, H.; Garcia-Manero, G. Phase II study of the histone deacetylase inhibitor panobinostat (LBH589) in patients with low or intermediate-1 risk myelodysplastic syndrome. Am. J. Hematol. 2012, 87, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; LoRusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.A.; Chambers, G.; Ma, A.; et al. Phase I first-in-human study of cudc-101, a multitargeted inhibitor of HDACS, egfr, and her2 in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Nemunaitis, J.J.; Bessudo, A.; Bauman, J.E.; Hamid, O.; Witta, S.E.; Dy, G.K.; Lai, C.; Laliberte, R.; Voi, M. A phase Ib study of CUDC-101, A Multitargeted Inhibitor of EGFR, HER2, and HDAC, in Patients with Advanced Head and Neck, Gastric, Breast, Liver, and Non-Small Cell Lung Cancer. In Proceedings of 2012 ASCO Annual Meeting, Chicago, IL, USA, 1–5 June 2012.
- Voi, M.; Fu, S.; Nemunaitis, J.; Bauman, J.; Bessudo, A.; Hamid, O.; Witta, S.; Dy, G.; Lai, C.; Laliberte, R. 590 Final results of a phase Ib study of CUDC-101, a multitargeted inhibitor of EGFR, HER2, and HDAC, in patients with advanced head and neck, gastric, breast, liver, and non-small cell lung cancer. Eur. J. Cancer 2012, 48. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.Gov (accessed on 1 September 2014).
- Fakih, M.G.; Groman, A.; McMahon, J.; Wilding, G.; Muindi, J.R. A randomized phase II study of two doses of vorinostat in combination with 5-fu/lv in patients with refractory colorectal cancer. Cancer Chemother. Pharmacol. 2012, 69, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Fakih, M.G.; Fetterly, G.; Egorin, M.J.; Muindi, J.R.; Espinoza-Delgado, I.; Zwiebel, J.A.; Litwin, A.; Holleran, J.L.; Wang, K.; Diasio, R.B. A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin. Cancer Res. 2010, 16, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Millward, M.; Price, T.; Townsend, A.; Sweeney, C.; Spencer, A.; Sukumaran, S.; Longenecker, A.; Lee, L.; Lay, A.; Sharma, G.; et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investig. New Drugs 2012, 30, 2303–2317. [Google Scholar] [CrossRef]
- Mazumder, A.; Vesole, D.H.; Jagannath, S. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: A case series illustrating utility in clinical practice. Clin. Lymphoma Myeloma Leuk. 2010, 10, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.M.; El-Khoueiry, A.; Iqbal, S.; Fazzone, W.; LaBonte, M.J.; Groshen, S.; Yang, D.; Danenberg, K.D.; Cole, S.; Kornacki, M.; et al. A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-fu-based chemotherapy. Cancer Chemother. Pharmacol. 2010, 65, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Gressette, M.; Vérillaud, B.; Jimenez-Pailhès, A.-S.; Lelièvre, H.; Lo, K.-W.; Ferrand, F.-R.; Gattolliat, C.-H.; Jacquet-Bescond, A.; Kraus-Berthier, L.; Depil, S. Treatment of nasopharyngeal carcinoma cells with the histone-deacetylase inhibitor abexinostat: Cooperative effects with cis-platin and radiotherapy on patient-derived xenografts. PLoS One 2014, 9, e91325. [Google Scholar] [CrossRef] [PubMed]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.T.; Trachy-Bourget, M.C.; Kalita, A.; Liu, J.; Lu, A.H.; Zhou, N.Z.; Robert, M.F.; et al. Mgcd0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Assouline, S.; Cortes, J.; Estrov, Z.; Kantarjian, H.; Yang, H.; Newsome, W.M.; Miller, W.H., Jr.; Rousseau, C.; Kalita, A.; et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor mgcd0103 in leukemia. Blood 2008, 112, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, J.; Zhao, J.; Yun, W.; Xie, C.; Taub, J.W.; Azmi, A.; Mohammad, R.M.; Dong, Y.; Kong, W.; et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS One 2012, 7, e52095. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Advani, A.; Fernandez, L.; van der Jagt, R.; Brandwein, J.; Kambhampati, S.; Kassis, J.; Davis, M.; Bonfils, C.; Dubay, M.; et al. Phase ii study of the histone deacetylase inhibitor mgcd0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2009, 147, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Pili, R.; Duran, I.; Messersmith, W.A.; Chen, E.X.; Sullivan, R.; MacLean, M.; King, S.; Brown, S.; Reid, G.K.; et al. Phase I study of mgcd0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J. Clin. Oncol. 2008, 26, 1940–1947. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Oki, Y.; Bociek, R.G.; Kuruvilla, J.; Fanale, M.; Neelapu, S.; Copeland, A.; Buglio, D.; Galal, A.; Besterman, J.; et al. Mocetinostat for relapsed classical hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011, 12, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Salumbides, B.; Zhao, M.; Altiok, S.; Qian, D.; Zwiebel, J.; Carducci, M.A.; Rudek, M.A. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br. J. Cancer 2012, 106, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Jotte, R.M.; Konduri, K.; Neubauer, M.A.; Spira, A.I.; Ruxer, R.L.; Varella-Garcia, M.; Bunn, P.A., Jr.; Hirsch, F.R. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012, 30, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Gore, L.; Rothenberg, M.L.; O’Bryant, C.L.; Schultz, M.K.; Sandler, A.B.; Coffin, D.; McCoy, C.; Schott, A.; Scholz, C.; Eckhardt, S.G. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, ms-275, in patients with refractory solid tumors and lymphomas. Clin. Cancer Res. 2008, 14, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Su, J.M.; Li, X.N.; Thompson, P.; Ou, C.N.; Ingle, A.M.; Russell, H.; Lau, C.C.; Adamson, P.C.; Blaney, S.M. Phase 1 study of valproic acid in pediatric patients with refractory solid or cns tumors: A children’s oncology group report. Clin. Cancer Res. 2011, 17, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, T.A.; Holen, K.D.; Jaskula-Sztul, R.; Mulkerin, D.; Lubner, S.J.; Schelman, W.R.; Eickhoff, J.; Chen, H.; Loconte, N.K. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist 2011, 16, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Janku, F.; Falchook, G.S.; Jackson, T.L.; Fu, S.; Naing, A.; Tsimberidou, A.M.; Moulder, S.L.; Hong, D.S.; Yang, H.; et al. Phase I study of anti-vegf monoclonal antibody bevacizumab and histone deacetylase inhibitor valproic acid in patients with advanced cancers. Cancer Chemother. Pharmacol. 2014, 73, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.F.; Karpenko, M.J.; Liu, Z.; Aimiuwu, J.; Villalona-Calero, M.A.; Chan, K.K.; Grever, M.R.; Otterson, G.A. Phase i study of 5-aza-2'-deoxycytidine in combination with valproic acid in non-small-cell lung cancer. Cancer Chemother. Pharmacol. 2013, 71, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, S.; Utsunomiya, T.; Imura, S.; Morine, Y.; Ikemoto, T.; Arakawa, Y.; Saito, Y.; Ishikawa, D.; Shimada, M. Effects of valproic acid in combination with s-1 on advanced pancreatobiliary tract cancers: Clinical study phases I/II. Anticancer Res. 2014, 34, 5187–5191. [Google Scholar] [PubMed]
- Bauman, J.; Shaheen, M.; Verschraegen, C.F.; Belinsky, S.A.; Houman Fekrazad, M.; Lee, F.C.; Rabinowitz, I.; Ravindranathan, M.; Jones, D.V., Jr. A phase i protocol of hydralazine and valproic acid in advanced, previously treated solid cancers. Trans. Oncol. 2014, 7, 349–354. [Google Scholar] [CrossRef]
- Prakash, S.; Foster, B.J.; Meyer, M.; Wozniak, A.; Heilbrun, L.K.; Flaherty, L.; Zalupski, M.; Radulovic, L.; Valdivieso, M.; LoRusso, P.M. Chronic oral administration of ci-994: A phase 1 study. Investig. New Drugs 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin a. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Fennell, K.A.; Mollmann, U.; Miller, M.J. Syntheses and biological activity of amamistatin b and analogs. J. Org. Chem. 2008, 73, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Fennell, K.A.; Miller, M.J. Syntheses of amamistatin fragments and determination of their HDAC and antitumor activity. Org. Lett. 2007, 9, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [PubMed]
- Duan, H.; Heckman, C.A.; Boxer, L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 2005, 25, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, M.; Kotani, J.; Usami, M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating gpr-41/gpr-43 pathways. Nutrition 2010, 26, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Petrella, A.; D’Acunto, C.W.; Rodriquez, M.; Festa, M.; Tosco, A.; Bruno, I.; Terracciano, S.; Taddei, M.; Paloma, L.G.; Parente, L. Effects of fr235222, a novel HDAC inhibitor, in proliferation and apoptosis of human leukaemia cell lines: Role of annexin a1. Eur. J. Cancer 2008, 44, 740–749. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Bruwiere, H.; Verhulst, T.; Steller, U.; Andries, L.; Wouters, W.; Janicot, M.; Arts, J.; van Heusden, J. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J. Pharmacol. Exp. Ther. 2003, 304, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.W.; Lee, H.W.; Han, J.W. Apicidin, a histone deacetylase inhibitor, induces apoptosis and fas/fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef]
- Nakao, Y.; Yoshida, S.; Matsunaga, S.; Shindoh, N.; Terada, Y.; Nagai, K.; Yamashita, J.K.; Ganesan, A.; van Soest, R.W.; Fusetani, N. Azumamides a-e: Histone deacetylase inhibitory cyclic tetrapeptides from the marine sponge mycale izuensis. Angew. Chem. Int. Ed. Engl. 2006, 45, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Villadsen, J.S.; Stephansen, H.M.; Maolanon, A.R.; Harris, P.; Olsen, C.A. Total synthesis and full histone deacetylase inhibitory profiling of azumamides a-e as well as beta(2)- epi-azumamide e and beta(3)-epi-azumamide e. J. Med. Chem. 2013, 56, 6512–6520. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Yoshida, M.; Horinouchi, S. Potent histone deacetylase inhibitors built from trichostatin a and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Taori, K.; Paul, V.J.; Luesch, H. Structure and activity of largazole, a potent antiproliferative agent from the floridian marine cyanobacterium symploca sp. J. Am. Chem. Soc. 2008, 130, 1806–1807. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Jawaid, P.; Yoshihisa, Y.; Li, P.; Zhao, Q.L.; Narita, K.; Katoh, T.; Kondo, T.; Shimizu, T. Spiruchostatin a and b, novel histone deacetylase inhibitors, induce apoptosis through reactive oxygen species-mitochondria pathway in human lymphoma u937 cells. Chem. Biol. Interact. 2014, 221, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Weinlander, E.; Somnay, Y.; Harrison, A.D.; Wang, C.; Cheng, Y.Q.; Jaskula-Sztul, R.; Yu, X.M.; Chen, H. The novel histone deacetylase inhibitor thailandepsin a inhibits anaplastic thyroid cancer growth. J. Surg. Res. 2014, 190, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, S.; Berger, A.; Bocker, A.; Busch, C.; Weiland, T.; Noor, S.; Leischner, C.; Schleicher, S.; Mayer, M.; Weiss, T.S.; et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS One 2013, 8, e73097. [Google Scholar] [CrossRef] [PubMed]
- Nian, H.; Delage, B.; Pinto, J.T.; Dashwood, R.H. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances sp3 binding on the p21waf1 promoter. Carcinogenesis 2008, 29, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Druesne, N.; Pagniez, A.; Mayeur, C.; Thomas, M.; Cherbuy, C.; Duee, P.H.; Martel, P.; Chaumontet, C. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis 2004, 25, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Clarke, J.D.; Dashwood, R.H. Dietary sulforaphane, a histone deacetylase inhibitor for cancer prevention. J. Nutr. 2009, 139, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Park, H.; Al-Awadhi, F.H.; Liu, Y.; Kim, B.; Zeller, S.L.; Chen, Q.Y.; Hong, J.; Luesch, H. Modulation of activity profiles for largazole-based HDAC inhibitors through alteration of prodrug properties. ACS Med. Chem. Lett. 2014, 5, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Hassig, C.A.; Symons, K.T.; Guo, X.; Nguyen, P.M.; Annable, T.; Wash, P.L.; Payne, J.E.; Jenkins, D.A.; Bonnefous, C.; Trotter, C.; et al. Kd5170, a novel mercaptoketone-based histone deacetylase inhibitor that exhibits broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.E.; Bonnefous, C.; Hassig, C.A.; Symons, K.T.; Guo, X.; Nguyen, P.M.; Annable, T.; Wash, P.L.; Hoffman, T.Z.; Rao, T.S.; et al. Identification of kd5170: A novel mercaptoketone-based histone deacetylase inhibitor. Bioorg. Med. Chem. Lett. 2008, 18, 6093–6096. [Google Scholar] [CrossRef] [PubMed]
- Baud, M.G.; Leiser, T.; Petrucci, V.; Gunaratnam, M.; Neidle, S.; Meyer-Almes, F.J.; Fuchter, M.J. Thioester derivatives of the natural product psammaplin a as potent histone deacetylase inhibitors. Beilstein J. Org. Chem. 2013, 9, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.A.; Luesch, H. Discovery and mechanism of natural products as modulators of histone acetylation. Curr. Drug Targets 2012, 13, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Owa, T.; Hassig, C.A.; Shimada, J.; Schreiber, S.L. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. Proc. Natl. Acad. Sci. USA 1998, 95, 3356–3361. [Google Scholar] [CrossRef] [PubMed]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar] [PubMed]
- Du, L.; Risinger, A.L.; King, J.B.; Powell, D.R.; Cichewicz, R.H. A potent HDAC inhibitor, 1-alaninechlamydocin, from a Tolypocladium sp. induces G2/M cell cycle arrest and apoptosis in MIA PaCa-2 cells. J. Nat. Prod. 2014, 77, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.R.; Wada, C.K.; Garland, R.B.; Curtin, M.L.; Michaelides, M.R.; Li, J.; Pease, L.J.; Glaser, K.B.; Marcotte, P.A.; Bouska, J.J.; et al. Trifluoromethyl ketones as inhibitors of histone deacetylase. Bioorg. Med. Chem. Lett. 2002, 12, 3443–3447. [Google Scholar] [CrossRef] [PubMed]
- Jose, B.; Oniki, Y.; Kato, T.; Nishino, N.; Sumida, Y.; Yoshida, M. Novel histone deacetylase inhibitors: Cyclic tetrapeptide with trifluoromethyl and pentafluoroethyl ketones. Bioorg. Med. Chem. Lett. 2004, 14, 5343–5346. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.S.; Kristensen, H.M.; Lanz, G.; Olsen, C.A. The effect of various zinc binding groups on inhibition of histone deacetylases 1–11. ChemMedChem 2014, 9, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Bishton, M.J.; Harrison, S.J.; Martin, B.P.; McLaughlin, N.; James, C.; Josefsson, E.C.; Henley, K.J.; Kile, B.T.; Prince, H.M.; Johnstone, R.W. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 2011, 117, 3658–3668. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Salmoiraghi, S.; Golay, J.; Gozzini, A.; Crippa, C.; Pescosta, N.; Rambaldi, A. A phase II multiple dose clinical trial of histone deacetylase inhibitor itf2357 in patients with relapsed or progressive multiple myeloma. Ann. Hematol. 2010, 89, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E.C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; de Francesco, R.; Gallinari, P.; et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem. 2013, 288, 26926–26943. [Google Scholar] [CrossRef]
- Kapustin, G.V.; Fejer, G.; Gronlund, J.L.; McCafferty, D.G.; Seto, E.; Etzkorn, F.A. Phosphorus-based saha analogues as histone deacetylase inhibitors. Org. Lett. 2003, 5, 3053–3056. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Nusinzon, I.; Smith, R.D., Jr.; Horvath, C.M.; Silverman, R.B. Carbonyl- and sulfur-containing analogs of suberoylanilide hydroxamic acid: Potent inhibition of histone deacetylases. Bioorg. Med. Chem. 2006, 14, 3320–3329. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, H.M.; Su, M.B.; Sheng, L.; Zhou, Y.B.; Li, J.; Lu, W. Synthesis and biological evaluation of piperamide analogues as HDAC inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4844–4846. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.; Cini, E.; Giannotti, L.; Giannini, G.; Battistuzzi, G.; Vignola, D.; Vesci, L.; Cabri, W. Lactam based 7-amino suberoylamide hydroxamic acids as potent HDAC inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Tashima, T.; Murata, H.; Kodama, H. Design and synthesis of novel and highly-active pan-histone deacetylase (pan-HDAC) inhibitors. Bioorg. Med. Chem. 2014, 22, 3720–3731. [Google Scholar] [CrossRef] [PubMed]
- Schrump, D.S.; Fischette, M.R.; Nguyen, D.M.; Zhao, M.; Li, X.; Kunst, T.F.; Hancox, A.; Hong, J.A.; Chen, G.A.; Kruchin, E.; et al. Clinical and molecular responses in lung cancer patients receiving romidepsin. Clin. Cancer Res. 2008, 14, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol. Cancer Ther. 2004, 3, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, C.; Wang, L.; Liu, P.; Zhao, L.; Zhu, C.; Tian, X. Inhibition of leukemic cells by valproic acid, an HDAC inhibitor, in xenograft tumors. Onco. Targets Ther. 2013, 6, 733–740. [Google Scholar] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.; Cardoso, B.A.; Belo, H.; Almeida, A.M. Vorinostat induces apoptosis and differentiation in myeloid malignancies: Genetic and molecular mechanisms. PLoS One 2013, 8, e53766. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.E.; Lee, S.W.; Moon, E.J.; Kim, H.S.; Lee, S.K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1α. Cancer Res. 2006, 66, 8814–8821. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Wang, Y.C.; Yang, H.C.; Huang, P.H.; Kulp, S.K.; Yang, C.C.; Lu, Y.S.; Matsuyama, S.; Chen, C.Y. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007, 67, 5318–5327. [Google Scholar] [CrossRef] [PubMed]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Marks, P.A. Thioredoxin in cancer—Role of histone deacetylase inhibitors. Semin. Cancer Biol. 2006, 16, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor saha arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, S.; Terracciano, S.; Bruno, I.; Rodriquez, M.; Riccio, R.; Taddei, M.; Bifulco, G. Molecular modeling studies toward the structural optimization of new cyclopeptide-based HDAC inhibitors modeled on the natural product FR235222. Bioorg. Med. Chem. 2008, 16, 8635–8642. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.; Wei, Y.; Xiu, Z.; Nishino, N. Discovery of potent HDAC inhibitors based on chlamydocin with inhibitory effects on cell migration. ChemMedChem 2014, 9, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Neelarapu, R.; Holzle, D.L.; Velaparthi, S.; Bai, H.; Brunsteiner, M.; Blond, S.Y.; Petukhov, P.A. Design, synthesis, docking, and biological evaluation of novel diazide-containing isoxazole- and pyrazole-based histone deacetylase probes. J. Med. Chem. 2011, 54, 4350–4364. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.E.; Wagner, F.F.; Kaya, T.; Gale, J.P.; Aidoud, N.; Davoine, E.L.; Lazzaro, F.; Weiwer, M.; Zhang, Y.L.; Holson, E.B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013, 56, 4816–4820. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; John, S.; Lee, K.W. Molecular dynamics simulation study explaining inhibitor selectivity in different class of histone deacetylases. J. Biomol. Struct. Dyn. 2012, 29, 677–698. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; John, S.; Lee, Y.; Arulalapperumal, V.; Lee, K.W. Molecular modeling study on tunnel behavior in different histone deacetylase isoforms. PLoS One 2012, 7, e49327. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Lugo, J.A.; Perez-Gonzalez, O.; Rosales-Hernandez, M.C.; Ilizaliturri-Flores, I.; Trujillo-Ferrara, J.; Correa-Basurto, J. Exploration of the valproic acid binding site on histone deacetylase 8 using docking and molecular dynamic simulations. J. Mol. Model. 2012, 18, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Estiu, G.; West, N.; Mazitschek, R.; Greenberg, E.; Bradner, J.E.; Wiest, O. On the inhibition of histone deacetylase 8. Bioorg. Med. Chem. 2010, 18, 4103–4110. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Ligand release mechanisms and channels in histone deacetylases. J. Comp. Chem. 2013, 34, 2270–2283. [Google Scholar] [CrossRef]
- Lu, H.; Chen, Y.-D.; Yang, B.; You, Q.-D. Design, synthesis and biological evaluation of novel histone deacetylase inhibitors based on virtual screening. Acta Pharm. Sin. B 2011, 1, 240–247. [Google Scholar] [CrossRef]
- Schlimme, S.; Hauser, A.T.; Carafa, V.; Heinke, R.; Kannan, S.; Stolfa, D.A.; Cellamare, S.; Carotti, A.; Altucci, L.; Jung, M.; et al. Carbamate prodrug concept for hydroxamate HDAC inhibitors. ChemMedChem 2011, 6, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, M.; Feng, J.; Fang, H.; Xu, W. Discovery of a novel histone deacetylase 8 inhibitor by virtual screening. Med. Chem. Res. 2012, 21, 152–156. [Google Scholar] [CrossRef]
- Park, H.; Kim, S.; Kim, Y.E.; Lim, S.J. A structure-based virtual screening approach toward the discovery of histone deacetylase inhibitors: Identification of promising zinc-chelating groups. ChemMedChem 2010, 5, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Kannan, S.; Hauser, A.T.; Moraes Mourao, M.; Caby, S.; Cura, V.; Stolfa, D.A.; Schmidtkunz, K.; Lancelot, J.; Andrade, L.; et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke schistosoma mansoni. PLoS Pathog. 2013, 9, e1003645. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhong, Q.; Jiang, Q.; Mottamal, M.; Zhang, Q.; Zhu, N.; Burow, M.E.; Worthylake, R.A.; Wang, G. Discovery of a series of thiazole derivatives as novel inhibitors of metastatic cancer cell migration and invasion. ACS Med. Chem. Lett. 2013, 4, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhong, Q.; Xi, Y.; Mottamal, M.; Zhang, Q.; Schroeder, R.L.; Sridhar, J.; He, L.; McFerrin, H.; Wang, G. Modification and biological evaluation of thiazole derivatives as novel inhibitors of metastatic cancer cell migration and invasion. J. Med. Chem. 2014, 57, 6653–6667. [Google Scholar] [CrossRef] [PubMed]
- Canela, M.D.; Perez-Perez, M.J.; Noppen, S.; Saez-Calvo, G.; Diaz, J.F.; Camarasa, M.J.; Liekens, S.; Priego, E.M. Novel colchicine-site binders with a cyclohexanedione scaffold identified through a ligand-based virtual screening approach. J. Med. Chem. 2014, 57, 3924–3938. [Google Scholar] [CrossRef] [PubMed]
- Svajger, U.; Brus, B.; Turk, S.; Sova, M.; Hodnik, V.; Anderluh, G.; Gobec, S. Novel toll-like receptor 4 (TLR4) antagonists identified by structure- and ligand-based virtual screening. Eur. J. Med. Chem. 2013, 70, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Energy based pharmacophore mapping of HDAC inhibitors against class I HDAC enzymes. Biochim. Biophys. Acta 2013, 1834, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Shanmugam, K.; Mahadevan, V. Energy-optimised pharmacophore approach to identify potential hotspots during inhibition of class II HDAC isoforms. J. Biomol. Struct. Dyn. 2015, 33, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Docking-enabled pharmacophore model for histone deacetylase 8 inhibitors and its application in anti-cancer drug discovery. J. Mol. Graph. Model. 2010, 29, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Chun, T.-G.; Nam, K.-Y.; Kim, H.M.; Han, G. Structure-activity relationship of novel lactam based histone deacetylase inhibitors as potential anticancer drugs. Bull. Korean Chem. Soc. 2012, 33, 2063–2066. [Google Scholar] [CrossRef]
- Choi, E.; Lee, C.; Park, J.E.; Seo, J.J.; Cho, M.; Kang, J.S.; Kim, H.M.; Park, S.K.; Lee, K.; Han, G. Structure and property based design, synthesis and biological evaluation of gamma-lactam based HDAC inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Choi, E.; Cho, M.; Lee, B.; Oh, S.J.; Park, S.K.; Lee, K.; Kim, H.M.; Han, G. Structure and property based design, synthesis and biological evaluation of gamma-lactam based HDAC inhibitors: Part II. Bioorg. Med. Chem. Lett. 2012, 22, 4189–4192. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D. Histone deacetylase inhibitors (HDACIs). Structure—Activity relationships: History and new qsar perspectives. Med. Res. Rev. 2012, 32, 1–165. [Google Scholar] [CrossRef] [PubMed]
- Sodji, Q.H.; Patil, V.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. Synthesis and structure-activity relationship of 3-hydroxypyridine-2-thione-based histone deacetylase inhibitors. J. Med. Chem. 2013, 56, 9969–9981. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Patil, V.; Guerrant, W.; Green, P.; Oyelere, A.K. Synthesis and structure-activity relationship of histone deacetylase (HDAC) inhibitors with triazole-linked cap group. Bioorg. Med. Chem. 2008, 16, 4839–4853. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules 2015, 20, 3898-3941. https://doi.org/10.3390/molecules20033898
Mottamal M, Zheng S, Huang TL, Wang G. Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules. 2015; 20(3):3898-3941. https://doi.org/10.3390/molecules20033898
Chicago/Turabian StyleMottamal, Madhusoodanan, Shilong Zheng, Tien L. Huang, and Guangdi Wang. 2015. "Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents" Molecules 20, no. 3: 3898-3941. https://doi.org/10.3390/molecules20033898
APA StyleMottamal, M., Zheng, S., Huang, T. L., & Wang, G. (2015). Histone Deacetylase Inhibitors in Clinical Studies as Templates for New Anticancer Agents. Molecules, 20(3), 3898-3941. https://doi.org/10.3390/molecules20033898