1. Introduction
The thieno[2,3-
b]thiophene moiety has been used in the design of a novel non-linear optical (NLO) properties system by incorporating this nucleus within an unsymmetrically functionalized cyclophane; this was first described by Mashraqui
et al. [
1,
2]. Additionally, thienothiophene building blocks have been used for the preparation of two series of human immunodeficiency virus (HIV) protease inhibitors possessing an α-hydroxyaminopentanamide transition state isoester. At low concentrations, these HIV protease inhibitors were found to be effective at halting the spread of the acquired immunodeficiency syndrome (AIDS) virus within the body [
3]. Egbertson
et al. [
4] reported on the synthesis and pharmacology of a potent thienothiophene non-peptide fibrinogen receptor antagonist. In 2003, Sasaki
et al. [
5] described the first potent and orally effective non-peptide antagonist for the human luteinizing hormone-releasing hormone receptor.
Due to their structural and therapeutic diversity, thienothiophene derivatives have attracted much synthetic interest because of their reactivity and biological activity, and have drawn considerable attention from researchers. They have been examined as potential antitumor, antiviral, antibacterial, anti-glaucoma drugs, and as inhibitors of platelet aggregation [
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18].
In 2010, Paek and coworkers have designed and synthesized six organic sensitizers containing 3,4-ethylenedioxythiophene and thienothiophene; these compounds have shown high efficiency and excellent stability [
19]. Recently, Mabkhot
et al. have reported for the first time on the anti-oxidant,
β-glucuronidase and
α-glucosidase inhibition potential of thieno[2,3-
b]thiophene derivatives [
20].
On the other hand, addition of the enaminone group to the thienothiophene nucleus has added new activities to these compounds; one derivative that contains both of these groups is used as a drug to treat epilepsy [
21,
22]. In view of the wide interest in the activity and profile of thienothiophenes and enaminones, and in the search for new therapeutic agents, we describe herein the synthesis and characterization of a number of new substituted thieno[2,3-
b]thiophenes which, to the best of our knowledge, have not previously been described in the literature. In addition, the bioactivity of the newly prepared compounds will be presented.
2. Results and Discussion
2.1. Chemistry
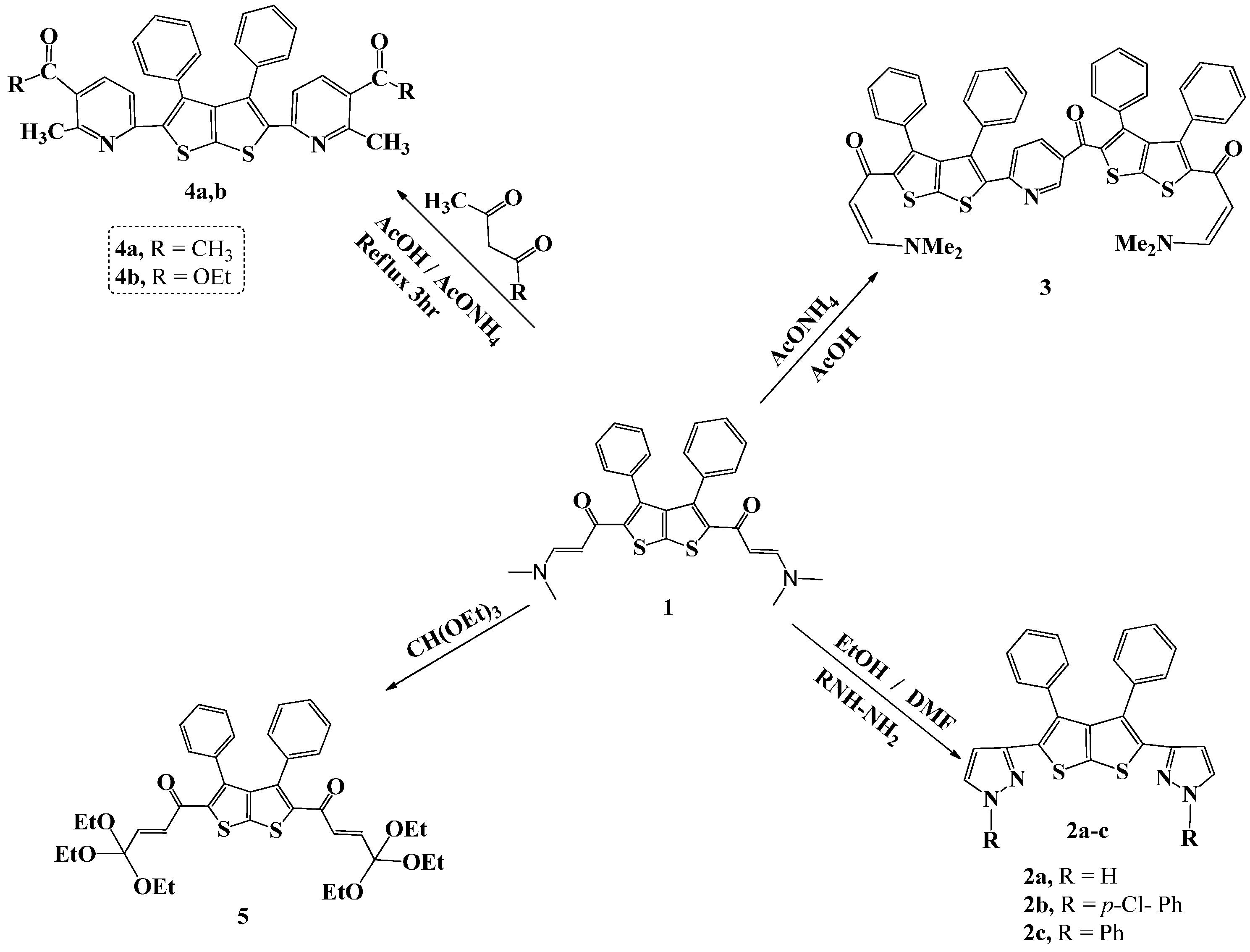
Compounds
1–
7 were synthesized as shown in
Scheme 1 and
Scheme 2. The newly synthesized compounds
1–
7 were characterized by elemental analyses, MS and NMR spectral data. These data, detailed in the experimental part, are consistent with the suggested structures. Thus, the mass spectra display the correct molecular ion peaks for which the measured high resolution (HRMS) data are in good agreement with the calculated values. DEPT and 2D (COSY, HMQC, HMBC) experiments showed correlations that helped in the
1H- and
13C-signal assignments to the different carbons and their attached, and/or neighboring hydrogens.
Compound
1, a synthon required in this study, was prepared by following a procedure outlined by Mabkhot,
et al. [
23]. Compounds
2a,
2b, and
2c were prepared through the reaction of compound
1 with the appropriate hydrazine in ethanol/DMF as solvent as shown in
Scheme 1. Compound
3, on the other hand, was synthesized by refluxing a mixture of the enaminone
1 with ammonium acetate in acetic acid as solvent for 5 h.
Scheme 1.
Synthesis of compounds 1–5.
Scheme 1.
Synthesis of compounds 1–5.
The IR spectrum of compound 1 exhibited an absorption band at 1619 cm−1 ascribed to the carbonyl group (C=O). Similarly, reaction of 1 with acetyl acetone and with ethyl acetoacetate in the presence of ammonium acetate in acetic acid as solvent, under reflux for 6 h afforded 4a and 4b, respectively. Compound 5 was prepared by fusing compound 1 with triethylorthoformate (TEOF) followed by the addition of ethanol/DMF (10 mL, 1:3) as solvent. The precipitate that resulted was filtered and its structure confirmed by spectroscopic methods. The IR spectrum showed absorption bands at 1626 and 3050 cm−1 due to C=O and C-H stretching, respectively. 1H-NMR spectrum of compound 5 showed a triplet at δ 1.05, and a quartet at 2.84–3.13 ppm, due to the methyl and CH2 of the ether group. In addition, two doublets for the CH=CH protons at 5.69 (J = 12.0 Hz) and 6.15 (J = 12.0 Hz) were observed in addition to a multiplet at δ 7.42–7.52 attributed to the phenyl protons. 13C-NMR spectrum agrees well with the suggested structure and showed the following signals: δ 22.4 (-CH3), 44.0 (-OCH2), 129.3, 129.9 (Ph), 130.5, 131.5, 133.3, 142.0 (Ar-C), 182.4 (C=O). The mass spectrum displayed the molecular ion [M]+ (19%) at m/z 694 corresponding to the molecular formula C38H46O8S2 in addition to fragments at 98 (100%) and 57 (89%).
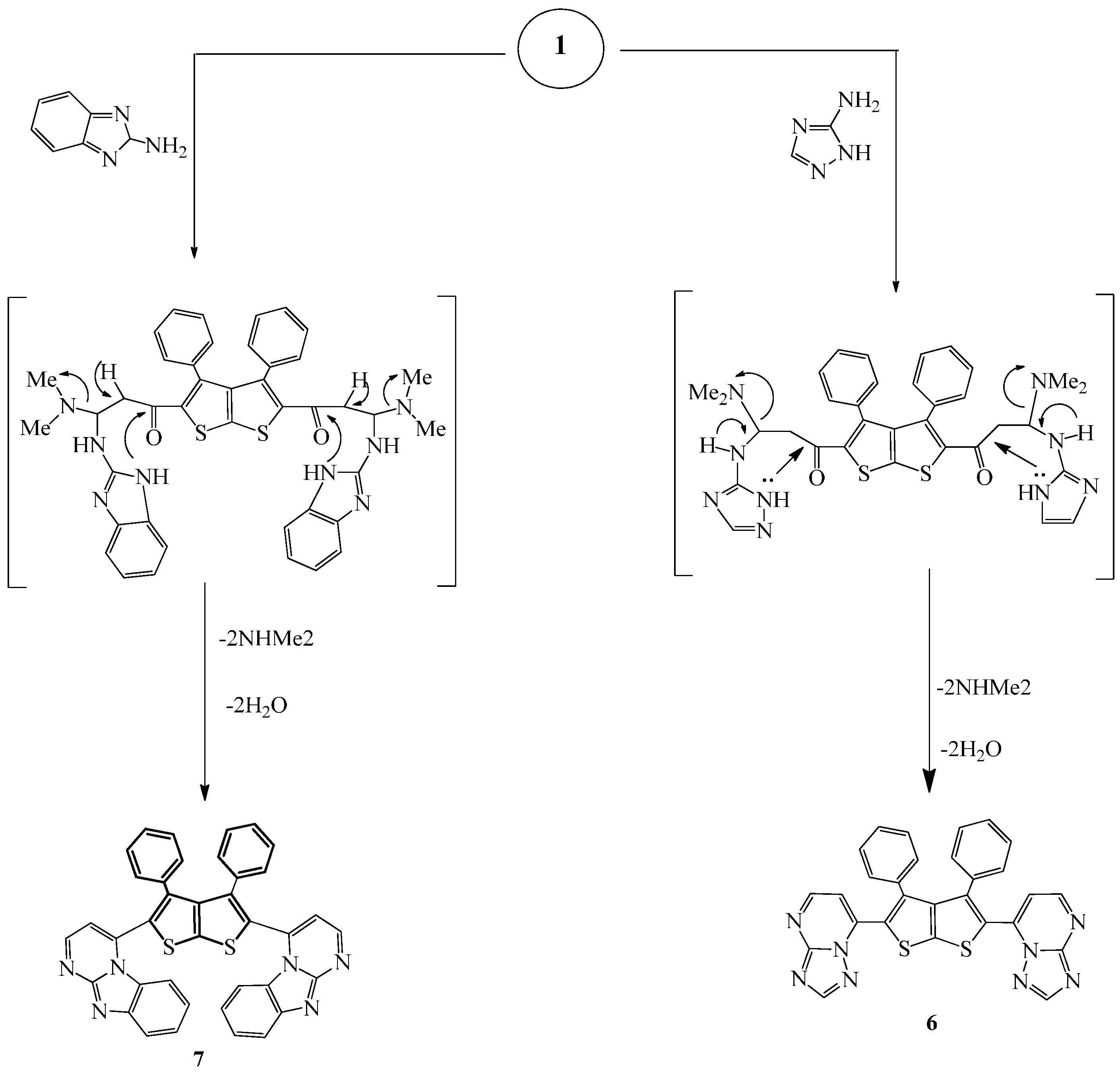
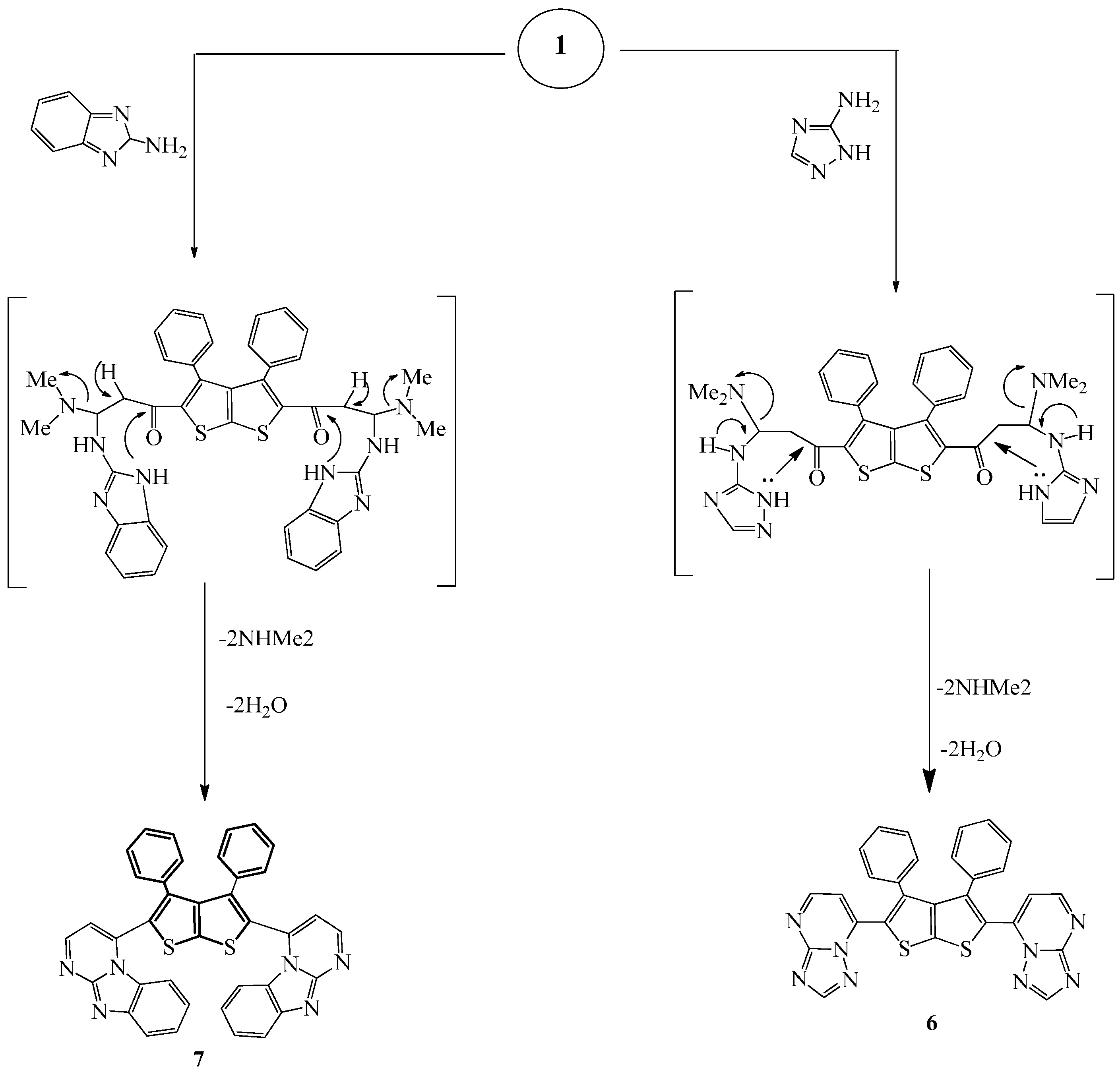
Compound
6 was prepared by refluxing a mixture of compound
1 and 3-amino-1
H-1,2,4-triazole in methanol/DMF (
Scheme 2). The IR spectrum of the prepared compound showed absorption bands at 1588 and 2924 cm
−1 due to C=N and C-H, respectively.
Scheme 2.
Synthesis of compounds 6 and 7.
Scheme 2.
Synthesis of compounds 6 and 7.
Its 1H-NMR spectrum recorded in DMSO-d6 showed a multiplet at δ 7.41–7.66 due to the aromatic hydrogens in addition to two doublets at δ 8.14 (J = 12.5 Hz) and 8.96 (J = 12.5 Hz) attributed to the two CH protons of the pyrimidine ring and to a singlet at 8.67 of the triazole hydrogen. Moreover, the 13C-NMR spectrum of the prepared compound is in agreement with the proposed structure and showed the following signals pertaining to the different carbon atoms: δ 115.4 (CH), 127.0, 129.1, 130.0 (Ph), 125.0, 130.2, 132.8, 135.8 (thiophene), 152.8, 154.4, 160.0, and 162.9 (Ar-C). Further support of the structure came from mass spectral data; mass spectrum of compound 6 displayed the correct molecular ion [M]+ at m/z = 530 corresponding to the molecular formula C28H18N8S2. Fragments at m/z 81 (86%), 67 (100%), and 55 (88%), in addition to other small ones have also appeared. Other prominent fragments at 368 (100%), 165 (98%), and 105 (40%) also appeared.
Finally, compound
7 was prepared by refluxing a mixture of
1 and 2-aminobenzimidazole in EtOH/DMF for 7 h (
Scheme 2). The IR spectrum of the prepared compound showed characteristic absorption bands at 1626 (C=N), 1539 cm
−1 (C=C) in addition to the disappearance of the C=O absorption band of compound
1.
1H-NMR spectrum in DMSO-
d6 displayed two doublets corresponding to the pyrimidine CH protons at δ 8.16 (
J = 12.5 Hz) and 8.80 (
J = 12.5 Hz) in addition to a multiplet corresponding to the phenyl and to the benzimidazole rings in the range δ 7.42–7.52.
13C-NMR spectrum is also in agreement with the suggested structure; it showed signals corresponding to the different carbon atoms in the compound as follows: δ 112.1, 115.1, 122.5, 127.1, 128.1, 129.9, 131.3 ,136.0, 139.1, 142.0, 148.1, 148.4, 156.0 (Ar-C). Similarly, the mass spectrum showed the correct molecular ion [M]
+ (25%) at
m/
z = 648 corresponding to the molecular formula C
38H
24N
6S
2 in addition to other related fragments at
m/
z = 439 (55%), 183 (100%) and at 57 (64%).
2.2. Antimicrobial Activity
To investigate the biological activity of the newly prepared compounds, the Cup-plate agar diffusion method was adopted by using discs of disinfected filter papers (6 mm in diameter). Tested compounds were dissolved in DMSO and were charged on the discs with concentrations of 5 mg/mL. The discs were then placed in Petri dishes and were loaded with different Gram-positive and Gram negative bacterial strains:
Pseudomoas aeruginosa (RCMB 010043) and
Escherichia coli (RCMB 010052) for Gram-negative bacteria and
Staphylococcus pneumonia (RCMB 010010),
Bacillis subtilis (RCMB 010067) for Gram-positive, and
Aspergillus fumigates (RCMB 02568) and
Candida albicans (RCMB 05036) for fungi. Results of the biological activity are displayed in
Table 1 as mm inhibition.
Table 1.
Antibacterial and antifungal activity of synthesized compounds.
Table 1.
Antibacterial and antifungal activity of synthesized compounds.
| Compd. | Fungi [a] | Gram (+) Bacteria [b] | Gram (−) Bacteria [c] |
|---|
| (A) | (B) | (C) | (D) | (E) | (F) |
|---|
| 1 | 17.3 ± 0.4 | 16.9 ± 0.3 | 16.3 ± 0.6 | 18.3 ± 0.3 | NA | NA |
| 2a | 20.2 ± 06 | 19.6 ± 0.3 | 23.8 ± 0.2 | 32.4 ± 0.3 | 17.3 ± 0.1 | 19.9 ± 0.3 |
| 2b | 16.7 ± 0.4 | 15.8 ± 0.5 | 10.8 ± 0.4 | 11.9 ± 0.3 | 10.8 ± 0.4 | 9.7 ± 0.5 |
| 3 | 15.9 ± 0.4 | 15.8 ± 0.5 | 10.8 ± 0.4 | 11.9 ± 0.3 | 10.8 ± 0.4 | 9.7 ± 0.5 |
| 4a | 18.1 ± 0.5 | 14.6 ± 0.5 | 17.9 ± 0.5 | 13.3 ± 0.4 | 11.4 ± 0.4 | 10.7 ± 0.3 |
| 4b | 14.6 ± 0.4 | 15.9 ± 0.5 | 10.2 ± 0.2 | 9.8 ± 0.3 | NA | 11.2 ± 0.3 |
| 5 | 13.9 ± 0.4 | 14.6 ± 0.5 | 16.3 ± 0.5 | 19.6 ± 0.6 | 12.5 ± 0.4 | 12.8 ± 0.4 |
| 6 | 12.8 ± 0.3 | 15.4 ± 0.5 | 14.1 ± 0.5 | 12.7 ± 0.4 | 11.6 ± 0.4 | 9.1 ± 0.4 |
| 7 | 18.1 ± 0.6 | 15.9 ± 0.5 | 12.6 ± 0.5 | 13.7 ± 0.6 | 12.1 ± 0.4 | 10.4 ± 0.2 |
| SD-1 [d] | 23.7 ± 0.1 | 25.4 ± 0.1 | 23.8 ± 0.2 | 32.4 ± 0.3 | --- | --- |
| SD-2 [e] | --- | --- | --- | --- | 17.3 ± 0.1 | 19.9 ± 0.3 |
Results shown in
Table 1 reveal that compound
2a has exhibited remarkable activity against both Gram-positive and negative bacteria while compounds
4a,
5 and
6 showed good activity against Gram-positive bacteria. On the other hand, compound
2a was the most active against tested fungus. Results also reveal that compound
2a was the most potent; it has antibacterial and antifungal activities due to the presence of electron-rich functional groups. These groups could bind with the microorganisms. In addition, the presence of acrylides bonded to the thionothiophene nucleus and the effect of thionothiophene itself against bacteria and fungi may explain the potency of these compounds; the tautomeric forms of these compounds penetrate the cell wall.
2.3. Molecular Docking Studies
Molecular docking is a technique which serves to verify binding integrity and interaction poses of ligands within the binding pocket of target proteins [
24]. To validate and specify the target for anti-fungal and anti-bacterial activity of newly synthesized thieno[2,3-
b]thiophene derivatives, nine different target proteins
i.e., dihydrofolate reductase (DHFR) (PDB ID 4HOF), secreted aspartic protease (PDB ID 3Q70), and N-myristoyl transferase (PDB ID 1IYL) from
C. albicans were selected as fungal targets, while for bacterial targets dihydrofolate reductase (PDB ID 3FYV), gyrase B (PDB ID 4URM) and sortase A (PDB ID 2MLM) from
S. aureus and rhomboid protease (PDB ID 3ZMI), methionine peptidase (PDB ID 4PNC) and undecaprenyl diphosphate synthase (PDB ID 4H2M) from
E. coli were fetched from the Protein Data Bank [
25]. Among all these nine target proteins, only two target proteins,
i.e., dihydrofolate reductase (DHFR) from
C. albicans and rhomboid protease from
E. coli were preferred as they showed good interactions and binding affinities with the synthesized compounds in docking simulation by MOE 2013 [
26], while the others have binding pockets that are too small to fit these large compounds. Before docking, structures of synthesized compounds were built and saved in their 3D conformation by MOE 2013. Moreover, for target protein preparation, all nine proteins were prepared, charged, protonated and minimized by MOE 2013. The 4HOF is a homotrimer (ABC chains) and 3ZMI is a monomer with bound inhibitors 18H and L6C, respectively. Chain A of 4HOF and a single chain of 3ZMI with one conserved water molecule (HOH2016) were selected for the evaluation of newly synthesized compounds. Both of these proteins were prepared, charged, protonated and minimized by MOE 2013. For docking, default MOE docking parameters
i.e., Triangle Matcher Algorithm with two rescoring functions London dG and GBVI/WSA dG were utilized to generate 30 poses of each compound. As a result, mdb output files were generated enclosing all docking results with scoring and multiple conformations of ligands. Results were finally inspected to determine the most potent and effective anti-fungal and anti-bacterial inhibitor by visualizing various interactions of ligands within binding pocket.
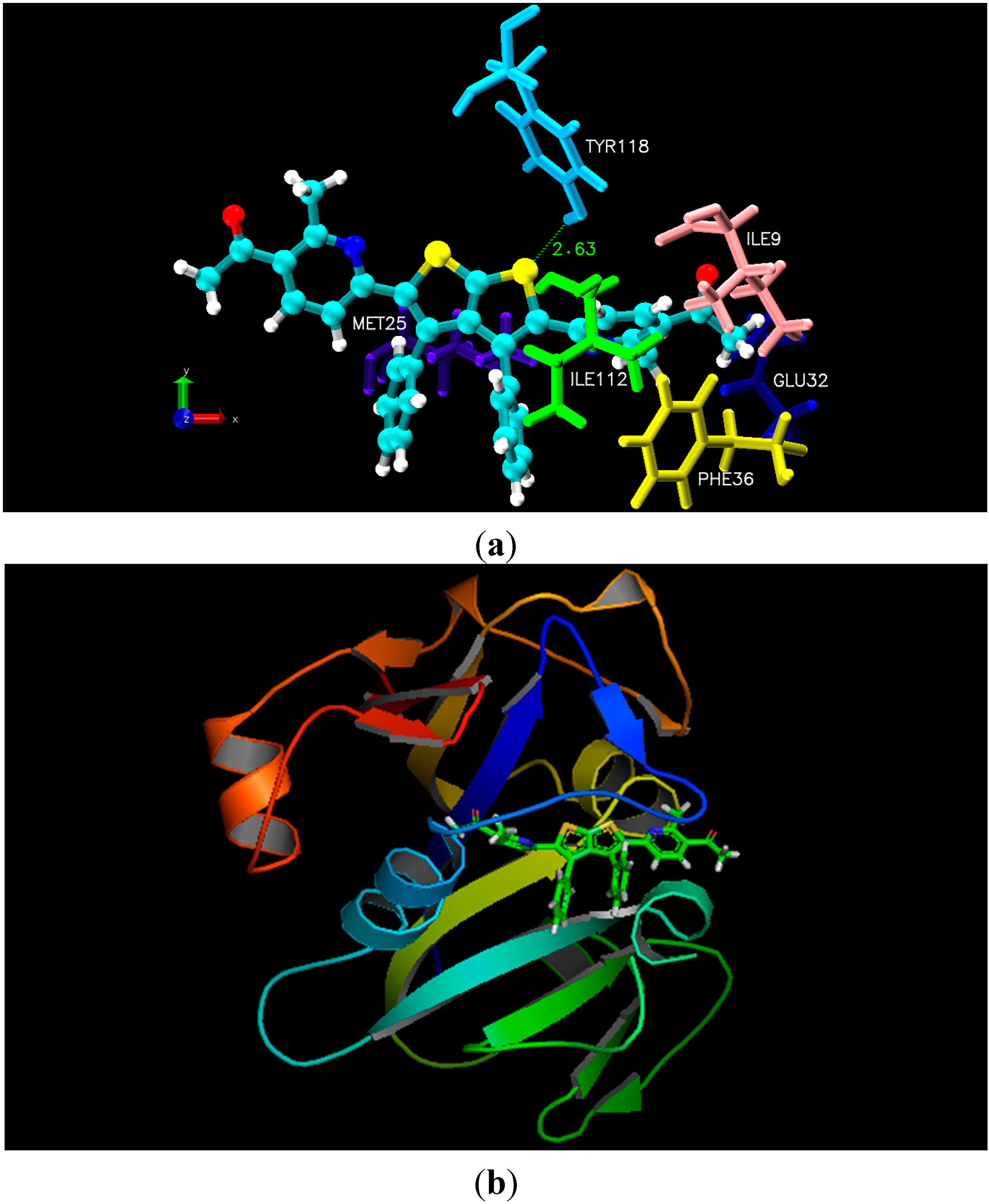
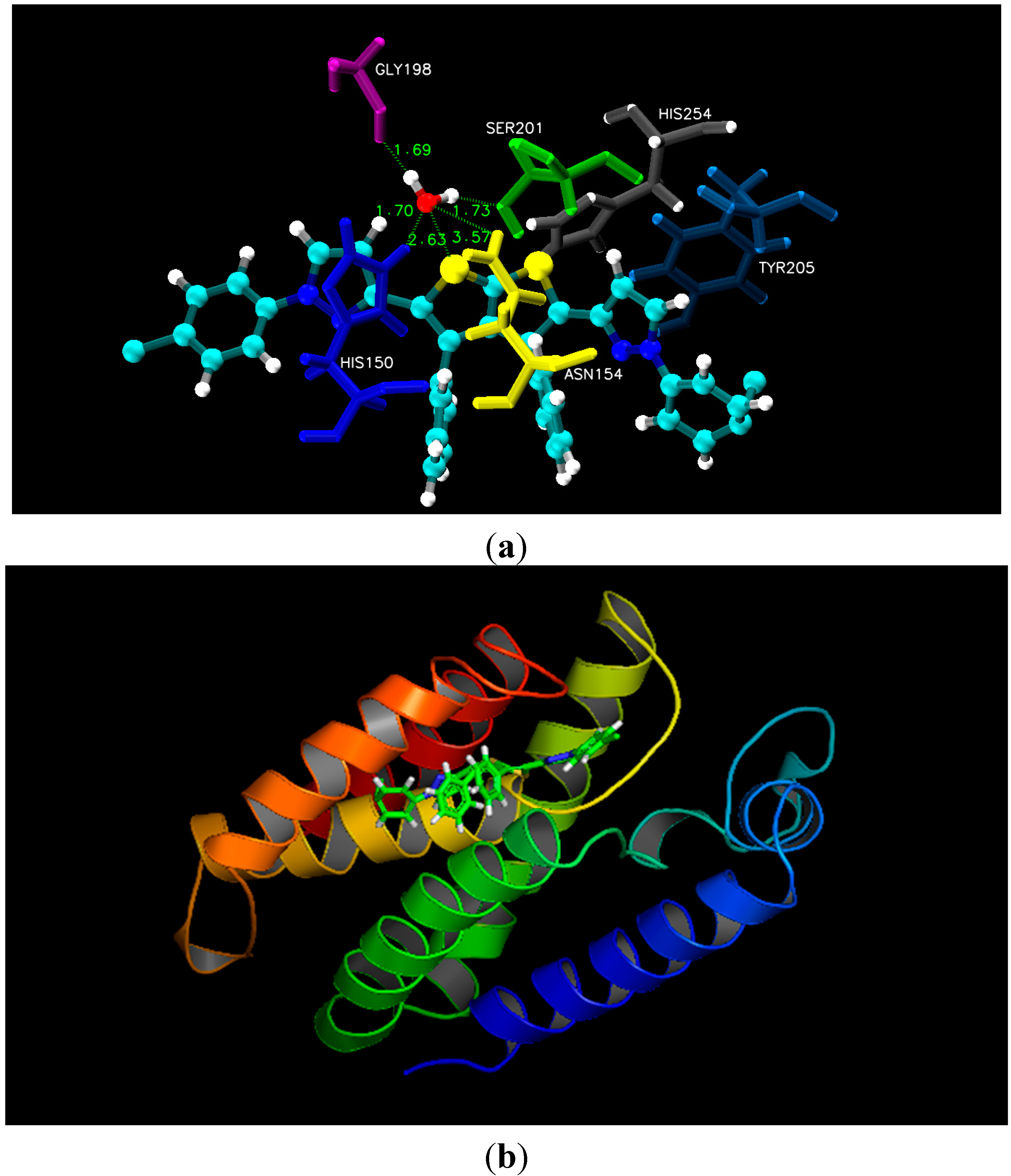
As illustrated in the experimental results, docking studies of all nine thieno[2,3-
b]thiophene derivatives unveiled their potency as anti-fungal and anti-bacterial inhibitors. All newly synthesized compounds contain large aromatic systems, exhibiting noticeable hydrophobic and Van der Waals (VDW) interactions along with few additional hydrogen bonding with the crucial residues
i.e., Ile9, Glu32, Phe36, Ile112, and Tyr118 of 4HOF (
C. albicans) [
27]. However, 3ZMI (
E. coli) His150, Asn154, Ser201, and His254 were observed to be the hotspot residues for their inhibitory activity [
28]. Docking results indicated that all newly synthesized compounds, especially
4a and
2b, displayed pronounced hydrophobic and Van der Waals interactions due to the presence of electronegative functionalities. Unfortunately, compounds
3,
5 and
6 being most potent inhibitors like
4a and
2b, did not show better interactions due to lack of fit in the cavity of the target proteins, due to the presence of bulky R-group that create steric hindrance whereas few compounds lack certain electronegative functionalities at R-group. Moreover, docking results with 4HOF presented hydrogen bonding of the thiol ring sulphur (compound
4a) with the hydroxyl group of Tyr118 at a distance of 2.63 Å, whereas the pyrimidine ring of
4a displayed hydrophobic interactions with the aromatic ring of Phe36. In addition, various Van der Waals interactions were observed with other prime residues
i.e., Glu32, Ile9 and Ile112 (
Figure 1). Alternatively, docking simulation with 3ZMI revealed that sulphur of the thiol ring of compound
2b was involved in hydrogen bonding with His150, Asn154 and Ser201, where conserved water molecule (HOH2016) acts as a bridging atom. Additionally, both pyrol rings of compound
2b displayed hydrophobic interaction with Tyr205 and His150 (
Figure 2). According to our docking simulation studies by MOE 2013, we concluded that all thieno[2,3-
b]thiophene derivatives, especially compounds
4a and
2b, exhibited marked hydrogen bonding, hydrophobic and Van der Waals interactions with vital residues of DHFR of
C. albicans and rhomboid protease of
E. coli and are the most active anti-fungal and anti-bacterial inhibitors, respectively.
Figure 1.
(a) Molecular docking interaction model for the most potent anti-fungal compound 4a with C. albicans dihydrofolate reductase protein (PDB ID.4HOF) showing hydrogen bonding, hydrophobic and Van der Waals interactions; (b) ribbon diagram of dihydrofolate reductase (C. albicans) complex with inhibitor 4a.
Figure 1.
(a) Molecular docking interaction model for the most potent anti-fungal compound 4a with C. albicans dihydrofolate reductase protein (PDB ID.4HOF) showing hydrogen bonding, hydrophobic and Van der Waals interactions; (b) ribbon diagram of dihydrofolate reductase (C. albicans) complex with inhibitor 4a.
Figure 2.
(a) Molecular docking interaction model for the most potent anti-bacterial compound 2b with rhomboid protease of E. coli (PDB ID.3ZMI) showing hydrogen bonding through conserved water molecule & hydrophobic interactions; (b) ribbon diagram of rhomboid protease (E. coli) complex with inhibitor 2b.
Figure 2.
(a) Molecular docking interaction model for the most potent anti-bacterial compound 2b with rhomboid protease of E. coli (PDB ID.3ZMI) showing hydrogen bonding through conserved water molecule & hydrophobic interactions; (b) ribbon diagram of rhomboid protease (E. coli) complex with inhibitor 2b.
2.4. POM Analyses of Compounds 1–7
POM theory is able to identify the type of pharmacophoric sites [
29]. POM has certainly become one of the best-known recent methods that are regularly used [
30] to produce two dimensional models to identify and to indicate the type of pharmacophoric sites that affect antimicrobial, antiviral, and antifungal activities with a change in the chemical substitution and partial charge distribution. In fact, the real advantages of POM theory are the ability to easily predict the biological activities of molecules and to show the relationship between steric/electrostatic properties and biological activity in the form of pharmacophoric sites; this gives key features not only on the ligand-receptor interaction, but also on the topology of the receptor with the coexistence of tautomerism, isomerism, and ring opening/closing processes [
31].
For a molecule to be a potential drug, besides having a good biological activity, it must have good pharmacokinetic properties in biological systems. To access the pharmacokinetic profile of synthesized molecules, we employed well established
in silico tools such as Osiris, Petra and Molinspiration validated with about 7000 drug molecules available on the market and created new economic and effecient bioinformatic platform called POM Analyses [
31]. Now POM theory is well verified and it is more and more popular by many other research groups involved in bioinformatics and drug design.
One of the practical problems associated with synthetic drugs is the existence of various side effects. Shown in
Table 2 are results of theoretical toxicity risks of compounds
1–
7 calculated with the aid of the Petra/Osiris/molinspiration (POM) program [
32,
33,
34,
35]. Our findings reveal that compounds
2,
4,
6 and
7, contrary to compounds
1,
3 and
5, are not toxic and can be utilized as therapeutic agents. In addition, results presented in
Table 2 show that structures of the investigated compounds are supposed to be non-mutagenic when run through the mutagenicity assessment of free system, and that these compounds are at low risk comparable with standard synthetic drugs as far as irritation and reproductive effects are concerned.
Table 2.
Osiris calculations of toxicity risks of compounds 1–7.
Table 2.
Osiris calculations of toxicity risks of compounds 1–7.
| Compd. | MW | Toxicity Risks [a] | Osiris Calculations [b] |
| MUT | TUM | IRRIT | REP | cLogP | Sol | DL | DS |
| 1 | 486 | +++ | +++ | ++ | ++ | 3.89 | −8.81 | 3.61 | 0.22 |
| 2a | 424 | +++ | +++ | +++ | ++ | 4.75 | −8.72 | 3.15 | 0.27 |
| 2b | 644 | +++ | +++ | +++ | ++ | 8.51 | −12.52 | 4.66 | 0.12 |
| 2c | 576 | +++ | +++ | +++ | ++ | 7.31 | 11.01 | 4.51 | 0.14 |
| 3 | 881 | +++ | +++ | ++ | ++ | 9.41 | −18.31 | 3.61 | 0.08 |
| 4a | 558 | +++ | +++ | +++ | ++ | 7.51 | −12.02 | 1.37 | 0.13 |
| 4b | 784 | +++ | +++ | +++ | ++ | 8.39 | −11.60 | −2.32 | 0.07 |
| 5 | 692 | +++ | +++ | ++ | ++ | 7.97 | −10.71 | −15.35 | 0.05 |
| 6 | 528 | +++ | +++ | +++ | ++ | 4.16 | −7.01 | 5.79 | 0.27 |
| 7 | 626 | +++ | +++ | +++ | ++ | 8.24 | −11.54 | 4.63 | 0.12 |
| SD-1 [c] | 581 | +++ | +++ | --- | +++ | 7.86 | −0.96 | 0.83 | 0.32 |
| SD-2 [d] | 344 | +++ | +++ | +++ | +++ | 5.37 | −7.72 | 0.92 | 0.30 |
The hydrophilicity character of each compound has been expressed in terms its cLog
P value since it has been established that the absorption or permeation is greatly affected by this quantity (value of cLog
P). Accordingly, when the value of cLog
P is higher than 5, the absorption or permeation decreases. Our results show that only 3/10 compounds (
1,
2a and
6) have cLog
P values within the acceptable criteria and are potentially active against various biotargets (GPCRL: GPCR ligand; ICM: Ion channel modulator; KI: Kinase inhibitor; NRL: Nuclear receptor ligand; PI: Protease inhibitor; EI: Enzyme inhibitor) as shown in
Table 2. Thus, the cLog
P parameter should be taken into consideration and serve as a guide for further enzymatic screening investigations.
Similarly, the geometrical conformation of pharmacophores in this context is important since it is not fixed for compounds
1–
7. Properties such as absorption, distribution characteristics, and bioactivity depend on the geometrical parameter and the aqueous solubility of each compound. Consequently, good absorption of tested compounds
1–
7 could presumably be due to their good solubility [
34,
35]. Furthermore,
Table 3 shows drug-likeness of compounds
1–
7, in the incomparable zone with standard known drugs (
SD-1: streptomycin for antimicrobial and
SD-2: clotrimazole as standard drug for fungi). For example, we have calculated an overall drug-score (DS) for compound
1 and compared it with that of compound
2a and concluded that
2a was better than rest of series
1–
7, except
6). The DS combines drug-likeness, cLog
P, log
S, molecular weight, and toxicity risks, in a one handy value that may be used to judge the compound’s overall potential to qualify for a drug. As shown in
Table 3, the reported compounds
1–
7 showed low to moderate DS (DS < 0.50).
Table 3.
Molinspirationcalculations of compounds (1–7).
Table 3.
Molinspirationcalculations of compounds (1–7).
| Compd. | Molinspiration Calculations [a] | Drug-Likeness [b] |
| TPSA | NONH | NV | VOL | GPCRL | ICM | KI | NRL | PI | EI |
| 1 | 41 | 0 | 1 | 437 | −0.04 | −0.34 | −0.21 | −0.35 | −0.06 | 0.10 |
| 2a | 57 | 2 | 1 | 357 | 0.09 | −0.02 | 0.12 | −0.28 | 0.01 | 0.13 |
| 2b | 36 | 0 | 2 | 527 | −0.08 | −0.64 | −0.38 | −0.51 | −0.10 | −0.29 |
| 2c | 36 | 0 | 2 | 500 | 0.01 | −0.43 | −0.22 | −0.34 | −0.04 | −0.13 |
| 3 | 91 | 1 | 2 | 754 | −2.72 | −3.66 | −3.32 | −3.57 | −2.11 | −2.92 |
| 4a | 60 | 0 | 2 | 491 | 0.08 | −0.35 | −0.10 | −0.20 | −0.03 | −0.04 |
| 4b | 78 | 0 | 2 | 543 | −0.16 | −0.72 | −0.32 | −0.43 | −0.11 | −0.27 |
| 5 | 90 | 0 | 2 | 632 | −0.47 | −1.28 | −0.91 | −0.96 | −0.23 | −0.64 |
| 6 | 86 | 0 | 2 | 429 | −0.01 | −0.37 | 0.07 | −0.53 | −0.10 | −0.05 |
| 7 | 60 | 0 | 2 | 525 | −0.19 | −0.82 | −0.38 | −0.87 | −0.24 | −0.50 |
| SD-1 [c] | 336 | 16 | 3 | 497 | 0.09 | −0.16 | −0.17 | −0.18 | 0.65 | 0.38 |
| SD-2 [d] | 18 | 0 | 1 | 310 | 0.17 | 0.30 | 0.14 | −0.21 | −0.13 | 0.42 |
Most importantly, the molinspiration calculations pertaining to compounds 1–7 show that most of them, could be good candidates to interact with various enzymatic targets (GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor and enzyme inhibitor) when the two central phenyl substituents are retrieved from principal skeleton (two methyl instead the two phenyl will be more interesting).
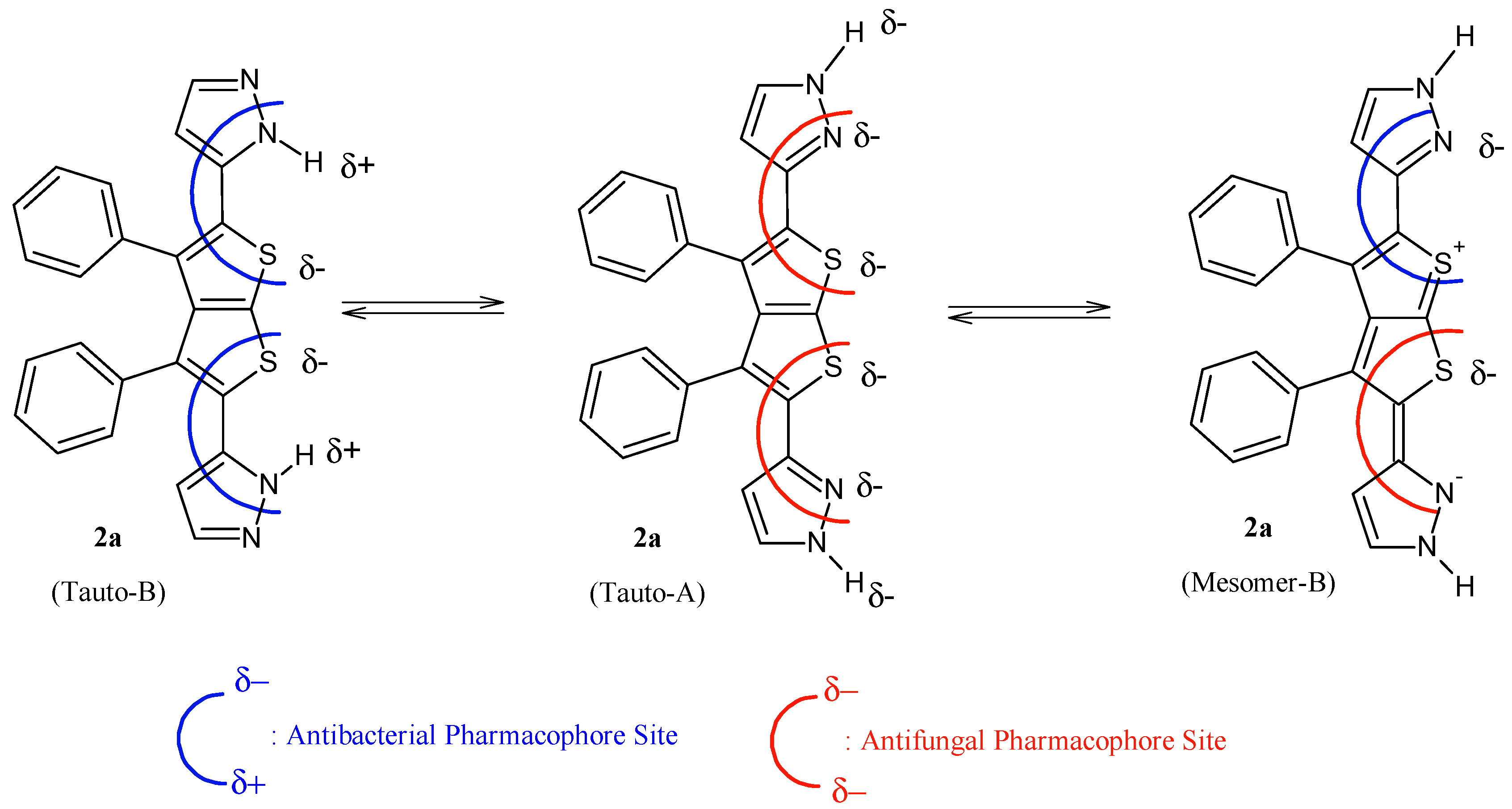
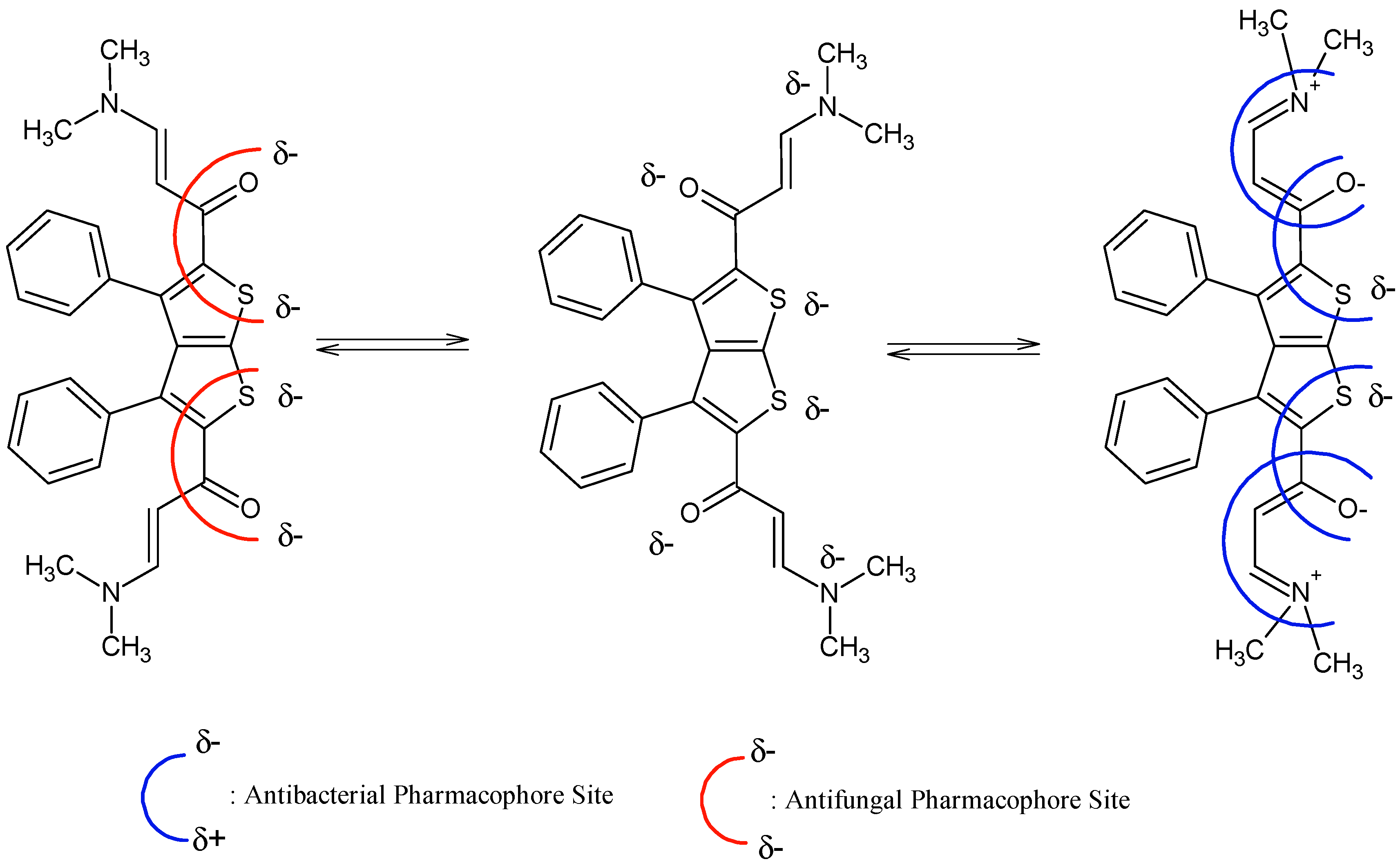
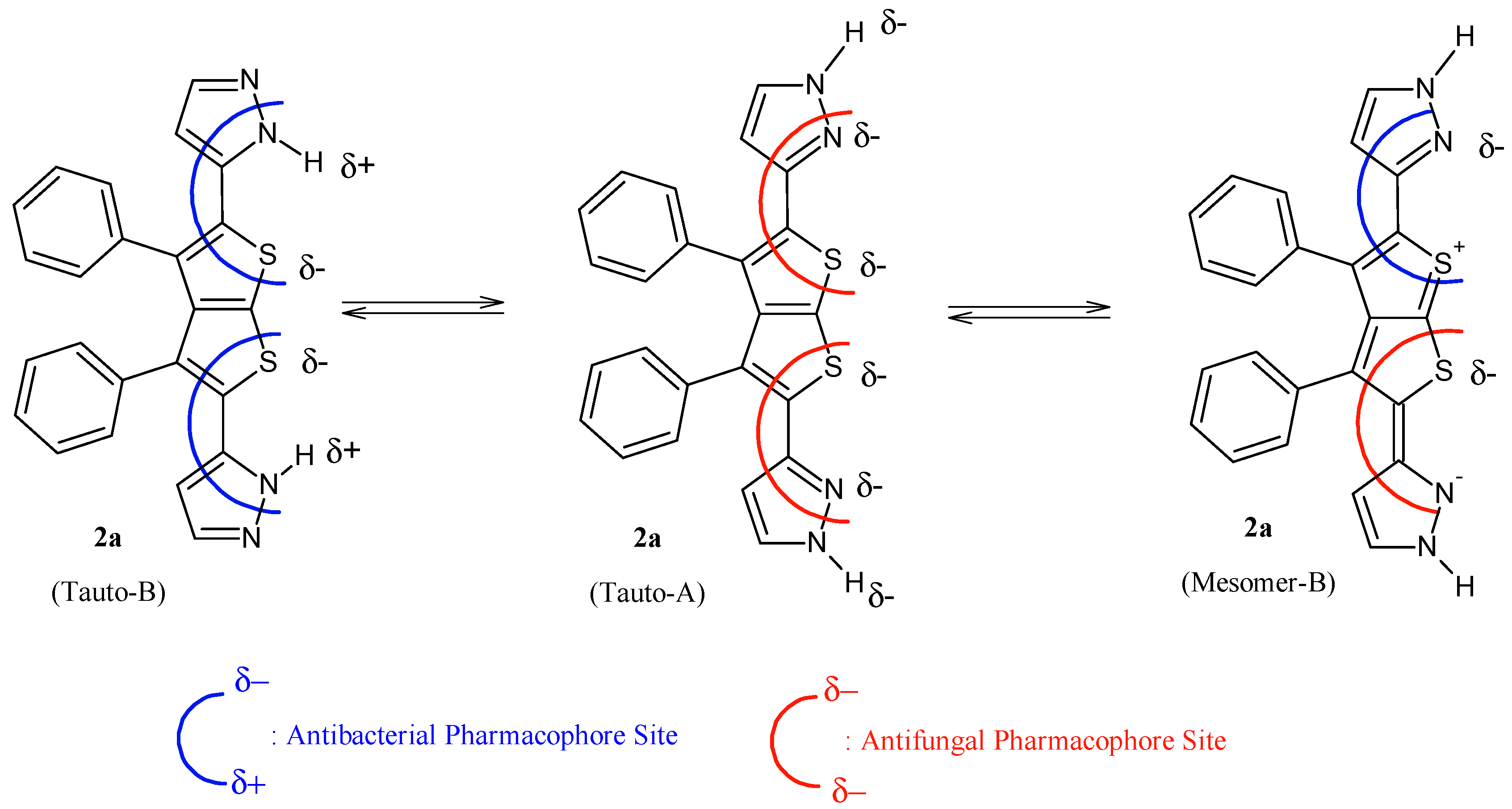
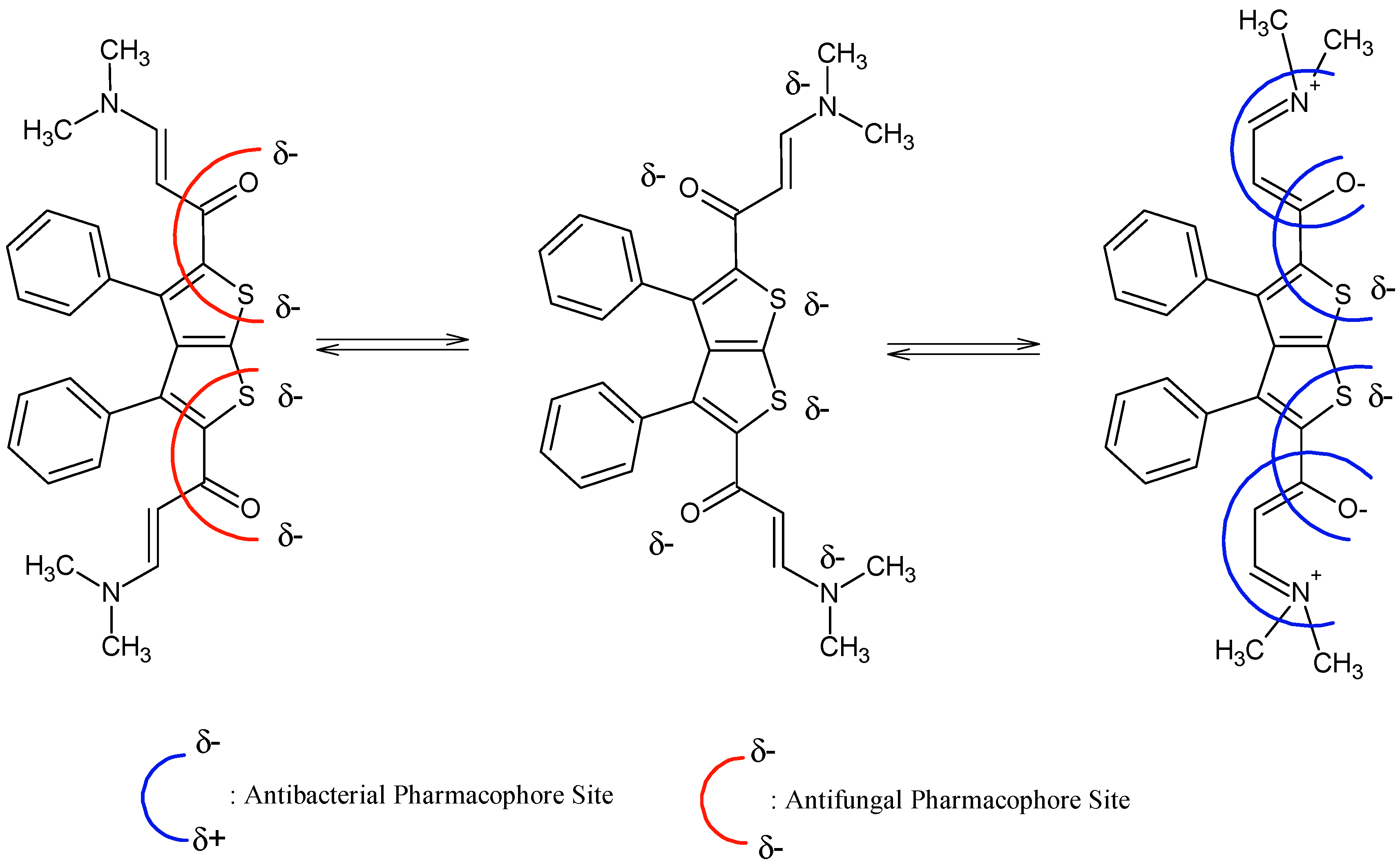
It is evident that compounds
1–
7 are subjected to important chemical processes of tautomerism/mesomerism, leading to regeneration of combined pharmacophoric sites as shown in
Figure 3 and
Figure 4 for compounds
1 and
2a, respectively.
In addition, results from
Figure 3 and
Figure 4 reveal that compound
2a has more chances to inhibit microorganisms because its pharmacophoric sites are more explicit than those of the polysubstituted analogue compound
1. POM analyses thus show the crucial role and impact of substituents on bioactivity and indicate the unfavorable structural parameters in actual drug design: more substitution doesn’t guarantee more efficiency in bioactivity.
Figure 3.
Identification of explicit and combined antibacterial/antifungal pharmacophoric sites of compound 2a.
Figure 3.
Identification of explicit and combined antibacterial/antifungal pharmacophoric sites of compound 2a.
Figure 4.
Identification of steric hindered antibacterial and antifungal pharmacophoric sites of compound 1.
Figure 4.
Identification of steric hindered antibacterial and antifungal pharmacophoric sites of compound 1.
3. Experimental Section
3.1. Materials and Equipment
Chemicals and materials used in this investigation were obtained from commercial sources and were used without further purification. Preparative TLC was performed on Merck Art. 5745, Kieselgel 60 F254/0.50 nm (Merck KGaA, 64271 Darmstadt, Germany). Melting points (uncorrected) were measured with a Electrothermal melting point apparatus (Bibby Scientific Limited, Beacon Road, Stone, Staffordshire, UK). Infrared spectra (IR) were recorded, as potassium bromide (KBr) discs, on a Nicolet 6700 FT-IR spectrophotometer (Madison, WI, USA). 1H- (400 MHz) and 13C-NMR (100 MHz) spectra were acquired with the aid of a JEOL-400 MHz spectrometer (Jeol, Tokyo, Japan) with DMSO-d6 as solvents and TMS as an internal standard. Chemical shifts are expressed in δ units; coupling constants (J-values) for 1H-1H are given in Hertz (Jeol, Tokyo, Japan). Mass spectra were recorded on a Jeol JMS-600 H instrument (Tokyo, Japan) at 70 eV. Elemental analyses were performed on a Euro Vector Elemental Analyzer (EA 3000 A, Via Tortona, Milan, Italy).
3.2. Preparation of 1,1'-(3,4-Diphenylthieno[2,3-b]thiene-2,5-diyl)bis(3-dimethyl-aminoprop-2-en-1-one) (1)
A mixture of 1,1'-(3,4-diphenylthieno[2,3-b]thiophene-2,5-diyl)diethanone (1.89 g, 5 mmol), DMF-DMA (1.19 mL, 0.01 mol) was refluxed in m-xylene (15 mL) for 7 h. After cooling, the resulting solid product was collected by filtration and recrystallized by using DMF/EtOH to give the desired product 1. Yield: 73%; m.p. 250 °C; IR (νmax): 1622 (C=O), 1539, 1457 cm−1; 1H-NMR (DMSO-d6) δ (ppm): 2.99 (s, 12H, CH3), 4.53 (d, 1H, J = 12 Hz, CH), 5.38 (d, 1H, J = 12 Hz, CH), 7.41–7.65 (m, 5H, C6H5); 13C-NMR (DMSO-d6) δ (ppm): 192.2, 153.2, 147.7, 145.8, 141.8, 138.8, 134.8, 129.8, 129.6, 129.2, 125.3, 30.6; MS m/z (%): 488 [M+, 30%], 386 (100), 368 (47), 213 (73), 43 (46); Anal. calcd. for C28H26N2O2S2: C, 69.11; H, 5.39; N, 5.76; Found: C, 69.15; H, 5.41; N, 5.78.
3.3. General Procedure for the Preparation of Compounds 2a–c
Compounds 2a–c were prepared using the following general procedure: compound 1 (0.244 g, 5.0 mmol) was added to hydrazine hydrate (2 mL), phenylhydrazine (1 mL), or 4-chlorophenyl hydrazine (0.5 g) (to make compounds 2a–c, respectively). The mixture of compound 1 and the appropriate hydrazine was refluxed until compound 1 is completely dissolved, followed by addition of the mixed solvent EtOH/DMF. The reaction mixture was then refluxed for 6–7 h and the product was filtered after cooling. The desired product was obtained in a pure form by recrystallization from EtOH/DMF. Using the general procedure, the following compounds were prepared.
3.3.1. 3,3'-(3,4-Diphenylthieno[2,3-b]thiophene-2,5-diyl)bis(1H-pyrazole) (2a)
Yellow crystals; yield: 89%; mp: 133–135 °C. IR (cm−1): 3192 (NH); 1508 (C=N). 1H-NMR (DMSO-d6) δ (ppm): 6.45 (d, J = 4.5 Hz, CH), 7.40–7.53 (m, 5H), 7.81 (d, J = 4.5 Hz, CH), 13.00 (s, 1H, NH). 13C-NMR δ: 103.0 (CH), 127.3 (CH), 128.6 (CH), 129.0 (C), 129.1 (CH), 130.0 (C), 130.4 (C), 131 (N-CH), 131.3 (C), 135.0 (C), 136.4 (C), 145.8 (C), 147.3 (Ar-C). MS m/z (70 eV): 580, M+ (4%); 279, [M−301]+ (100%); 189, [M−391]+ (46%); 104, [M−476]+ (33%). Anal. Calcd for C24H16N4S2: C, 67.90; H, 3.80; N, 13.20; S, 15.10. Found: C, 67.78; H, 4.21; N, 13.00; S, 15.19.
3.3.2. 3,3'-(3,4-Diphenylthieno[2,3-b]thiophene-2,5-diyl)bis(1-chloro phenyl-1H-pyrazole) (2b)
Red powder; yield: 67%; mp: 339–341 °C. IR (cm−1): 1610. 1H-NMR (DMSO-d6): δ, 6.48 (d, J = 4.5 Hz, CH), 6.53–7.20 (m, 9H, C6H5), 7.55 (d, J = 4.5 Hz, 1H). 13C-NMR: δ, 102.0 (CH), 127.3 (CH), 128.5 (CH), 132.0 (N-CH), 129.2 (CH), 129.9 (C), 130.4 (C), 130.5, 136.4 (C), 145.8 (C), 147.3 (C), 152.8 (C). MS m/z (70 eV): 646, [M−301]+ (100%); 189, [M−391]+ (46%); 104, [M−476]+ (33%). Anal. Calcd for C36H22Cl2N4S2: C, C, 66.97; H, 3.43; Cl, 10.98; N, 8.68; S, 9.93. Found: C, 66.99; H, 3.47; Cl, 11.00; N, 8.55; S, 9.78.
3.3.3. 3,3'-(3,4-Diphenylthieno[2,3-b]thiophene-2,5-diyl)bis(1-phenyl-1H-pyrazole) (2c)
Brown powder; yield: 64%; mp: 194–196 °C. IR (cm−1): 1618. 1H-NMR (DMSO-d6): δ, 6.50 (d, J = 4.5 Hz, 1H), 6.53–7.20 (m, 9H, C6H5), 7.57 (d, J = 4.5 Hz, 1H). 13C-NMR: δ, 102.1 (CH), 132.0 (N-CH), 128.4, 190.4, 130.5, 131.5, 132.0, 132.9, 133.50, 141.4, 153.3, 160.8 (Ar-C). MS m/z (70 eV): 576, M+ (694%); Anal. Calcd for C36H24N4S2 C, 74.97; H, 4.19; N, 9.71; S, 11.12. Found: C, 74.87; H, 4.00; N, 9.83; S, 10.92.
3.4. Synthesis of 3-(Dimethylamino)-1-(5-(6-(5-((E)-3-(dimethylamino)acryloyl)-3,4-diphenylthieno[2,3-b]thiophen-2-yl)nicotinoyl)-3-methyl-4-phenylthieno[2,3-b]thiophen-2-yl)prop-2-en-1-one (3)
Compound 1 (0.244 g, 0.5 mmol) was dissolved in glacial acetic acid (10 mL) containing ammonium acetate (1.0 g) in a 100-mL round bottomed flask connected to a condenser. The mixture was refluxed for 5 h and after cooling, the precipitate was filtered and recrystallized from DMF/EtOH to afford the desired product as yellow powder. Yield: 75%; mp: 298–300 °C. IR (cm−1): 1618. The product was insoluble in most organic solvents, which prevented us from recording its 1H- and 13C-NMR spectra. MS m/z (70 eV): 821, M+ (2%); 262, [M−559]+ (27%); 67, [M−754]+ (100%). Anal. Calcd for C52H39N3O3S4: C, 70.80; H, 4.46; N, 4.76; S, 14.54; Found: C, 70.81; H, 4.46; N, 4.78; S, 14.50.
3.5. Synthesis of Compounds 4a and 4b
Compounds 4a and 4b were prepared according to the following general procedure: a mixture of compound 1 (0.244 g, 0.5 mmol), acetyl acetone (0.56 g, 1 mmol), and ammonium acetate (1.0 g) in glacial acetic acid (10 mL) was refluxed for 6 h. The solid product was filtered and recrystallized from DMF/EtOH to afford the desired product in pure form.
3.5.1. 1,1'-(6,6'-(3,4-Diphenylthieno[2,3-b]thiophene-2,5-diyl)bis(2-methylpyridine-6,3-diyl))-diethanone (4a)
Obtained as a deep yellow powder. Yield 77%; mp: >360 °C. IR (cm−1): 1573 (C=N), 1671 (C=O). 1H-NMR (DMSO-d6) δ (ppm): 2.47 (s, 3H, COCH3), 2.62 (s, 3H, CH3), 7.29–7.59 (m, 5H), 7.95 (d, J = 8.0 Hz, 1H, CH pyridine), 8.26 (d, J = 8.0 Hz, 1H, CH pyridine). 13C-NMR (DMSO-d6) δ (ppm): 29.4 (CH3), 30.6 (COCH3), 119.0 (CH), 129.2 (CH), 129.5 (CH), 129.9 (CH), 134.8 (CH), 138.8 (C), 141.8 (C), 147.7 (C), 151.2 (C), 166.1 (C), 194.1 (C, C=O). MS m/z (70 eV): 560, M+ (3%); 522, [M−38]+ (25%); 431, [M−1291]+ (100%); 326, [M−234]+ (94%). Anal. Calcd for C34H26N2O2S2: C, 73.09; H, 4.69; N, 5.01; S, 11.48; Found: C, 73.20; H, 4.74; N, 4.91; S, 11.40.
3.5.2. Diethyl 6,6'-(3,4-diphenylthieno[2,3-b]thiophene-2,5-diyl)bis(2-methylnicotinate) (4b)
Obtained as a brown powder; yield: 74%; mp: 245–247 °C. IR (cm−1): 1568, 11718. 1H-NMR (DMSO-d6) δ (ppm): 1.29 (t, J = 12 Hz, 3H, CH2CH3), 2.57 (s, 3H, CH3), 4.21 (q, J = 12.0 Hz, 2H, CH2CH3), 7.29–7.59 (m, 5H), 8.20 (d, J = 8.0 Hz, 1H, CH pyridine), 8.26 (d, J = 8.0 Hz, 1H, CH pyridine). 13C-NMR (DMSO-d6) δ (ppm): 29.4 (CH3), 30.6 (COCH3), 62.17 (CH2), 119.1 (CH), 129.2 (CH), 129.6 (CH), 129.9 (CH), 134.8 (CH), 138.8 (C), 141.8 (C), 147.7 (C), 151.2 (C), 166.1 (C), 194.1 (C, C=O). MS data: m/z (70 eV) 580, M+ (4%); 279, [M−301]+ (100%); 189, [M−391]+ (46%); 104, [M−476]+ (33%). Anal. Calcd for C36H30N2O4S2: C, 69.88; H, 4.89; N, 4.53; S, 10.36; Found: C, 69.89; H, 5.09; N, 4.70; S, 10.17.
3.6. Synthesis of 1,1′-(3,4-Diphenylthieno[2,3-b]thiophene-2,5-diyl)bis(4,4,4-triethoxybut-2-en-1-one) (5)
The title compound was prepared by mixing compound 1 (0.244 g, 0.5 mmol) and triethyl orthoformate (0.148 g, 1 mmol) in presence of ZnCl2 (0.5 g) as a catalyst. The mixture was stirred for 4 h followed by addition of DMF/EtOH. The solid product was collected by filtration and recrystallized from DMF/EtOH to afford compound 5 as a brown solid. Yield: 56%; mp: >360 °C. IR (cm−1): 1626. 1H-NMR (DMSO-d6): δ (ppm): 1.05 (t, J = 12.0 Hz, 3H, CH3), 2.95(q, J = 12.0 Hz, 2H, CH2CH3), 5.69 (d, J = 12.0 Hz, 1H), 6.53 (d, J = 12.0 Hz, 1H), 7.42–7.52 (m, 5H). 13C-NMR (DMSO-d6) δ (ppm): δ, 22.4 (CH3), 44.0 (OCH2), 127.8 (C) 129.3 (CH), 129.9 (CH), 130.5 (CH), 131.5 (C), 133.3 (C), 136.5 (C), 142.0 (C), 151.0 (C), 182.4 (C=O). MS data: m/z (70 eV) 694, M+ (19%); 480, [M−205]+ (32%); 98, [M−587]+ (100%). Anal. Calcd for C38H44O8S2: C, C, 65.87; H, 6.40; S, 9.26; Found: C, 65.89; H, 6.59; S, 9.40.
3.7. Synthesis of 7,7′-(3,4-Diphenylthieno[2,3-b]thiophene-2,5-diyl)di-[1,2,4]triazolo[1,5-a] pyrimidine (6)
Compound 6 was prepared according to the following procedure: compound 1 (0.244 g, 0.5 mmol) was dissolved in DMF/EtOH (10 mL, 1:3) followed by addition of 3-amino-1H-1,2,4-triazole (0.084 g, 1 mmol). The mixture was then refluxed for 6 h and the solid that formed was filtered while hot to afford the desired compound which was recrystallized from DMF/EtOH to afford the title compound in a pure form as pale brown crystals; yield: 68%; mp: 349–351 °C. IR (cm−1): 1588. 1H-NMR (DMSO-d6) δ (ppm): 7.41–7.66 (m, 5H), 8.14 (d, J = 12.5 Hz, 1H), 8.67 (s, 1H), 8.96 (d, J = 12.5 Hz, 1H). 13C-NMR (DMSO-d6) δ (ppm): 115.4 (CH), 125.0 (C), 127.0 (C), 127.3 (CH), 129.1 (CH), 129.9 (CH), 130.2 (C), 132.8 (C), 135.8 (C), 152.8 (C), 154.4 (CH), 159.9 (CH), 162.9 (C). MS data: m/z (70 eV) 530, M+ (1%); 368, [M−162]+ (100%); 165, [M−165]+ (98%); 105, [M−425]+ (40%). Anal. Calcd for C28H16N8S2: C, 63.62; H, 3.05; N, 21.20; S, 12.13; Found: C, 63.63; H, 5.31; N, 21.30; S, 12.18.
3.8. Synthesis of 4,4′-(3,4-Diphenylthieno[2,3-b]thiophene-2,5diyl)bis(benzo[4,5]imidazo[1,2-a] pyrimidine) (7)
The title compound was prepared by the following procedure: Compound 1 (0.244 g, 0.5 mmol) was dissolved in ethanol (15 mL) followed by addition of 2-aminobenzimidazole (0.133 g, 1.0 mmol) in DMF/EtOH. The mixture was heated under reflux for 7 h and the solid product was filtered while hot to afford the desired product in a pure form as a white powder. Yield: 70%; mp: 248–350 °C. IR (cm−1): 1626. 1H-NMR (DMSO-d6): δ (ppm): 7.42–7.52 (m, 9H), 8.16 (d, J = 12.5 Hz, 1H), 8.80 (d, J = 12.5 Hz, 1H). 13C-NMR (DMSO-d6) δ (ppm): 112.1 (CH), 115.1 (CH), 122.6 (CH), 125 (C), 127.0 (C), 127.1 (CH), 128.1 (CH), 129.9 (CH), 131.3(C), 136.0 (C), 139.1 (C), 142.0, 148.2 (C), 148.4 (CH), 156 (C). MS data: m/z (70 eV) 622, M+ (25%); 567, [M−45]+ (56%); 439, [M−183]+ (55%); 183, [M−439]+ (100%). Anal. Calcd for C38H22N6S2: C, C, 72.82; H, 3.54; N, 13.41; S, 10.23. Found: C, 72.82; H, 3.54; N, 13.41; S, 10.23.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}