
Unfolded Protein Response and Macroautophagy in Alzheimer’s, Parkinson’s and Prion Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
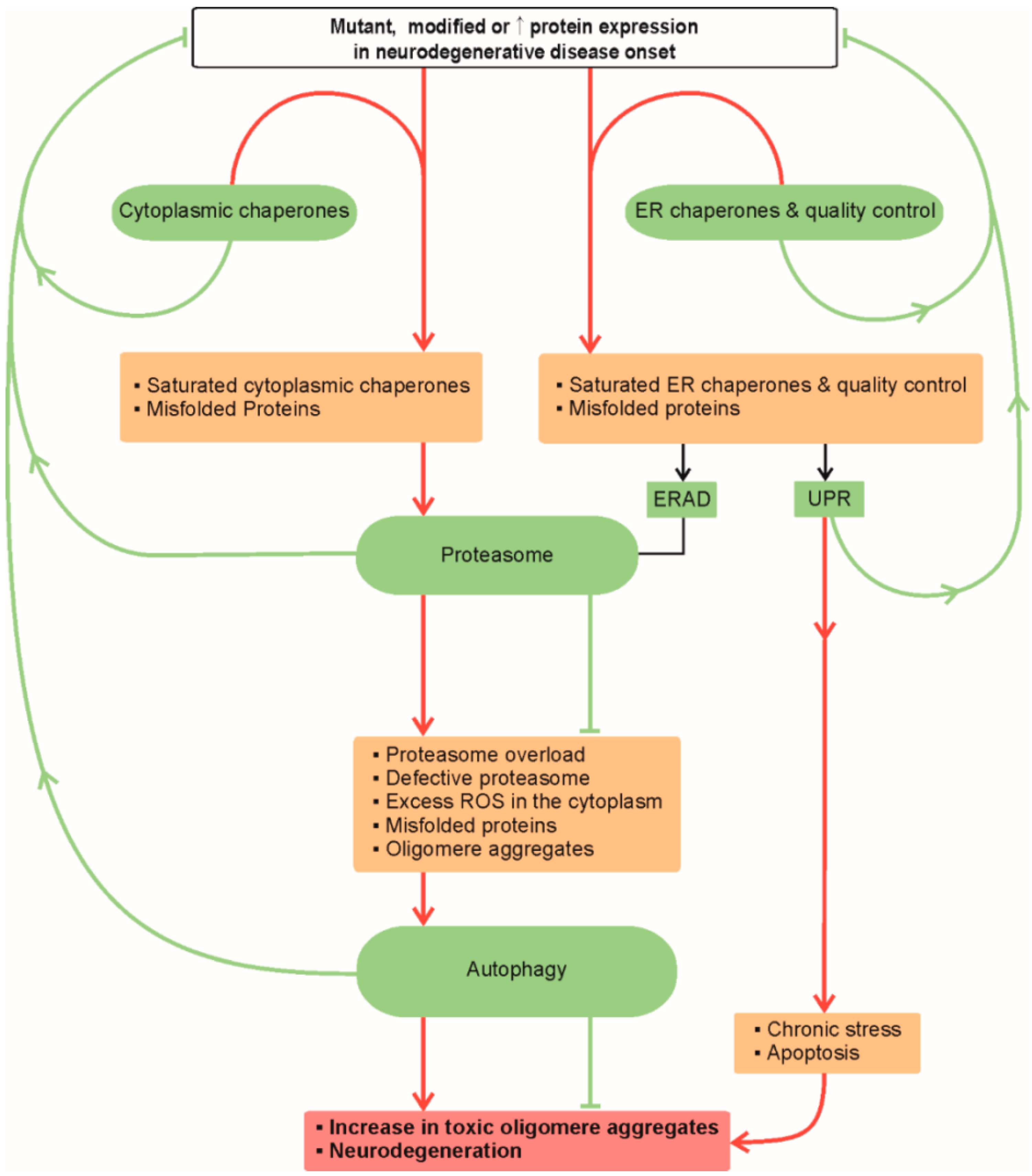
:1. Proteostasis in Neurodegeneration
1.1. Endoplasmic Reticulum
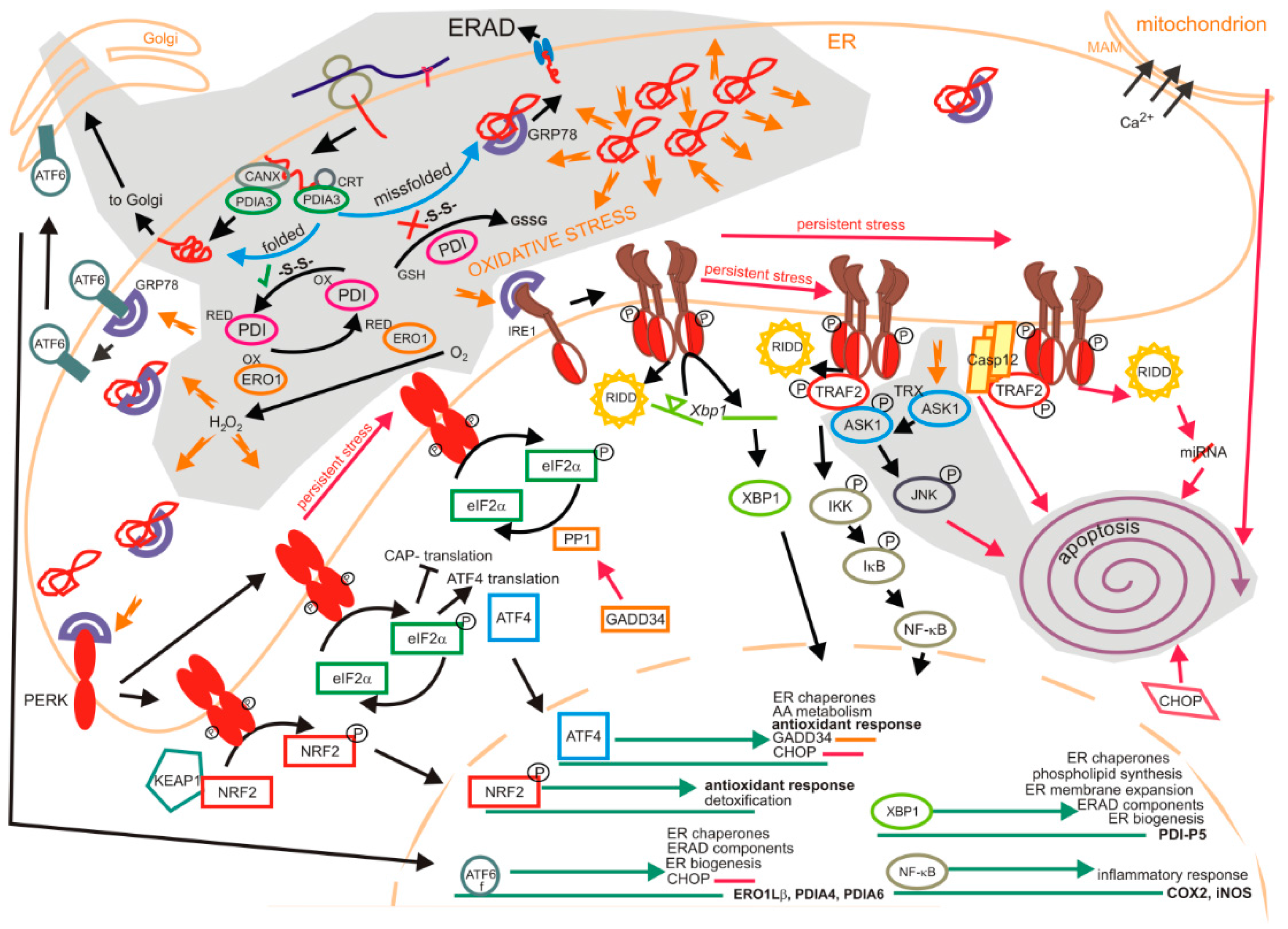
1.2. Unfolded Protein Response
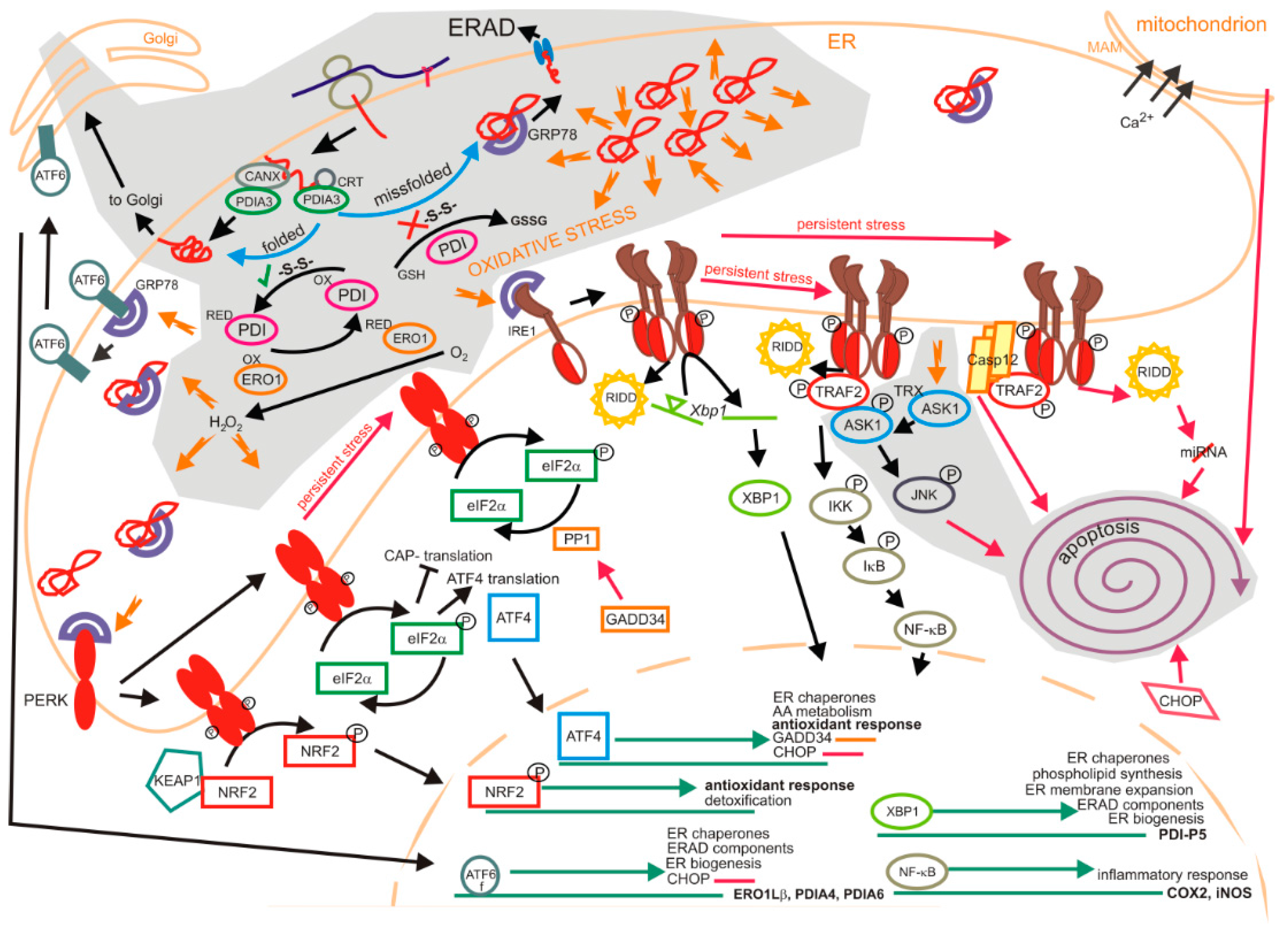
1.2.1. Impact of Oxidation on the Unfolded Protein Response
1.2.2. Degradation of Oxyproteins by the Unfolded Protein Response
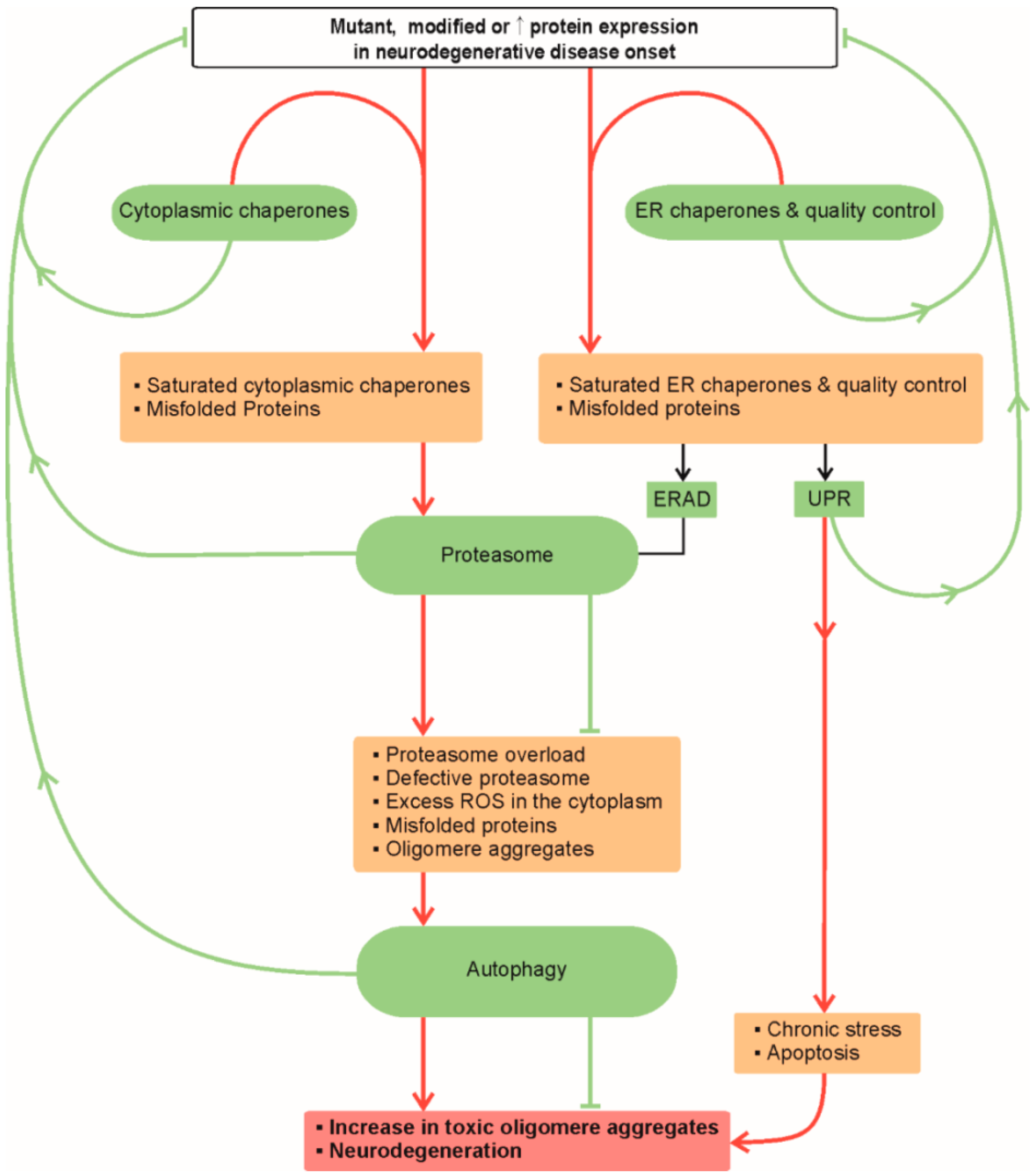
1.2.3. Unfolded Protein Response in Neurodegenerative Disorders
1.3. Macroautophagy
2. Alzheimer’s Disease
2.1. Amyloid β (Aβ) and Tau
- (a)
- Reducing Ca2+/Calmodulin-dependent protein kinase II synaptic distribution and thus decreasing the density of AMPA synaptic receptors [137];
- (b)
- Uncoupling metabotropic glutamate receptors’ (mGluR5) dependent activation of PKC [138];
- (c)
- (d)
- Binding with PrPc to N-methyl-D-aspartate receptors (NMDAR) and
- (e)
- Reducing glutamate reuptake thus promoting an increased NMDAR and mGluR5 mediated entry of Ca2+ [148].
2.2. Unfolded Protein Response in Alzheimer’s Disease
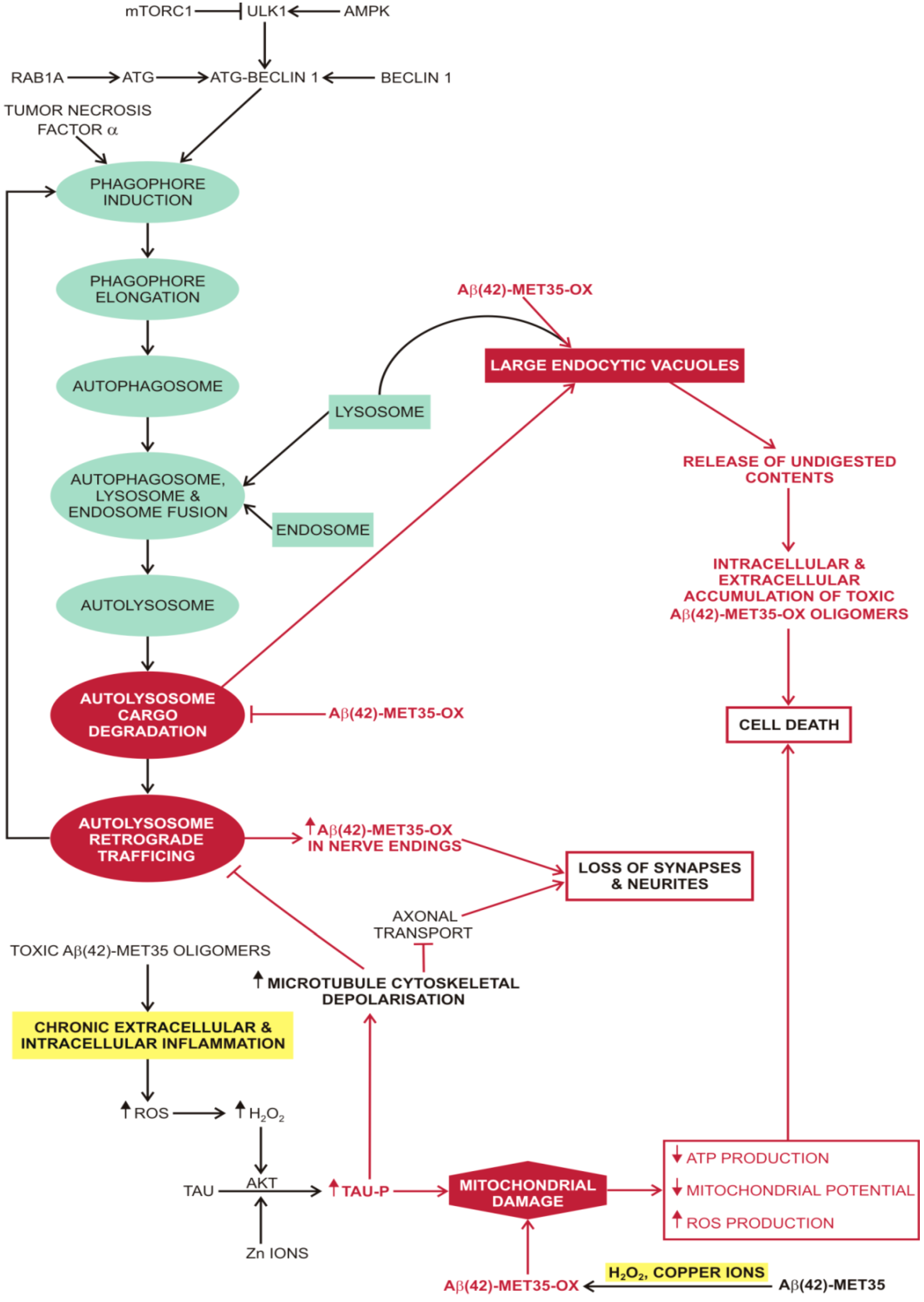
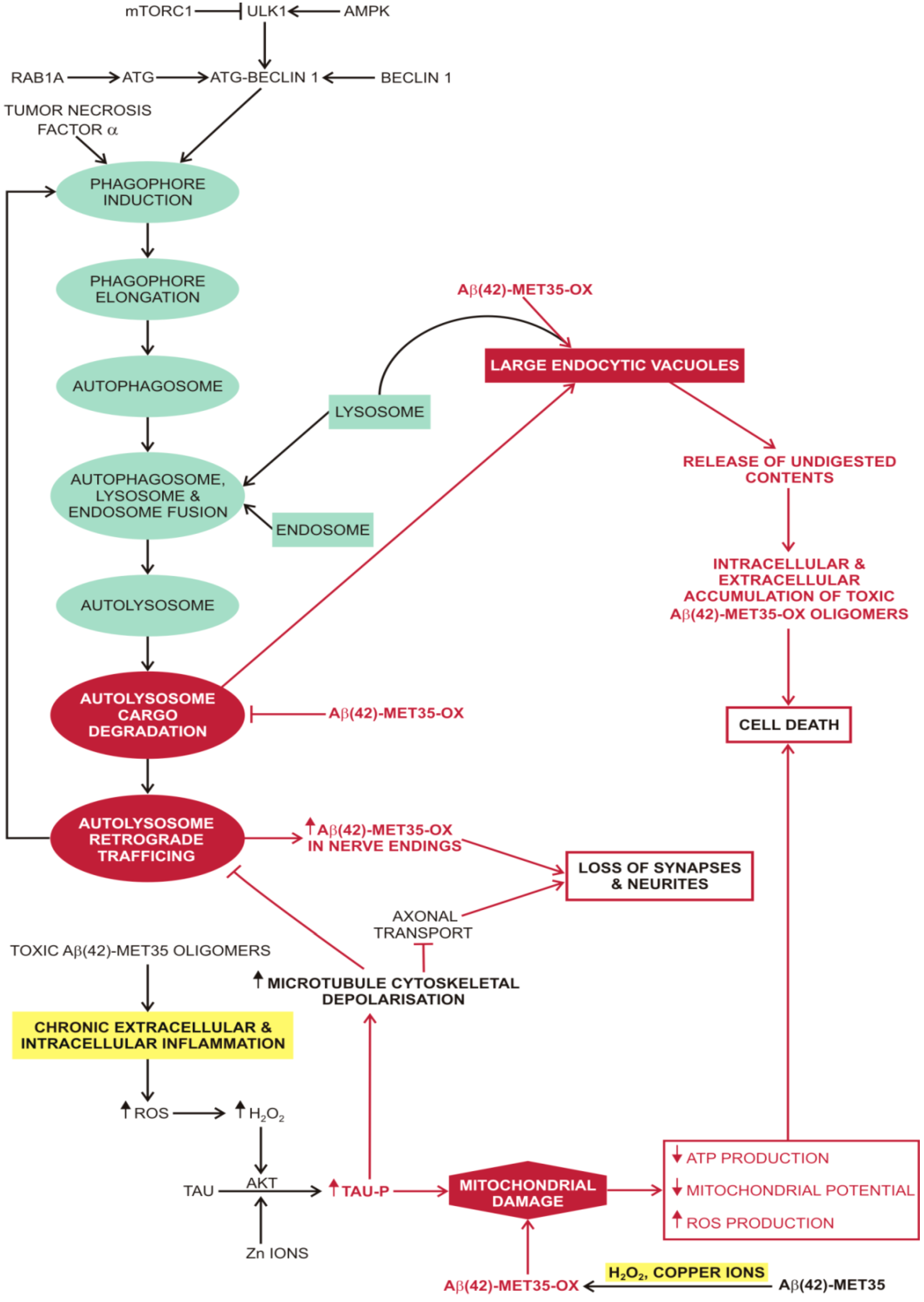
2.3. Macroautophagy in Alzheimer’s disease
3. Parkinson’s Disease
3.1. α-Synuclein (αSYN)
3.2. Unfolded Protein Response in Parkinson’s Disease
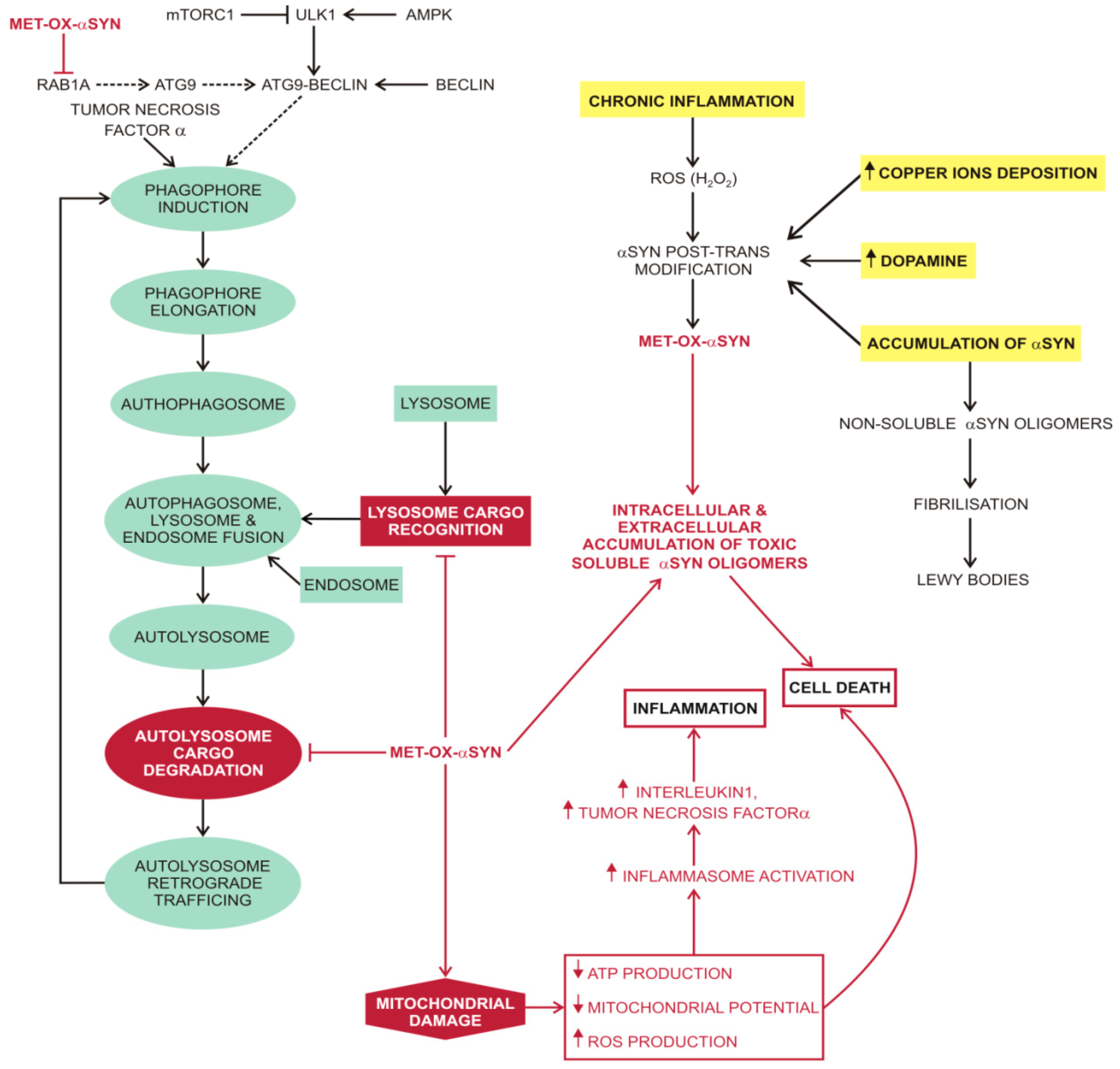
3.3. Macroautophagy in Parkinson’s Disease
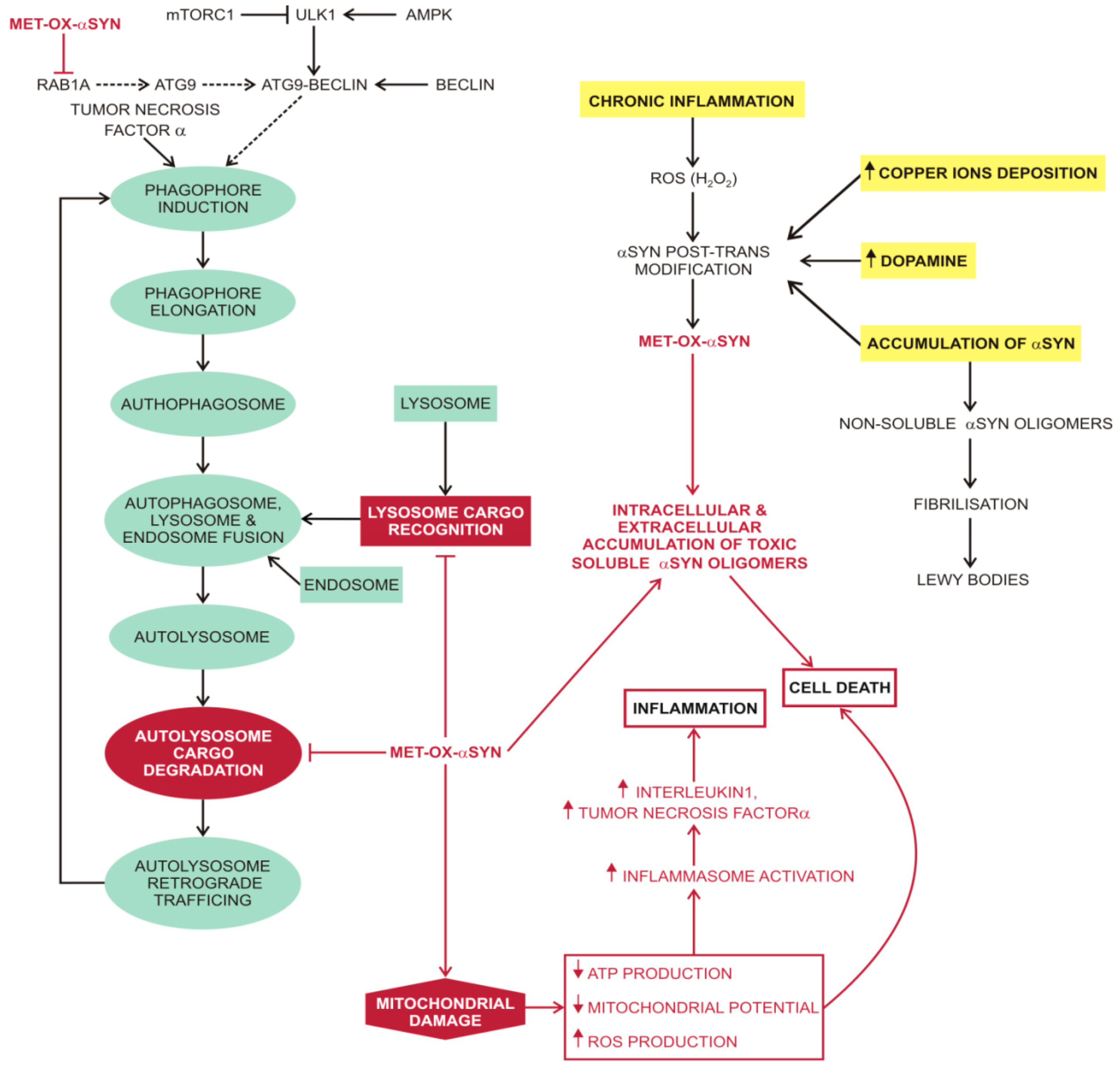
4. Prion Diseases
4.1. Normal Form of Prion Protein (PrPc)
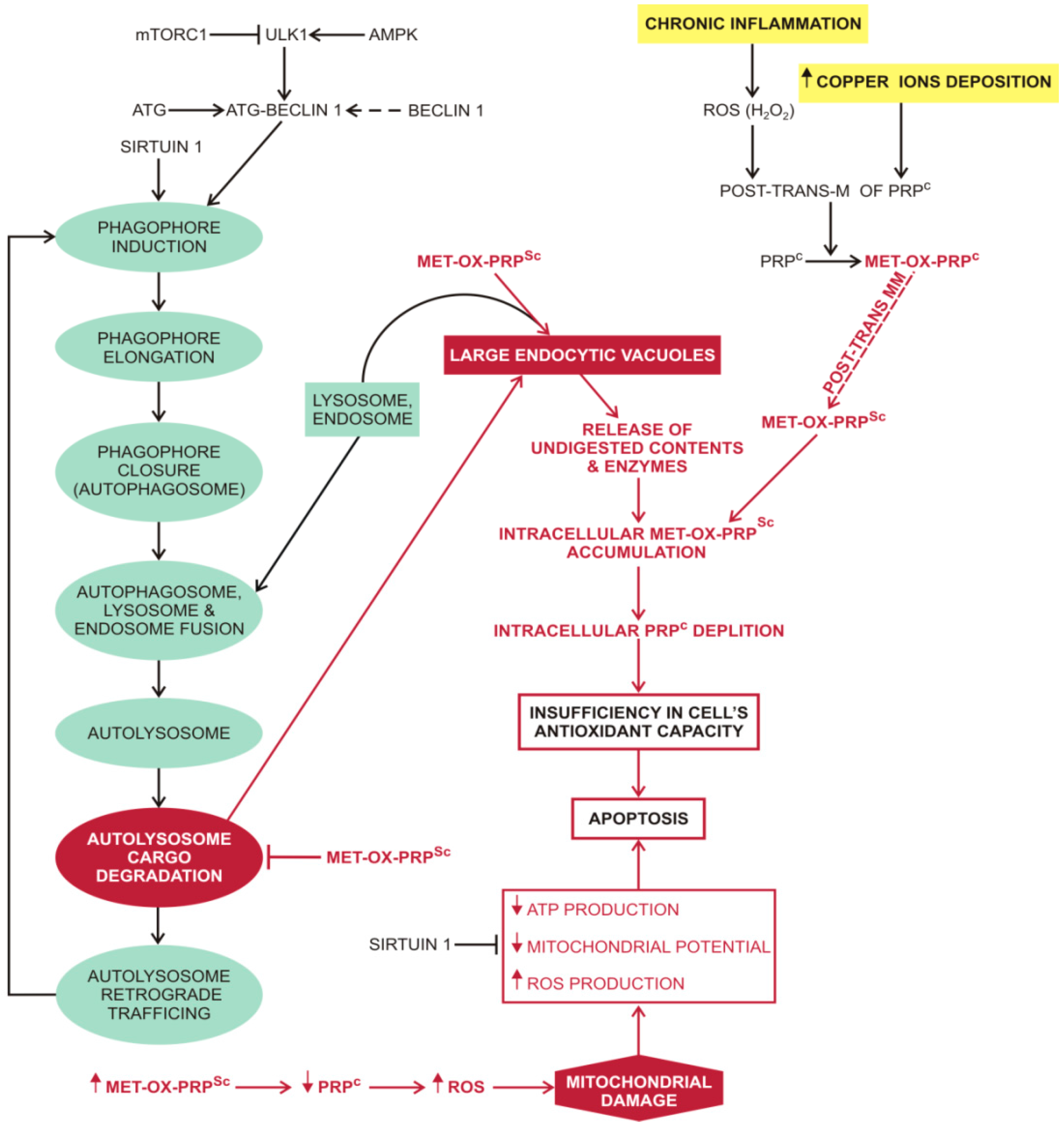
4.2. Unfolded Protein Response in Prion Diseases
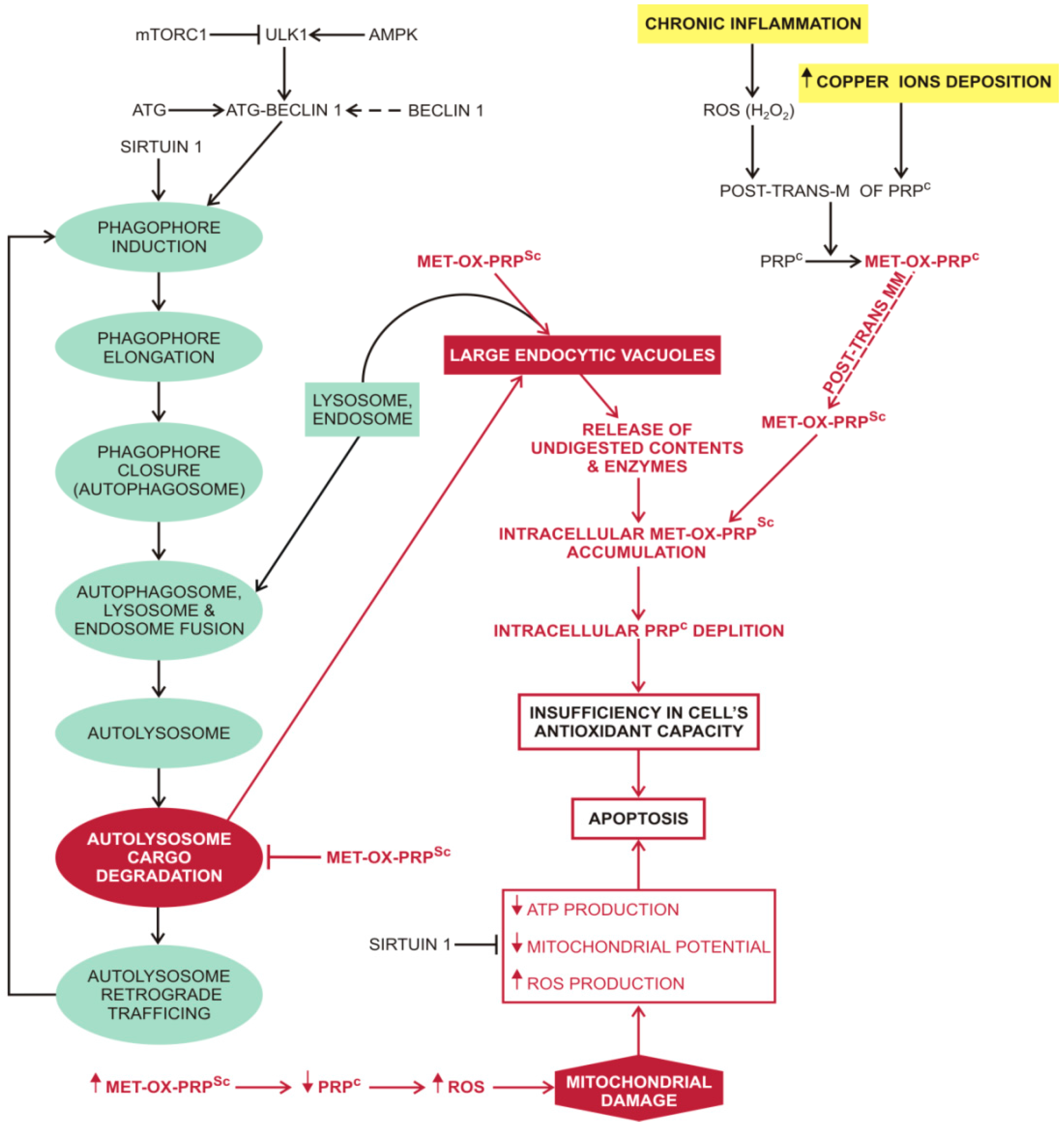
4.3. Macroautophagy in Prion Diseases
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef]
- Heiseke, A.; Aguib, Y.; Schatzl, H.M. Autophagy, prion infection and their mutual interactions. Curr. Issues Mol. Biol. 2010, 12, 87–97. [Google Scholar] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 904, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Szul, T.; Sztul, E. COPII and COPI traffic at the ER-Golgi interface. Physiology 2011, 26, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Böhm, K.J.; Winner, B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; Agostinis, P. PERK is required at the ER–mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; Patterson, R.L.; Snyder, S.H. Apoptosis and calcium: New roles for cytochrome c and inositol 1,4,5-trisphosphate. Cell Cycle 2004, 3, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Boehning, D.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H. A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc. Natl. Acad. Sci. USA 2005, 102, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Hoozemans, J.J. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [PubMed]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death—Mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Kitamura, A.; Kinjo, M.; Iwawaki, T. Direct association of unfolded proteins with mammalian ER stress sensor, IRE1β. PLoS ONE 2012, 7, e51290. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [PubMed]
- Deldicque, L. Endoplasmic reticulum stress in human skeletal muscle: Any contribution to sarcopenia? Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Dufey, E.; Sepúlveda, D.; Rojas-Rivera, D.; Hetz, C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am. J. Physiol. Cell Physiol. 2014, 307, C582–C594. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Alzheimer’s disease causation by copper toxicity and treatment with zinc. Front. Aging Neurosci. 2014, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional andmorphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [PubMed]
- Ameri, K.; Harris, A.L. Activating transcription factor 4. Int. J. Biochem. Cell Biol. 2008, 40, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C. Crosstalk between endoplasmic reticulum stress and protein misfolding in neurodegenerative diseases. ISRN Cell Biol. 2013, 2013, 22. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Averous, J.; Bruhat, A.; Jousse, C.; Carraro, V.; Thiel, G.; Fafournoux, P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J. Biol. Chem. 2004, 279, 5288–5297. [Google Scholar] [CrossRef] [PubMed]
- Nguyên, D.T.; Kebache, S.; Fazel, A.; Wong, H.N.; Jenna, S.; Emadali, A.; Lee, E.H.; Bergeron, J.J.; Kaufman, R.J.; Larose, L. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol. Biol. Cell 2004, 15, 4248–4260. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Lynes, E.M.; Gesson, K.; Thomas, G. Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2010, 1798, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Satpute-Krishnan, P.; Ajinkya, M.; Bhat, S.; Itakura, E.; Hegde, R.S.; Lippincott-Schwartz, J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 2014, 158, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Couve, A.; Hetz, C. RESETing ER proteostasis: Selective stress pathway hidden in the secretory route. EMBO J. 2014, 33, 2444–2446. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Chen, Q.; Tay, A.S.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Horelli-Kuitunen, N.; Helaakoski, T.; Karvonen, P.; Jaakkola, M.; Palotie, A.; Kivirikko, K.I. Structures of the human gene for the protein disulfide isomerase-related polypeptide ERp60 and a processed gene and assignment of these genes to 15q15 and 1q21. Genomics 1997, 42, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Higa, A.; Chevet, E. Redox signaling loops in the unfolded protein response. Cell Signal. 2012, 24, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5, a013185. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta 2008, 1783, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Benham, A.M. The protein disulfide isomerase family: Key players in health and disease. Antioxid. Redox Signal. 2012, 16, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Pollock, S.; Kozlov, G.; Pelletier, M.F.; Trempe, J.F.; Jansen, G.; Sitnikov, D.; Bergeron, J.J.; Gehring, K.; Ekiel, I.; Thomas, D.Y. Specific interaction of ERp57 and calnexin determined by NMR spectroscopy and an ER two-hybrid system. EMBO J. 2004, 23, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Bridges, J.P.; Apsley, K.; Xu, Y.; Weaver, T.E. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol. Biol. Cell 2008, 19, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Tavender, T.J.; Bulleid, N.J. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 2010, 123, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Ramming, T.; Appenzeller-Herzog, C. The physiological functions of mammalian endoplasmic oxidoreductin 1: On disulfides and more. Antioxid. Redox Signal. 2012, 16, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Molinari, M.; Calanca, V.; Galli, C.; Lucca, P.; Paganetti, P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 2003, 299, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Nijholt, D.A.; de Graaf, T.R.; van Haastert, E.S.; Oliveira, A.O.; Berkers, C.R.; Zwart, R.; Ovaa, H.; Baas, F.; Hoozemans, J.J.; Scheper, W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: Implications for Alzheimer’s disease. Cell Death Differ. 2011, 18, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Elfrink, H.L.; Zwart, R.; Baas, F.; Scheper, W. Inhibition of endoplasmic reticulum associated degradation reduces endoplasmic reticulum stress and alters lysosomal morphology and distribution. Mol. Cells 2013, 35, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kaniuk, N.A.; Kiraly, M.; Bates, H.; Vranic, M.; Volchuk, A.; Brumell, J.H. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007, 56, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A. How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol. Biol. Cell 1994, 5, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Groisman, B.; Shenkman, M.; Ron, E.; Lederkremer, G.Z. Mannose Trimming Is Required for Delivery of a Glycoprotein from EDEM1 to XTP3-B and to Late Endoplasmic Reticulum-associated Degradation Steps. J. Biol. Chem. 2011, 286, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Lederkremer, G.Z.; Glickman, M.H. A window of opportunity: Timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem. Sci. 2005, 30, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Nagata, K. Protein Folding and Quality Control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Maegawa, K.; Suzuki, M.; Ushioda, R.; Araki, K.; Matsumoto, Y.; Hoseki, J.; Nagata, K.; Inaba, K. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 2011, 41, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, R.; Galli, C.; Calanca, V.; Nakajima, T.; Molinari, M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 2010, 188, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Riemer, J.; Hansen, H.G.; Appenzeller-Herzog, C.; Johansson, L.; Ellgaard, L. Identification of the PDIfamily member ERp90 as an interaction partner of ERFAD. PLoS ONE 2011, 6, e17037. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Ron, E.; Shenkman, M.; Groisman, B.; Izenshtein, Y.; Leitman, J.; Lederkremer, G.Z. Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol. Biol. Cell 2011, 22, 3945–3954. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kunz, J.B.; Schwarz, H.; Mayer, A. Determination of four sequential stages during microautophagy in vitro. J. Biol. Chem. 2004, 279, 9987–9996. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cuervo, A.M.; Dunn, W.A., Jr.; Levine, B.; van der Klei, I.; Seglen, P.O. How shall I eat thee? Autophagy 2007, 3, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Huang, W.-P.; Stromhaug, P.E.; Klionsky, D.J. Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev. Cell 2002, 3, 825–837. [Google Scholar] [CrossRef]
- Kanki, T.; Wang, K.; Cao, Y.; Baba, M.; Klionsky, D.J. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev. Cell 2009, 17, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, M.U.; Klionsky, D.J. Vacuolar localization of oligomeric a-mannosidase requires the cytoplasm to vacuole targeting and autophagy pathway components in Saccharomyces cerevisiae. J. Biol. Chem. 2001, 276, 20491–20498. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Cueva, R.; Yaver, D.S. Aminopeptidase I of Saccharomyces cerevisiae is localized to the vacuole independent of the secretory pathway. J. Cell Biol. 1992, 119, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Yuga, M.; Gomi, K.; Klionsky, D.J.; Shintani, T. Aspartyl aminopeptidase is imported from the cytoplasm to the vacuole by selective autophagy in Saccharomyces cerevisiae. J. Biol. Chem. 2011, 286, 13704–13713. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immunoelectron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.; Larsen, K.E.; Behr, G.G.; Romero, N.; Przedborski, S.; Brundin, P.; Sulzer, D. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum. Mol. Genet. 2001, 10, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTORindependent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schatzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schatzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in communitydwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Bennett, D.A.; Boyle, P.A.; Leurgans, S.; Schneider, J.A. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA 2012, 307, 1798–800. [Google Scholar] [CrossRef] [PubMed]
- Davidson, Y.S.; Raby, S.; Foulds, P.G.; Robinson, A.; Thompson, J.C.; Sikkink, S.; Yusuf, I.; Amin, H.; DuPlessis, D.; Troakes, C.; et al. TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: Association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol. 2011, 122, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Toledo, J.B.; Cairns, N.J.; Da, X.; Chen, K.; Carter, D.; Fleisher, A.; Householder, E.; Ayutyanont, N.; Roontiva, A.; Bauer, R.J.; et al. Clinical and multimodal biomarker correlates of ADNI neuropathological findings. Acta Neuropathol. Commun. 2013, 1. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Attems, J. Prevalence and pathology of dementia with Lewy bodies in the oldest old: A comparison with other dementing disorders. Dement. Geriatr. Cogn. Disord. 2011, 31, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.A.; Masliah, E.; Galasko, D.; Terry, R.D. Plaque-only Alzheimer disease is usually the lewy body variant, and vice versa. J. Neuropathol. Exp. Neurol. 1993, 52, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Leurgans, S.E.; Bennett, D.A. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 2009, 66, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Olichney, J.M.; Galasko, D.; Salmon, D.P.; Hofstetter, C.R.; Hansen, L.A.; Katzman, R.; Thal, L.J. Cognitive decline is faster in Lewybody variant than in Alzheimer’s disease. Neurology 1998, 51, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Mathis, C.A.; Mason, N.S.; Lopresti, B.J.; Klunk, W.E. Development of positron emission tomography beta-amyloid plaque imaging agents. Semin. Nucl. Med. 2012, 42, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Jagust, W. Time for tau. Brain 2014, 137, 1570–1571. [Google Scholar] [CrossRef] [PubMed]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of beta-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Xiong, C.; Jasielec, M.S.; Bateman, R.J.; Goate, A.M.; Benzinger, T.L.; Ghetti, B.; Martins, R.N.; Masters, C.L.; Mayeux, R.; et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Takwoingi, Y.; Flicker, L.; Mason, S.E.; McShane, R. Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2014, 6. [Google Scholar]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer’s disease a systemic disease? Biochim. Biophys. Acta 2014, 842, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.X.; Manly, J.J.; Mayeux, R. Relation of diabetes tomild cognitive impairment. Arch. Neurol. 2007, 64, 570–575. [Google Scholar]
- Xu, W.; Qiu, C.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Mid- and late-life diabetes in relation to the risk of dementia: A population-based twin study. Diabetes 2009, 58, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Holtzman, D.M.; Fagan, A.M.; Shaw, L.M.; Perrin, R.; Arnold, S.E.; Grossman, M.; Xiong, C.; Craig-Schapiro, R.; Clark, C.M.; et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 2012, 79, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Tan, D.X.; Manchester, L.; Reiter, R.J. Diabetes and Alzheimer disease, two overlapping pathologies with the same background: Oxidative stress. Oxid. Med. Cell Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Choi, J.K.; Surewicz, K.; Surewicz, W.K. Soluble Prion Protein Binds Isolated Low Molecular Weight Amyloid-β Oligomers Causing Cytotoxicity Inhibition. ACS Chem. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef] [PubMed]
- Barger, S.W.; Harmon, A.D. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 1997, 388, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Dodart, J.C.; Mathis, C.; Little, S.; Clemens, J.; Paul, S.M.; Ungerer, A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12683–12688. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Hussain, I.; Karran, E.H.; Turner, A.J.; Hooper, N.M. Characterization of detergent-insoluble complexes containing the familial Alzheimer’s disease-associated presenilins. J. Neurochem. 1999, 72, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Turner, A.J.; Hooper, N.M. Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem. J. 1999, 344, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, G.L.; Gandy, S.E.; Buxbaum, J.D.; Ramabhadran, T.V.; Greengard, P. Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1992, 89, 3055–3059. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Trusko, S.P.; Howland, D.S.; Pinsker, L.R.; Mistretta, S.; Reaume, A.G.; Greenberg, B.D.; Siman, R.; Scott, R.W. Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci. 1998, 18, 1743–1752. [Google Scholar] [PubMed]
- Van Nostrand, W.E.; Wagner, S.L.; Shankle, W.D.; Farrow, J.S.; Dick, M.; Rozemuller, J.M.; Kuiper, M.A.; Wolters, E.C.; Zimmerman, J.; Cotman, C.W.; et al. Decreased levels of soluble amyloid beta-protein precursor in cerebrospinal fluid of live Alzheimer disease patients. Proc. Natl. Acad. Sci. USA 1992, 89, 2552–2555. [Google Scholar] [CrossRef]
- Lannfelt, L.; Basun, H.; Wahlund, L.O.; Rowe, B.A.; Wagner, S.L. Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimer’s disease. Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimer’s disease. Nat. Med. 1995, 1, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Hooper, N.M. Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem. Soc. Trans. 2005, 33, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Schellenberg, G.D.; Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, N.; Tomita, T.; Hayashi, I.; Tsuruoka, M.; Niimura, M.; Takahashi, Y.; Thinakaran, G.; Iwatsubo, T. The role of presenilin cofactors in the γ-secratase complex. Nature 2003, 422, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin I variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Funk, K.E.; Kuret, J. Lysosomal fusion dysfunction as a unifying hypothesis for Alzheimer’s disease pathology. Int. J. Alzheimers Dis. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, W.; Yan, Z. β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 2009, 284, 1063–10649. [Google Scholar] [CrossRef] [PubMed]
- Tyszkiewicz, J.P.; Yan, Z. β-Amyloid peptides impair PKC-dependent functions of metabotropic glutamate receptors in prefrontal cortical neurons. J. Neurophysiol. 2005, 93, 3102–3111. [Google Scholar] [CrossRef] [PubMed]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Stephen, M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-B oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Laurén, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived Amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2010, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Cissé, M.; Sanchez, P.E.; Kim, D.H.; Ho, K.; Yu, G.Q.; Mucke, L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J. Neurosci. 2011, 31, 10427–10431. [Google Scholar] [CrossRef] [PubMed]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Müller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Freir, D.B.; Nicoll, A.J.; Klyubin, I.; Panico, S.; Mc Donald, J.M.; Risse, E.; Asante, E.A.; Farrow, M.A.; Sessions, R.B.; Saibil, H.R.; et al. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Zamponi, G.W.; Ferguson, S.S. Glutamate receptors function as scaffolds for the regulation of β-amyloid and cellular prion protein signaling complexes. Mol. Brain 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; You, H.; Zamponi, G.W. Copper-dependent regulation of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012, 590, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J. Clin. Investig. 2005, 115, 1121–1129. [Google Scholar] [PubMed]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Galvan, V.; Lange, M.B.; Tang, H.; Sowell, R.A.; Spilman, P.; Fombonne, J.; Gorostiza, O.; Zhang, J.; Sultana, R.; et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic. Biol. Med. 2010, 48, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Al-Hilaly, Y.K.; Williams, T.L.; Stewart-Parker, M.; Ford, L.; Skaria, E.; Cole, M.; Bucher, W.G.; Morris, K.L.; Sada, A.A.; Thorpe, J.R.; et al. A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 83. [Google Scholar] [CrossRef] [PubMed]
- Drewes, G.; Ebneth, A.; Mandelkow, E.M. MAPs, MARKs and microtubule dynamics. Trends Biochem. Sci. 1998, 23, 307–311. [Google Scholar] [CrossRef]
- Kosik, K.S.; McConlogue, L. Microtubule-associated protein function: Lessons from expression in Spodoptera frugiperda cells. Cell Motil. Cytoskeleton. 1994, 28, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Illenberger, S.; Zheng-Fischhöfer, Q.; Preuss, U.; Stamer, K.; Baumann, K.; Trinczek, B.; Biernat, J.; Godemann, R.; Mandelkow, E.M.; Mandelkow, E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: Implications for Alzheimer’s disease. Mol. Biol. Cell 1998, 9, 1495–1512. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Biernat, J.; Drewes, G.; Gustke, N.; Trinczek, B.; Mandelkow, E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol. Aging 1995, 16, 355–362. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Lee, V.M. Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: Focusing on phosphatases. FASEB J. 1995, 9, 1570–1576. [Google Scholar] [PubMed]
- Delacourte, A.; Buée, L. Normal and pathological Tau proteins as factors for microtubule assembly. Int. Rev. Cytol. 1997, 171, 167–224. [Google Scholar] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [PubMed]
- Yan, S.D.; Chen, X.; Schmidt, A.M.; Brett, J.; Godman, G.; Zou, Y.S.; Scott, C.W.; Caputo, C.; Frappier, T.; Smith, M.A.; et al. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Marzloff, K.; Arriagada, P.V. The lack of accumulation of senile plaques or amyloid burden in Alzheimer’s disease suggests a dynamic balance between amyloid deposition and resolution. J. Neuropathol. Exp. Neurol. 1993, 52, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Ishiki, A.; Okamura, N.; Furukawa, K.; Furumoto, S.; Harada, R.; Tomita, N.; Hiraoka, K.; Watanuki, S.; Ishikawa, Y.; Tago, T.; et al. Longitudinal Assessment of Tau Pathology in Patients with Alzheimer’s Disease Using [18F]THK-5117 Positron Emission Tomography. PLoS ONE 2015, 10, e0140311. [Google Scholar] [CrossRef] [PubMed]
- Plácido, A.I.; Pereira, C.M.; Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Oliveira, C.R.; Moreira, P.I. The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: Implications for Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Wong, A.K.; Ng, H.K.; Hugon, J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 2002, 13, 2429–2432. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, L. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Honjo, Y.; Ito, H.; Horibe, T.; Takahashi, R.; Kawakami, K. Protein disulfide isomerase-immunopositive inclusions in patients with Alzheimer disease. Brain Res. 2010, 1349, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Veerhuis, R.; van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Hamos, J.E.; Oblas, B.; Pulaski-Salo, D.; Welch, W.J.; Bole, D.G.; Drachman, D.A. Expression of heat shock proteins in Alzheimer’s disease. Neurology 1991, 41, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Unterberger, U.; Hoftberger, R.; Gelpi, E.; Flicker, H.; Budka, H.; Voigtlander, T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J. Neuropathol. Exp. Neurol. 2006, 65, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.O.; Ferreiro, E.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M. ER stressmediated apoptotic pathway induced by Abeta peptide requires the presence of functional mitochondria. J. Alzheimers Dis. 2010, 20, 625–636. [Google Scholar] [PubMed]
- Resende, R.; Ferreiro, E.; Pereira, C.; Oliveira, C.R. ER stress is involved in Abetainduced GSK-3beta activation and tau phosphorylation. J. Neurosci. Res. 2008, 86, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lee, H.; Kam, T.I.; Tai, M.L.; Lee, J.Y.; Noh, J.Y.; Shim, S.M.; Seo, S.J.; Kong, Y.Y.; Nakagawa, T.; et al. E2–25K/Hip-2 regulates caspase-12 in ER stress-mediated Abeta neurotoxicity. J. Cell Biol. 2008, 182, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Milhavet, O.; Martindale, J.L.; Camandola, S.; Chan, S.L.; Gary, D.S.; Cheng, A.; Holbrook, N.J.; Mattson, M.P. Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations. J. Neurochem. 2002, 83, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Raza, S.; Prasanthi, J.R.; Ghribi, O. Gadd153 and NF-kappaB crosstalk regulates 27-hydroxycholesterol-induced increase in BACE1 and beta-amyloid production in human neuroblastoma SH-SY5Y cells. PLoS ONE 2013, 8, e70773. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.L.; Shen, Y.X.; Zhu, H.Y.; Sun, H.; Yan, X.B.; Fang, H.; Zhou, J.N. Alterations of hHrd1 expression are related to hyperphosphorylated tau in the hippocampus in Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Van der Harg, J.M.; Nölle, A.; Zwart, R.; Boerema, A.S.; van Haastert, E.S.; Strijkstra, A.M.; Hoozemans, J.J.; Scheper, W. The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis. 2014, 5, e1393. [Google Scholar] [CrossRef] [PubMed]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., 3rd; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.G.; Qureshi, Y.H.; Yu, W.H. Promoting Autophagic Clearance: Viable Therapeutic Targets in Alzheimer’s Disease. Neurotherapeutics 2014, 12, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, S.; Xiong, J.; Li, Y.; Qu, L. Luteolin reduces zinc-induced tau phosphorylation at Ser262/356 in an ROS-dependent manner in SH-SY5Y cells. Biol. Trace Elem. Res. 2012, 149, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March separate, strike together--role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Maurer, I.; Zierz, S.; Moller, H.J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Kokhan, V.S.; Afanasyeva, M.A.; Van’kin, G.I. α-Synuclein knockout mice have cognitive impairments. Behav. Brain Res. 2012, 231, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Yu, L.; Boyle, P.A.; Leurgans, S.E.; Bennett, D.A. Cognitive impairment, decline and fluctuations in older community-dwelling subjectswith Lewy bodies. Brain 2012, 135, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S. Ubiquitination of alpha-synuclein and autophagy in Parkinson’s disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, T.; Mattila, P.; Davies, P.; Wang, D.; Dickson, D.W. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J. Neuropathol. Exp. Neurol. 2003, 62, 389–397. [Google Scholar] [PubMed]
- Arima, K.; Hirai, S.; Sunohara, N.; Aoto, K.; Izumiyama, Y.; Uéda, K.; Ikeda, K.; Kawai, M.; Arima, K.; Hirai, S.; et al. Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in Lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res. 1999, 843, 53–61. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in a-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.L.; Nemani, V.M.; Voglmaier, S.M.; Anthony, M.D.; Ryan, T.A.; Edwards, R.H. Neural activity controls the synaptic accumulation of a-synuclein. J. Neurosci. 2005, 25, 10913–10921. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, X.; Liu, G.; Han, J.; Zhang, C.; Li, Y.; Xu, S.; Liu, C.; Gao, Y.; Yang, H.; et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 2007, 145, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Uéda, K.; Chan, P.; Yu, S. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Alim, M.A.; Hossain, M.S.; Arima, K.; Takeda, K.; Izumiyama, Y.; Nakamura, M.; Kaji, H.; Shinoda, T.; Hisanaga, S.; Ueda, K. Tubulin seeds alpha-synuclein fibril formation. J. Biol. Chem. 2002, 277, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Alim, M.A.; Ma, Q.L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, M.; Asada, A.; Saito, T.; Kaji, H.; Yoshii, M. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 2004, 6, 435–442. [Google Scholar] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified a-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduíno, D.M.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Microtubule depolymerization potentiates α-synuclein oligomerization. Front. Aging Neurosci. 2010, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jung, W.; Lee, I.H.; Bhak, G.; Paik, S.R.; Hahn, J.S. Impairment of microtubule system increases α-synuclein aggregation and toxicity. Biochem. Biophys. Res. Commun. 2008, 365, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Suzuki, Y.; Yazawa, I. Microtubule depolymerization suppresses α-synuclein accumulation in a mouse model of multiple system atrophy. Am. J. Pathol. 2009, 174, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Suzuki, Y.; Yazawa, I. Binding of neuronal α-synuclein to β-III tubulin and accumulation in a model of multiple system atrophy. Biochem. Biophys. Res. Commun. 2012, 417, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.M.; Huang, Y.X.; Li, X.L.; Chen, C.; Shi, Q.; Wang, G.R.; Tian, C.; Wang, Z.Y.; Jing, Y.Y.; Gao, C.; Dong, X.P. Molecular interaction of α-synuclein with tubulin influences on the polymerization of microtubule in vitro and structure of microtubule in cells. Mol. Biol. Rep. 2010, 37, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, V.R.; Spinelli, K.J.; Weston, L.J.; Luk, K.C.; Woltjer, R.L.; Unni, V.K. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.E. Prions and prion-like proteins. J. Biol. Chem. 2014, 289, 19839–19840. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Spillantini, M.G. Physiological and pathological properties of alpha-synuclein. Cell Mol. Life Sci. 2007, 64, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann. Neurol. 2008, 63, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Hashida, K.; Kitao, Y.; Sudo, H.; Awa, Y.; Maeda, S.; Mori, K.; Takahashi, R.; Iinuma, M.; Hori, O. ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson’s disease. PLoS ONE 2012, 7, e47950. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Yamamoto, K.; Inoue, H.; Hikawa, R.; Nishi, K.; Mori, K.; Takahashi, R. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem. 2011, 286, 7947–7957. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; Forcelli, P.A.; Delannoy, M.; Oaks, A.W.; Permaul, E.; Berry, D.L.; Duka, V.; Wills, J.; Sidhu, A. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol. Dis. 2015, 76, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; van Haastert, E.S.; Eikelenboom, P.; de Vos, R.A.; Rozemuller, J.M.; Scheper, W. Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Makioka, K.; Yamazaki, T.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Involvement of endoplasmic reticulum stress defined by activated unfolded protein response in multiple system atrophy. J. Neurol. Sci. 2010, 297, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Hokenson, M.J.; Uversky, V.N.; Goers, J.; Yamin, G.; Munishkina, L.A.; Fink, A.L. Role of individual methionines in the fibrillation of methionine-oxidized alphasynuclein. Biochemistry 2004, 43, 4621–4633. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.L.; Pham, C.L.; Galatis, D.; Fodero-Tavoletti, M.T.; Perez, K.; Hill, A.F.; Masters, C.L.; Ali, F.E.; Barnham, K.J.; Cappai, R. Formation of dopamine-mediated alpha-synuclein-soluble oligomers requires methionine oxidation. Free Radic. Biol. Med. 2009, 46, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 2002, 22, 7006–7015. [Google Scholar] [PubMed]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W., II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Mitsui, T.; Kunishige, M.; Shono, M.; Akaike, M.; Azuma, H.; Matsumoto, T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 2006, 15, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, O.; Fischer, H.; Hasegawa, T.; Maisel, M.; Leitner, P.; Miesel, F.; Sharma, M.; Bornemann, A.; Berg, D.; Gasser, T.; Patenge, N. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009, 18, 3832–3850. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Narendra, D.P.; Tanaka, A.; Manfredi, G.; Youle, R.J. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11835–11840. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [PubMed]
- van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; de Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed]
- Hickson-Bick, D.L.; Jones, C.; Buja, L.M. Stimulation of mitochondrial biogenesis and autophagy by lipopolysaccharide in the neonatal rat cardiomyocyte protects against programmed cell death. J. Mol. Cell Cardiol. 2008, 44, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Zhu, J.; Kulich, S.M.; Chu, C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: Implications for Parkinson’s disease. Autophagy 2008, 4, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, A.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Feng, Z.; Liu, J.; Shen, W.; Wang, Y.; Wertz, K.; Weber, P.; Long, J. Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: Ameliorating effects of (−)-epigallocatechin-3-gallate. J. Nutr. Biochem. 2012, 23, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Horbinski, C.; Guo, F.; Watkins, S.; Uchiyama, Y.; Chu, C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4- phenylpyridinium-induced cell death. Am. J. Pathol. 2007, 170, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Budka, H. Prion diseases: From protein to cell pathology. Am. J. Pathol. 2008, 172, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Cuille, J.; Chelle, P.L. Pathologie animale, la maladie dite tremblant du mouton est-elle inoculable. Compt. Rend. Acad. Sci. 1936, 203, 1552–1554. [Google Scholar]
- Dickinson, A.G. Scrapie in sheep and goats. Front. Biol. 1976, 44, 209–241. [Google Scholar] [PubMed]
- Wells, G.A.; Scott, A.; Johnson, C.; Gunning, R.; Hancock, R.; Jeffrey, M.; Dawson, M.; Bradley, R. A novel progressive spongiform encephalopathy in cattle. Vet. Rec. 1987, 121, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.S.; Young, S. Spongiform encephalopathy of Rocky Mountain elk. J. Wildl. Dis. 1982, 18, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Liberski, P.P.; Guiroy, D.C.; Williams, E.S.; Walis, A.; Budka, H. Deposition patterns of disease-associated prion protein in captive mule deer brains with chronic wasting disease. Acta Neuropathol. 2001, 102, 496–500. [Google Scholar] [PubMed]
- Wyatt, J.M.; Pearson, G.R.; Smerdon, T.N.; Gruffydd-Jones, T.J.; Wells, G.; Wilesmith, J.W. Naturally occurring scrapie-like spongiform encephalopathy in five domestic cats. Vet. Rec. 1991, 129, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Gajdusek, D.C.; Gibbs, C.J.; Alpers, M. Experimental transmission of a Kuru-like syndrome to chimpanzees. Nature 1966, 209, 794–796. [Google Scholar] [CrossRef] [PubMed]
- Liberski, P.P.; Gajdusek, D.C. Kuru: Forth years later, a historical note. Brain Pathol. 1997, 7, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C., Jr.; Gajdusek, D.C.; Asher, D.M.; Alpers, M.; Beck, E.; Daniel, P.M.; Matthews, W.B. Creutzfeldt-Jakob disease (spongiform encephalopathy): Transmission to the chimpanzee. Science 1968, 161, 388–389. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Gajdusek, D.C.; Gibbs, C.J., Jr. Creutzfeldt-Jakob disease virus isolations from the Gerstmann–Stra¨ussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain 1981, 104, 559–588. [Google Scholar] [CrossRef] [PubMed]
- Lugaresi, E.; Medori, R.; Montagna, P.; Baruzzi, A.; Cortelli, P.; Lugaresi, A.; Tinuper, P.; Zucconi, M.; Gambetti, P. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N. Engl. J. Med. 1986, 315, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Llewelyn, C.A.; Hewitt, P.E.; Knight, R.S.; Amar, K.; Cousens, S.; Mackenzie, J.; Will, R.G. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004, 363, 417–421. [Google Scholar] [CrossRef]
- Ironside, J.W. Variant Creutzfeldt-Jakob disease: Risk of transmission by blood transfusion and blood therapies. Haemophilia 2006, 12, 8–15, discussion 26–28. [Google Scholar] [CrossRef] [PubMed]
- Godsave, S.F.; Peters, P.J.; Wille, H. Subcellular distribution of the prion protein in sickness and in health. Virus Res. 2015, 207, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary forscrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spon-giosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesne, S.E. The complex PrP(c)-Fyn couples human oligomericAbeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A. Unraveling prion strains with cell biology and organic chemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Scott, M.; Taraboulos, A.; Stahl, N.; Prusiner, S.B. Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured-cells. J. Cell Biol. 1990, 110, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Raymond, G.J. The scrapie-associated form of PrP is made from a cellsurface precursor that is both protease-sensitive and phospholipase-sensitive. J. Biol. Chem. 1991, 266, 18217–18223. [Google Scholar] [PubMed]
- Goold, R.; Rabbanian, S.; Sutton, L.; Andre, R.; Arora, P.; Moonga, J.; Clarke, A.R.; Schiavo, G.; Jat, P.; Collinge, J.; et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Beranger, F.; Mange, A.; Goud, B.; Lehmann, S. Stimulation of PrPC retrograde transport toward the endoplasmic reticulum increases accumulation of PrPSc in prioninfected cells. J. Biol. Chem. 2002, 277, 38972–38977. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar] [PubMed]
- Caughey, B.; Raymond, G.J.; Ernst, D.; Race, R.E. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s)—Implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 1991, 65, 6597–6603. [Google Scholar] [PubMed]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Baron, G.S.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Characterization of intracellular dynamics of inoculated PrP-res and newly generated PrPSc during early stage prion infection in Neuro2a cells. Virology 2014, 450, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C.; Scott, M.; Mastrianni, J.; Gabizon, R.; Torchia, M.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995, 83, 79–90. [Google Scholar] [CrossRef]
- Basler, K.; Oesch, B.; Scott, M.; Westaway, D.; Walchli, M.; Groth, D.F.; McKinley, M.P.; Prusiner, S.B.; Weissmann, C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 1986, 46, 417–428. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wuthrich, K. NMR structure of the mouse prion protein domain PrP (121–231). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol. 1995, 129, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinctsignaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggersneuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion proteinrecruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and toenhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Preferential Cu2+ coor-dination by His96 and His111 induces beta-sheet formation in the unstructuredamyloidogenic region of the prion protein. J. Biol. Chem. 2004, 279, 32018–32027. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zou, W.; Wang, G. Cellular prion protein (PrP(C)) and its role in stress responses. Int. J. Clin. Exp. Med. 2015, 8, 8042–8050. [Google Scholar] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; Knight, R.; Haswell, S.J.; Bujdoso, R.; Brown, D.R. Metal imbalance and compromised antioxidant function are early changes in prion disease. Biochem. J. 2002, 362, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A. Prion protein and its role in signal transduction. Cell. Mol. Biol. Lett. 2013, 18, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Martins, V.R.; Beraldo, F.H.; Hajj, G.N.; Lopes, M.H.; Lee, K.S.; Prado, M.A.; Linden, R. Prion protein: Orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 2010, 12, 63–86. [Google Scholar] [PubMed]
- Sunyach, C.; Jen, A.; Deng, J.; Fitzgerald, K.T.; Frobert, Y.; Grassi, J.; McCaffrey, M.W.; Morris, R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003, 22, 3591–3601. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prionprotein trafficking. Trends Cell Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Hegde, R.S. Retrotranslocation of prion proteins from the endo-plasmic reticulum by preventing GPI signal transamidation. Mol. Biol. Cell 2008, 19, 3463–3476. [Google Scholar] [CrossRef] [PubMed]
- Yedidia, Y.; Horonchik, L.; Tzaban, S.; Yanai, A.; Taraboulos, A. Proteasomesand ubiquitin are involved in the turnover of the wild-type prion protein. EMBOJ. 2001, 20, 5383–5391. [Google Scholar] [CrossRef] [PubMed]
- Drisaldi, B.; Stewart, R.S.; Adles, C.; Stewart, L.R.; Quaglio, E.; Biasini, E.; Fior-iti, L.; Chiesa, R.; Harris, D.A. Mutant PrP is delayed in its exit fromthe endoplasmic reticulum, but neither wild-type nor mutant PrP under-goes retrotranslocation prior to proteasomal degradation. J. Biol. Chem. 2003, 278, 21732–21743. [Google Scholar] [CrossRef] [PubMed]
- Orsi, A.; Fioriti, L.; Chiesa, R.; Sitia, R. Conditions of endoplasmic reticulumstress favor the accumulation of cytosolic prion protein. J. Biol. Chem. 2006, 281, 30431–30438. [Google Scholar] [CrossRef] [PubMed]
- Levine, C.G.; Mitra, D.; Sharma, A.; Smith, C.L.; Hegde, R.S. The efficiency ofprotein compartmentalization into the secretory pathway. Mol. Biol. Cell 2005, 16, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Rane, N.S.; Kang, S.W.; Chakrabarti, O.; Feigenbaum, L.; Hegde, R.S. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev. Cell 2008, 15, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Guo, Q.; Zhang, Y.; Goodyer, C.G.; Le Blanc, A.C. Cytosolic prion pro-tein is not toxic and protects against Bax-mediated cell death in human primaryneurons. J. Biol. Chem. 2003, 278, 40877–40881. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Gains, M.; LeBlanc, A.C. Neuroprotective functions of prion protein. J. Neurosci. Res. 2004, 75, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Roucou, X.; Giannopoulos, P.N.; Zhang, Y.; Jodoin, J.; Goodyer, C.G.; LeBlanc, A. Cellular prion protein inhibits proapoptotic Bax conformational changein human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005, 12, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Vanderperre, B.; Staskevicius, A.B.; Tremblay, G.; McCoy, M.; O’Neill, M.A.; Cashman, N.R.; Roucou, X. An overlapping reading frame in the PRNP gene encodesa novel polypeptide distinct from the prion protein. FASEB J. 2011, 25, 2373–2386. [Google Scholar] [CrossRef] [PubMed]
- Milhavet, O.; Lehmann, S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res. Brain Res. Rev. 2002, 38, 328–339. [Google Scholar] [CrossRef]
- Brown, D.R. Neurodegeneration and oxidative stress: Prion disease results from loss of antioxidant defence. Folia Neuropathol. 2005, 43, 229–243. [Google Scholar] [PubMed]
- Nadal, R.C.; Abdelraheim, S.R.; Brazier, M.W.; Rigby, S.E.; Brown, D.R.; Viles, J.H. Prion protein does not redox-silence Cu2+, but is a sacrificial quencher of hydroxyl radicals. Free Radic. Biol. Med. 2007, 42, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Protein oxidation in aging and age-related diseases. Ann. N. Y. Acad. Sci. 2001, 928, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.J.; Han, Z.Q.; Li, Z.Y. Modulating protein activity and cellular function by methionine residue oxidation. Amino Acids 2012, 43, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, J.; Bar-Noy, S.; Williams, W.M.; Requena, J.; Berlett, B.S.; Stadtman, E.R. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. USA 2001, 98, 12920–12925. [Google Scholar] [CrossRef] [PubMed]
- Elmallah, M.I.; Borgmeyer, U.; Betzel, C.; Redecke, L. Impact of methionine oxidation as an initial event on the pathway of human prion protein conversion. Prion 2013, 7, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Younan, N.D.; Nadal, R.C.; Davies, P.; Brown, D.R.; Viles, J.H. Methionine oxidation perturbs the structural core of the prion protein and suggests a generic misfolding pathway. J. Biol. Chem. 2012, 287, 28263–28275. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Abid, K.; Soto, C. The intriguing prion disorders. Cell Mol. Life Sci. 2006, 63, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Corsaro, A.; Villa, V.; Simi, A.; Vella, S.; Pagano, A.; Florio, T. Human PrP90–231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death Dis. 2011, 2, e138. [Google Scholar] [CrossRef] [PubMed]
- Chiovitti, K.; Corsaro, A.; Thellung, S.; Villa, V.; Paludi, D.; D’Arrigo, C.; Russo, C.; Perico, A.; Ianieri, A.; di Cola, D.; et al. Intracellular accumulation of a mild-denatured monomer of the human PrP fragment 90–231, as possible mechanism of its neurotoxic effects. J. Neurochem. 2007, 103, 2597–2609. [Google Scholar] [CrossRef] [PubMed]
- Borger, E.; Aitken, L.; Muirhead, K.E.; Allen, Z.E.; Ainge, J.A.; Conway, S.J.; Gunn-Moore, F.J. Mitochondrial beta-amyloid in Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 868–873. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.W.; Marciniack, C.; LaFerla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 2012, 1820, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Siskova, Z.; Mahad, D.J.; Pudney, C.; Campbell, G.; Cadogan, M.; Asuni, A.; O’Connor, V.; Perry, V.H. Morphological and functional abnormalities in mitochondria associated with synaptic degeneration in prion disease. Am. J. Pathol. 2010, 177, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of prion protein aggregation in neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648–8669. [Google Scholar] [CrossRef] [PubMed]
- Belay, E.D. Transmissible spongiform encephalopathies in humans. Annu. Rev. Microbiol. 1999, 53, 283–314. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Wälchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 2005, 25, 2793–2802. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Lee, A.H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Hetz, C.; Yi, C.H.; Jackson, W.S.; Borkowski, A.W.; Yuan, J.; Wollmann, R.H.; Lindquist, S. Prion pathogenesis is independent of caspase-12. Prion 2007, 1, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Mathison, J.C.; Wolinski, M.K.; Bensinger, S.J.; Fitzgerald, P.; Droin, N.; Ulevitch, R.J.; Green, D.R.; Nicholson, D.W. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 2006, 440, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Vaillancourt, J.P.; Graham, R.K.; Huyck, M.; Srinivasula, S.M.; Alnemri, E.S.; Steinberg, M.H.; Nolan, V.; Baldwin, C.T.; Hotchkiss, R.S.; et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 2004, 429, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; Mallucci, G.R. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Nunziante, M.; Ackermann, K.; Dietrich, K.; Wolf, H.; Gädtke, L.; Gilch, S.; Vorberg, I.; Groschup, M.; Schätzl, H.M. Proteasomal dysfunction and endoplasmic reticulum stress enhance traffi cking of prion protein aggregates through the secretory pathway and increase accumulation of pathologic prion protein. J. Biol. Chem. 2011, 286, 33942–33953. [Google Scholar] [CrossRef] [PubMed]
- Boellaard, J.W.; Kao, M.; Schlote, W.; Diringer, H. Neuronal autophagy in experimental scrapie. Acta Neuropathol. 1991, 82, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Boellaard, J.W.; Schlote, W.; Tateishi, J. Neuronal autophagy in experimental Creutzfeldt-Jakob’s disease. Acta Neuropathol. 1989, 78, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Schatzl, H.M.; Laszlo, L.; Holtzman, D.M.; Tatzelt, J.; DeArmond, S.J.; Weiner, R.I.; Mobley, W.C.; Prusiner, S.B. A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 1997, 71, 8821–8831. [Google Scholar] [PubMed]
- Liberski, P.P.; Sikorska, B.; Bratosiewicz-Wasik, J.; Gajdusek, D.C.; Brown, P. Neuronal cell death in transmissible spongiform encephalopathies (prion diseases) revisited: From apoptosis to autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2473–2490. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, B.; Liberski, P.P.; Giraud, P.; Kopp, N.; Brown, P. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: A brain biopsy study. Int. J. Biochem. Cell Biol. 2004, 36, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Dandoy-Dron, F.; Benboudjema, L.; Guillo, F.; Jaegly, A.; Jasmin, C.; Dormont, D.; Tovey, M.G.; Dron, M. Enhanced levels of scrapie responsive gene mRNA in BSE-infected mouse brain. Brain Res. 2000, 76, 173–179. [Google Scholar] [CrossRef]
- Dandoy-Dron, F.; Guillo, F.; Benboudjema, L.; Deslys, J.P.; Lasmezas, C.; Dormont, D.; Tovey, M.G.; Dron, M. Gene expression in scrapie. Cloning of a new scrapie-responsive gene and the identification of increased levels of seven other mRNA transcripts. J. Biol. Chem. 1998, 273, 7691–7697. [Google Scholar] [CrossRef] [PubMed]
- Dron, M.; Dandoy-Dron, F.; Guillo, F.; Benboudjema, L.; Hauw, J.J.; Lebon, P.; Dormont, D.; Tovey, M.G. Characterization of the human analogue of a Scrapie-responsive gene. J. Biol. Chem. 1998, 273, 18015–18018. [Google Scholar] [CrossRef] [PubMed]
- Dron, M.; Bailly, Y.; Beringue, V.; Haeberle, A.M.; Griffond, B.; Risold, P.Y.; Tovey, M.G.; Laude, H.; Dandoy-Dron, F. Scrg1 is induced in TSE and brain injuries, and associated with autophagy. Eur. J. Neurosci. 2005, 22, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Dron, M.; Bailly, Y.; Beringue, V.; Haeberle, A.M.; Griffond, B.; Risold, P.Y.; Tovey, M.G.; Laude, H.; Dandoy-Dron, F. SCRG1, a potential marker of autophagy in transmissible spongiform encephalopathies. Autophagy 2006, 2, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Shin, H.Y.; Park, S.J.; Kim, B.H.; Choi, J.K.; Choi, E.K.; Carp, R.I.; Kim, Y.S. The involvement of cellular prion protein in the autophagy pathway in neuronal cells. Mol. Cell Neurosci. 2008, 39, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Liberski, P.P.; Brown, D.R.; Sikorska, B.; Caughey, B.; Brown, P. Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol 2008, 46, 1–25. [Google Scholar] [PubMed]
- Liberski, P.P.; Gajdusek, D.C.; Brown, P. How do neurons degenerate in prion diseases or transmissible spongiform encephalopathies (TSEs): Neuronal autophagy revisited. Acta Neurobiol. Exp. 2002, 62, 141–147. [Google Scholar]
- Mok, S.W.; Riemer, C.; Madela, K.; Hsu, D.K.; Liu, F.T.; Gultner, S.; Heise, I.; Baier, M. Role of galectin-3 in prion infections of the CNS. Biochem. Biophys. Res. Commun. 2007, 359, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Ertmer, A.; Gilch, S.; Yun, S.W.; Flechsig, E.; Klebl, B.; Stein-Gerlach, M.; Klein, M.A.; Schatzl, H.M. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J. Biol. Chem. 2004, 279, 41918–41927. [Google Scholar] [CrossRef] [PubMed]
- Ertmer, A.; Huber, V.; Gilch, S.; Yoshimori, T.; Erfle, V.; Duyster, J.; Elsasser, H.P.; Schatzl, H.M. The anticancer drug imatinib induces cellular autophagy. Leukemia 2007, 21, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.W.; Ertmer, A.; Flechsig, E.; Gilch, S.; Riederer, P.; Gerlach, M.; Schatzl, H.M.; Klein, M.A. The tyrosine kinase inhibitor imatinib mesylate delays prion neuroinvasion by inhibiting prion propagation in the periphery. J. Neurovirol. 2007, 13, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Aguib, Y.; Gilch, S.; Krammer, C.; Ertmer, A.; Groschup, M.H.; Schatzl, H.M. Neuroendocrine cultured cells counteract persistent prion infection by downregulation of PrPc. Mol. Cell Neurosci. 2008, 38, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Schobel, S.; Lichtenthaler, S.F.; Vorberg, I.; Groschup, M.H.; Kretzschmar, H.; Schatzl, H.M.; Nunziante, M. The novel sorting nexin SNX33 interferes with cellular PrP formation by modulation of PrP shedding. Traffic 2008, 9, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Marella, M.; Lehmann, S.; Grassi, J.; Chabry, J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J. Biol. Chem. 2002, 277, 25457–25464. [Google Scholar] [CrossRef] [PubMed]
- Parkin, E.T.; Watt, N.T.; Turner, A.J.; Hooper, N.M. Dual mechanisms for shedding of the cellular prion protein. J. Biol. Chem. 2004, 279, 11170–11178. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.C.; Baron, G.S.; Lee, K.S.; Steele-Mortimer, O.; Dorward, D.; Prado, M.A.M.; Caughey, B. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J. Neurosci. 2005, 25, 5207–5216. [Google Scholar] [CrossRef] [PubMed]
- Rouvinski, A.; Karniely, S.; Kounin, M.; Moussa, S.; Goldberg, M.D.; Warburg, G.; Lyakhovetsky, R.; Papy-Garcia, D.; Kutzsche, J.; Korth, C.; et al. Live imaging of prions reveals nascent PrPSc in cell-surface, raft-associated amyloid strings and webs. J. Cell Biol. 2014, 204, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Veith, N.M.; Plattner, H.; Stuermer, C.A.O.; Schulz-Schaeffer, W.J.; Burkle, A. Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 2009, 88, 45–63. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Zhao, D.M.; Khan, S.H.; Yang, L.F. Role of autophagy in prion proteininduced neurodegenerative diseases. Acta Biochim. Biophys. Sin. 2013, 45, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Hegde, R.S. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009, 5, e1000479. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.; McKinnon, C.; Rabbanian, S.; Collinge, J.; Schiavo, G.; Tabrizi, S.J. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J. Cell Sci. 2013, 126, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.; McKinnon, C.; Tabrizi, S.J. Prion degradation pathways: Potential for therapeutic intervention. Mol. Cell Neurosci. 2015, 66, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Grenier, C.; Bissonnette, C.; Volkov, L.; Roucou, X. Molecular morphology and toxicity of cytoplasmic prion protein aggregates in neuronal and non-neuronal cells. J. Neurochem. 2006, 97, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Messenger, M.J.; Klohn, P.C.; Brandner, S.; Wadsworth, J.D.F.; Collinge, J.; Tabrizi, S.J. Disease-related prion protein forms aggresomes in neuronal cells leading to caspase activation and apoptosis. J. Biol. Chem. 2005, 280, 38851–38861. [Google Scholar] [CrossRef] [PubMed]
- Micsenyi, M.C.; Sikora, J.; Stephney, G.; Dobrenis, K.; Walkley, S.U. Lysosomal membrane permeability stimulates protein aggregate formation in neurons of a lysosomal disease. J. Neurosci. 2013, 33, 10815–10827. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.C.; Gu, Y.P.; Zanusso, G.; Sy, M.S.; Kumar, A.; Cohen, M.; Gambetti, P.; Singh, N. The chaperone protein BiP binds to amutant prion protein and mediates its degradation by the proteasome. J. Biol. Chem. 2000, 275, 38699–38704. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Petersen, R.B.; Jin, T.C.; Jing, Y.; Kanoush, R.; Ferrari, S.; Gambetti, P.; Singh, N. Proteasomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. J. Biol. Chem. 1999, 274, 23396–23404. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milisav, I.; Šuput, D.; Ribarič, S. Unfolded Protein Response and Macroautophagy in Alzheimer’s, Parkinson’s and Prion Diseases. Molecules 2015, 20, 22718-22756. https://doi.org/10.3390/molecules201219865
Milisav I, Šuput D, Ribarič S. Unfolded Protein Response and Macroautophagy in Alzheimer’s, Parkinson’s and Prion Diseases. Molecules. 2015; 20(12):22718-22756. https://doi.org/10.3390/molecules201219865
Chicago/Turabian StyleMilisav, Irina, Dušan Šuput, and Samo Ribarič. 2015. "Unfolded Protein Response and Macroautophagy in Alzheimer’s, Parkinson’s and Prion Diseases" Molecules 20, no. 12: 22718-22756. https://doi.org/10.3390/molecules201219865
APA StyleMilisav, I., Šuput, D., & Ribarič, S. (2015). Unfolded Protein Response and Macroautophagy in Alzheimer’s, Parkinson’s and Prion Diseases. Molecules, 20(12), 22718-22756. https://doi.org/10.3390/molecules201219865