
Asymmetric Synthesis and Absolute Configuration Assignment of a New Type of Bedaquiline Analogue

Abstract
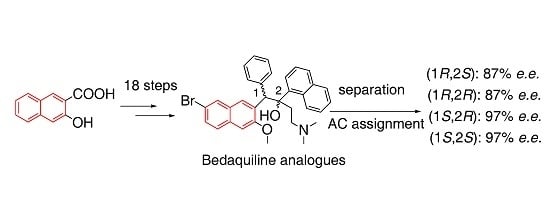
:
1. Introduction
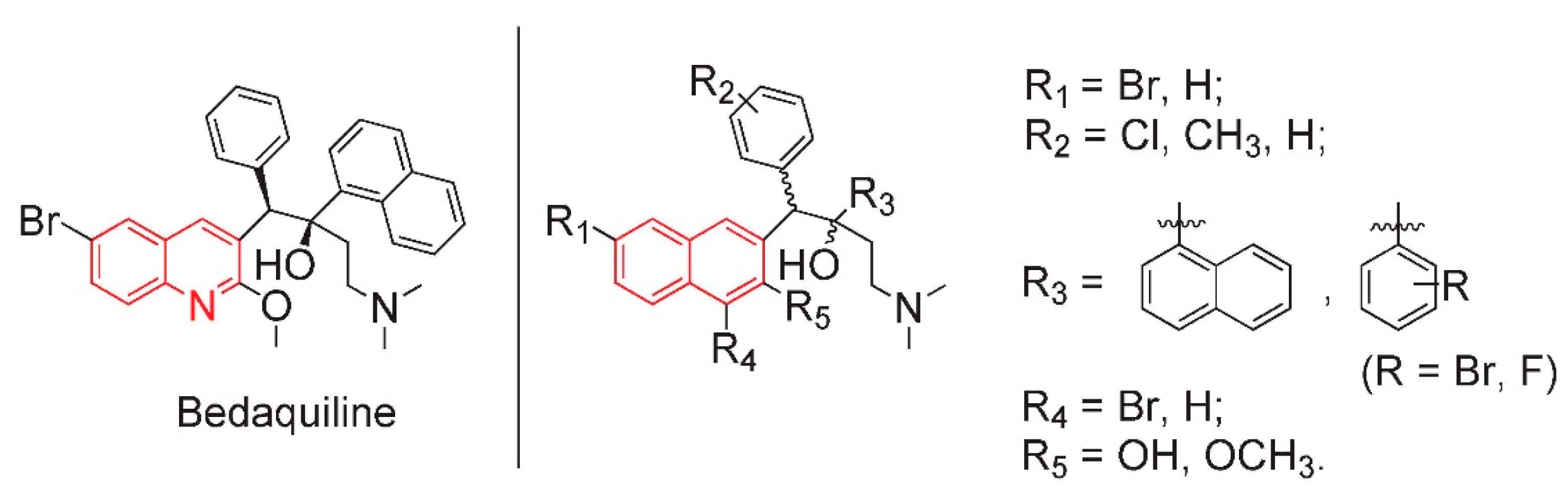
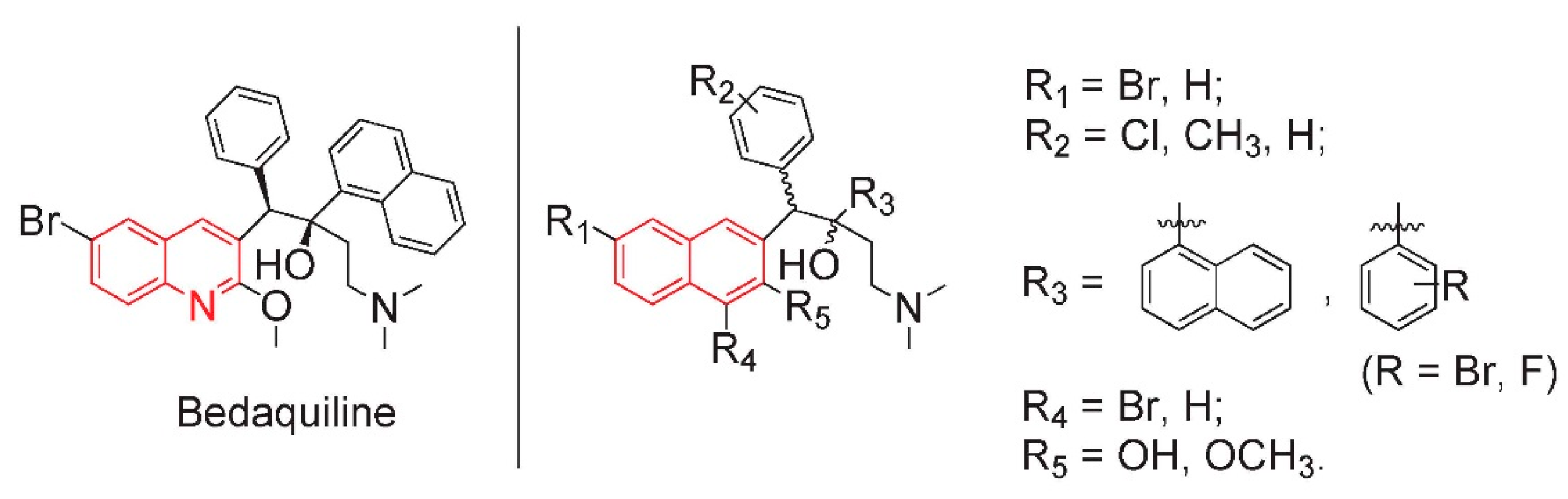
2. Results and Discussion
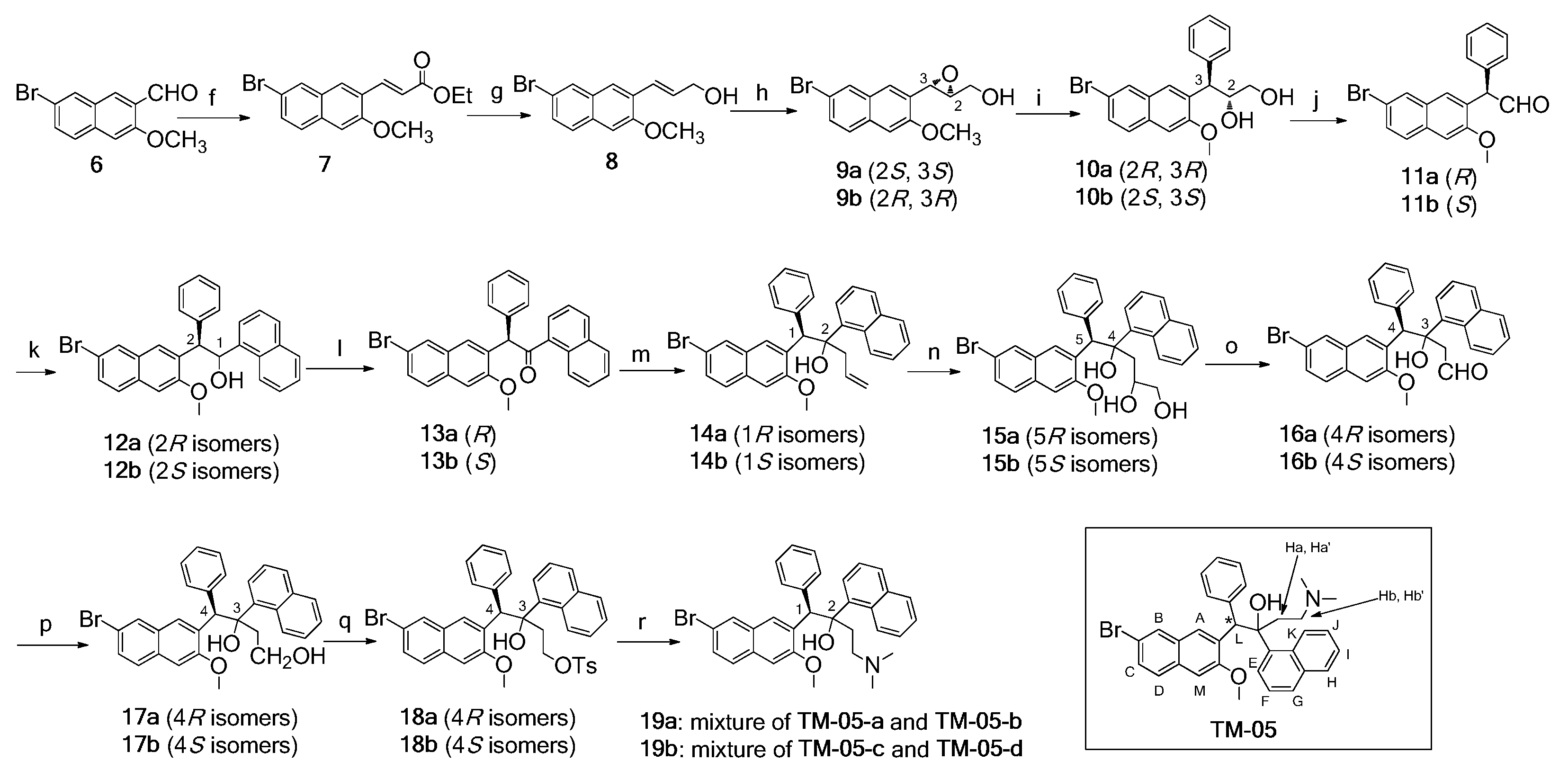
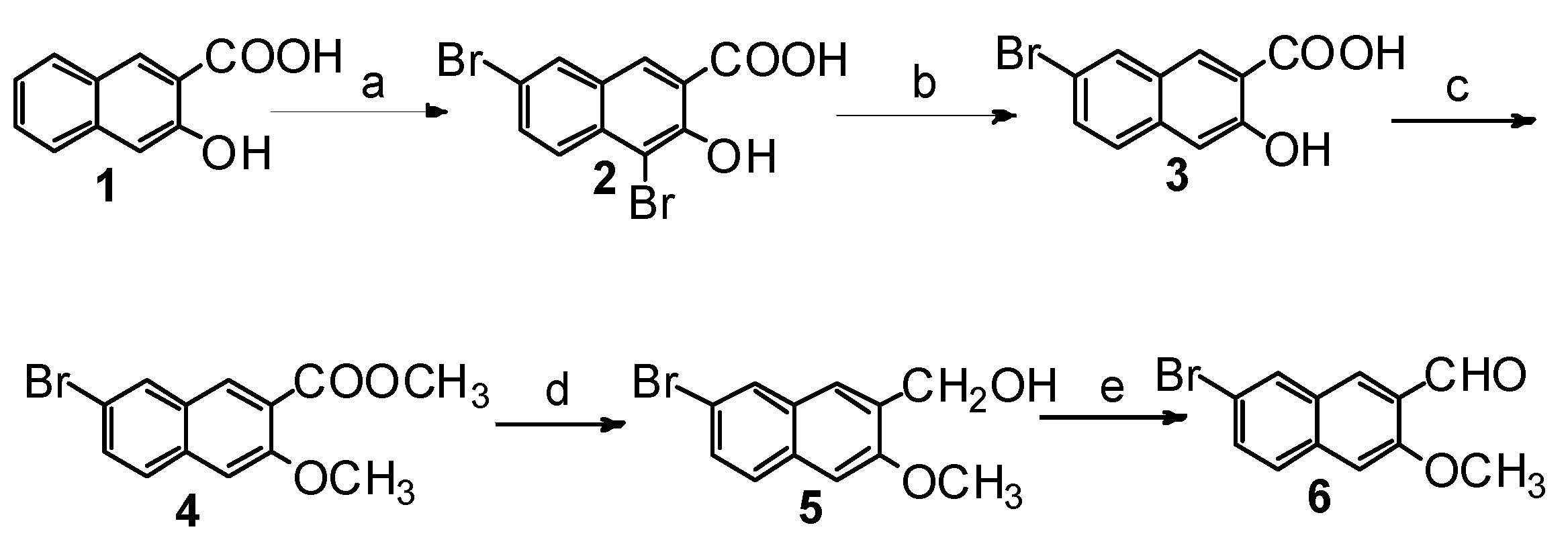
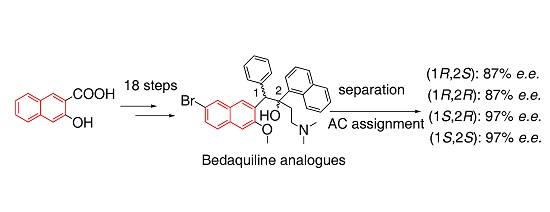
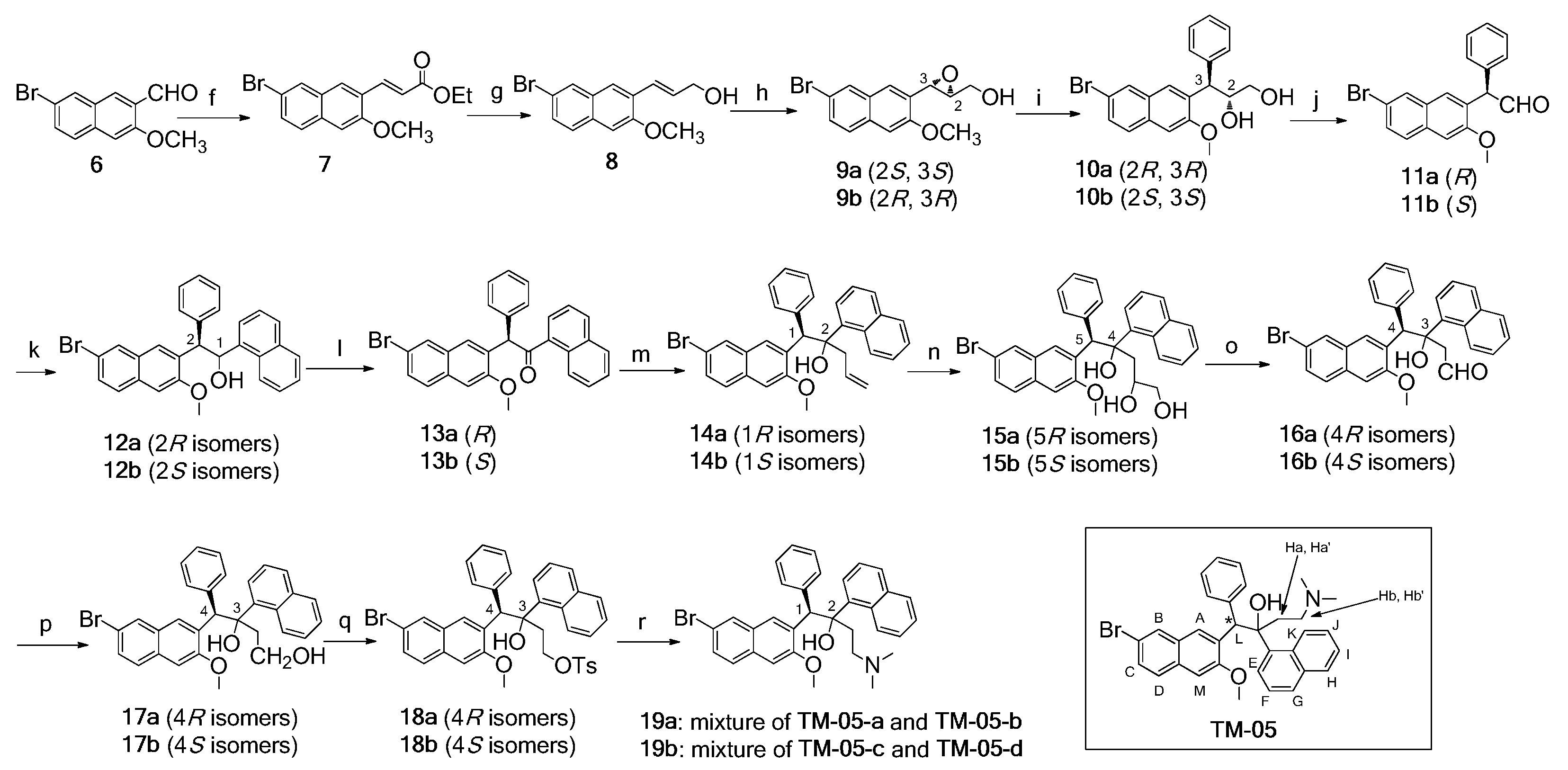
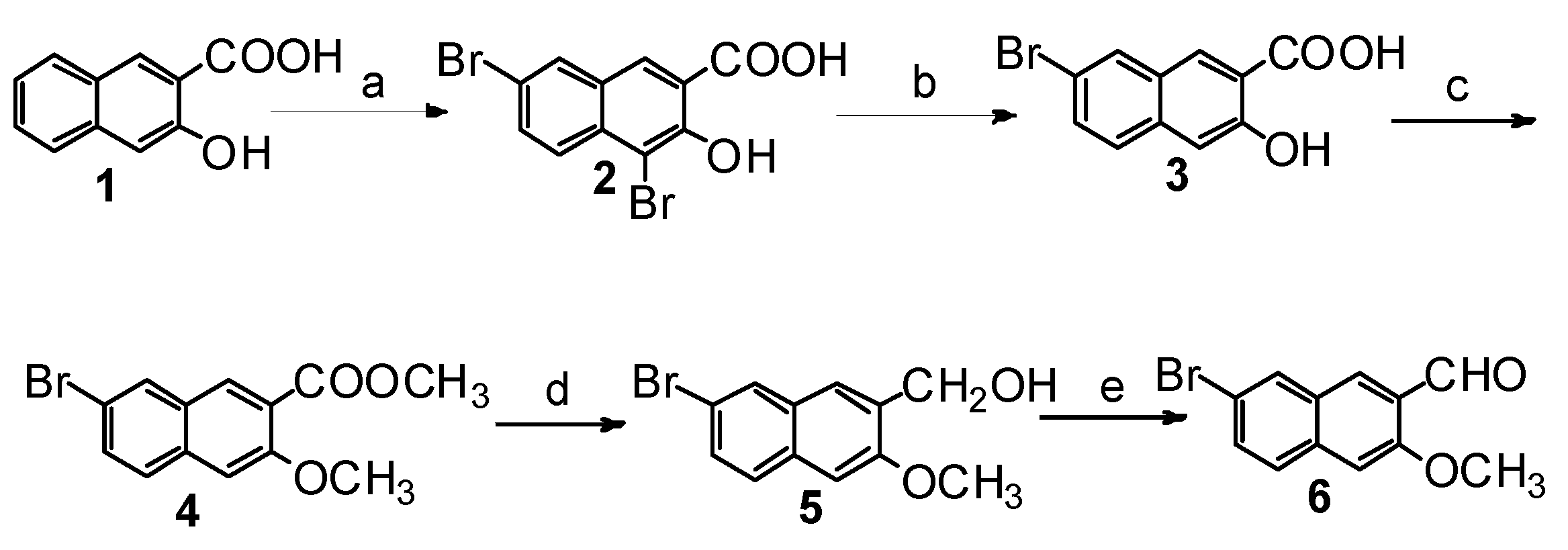
2.1. Synthesis of Four Optical Isomers
2.2. Determination of Optical Purity
2.3. Absolute Configuration Assignment
2.3.1. AC Assignment by Electronic Circular Dichroism (ECD) Spectroscopy
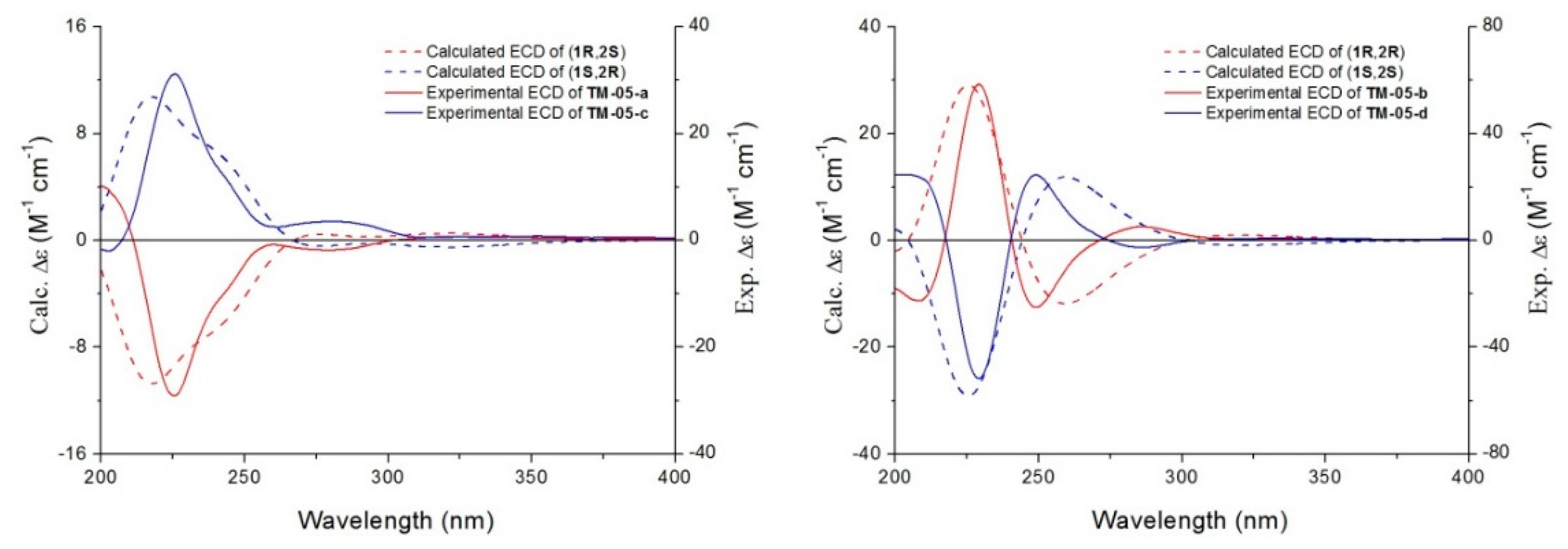
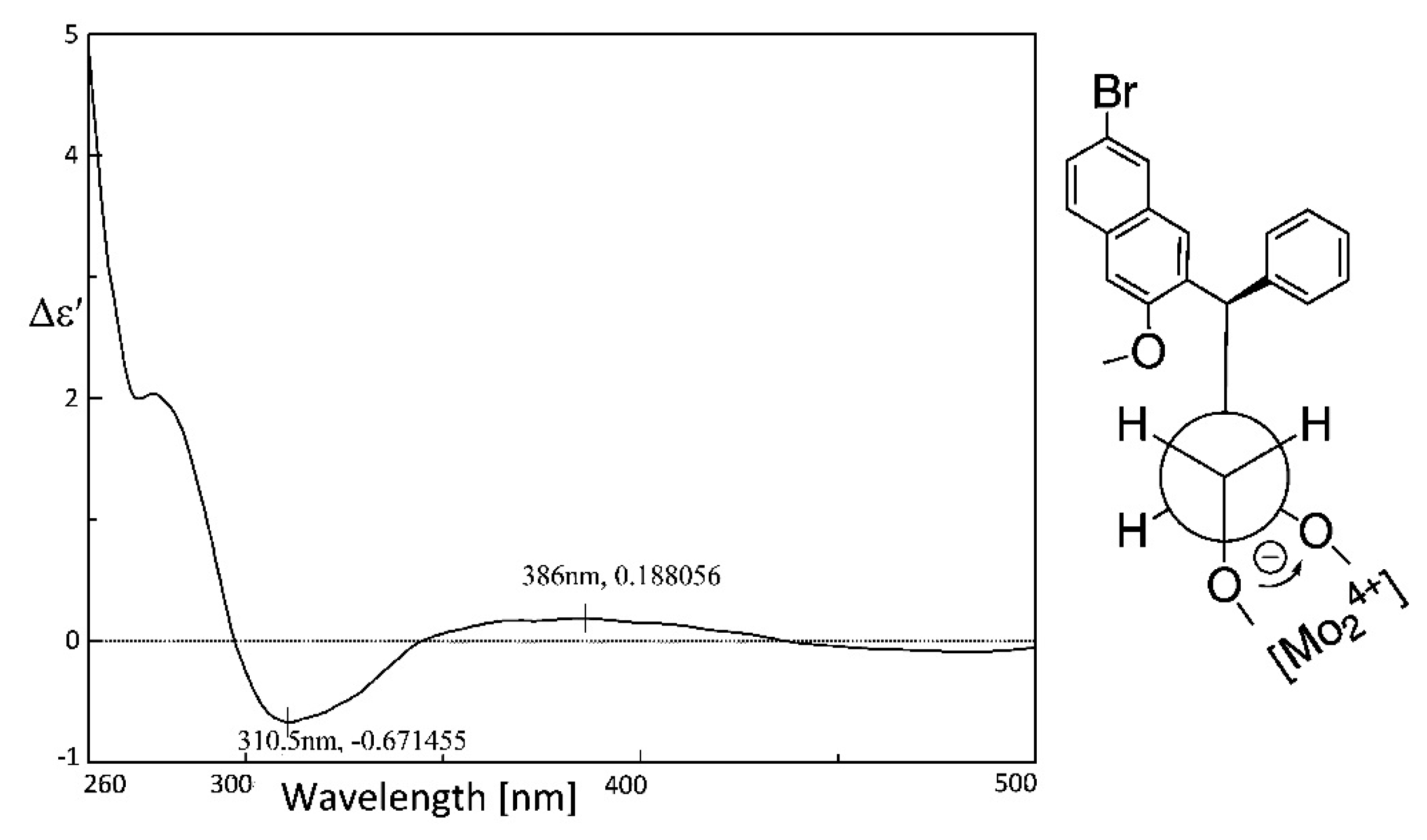
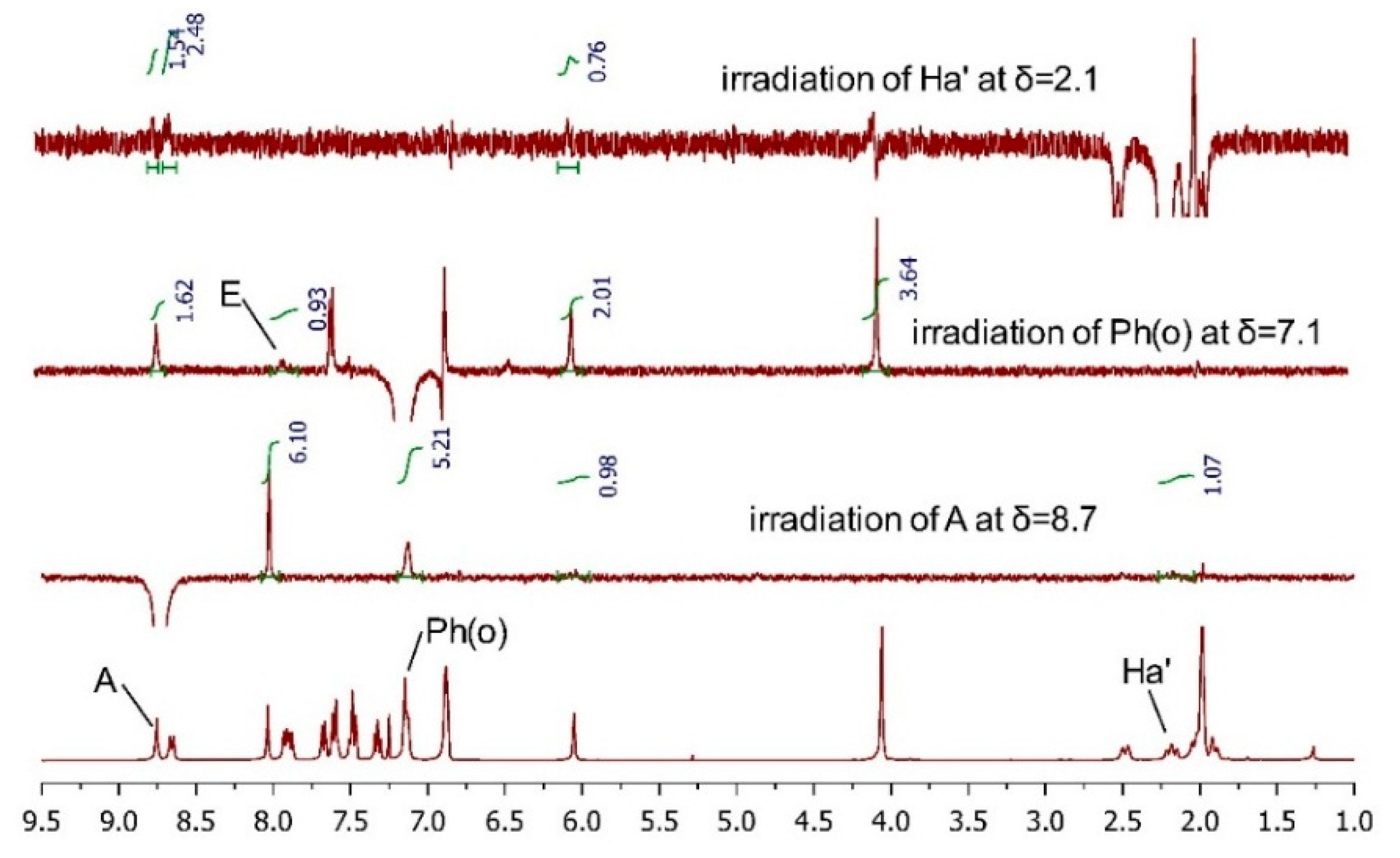
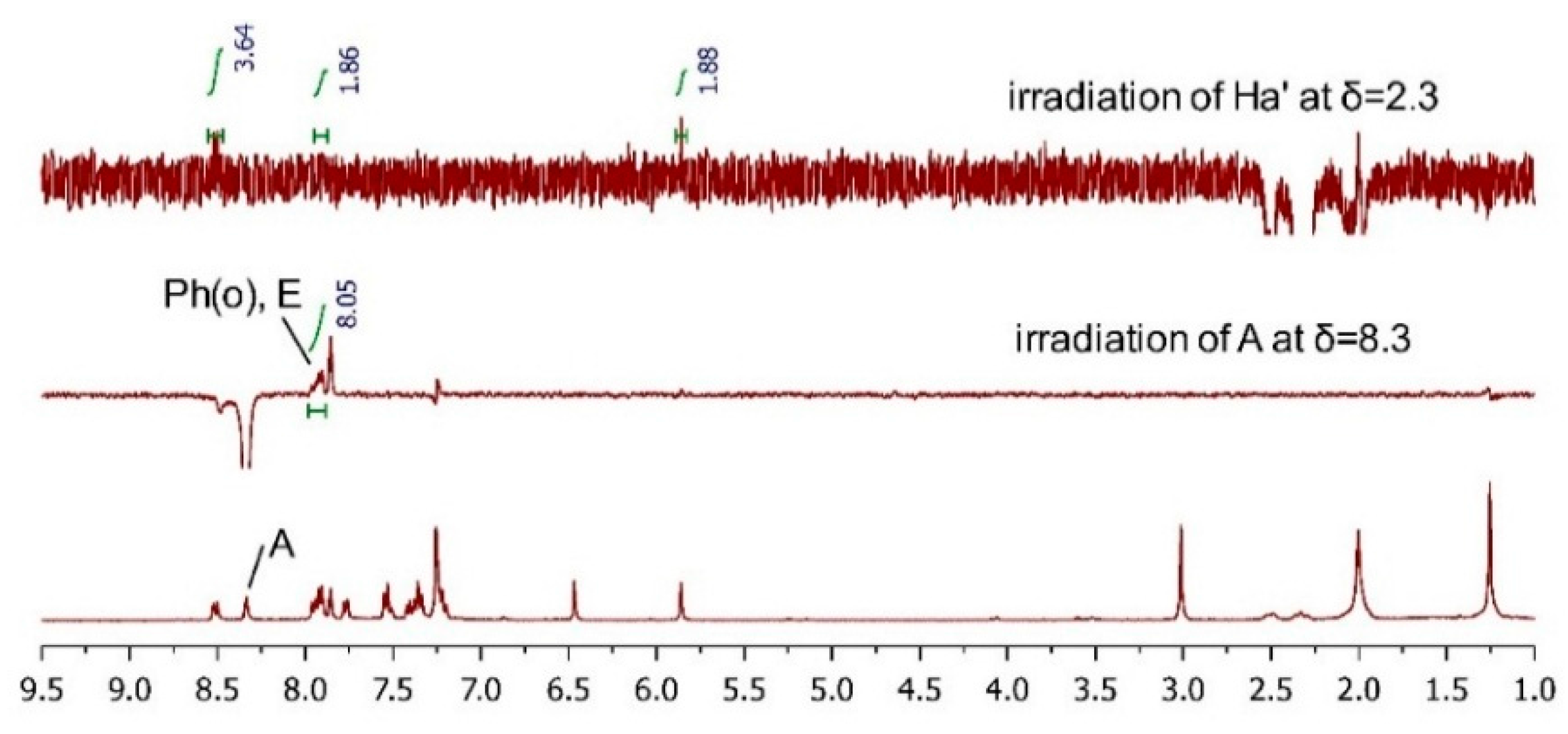
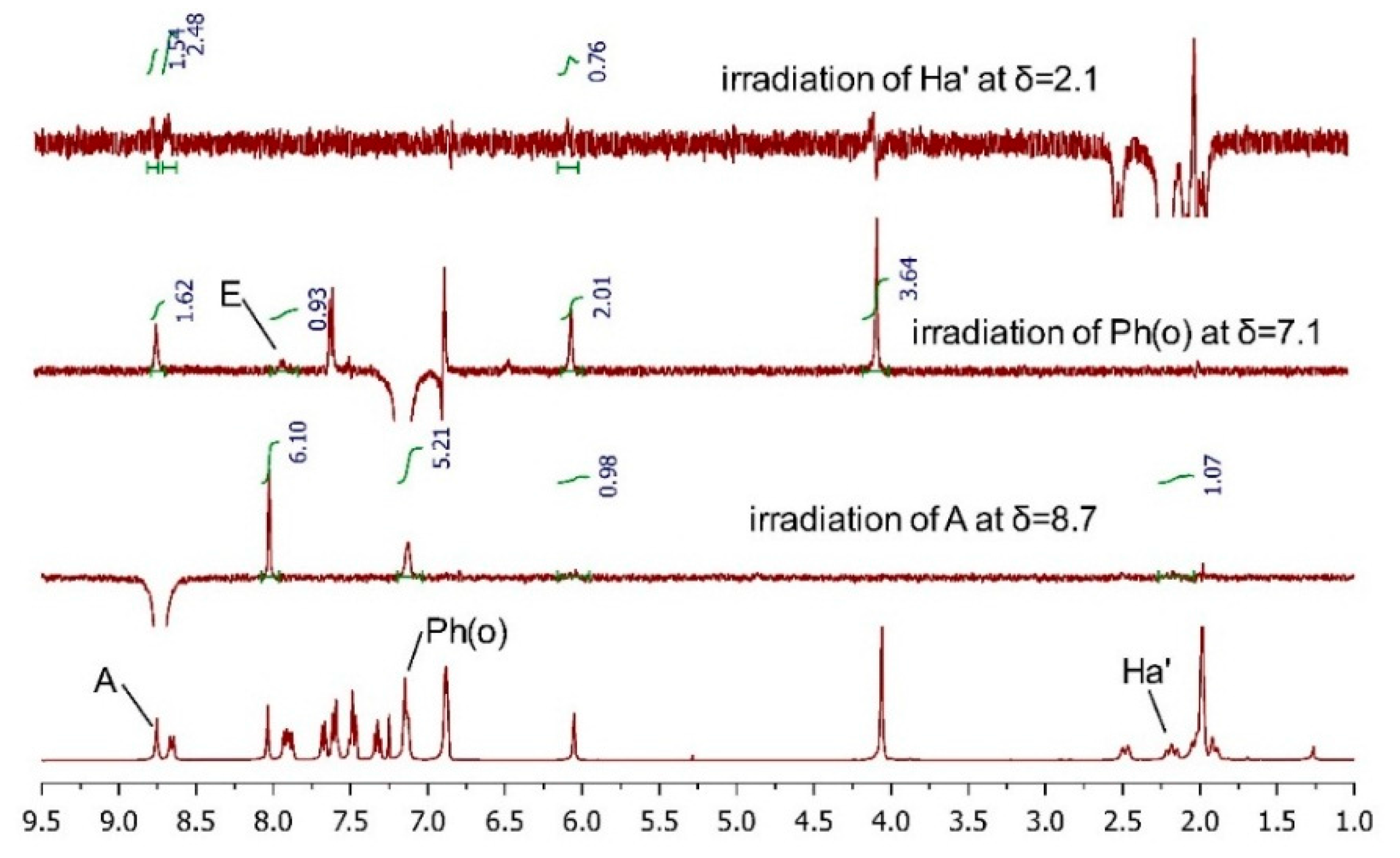
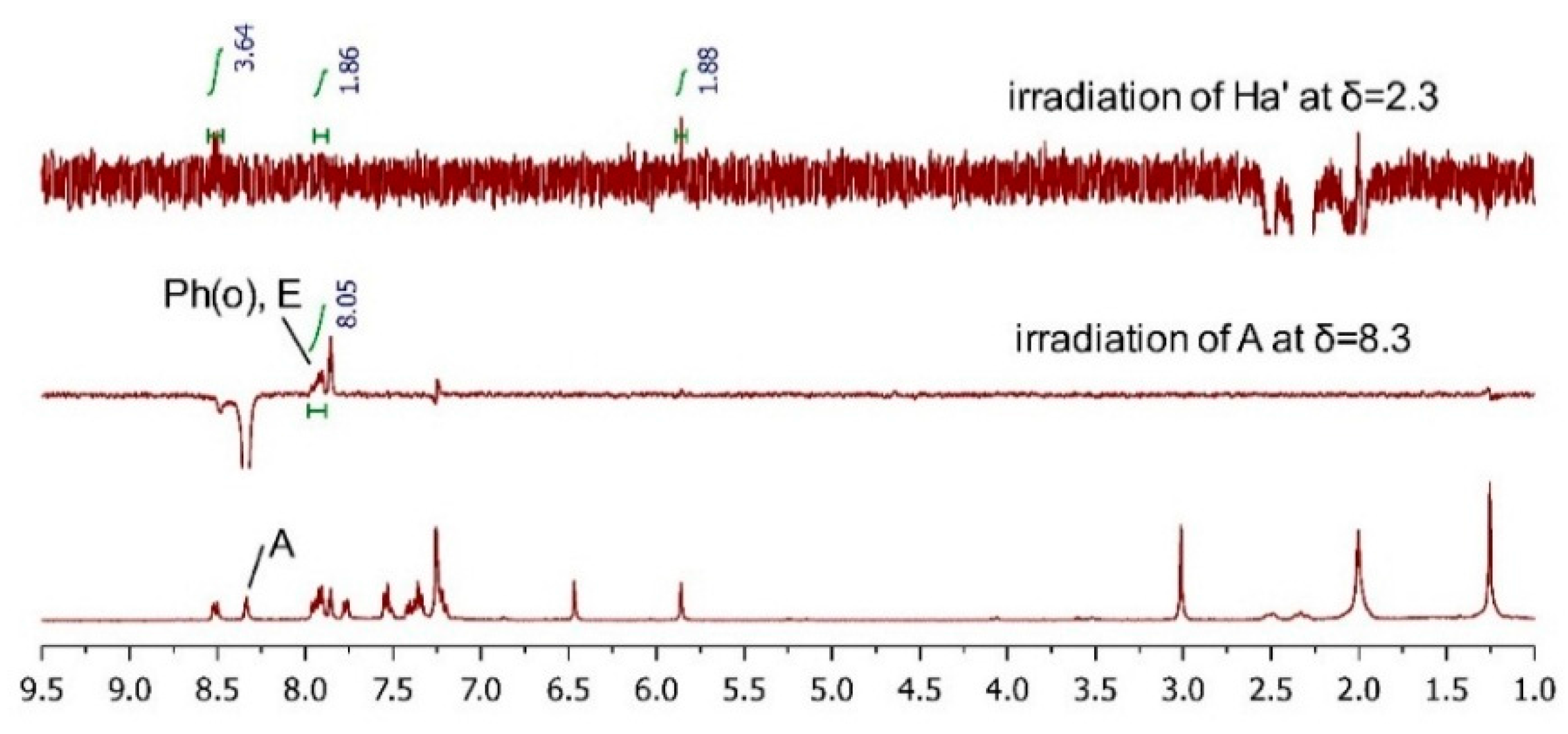
2.3.2. AC Corroboration by the Combined Use of Circular Dichroism and NMR Spectroscopy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound TM-05-c | Compound TM-05-d | ||||
|---|---|---|---|---|---|
| Proton | δ (ppm) | J (Hz) | Proton | δ (ppm) | J (Hz) |
| N(CH3)2,Hb,Hb’ | 1.88–2.04 | m | N(CH3)2,Hb,Hb’ | 1.93–2.06 | m |
| Ha′ | 2.17 | t (12.9) | Ha’ | 2.32 | t (12.0) |
| Ha | 2.47 | d (14.3) | Ha | 2.49 | d (14.3) |
| OCH3 | 4.05 | s | OCH3 | 3.00 | s |
| L | 6.04 | s | L | 5.86 | s |
| Ph(m, p) | 6.86–6.88 | m | M | 6.46 | s |
| Ph(o),M | 7.12–7.14 | m | Ph(p),F,G,D | 7.20–7.25 | m |
| F | 7.31 | t (7.6) | Ph(m),H | 7.34–7.42 | m |
| I,C | 7.45–7.50 | m | C,J | 7.51–7.55 | m |
| D,J | 7.57–7.61 | m | I | 7.77 | d (8.1) |
| G | 7.67 | d (8.1) | B | 7.86 | s |
| E,H | 7.87–7.92 | m | Ph(o),E | 7.91–7.97 | m |
| B | 8.03 | s | A | 8.34 | s |
| K | 8.65 | d (8.7) | K | 8.52 | d (8.7) |
| A | 8.75 | s | |||
3. Experimental Section
3.1. General Information
3.2. Computational Details
3.3. Synthesis and Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keshavjee, S.; Farmer, P.E. Tuberculosis, drug resistance, and the history of modern medicine. N. Engl. J. Med. 2012, 367, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent mycobacterium tuberculosis infection. N. Engl. J. Med. 2015, 372, 2127–2135. [Google Scholar] [PubMed]
- Global Tuberculosis Report 2015. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 8 December 2015).
- Andries, K.; Verhasselt, P.; Guillemont, J.; Gohlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit V of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, A.C.; Podasca, I.; Koul, A.; Andries, K.; Guillemont, J.; Lill, H.; Bald, D. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS ONE 2011, 6, e23575. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.R.; Koymans, L.H.; Guillemont, J.E.; Koul, A.; Andries, K. A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910. Proteins Struct. Funct. Bioinform. 2007, 67, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Segala, E.; Sougakoff, W.; Nevejans-Chauffour, A.; Jarlier, V.; Petrella, S. New mutations in the mycobacterial atp synthase: New insights into the binding of the diarylquinoline TMC207 to the ATP synthase C-ring structure. Antimicrob. Agents Chemother. 2012, 56, 2326–2334. [Google Scholar] [CrossRef] [PubMed]
- Biukovic, G.; Basak, S.; Manimekalai, M.S.; Rishikesan, S.; Roessle, M.; Dick, T.; Rao, S.P.; Hunke, C.; Gruber, G. Variations of subunit ε of the Mycobacterium tuberculosis F1FO ATP synthase and a novel model for mechanism of action of the tuberculosis drug TMC207. Antimicrob. Agents Chemother. 2013, 57, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Preiss, L.; Langer, J.D.; Yildiz, Ö.; Eckhardt-Strelau, L.; Guillemont, J.E.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase of rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef] [PubMed]
- Guillemont, J.; Meyer, C.; Poncelet, A.; Bourdrez, X.; Andries, K. Diarylquinolines, synthesis pathways and quantitative structure–activity relationship studies leading to the discovery of TMC207. Future Med. Chem. 2011, 3, 1345–1360. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Vranckx, L.; Lounis, N.; Pop, O.; Guillemont, J.; Vergauwen, K.; Mol, S.; Gilissen, R.; Motte, M.; Lancois, D.; et al. Novel antibiotics targeting respiratory ATP synthesis in Gram-positive pathogenic bacteria. Antimicrob. Agents Chemother. 2012, 56, 4131–4139. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Kulkarni, G.M.; Vasireddy, N.R.; Vandavasi, J.K.; Dixit, S.S.; Sharma, V.; Chattopadhyaya, J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 17, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Vandavasi, J.K.; Vasireddy, N.R.; Sharma, V.; Dixit, S.S.; Chattopadhyaya, J. Design, synthesis, biological evaluation and molecular modelling studies of novel quinoline derivatives against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Lahore, S.V.; Sayyed, A.Y.; Dixit, S.S.; Shinde, P.D.; Chattopadhyaya, J. Conformationally-constrained indeno[2,1-c]quinolines—A new class of anti-mycobacterial agents. Org. Biomol. Chem. 2010, 8, 2180–2197. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Shinde, P.D.; Sayyed, A.Y.; Kadam, S.A.; Bawane, A.N.; Poddar, A.; Plashkevych, O.; Foldesi, A.; Chattopadhyaya, J. Synthesis and structure of azole-fused indeno[2,1-c]quinolines and their anti-mycobacterial properties. Org. Biomol. Chem. 2010, 8, 5661–5673. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Vandavasi, J.K.; Kardile, R.A.; Lahore, S.V.; Dixit, S.S.; Deokar, H.S.; Shinde, P.D.; Sarmah, M.P.; Chattopadhyaya, J. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur. J. Med. Chem. 2010, 45, 1854–1867. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhong, W.; Liu, P.; Xiao, J.; Zheng, Z.; Xie, Y.; Zhao, G.; Wang, X.; Wang, L.; Li, X. 2013. Aromatic butan-2-ol Compounds, Preparation Methods and Uses Thereof. U.S. Patent 8,674,136, 18 March 2014. [Google Scholar]
- Saga, Y.; Motoki, R.; Makino, S.; Shimizu, Y.; Kanai, M.; Shibasaki, M. Catalytic asymmetric synthesis of R207910. J. Am. Chem. Soc. 2010, 132, 7905–7907. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, S.; Babu, G.S.K.; Mohapatra, D.K. Practical syntheses of (2S)-R207910 and (2R)-R207910. Eur. J. Org. Chem. 2011, 2011, 2057–2061. [Google Scholar] [CrossRef]
- Murphy, R.A.; Kung, H.F.; Kung, M.P.; Billings, J. Synthesis and characterization of iodobenzamide analogues: Potential D-2 dopamine receptor imaging agents. J. Med. Chem. 1990, 33, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Klunder, J.M.; Hanson, R.M.; Masamune, H.; Ko, S.Y.; Sharpless, K.B. Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar] [CrossRef]
- Dieter, R.K.; Silks, L.A.; Fishpaugh, J.A.; Kastner, M.E. Control of chemo- and stereoselectivity in the reactions of organocuprates with α-oxoketene dithioacetals. J. Am. Chem. Soc. 1985, 107, 4679–4692. [Google Scholar] [CrossRef]
- Zhong, Y.L.; Shing, T.K.M. Efficient and facile glycol cleavage oxidation using improved silica gel-supported sodium metaperiodate. J. Org. Chem. 1997, 62, 2622–2624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Jia, X.F.; Wang, J.X. The solvent-free addition reaction of allylzinc bromide and carbonyl compounds. Eur. J. Org. Chem. 2009, 2983–2986. [Google Scholar] [CrossRef]
- Knochel, P.; Yeh, M.C.P.; Berk, S.C.; Talbert, J. Synthesis and reactivity toward acyl chlorides and enones of the new highly functionalized copper reagents RCu(CN)Zni. J. Org. Chem. 1988, 53, 2390–2392. [Google Scholar] [CrossRef]
- Pappo, R.; Allen, J.D.S.; Lemieux, R.U.; Johnson, W.S. Notes—Osmium tetroxide-catalyzed periodate oxidation of olefinic bonds. J. Org. Chem. 1956, 21, 478–479. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Pescitelli, G.; Di Bari, L.; Berova, N. Conformational aspects in the studies of organic compounds by electronic circular dichroism. Chem. Soc. Rev. 2011, 40, 4603–4625. [Google Scholar] [CrossRef] [PubMed]
- McCann, D.M.; Stephens, P.J. Determination of absolute configuration using density functional theory calculations of optical rotation and electronic circular dichroism: Chiral alkenes. J. Org. Chem. 2006, 71, 6074–6098. [Google Scholar] [CrossRef] [PubMed]
- Di Bari, L.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke's method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef] [PubMed]
- Snatzke, G.; Wagner, U.; Wolff, H.P. Circular dichroism—LXXV: Cottonogenic derivatives of chiral bidentate ligands with the complex [Mo2(O2CCH3)4]. Tetrahedron 1981, 37, 349–361. [Google Scholar] [CrossRef]
- Gaurrand, S.; Desjardins, S.; Meyer, C.; Bonnet, P.; Argoullon, J.M.; Oulyadi, H.; Guillemont, J. Conformational analysis of R207910, a new drug candidate for the treatment of tuberculosis, by a combined NMR and molecular modeling approach. Chem. Biol. Drug Des. 2006, 68, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2–19 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, C.-J.; Wang, X.-K.; Xie, F.; Zhong, W.; Li, S. Asymmetric Synthesis and Absolute Configuration Assignment of a New Type of Bedaquiline Analogue. Molecules 2015, 20, 22272-22285. https://doi.org/10.3390/molecules201219846
Qiao C-J, Wang X-K, Xie F, Zhong W, Li S. Asymmetric Synthesis and Absolute Configuration Assignment of a New Type of Bedaquiline Analogue. Molecules. 2015; 20(12):22272-22285. https://doi.org/10.3390/molecules201219846
Chicago/Turabian StyleQiao, Chang-Jiang, Xiao-Kui Wang, Fei Xie, Wu Zhong, and Song Li. 2015. "Asymmetric Synthesis and Absolute Configuration Assignment of a New Type of Bedaquiline Analogue" Molecules 20, no. 12: 22272-22285. https://doi.org/10.3390/molecules201219846
APA StyleQiao, C.-J., Wang, X.-K., Xie, F., Zhong, W., & Li, S. (2015). Asymmetric Synthesis and Absolute Configuration Assignment of a New Type of Bedaquiline Analogue. Molecules, 20(12), 22272-22285. https://doi.org/10.3390/molecules201219846