
7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment



 ,
,
Abstract
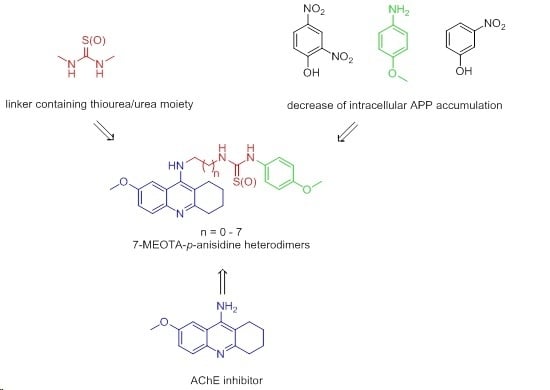
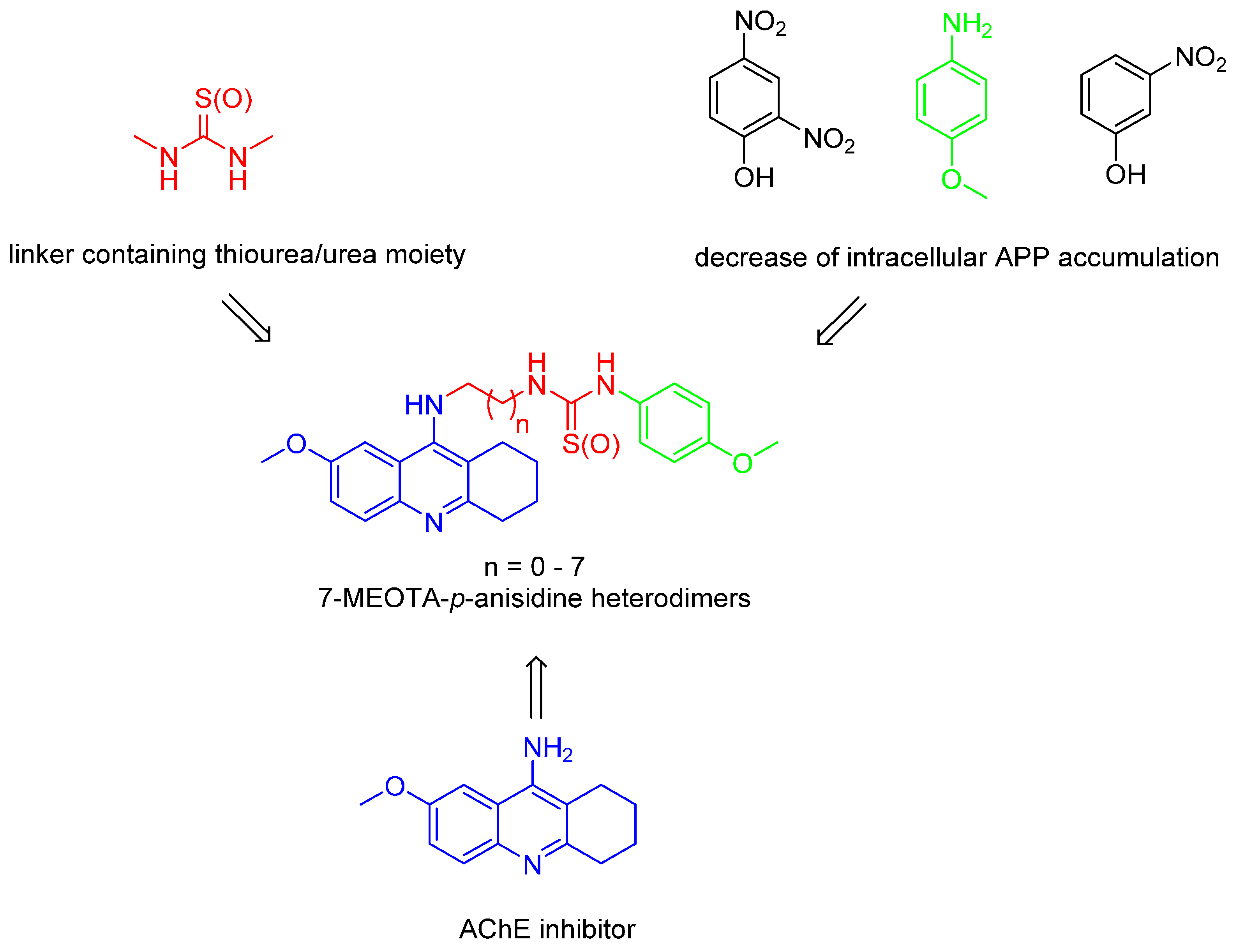
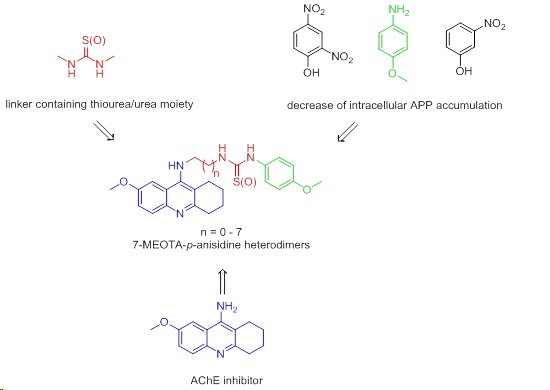
:
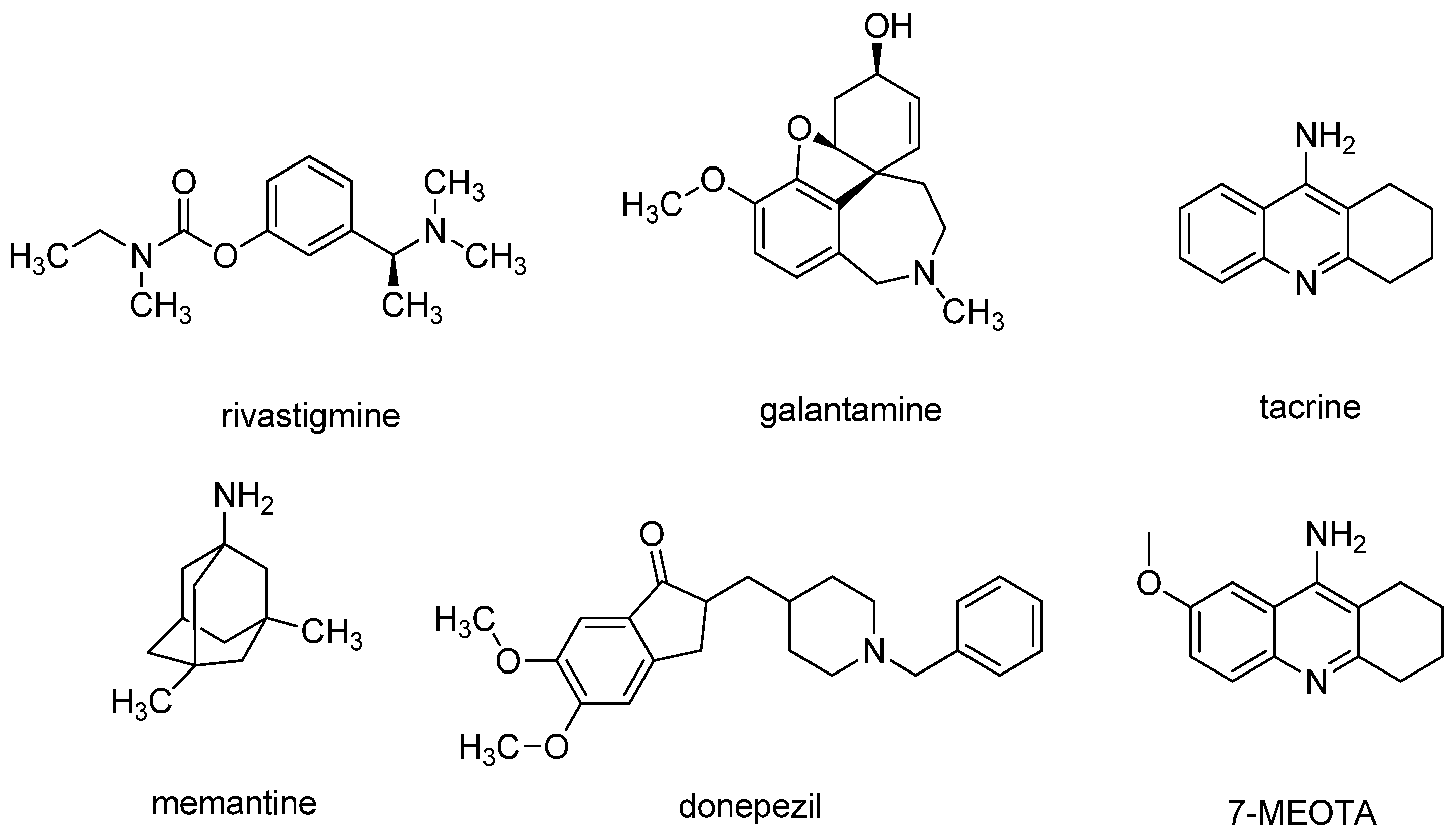
1. Introduction

2. Results and Discussion
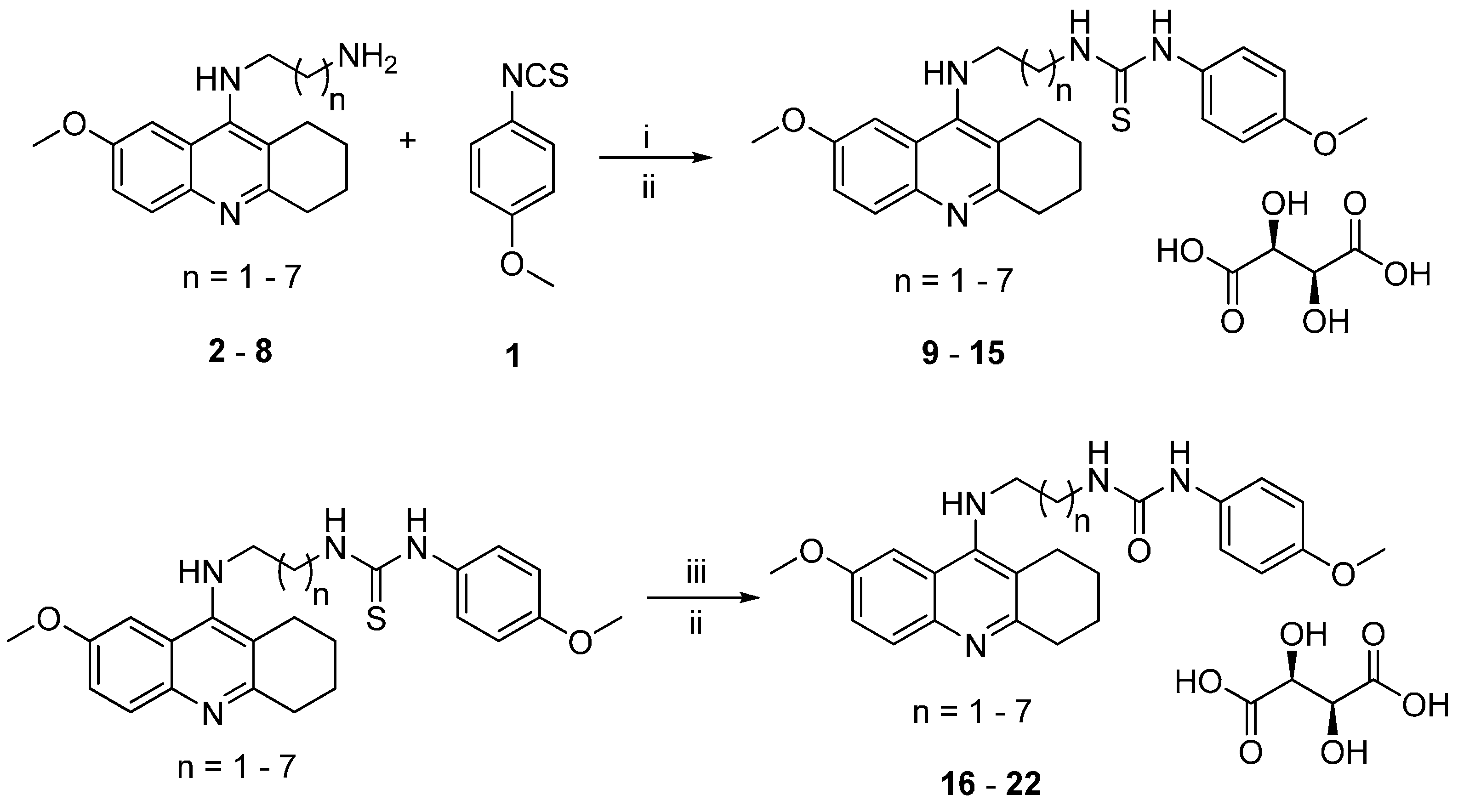
2.1. Chemistry
2.2. Biological Evaluation of AChE/BChE Activity
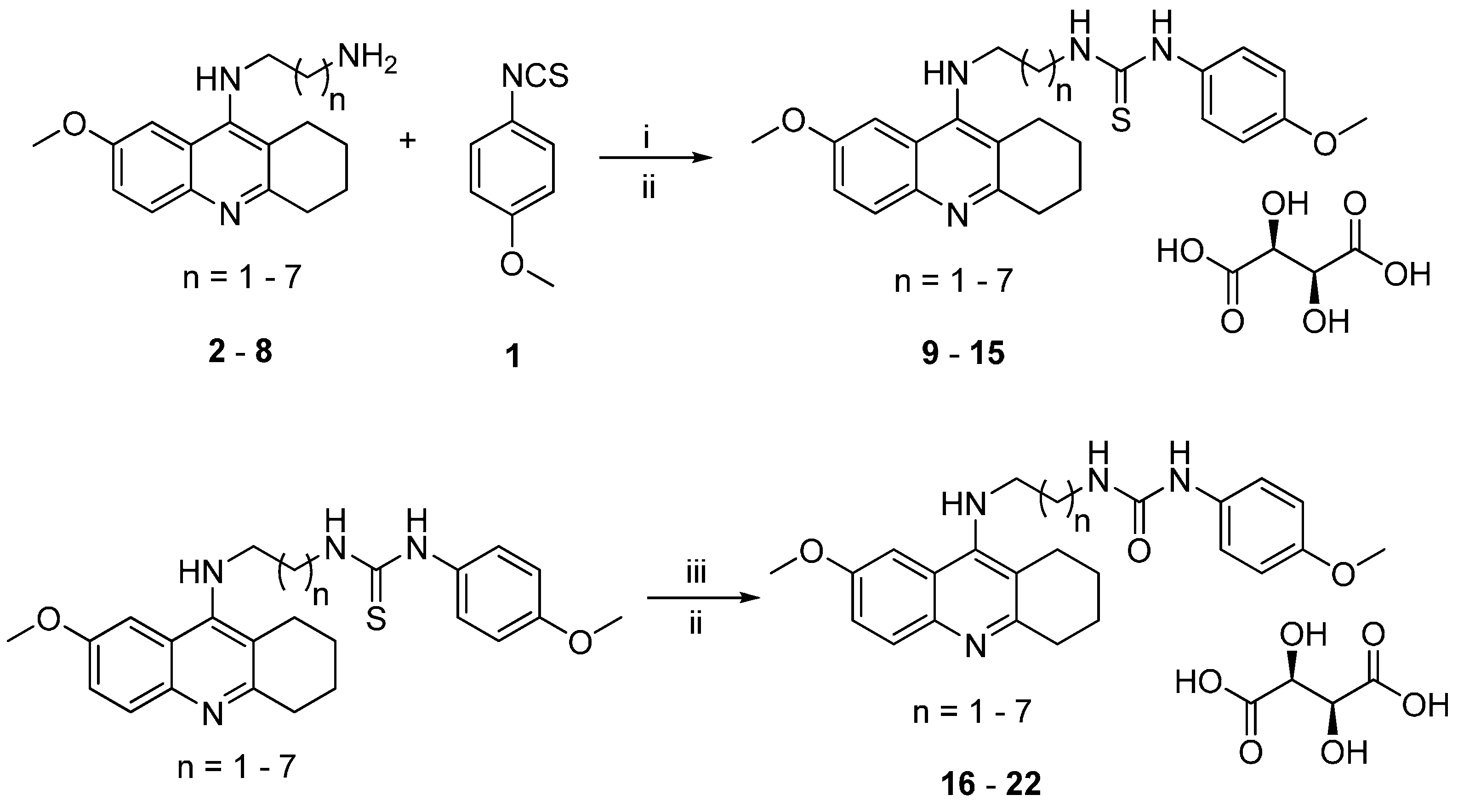

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | n | hAChE IC50 ± SEM (μM) a | hBChE IC50 ± SEM (μM) a | Selectivity for hAChE b |
|---|---|---|---|---|
| 9 | 1 | 43.6 ± 2.1 | 1.03 ± 0.1 | 0.02 |
| 10 | 2 | 6.36 ± 0.5 | 8.73 ± 0.1 | 1.37 |
| 11 | 3 | 32.8 ± 9.9 | 6.04 ± 0.1 | 0.18 |
| 12 | 4 | 4.9 ± 0.3 | 13.5 ± 0.7 | 2.71 |
| 13 | 5 | 10.3 ± 1.3 | 9.35 ± 0.1 | 0.90 |
| 14 | 6 | 3.96 ± 0.1 | 3.13 ± 0.3 | 0.79 |
| 15 | 7 | 1.36 ± 0.4 | 10.2 ± 4.7 | 7.53 |
| 16 | 1 | 44.9 ± 1.4 | 11.9 ± 2.3 | 0.27 |
| 17 | 2 | 26.9 ± 5.9 | 15.9 ± 4.1 | 0.59 |
| 18 | 3 | 13.8 ± 3.9 | 9.34 ± 0.1 | 0.68 |
| 19 | 4 | 1.35 ± 0.3 | 10.9 ± 1.6 | 8.07 |
| 20 | 5 | 4.56 ± 0.9 | 5.75 ± 0.4 | 1.26 |
| 21 | 6 | 1.72 ± 0.3 | 1.69 ± 0.2 | 0.98 |
| 22 | 7 | 2.14 ± 0.6 | 1.34 ± 0.2 | 0.63 |
| Tacrine | - | 0.32 ± 0.01 | 0.08 ± 0.001 | 0.68 |
| 7-MEOTA | - | 10.0 ± 0.9 | 17.6 ± 0.8 | 1.76 |
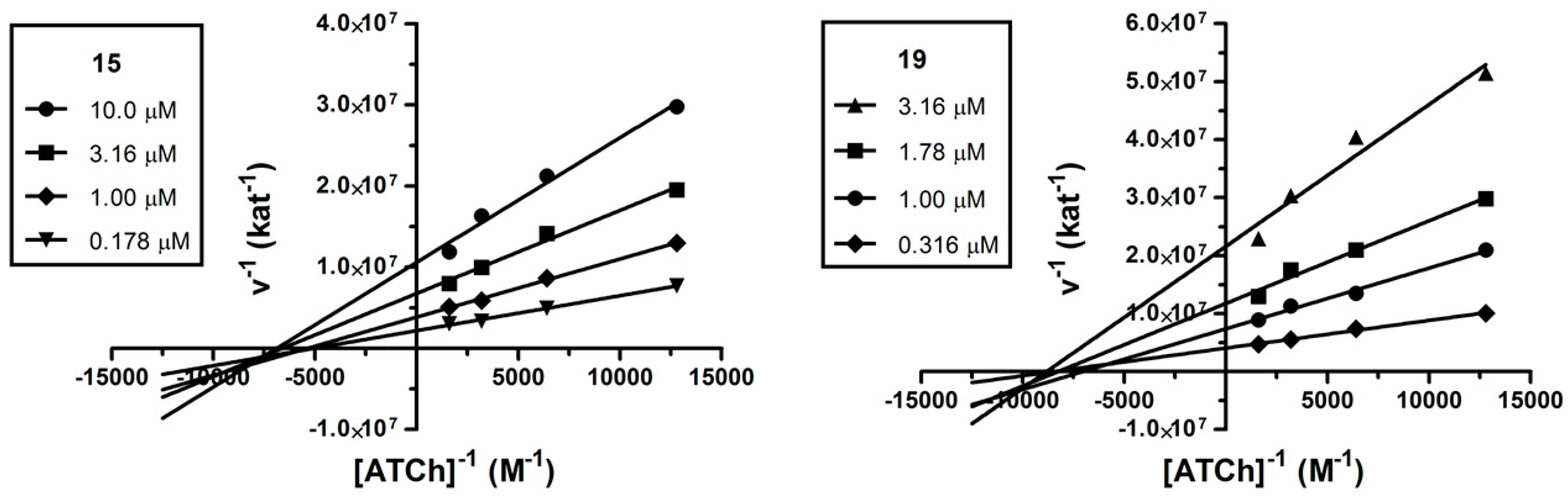
2.3. Kinetic Analysis
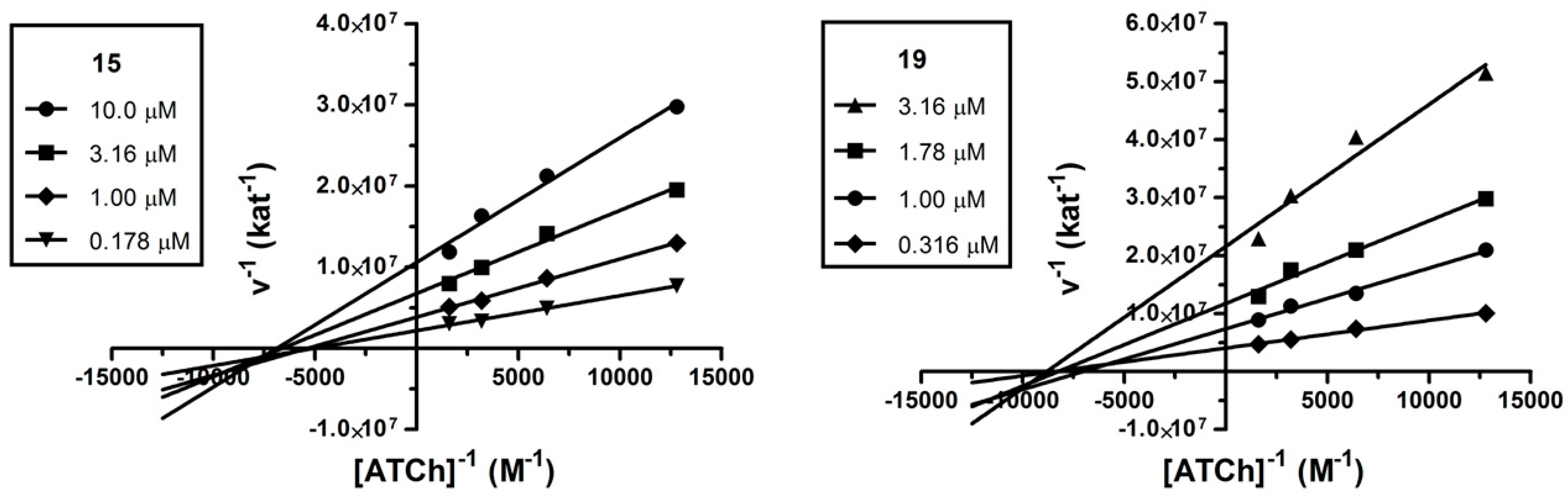
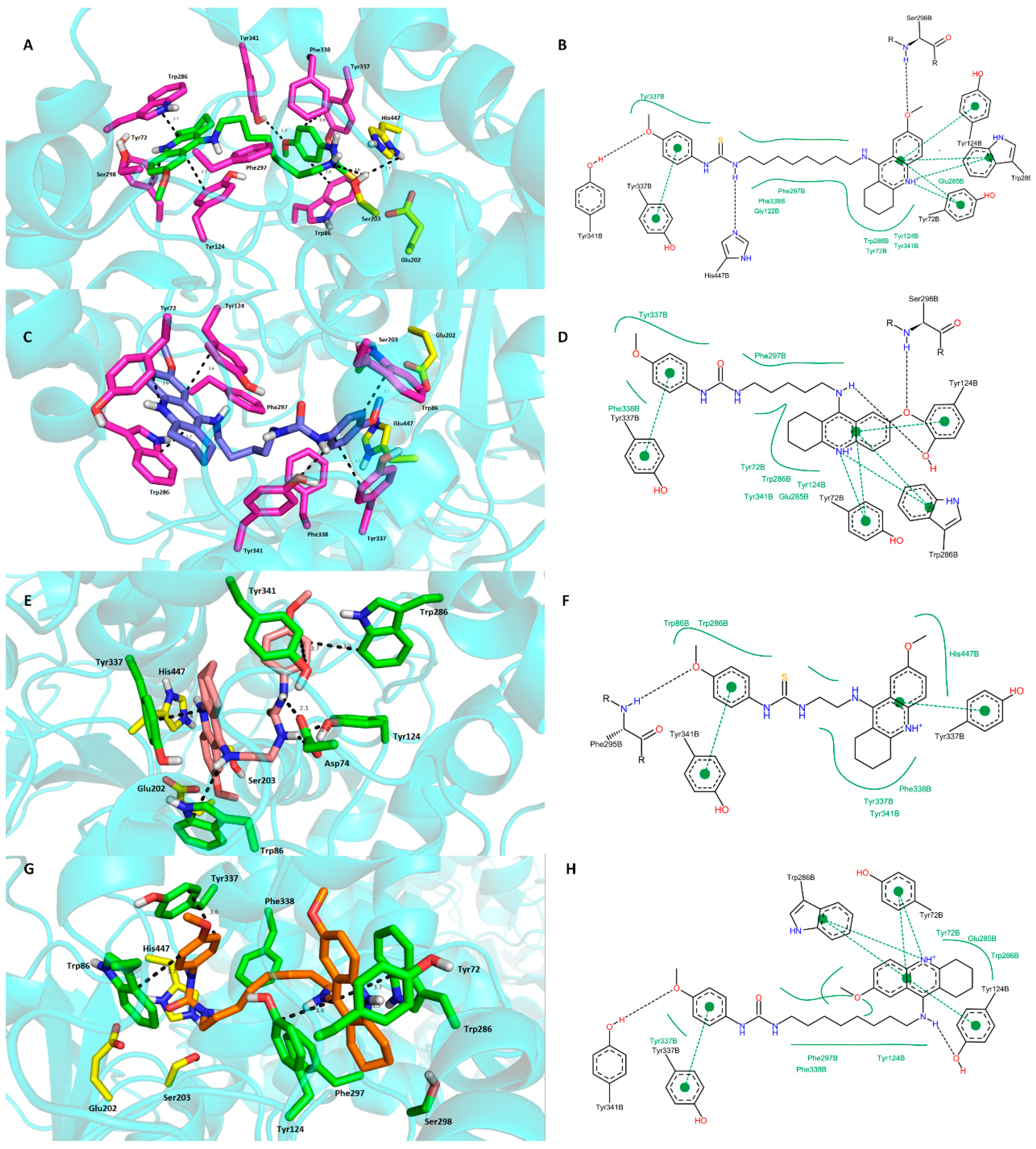
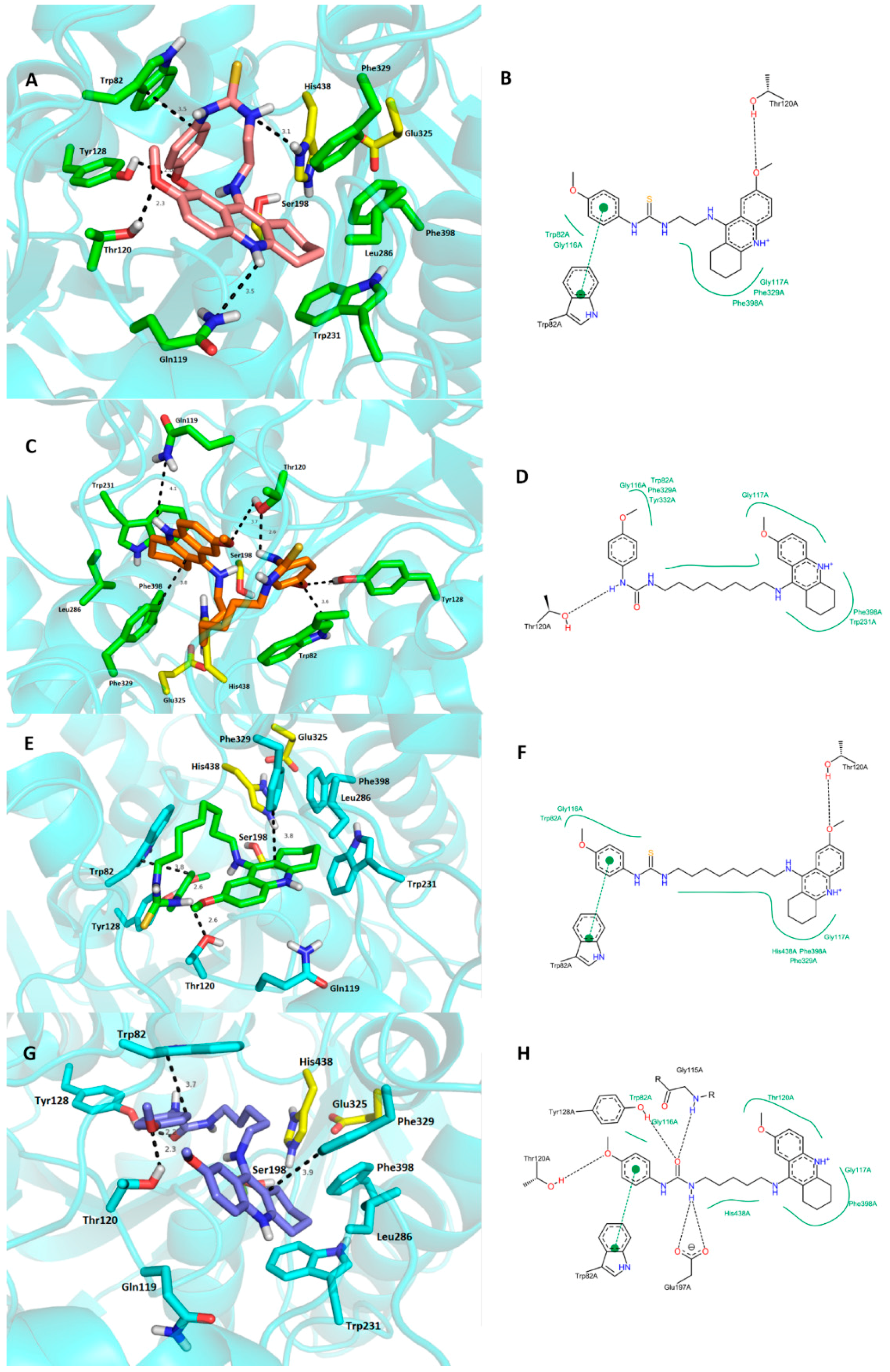
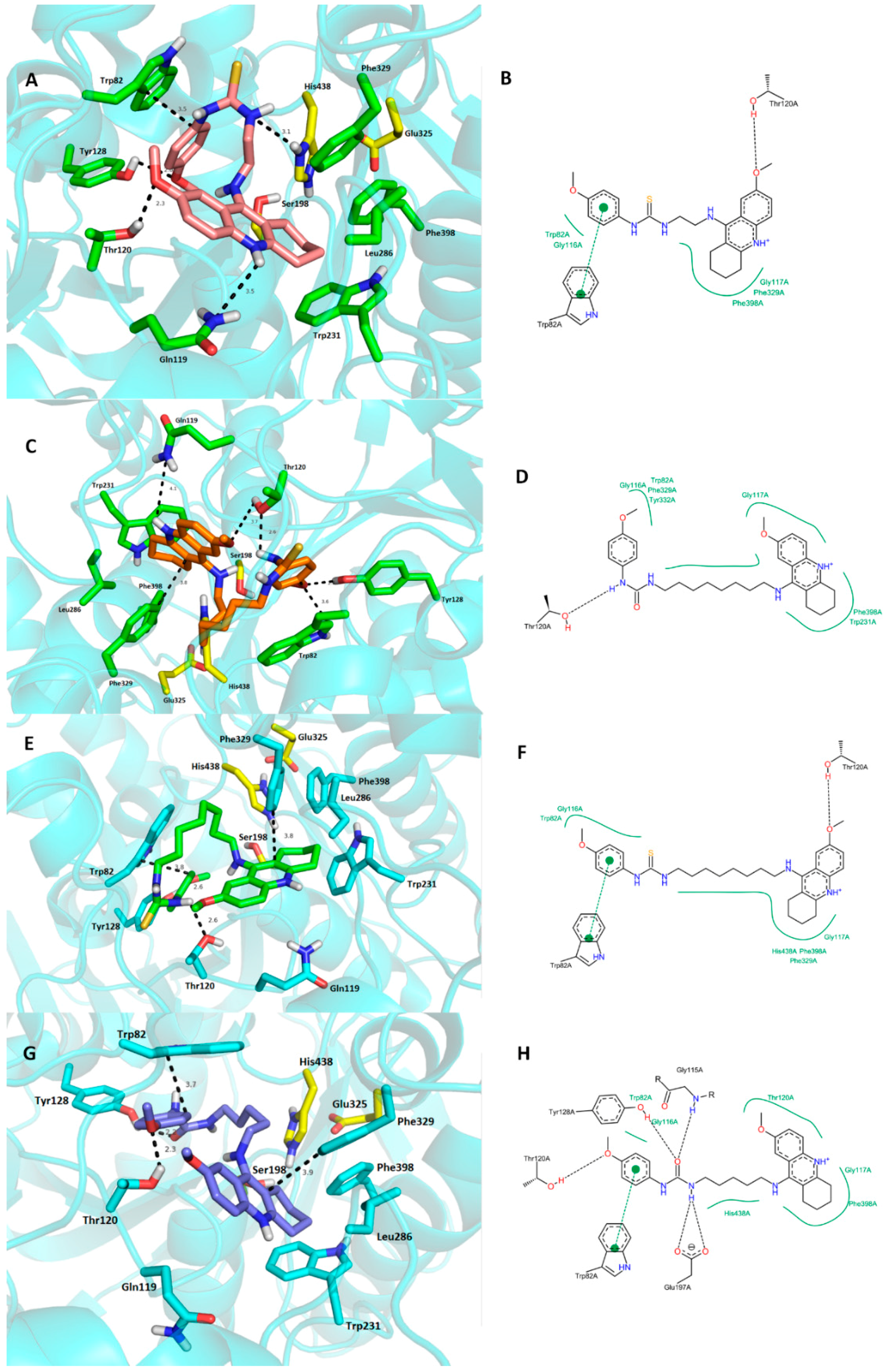
2.4. Molecular Modeling Studies
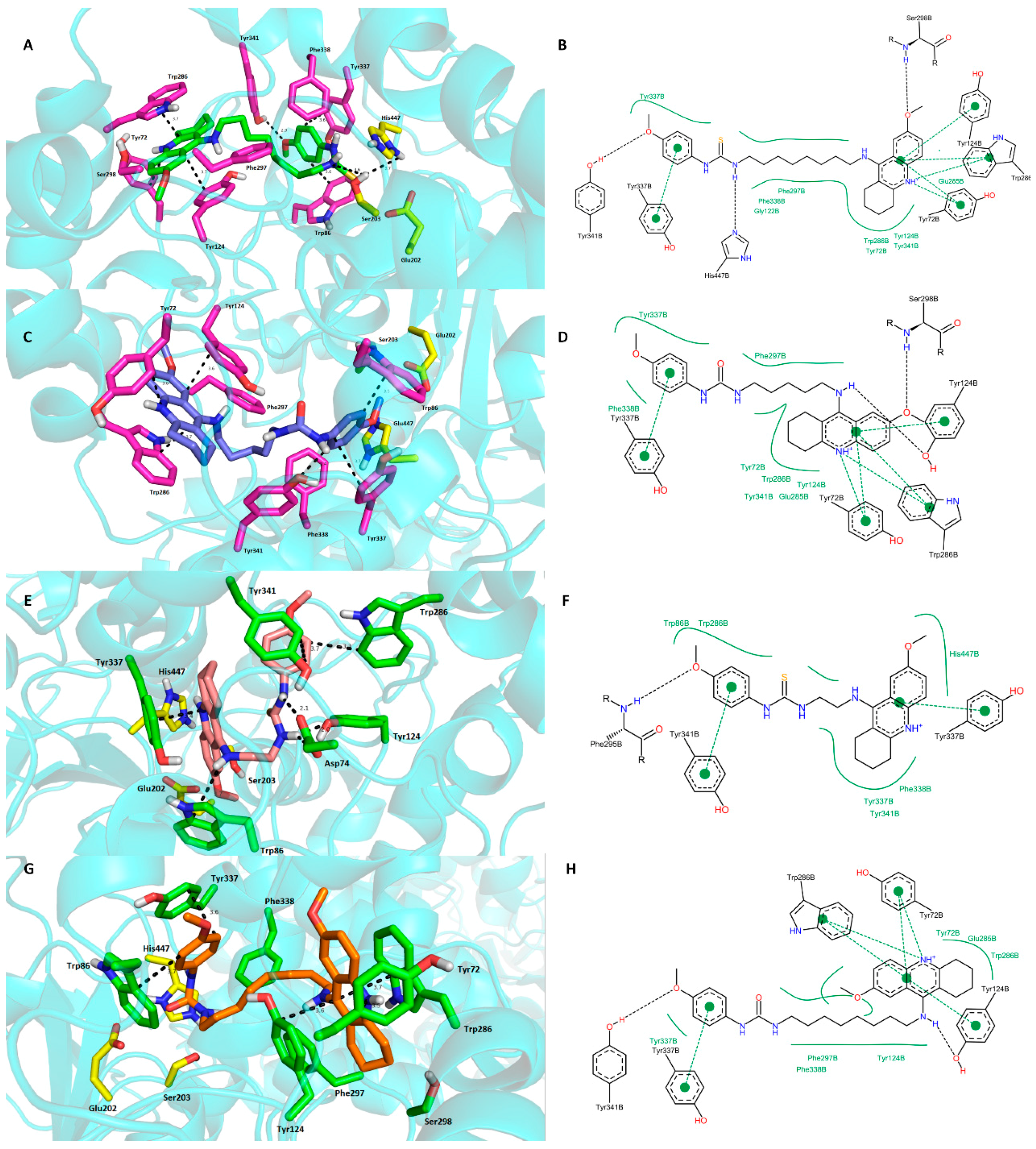
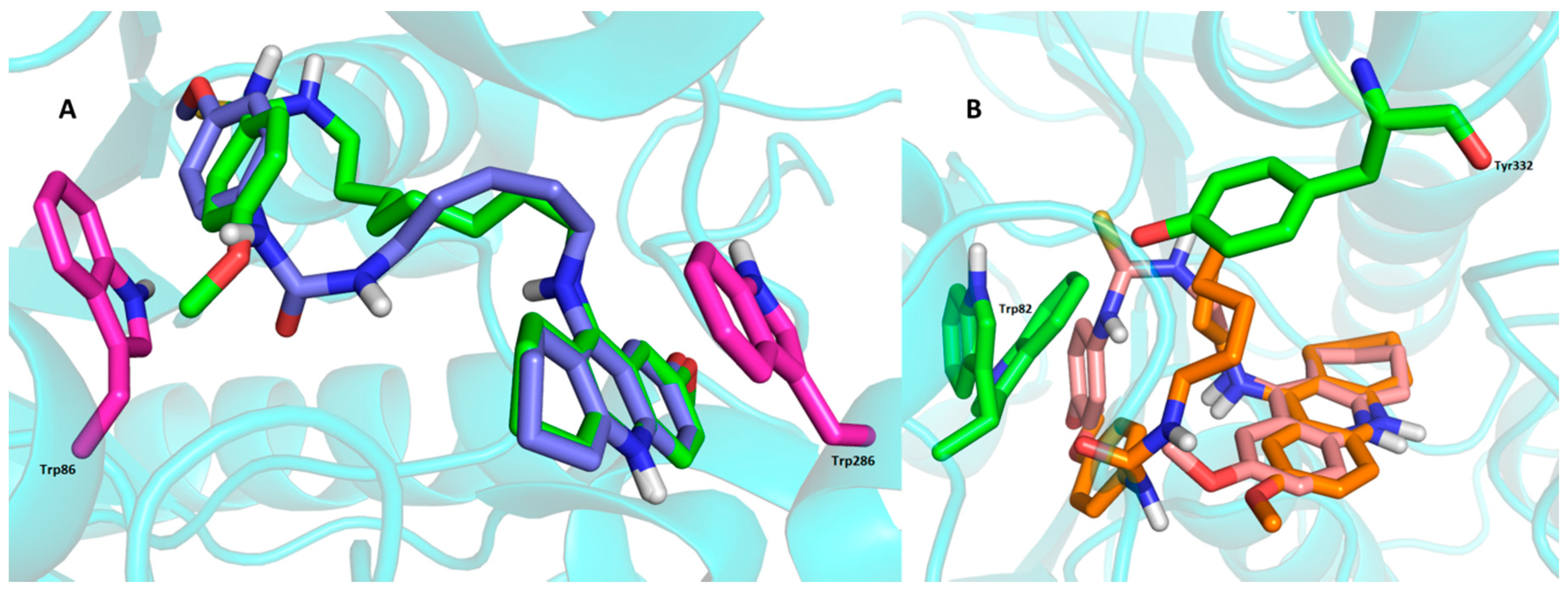
3. Experimental Section
3.1. General Chemistry
3.1.1. General Synthetic Procedure for 7-MEOTA-p-anisidine Thiourea 2,3-Dihydroxysuccinate Hybrids 9–15
3.1.2. General Synthetic Procedure for 7-MEOTA-p-anisidine Urea 2,3-Dihydroxysuccinate Hybrids 16–22
3.2. Biochemical Studies
3.2.1. In Vitro Anti-Cholinesterase Assay
3.2.2. Kinetic Study of AChE Inhibition
3.3. Molecular Modeling Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332–384. [Google Scholar]
- Maresova, P.; Mohelska, H.; Dolejs, J.; Kuca, K. Socio-economic aspects of Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; Mercken, M.; de Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Schrag, M.; Mueller, C.; Oyoyo, U.; Smith, M.A.; Kirsch, W.M. Iron, zinc and copper in the Alzheimer’s disease brain: A quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog. Neurobiol. 2011, 94, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [PubMed]
- Johnson, J.W.; Glasgow, N.G.; Povysheva, N.V. Recent insights into the mode of action of memantine and ketamine. Curr. Opin. Pharmacol. 2015, 20, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.K.; Majovski, L.V.; Marsh, G.M.; Tachiki, K.; Kling, A. Oral tetrahydroaminoacridine in long-term treatment of senile dementia, Alzheimer type. N. Engl. J. Med. 1986, 315, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Patocka, J.; Jun, D.; Kuca, K. Possible role of hydroxylated metabolites of tacrine in drug toxicity and therapy of Alzheimer’s disease. Curr. Drug Metab. 2008, 9, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Soukup, O.; Jun, D.; Zdarova-Karasova, J.; Patocka, J.; Musilek, K.; Korabecny, J.; Krusek, J.; Kaniakova, M.; Sepsova, V.; Mandikova, J.; et al. A resurrection of 7-MEOTA: A comparison with tacrine. Curr. Alzheimer Res. 2013, 10, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Sepsova, V.; Karasova, J.Z.; Tobin, G.; Jun, D.; Korabecny, J.; Cabelova, P.; Janska, K.; Krusek, J.; Skrenkova, K.; Kuca, K.; et al. Cholinergic properties of new 7-methoxytacrine-donepezil derivatives. Gen. Physiol. Biophys. 2015, 34, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Misik, J.; Korabecny, J.; Nepovimova, E.; Cabelova, P.; Kassa, J. The effects of novel 7-MEOTA-donepezil like hybrids and N-alkylated tacrine analogues in the treatment of quinuclidinyl benzilate-induced behavioural deficits in rats performing the multiple T-maze test. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czechoslov. 2015. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget drug design strategy: Quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Dolezal, R.; Cabelova, P.; Horova, A.; Hruba, E.; Ricny, J.; Sedlacek, L.; Nepovimova, E.; Spilovska, K.; Andrs, M.; et al. 7-MEOTA–donepezil like compounds as cholinesterase inhibitors: Synthesis, pharmacological evaluation, molecular modeling and QSAR studies. Eur. J. Med. Chem. 2014, 82, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Binder, J.; Zemek, F.; Marek, J.; Pohanka, M.; Opletalova, V.; Dohnal, V.; Kuca, K. Synthesis and in vitro evaluation of N-alkyl-7-methoxytacrine hydrochlorides as potential cholinesterase inhibitors in Alzheimer disease. Bioorg. Med. Chem. Lett. 2010, 20, 6093–6095. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Zemek, F.; Horova, A.; Holas, O.; Nepovimova, E.; Opletalova, V.; Hroudova, J.; Fisar, Z.; Jung, Y.-S.; et al. Synthesis and in vitro evaluation of 7-methoxy-N-(pent-4-enyl)-1,2,3,4-tetrahydroacridin-9-amine—New tacrine derivate with cholinergic properties. Bioorg. Med. Chem. Lett. 2011, 21, 6563–6566. [Google Scholar] [CrossRef] [PubMed]
- Hamulakova, S.; Janovec, L.; Hrabinova, M.; Spilovska, K.; Korabecny, J.; Kristian, P.; Kuca, K.; Imrich, J. Synthesis and biological evaluation of novel tacrine derivatives and tacrine-coumarin hybrids as cholinesterase inhibitors. J. Med. Chem. 2014, 57, 7073–7084. [Google Scholar] [CrossRef] [PubMed]
- Korabecny, J.; Musilek, K.; Holas, O.; Nepovimova, E.; Jun, D.; Zemek, F.; Opletalova, V.; Patocka, J.; Dohnal, V.; Nachon, F.; et al. Synthesis and in vitro evaluation of N-(Bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a cholinesterase inhibitor with regard to Alzheimer’s disease treatment. Mol. Basel Switz. 2010, 15, 8804–8812. [Google Scholar] [CrossRef] [PubMed]
- Minarini, A.; Milelli, A.; Simoni, E.; Rosini, M.; Bolognesi, M.L.; Marchetti, C.; Tumiatti, V. Multifunctional tacrine derivatives in Alzheimer’s disease. Curr. Top. Med. Chem. 2013, 13, 1771–1786. [Google Scholar] [CrossRef] [PubMed]
- Carlier, P.R.; Chow, E.S.; Han, Y.; Liu, J.; el Yazal, J.; Pang, Y.P. Heterodimeric tacrine-based acetylcholinesterase inhibitors: Investigating ligand-peripheral site interactions. J. Med. Chem. 1999, 42, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Park, M.K.; Jhang, H.E.; Yi, J.; Nahm, K.; Cho, D.W.; Ra, C.S.; Musilek, K.; Horova, A.; Korabecny, J.; et al. Preparation of 7-Methoxy Tacrine Dimer Analogs and Their in vitro/in silico Evaluation as Potential Cholinesterase Inhibitors. Bull. Korean Chem. Soc. 2015, 36, 1654–1660. [Google Scholar] [CrossRef]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: How is structure related to function? Chem. Biol. Interact. 2008, 175, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Kral, J.; Horova, A.; Musilek, K.; Soukup, O.; Drtinova, L.; Gazova, Z.; Siposova, K.; Kuca, K. 7-Methoxytacrine-adamantylamine heterodimers as cholinesterase inhibitors in Alzheimer’s disease treatment—Synthesis, biological evaluation and molecular modeling studies. Mol. Basel Switz. 2013, 18, 2397–2418. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Horova, A.; Musilek, K.; Nepovimova, E.; Drtinova, L.; Gazova, Z.; Siposova, K.; Dolezal, R.; Jun, D.; et al. Design, synthesis and in vitro testing of 7-methoxytacrine-amantadine analogues: A novel cholinesterase inhibitors for the treatment of Alzheimer’s disease. Med. Chem. Res. 2015, 24, 2645–2655. [Google Scholar] [CrossRef]
- Recanatini, M.; Cavalli, A.; Hansch, C. A comparative QSAR analysis of acetylcholinesterase inhibitors currently studied for the treatment of Alzheimer’s disease. Chem. Biol. Interact. 1997, 105, 199–228. [Google Scholar] [CrossRef]
- Iqbal, J.; Zaib, S.; Saeed, A.; Muddassar, M. Biological Evaluation of Halogenated Thioureas as Cholinesterases Inhibitors against Alzheimer’s Disease & Molecular Modeling Studies. Lett. Drug Des. Discov. 2015, 12, 488–494. [Google Scholar]
- Paula Lima, A.C.; Arriagada, C.; Toro, R.; Cárdenas, A.M.; Caviedes, R.; Ferreira, S.T.; Caviedes, P. Small-molecule aggregation inhibitors reduce excess amyloid in a trisomy 16 mouse cortical cell line. Biol. Res. 2008, 41, 129–136. [Google Scholar] [PubMed]
- Munch, H.; Hansen, J.S.; Pittelkow, M.; Christensen, J.B.; Boas, U. A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 2008, 49, 3117–3119. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Pohanka, M.; Karasova, J.Z.; Kuca, K.; Pikula, J.; Holas, O.; Korabecny, J.; Cabal, J. Colorimetric dipstick for assay of organophosphate pesticides and nerve agents represented by paraoxon, sarin and VX. Talanta 2010, 81, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Geula, C.; Mesulam, M.M. Cholinesterases and the pathology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1995, 9 (Suppl. 2), 23–28. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.G. The pharmacology of donepezil: A new treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 1999, 1, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P. Crystal Structures of Human Cholinesterases in Complex with Huprine W and Tacrine: Elements of Specificity for Anti-Alzheimer’S Drugs Targeting Acetyl- and Butyrylcholinesterase. Biochem.J. 2012, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, R.; Sobeslav, V.; Hornig, O.; Balik, L.; Korabecny, J.; Kuca, K. HPC Cloud Technologies for Virtual Screening in Drug Discovery. In Intelligent Information and Database Systems; Nguyen, N.T., Trawiński, B., Kosala, R., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2015; pp. 440–449. [Google Scholar]
- Dolezal, R.; Korabecny, J.; Malinak, D.; Honegr, J.; Musilek, K.; Kuca, K. Ligand-based 3D QSAR analysis of reactivation potency of mono- and bis-pyridinium aldoximes toward VX-inhibited rat acetylcholinesterase. J. Mol. Graph. Model. 2015, 56, 113–129. [Google Scholar] [CrossRef] [PubMed]
- PoseView. Available online: http://poseview.zbh.uni-hamburg.de/poseview/wizard (accessed on 16 July 2012).
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine—Trolox Hybrids: A Novel Class of Centrally Active, Non-Hepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low in Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, L.; Jin, Y.-H. An effective PSO-based memetic algorithm for flow shop scheduling. IEEE Trans. Syst. Man Cybern. B Cybern. Cybern. 2007, 37, 18–27. [Google Scholar] [CrossRef]
- Korábecný, J.; Hrubá, E.; Soukup, O.; Zemek, F.; Musílek, K.; Nepovímová, E.; Spilovská, K.; Opletalová, V.; Kuca, K. Intended pharmacotherapeutical approaches of Alzheimer’s disease therapy. Ceská Slov. Farm. Cas. Ceské Farm. Spolecnosti Slov. Farm. Spolecnosti 2012, 61, 4–10. [Google Scholar]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- León, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korabecny, J.; Andrs, M.; Nepovimova, E.; Dolezal, R.; Babkova, K.; Horova, A.; Malinak, D.; Mezeiova, E.; Gorecki, L.; Sepsova, V.; et al. 7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment. Molecules 2015, 20, 22084-22101. https://doi.org/10.3390/molecules201219836
Korabecny J, Andrs M, Nepovimova E, Dolezal R, Babkova K, Horova A, Malinak D, Mezeiova E, Gorecki L, Sepsova V, et al. 7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment. Molecules. 2015; 20(12):22084-22101. https://doi.org/10.3390/molecules201219836
Chicago/Turabian StyleKorabecny, Jan, Martin Andrs, Eugenie Nepovimova, Rafael Dolezal, Katerina Babkova, Anna Horova, David Malinak, Eva Mezeiova, Lukas Gorecki, Vendula Sepsova, and et al. 2015. "7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment" Molecules 20, no. 12: 22084-22101. https://doi.org/10.3390/molecules201219836
APA StyleKorabecny, J., Andrs, M., Nepovimova, E., Dolezal, R., Babkova, K., Horova, A., Malinak, D., Mezeiova, E., Gorecki, L., Sepsova, V., Hrabinova, M., Soukup, O., Jun, D., & Kuca, K. (2015). 7-Methoxytacrine-p-Anisidine Hybrids as Novel Dual Binding Site Acetylcholinesterase Inhibitors for Alzheimer’s Disease Treatment. Molecules, 20(12), 22084-22101. https://doi.org/10.3390/molecules201219836