
Mitochondrion-Targeted Peptide SS-31 Inhibited Oxidized Low-Density Lipoproteins-Induced Foam Cell Formation through both ROS Scavenging and Inhibition of Cholesterol Influx in RAW264.7 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
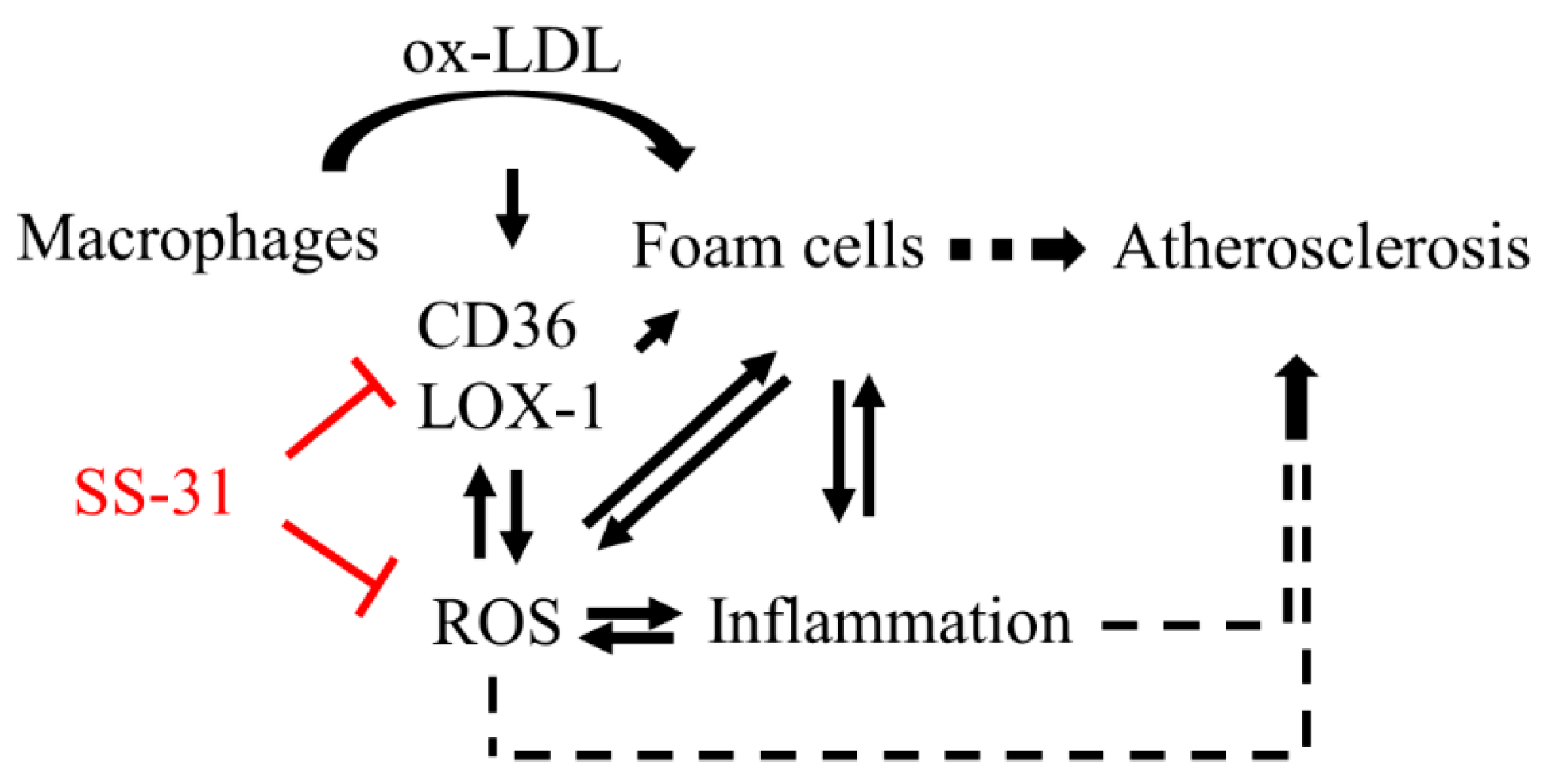
:1. Introduction
2. Results
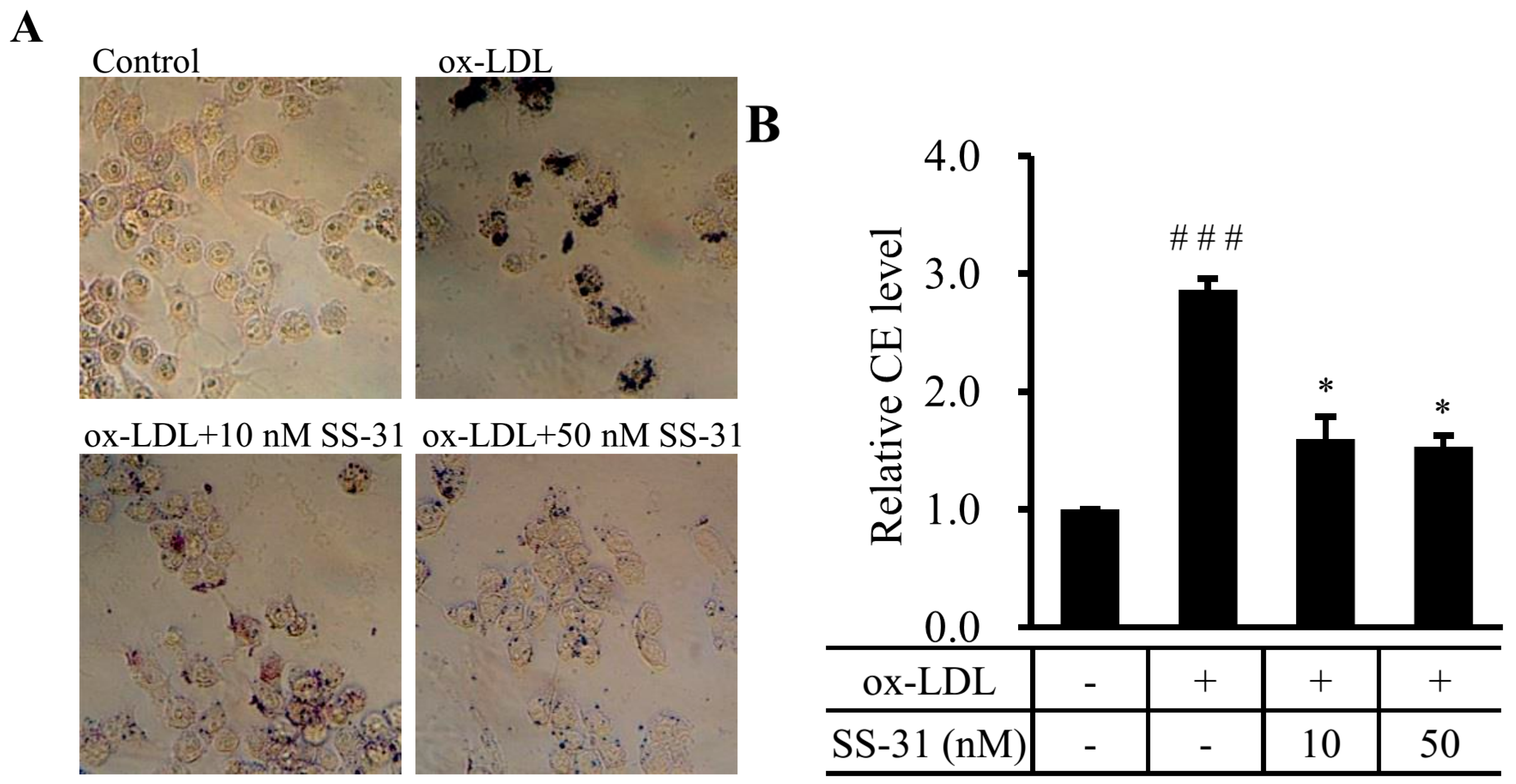
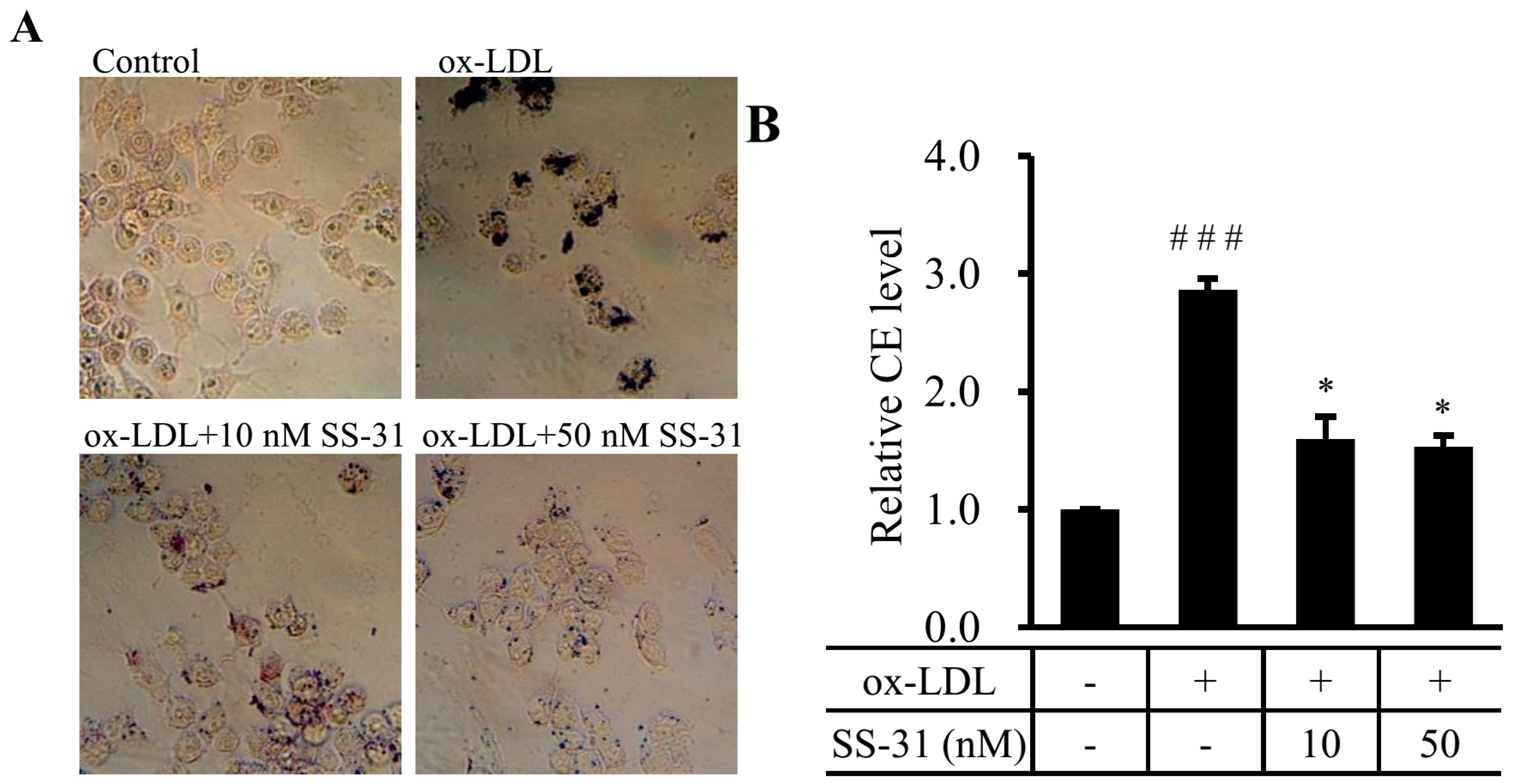
2.1. SS-31 Reduces Ox-LDL-Induced Cholesterol Accumulation in RAW264.7 Cells
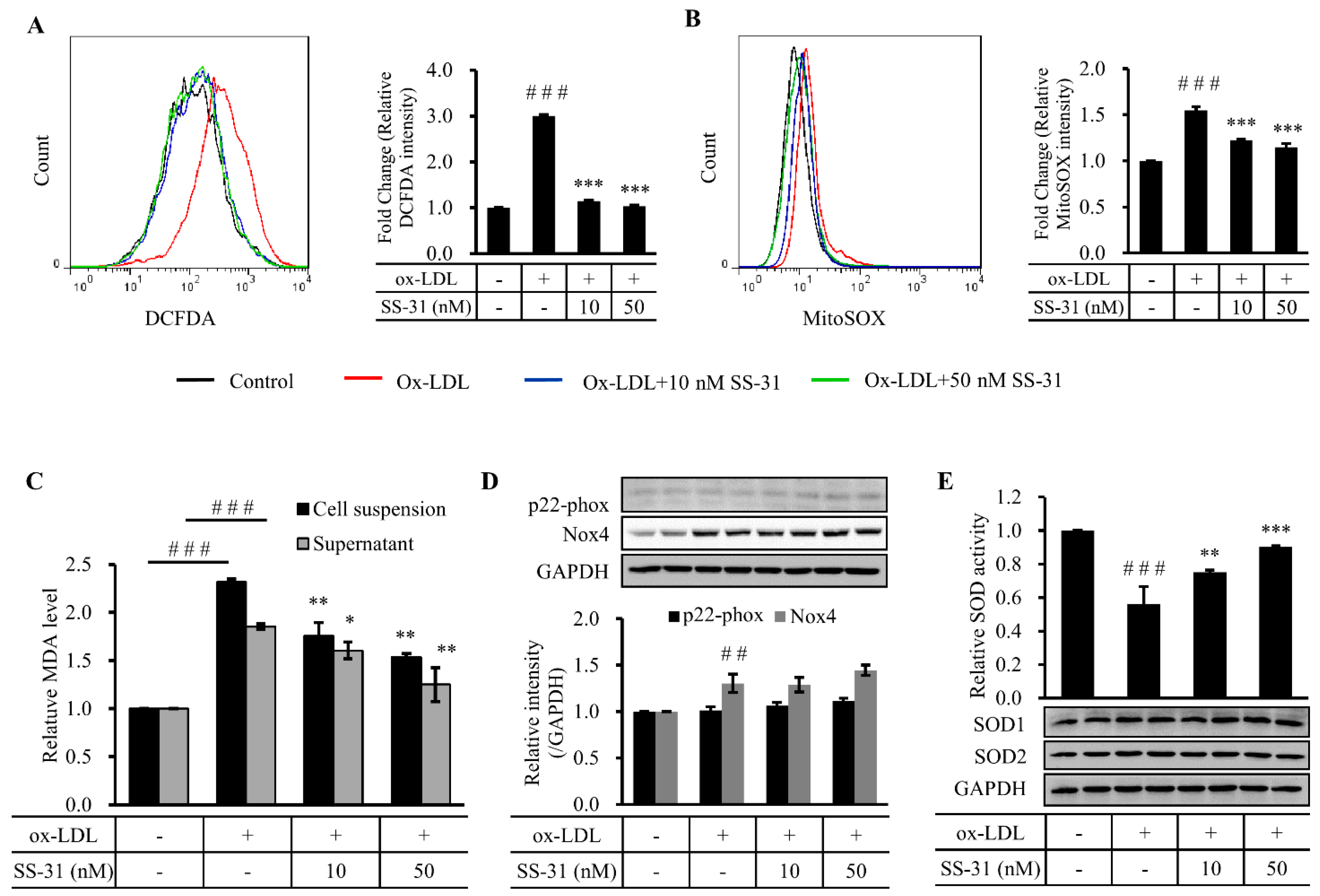
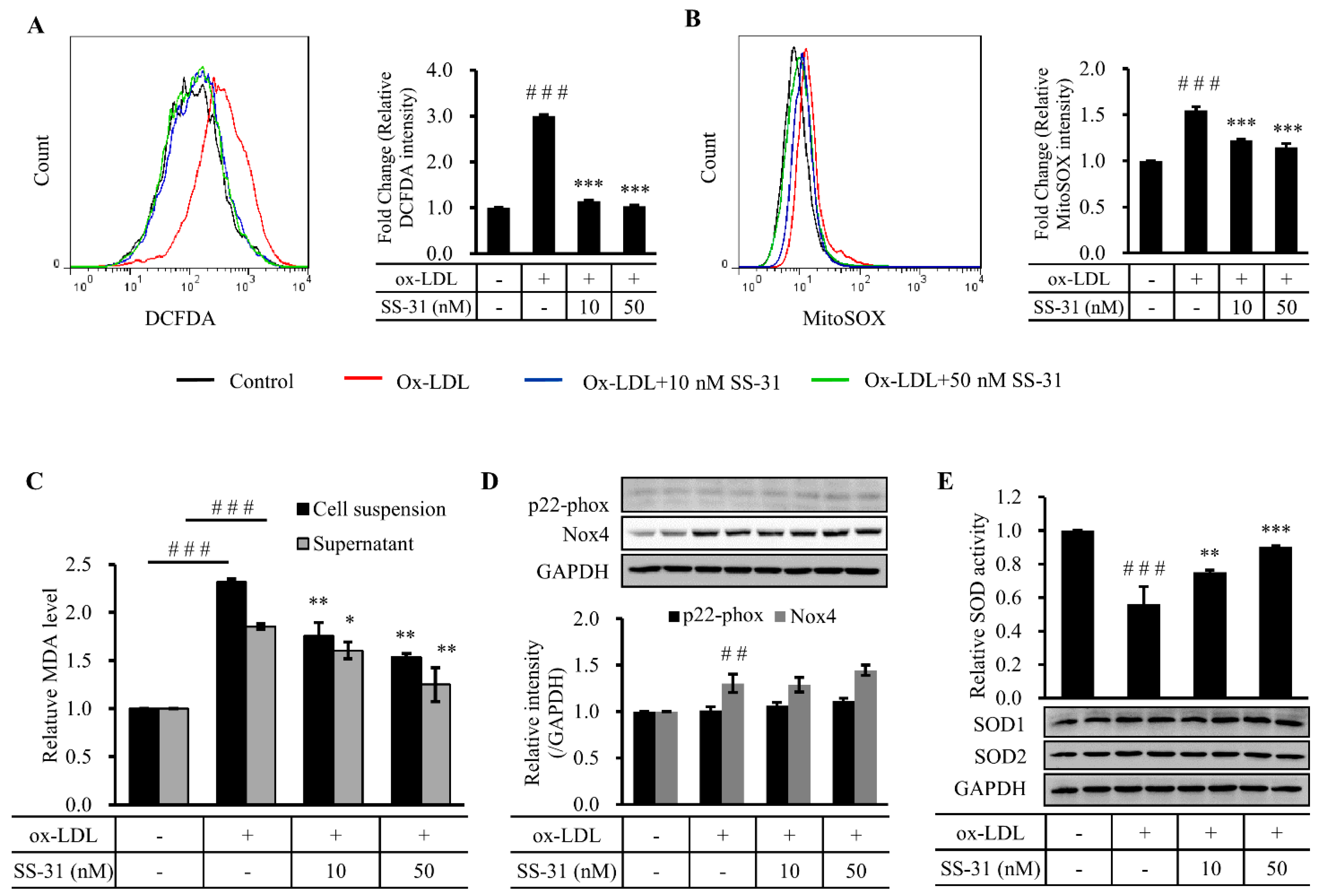
2.2. SS-31 Suppresses Ox-LDL-Induced Oxidative Stress in RAW264.7 Cells
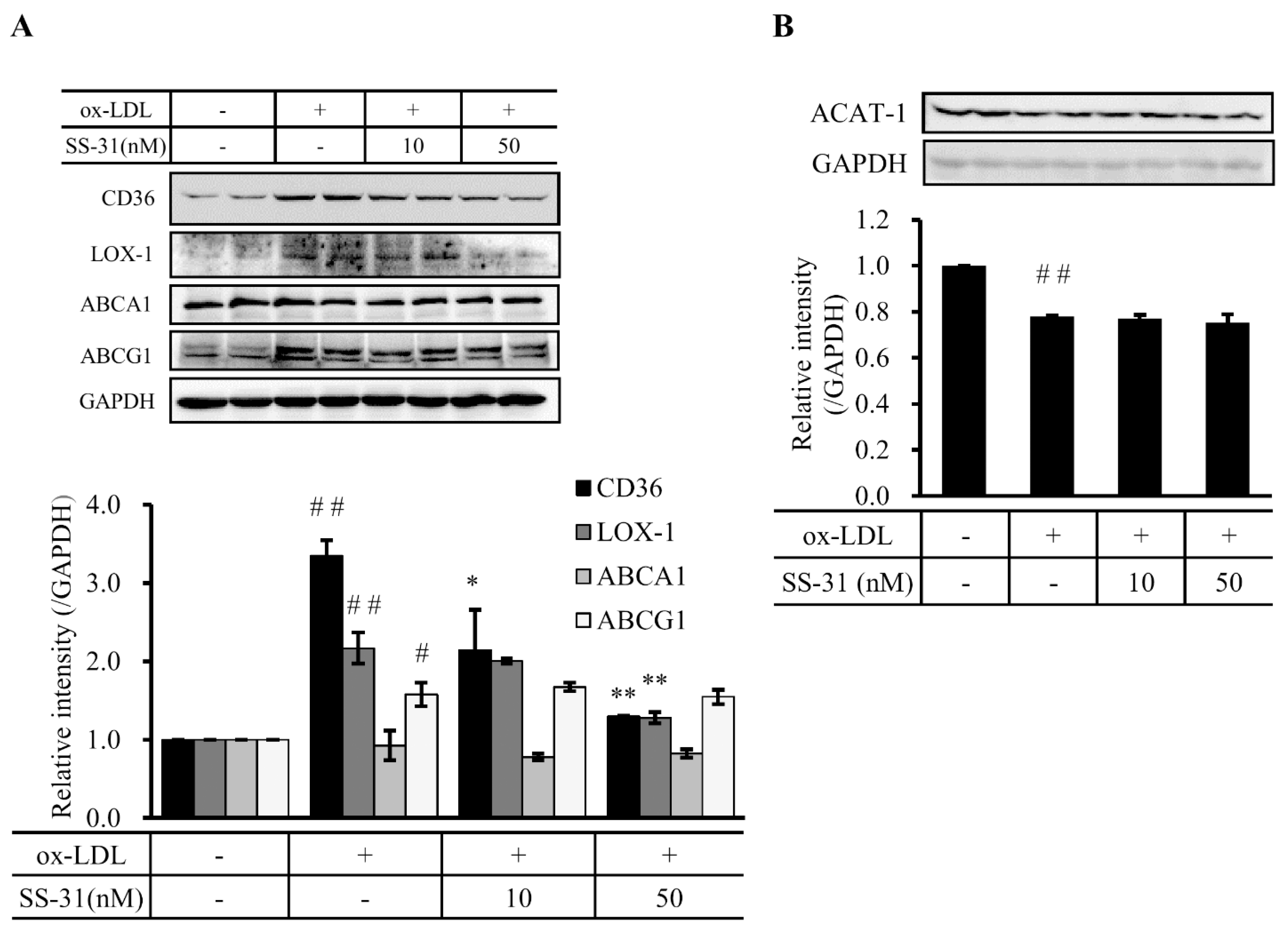
2.3. SS-31 Down-Regulates Ox-LDL-Induced Expression of CD36 and LOX-1 in RAW264.7 Cells
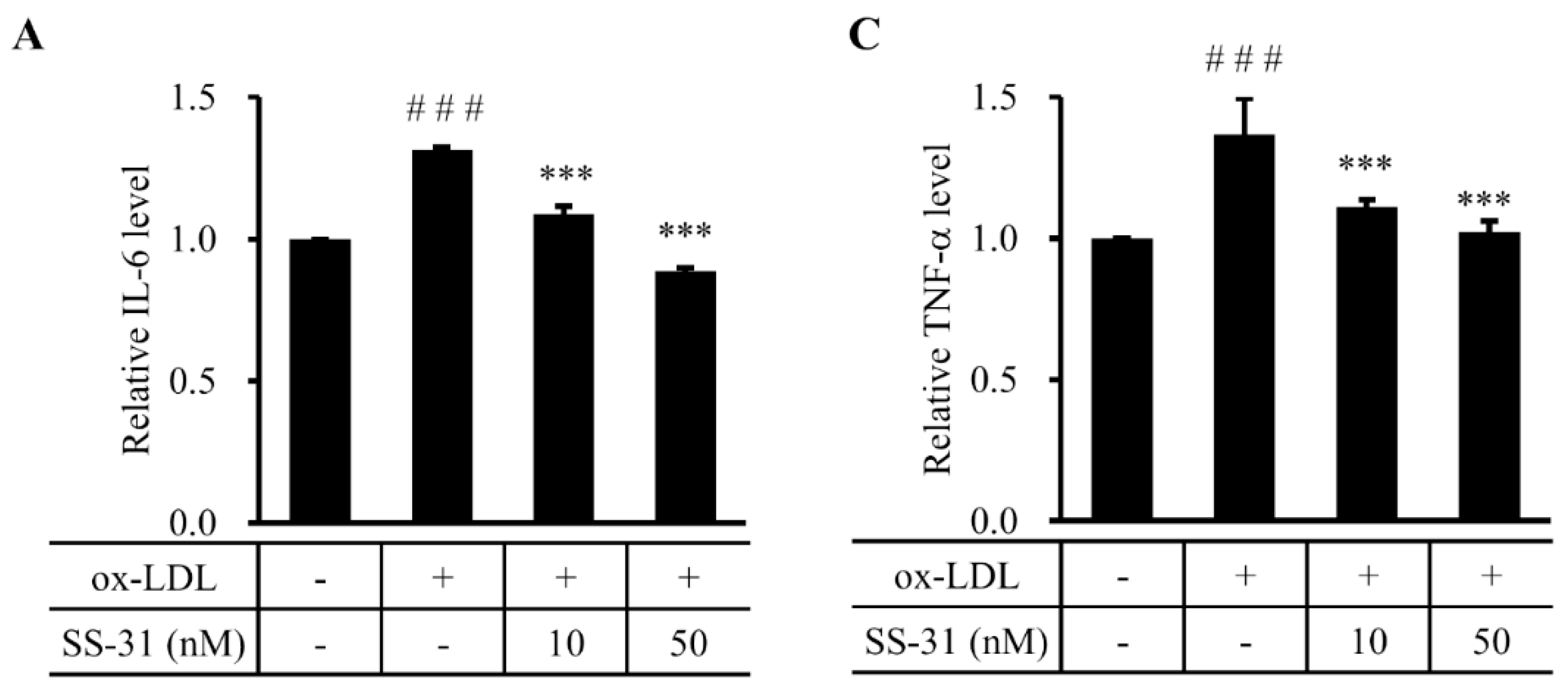
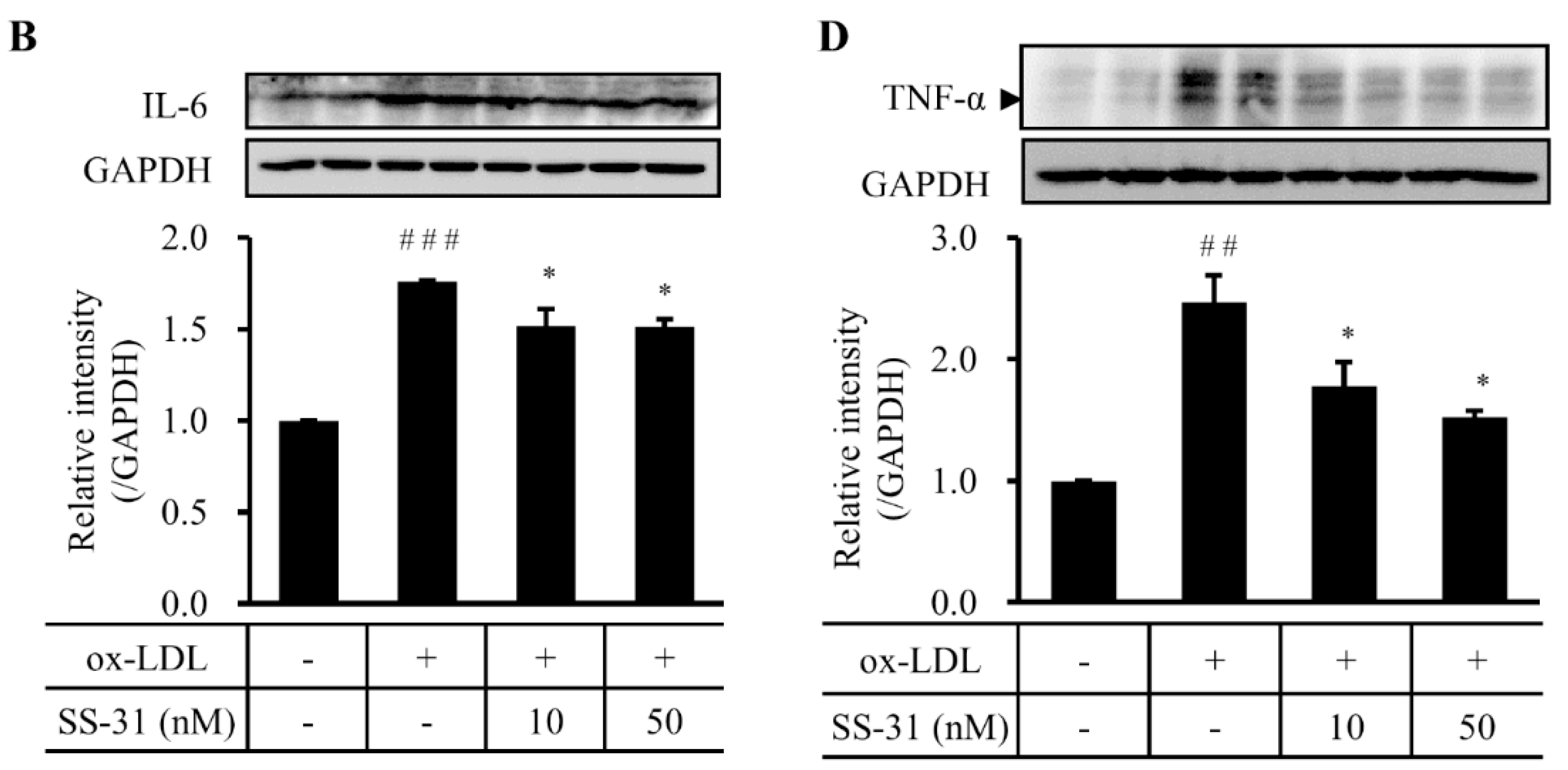
2.4. SS-31 Inhibits Ox-LDL-Induced Inflammatory Response in RAW264.7 Cells
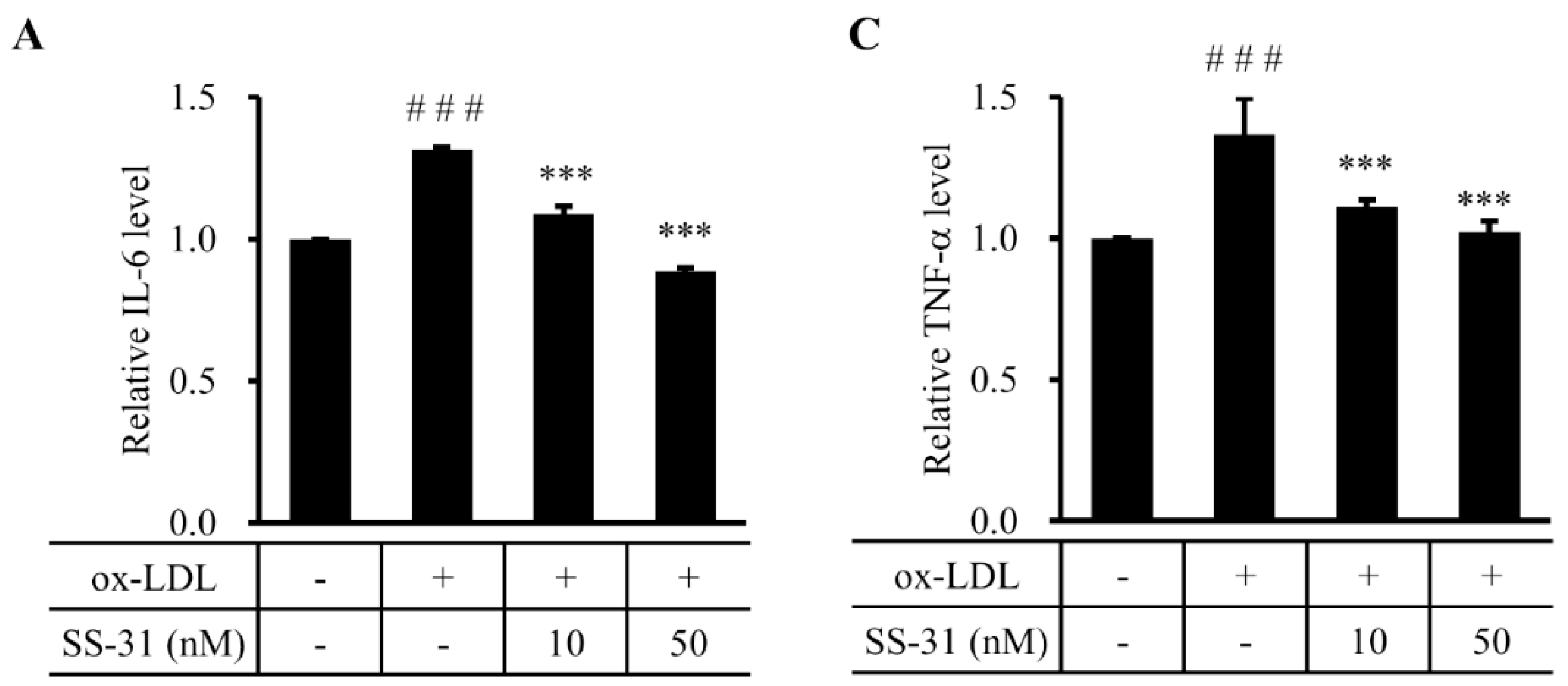
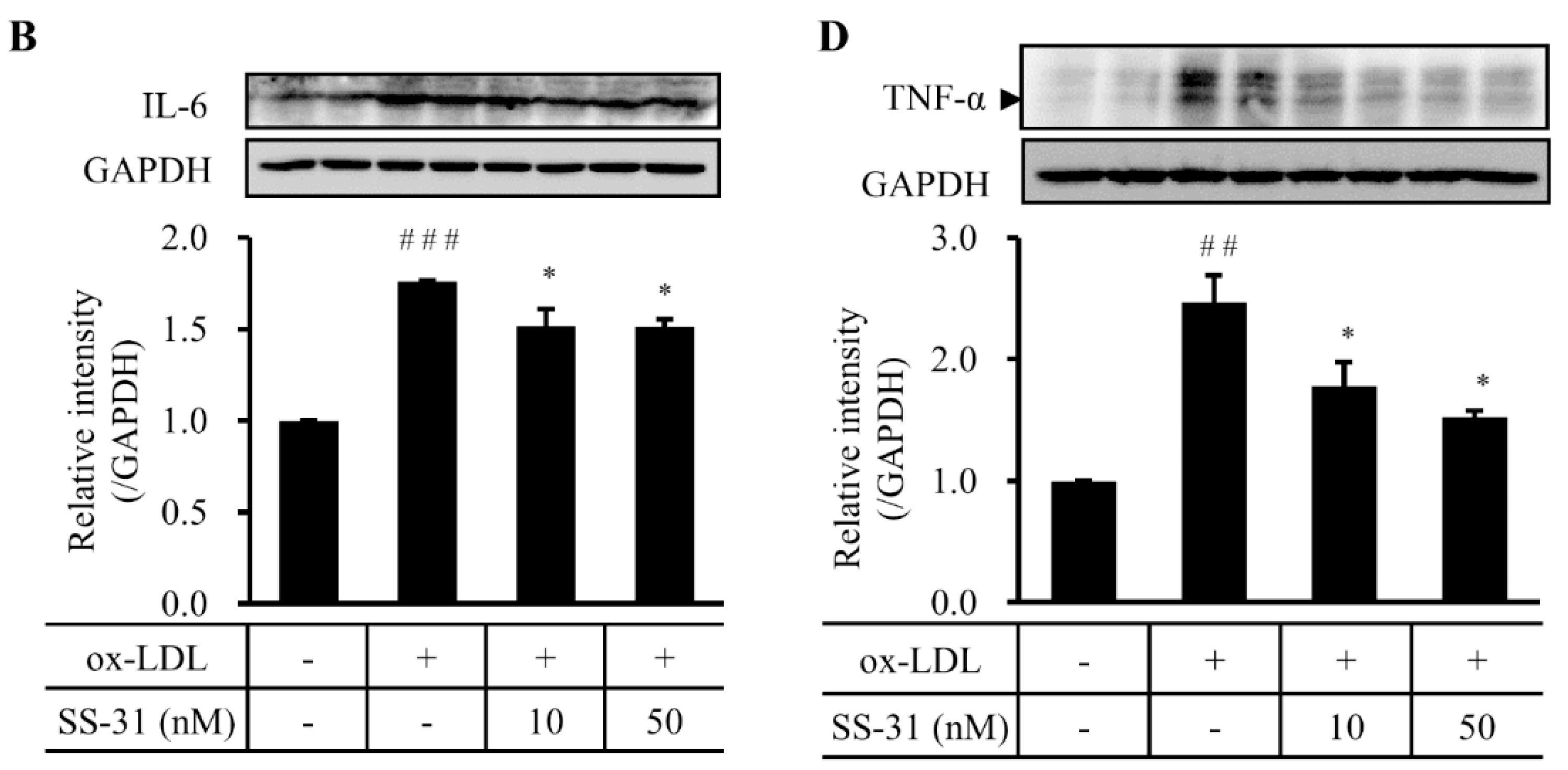
3. Discussion
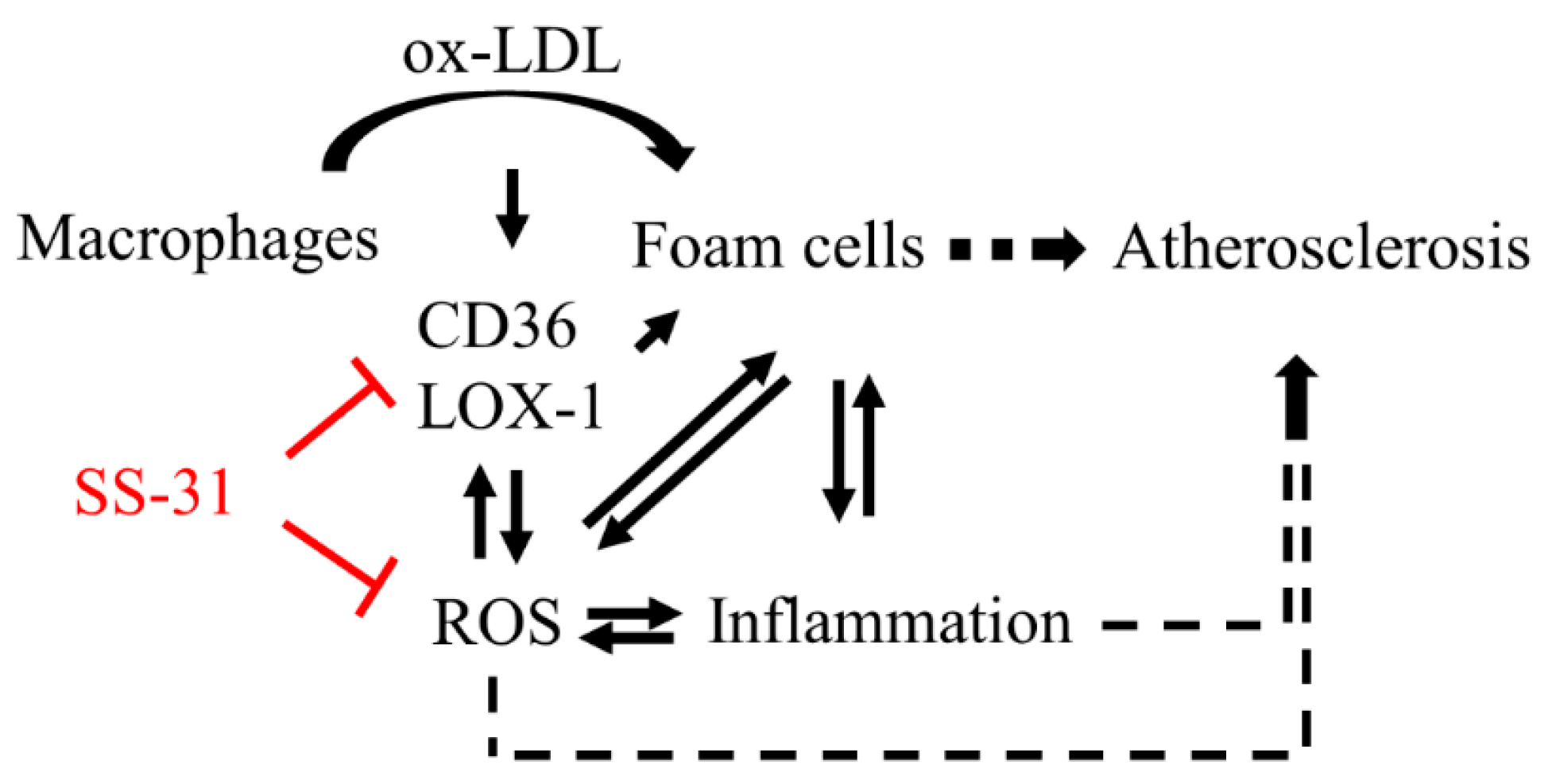
4. Experimental Section
4.1. Cell Culture and Treatment
4.2. Cholesterol Content
4.3. Oil Red O Staining
4.4. Western Blot Analysis
4.5. Measurement of Cellular ROS Production, SOD Activity and MDA Level
4.6. Statistics Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fortuno, A.; San Jose, G.; Moreno, M.U.; Diez, J.; Zalba, G. Oxidative stress and vascular remodelling. Exp. Physiol. 2005, 90, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Psaltopoulou, T.; Androulakis, E.; Papageorgiou, N.; Papaioannou, S.; Oikonomou, E.; Synetos, A.; Stefanadis, C. Oxidative stress and early atherosclerosis: Novel antioxidant treatment. Cardiovasc. Drug. Ther. 2015, 29, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Sakaguchi, M.; Miwa, K.; Furukado, S.; Yamagami, H.; Yagita, Y.; Mochizuki, H.; Kitagawa, K. Association of interleukin-6 with the progression of carotid atherosclerosis a 9-year follow-up study. Stroke 2014, 45, 2924–2929. [Google Scholar] [CrossRef] [PubMed]
- Scott, J. The liver X receptor and atherosclerosis. N. Engl. J. Med. 2007, 357, 2195–2197. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Hajjar, D.P.; Febbraio, M.; Nicholson, A.C. Native and modified low density lipoproteins increase the functional expression of the macrophage class B scavenger receptor, CD36. J. Biol. Chem. 1997, 272, 21654–21659. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Febbraio, M.; Sheibani, N.; Schmitt, D.; Silverstein, R.L.; Hajjar, D.P.; Cohen, P.A.; Frazier, W.A.; Hoff, H.F.; Hazen, S.L. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J. Clin. Investig. 2000, 105, 1483–1483. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDl, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscl. Throm. Vas. 2010, 30, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Bai, W.L.; Li, S.; Zhang, Y.Q.; Han, Y.; Gu, Y.; Meng, G.L.; Xie, L.P.; Wang, J.; Xiao, Y.J.; et al. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2(−/−) mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Simon, D.I. Inflammation and thrombosis: The clot thickens. Circulation 2001, 103, 1718–1720. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.H.; Tripathi, J.; Mishra, N.K.; Cai, Y.; Tripathi, S.; Wang, X.P.; Imes, S.; Fishbein, M.C.; Clinton, S.K.; Libby, P.; et al. Role of macrophage colony-stimulating factor in atherosclerosis—Studies of osteopetrotic mice. Am. J. Pathol. 1997, 150, 1687–1699. [Google Scholar] [PubMed]
- Zhao, K.S.; Zhao, G.M.; Wu, D.L.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Liu, S.Y.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.Y.; Soong, Y.; Wu, D.L.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.H.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates atp recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Mao, P.Z.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J. Alzheimers Dis. 2010, 20, S609–S631. [Google Scholar] [PubMed]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintron, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.C.; Zhao, K.S.; Calingasan, N.Y.; Luo, G.X.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Sign. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Sign. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.Y.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Schiller, P.W. Novel therapies targeting inner mitochondrial membrane—From discovery to clinical development. Pharm. Res. 2011, 28, 2669–2679. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://clinicaltrials.gov/ct2/results?term=Bendavia&Search=Search (accessed on 26 November 2015).
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Brit. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Matsebatlela, T.M.; Anderson, A.L.; Gallicchio, V.S.; Elford, H.; Rice, C.D. 3,4-Dihydroxy-benzohydroxamic acid (Didox) suppresses pro-inflammatory profiles and oxidative stress in TLR4-activated RAW264.7 murine macrophages. Chem.-Biol. Interact. 2015, 233, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.R.; Yu, E.M.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM(+/−)/Apoe(−/−) mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.T.; Lu, M.L.; Zhang, S.P.; Mei, M.; Wang, T.Q.; Liu, P.Q.; Wang, H.X. Vitamin e conditionally inhibits atherosclerosis in apoe knockout mice by anti-oxidation and regulation of vasculature gene expressions. Lipids 2014, 49, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.W.; Han, J.H.; Pearce, S.F.A.; Silverstein, R.L.; Gotto, A.M.; Hajjar, D.P.; Nicholson, A.C. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J. Lipid. Res. 2000, 41, 688–696. [Google Scholar] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.A.; Chen, H.W.; Evans, R.M. Oxidized ldl regulates macrophage gene expression through ligand activation of PPAR gamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Fuhrman, B.; Judith, O.; Keidar, S.; BenYaish, L.; Kaplan, M.; Aviram, M. Increased uptake of LDL by oxidized macrophages is the result of an initial enhanced LDL receptor activity and of a further progressive oxidation of LDL. Free Radic. Biol. Med. 1997, 23, 34–46. [Google Scholar] [CrossRef]
- Fuhrman, B.; Volkova, N.; Aviram, M. Oxidative stress increases the expression of the CD36 scavenger receptor and the cellular uptake of oxidized low-density lipoprotein in macrophages from atherosclerotic mice: Protective role of antioxidants and of paraoxonase. Atherosclerosis 2002, 161, 307–316. [Google Scholar] [CrossRef]
- Hashizume, M.; Mihara, M. Atherogenic effects of TNF-alpha and IL-6 via up-regulation of scavenger receptors. Cytokine 2012, 58, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Mihara, M. Blockade of IL-6 and TNF-alpha inhibited oxldl-induced production of MCP-1 via scavenger receptor induction. Eur. J. Pharmacol. 2012, 689, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Zhou, M.L.; You, W.C.; Zhu, L.; Ma, C.Y.; Sun, X.J.; Shi, J.X. Hydrogen-rich saline alleviates early brain injury via reducing oxidative stress and brain edema following experimental subarachnoid hemorrhage in rabbits. BMC. Neurosci. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compound SS-31 are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, S.; Ji, J.; Zhao, H.; Shang, L.; Wu, J.; Li, H.; Qiao, T.; Li, K. Mitochondrion-Targeted Peptide SS-31 Inhibited Oxidized Low-Density Lipoproteins-Induced Foam Cell Formation through both ROS Scavenging and Inhibition of Cholesterol Influx in RAW264.7 Cells. Molecules 2015, 20, 21287-21297. https://doi.org/10.3390/molecules201219764
Hao S, Ji J, Zhao H, Shang L, Wu J, Li H, Qiao T, Li K. Mitochondrion-Targeted Peptide SS-31 Inhibited Oxidized Low-Density Lipoproteins-Induced Foam Cell Formation through both ROS Scavenging and Inhibition of Cholesterol Influx in RAW264.7 Cells. Molecules. 2015; 20(12):21287-21297. https://doi.org/10.3390/molecules201219764
Chicago/Turabian StyleHao, Shuangying, Jiajie Ji, Hongting Zhao, Longcheng Shang, Jing Wu, Huihui Li, Tong Qiao, and Kuanyu Li. 2015. "Mitochondrion-Targeted Peptide SS-31 Inhibited Oxidized Low-Density Lipoproteins-Induced Foam Cell Formation through both ROS Scavenging and Inhibition of Cholesterol Influx in RAW264.7 Cells" Molecules 20, no. 12: 21287-21297. https://doi.org/10.3390/molecules201219764
APA StyleHao, S., Ji, J., Zhao, H., Shang, L., Wu, J., Li, H., Qiao, T., & Li, K. (2015). Mitochondrion-Targeted Peptide SS-31 Inhibited Oxidized Low-Density Lipoproteins-Induced Foam Cell Formation through both ROS Scavenging and Inhibition of Cholesterol Influx in RAW264.7 Cells. Molecules, 20(12), 21287-21297. https://doi.org/10.3390/molecules201219764