3.2. Synthesis and Characterization
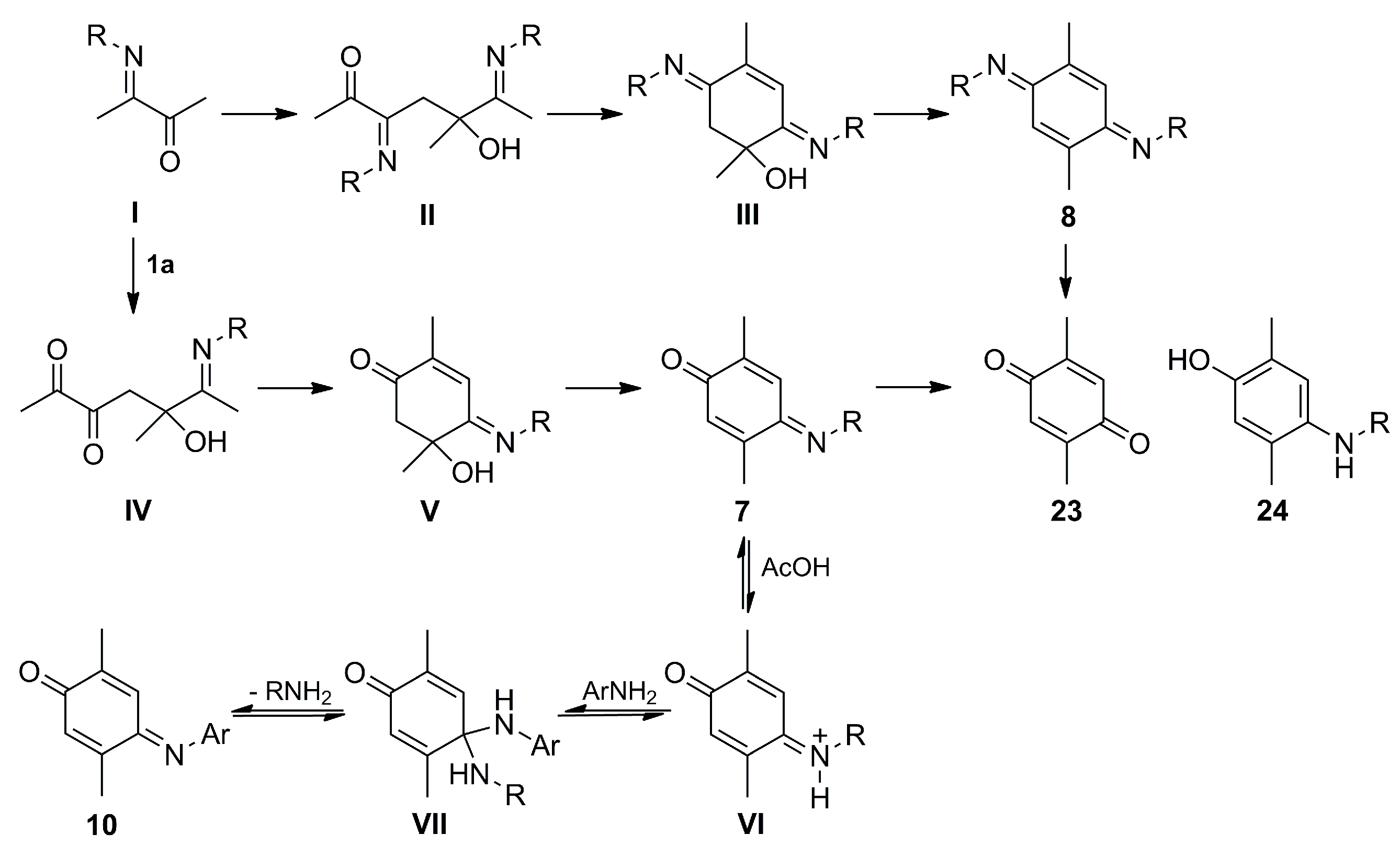
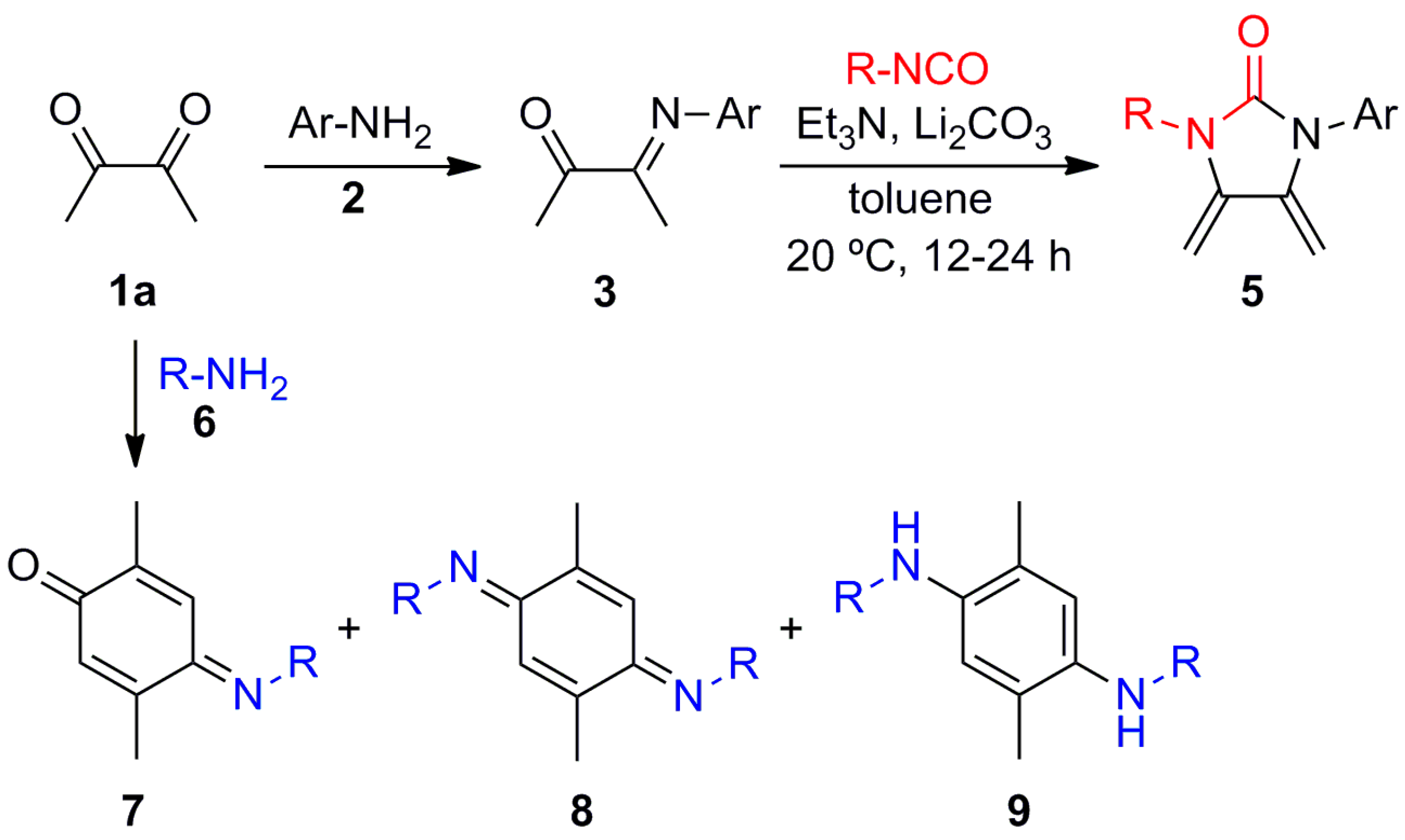
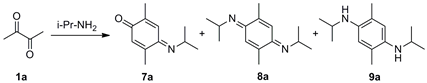
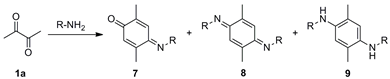
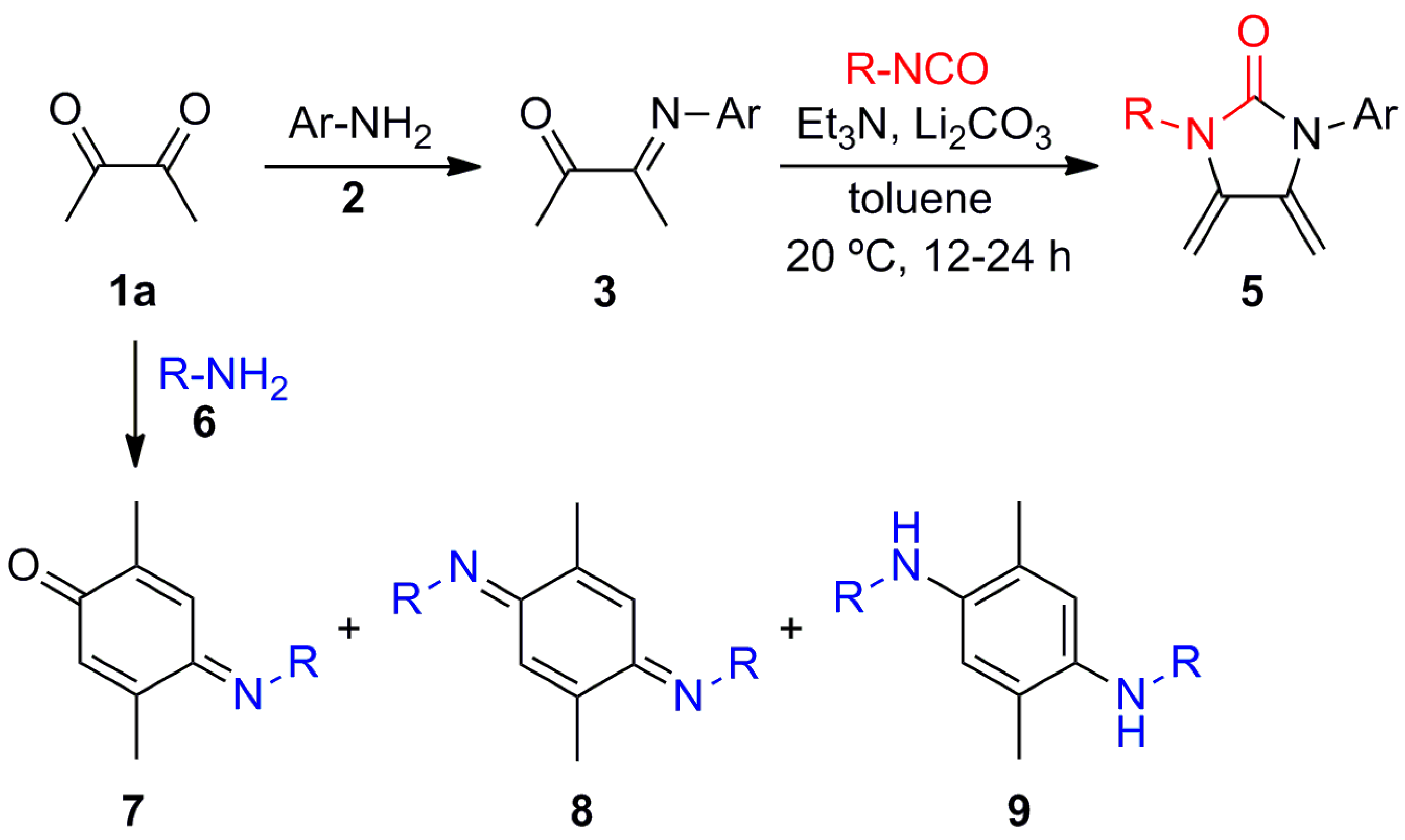
(E)-4-(Isopropylimino)-2,5-dimethylcyclohexa-2,5-dien-1-one (7a), (E,E)-N,N′-Diisopropyl-2,5-dimethylcyclohexa-2,5-diene-1,4-diimine (8a) and N,N′-Diisopropyl-2,5-dimethylbenzene-1,4-diamine (9a). A mixture of 2,3-butanedione (1a) (1.971 g, 22.91 mmol) and isopropylamine (6a) (2.707 g, 45.88 mmol) in MeOH (400 mL) was stirred at room temperature for 48 h. The crude reaction mixture was concentrated under vacuum, and the residue was purified by column chromatography over silica gel impregnated with triethylamine (10%) in hexane (40 g/g of crude, hexane) to give 7a (0.610 g, 30%) as a pale green-yellow solid, 8a (0.524 g, 21%) as a dark red solid, and 9a (1.04 g, 41%) as a red solid.
Data of 7a: Rf 0.53 (hexane/EtOAc, 8:2); mp 64–65 °C. IR (film): νmax 2969, 2925, 1652, 1631, 1604, 1518, 1461, 1376, 1256, 1169, 894 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.25 (d, J = 6.0 Hz, 6H, (CH3)2CH), 2.00 (d, J = 1.5 Hz, 3H, CH3-C2), 2.15 (d, J = 1.5 Hz, 3H, CH3-C5), 4.18 (sept, J = 6.5 Hz, 1H, (CH3)2CH), 6.41 (q, J = 1.5 Hz, 1H, H-6), 7.10 (dq, J = 1.5, 0.5 Hz, 1H, H-3). 13C-NMR (125 MHz, CDCl3): δ = 16.1 (CH3-C2), 17.7 (CH3-C5), 24.2 ((CH3)2CH), 51.8 ((CH3)2CH), 122.2 (C-3), 129.3 (C-6), 140.0 (C-2), 150.9 (C-5), 156.1 (C-4), 188.3 (C-1). MS (70 eV): m/z (%) 177 (M+, 100), 162 (73), 149 (45), 134 (58), 117 (43), 106 (35), 91 (52), 77 (19). Anal. calcd for C11H15NO: C, 74.54; H, 8.53; N, 7.90. Found: C, 74.56; H, 8.49; N, 7.94.
Data of 8a: Rf 0.64 (hexane/EtOAc, 8:2); mp 94–95 °C. IR (film): νmax 2967, 2924, 1599, 1581, 1519, 1377, 1359, 1345, 1258, 1116, 875 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.20 (d, J = 6.0 Hz, 6H, (CH3)2CH), 2.00 (d, J = 1.5 Hz, 6H, CH3-C2, CH3-C5), 4.08 (sept, J = 6.0 Hz, 2H, (CH3)2CH), 6.79 (br d, J = 1.5 Hz, 2H, H-3, H-6). 13C-NMR (125 MHz, CDCl3): δ = 18.5 (CH3-C2, CH3-C5), 24.2 (2(CH3)2CH), 50.4 (2(CH3)2CH), 118.4 (C-3, C-6), 143.3 (C-2, C-5), 156.9 (C-1, C-4). MS (70 eV): m/z (%) 218 (M+, 20), 203 (95), 161 (34), 146 (100), 145 (43), 132 (20), 117 (6), 91 (7). Anal. calcd for C14H22N2: C, 77.01; H, 10.16; N, 12.83. Found: C, 77.00; H, 10.21; N, 12.78.
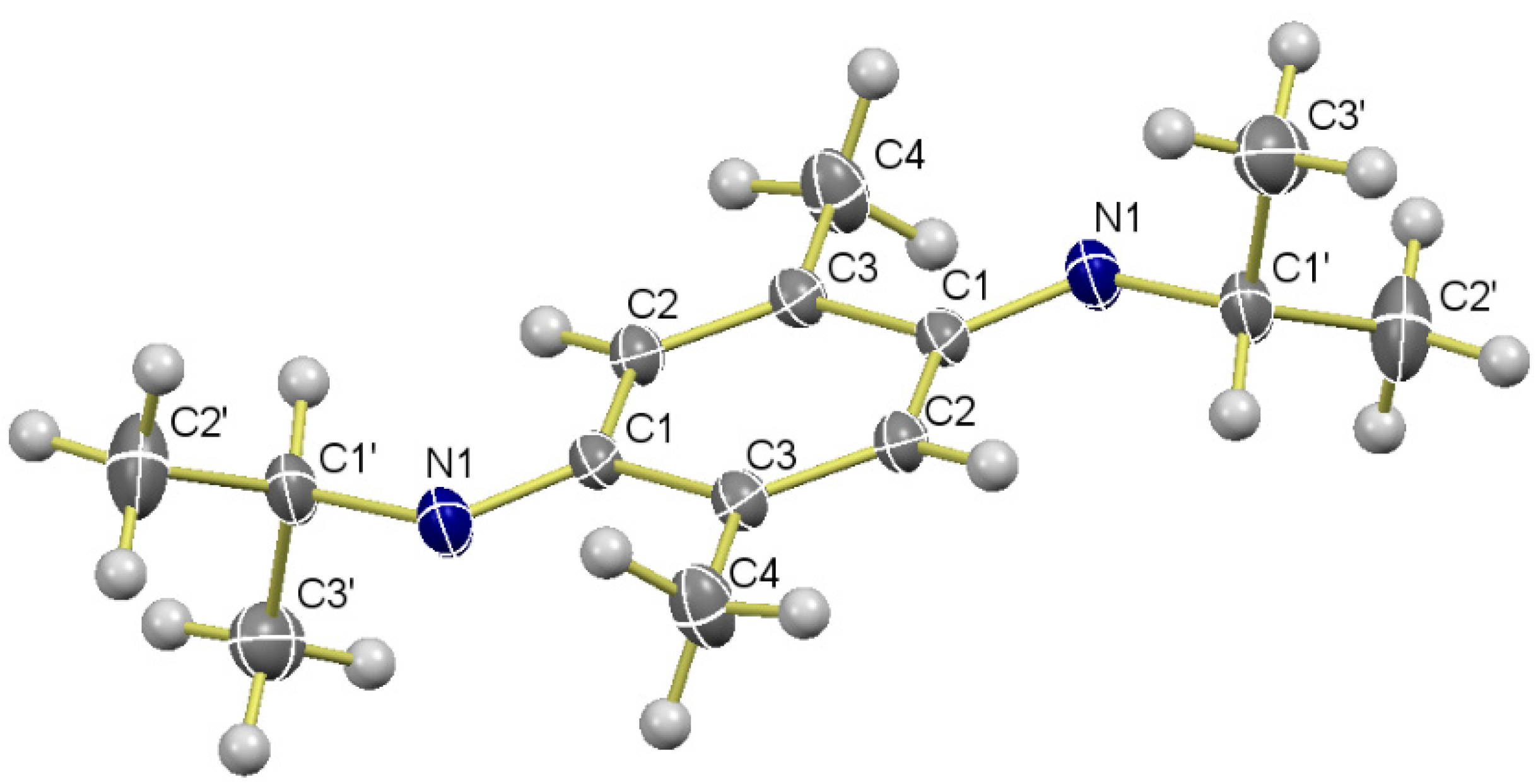
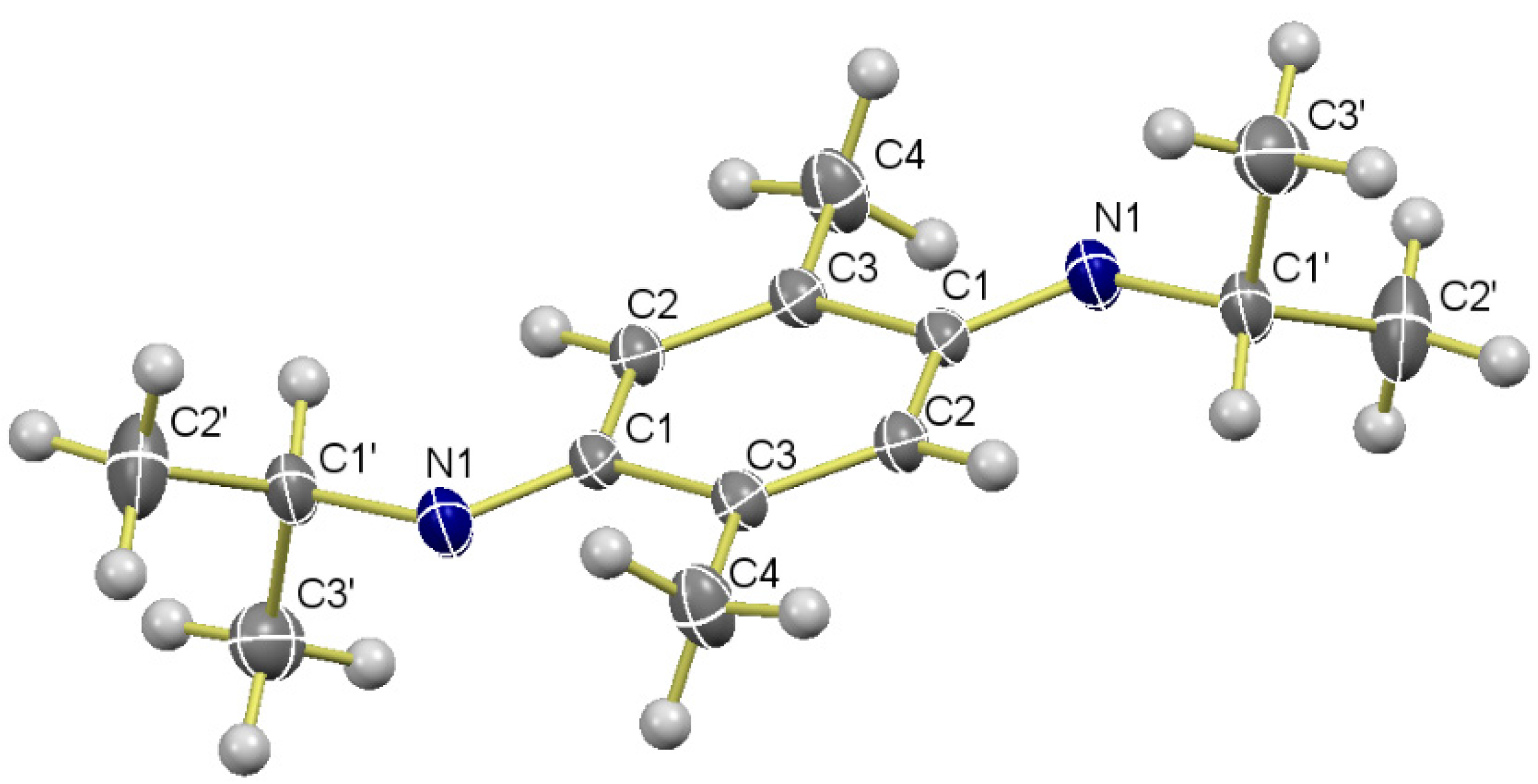
Data of 9a: Rf 0.22 (hexane/EtOAc, 8:2); mp 53–54 °C. IR (film): νmax 3382, 2965, 2928, 1520, 1463, 1413, 1381, 1218, 1166, 1125, 1004, 857 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.19 (d, J = 6.5 Hz, 6H, (CH3)2CH), 2.10 (s, 6H, CH3-C2, CH3-C5), 2.70 (br s, 2H, NH), 3.53 (sept, J = 6.5 Hz, 2H, (CH3)2CH), 6.46 (s, 2H, H-3, H-6). 13C-NMR (125 MHz, CDCl3): δ = 17.6 (CH3-C2, CH3-C5), 23.5 (2(CH3)2CH), 45.4 (2(CH3)2CH), 115.6 (C-3, C-6), 121.7 (C-2, C-5), 137.3 (C-1, C-4). MS (70 eV): m/z (%) 220 (M+, 98), 205 (100), 177 (99), 148 (20), 135 (56), 95 (15), 75 (6). HRMS (EI): m/z [M+] calcd for C14H24N2: 220.1940; found: 220.1938.
(E)-4-(Cyclohexylimino)-2,5-dimethylcyclohexa-2,5-dien-1-one (7b), (E,E)-N,N′-Dicyclohexyl-2,5-dimethylcyclohexa-2,5-diene-1,4-diimine (8b) and N,N′-Dicyclohexyl-2,5-dimethylbenzene-1,4-diamine (9b). The procedure for the preparation of 7a–9a was followed using a mixture of 1a (1.971 g, 22.91 mmol) and cyclohexylamine (6b) (4.53 g, 45.8 mmol) in MeOH (400 mL) to give 7b (0.721 g, 29%) as a pale green-yellow solid, 8b (0.784 g, 23%) as a yellow solid, and 9b (1.34 g, 39%) as a dark brown solid.
Data of 7b: Rf 0.51 (hexane/EtOAc, 9:1); mp 63–64 °C. IR (film): νmax 2925, 2853, 1647, 1620, 1370, 1268, 1161, 900 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.33 (qt, J = 12.0, 3.3 Hz, 1H, H-Cy), 1.43 (qt, J = 12.0, 3.3 Hz, 2H, H-Cy), 1.55–1.62 (m, 2H, H-Cy), 1.64–1.72 (m, 3H, H-Cy), 1.82–1.87 (m, 2H, H-Cy), 2.01 (d, J = 1.5 Hz, 3H, CH3-C2), 2.14 (d, J = 1.5 Hz, 3H, CH3-C5), 3.80–3.87 (m, 1H, NCH-Cy), 6.41 (br s, 1H, H-6), 7.08 (br s, J = 1.5 Hz, 1H, H-3). 13C-NMR (125 MHz, CDCl3): δ = 16.1 (CH3-C2), 17.7 (CH3-C5), 24.3 (2C-3′), 25.6 (C-4′), 34.3 (2C-2′), 60.3 (C-1′), 122.3 (C-3), 129.3 (C-6), 139.9 (C-2), 150.9 (C-5), 156.3 (C-4), 188.4 (C-1). HRMS (EI): m/z [M+] calcd for C14H19NO: 217.1467; found: 217.1470.
Data of
8b: R
f 0.68 (hexane/EtOAc, 9:1); mp 147–148 °C [Lit. [
33] 145.6–147 °C]. IR (film): ν
max 2923, 2850, 1598, 1575, 1454, 1351, 1166, 962, 889, 871 cm
−1.
1H-NMR (500 MHz, CDCl
3): δ = 1.28 (qt,
J = 13.0, 2.5 Hz, 2H, H-Cy), 1.35 (qt,
J = 13.0, 2.5 Hz, 4H, H-Cy), 1.40–1.48 (m, 4H, H-Cy), 1.56–1.61 (m, 6H, H-Cy), 1.72–1.79 (m, 4H, H-Cy), 2.02 (s, 6H, C
H3-C2, C
H3-C5), 3.61–3.68 (m, 2H, NCH-Cy), 6.70 (br s, 2H, H-3, H-6).
13C-NMR (125 MHz, CDCl
3): δ = 18.4 (
CH
3-C2,
CH
3-C5), 24.7 (4C-3′), 25.8 (2C-4′), 34.2 (4C-2′), 59.0 (2C-1′), 118.5 (C-3, C-6), 143.3 (C-2, C-5), 157.1 (C-1, C-4). HRMS (EI):
m/z [M
+] calcd for C
20H
30N
2: 298.2409; found: 298.2402.
Data of 9b: Rf 0.21 (hexane/EtOAc, 9:1); mp 108–109 °C. IR (film): νmax 3383, 2927, 2850, 1519, 1445, 1412, 1211, 1107, 850 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.06–1.44 (m, 10H, H-Cy), 1.57–1.67 (m, 2H, H-Cy), 1.70–1.80 (m, 4H, H-Cy), 2.00–2.08 (m, 2H, H-Cy), 2.10 (s, 6H, CH3-C2, CH3-C5), 2.76 (br s, 2H, NH), 3.08–3.20 (m, 2Hz, NCH-Cy), 6.45 (s, 2H, H-3, H-6). 13C-NMR (75 MHz, CDCl3): δ = 17.7 (CH3-C2, CH3-C5), 25.1 (4C-3′), 26.0 (2C-4′), 34.0 (4C-2′), 53.0 (2C-1′), 115.4 (C-3, C-6), 121.5 (C-2, C-5), 137.0 (C-1, C-4). HRMS (EI): m/z [M+] calcd for C20H32N2: 300.2566; found: 300.2570.
(E,E)-2,5-Dimethyl-N,N′-bis((S)-1-phenylethyl)cyclohexa-2,5-diene-1,4-diimine (8c) and 2,5-Dimethyl-N,N′-bis((S)-1-phenylethyl)benzene-1,4-diamine (9c). The procedure for the preparation of 7a–9a was followed using a mixture of 1a (0.098 g, 1.14 mmol) and (S)-phenylethylamine (6c) (0.278 g, 2.3 mmol) in MeOH (10 mL) to give 8c (0.031 g, 16%) as a yellow solid and 9c (0.059 g, 30%) as a dark brown oil.
Data of 8c: Rf 0.65 (hexane/EtOAc, 8:2); mp 108–109 °C; = −44.28 (c 0.473, CHCl3). IR (film): νmax 1699, 1493, 1450, 1354, 1125, 760, 699 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.49 (d, J = 6.5 Hz, 6H, CH3CH), 2.17 (d, J = 1.5 Hz, 6H, CH3-C2, CH3-C5), 5.08 (q, J = 6.5 Hz, 2H, CH3CH), 6.89 (br s, 1H, H-3, H-6), 7.19–7.24 (m, 2H, H-4′′), 7.30–7.34 (m, 4H, H-3′′), 7.43–7.46 (m, 4H, H-2′′). 13C-NMR (125 MHz, CDCl3): δ = 18.5 (CH3-C2, CH3-C5), 25.7 (2CH3CH), 58.9 (2CH3CH), 118.5 (C-3, C-6), 126.58 (2C-4′′), 126.61 (4C-2′′), 128.4 (4C-3′′), 144.1 (C-2, C-5), 146.2 (2C-1′′), 157.5 (C-1, C-4). MS (70 eV): m/z (%) 342 (M+, 22), 328 (23), 327 (100), 300 (14), 237 (12), 222 (24), 208 (26), 105 (20), 97 (24), 86 (38), 71 (43), 57 (45). HRMS (EI): m/z [M+] calcd for C24H26N2: 342.2096; found: 342.2090.
Data of 9c: Rf 0.32 (hexane/EtOAc, 9:1); = +10.75 (c 0.282, CHCl3). IR (film): νmax 3417, 2969, 1448, 1414, 1520, 1371, 1224, 769, 698 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.47 (d, J = 6.6 Hz, 6H, CH3CH), 2.02 (s, 6H, CH3-C2, CH3-C5), 3.30 (br s, 2H, NH), 4.39 (q, J = 6.6 Hz, 2H, CH3CH), 6.21 (s, 2H, H-3, H-6), 7.22–7.37 (m, 10H, PhH). 13C-NMR (75 MHz, CDCl3): δ = 17.6 (2CH3Ar), 25.1 (2CH3CH), 54.0 (2CH3CH), 114.7 (C-3, C-6), 120.6 (C-2, C-5), 125.8 (4C-2′′), 126.5 (2C-4′′), 128.4 (4C-3′′), 136.9 (C-1, C-4), 145.9 (2C-1ʹʹ). HRMS (EI): m/z [M+] calcd for C24H28N2: 344.2253; found: 344.2257.
N,N′-Dibutyl-2,5-dimethylbenzene-1,4-diamine (9d). The procedure for the preparation of 7a–9a was followed using a mixture of 1a (1.971 g, 22.91 mmol) and n-butylamine (6d) (3.34 g, 45.8 mmol) in MeOH (400 mL) to give 9d (0.512 g, 18%) as a dark brown oil. Rf 0.44 (hexane/EtOAc, 8:2). IR (film): νmax 3369, 2955, 2925, 1518, 1469, 1413, 1222, 1214, 994, 855, 742 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 0.96 (t, J = 7.5 Hz, 6H, N(CH2)3CH3), 1.44 (sext, J = 7.5 Hz, 4H, N(CH2)2CH2CH3), 1.62 (qu, J = 7.0 Hz, 4H, NCH2CH2CH2CH3), 2.12 (s, 6H, CH3-C2, CH3-C5), 2.98 (br s, 2H, NH), 3.08 (br t, J = 7.0 Hz, 4H, NCH2(CH2)2CH3), 6.44 (s, 2H, H-3, H-6). 13C-NMR (125 MHz, CDCl3): δ = 13.9 (2N(CH2)3CH3), 17.4 (CH3-C2, CH3-C5), 20.4 (2N(CH2)2CH2CH3), 32.0 (2NCH2CH2CH2CH3), 45.0 (2NCH2(CH2)2CH3), 114.1 (C-3, C-6), 121.1 (C-2, C-5), 138.3 (C-1, C-4). MS (70 eV): m/z (%) 248 (M+, 70), 205 (100), 191 (32), 135 (10), 120 (11), 81 (26). HRMS (EI): m/z [M+] calcd for C16H28N2: 248.2253; found: 248.2260.
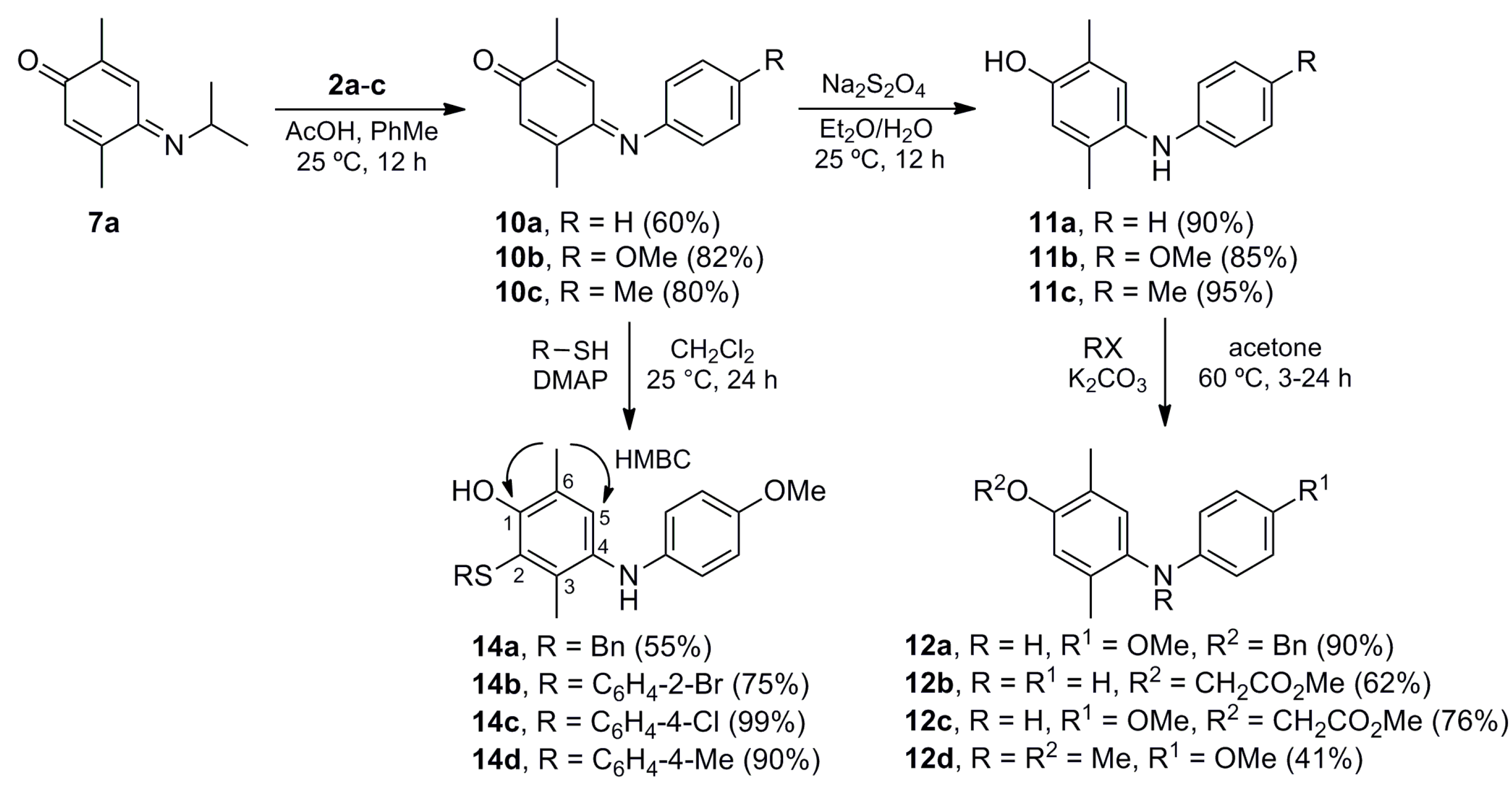
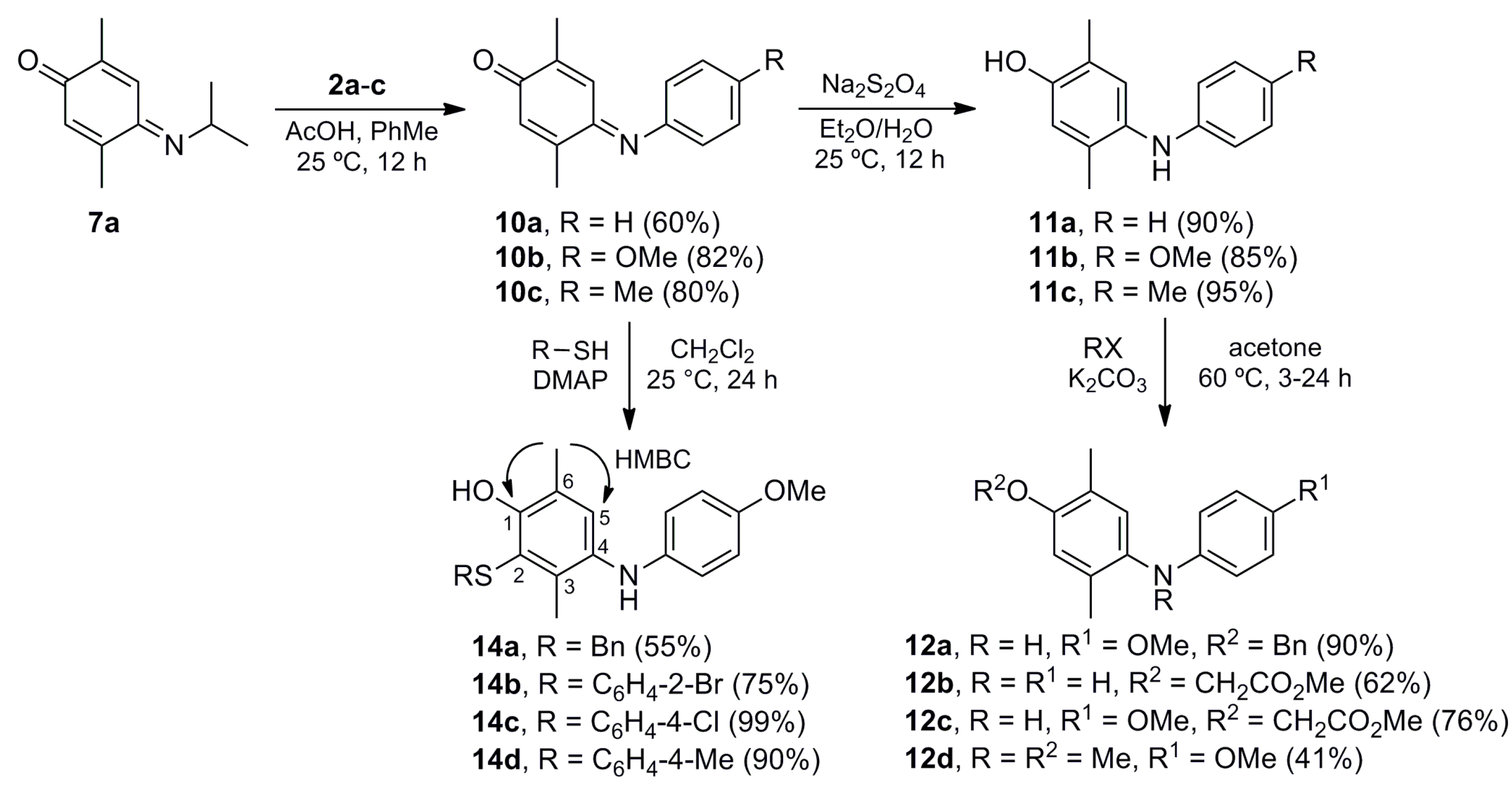
(E)-2,5-Dimethyl-4-(phenylimino)cyclohexa-2,5-dien-1-one (10a). A mixture of 7a (0.442 g, 2.50 mmol) and aniline (2a) (0.233 g, 2.50 mmol) in toluene (40 mL) was stirred at room temperature for 10 min. Then, glacial acetic acid (0.524 g, 8.74 mmol) was added dropwise and the mixture was stirred at room temperature for 12 h. The crude mixture was concentrated under vacuum and purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 8:2) to give 10a (0.316 g, 60%) as a dark red solid. Rf 0.66 (hexane/EtOAc, 8:2); mp 73–74 °C. IR (film): νmax 1649, 1633, 1603, 1497, 1482, 1262, 1204, 1159, 900, 762, 697 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.93 (d, J = 1.5 Hz, 3H, CH3-C2), 2.27 (d, J = 1.5 Hz, 3H, CH3-C5), 6.55 (q, J = 1.5 Hz, 1H, H-6), 6.78 (q, J = 1.5 Hz, 1H, H-3), 6.80–6.85 (m, 2H, H-2′), 7.17-7.22 (m, 1H, H-4′), 7.34–7.44 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 16.1 (CH3-C2), 18.0 (CH3-C5), 120.2 (C-2′), 125.4 (C-4′), 125.6 (C-3), 129.2 (C-3′), 131.1 (C-6), 141.5 (C-2), 149.5 (C-4), 150.2 (C-1′), 158.5 (C-5), 188.4 (C-1). HRMS (EI): m/z [M+] calcd for C14H13NO: 211.0997; found: 211.0989.
(E)-4-((4-Methoxyphenyl)imino)-2,5-dimethylcyclohexa-2,5-dien-1-one (10b). The procedure for the preparation of 10a was followed using a mixture of 7a (0.202 g, 1.14 mmol) and 4-methoxyaniline (2b) (0.140 g, 1.14 mmol) and glacial acetic acid (0.524 g, 8.74 mmol) in toluene (20 mL) to give 10b (0.227 g, 82%) as a dark red solid. Rf 0.40 (hexane/EtOAc, 1:1); mp 79–80 °C. IR (film): νmax 1648, 1628, 1599, 1499, 1246, 1164, 1033, 902, 843, 762 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.95 (d, J = 1.5 Hz, 3H, CH3-C2), 2.26 (d, J = 1.5 Hz, 3H, CH3-C5), 3.84 (s, 3H, CH3O), 6.52 (br d, J = 1.5 Hz, 1H, H-6), 6.83–6.86 (m, 2H, H-2′), 6.90 (br d, J = 1.5 Hz, 1H, H-3), 6.93–6.97 (m, 2H, H-3′). 13C-NMR (125 MHz, CDCl3): δ = 15.8 (CH3-C2), 17.7 (CH3-C5), 55.4 (CH3O), 114.2 (C-3′), 122.4 (C-2′), 125.3 (C-3), 130.4 (C-6), 140.7 (C-2), 143.1 (C-1′), 149.4 (C-5), 157.7 (C-4), 157.9 (C-4′), 188.1 (C-1). MS (70 eV): m/z (%) 241 (M+, 70), 226 (36), 210 (100), 198 (22), 182 (40), 167 (26), 155 (18). HRMS (EI): m/z [M+] calcd for C15H15NO2: 241.1103; found: 241.1090.
(E)-2,5-Dimethyl-4-(p-tolylimino)cyclohexa-2,5-dien-1-one (10c). The procedure of preparation for 10a was followed using a mixture of 7a (0.300 g, 1.69 mmol), 4-methylaniline (2c) (0.208 g, 1.69 mmol) and glacial acetic acid (0.524 g, 8.73 mmol) in toluene (20 mL) to give 10c (0.305 g, 80%) as a dark red solid. Rf 0.68 (hexane/EtOAc, 7:3); mp 88–89 °C. IR (film): νmax 2922, 1628, 1508, 1446, 1259, 1110, 1091, 1006, 903, 842, 804 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.93 (d, J = 1.4 Hz, 3H, CH3-C2), 2.27 (d, J = 1.4 Hz, 3H, CH3-C5), 2.38 (s, 3H, CH3Ar), 6.53 (q, J = 1.4 Hz, 1H, H-6), 6.72–6.78 (m, 2H, H-2′), 6.84 (q, J = 1.4 Hz, 1H, H-3), 7.17–7.24 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 15.8 (CH3-C2), 17.8 (CH3-C5), 20.8 (CH3Ar), 120.3 (C-2′), 125.4 (C-3), 129.5 (C-3′), 130.6 (C-6), 135.1 (C-4′), 140.9 (C-2), 147.4 (C-1′), 149.4 (C-5), 158.1 (C-4), 188.3 (C-1). HRMS (EI): m/z [M+] calcd for C15H15NO: 225.1154; found: 225.1155.
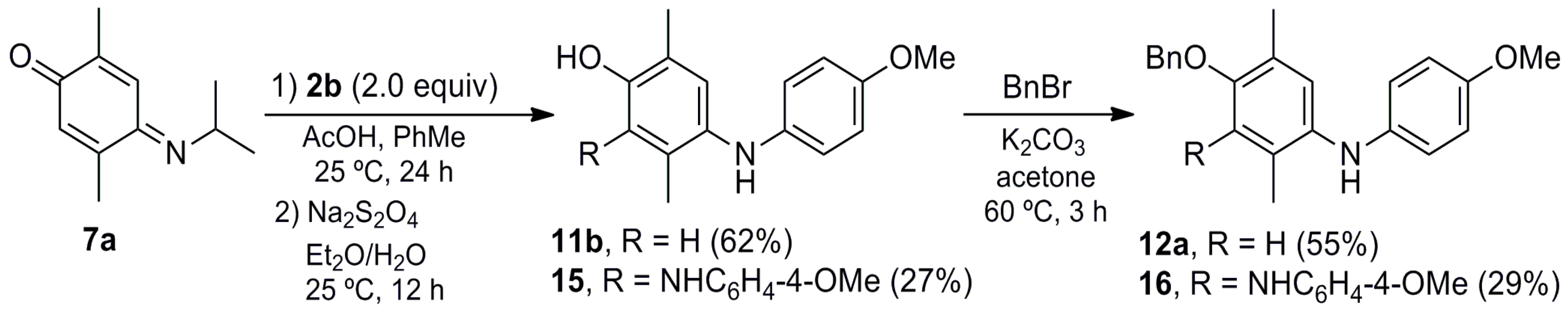
2,5-Dimethyl-4-(phenylamino)phenol (11a). To a solution of 10a (0.100 g, 0.47 mmol) in Et2O (10 mL) a saturated aqueous solution of sodium hydrosulfite (30 mL) was added, and the mixture was stirred at room temperature for 12 h. The crude mixture was washed with CH2Cl2 (3 × 10 mL) and the organic layer was dried (Na2SO4), concentrated under vacuum and purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 9:1) to give 11a (0.090 g, 90%) as a pink oil. Rf 0.49 (hexane/EtOAc, 8:2). IR (film): νmax 3382, 2923, 1600, 1497, 1462, 1407, 1200, 864, 745, 694 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.14 (s, 3H, CH3-C5), 2.18 (s, 3H, CH3-C2), 4.50–5.30 (br, 2H, NH, OH), 6.65 (s, 1H, H-6), 6.66–6.73 (m, 2H, H-2′), 6.77 (t, J = 7.3 Hz, 1H, H-4′), 6.98 (s, 1H, H-3), 7.14–7.22 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 15.4 (CH3-C2), 17.5 (CH3-C5), 114.6 (C-2′), 117.1 (C-6), 118.6 (C-4′), 121.9 (C-2), 126.8 (C-3), 129.2 (C-3′), 131.6 (C-5), 133.1 (C-4), 146.4 (C-1′), 150.4 (C-1). HRMS (EI): m/z [M+] calcd for C14H15NO: 213.1154; found: 213.1144.
4-((4-Methoxyphenyl)amino)-2,5-dimethylphenol (11b). The procedure for the preparation of 11a was followed using a mixture of 10b (0.099 g, 0.41 mmol) in Et2O (10 mL) and a saturated aqueous solution of sodium hydrosulfite (30 mL) to give 11b (0.085 g, 85%) as a red solid. Rf 0.25 (hexane/EtOAc, 8:2); mp 86–87 °C. IR (KBr): νmax 3406, 1511, 1466, 1246, 1193, 1179, 1036, 825 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.13 (br s, 3H, CH3Ar), 2.16 (br s, 3H, CH3Ar), 3.76 (s, 3H, CH3O), 4.53–4.92 (br s, 2H, OH, NH), 6.61 (br s, 1H, H-6), 6.72–6.76 (m, 2H, H-2′), 6.78–6.81 (m, 2H, H-3′), 6.85 (br s, 1H, H-3). 13C-NMR (75 MHz, CDCl3): δ = 15.4 (CH3Ar), 17.4 (CH3Ar), 55.7 (CH3O), 114.8 (C-3′), 117.3 (C-6), 118.0 (C-2′), 121.9 (C-2), 123.8 (C-3), 129.0 (C-5), 135.0 (C-1′), 139.4 (C-4), 149.3 (C-1), 153.4 (C-4′). MS (70 eV): m/z (%) 243 (M+, 91), 228 (100), 200 (11), 185 (9), 168 (7), 134 (5), 77 (7). HRMS (EI): m/z [M+] calcd for C15H17NO2: 243.1259; found: 243.1260.
2,5-Dimethyl-4-((4-tolyl)amino)phenol (11c). The procedure for the preparation of 11a was followed using a mixture of 10c (0.200 g, 0.89 mmol) in Et2O (10 mL) and a saturated aqueous solution of sodium hydrosulfite (30 mL) to give 11c (0.193 g, 95%) as a brown solid. Rf 0.45 (hexane/EtOAc, 7:3); mp 104–105 °C. IR (film): νmax 3388, 2922, 1614, 1511, 1461, 1196, 993, 811, 738 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.12 (s, 3H, CH3-C5), 2.16 (s, 3H, CH3-C2), 2.25 (s, 3H, CH3Ar), 4.70–5.50 (br, 2H, OH, NH), 6.62–6.67 (m, 2H, H-2′), 6.65 (br s, 1H, H-6), 6.93 (br s, 1H, H-3), 6.96–7.02 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 15.5 (CH3-C2), 17.5 (CH3-C5), 20.5 (CH3Ar), 115.3 (C-2′), 117.2 (C-6), 121.8 (C-2), 125.6 (C-3), 128.1 (C-4′), 129.7 (C-3′), 130.6 (C-5), 133.6 (C-4), 143.7 (C-1′), 150.1 (C-1). HRMS (EI): m/z [M+] calcd for C15H17NO: 227.1310; found: 227.1314.
4-(Benzyloxy)-N-(4-methoxyphenyl)-2,5-dimethylaniline (12a). To a mixture of 11b (0.137 g, 0.56 mmol) in acetone (15 mL) K2CO3 (0.116 g, 0.84 mmol) and benzyl bromide (0.115 g, 0.67 mmol) were added, and the mixture was stirred at reflux for 3 h. The crude mixture was filtered over Celite, concentrated under vacuum, and purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 8:2) to give 12a (0.169 g, 90%) as a white powder. Rf 0.44 (hexane/EtOAc, 8:2); mp 55–56 °C. IR (KBr): νmax 3412, 2961, 2916, 1521, 1465, 1382, 1293, 1249, 1197, 1098, 1037, 1014, 825, 744, 698 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.16 (s, 3H, CH3-C5), 2.19 (s, 3H, CH3-C2), 3.72 (s, 3H, CH3O), 5.00 (s, 2H, CH2Ph), 6.72–6.80 (m, 4H, H-2′, H-3′), 6.74 (s, 1H, H-6), 6.89 (s, 1H, H-3), 7.29 (t, J = 7.5 Hz, 1H, H-4′′), 7.36 (t, J = 7.5 Hz, 2H, H-3′′), 7.43 (d, J = 7.5 Hz, 2H, H-2′′). 13C-NMR (125 MHz, CDCl3): δ = 16.0 (CH3-C2), 17.8 (CH3-C5), 55.5 (CH3O), 70.4 (CH2Ph), 114.6 (C-6), 114.64 (2C-3′), 118.2 (2C-2′), 123.3 (C-3), 125.3 (C-2), 127.1 (2C-2′′), 127.6 (C-4′′), 127.8 (C-5), 128.4 (2C-3′′), 135.1 (C-1′), 137.7 (C-1′′), 139.1 (C-4), 152.4 (C-1), 153.5 (C-4′). MS (70 eV): m/z (%) 333 (M+, 16), 243 (87), 228 (100), 212 (61), 197 (80), 179 (28), 135 (36), 108 (53), 91 (48), 77 (21). HRMS (EI): m/z [M+] calcd for C22H23NO2: 333.1729; found: 333.1715.
Methyl 2-(2,5-dimethyl-4-(phenylamino)phenoxy)acetate (12b). The procedure for the preparation of 12a was followed with a mixture of 11a (0.104 g, 0.49 mmol), K2CO3 (0.102 g, 0.74 mmol) and methyl bromoacetate (0.083 g, 0.54 mmol) to give 12b (0.086 g, 62%) as a brown powder. Rf 0.41 (hexane/EtOAc, 8:2); mp 102–103 °C. IR (film): νmax 3387, 2923, 1600, 1497, 1406, 1196, 994, 862, 747, 694 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.17 (s, 3H, CH3-C5′), 2.22 (s, 3H, CH3-C2′), 3.80 (s, 3H, CO2CH3), 4.63 (s, 2H, CH2CO2Me), 5.19 (br s, 1H, NH), 6.58 (s, 1H, H-6′), 6.69–6.75 (m, 2H, H-2′′), 6.78 (t, J = 7.2 Hz, 1H, H-4′′), 7.02 (s, 1H, H-3′), 7.14–7.24 (m, 2H, H-3′′). 13C-NMR (75 MHz, CDCl3): δ = 15.8 (CH3-C2′), 17.9 (CH3-C5′), 52.1 (CH2CO2CH3), 66.0 (CH2CO2Me), 114.1 (C-6′), 115.0 (2C-2′′), 118.8 (C-4′′), 125.6 (C-2′), 125.8 (C-3′), 129.2 (2C-3′′), 130.0 (C-5′), 134.0 (C-4′), 145.8 (C-1′′), 152.5 (C-1′), 169.8 (CO2Me). HRMS (EI): m/z [M+] calcd for C17H19NO3: 285.1365; found: 285.1377.
Methyl 2-(4-((4-methoxyphenyl)amino)-2,5-dimethylphenoxy)acetate (12c). The procedure for the preparation of 12a was followed using a mixture of 11b (0.081 g, 0.33 mmol), K2CO3 (0.066 g, 0.48 mmol) and methyl bromoacetate (0.057 g, 0.37 mmol) to give 12c (0.08 g, 76%) as a brown solid. Rf 0.47 (hexane/EtOAc, 8:2); mp 124–125 °C. IR (film): νmax 3340, 2922, 1755, 1508, 1438, 1233, 1198, 1116, 1034, 822 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.17 (s, 3H, CH3-C5′), 2.20 (s, 3H, CH3-C2′), 3.76 (s, 3H, CH3O), 3.80 (s, 3H, CO2CH3), 4.61 (s, 2H, CH2CO2Me), 4.99 (br s, 1H, NH), 6.58 (s, 1H, H-6′), 6.79-6.82 (br s, 4H, H-2′′, H-3′′), 6.88 (s, 1H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 15.8 (CH3-C2′), 17.8 (CH3-C5′), 52.1 (CH2CO2CH3), 55.6 (CH3O), 66.4 (CH2CO2Me), 114.6 (2C-3′′), 114.7 (C-6′), 118.9 (2C-2′′), 122.3 (C-3′), 125.6 (C-2′), 126.9 (C-5′), 136.3 (C-4′), 138.4 (C-1′′), 151.2 (C-1′), 153.7 (C-4′′), 169.9 (CO2Me). HRMS (EI): m/z [M+] calcd for C18H21NO4: 315.1471; found: 315.1464.
4-Methoxy-N-(4-methoxyphenyl)-N,2,5-trimethylaniline (12d). A mixture of 11b (0.180 g, 0.74 mmol), iodomethane (0.210 g, 1.48 mmol) and K2CO3 (0.204 g, 1.48 mmol) in acetone (2 mL) was stirred at 60 °C for 24 h. Then, the crude mixture was filtered over Celite and concentrated under vacuum, and the residue was purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 99:1) to give 12d (0.082 g, 41%) as a pink oil. Rf 0.59 (hexane/EtOAc, 8:2). IR (film): νmax 2929, 1508, 1465, 1240, 1153, 1065, 1038, 818 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.11 (s, 3H, CH3-C2), 2.15 (s, 3H, CH3-C5), 3.13 (s, 3H, NCH3), 3.72 (s, 3H, CH3O-C4′), 3.82 (s, 3H, CH3O-C4), 6.45–6.51 (m, 2H, H-2′), 6.70 (s, 1H, H-3), 6.74–6.79 (m, 2H, H-3′), 6.87 (s, 1H, H-6). 13C-NMR (125 MHz, CDCl3): δ = 15.7 (CH3-C5), 17.8 (CH3-C2), 39.5 (NCH3), 55.4 (CH3O-C4), 55.7 (CH3O-C4′), 112.3 (C-3), 113.8 (C-2′), 114.6 (C-3′), 125.4 (C-5), 130.0 (C-6), 134.7 (C-2), 139.8 (C-1), 144.4 (C-1′), 151.3 (C-4′), 155.6 (C-4). HRMS (EI): m/z [M+] calcd for C17H21NO2: 271.1572; found: 271.1573.
2-(Benzylthio)-4-((4-methoxyphenyl)amino)-3,6-dimethylphenol (14a). A mixture of 10b (0.096 g, 0.40 mmol), benzylthiol (13a) (0.055 g, 0.044 mmol) and DMAP (0.054 g, 0.44 mmol) in CH2Cl2 (20 mL) was stirred at room temperature for 24 h. Then the mixture was concentrated under vacuum and purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 95:5) to give 14a (0.080 g, 55%) as a purple oil. Rf 0.75 (hexane/EtOAc, 7:3). IR (film): νmax 3377, 2924, 1507, 1455, 1409, 1232, 1178, 1033, 1007, 820, 697 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.15 (s, 3H, CH3-C3), 2.17 (s, 3H, CH3-C6), 3.76 (s, 3H, CH3O), 3.77 (s, 2H, CH2Ph), 4.91 (br s, 1H, NH), 6.60–6.64 (m, 2H, H-2′′), 6.76–6.80 (m, 2H, H-3′′), 6.88 (s, 1H, OH), 6.92 (s, 1H, H-5), 7.06–7.10 (m, 2H, H-2′), 7.20–7.26 (m, 3H, H-3′, H-4′). 13C-NMR (125 MHz, CDCl3): δ = 15.9 (CH3-C3), 16.3 (CH3-C6), 40.0 (CH2Ph), 55.7 (CH3O), 114.8 (C-3′′), 117.2 (C-2′′), 118.2 (C-2), 121.7 (C-6), 127.1 (C-5), 127.4 (C-4′), 128.5 (C-3′), 128.8 (C-2′), 134.0 (C-4), 134.1 (C-3), 137.3 (C-1′), 139.8 (C-1′′), 152.0 (C-1), 153.3 (C-4′′). HRMS (EI): m/z [M+] calcd for C22H23NO2S: 365.1449; found: 365.1448.
2-((2-Bromophenyl)thio)-4-((4-methoxyphenyl)amino)-3,6-dimethylphenol (14b). The procedure for the preparation of 14a was followed using a mixture of 10b (0.048 g, 0.20 mmol), o-bromobenzenethiol (13b) (0.038 g, 0.20 mmol) and DMAP (0.025 g, 0.20 mmol) to give 14b (0.064 g, 75%) as a purple oil. Rf 0.69 (hexane/EtOAc, 7:3). IR (film): νmax 3402, 2924, 1507, 1461, 1444, 1232, 1176, 1035, 1016, 820, 744 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.24 (s, 3H, CH3-C6), 2.26 (s, 3H, CH3-C3), 3.77 (s, 3H, CH3O), 5.03 (br s, 1H, NH), 6.51 (d, J = 8.0 Hz, 1H, H-6′), 6.56 (s, 1H, HO), 6.70–6.75 (m, 2H, H-2′′), 6.77–6.83 (m, 2H, H-3′′), 6.99 (dd, J = 8.0, 7.5 Hz, 1H, H-4′), 7.08 (s, 1H, H-5), 7.10 (dd, J = 8.0, 7.5 Hz, 1H, H-5′), 7.53 (d, J = 8.0 Hz, 1H, H-3′). 13C-NMR (125 MHz, CDCl3): δ = 15.9 (CH3-C3), 16.4 (CH3-C6), 55.7 (CH3O), 114.8 (2C-3′′), 115.5 (C-2), 117.9 (2C-2′′), 121.0 (C-2′), 122.8 (C-6), 125.9 (C-6′), 126.7 (C-4′), 127.4 (C-5), 128.0 (C-5′), 133.0 (C-3′), 133.4 (C-4), 135.1 (C-3), 136.6 (C-1′), 139.1 (C-1′′), 152.1 (C-1), 153.7 (C-4′′). HRMS (EI): m/z [M+] calcd for C21H20BrNO2S: 429.0398; found: 429.0386.
2-((4-Chlorophenyl)thio)-4-((4-methoxyphenyl)amino)-3,6-dimethylphenol (14c). The procedure for the preparation of 14a was followed using a mixture of 10b (0.146 g, 0.61 mmol), p-chlorobenzenethiol (13c) (0.088 g, 0.61 mmol) and DMAP (0.074 g, 0.61 mmol) to give 14c (0.232 g, 99%) as a purple oil. Rf 0.58 (hexane/EtOAc, 7:3). IR (film): νmax 3386, 2923, 1508, 1461, 1241, 1181, 1090, 1031, 1009, 816 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.23 (s, 3H, CH3-C6), 2.25 (s, 3H, CH3-C3), 3.75 (s, 3H, CH3O), 5.02 (br s, 1H, HN), 5.26 (s, 1H, HO), 6.65–6.74 (m, 2H, H-2′′), 6.75–6.82 (m, 2H, H-3′′), 6.90–6.97 (m, 2H, H-2′), 7.04 (s, 1H, H-5), 7.13–7.22 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 15.9 (CH3-C6), 16.4 (CH3-C3), 55.6 (CH3O), 114.7 (C-3′′), 115.4 (Ar), 117.7 (C-2′′), 122.5 (Ar), 127.2 (C-5), 127.3 (2C-2′), 129.2 (2C-3′), 131.6 (Ar), 133.2 (Ar), 134.0 (Ar), 134.9 (Ar), 139.0 (C-1′′), 151.8 (C-1), 153.5 (C-4′′). HRMS (EI): m/z [M+] calcd for C21H20ClNO2S: 385.0903; found: 385.0900.
4-((4-Methoxyphenyl)amino)-3,6-dimethyl-3-((4-tolyl)thio)phenol (14d). The procedure for the preparation of 14a was followed using a mixture of 10b (0.200 g, 0.83 mmol), p-tolylthiol (13d) (0.103 g, 0.84 mmol) and DMAP (0.101 g, 0.83 mmol) to give 14d (0.274 g, 90%) as a purple oil. Rf 0.73 (hexane/EtOAc, 7:3). IR (film): νmax 3394, 2919, 1508, 1473, 1233, 1179, 1090, 1031, 1010, 819 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.23 (s, 3H, CH3-C6), 2.26 (s, 3H, CH3Ar), 2.27 (s, 3H, CH3-C3), 3.74 (s, 3H, CH3O), 4.99 (br s, 1H, HN), 6.66–6.71 (m, 2H, H-2′′), 6.75–6.80 (m, 2H, H-3′′), 6.92–6.96 (m, 2H, H-3′), 7.01 (s, 1H, H-5), 7.00–7.04 (m, 2H, H-2′). 13C-NMR (75 MHz, CDCl3): δ = 15.9 (CH3-C3), 16.4 (CH3-C6), 20.8 (CH3Ar), 55.6 (CH3O), 114.7 (C-3′′), 116.6 (C-2), 117.5 (C-2′′), 122.2 (C-6), 126.5 (C-3′), 127.2 (C-5), 129.9 (C-2′), 131.8 (C-4′), 133.7 (C-3), 134.6 (C-4), 135.7 (C-1′), 139.5 (C-1′′), 152.1 (C-1), 153.4 (C-4′′). HRMS (EI): m/z [M+] calcd for C22H23NO2S: 365.1449; found: 365.1440.
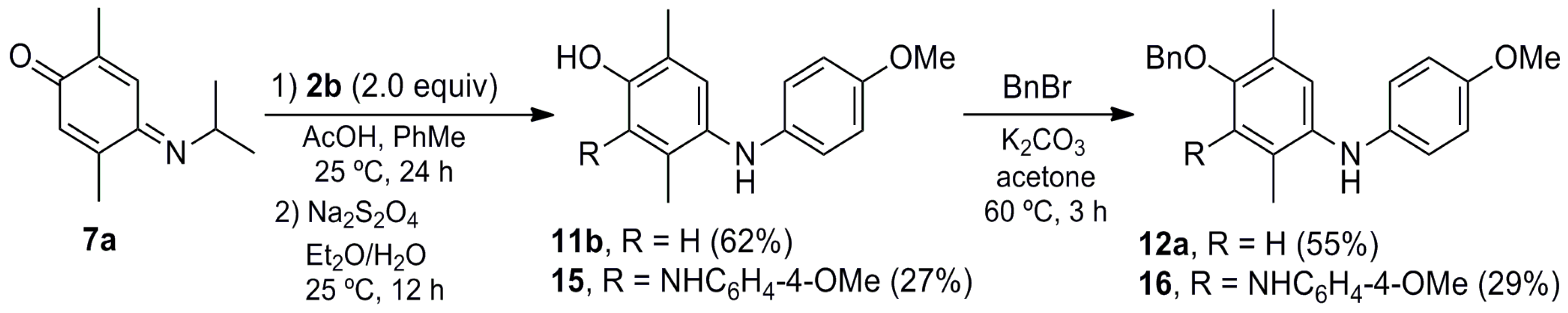
2,4-Bis((4-Methoxyphenyl)amino)-3,6-dimethylphenol (15). A mixture of 7a (0.500 g, 2.82 mmol), 2b (0.694 g, 5.64 mmol) and AcOH (0.051 g, 0.85 mmol) in toluene (30 mL) was stirred at room temperature for 24 h. The crude mixture was concentrated under vacuum, and then Et2O (30 mL) and a saturated aqueous solution of sodium hydrosulfite (30 mL) were added, followed by stirring at room temperature for 12 h. The crude mixture was washed with CH2Cl2 (3 × 10 mL) and the organic layer dried (Na2SO4), concentrated under vacuum and purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 9:1) to give 11b (0.426 g, 62%) and 15 (0.278 g, 27%) as a purple solid.
Data of 15a: Rf 0.32 (hexane/EtOAc, 1:1); mp 70–71 °C. IR (KBr): νmax 3344, 2932, 1629, 1510, 1239, 1033, 825 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.97 (s, 3H, CH3-C3), 2.24 (s, 3H, CH3-C6), 3.73 (s, 3H, CH3O), 3.75 (s, 3H, CH3O), 4.79 (br, 1H, NH), 4.95 (br, 1H, OH), 6.35 (br, 1H, NH), 6.54–6.58 (m, 2H, 2ArH), 6.65–6.69 (m, 2H, 2ArH), 6.74–6.79 (m, 4H, 4ArH), 6.88 (s, 1H, H-5). 13C-NMR (125 MHz, CDCl3): δ = 12.6 (CH3-C2), 15.8 (CH3-C5), 55.6 (CH3O), 55.7 (CH3O), 114.7 (2ArH), 114.9 (2ArH), 115.3 (2ArH), 117.3 (2ArH), 121.5 (Ar), 123.6 (C-5), 127.0 (Ar), 127.5 (Ar), 134.1 (Ar), 139.8 (Ar), 140.1 (Ar), 148.6 (C-4), 153.3 (ArO), 153.4 (ArO). MS (70 eV): m/z (%) 348 (M+, 100), 333 (68), 273 (70), 243 (44), 228 (60), 160 (36), 146 (18), 122 (16), 77 (14). HRMS (EI): m/z [M+] calcd for C22H24N2O3: 364.1787; found: 364.1786.
4-(Benzyloxy)-N,N′-bis(4-methoxyphenyl)-2,5-dimethylbenzene-1,2-diamine (16). A mixture of 7a (0.618 g, 3.49 mmol), 2b (1.073 g, 8.72 mmol) and AcOH (0.063 g, 1.05 mmol) in toluene (40 mL) was stirred at room temperature for 24 h, then concentrated under vacuum and suspended in Et2O (30 mL). A saturated aqueous solution of sodium hydrosulfite (30 mL) was added and the mixture was stirred at room temperature for 12 h. The crude mixture was washed with CH2Cl2 (3 × 10 mL) and the organic layer dried (Na2SO4) and concentrated under vacuum. The crude mixture was suspended in acetone (30 mL) and K2CO3 (0.723 g, 5.24 mmol), followed by the addition of benzyl bromide (0.718 g, 4.20 mmol) and stirring at reflux for 3 h. The mixture was filtered over Celite and concentrated under vacuum, then purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 8:2) to give 12a (0.640 g, 55%) and 16 (0.460 g, 29%) as a purple solid.
Data of 16: Rf 0.26 (hexane/EtOAc, 8:2); mp 60–61 °C. IR (film): νmax 3377, 2930, 1508, 1457, 1235, 1178, 1034, 823 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.99 (s, 3H, CH3-C2), 2.24 (s, 3H, CH3-C5), 3.77 (s, 3H, CH3O), 3.80 (s, 3H, CH3O), 4.61 (s, 2H, CH2Ph), 4.80–6.00 (br, 2H, NH), 6.59–6.64 (m, 2H, 2ArH), 6.70 (s, 1H, H-6), 6.77–6.80 (m, 2H, 2ArH), 6.82–6.89 (m, 2H, 2ArH), 6.93–6.99 (m, 2H, 2ArH), 7.23–7.34 (m, 5H, PhH). 13C-NMR (75 MHz, CDCl3): δ = 13.2 (CH3-C2), 16.3 (CH3-C5), 55.61 (CH3O), 55.62 (CH3O), 75.5 (CH2Ph), 114.5 (2ArH), 114.7 (C-6), 116.7 (2ArH), 120.6 (C-2), 120.8 (4ArH), 128.1 (PhH), 128.2 (2PhH), 128.5 (2PhH), 129.0 (C-5), 135.5 (C-3), 137.3 (Ar), 137.4 (Ar), 139.7 (Ar), 140.0 (Ar), 145.6 (C-4), 153.2 (ArO), 154.5 (ArO). HRMS (EI): m/z [M+] calcd for C29H30N2O3: 454.2257; found: 454.2221.
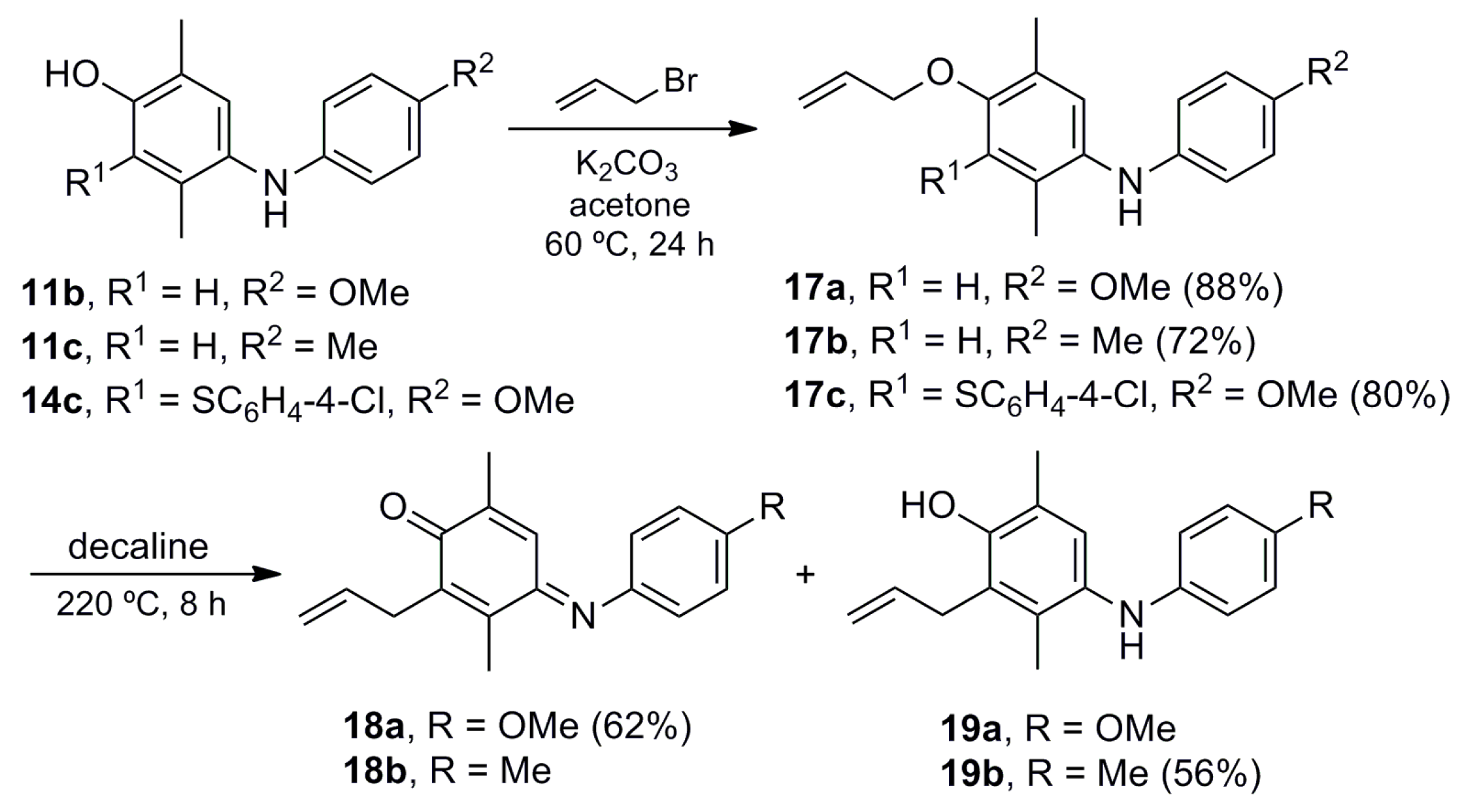
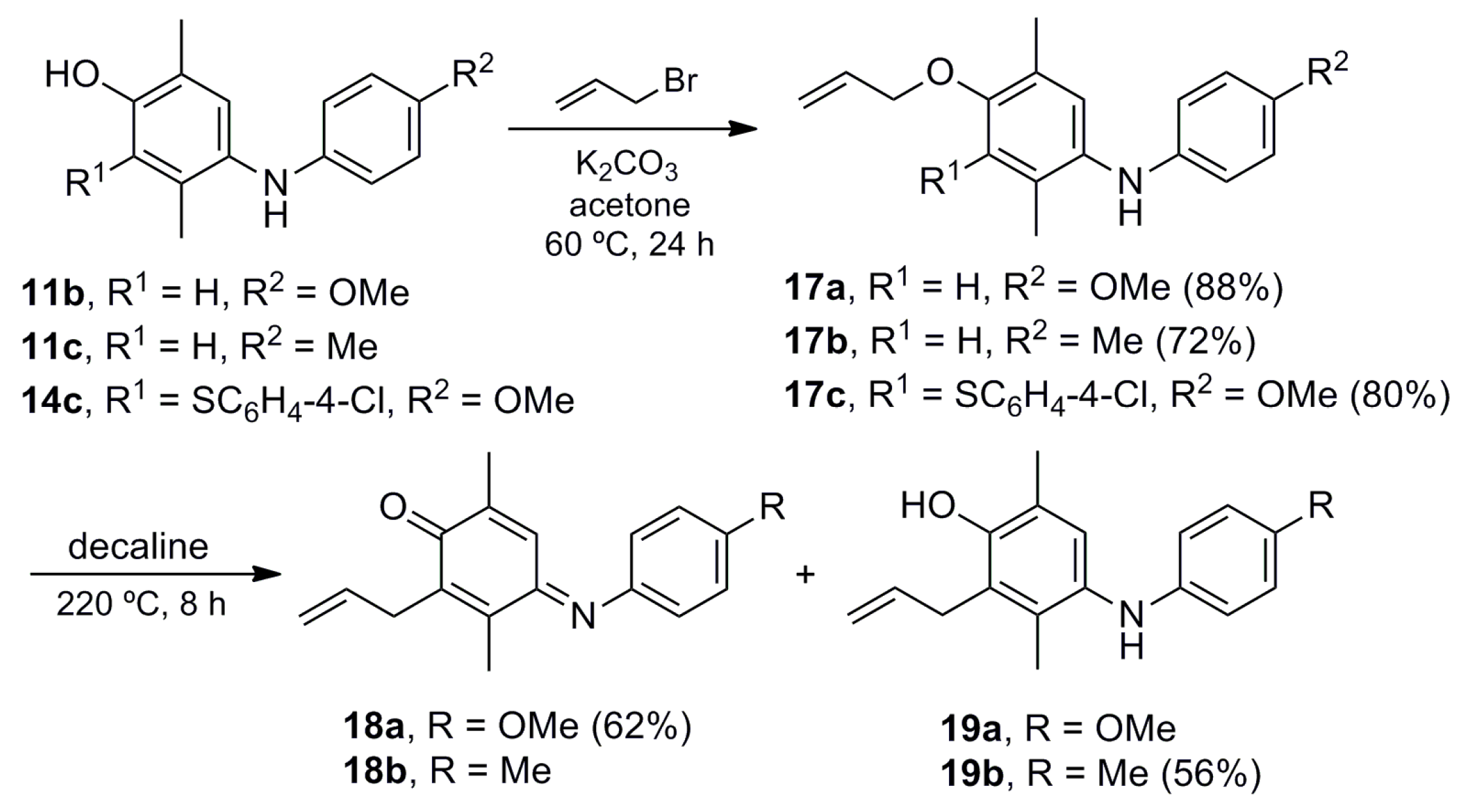
4-(Allyloxy)-N-(4-methoxyphenyl)-2,5-dimethylaniline (17a). A mixture of 11b (0.100 g, 0.41 mmol), potassium carbonate (0.062 g, 0.45 mmol) and allyl bromide (0.075 g, 0.62 mmol) in acetone (20 mL) was stirred at 60 °C for 24 h. The crude mixture was filtered over Celite, concentrated under vacuum and purified by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 95:5) to give 17a (0.102 g, 88%) as a pink solid. Rf 0.76 (hexane/EtOAc, 7:3); mp 89–90 °C. IR (film): νmax 1506, 1480, 1239, 1212, 1098, 1034, 1010, 947, 815 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.17 (s, 3H, CH3-C5), 2.19 (s, 3H, CH3-C2), 3.77 (s, 3H, CH3O), 4.50 (dt, J = 5.0, 1.5 Hz, 2H, CH2CH=), 4.97 (br, 1H, NH), 5.27 (dq, J = 10.5, 1.5 Hz, 1H, CH2=), 5.43 (dq, J = 17.4, 1.5 Hz, 1H, CH2=), 6.01–6.15 (m, 1H, CH=), 6.68 (br s, 1H, H-3), 6.73–6.83 (m, 4H, Ar-H), 6.89 (br s, 1H, H-6). 13C-NMR (75 MHz, CDCl3): δ = 15.9 (CH3-C5), 17.9 (CH3-C2), 55.6 (CH3O), 60.3 (CH2CH=), 114.4 (C-3), 114.6 (C-3′), 116.8 (CH2=), 118.2 (C-2′), 123.2 (C-6), 125.2 (C-5), 127.7 (C-2), 133.8 (CH=), 134.9 (C-1), 139.1 (C-1′), 152.2 (C-4), 153.5 (C-4′). HRMS (EI): m/z [M+] calcd for C18H21NO2: 283.1572; found: 283.1570.
4-(Allyloxy)-2,5-dimethyl-N-(p-tolyl)aniline (17b). The procedure for the preparation of 17a was followed using a mixture of 11c (0.162 g, 0.71 mmol), potassium carbonate (0.108 g, 0.78 mmol) and allyl bromide (0.129 g, 1.07 mmol) in acetone (20 mL) to give 17b (0.137 g. 72%) as a pink solid. Rf 0.71 (hexane/EtOAc, 8:2); mp 59–60 °C. IR (film): νmax 3405, 2918, 1612, 1510, 1410, 1391, 1283, 1196, 1093, 1012, 997, 917, 814 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.22 (s, 6H, CH3-C2, CH3-C5), 2.29 (s, 3H, CH3Ar), 4.53–4.62 (m, 2H, CH2CH=), 5.10 (br s, 1H, NH), 5.30 (dm, J = 10. 5 Hz, 1H, CH2=), 5.47 (dm, J = 17.3 Hz, 1H, CH2=), 6.05–6.21 (m, 1H, CH=), 6.67–6.75 (m, 2H, H-2′), 6.72 (br s, 1H, H-3), 7.01 (br s, 1H, H-6), 7.02–7.10 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 15.9 (CH3-C2 or CH3-C5), 17.9 (CH3-C5 or CH3-C2), 20.5 (CH3Ar), 69.2 (CH2CH=), 114.1 (C-3), 115.4 (C-2′), 116.8 (CH2=), 125.1 (C-5), 125.2 (C-6), 128.2 (C-2), 129.5 (C-4′), 129.7 (C-3′), 133.6 (CH=), 133.8 (C-1), 143.5 (C-1′), 152.9 (C-4). HRMS (EI): m/z [M+] Calcd for C18H21NO: 267.1623; found: 267.1627.
4-(Allyloxy)-3-((4-chlorophenyl)thio)-N-(4-methoxyphenyl)-2,5-dimethylaniline (17c). The procedure for the preparation of 17a was followed using a mixture of 14c (0.200 g, 0.52 mmol), potassium carbonate (0.079 g, 0.57 mmol) and allyl bromide (0.094 g, 0.78 mmol) in acetone (20 mL) to give 17c (0.179 g, 80%) as a pink oil; Rf 0.79 (hexane/EtOAc, 7:3). IR (film): νmax 2920, 1709, 1508, 1473, 1233, 1089, 1030, 1010, 816 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.21 (s, 3H, CH3-C2), 2.26 (s, 3H, CH3-C5), 3.75 (s, 3H, CH3O), 4.33 (br d, J = 5.5 Hz, 2H, CH2CH=), 5.14 (br s, 1H, NH), 5.15 (br d, J = 11.0 Hz, 1H, CH2=), 5.28 (br d, J = 17.5 Hz, 1H, CH2=), 5.98–6.08 (m, 1H, CH=), 6.81–6.82 (m, 2H, H-3′), 6.86–6.88 (m, 2H, H-2′), 6.93–6.97 (m, 2H, H-2′′), 6.96 (br s, 1H, H-5), 7.09–7.13 (m, 2H, H-3′′). 13C-NMR (125 MHz, CDCl3): δ = 15.5 (CH3-C5), 16.7 (CH3-C2), 55.4 (CH3O), 74.3 (CH2CH=), 114.6 (2C-3′), 117.1 (CH2=), 120.8 (2C-2′), 121.5 (C-6), 125.8 (C-3), 127.3 (2C-2′′), 128.7 (2C-3′′), 130.0 (C-5), 130.3 (C-4′′), 130.4 (C-2), 133.9 (CH=), 136.9 (C-1′′), 137.1 (C-1′), 139.9 (C-1), 153.3 (C-4), 154.6 (C-4′). HRMS (EI): m/z [M+] calcd for C24H24ClNO2S: 425.1216; found: 425.1212.
(E)-2-Allyl-4-((4-methoxyphenyl)imino)-3,6-dimethylcyclohexa-2,5-dienone (18a). A solution of 17a (0.050 g, 0.18 mmol) in decaline (1.0 mL) was stirred at 220 °C for 8 h. The crude mixture was concentrated by azeotropic distillation with toluene (3 × 20 mL) under vacuum, and purified by column chromatography over silica gel (10 g/g crude, hexane) to give 18a (0.032 g, 62%) as a red oil. Rf 0.77 (hexane/EtOAc, 7:3). IR (film): νmax 2922, 1627, 1601, 1498, 1464, 1440, 1288, 1243, 1035, 840, 723 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.96 (s, 3H, CH3-C6), 2.29 (s, 3H, CH3-C3), 3.35 (br d, J = 6.0 Hz, 2H, CH2CH=), 3.85 (s, 3H, CH3O), 5.03 (br d, J = 10.0 Hz, 1H, CH2=), 5.08 (br d, J = 17.0 Hz, 1H, CH2=), 5.78–5.88 (m, 1H, CH=), 6.79–6.84 (m, 2H, H-2′), 6.87 (br s, 1H, H-5), 6.93–6.97 (m, 2H, H-3′). 13C-NMR (125 MHz, CDCl3): δ = 13.5 (CH3-C3), 16.3 (CH3-C6), 30.7 (CH2CH=), 55.5 (CH3O), 114.3 (C-3′), 115.6 (CH2=), 122.3 (C-2′), 125.1 (C-5), 134.4 (CH=), 138.1 (C-3), 140.2 (C-6), 143.5 (C-1′), 145.0 (C-2), 157.6 (C-4), 157.7 (C-4′), 187.2 (C-1). HRMS (EI): m/z [M+] calcd for C18H21NO2: 283.1572; found: 283.1576.
2-Allyl-3,6-dimethyl-4-(p-tolylamino)phenol (19b). The procedure for the preparation of 18a was followed using a mixture of 17b (0.300 g, 1.12 mmol) in decaline (2 mL) to give 19b (0.168 g. 56%) as a dark red oil. Rf 0.67 (hexane/EtOAc, 8:2). IR (film): νmax 3542, 3384, 2920, 1635, 1614, 1514, 1473, 1285, 1242, 1180, 910, 810 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.13 (s, 3H, CH3-C3), 2.18 (s, 3H, CH3-C6), 2.25 (s, 3H, CH3Ar), 3.47 (dq, J = 5.7, 1.8 Hz, 2H, CH2CH=), 4.70 (br s, 1H, NH), 5.00–5.13 (m, 2H, CH2=), 5.98 (ddt, J = 17.1, 10.2, 5.7 Hz, 1H, CH=), 6.54–6.60 (m, 2H, H-2′), 6.88 (s, 1H, H-5), 6.95–7.01 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 13.8 (CH3-C3), 15.8 (CH3-C6), 20.4 (CH3Ar), 31.4 (CH2CH=), 114.8 (C-2′), 115.5 (CH2=), 121.7 (C-6), 124.3 (C-2), 125.0 (C-5), 127.7 (C-4′), 129.7 (C-3′), 130.5 (C-3), 133.5 (C-4), 135.5 (CH=), 144.3 (C-1′), 149.2 (C-1). HRMS (EI): m/z [M+] Calcd for C18H21NO: 267.1623; found: 267.1626.
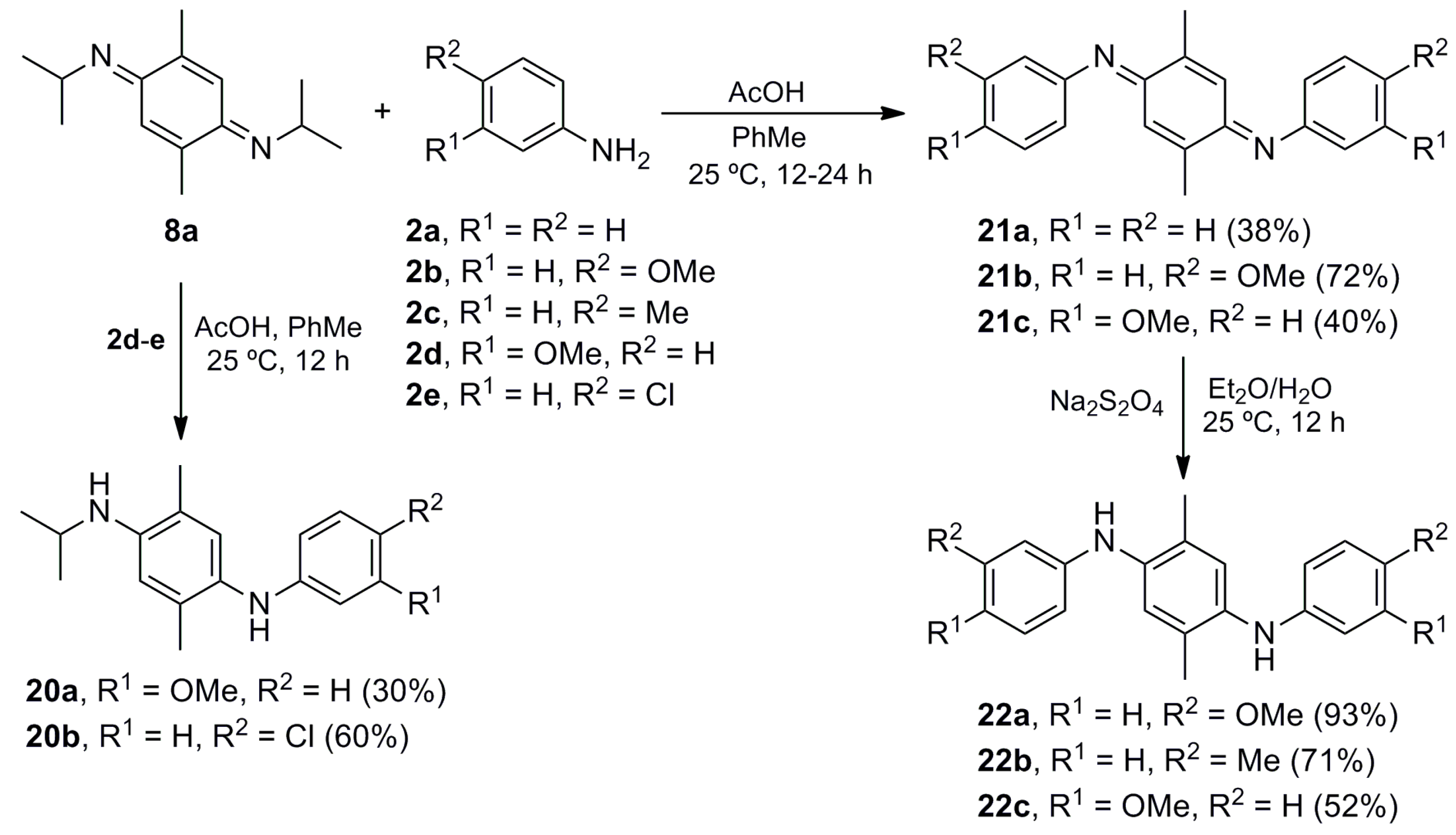
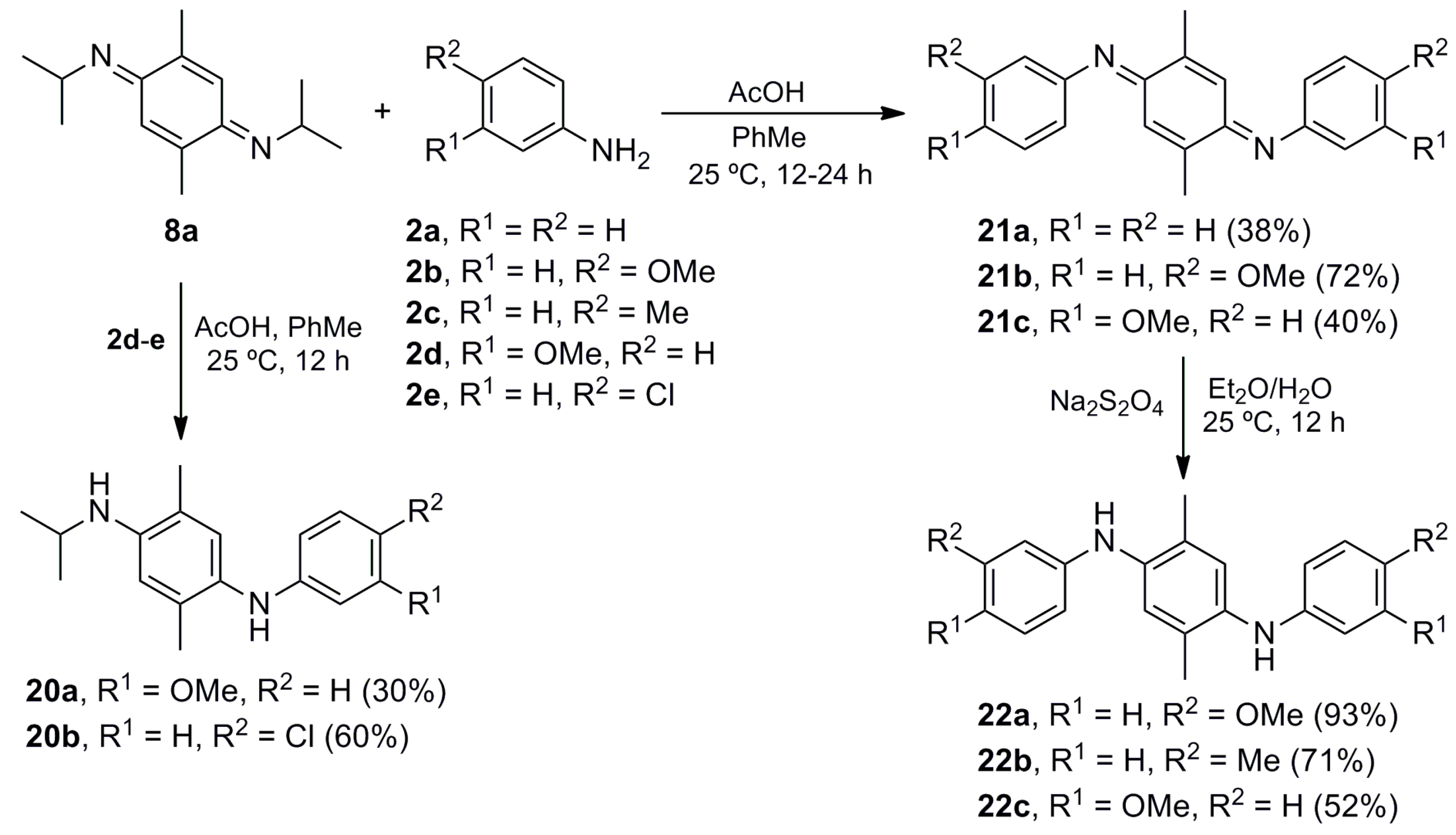
N-Isopropyl-N′-(3-methoxyphenyl)-2,5-dimethylbenzene-1,4-diamine (20a). The procedure for the preparation of 10a was followed using a mixture of 8a (0.299 g, 1.37 mmol), m-anisidine (2d) (0.169 g, 1.37 mmol) and glacial acetic acid (1.419 g, 23.64 mmol) in toluene (40 mL) to give 20a (0.117 g, 30%) as a brown solid. Rf 0.54 (hexane/EtOAc, 8:2); mp 83–84 °C. IR (film): νmax 3381, 2963, 1614, 1598, 1519, 1500, 1462, 1410, 1219, 1156, 1041, 839, 756, 688 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.25 (d, J = 6.0 Hz, 6H, (CH3)2CH), 2.06 (s, 3H, CH3-C5), 2.17 (s, 3H, CH3-C2), 3.66 (sept, J = 6.3 Hz, 1H, (CH3)2CH), 3.73 (s, 3H, CH3O), 5.14 (br s, 1H, NH), 6.17 (t, J = 2.1 Hz, 1H, H-2′), 6.22–6.30 (m, 2H, H-4′, H-6′), 6.49 (s, 1H, H-3), 6.91 (s, 1H, H-6), 7.05 (t, J = 8.3 Hz, 1H, H-5′). 13C-NMR (75 MHz, CDCl3): δ = 17.1 (CH3-C5), 18.0 (CH3-C2), 23.3 ((CH3)2CH), 44.3 ((CH3)2CH), 55.0 (CH3O), 99.6 (C-2′), 102.9 (C-6′), 106.8 (C-4′), 112.6 (C-3), 120.3 (C-5), 128.3 (C-6), 128.8 (C-1), 129.8 (C-5′), 132.8 (C-2), 143.0 (C-4), 149.0 (C-1′), 160.8 (C-3′). HRMS (EI): m/z [M+] calcd for C18H24N2O: 284.1889; found: 284.1886.
N-(4-Chlorophenyl)-N′-isopropyl-2,5-dimethylbenzene-1,4-diamine (20b). The procedure for the preparation of 10a was followed using a mixture of 8a (0.200 g, 0.92 mmol), p-chloroaniline (2e) (0.116 g, 0.91 mmol) and glacial acetic acid (0.943 g, 15.74 mmol) in toluene (40 mL) to give 20b (0.159 g, 60%) as a brown solid. Rf 0.53 (hexane/EtOAc, 9:1); mp 93–94 °C. IR (film): νmax 3401, 2965, 1596, 1519, 1492, 1303, 1250, 1215, 1171, 999, 818 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 1.24 (d, J = 6.3 Hz, 6H, (CH3)2CH), 2.06 (s, 3H, CH3-C5), 2.14 (s, 3H, CH3-C2), 3.16 (br s, 1H, NH), 3.66 (sept, J = 6.3 Hz, 1H, (CH3)2CH), 5.10 (br s, 1H, NH), 6.49 (s, 1H, H-3), 6.48–6.54 (m, 2H, H-2′), 6.85 (s, 1H, H-6), 7.03–7.09 (m, 2H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 17.1 (CH3-C5), 17.9 (CH3-C2), 23.2 ((CH3)2CH), 44.2 ((CH3)2CH), 112.5 (C-3), 114.8 (2C-2′), 120.4 (C-5), 122.0 (C-4′), 128.3 (C-6), 128.4 (C-5), 128.9 (2C-3′), 132.9 (C-2), 143.2 (C-4), 146.2 (C-1′). HRMS (EI): m/z [M]+calcd for C17H21ClN2: 288.1393; found: 288.1401.
(E,E)-2,5-Dimethyl-N,N′-(diphenyl)cyclohexa-2,5-diene-1,4-diimine (21a). The procedure for the preparation of 10a was followed with a mixture of 8a (0.100 g, 0.46 mmol), 2a (0.093 g, 1.00 mmol) and glacial acetic acid (0.524 g, 8.74 mmol) in toluene (20 mL) to give 21a (0.050 g, 38%) as a red solid. Rf 0.79 (hexane/EtOAc, 8:2); mp 194-195 °C. IR (film): νmax 1594, 1573, 1481, 1384, 1268, 1164, 895, 821, 759, 697 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.13 (d, J = 1.2 Hz, 6H, CH3-C2, CH3-C5), 6.64 (br d, J = 1.2 Hz, 2H, H-3, H-6), 6.58–6.74 (m, 4H, H-2′), 7.10–7.17 (m, 2H, H-4′), 7.33–7.42 (m, 4H, H-3′). 13C-NMR (75 MHz, CDCl3): δ = 17.9 (2CH3Ar), 120.1 (4C-2′), 122.8 (C-3, C-6), 124.2 (2C-4′), 128.9 (4C-3′), 143.8 (C-2, C-5), 150.7 (2C-1′), 158.9 (C-1, C-4). Anal. calcd for C20H18N2: C, 83.88; H, 6.34; N, 9.78. Found: C, 83.89; H, 6.33; N, 9.75.
(E,E)-N,N′-Bis(4-methoxyphenyl)-2,5-dimethylcyclohexa-2,5-diene-1,4-diimine (21b). The procedure for the preparation of 10a was followed with a mixture of 8a (0.100 g, 0.46 mmol), 2b (0.123 g, 1.00 mmol) and glacial acetic acid (0.524 g, 8.74 mmol) in toluene (20 mL) to give 21b (0.114 g, 72%) as a red solid. Rf 0.42 (hexane/EtOAc, 8:2); mp 144–145 °C. IR (KBr): νmax 2959, 1599, 1572, 1499, 1463, 1440, 1243, 1165, 1034, 844, 815, 764 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.14 (d, J = 1.5 Hz, 6H, CH3-C2, CH3-C5), 3.84 (s, 6H, 2CH3O), 6.73 (br d, J = 1.5 Hz, 2H, H-3, H-6), 6.82–6.86 (m, 4H, H-2′), 6.92–6.96 (m, 4H, H-3′). 13C-NMR (125 MHz, CDCl3): δ = 18.1 (2CH3Ar), 55.5 (2CH3O), 114.2 (4C-3′), 122.0 (4C-2′), 122.5 (C-3, C-6), 143.5 (C-2, C-5), 144.0 (2C-1′), 157.0 (2C-4′), 159.0 (C-1, C-4). MS (70 eV): m/z (%) 346 (M+, 5), 284 (45), 194 (55), 150 (100), 123 (98), 108 (98), 77 (41). HRMS (EI): m/z [M+] calcd for C22H22N2O2: 346.1681; found: 346.1682.
(E,E)-N,N′-Bis(3-Methoxyphenyl)-2,5-dimethylcyclohexa-2,5-diene-1,4-diimine (21c). The procedure for the preparation of 10a was followed with a mixture of 8a (0.100 g, 0.46 mmol), 2d (0.123 g, 1.00 mmol) and glacial acetic acid (0.477 g, 7.94 mmol) in toluene (20 mL) to give 21c (0.064 g, 40%) as an orange oil; Rf 0.40 (hexane/EtOAc, 8:2). IR (film): νmax 2921, 1579, 1478, 1280, 1145, 1042, 856, 776, 697 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.12 (d, J = 1.5 Hz, 6H, CH3-C2, CH3-C5), 3.82 (s, 6H, CH3O), 6.36–6.44 (m, 4H, H-2′, H-4′), 6.65 (br d, J = 1.3 Hz, 2H, H-3, H-6), 6.70 (ddd, J = 8.4, 2.4, 0.9 Hz, 2H, H-6′), 7.27 (t, J = 8.4 Hz, 2H, H-5′). 13C-NMR (75 MHz, CDCl3): δ = 17.9 (2CH3Ar), 55.3 (2CH3O), 105.5 (2C-2′), 109.9 (2C-6′), 112.2 (2C-4′), 123.0 (C-3, C-6), 129.7 (2C-5′), 143.7 (C-2, C-5), 152.1 (2C-1′), 159.0 (C-1, C-4), 160.1 (2C-3′). HRMS (EI): m/z [M+] calcd for C22H22N2O2: 346.1681; found: 346.1686.
N,N′-Bis(4-Methoxyphenyl)-2,5-dimethylbenzene-1,4-diamine (22a). The procedure for the preparation of 11a was followed using a mixture of 21b (0.121 g, 0.35 mmol) in Et2O (10 mL) to give 22a (0.113 g, 93%) as a dark brown solid. Rf 0.56 (hexane/EtOAc, 8:2); mp 145–146 °C. IR (KBr): νmax 3407, 1527, 1509, 1464, 1440, 1390, 1285, 1246, 1178, 1117, 1036, 1002, 825 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.13 (s, 6H, 2CH3Ar), 3.78 (s, 6H, 2CH3O), 4.88 (br s, 2H, 2NH), 6.80-6.88 (m, 8H, 2H-2′, 2H-3′), 6.90 (br s, 2H, H-3, H-6). 13C-NMR (125 MHz, CDCl3): δ = 17.5 (2CH3Ar), 55.6 (2CH3O), 114.7 (4C-3′), 119.2 (4C-2′), 121.5 (C-3, C-6), 127.0 (C-2, C-5), 136.6 (2C-1′), 138.4 (C-1, C-4), 153.9 (2C-4′). MS (70 eV): m/z (%) 348 (M+, 5), 284 (100), 269 (98), 241 (98), 210 (27), 127 (20), 122 (53). HRMS (EI): m/z [M+] calcd for C22H24N2O2: 348.1838; found: 348.1846.
2,5-Dimethyl-N,N′-di-p-tolylbenzene-1,4-diamine (22b). A mixture of 8a (0.100 g, 0.46 mmol), p-toluidine (2c) (0.107 g, 1.00 mmol) and AcOH (0.477 g, 7.94 mmol) in toluene (20 mL) was stirred at room temperature for 24 h. The crude mixture was concentrated under vacuum and suspended in Et2O (30 mL). A saturated aqueous solution of sodium hydrosulfite (30 mL) was added and the mixture stirred at room temperature for 12 h. The crude mixture was washed with CH2Cl2 (3 × 10 mL), and the organic layer dried (Na2SO4) and concentrated under vacuum, before purifying by column chromatography over silica gel (10 g/g crude, hexane/EtOAc, 8:2) to give 22b (0.11 g, 71%) as a brown solid. Rf 0.83 (hexane/EtOAc, 8:2); mp 134-135 °C. IR (film): νmax 3406, 3053, 1613, 1510, 1459, 1265, 1126, 1039, 896, 813, 738, 704 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.19 (s, 6H, CH3-C2, CH3-C5), 2.32 (s, 6H, 2CH3Ar), 4.90–5.30 (br, 2H, NH), 6.79–6.85 (m, 4H, 2H-2′), 7.06 (s, 2H, H-3, H-6), 7.05–7.10 (m, 4H, 2H-3′). 13C-NMR (75 MHz, CDCl3): δ = 17.6 (CH3-C2, CH3-C5), 20.5 (2CH3Ar), 116.7 (4C-2′), 122.8 (C-3, C-6), 128.1 (2C-4′), 129.0 (C-2, C-5), 129.8 (4C-3′), 136.1 (C-1, C-4), 142.6 (2C-1′). Anal. calcd for C22H24N2: C, 83.50; H, 7.64; N, 8.85. Found: C, 83.50; H, 7.68; N, 8.81.
N,N′-Bis(3-Methoxyphenyl)-2,5-dimethylbenzene-1,4-diamine (22c). The procedure for the preparation of 22b was followed using a mixture of 8a (0.100 g, 0.46 mmol) and 2d (0.123 g, 1.00 mmol) to give 22c (0.083 g, 52%) as a brown solid. Rf 0.66 (hexane/EtOAc, 8:2); mp 97–98 °C. IR (film): νmax 3381, 2956, 2924, 1599, 1497, 1215, 1156, 1042, 841, 763, 689 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.17 (s, 6H, 2CH3Ar), 3.76 (s, 6H, 2CH3O), 4.70–5.60 (br, 2H, NH), 6.38-6.42 (m, 4H, H-2′, H-4′), 6.42–6.46 (m, 2H, H-6′), 7.09 (s, 2H, H-3, H-6), 7.12 (t, J = 9.0 Hz, 2H, H-5′). 13C-NMR (125 MHz, CDCl3): δ = 17.6 (2CH3Ar), 55.1 (2CH3O), 101.8 (2C-2′), 104.5 (2C-4′), 108.7 (2C-6′), 124.5 (C-3, C-6), 129.3 (C-2, C-5), 130.0 (2C-5′), 135.9 (C-1, C-4), 146.8 (2C-1′), 160.8 (2C-3′). HRMS (EI): m/z [M+] calcd for C22H24N2O2: 348.1838; found: 348.1840.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}