
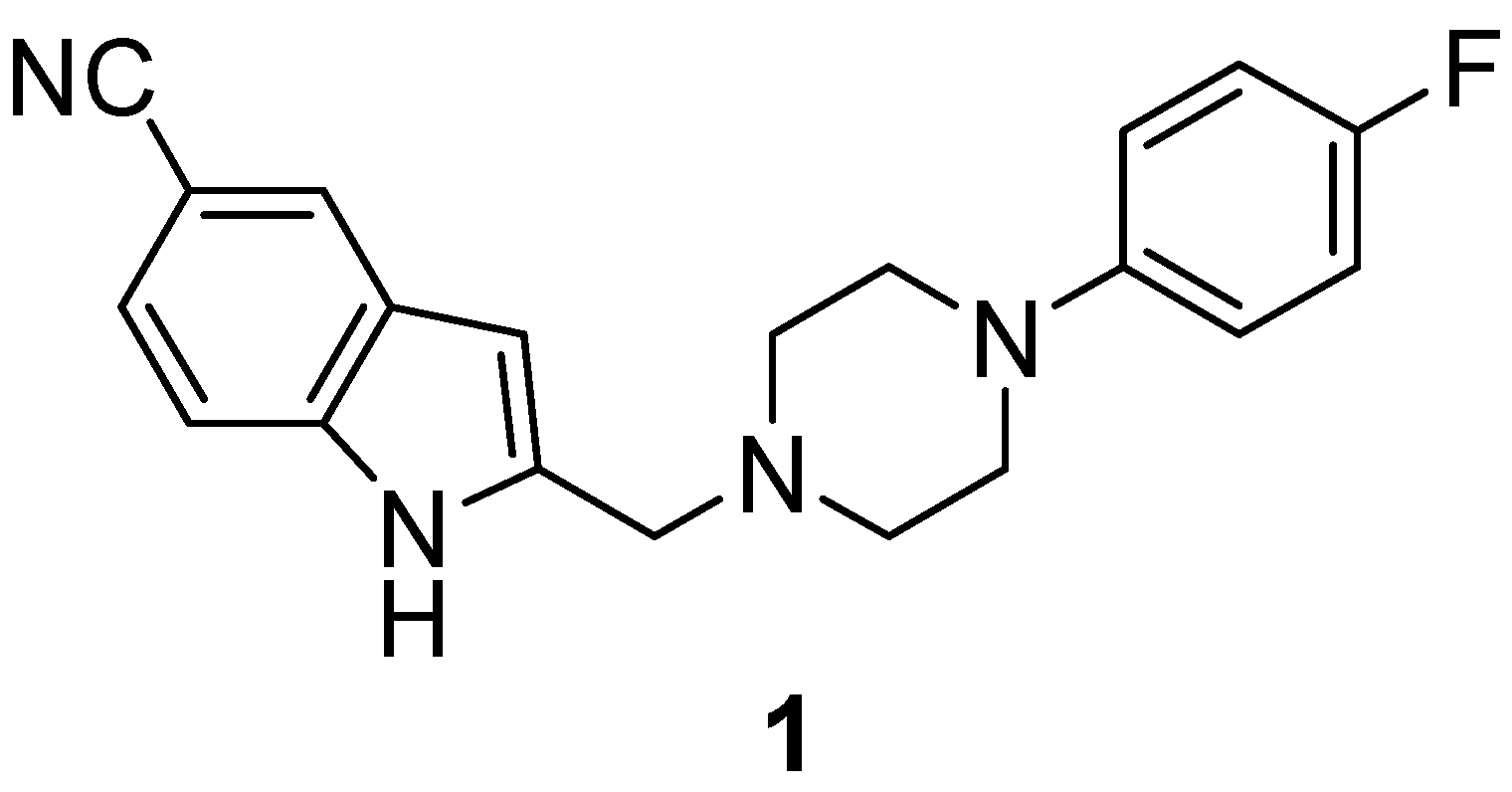
4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-Arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to Build-Up of the Dopamine D4 Ligand [18F]FAUC 316


Abstract
:1. Introduction
- Whenever possible, multi-step reactions are avoided in radiosyntheses with short-lived nuclides.
- Syntheses with many reactants often lead to problems of reproducibility and optimization under no-carrier-added (n.c.a.) conditions and complicate a rapid on-line separation of products.
- The typical use of non-polar solvents and dry reaction conditions for the coupling reaction necessitate an extensive pre-separation of labelled intermediates which is often time and yield consuming.
- Insoluble compounds (mixture of organic and inorganic reactants) complicate the applicability of the method in remotely controlled synthesis devices.
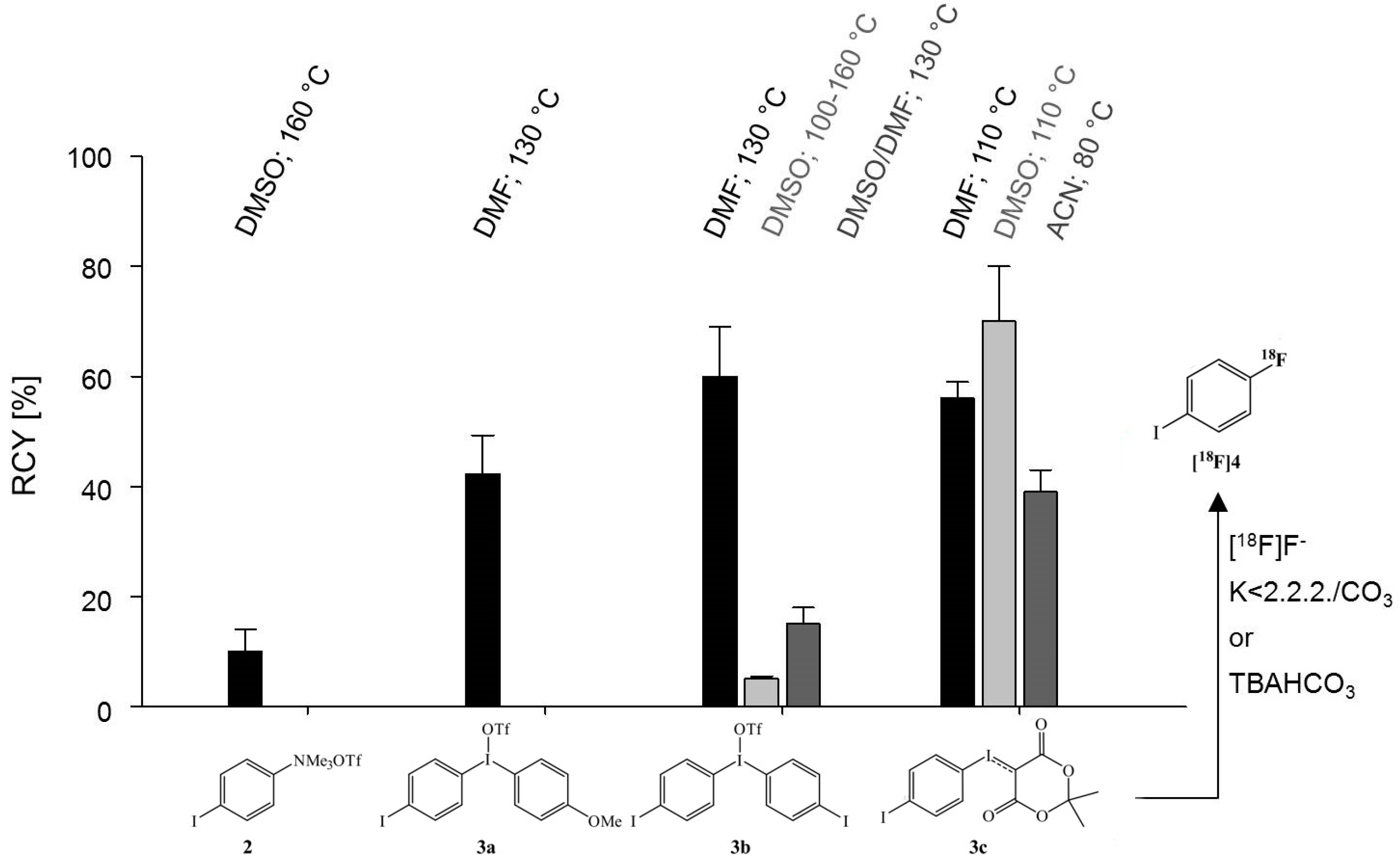
2. Results and Discussion
2.1. Palladium-Mediated N-Arylation

{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Catalyst System | Base | Solvent | Radiochemical Conversion Yield/Reaction Time |
| 1 | Pd2(dba)3/DavePhos | NaO tBu | Toluene | 70%/15 min |
| 2 | Pd2(dba)3/RuPhos | NaO tBu | Toluene | 73%/15 min |
| 3 | [CinnamylPdCl]2/BrettPhos | NaO tBu | Toluene | 25%/30 min |
| 4 | Pd(OAc)2/RuPhos | NaO tBu | Toluene | 60%/5 min 74%/10 min |
| 5 | Pd2(dba)3/DavePhos | NaO tBu | 1,4-dioxane | 40%/30 min |
| 6 | Pd2(dba)3/DavePhos | NaO tBu | DMF | 0%/60 min |
| 7 | Pd(OAc)2/RuPhos | NaO tBu | DMF | 0%/60 min |
| 8 | [P(tBu)3PdBr]2 | K3PO4 | DMF | 0%/60 min |
| 9 | [P(tBu)3PdBr]2 | K2CO3 | DMF | 0%/60 min |
| 10 | Pd2(dba)3/RuPhos | K3PO4 | DMF | 50%/6 min 88%/15 min |
| 11 | Pd[P(tBu)3]2 | K3PO4 | DMF | 31%/15 min |
| 12 | Pd2(dba)3/JohnPhos | K3PO4 | DMF | 67%/15 min |
| 13 | Pd2(dba)3/JohnPhos | Cs2CO3 | DMF | 51%/15 min |
| 14 | Pd2(dba)3/XPhos | K3PO4 | DMF | 28%/15 min |
| 15 | Pd2(dba)3/XPhos | Cs2CO3 | DMF | 11%/15 min |
| 16 | Pd2(dba)3/RuPhos | K3PO4 | DMSO | 60%/15 min |
| 17 | Pd2(dba)3/RuPhos | Cs2CO3 | DMSO | ~95%/15 min |
| 18 | Pd2(dba)3/JohnPhos | K3PO4 | DMSO | 82%/15 min |
| 19 | Pd2(dba)3/XPhos | K3PO4 | DMSO | 77%/15 min |
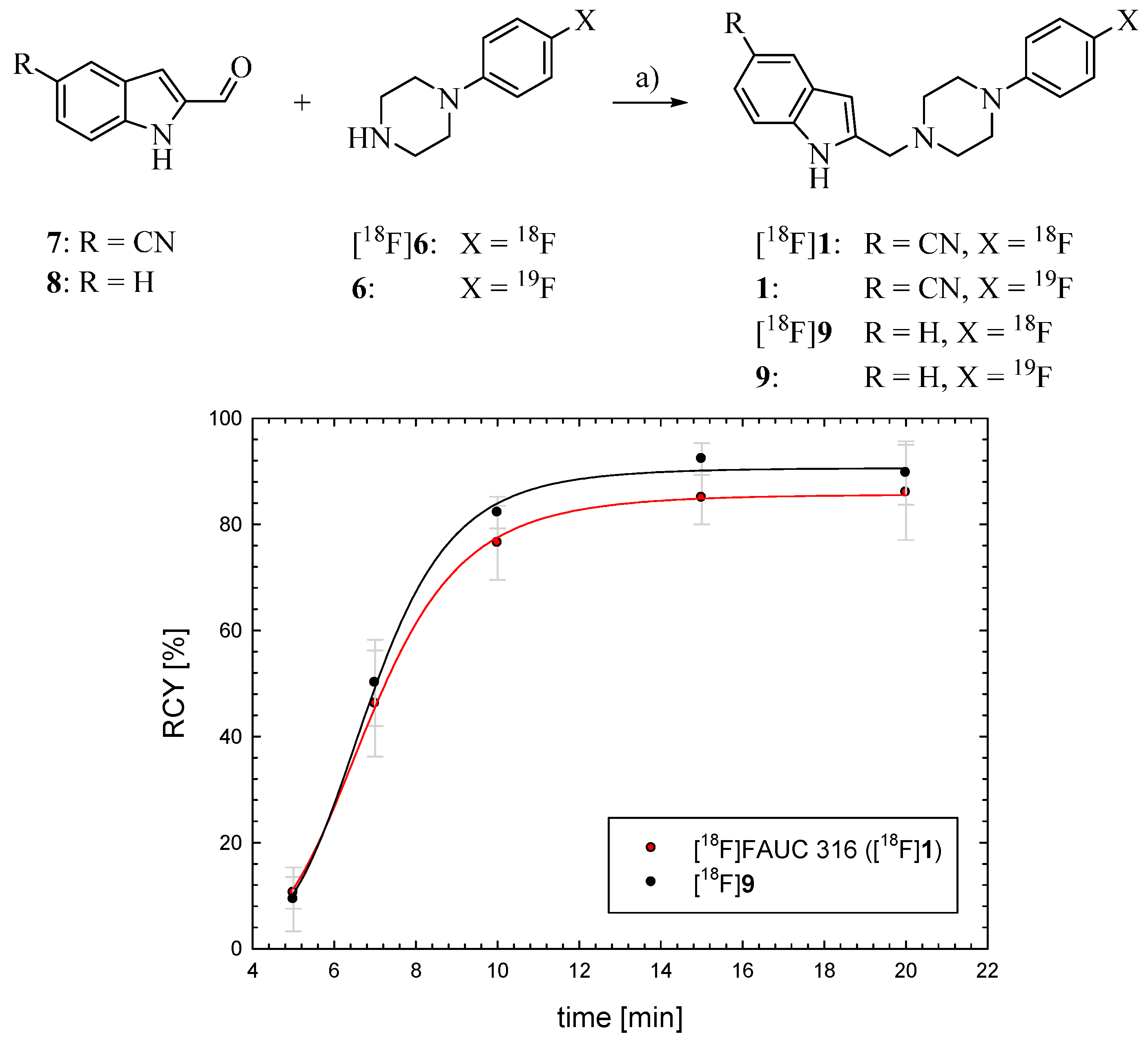
2.2. Synthesis and Evaluation of the D4 Agonist [18F]FAUC 316
| Structures | Time (min) | RCY (%) | Compounds and Conditions |
|---|---|---|---|
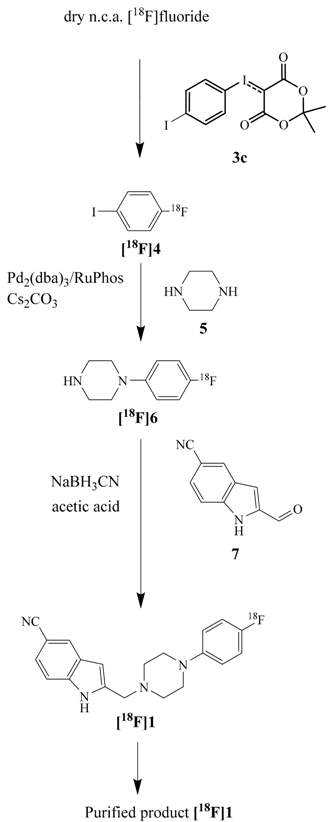 | 0 | Drying process | |
| 5 | 100 | ||
| Addition of iodonium ylide 3c | |||
| 15 | 70 | Heating at 110 °C | |
| 60 | Purification step | ||
| Addition of piperazine and HBC reagents | |||
| 30 | 47 | Heating at 100 °C | |
| 37 | Purification step | ||
| Addition of 2-formyl-1 H-indole-5-carbonitrile and NaBH3CN | |||
| Stirring | |||
| 45 | 28 | ||
| Pre-purification | |||
| HPLC-separation | |||
| 80 | 10 | Formulation |
3. Experimental Section
3.1. General
3.2. Chemistry
3.3. Radiochemistry
3.3.1. Reactive n.c.a [18F]Fluoride
3.3.2. General Preparation of 1-[18F]Fluoro-4-iodobenzene ([18F]4)
3.3.3. General Procedure of N-Arylation via Hartwig-Buchwald Coupling
3.3.4. Pre-Purification of 4-[18F]Fluorophenylpiperazine ([18F]6)
3.3.5. General Preparation of [18F]1 and [18F]9 by Reductive Amination
3.3.6. In Vitro Evaluation of [18F]FAUC 316 ([18F]1)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heck, R.F. Arylation, methylation, and carboxyalkylation of olefins by group VIII metal derivatives. J. Am. Chem. Soc. 1968, 90, 5518–5526. [Google Scholar] [CrossRef]
- Wu, X.F.; Anbarasan, P.; Neumann, H.; Beller, M. From Noble Metal to Nobel Prize: Palladium-Catalyzed Coupling Reactions as Key Methods in Organic Synthesis. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef]
- Allard, M.; Fouquet, E.; James, D.; Szlosek-Pinaud, M. State of art in 11C labelled radiotracers synthesis. Curr. Med. Chem. 2008, 15, 235–277. [Google Scholar] [CrossRef] [PubMed]
- Ermert, J. 18F-Labelled Intermediates for Radiosynthesis by Modular Build-Up Reactions: Newer Developments. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Way, J.; Bouvet, V.; Wuest, F. Synthesis of 4-[18F]fluorohalobenzenes and Palladium-mediated Cross-coupling Reactions for the Synthesis of 18F-labeled Radiotracers. Curr. Org. Chem. 2013, 17, 2138–2152. [Google Scholar] [CrossRef]
- Elizarov, A.M. Microreactors for radiopharmaceutical synthesis. Lab Chip 2009, 9, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.Y.; Pike, V.W. Micro-reactors for PET tracer labeling. In PET Chemistry: The Driving Force in Molecular Imaging; Schubiger, P.A., Lehmann, L., Friebe, M., Eds.; Springer-Verlag: Berlin-Heidelberg, Germany, 2007; Volume 62, pp. 271–287. [Google Scholar]
- Ermert, J.; Hocke, C.; Ludwig, T.; Gail, R.; Coenen, H. Comparison of pathways to the versatile synthon of no-carrier-added 1-bromo-4-[18F]fluorobenzene. J. Label. Compd. Radiopharm. 2004, 47, 429–441. [Google Scholar] [CrossRef]
- Ross, T.L.; Ermert, J.; Hocke, C.; Coenen, H.H. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride. J. Am. Chem. Soc. 2007, 129, 8018–8025. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Pike, V.W.; Widdowson, D.A. The synthesis of [18F]fluoroarenes from the reaction of cyclotron-produced [18F]fluoride ion with diaryliodonium salts. J. Chem. Soc. Perkin Trans. 1 1998, 2043–2046. [Google Scholar] [CrossRef]
- Allain-Barbier, L.; Lasne, M.C.; Perrio-Huard, C.; Moreau, B.; Barré, L. Synthesis of 4-[18F]fluorophenyl-alkenes and -arenes via palladium-catalyzed coupling of 4-[18F]fluoroiodobenzene with vinyl and aryl tin reagents. Acta Chem. Scand. 1998, 52, 480–489. [Google Scholar] [CrossRef]
- Wüst, F.R.; Kniess, T. No-carrier added synthesis of 18F-labelled nucleosides using Stille cross-coupling reactions with 4-[18F]fluoroiodobenzene. J. Label. Compd. Radiopharm. 2004, 47, 57–468. [Google Scholar] [CrossRef]
- Steiniger, B.; Wuest, F.R. Synthesis of 18F-labelled biphenyls via SUZUKI cross-coupling with 4-[18F]fluoroiodobenzene. J. Label. Compd. Radiopharm. 2006, 49, 817–827. [Google Scholar] [CrossRef]
- Marrière, E.; Rouden, J.; Tadino, V.; Lasne, M.C. Synthesis of analogues of (-)-cytisine for in vivo studies of nicotinic receptors using positron emission tomography. Org. Lett. 2000, 2, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.J. Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley-lnterscience: New York, NY, USA, 1979. [Google Scholar]
- Collins, M.; Lasne, M.C.; Barré, L. Rapid synthesis of N,N′-disubstituted piperazines. Application to the preparation of no carrier added 1-(4-[18F]fluorophenyl)piperazine and of an [18F]-selective ligand of serotoninergic receptors (5HT2 antagonist). J. Chem. Soc. Perkin Trans. 1 1992, 23, 3185–3188. [Google Scholar] [CrossRef]
- Mishani, E.; Cristel, M.E.; Dence, C.S.; McCarthy, T.J.; Welch, M.J. Application of a novel phenylpiperazine formation reaction to the radiosynthesis of a model fluorine-18-labeled radiopharmaceutical (18FTFMPP). Nucl. Med. Biol. 1997, 24, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Wüst, F.R.; Kniess, T. N-Arylation of indoles with 4-[18F]fluoroiodobenzene: Synthesis of 18F-labelled σ2 receptor ligands for positron emission tomography (PET). J. Label. Compd. Radiopharm. 2005, 48, 31–43. [Google Scholar] [CrossRef]
- Betts, H.M.; Robins, E.G. 2-Bromo-6-[18F]fluoropyridine: Two-step fluorine-18 radiolabelling via transition metal-mediated chemistry. J. Label. Compd. Radiopharm. 2014, 57, 215–218. [Google Scholar] [CrossRef]
- Scherman, D.; Hamon, M.; Gozlan, H.; Henry, J.P.; Lesage, A.; Masson, M.; Rumigny, J.F. Molecular pharmacology of niaprazine. Prog. Neuropsychopharmacol. Biol. Psychiatry 1988, 12, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Glennon, R.A.; Yousif, M.Y.; Ismaiel, A.M.; el-Ashmawy, M.B.; Herndon, J.L.; Fischer, J.B.; Server, A.C.; Howie, K.J.B. Novel 1-phenylpiperazine and 4-phenylpiperidine derivatives as high-affinity.sigma. ligands. J. Med. Chem. 1991, 34, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.N.; McKown, L.A.; Kuo, G.H. Hepatic metabolism of two α-1A-adrenergic receptor antagonists, phthalimide-phenylpiperazine analogs (RWJ-69205 and RWJ-69471), in the rat, dog and human. Eur. J. Drug Metab. Pharmacokinet. 2006, 31, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Mockett, B.G.; Guévremont, D.; Wutte, M.; Hulme, S.R.; Williams, J.M.; Abraham, W.C. Calcium/Calmodulin-dependent protein kinase II mediates group I metabotropic glutamate receptor-dependent protein synthesis and long-term depression in rat hippocampus. J. Neurosci. 2011, 31, 7380–7391. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.M.D. Important fluorinated drugs in experimental and clinical use. J. Fluor. Chem. 2002, 118, 27–33. [Google Scholar] [CrossRef]
- Gakh, A.A.; Burnett, M.N. Extreme modulation properties of aromatic fluorine. J. Fluor. Chem. 2011, 132, 88–93. [Google Scholar] [CrossRef]
- Hübner, H.; Kraxner, J.; Gmeiner, P. Cyanoindole Derivatives as Highly Selective Dopamine D4 Receptor Partial Agonists: Solid-Phase Synthesis, Binding Assays, and Functional Experiments. J. Med. Chem. 2000, 43, 4563–4569. [Google Scholar] [CrossRef] [PubMed]
- Tietze, R.; Hocke, C.; Löber, S.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Syntheses and radiofluorination of two derivatives of 5-cyano-indole as selective ligands for the dopamine subtype-4 receptor. J. Label. Compd. Radiopharm. 2006, 49, 55–70. [Google Scholar] [CrossRef]
- Prante, O.; Tietze, R.; Hocke, C.; Löber, S.; Hübner, H.; Kuwert, T.; Gmeiner, P. Synthesis, Radiofluorination, and In Vitro Evaluation of Pyrazolo[1,5-a]pyridine-Based Dopamine D4 Receptor Ligands: Discovery of an Inverse Agonist Radioligand for PET. J. Med. Chem. 2008, 51, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Primus, R.J.; Thurkauf, A.; Xu, J.; Yevich, E.; McInerney, S.; Shaw, K.; Tallman, J.F.; Gallager, D.W., II. Localization and characterization of dopamine D4 binding sites in rat and human brain by use of the novel, D4 receptor-selective ligand [3H]NGD 94-1. J. Pharmacol. Exp. Ther. 1997, 282, 1020–1027. [Google Scholar] [PubMed]
- Banerjee, A.; Prante, O. Subtype-selective dopamine receptor radioligands for PET imaging: Current status and recent developments. Curr. Med. Chem. 2012, 19, 3957–3966. [Google Scholar] [CrossRef] [PubMed]
- Prante, O.; Maschauer, S.; Banerjee, A. Radioligands for the dopamine receptor subtypes. J. Label. Compd. Radiopharm. 2013, 56, 130–148. [Google Scholar] [CrossRef]
- Ye, N.; Neumeyer, J.L.; Baldessarini, R.J.; Zhen, X.; Zhang, A. Update 1 of: Recent Progress in Development of Dopamine Receptor Subtype-Selective Agents: Potential Therapeutics for Neurological and Psychiatric Disorders. Chem. Rev. 2013, 113, PR123–PR178. [Google Scholar] [CrossRef] [PubMed]
- Löber, S.; Hübner, H.; Tschammer, N.; Gmeiner, P. Recent advances in the search for D3- and D4-selective drugs: probes, models and candidates. Trends Pharmacol. Sci. 2011, 32, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Grushin, V.V.; Demkina, I.I.; Tolstaya, T.P. Unified mechanistic analysis of polar reactions of diaryliodonium salts. J. Chem. Soc. Perkin Trans. 2 1992, 505–511. [Google Scholar] [CrossRef]
- Stang, P.J.; Olenyuk, B.; Chen, K. Preparation of nitrogen-containing bis(heteroaryl)iodonium salts. Synthesis 1995, 937–938. [Google Scholar] [CrossRef]
- Bielawski, M.; Malmgren, J.; Pardo, L.M.; Wikmark, Y.; Olofsson, B. One-Pot Synthesis and Applications of N-Heteroaryl Iodonium Salts. ChemistryOpen 2014, 3, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.H.; Pike, V.W. Selective syntheses of no-carrier-added 2- and 3-[18F]fluorohalopyridines through the radiofluorination of halopyridinyl(4[prime or minute]-methoxyphenyl)iodonium tosylates. Chem. Commun. 2012, 48, 9921–9923. [Google Scholar] [CrossRef]
- Jang, K.S.; Jung, Y.W.; Gu, G.; Koeppe, R.A.; Sherman, P.S.; Quesada, C.A.; Raffel, D.M. 4-[18F]Fluoro-m-hydroxyphenethylguanidine: A Radiopharmaceutical for Quantifying Regional Cardiac Sympathetic Nerve Density with Positron Emission Tomography. J. Med. Chem. 2013, 56, 7312–7323. [Google Scholar] [CrossRef] [PubMed]
- Moon, B.S.; Kil, H.S.; Park, J.H.; Kim, J.S.; Park, J.; Chi, D.Y.; Lee, B.C.; Kim, S.E. Facile aromatic radiofluorination of [18F]flumazenil from diaryliodonium salts with evaluation of their stability and selectivity. Org. Biomol. Chem. 2011, 9, 8346–8355. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, K.C.; Lee, H.; Mach, R.H.; Katzenellenbogen, J.A. Strategies for the Labeling of Halogen-Substituted Peroxisome Proliferator-Activated Receptor γ Ligands: Potential Positron Emission Tomography and Single Photon Emission Computed Tomography Imaging Agents. Bioconjug. Chem. 2007, 18, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.; Ermert, J.; Coenen, H.H. Convenient preparation of (4-iodophenyl)aryliodonium salts. Tetrahedron 2012, 68, 4112–4116. [Google Scholar] [CrossRef]
- Carroll, M.A.; Nairne, J.; Smith, G.; Widdowson, D.A. Radical scavengers: A practical solution to the reproducibility issue in the fluoridation of diaryliodonium salts. J. Fluor. Chem. 2007, 128, 127–132. [Google Scholar] [CrossRef]
- Cardinale, J.; Ermert, J. Simplified synthesis of aryliodonium ylides by a one-pot procedure. Tetrahedron Lett. 2013, 54, 2067–2069. [Google Scholar] [CrossRef]
- Mu, L.; Fischer, C.R.; Holland, J.P.; Becaud, J.; Schubiger, P.A.; Schibli, R.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Dinkelborg, L.M.; et al. 18F-radiolabeling of aromatic compounds using triarylsulfonium salts. Eur. J. Org. Chem. 2012, 2012, 889–892. [Google Scholar] [CrossRef]
- Way, J.D.; Wuest, F. Automated radiosynthesis of no-carrier-added 4-[18F]fluoroiodobenzene: A versatile building block in 18F radiochemistry. J. Label. Compd. Radiopharm. 2014, 57, 104–109. [Google Scholar] [CrossRef]
- Surry, D.S.; Buchwald, S.L. Biaryl phosphane ligands in palladium-catalyzed amination. Angew. Chem. Int. Ed. 2008, 47, 6338–6361. [Google Scholar] [CrossRef]
- Surry, D.S.; Buchwald, S.L. Dialkylbiaryl phosphines in Pd-catalyzed amination: A user’s guide. Chem. Sci. 2011, 2, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Christensen, H.; Kiil, S.; Dam-Johansen, K.; Nielsen, O.; Sommer, M.B. Effect of solvents on the product distribution and reaction rate of a Buchwald-Hartwig amination reaction. Org. Process Res. Dev. 2006, 10, 762–769. [Google Scholar] [CrossRef]
- Ohshita, K.; Ishiyama, H.; Oyanagi, K.; Nakata, H.; Kobayashi, J. Synthesis of hybrid molecules of caffeine and eudistomin D and its effects on adenosine receptors. Bioorg. Med. Chem. 2007, 15, 3235–3240. [Google Scholar] [CrossRef] [PubMed]
- Tasler, S.; Mies, J.; Lang, M. Applicability aspects of transition metal-catalyzed aromatic amination protocols in medicinal chemistry. Adv. Synth. Catal. 2007, 349, 2286–2300. [Google Scholar] [CrossRef]
- Leadbeater, N.E. Fast, easy, clean chemistry by using water as a solvent and microwave heating: The Suzuki coupling as an illustration. Chem. Commun. 2005, 2881–2902. [Google Scholar] [CrossRef]
- Foreman, J.C.; Johansen, T. Textbook of Receptor Pharmacology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Gail, R.; Coenen, H.H. A one step preparation of the n.c.a. fluorine-18 labelled synthons: 4-Fluorobromobenzene and 4-fluoroiodobenzene. Appl. Radiat. Isot. 1994, 45, 105–111. [Google Scholar] [CrossRef]
- Ambrogio, I.; Cacchi, S.; Fabrizi, G. Palladium-Catalyzed Synthesis of 2-(Aminomethyl)indoles from Ethyl 3-(o-Trifluoroacetamidophenyl)-1-propargyl Carbonate. Org. Lett. 2006, 8, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Qaim, S.M.; Stöcklin, G. Production of Some Medically Important Short-Lived Neutron-Deficient Radioisotopes of Halogens. Radiochim. Acta 1983, 34, 25–40. [Google Scholar]
- Hamacher, K.; Hirschfelder, T.; Coenen, H.H. Electrochemical cell for separation of [18F]fluoride from irradiated 18O-water and subsequent no carrier added nucleophilic fluorination. Appl. Radiat. Isot. 2002, 56, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Coenen, H.H.; Klatte, B.; Knöchel, A. Preparation of n.c.a. [17-18F]-fluoroheptadecanoic acid in high yields via aminopolyether supported, nucleophilic fluorination. J. Label. Compd. Radiopharm. 1986, 23, 455–466. [Google Scholar] [CrossRef]
- Ermert, J.; Stüsgen, S.; Lang, M.; Roden, W.; Coenen, H.H. High molar activity of [11C]TCH346 via [11C]methyl triflate using the “wet” [11C]CO2reduction method. Appl. Radiat. Isot. 2008, 66, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Weiss, N.T.; Tarazi, F.I.; Kula, N.S.; Baldessarini, R.J. Effects of alkylating agents on dopamine D3 receptors in rat brain: Selective protection by dopamine. Brain Res. 1999, 847, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: All compounds used in the study are commercially available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kügler, F.; Ermert, J.; Kaufholz, P.; Coenen, H.H. 4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-Arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to Build-Up of the Dopamine D4 Ligand [18F]FAUC 316. Molecules 2015, 20, 470-486. https://doi.org/10.3390/molecules20010470
Kügler F, Ermert J, Kaufholz P, Coenen HH. 4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-Arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to Build-Up of the Dopamine D4 Ligand [18F]FAUC 316. Molecules. 2015; 20(1):470-486. https://doi.org/10.3390/molecules20010470
Chicago/Turabian StyleKügler, Fabian, Johannes Ermert, Peter Kaufholz, and Heinz H. Coenen. 2015. "4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-Arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to Build-Up of the Dopamine D4 Ligand [18F]FAUC 316" Molecules 20, no. 1: 470-486. https://doi.org/10.3390/molecules20010470
APA StyleKügler, F., Ermert, J., Kaufholz, P., & Coenen, H. H. (2015). 4-[18F]Fluorophenylpiperazines by Improved Hartwig-Buchwald N-Arylation of 4-[18F]fluoroiodobenzene, Formed via Hypervalent λ3-Iodane Precursors: Application to Build-Up of the Dopamine D4 Ligand [18F]FAUC 316. Molecules, 20(1), 470-486. https://doi.org/10.3390/molecules20010470