20 Years of Fatty Acid Analysis by Capillary Electrophoresis

Abstract
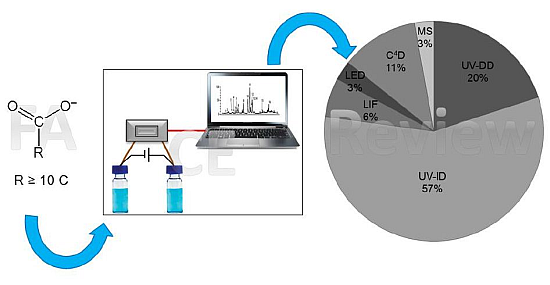
:
1. Introduction

2. Separations in Different CE and Detection Modes
2.1. CE with Direct UV Detection
2.2. CE with Indirect UV Detection

{kind=link}
{kind=link}
| Samples | FA | BGE and Experimental Condition | CE Mode and Detection | Year | Ref. |
|---|---|---|---|---|---|
| Mix standard | Saturated C2 at C20 | heptane (0.66% w/w), cholate (4.87% w/w), n-BuOH (6.55% w/w) and 10 mmol·L−1 borate buffer (87.93% w/w); 30 °C, 15 kV | MEEKC-UV-DD (λ = 243 nm) | 1998 | [34] |
| Seed oils | CTFAs | 24 mmol·L−1 SDS, 20% ACN, 40 mmol·L−1 borate (pH 9.2), 4 mmol·L−1 urea, 5 mmol·L−1 de [(R)-N-DOCV], 1 mmol·L−1 HS-β-CD and 10 mmol·L−1 β-CD; 15 °C, 30 kV | CZE-UV-DD (λ = 268 nm) | 2003 | [35] |
| Fish oil and enriched milk | C16:0, C18:1c, C18:2cc, C18:3ccc, C20:5ccccc and C22:6cccccc | 10 mmol·L−1 tetraborate (pH 9.2), 10 mmol·L−1 of Brij 35, 25% ACN and 25% MeOH; 25 °C, 30 kV for DD | CZE-UV-DD (λ = 200 nm for DD) | 2003 | [36] |
| Mix standard | 9c, 11c-CLA, 9c, 11t-CLA, 9t, 11t-CLA, 10t, 12c-CLA, 11c, 13c-CLA, 11c, 13t-CLA and 11t, 13t-CLA | 4% (w/v) β-CD, 54 mmol·L−1 SDS, 80 mmol·L−1 borate (pH 9.0), 8 mol L−1 urea and 4% (v/v ) EtOH; 15 °C, 30 kV | CE-DAD-DD (λ = 231 nm) | 2005 | [37] |
| Enriched chicken eggs | C18:1 c, C18:2 cc, C18:3ccc, C20:5 ccccc and C22:6 cccccc | 12 mmol·L−1 tetraborate (pH 9.2), 12 mmol·L−1 of Brij 35, 17% ACN and 33% MeOH; 27 °C, 27 kV | CZE-UV-DD (λ = 200 nm) | 2011 | [29] |
| Bacterial strains | 9c, 11c-CLA, 9c, 11t-CLA, 9t, 11t-CLA, 10t, 12c-CLA, 11c, 13c-CLA, 11c, 13t-CLA and 11t, 13t-CLA | 4% (w/v) β-CD, 54 mmol·L−1 SDS, 80 mmol·L−1 borate (pH 9.0), 8 mol L−1 urea and 4% (v/v) EtOH; 15 °C, 30 kV | CE-UV-DD (λ = 231 nm) | 2012 | [38] |
| Oil and meat | C18:3 n-3, C22:3 n-3, C20:5 n-3, C22:5 n-3, C22:6 n-3, C18:2 n-6, C18:3 n-6, C19:2 n-6, C20:2 n-6, C20:3 n-6, C20:4 n-6, C22:2 n-6, C22:4 n-6, C22:5 n-6 and C20:3 n-3 | 50 mmol·L−1 SDS, 10% ACN, 40 mmol·L−1 borate (pH 9.5), 6 mmol·L−1 urea, 5 mmol·L−1 [(R)-N-COAD] and 10 mmol·L−1 β-CD; 25 kV | CZE-UV-DD (λ = 214 nm) | 2013 | [39] |
2.3. CE with Laser-Induced Fluorescence Detection
2.4. CE with Light-Emitting Diode Detection
| Samples | FA | BGE and Experimental Condition | CE Mode and Detection | Year | Ref. |
|---|---|---|---|---|---|
| Butter and palm oil | Saturated C12 at C18 | 30% acetone, a cationic surfactant and 3,5-dinitrobenzoic acid | CZE-UV-ID | 1994 | [48] |
| Butter | Saturated C8 at C20 | 10 mmol·L−1 SDBS, 50% ACN and 30 mmol·L−1 Brij; 23 °C, 20 kV | NACE-UV-ID (λ = 198 nm) | 1995 | [41] |
| Coconut oil | Saturated C2 at C18 | 20 mmol·L−1 Tris, 10 mmol·L−1 p-anisate, 1.0 mmol·L−1 trimethyl-β-CD in water:methanol 40:60 and pH ~ 8.2; 30 °C, 30 kV | CZE-UV-ID (λ = 270 nm) | 1995 | [40] |
| Hydrogenated fish oil | Saturated C14 at C26 | 2.5 mmol·L−1 antraquinone-2-carboxylic acid and 40 mmol·L−1 Tris in NMF-dioxane (3:1, v:v); the temperature stabilized at 6–7 °C above the room temperature, 20 kV | NACE-UV-ID (λ = 264 nm) | 1997 | [52] |
| Mix standard | Saturated C14 at C20 | 10 mmol·L−1 tris, 5 mmol·L−1 p- methoxybenzoate at pH 8.1, 10 mmol·L−1 Brij in MeOH-H2O (50:50, v/v) | MEKC-UV-ID (λ = 254 nm) | 1997 | [59] |
| Mix standard | Saturated C12 at C31 | NMF-dioxane (3:2), 40 mmol·L−1 Tris, 2.5 mmol·L−1 AMP and 0.5% (w/v) Brij; 40 °C, 20 kV | CZE-UV-ID | 1999 | [46] |
| Soy and olive oils and fatty acid enriched margarine | C10 at C18:0, C16:1c, C18:1c, C18:2cc and C20 | 5.0 mmol·L−1 phosphate buffer, 4.0 mmol·L−1 SDBS, 4.0 mmol·L−1 DM-β-CD and 45% CAN; 25 °C, 25 kV | CZE-UV-ID (λ = 224 nm) | 2001 | [31] |
| Parenteral feed formulation, fish oil | C16:0, C18:1c, C18:2cc, C18:3ccc, C20:5ccccc and C22:6cccccc | 5.0 mmol·L−1 phosphate buffer, 4.0 mmol·L−1 SDBS, 4.0 mmol·L−1 DM-β-CD and 45% ACN; 25 °C, 25 kV for ID | CZE-UV-ID (λ = 224 nm for ID) | 2003 | [36] |
| Olive, rice and linseed oils and salmon fish | C16:0, C18:0, C18:1c, C18:2 and C18:3 in the vegetable oils and C22:5, C18:0, C18:1, C22:6, C20:5 + C16:0, C16:1 e C14:0 in the salmon fish | 40 mmol·L−1 Tris buffer and 2.5 mmol·L−1 AMP in NMF-dioxane-water (5:4:1 v:v:v for vegetable oil FA and 4:6:1 v:v:v for fish oil FA); 25 °C, 25 kV | CZE-UV-ID (λ = 254 nm) | 2003 | [47] |
| Hydrogenated of Brazilnut | C12 at C18 and C18:1c, C18:1t, C18:2cc, C18:2tt and C18:3ccc | 15 mmol·L−1 phosphate buffer, 4.0 mmol·L−1 sodium dodecylbenzensulfonate (SDBS), 10 mmol·L−1 Brij, 2% 1-octanol, 45% ACN and pH ~ 7.0; 25 °C, 20 kV | CZE-UV-ID (λ = 224 nm) | 2003 | [53] |
| Olive oil | C18: 1c ,C18:2cc and C16:0 | 15 mmol·L−1 phosphate buffer solution at pH 6.86 containing 4 mmol·L−1 SDBS, 10 mmol·L−1 Brij 35, 45% v/v ACN and 2% v/v 1-octanol; 25 °C, 20 kV | CZE-UV-ID (λ = 224 nm) | 2007 | [43] |
| Peanut seeds | C18.1 and C18.3 | 40 mmol·L−1 Tris, 2.5 mmol·L−1 AMP and 7.0 mmol·L−1 α-CD in NMF-dioxane-water (5:3:2), pH 8–9; 25 °C, 28 kV | CZE-UV-ID (λ = 254 nm) | 2008 | [27] |
| Hydrogenated vegetable fat and spreadable cheese | C18:1c and C18:1t | 15.0 mmol·L-1 KH2PO4/Na2HPO4 buffer (pH∼7.0), 4.0 mmol·L-1 SDBS, 8.0 mmol·L-1 Brij35, 45%v/v ACN, 8% methanol, and 1.5% v/v n-octanol; 25 °C, 28 kV | CZE-UV-ID (λ = 224 nm) | 2010 | [42] |
| Oils from avocado, corn, extra virgin olive, hazelnut, and soybean | C 18:0;, C 18:1; C 16:0; , C18:2; C 16:1; C 18:3 and C 14:0 | 10 mmol·L−1 p-hydroxybenzoate, 5 mmol·L−1 Tris, 80 mmol·L−1 Brij 98, 40% acetonitrile, 10% 2-propanol and pH 8.8; 45 °C, 25 kV | CZE-UV-ID (λ = 254 nm) | 2011 | [51] |
| Cosmetic formulations containing Brazilian nut oil | C18.0, C17.0, C16.0, C18.1c, C18.2cc and C18.3 | 12.5 mmol·L−1 sodium tetraborate buffer, 12.5 mmol·L−1Brij, 7.5 mmol·L−1SDBS and 35% ACN pH 7.0; 25 °C, 20 kV | CZE-UV-ID (λ = 254 nm) | 2011 | [45] |
| Spreadable cheese | C18: 2 cis -9 cis-12, C18: 1 cis- 9, C18: 1 trans-9, C18.0 | 15 mmol·L−1 NaH2PO4/Na2HPO4 at pH 6.86, 4 mmol·L−1 SDBS , 8.3 mmol·L−1 Brij 35, 45% v/v ACN and 2.1% n-octanol; 25 °C, 19 kV | CZE-UV-ID (λ = 224 nm) | 2012 | [28] |
| Brachiaria ruzizienses forages | C18.2 n-6 and C18.3 n-3 | 15 mmol·L−1 NaH2PO4/Na2HPO4, 4.0 mmol·L−1 SDBS , 10 mmol·L−1 Brij 35, 43.5% ACN and 2.2% of 1-octanol; 25 °C, 19 kV | CZE-UV-ID (λ = 224 nm) | 2013 | [30] |
| Soybean oil, olive oil, butter, margarine, cookie filling, hydrogenated vegetable fat and beef liver | C18,0,C18.1t, C18.1c,C16.0, C18.2cc , C13.0 and C18.3ccc | 15 mmol·L−1 NaH2PO4/Na2HPO4 at pH 6.86, 4.0 mmol·L−1 SDBS , 8.3 mmol·L−1 Brij 35, 45% v/v ACN and 2.1% of 1 – octanol; 25 °C, 19 kV | CZE-UV-ID (λ = 224 nm) | 2013 | [44] |
| Soybean oil, virgin and extra virgin olive oil | C18: 2cc, C18:1c, C16:0 | 15 mmol·L−1 NaH2PO4/Na2HPO4, 4.0 mmol·L−1 SDBS , 10 mmol·L−1 Brij 35, 43.5% ACN and 2.2% of 1-octanol; 25 °C, 19 kV | CZE-UV-ID (λ = 224 nm) | 2014 | [54] |
| Soybean oil and rat liver | C18:2cc, C18:1c,C18:0, C16.0, C18:3ccc | 15 mmol·L−1 NaH2PO4/Na2HPO4, 4.0 mmol·L−1 SDBS , 10 mmol·L−1 Brij 35, 43.5% ACN and 2.2% of 1-octanol; 25 °C, 19 kV | CZE-UV-ID (λ = 224 nm) | 2014 | [55] |
2.5. CE with Capacitively Coupled Contactless Conductivity Detection
2.6. CE with Mass Spectrometric Detection
| Samples | FA | BGE and Experimental Condition | CE Mode and Detection | Year | Ref. |
|---|---|---|---|---|---|
| Mix standard | C3:0, C5:0, C6:0, C10:0, C12:0, C18:1 and C16:0 | methanol with 12.5 mmol·L−1; tetraethylammonium chloride; ambient temperature, 25 kV | CE-LIF (λ = 780 nm) | 1999 | [56] |
| Mycobacterium tuberculosis fatty acids | C19:0, C18:0, C18:1c, C16:0 with C14:0 (internal standard) | 25 mmol·L−1 aqueous sodium borate and acetonitrile 30% (v/v) at pH 9; 25 °C, 30 kV | CE-LIF (λ = 488 nm) | 2002 | [57] |
| Mix standard | Saturated C8-C20 | 25 mmol·L−1 phosphate buffer, pH 2.2, 1% w/v SDS, 7% v/v n-BuOH, 10% v/v 2-propanol; 25 °C, −25 kV for MECK and 100 mM ammonium acetate, pH 2.2, 10% v/v glacial acetic acid in ACN; 25 °C, +20 kV for NACE | MECK-LED and NACE-LED | 2007 | [58] |
| Coconut vegetable oil | C8:0, C10:0, C12:0, C14:0, C18:2, C18:1, C16:0 and C18:0 | 5.0 mmol·L−1 phosphate buffer at pH 7, 4.0 mmol·L−1 dimethyl-β-cyclodextrin, 2.0 mmol·L−1 trimethyl-β-cyclodextrin, acetonitrile 50% , and methanol 20%; 20 kV | CE-C4D | 2003 | [62] |
| Recent and aged linseed oil, aged walnut oil and oil paintings from the 19th century | C18:0, C18:1, C16:0, C18:2 and C18:3 | 15 mmol·L−1 phosphate buffer at pH 6.86, 4 mmol·L−1 SDBS, 10 mmol·L−1 Brij 35, 1-octanol 2% and acetonitrile45%; 25 °C, 20 kV | CE-C4D | 2004 | [63] |
| Homemade olive oil, commercial olive oil and sunflower oil | C16:0, C18:2, C18:1 and C18:0 | buffer, 10 mmol·L−1 deoxycholate and 100% methanol; −30 kV | NACE-C4D | 2013 | [64] |
| Margarine | C18:0, C18:1t, C18:1c, C16:0, C18:2, C18:3, C14:0 and C12:0. | 6 mmol·L−1 methyl-β-cyclodextrin, 8 mmol·L−1 heptakis-(2,3,6-tri-O-methyl)-β-cyclodextrin, 5 mmol·L−1 Na2HPO4/KH2PO4 at pH 7.4, acetonitrile 40%, methanol 25% and 1-octanol 5%; 20 °C, 30 kV | CE-C4D | 2013 | [3] |
| Mix standard | Saturated C12, C14, C16 and C18 | 2 mmol·L−1 acetic acid ammonium acetate in 50% (v/v) acetonitrile in water at pH 5.2; 32 kV | CZE-MS | 1999 | [66] |
3. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Otieno, A.C.; Mwongela, S.M. Capillary electrophoresis-based methods for the determination of lipids-A review. Anal. Chim. Acta 2008, 624, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Christie, W.W.; X.; Han, X. Lipid Analysis-Isolation, Separation, Identification and Lipidomic Analysis, 4th ed.; Oily Press: Cambridge, UK, 2010. [Google Scholar]
- Wong, Y.F.; Saad, B.; Makahleh, A. Capillary electrophoresis with capacitively coupled contactless conductivity detection for the determination of cis/trans isomers of octadec-9-enoic acid and other long chain fatty acids. J. Chromatogr. A 2013, 1290, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gurr, M.I.; Harwood, J.L. Fatty Acid Structure and Metabolism; Chapman and Hall: London, UK, 1991. [Google Scholar]
- Destaillats, F.; Sébédio, J.L.; Dionisi, F.; Chardigny, J.M. Trans Fatty Acids in Human Nutrition; Oily Press: Cambridge, UK, 2009. [Google Scholar]
- Albuquerque, T.G.; Costa, H.S.; Castilho, M.C.; Sanches-Silva, A. Trends in the analytical methods for the determination of trans fatty acids content in foods. Trends Food Sci. Technol. 2011, 22, 543–560. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology (the “Gold Book”), 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Tholstrup, T.; Ehnholm, C.; Jauhiainen, M.; Petersen, M.; Hoy, C.E.; Lund, P.; Sandström, B. Effects of medium-chain fatty acids and oleic acid on blood lipids, lipoproteins, glucose, insulin, and lipid transfer protein activities. Am. J. Clin. Nutr. 2004, 79, 564–569. [Google Scholar] [PubMed]
- Zhou, L.; Nilsson, A. Sources of eicosanoid precursor fatty pools in tissue. J. Lipid Res. 2001, 42, 1521–1542. [Google Scholar] [PubMed]
- Connor, W.E.; Neuringer, M.; Reisbick, S. Essential fatty acids: The importance of n-3 fatty acids in the retina and brain. Nutr. Rev. 1992, 50, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.D.; Campos, P.R.B.; Noffs, M.D.; de Oliveira, J.G.; Shimizu, M.T.; da Silva, D.M. Application of microbial lipases to concentrate polyunsaturated fatty acids. Química Nova 2003, 26, 75–80. [Google Scholar]
- Jörgensen, J.A.; Zadravec, D.; Jacobsson, A. Norepinephrine and rosiglitazone synergistically induce Elovl3 expression in brown adipocytes. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1159–1168. [Google Scholar]
- Golay, P.A.; Giuffrida, F.; Dionisi, F.; Destaillats, F. Streamlined Methods for the Resolution and Quantification of Fatty Acids Including Trans Fatty Acid Isomers in Food Products by Gas Chromatography. J. AOAC Int. 2009, 92, 1301–1309. [Google Scholar] [PubMed]
- Sebedio, J.L.; Ratnayake, W.M. Analysis of Trans Mono- and Polyunsaturated Fatty Acids. In Trans Fatty Acids; Blackwell Publishing: Oxford, UK, 2008; pp. 102–131. [Google Scholar]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Bligh, E.; Dyer, W. Rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, A.J. Suitability of lipid extraction procedures for gas-liquid chromatography. J. Am. Oil Chem. Soc. 1963, 40, 545–548. [Google Scholar] [CrossRef]
- Hara, A.; Radin, N.S. Lipid Extraction of Tissues with a Low-Toxicity Solvent. Anal. Biochem. 1978, 90, 420–426. [Google Scholar] [CrossRef] [PubMed]
- AOCS. Official Method Cd 14d-99. In Official Methods and Recommended Practices, 5th ed.; Firestone, D., Ed.; American Oil Chemists’ Society: Champaign, IL, USA, 1999. [Google Scholar]
- Mossoba, M.M.; Adam, M.; Lee, T. Rapid determination of total trans fat content-An attenuated total reflection infrared spectroscopy international collaborative study. J. AOAC Int. 2001, 84, 1144–1150. [Google Scholar] [PubMed]
- Mossoba, M.M.; Milosevic, V.; Milosevic, M.; Kramer, J.K. G.; Azizian, H. Determination of total trans fats and oils by infrared spectroscopy for regulatory compliance. Anal. Bioanal. Chem. 2007, 389, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Delmonte, P.; Rader, J.I. Evaluation of gas chromatographic methods for the determination of trans fat. Anal. Bioanal. Chem. 2007, 389, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Delmonte, P.; Yurawecz, M.P.; Mossoba, M.M.; Cruz-Hernandez, C.; Kramer, J.K. G. Improved identification of conjugated linoleic acid isomers using silver-ion HPLC separations. J. AOAC Int. 2004, 87, 563–568. [Google Scholar] [PubMed]
- Huang, Z.; Wang, B.; Crenshaw, A.A. A simple method for the analysis of trans fatty acid with GC-MS and ATTM-Silar-90 capillary column. Food Chem. 2006, 98, 593–598. [Google Scholar] [CrossRef]
- Bannore, Y.C.; Chenault, K.D.; Melouk, H.A.; Rassi, Z. Capillary electrophoresis of some free fatty acids using partially aqueous electrolyte systems and indirect UV detection. Application to the analysis of oleic and linoleic acids in peanut breeding lines. J. Sep. Sci. 2008, 31, 2667–2676. [Google Scholar]
- Barra, P.M.D.; Barra, M.M.; Azevedo, M.S.; Fett, R.; Micke, G.A.; Costa, A.C.O.; de Oliveira, M.A.L. A rapid method for monitoring total trans fatty acids (TTFA) during industrial manufacturing of Brazilian spreadable processed cheese by capillary zone electrophoresis. Food Control 2012, 23, 456–461. [Google Scholar]
- Porto, B.L.S.; de Souza, M.V.N.; de Oliveira, M.A.L. Analysis of Omega 3 Fatty Acid in Natural and Enriched Chicken Eggs by Capillary Zone Electrophoresis. Anal. Sci. 2011, 27, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Castro, R.D.C.; Sobrinho, F.S.; da Gama, M.A.S.; Barra, P.M.D.; Antoniassi, R.; de Oliveira, M.A.L. Analysis of the main conjugated linoleic acid (CLA) precursors (C18:2 n-6 and C18:3 n-3) in Brachiaria ruzizienses by capillary zone electrophoresis. Cent. Eur. J. Chem. 2013, 11, 1286–1296. [Google Scholar] [CrossRef]
- De Oliveira, M.A.L.; Micke, G.A.; Bruns, R.; Tavares, M.F.M. Factorial design of electrolyte systems for the separation of fatty acids by capillary electrophoresis. J. Chromatogr. A 2001, 924, 533–539. [Google Scholar]
- Del Valle, E.M.M. Cyclodextrins and their uses: a review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar]
- Schmitz, O.; Gab, S. Separation of unsaturated fatty acids and related isomeric hydroperoxides by micellar electrokinetic chromatography. J. Chromatogr. A 1997, 781, 215–221. [Google Scholar] [CrossRef]
- Miksik, I.; Deyl, Z. Microemulsion electrokinetic chromatography of fatty acids as phenacyl esters. J. Chromatogr. A 1998, 807, 111–119. [Google Scholar] [CrossRef]
- Bohlin, M.E.; Ohman, M.; Hamberg, M.; Blomberg, L.G. Separation of conjugated trienoic fatty acid isomers by capillary electrophoresis. J. Chromatogr. A 2003, 985, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Tavares, M.; Jager, A.; Silva, C.; Moraes, E.; Pereira, E.; ECLima; Fonseca, F.; Tonin, F.; Micke, G.; Santos, M.; Oliveira, M.; Moraes, M.; MHKampen; Fujiya, N. Applications of capillary electrophoresis to the analysis of compounds of clinical, forensic, cosmetological, environmental, nutritional and pharmaceutical importanceal, nutritional and pharmaceutical importance. J. Braz. Chem. Soc. 2003, 14, 281–290. [Google Scholar] [CrossRef]
- Liu, X.H.; Cao, Y.S.; Chen, Y. Separation of conjugated linoleic acid isomers by cyclodextrin-modified micellar electrokinetic chromatography. J. Chromatogr. A 2005, 1095, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, H.; Chen, Y.; Cao, Y. Method for screening of bacterial strains biosynthesizing specific conjugated linoleic acid isomers. J. Agric. Food Chem. 2012, 60, 9705–9710. [Google Scholar] [CrossRef] [PubMed]
- Soliman, L.C.; Donkor, K.K.; Church, J.S.; Cinel, B.; Prema, D.; Dugan, M.E.R. Separation of dietary omega-3 and omega-6 fatty acids in food by capillary electrophoresis. J. Sep. Sci. 2013, 36, 3440–3448. [Google Scholar] [PubMed]
- Roldanassad, R.; Gareil, P. Capillary zone electrophoretic determination of C-2-C-18 linear saturated free fatty-acids with indirect absorbency detection. J. Chromatogr. A 1995, 708, 339–350. [Google Scholar] [CrossRef]
- Erim, F.B.; Xu, X.; Kraak, J.C. Application of micellar electrokinetic chromatography and indirect UV detection for the analysis of fatty acids. J. Chromatogr. A 1995, 694, 471–479. [Google Scholar] [CrossRef]
- De Castro, P.M.; Barra, M.M.; Ribeiro, M.C.C.; Aued-Pimentel, S.; da Silva, S.A.; de Oliveira, M.A.L. Total Trans Fatty Acid Analysis in Spreadable Cheese by Capillary Zone Electrophoresis. J. Agric. Food Chem. 2010, 58, 1403–1409. [Google Scholar]
- Balesteros, M.R.; Tavares, M.F.M.; Ribeiro, S.J.L.; Polachini, F.C.; Messaddeq, Y.; de Oliveira, M.A.L. Determination of olive oil acidity by CE. Electrophoresis 2007, 28, 3731–3736. [Google Scholar] [CrossRef] [PubMed]
- Barra, P.M.D.; Castro, R.D.C.; de Oliveira, P.L.; Aued-Pimentel, S.; da Silva, S.A.; de Oliveira, M.A.L. An alternative method for rapid quantitative analysis of majority cis-trans fatty acids by CZE. Food Res. Int. 2013, 52, 33–41. [Google Scholar] [CrossRef]
- Lima, C.R.R.C.; López-García, P.; Tavares, V.F.; Almeida, M.M.; Zanolini, C.; Aurora-Prado, M.S.; Santoro, M.I.R.M.; Kedor-Hackamnn, E.R.M. Separation and identification of fatty acids in cosmetic formulations containing Brazil nut oil by capillary electrophoresis. Revista de Ciências Farmacêuticas Básica e Aplicada 2011, 32, 341–348. [Google Scholar]
- Haddadian, F.; Shamsi, S.A.; Warner, I.M. Separation of saturated and unsaturated free fatty acids using capillary electrophoresis with indirect photometric detection. J. Chromatogr. Sci. 1999, 37, 103–107. [Google Scholar] [CrossRef]
- Tadaumi, S.; Endo, S.; Akagawa, H.; Itabashi, Y. Analysis of fatty acids by capillary electrophoresis with indirect UV detection. Bunseki Kagaku 2003, 52, 847–850. [Google Scholar] [CrossRef]
- Gutnikov, G.; Beck, W.; Engelhardt, H. Separation of Homologous Fatty-Acids by Capillary Electrophoresis. J. Microcolumn Sep. 1994, 6, 565–570. [Google Scholar] [CrossRef]
- Chen, M.J.; Chen, H.S.; Lin, C.Y.; Chang, H.T. Indirect detection of organic acids in non-aqueous capillary electrophoresis. J. Chromatogr. A 1999, 853, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.; Gareil, P. Micellar electrokinetic chromatography of long chain saturated and unsaturated free fatty acids with neutral micelles-Considerations regarding selectivity and resolution optimization. J. Chromatogr. A 1997, 792, 165–177. [Google Scholar] [CrossRef]
- Vergara-Barberan, M.; Escrig-Domenech, A.; Lerma-Garcia, M.J.; Simo-Alfonso, E.F.; Herrero-Martinez, J.M. Capillary Electrophoresis of Free Fatty Acids by Indirect Ultraviolet Detection: Application to the Classification of Vegetable Oils According to Their Botanical Origin. J. Agric. Food Chem. 2011, 59, 10775–10780. [Google Scholar]
- Drange, E.; Lundanes, E. Determination of long-chained fatty acids using non-aqueous capillary electrophoresis and indirect UV detection. J. Chromatogr. A 1997, 771, 301–309. [Google Scholar] [CrossRef]
- De Oliveira, M.A.L.; Solis, V.E.S.; Gioielli, L.A.; Polakiewicz, B.; Tavares, M.F.M. Method development for the analysis of trans-fatty acids in hydrogenated oils by capillary electrophoresis. Electrophoresis 2003, 24, 1641–1647. [Google Scholar]
- Sato, R.T.; de Jesus, R.C.C.; de Barra, C.P.M.B.; de Oliveira, M.A.L. Rapid Separation of Free Fatty Acids in Vegetable Oils by Capillary Zone Electrophoresis. Phytochem. Anal. 2014, 25, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Barra, P.M.C.; Oliveira, P.L.; Aragão, D.M.O.; Sabarense, C.M.; Aarestrup, B.J.V.; Azevedo, M.S.; Costa, A.C.O.; Micke, G.A.; Oliveira, M.A.L. Study of fatty acids profile in biological sample by capillary zone electrophoresis associate to chemometric approach. J. Braz. Chem. Soc. 2014, 25, 675–685. [Google Scholar]
- Gallaher, D.L.; Johnson, M.E. Development of near-infrared fluorophoric labels for the determination of fatty acids separated by capillary electrophoresis with diode laser induced fluorescence detection. Analyst 1999, 124, 1541–1546. [Google Scholar] [CrossRef]
- Brando, T.; Pardin, C.; Prandi, J.; Puzo, G. Analysis of aminofluorescein-fatty acid derivatives by capillary electrophoresis with laser-induced fluorescence detection at the attomole level: Application to mycobacterial fatty acids. J. Chromatogr. A 2002, 973, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Breadmore, M.C.; Henderson, R.D.; Fakhari, A.R.; Macka, M.; Haddad, P.R. Separation of nile blue-labelled fatty acids by CE with absorbance detection using a red light-emitting diode. Electrophoresis 2007, 28, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Otegui, M.; Lima, C.; Maldonado, S.; de Lederkremer, R.M. Histological and chemical characterization of Myrsine laetevirens seed. Int. J. Plant Sci. 1998, 159, 762–772. [Google Scholar] [CrossRef]
- Kubáñ, P.; Hauser, P.C. A review of the recent achievements in capacitively coupled contactless conductivity detection. Anal. Chim. Acta 2008, 607, 15–29. [Google Scholar]
- Da Silva, F.J.A. Contactless Conductivity Detection (Oscillometric) for Capillary Zone Electrophoresis and Micellar Electrokinetic Chromatography; University of São Paulo: São Paulo, Brazil, 2001. [Google Scholar]
- Oliveira, M.A.L.; do Lago, C.L.; Tavares, M.F.M.; da Silva, J.A.F. Analysis of fatty acids by capillary electrophoresis with contactless conductivity detection. Quim. Nova 2003, 26, 821–824. [Google Scholar] [CrossRef]
- Surowiec, I.; Kaml, I.; Kenndler, E. Analysis of drying oils used as binding media for objects of art by capillary electrophoresis with indirect UV and conductivity detection. J. Chromatogr. A 2004, 1024, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Buglione, L.; See, H.H.; Hauser, P.C. Rapid separation of fatty acids using a poly(vinyl alcohol) coated capillary in nonaqueous capillary electrophoresis with contactless conctivity detection. Electrophoresis 2013, 34, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.Y.; Henion, J. Capillary electrophoresis mass-spectrometry. J. Chromatogr. A 1995, 703, 667–692. [Google Scholar] [CrossRef]
- Petersson, M.A.; Hulthe, G.; Fogelqvist, E. New sheathless interface for coupling capillary electrophoresis to electrospray mass spectrometry evaluated by the analysis of fatty acids and prostaglandins. J. Chromatogr. A 1999, 854, 141–154. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Oliveira, M.A.L.; Porto, B.L.S.; Faria, I.D.L.; De Oliveira, P.L.; De Castro Barra, P.M.; Castro, R.D.J.C.; Sato, R.T. 20 Years of Fatty Acid Analysis by Capillary Electrophoresis. Molecules 2014, 19, 14094-14113. https://doi.org/10.3390/molecules190914094
De Oliveira MAL, Porto BLS, Faria IDL, De Oliveira PL, De Castro Barra PM, Castro RDJC, Sato RT. 20 Years of Fatty Acid Analysis by Capillary Electrophoresis. Molecules. 2014; 19(9):14094-14113. https://doi.org/10.3390/molecules190914094
Chicago/Turabian StyleDe Oliveira, Marcone Augusto Leal, Brenda Lee Simas Porto, Isaura Daniele Leite Faria, Patricia Lopes De Oliveira, Patricia Mendonça De Castro Barra, Renata De Jesus Coelho Castro, and Renata Takabayashi Sato. 2014. "20 Years of Fatty Acid Analysis by Capillary Electrophoresis" Molecules 19, no. 9: 14094-14113. https://doi.org/10.3390/molecules190914094
APA StyleDe Oliveira, M. A. L., Porto, B. L. S., Faria, I. D. L., De Oliveira, P. L., De Castro Barra, P. M., Castro, R. D. J. C., & Sato, R. T. (2014). 20 Years of Fatty Acid Analysis by Capillary Electrophoresis. Molecules, 19(9), 14094-14113. https://doi.org/10.3390/molecules190914094