Molecular Processes Studied at a Single-Molecule Level Using DNA Origami Nanostructures and Atomic Force Microscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
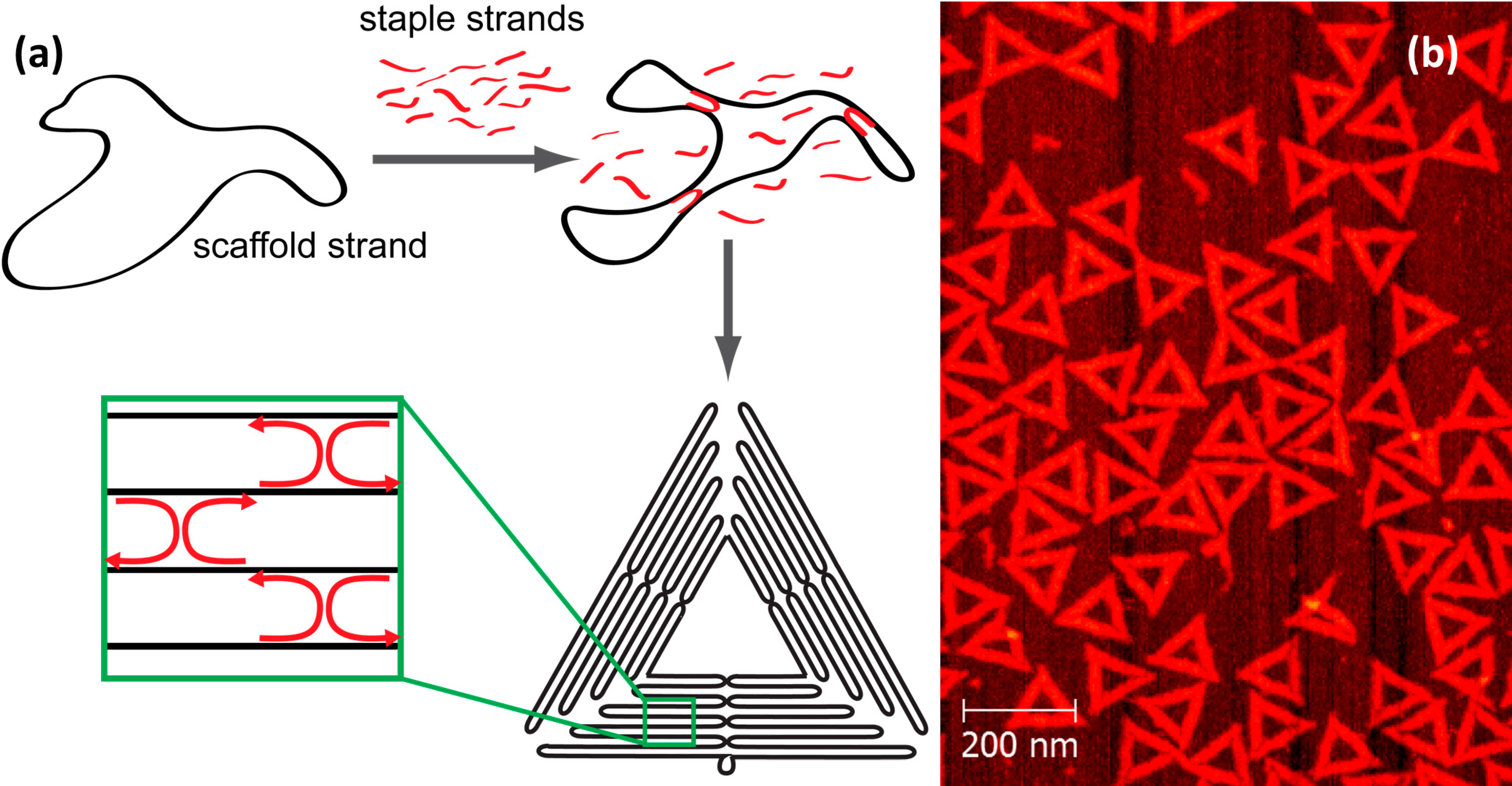
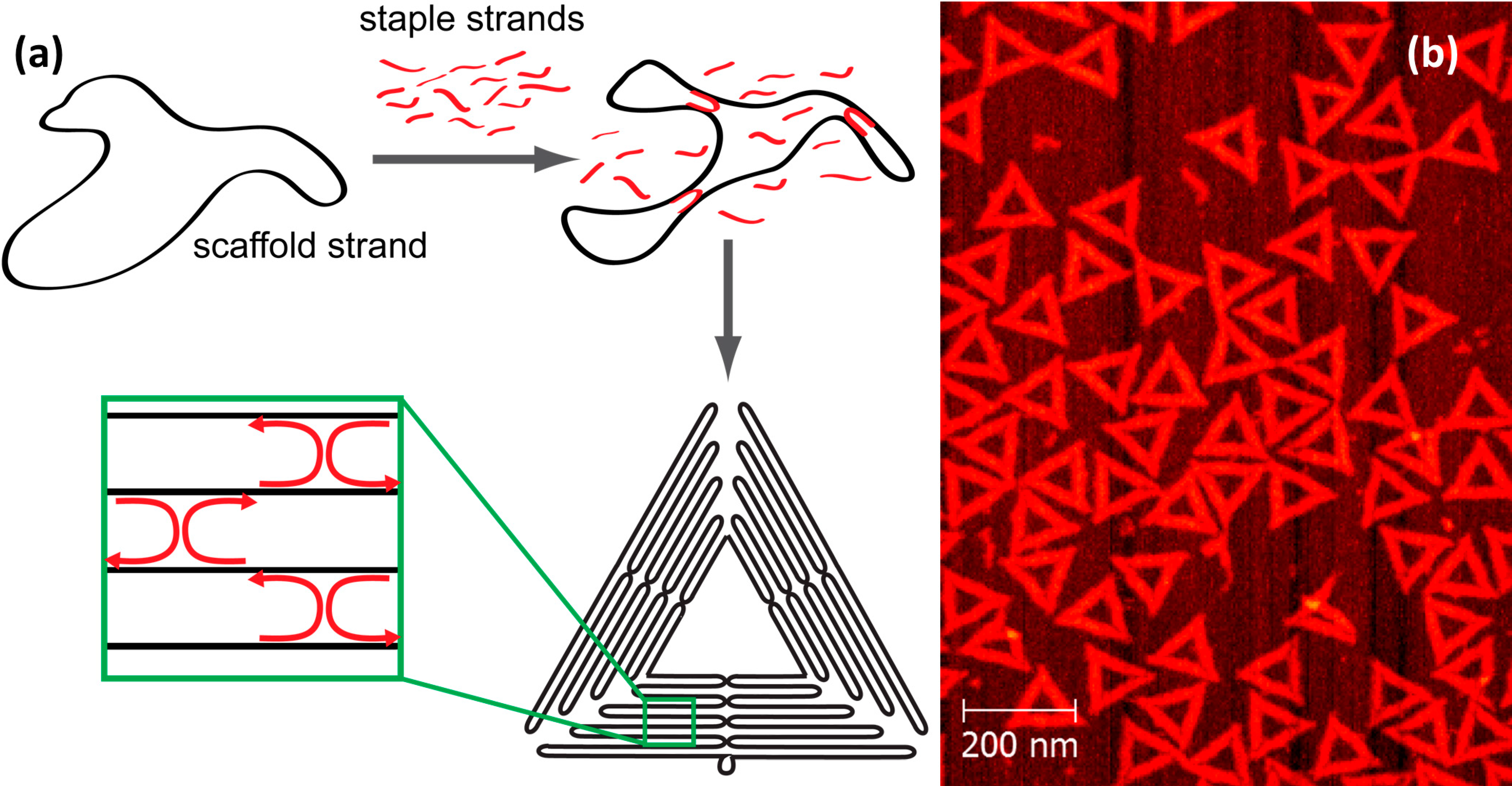
:1. Introduction
2. Chemical Reactions
2.1. Single-Molecule Chemical Reactions
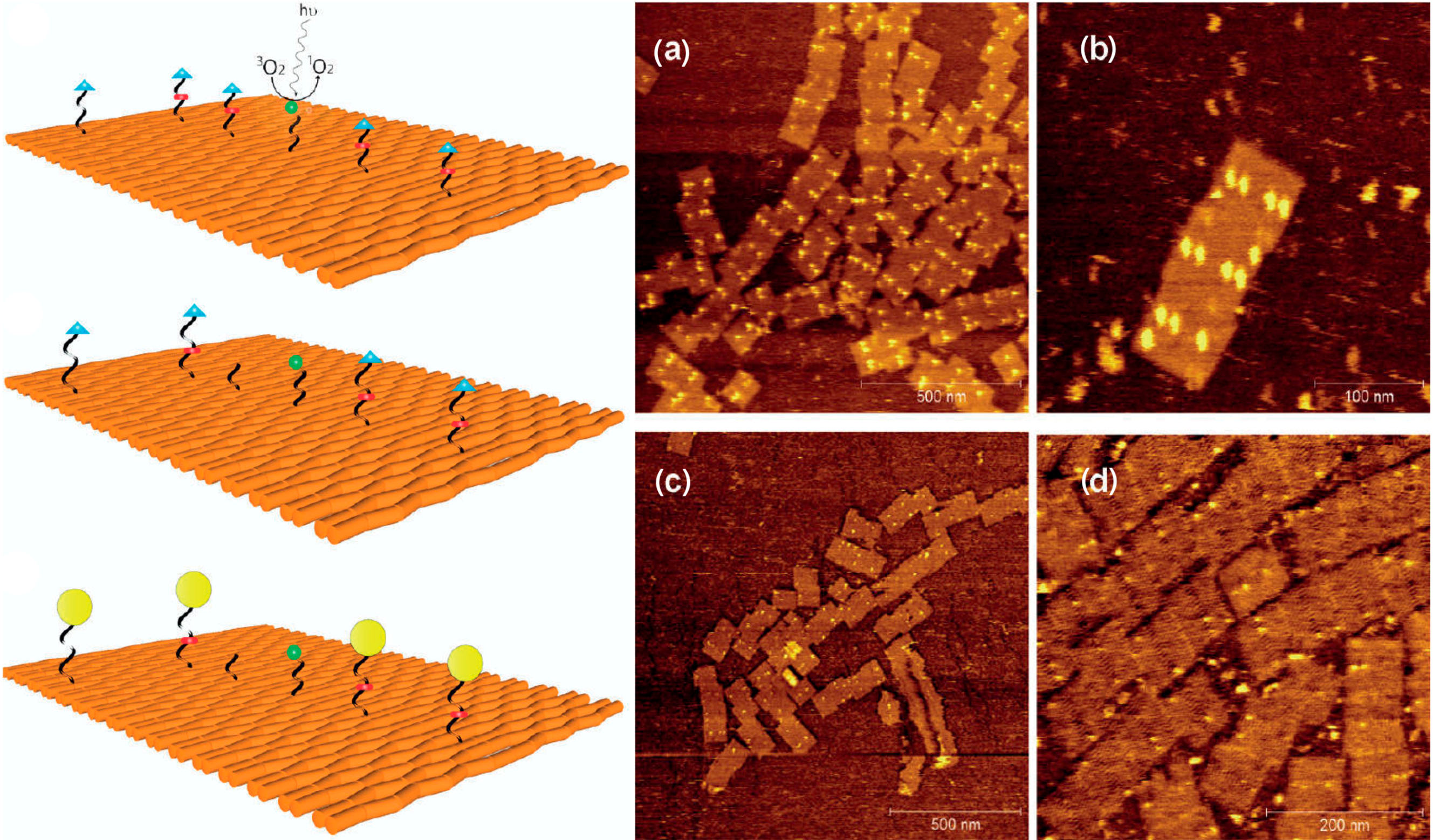
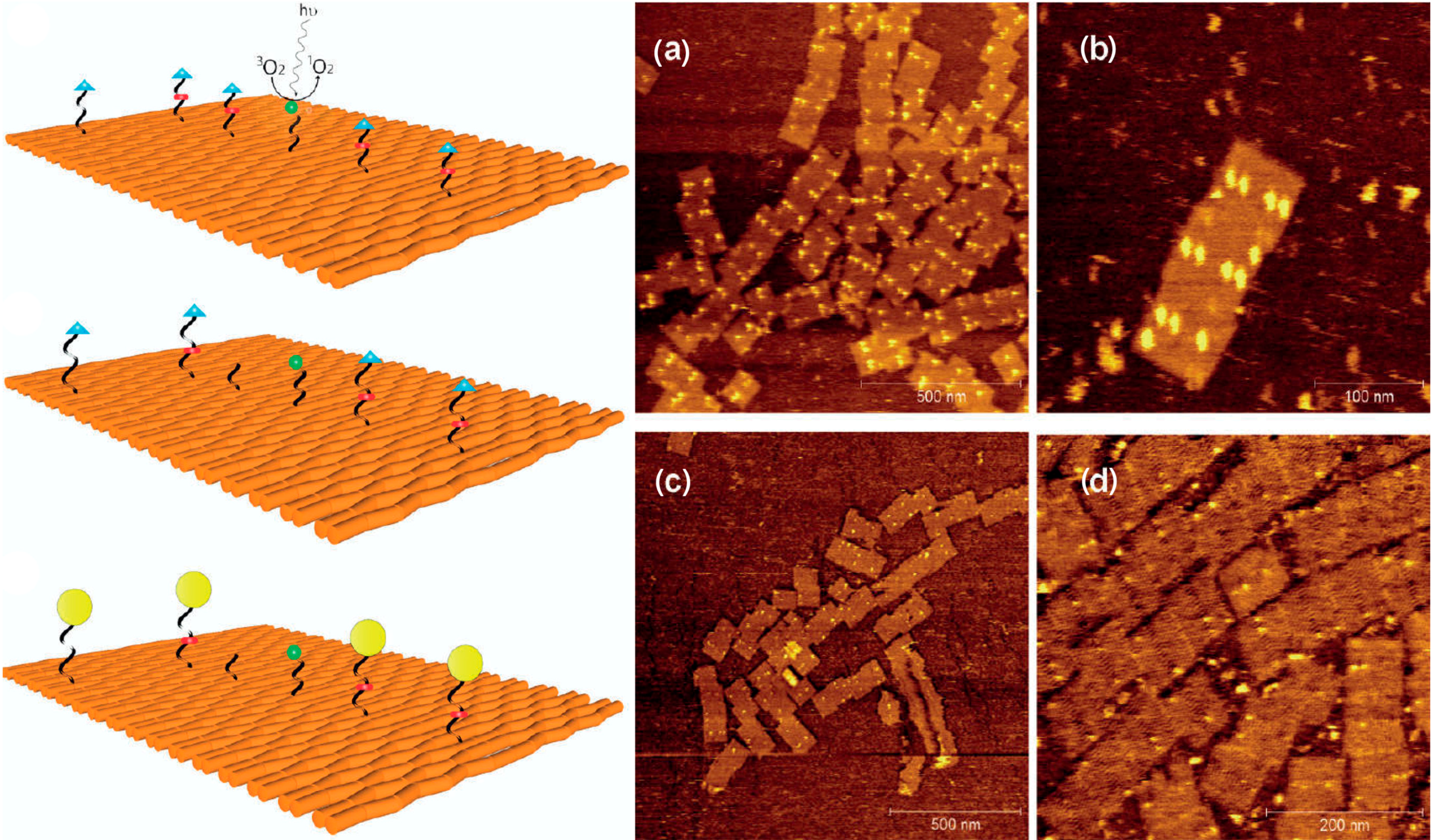
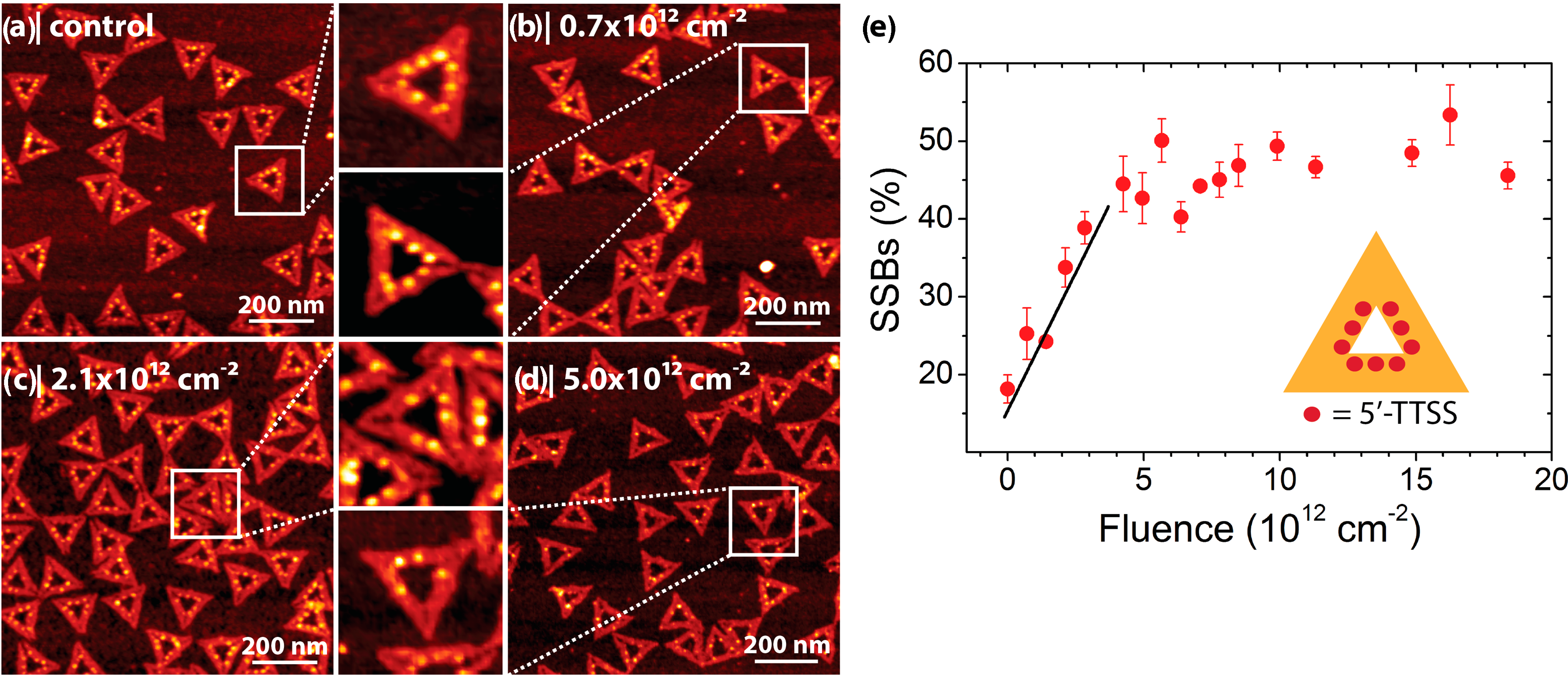
2.2. Electron-Induced Processes
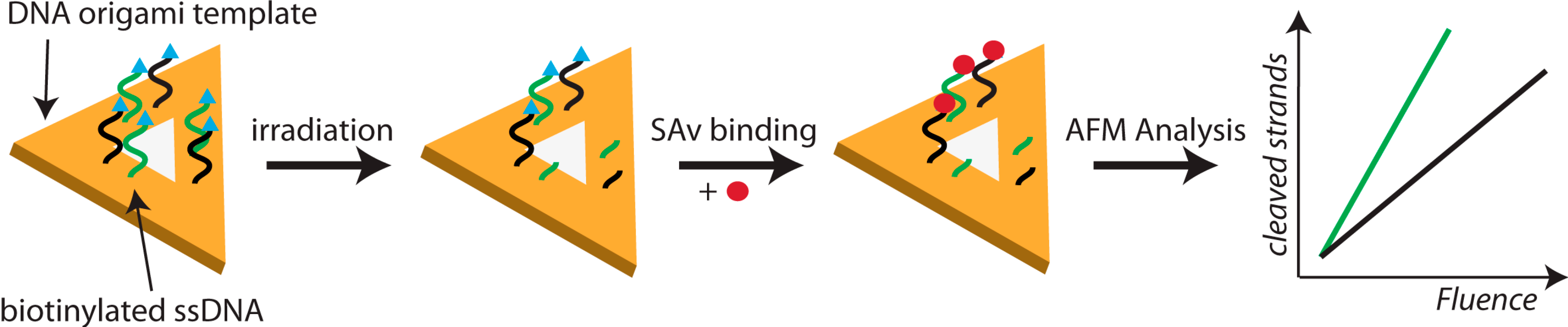
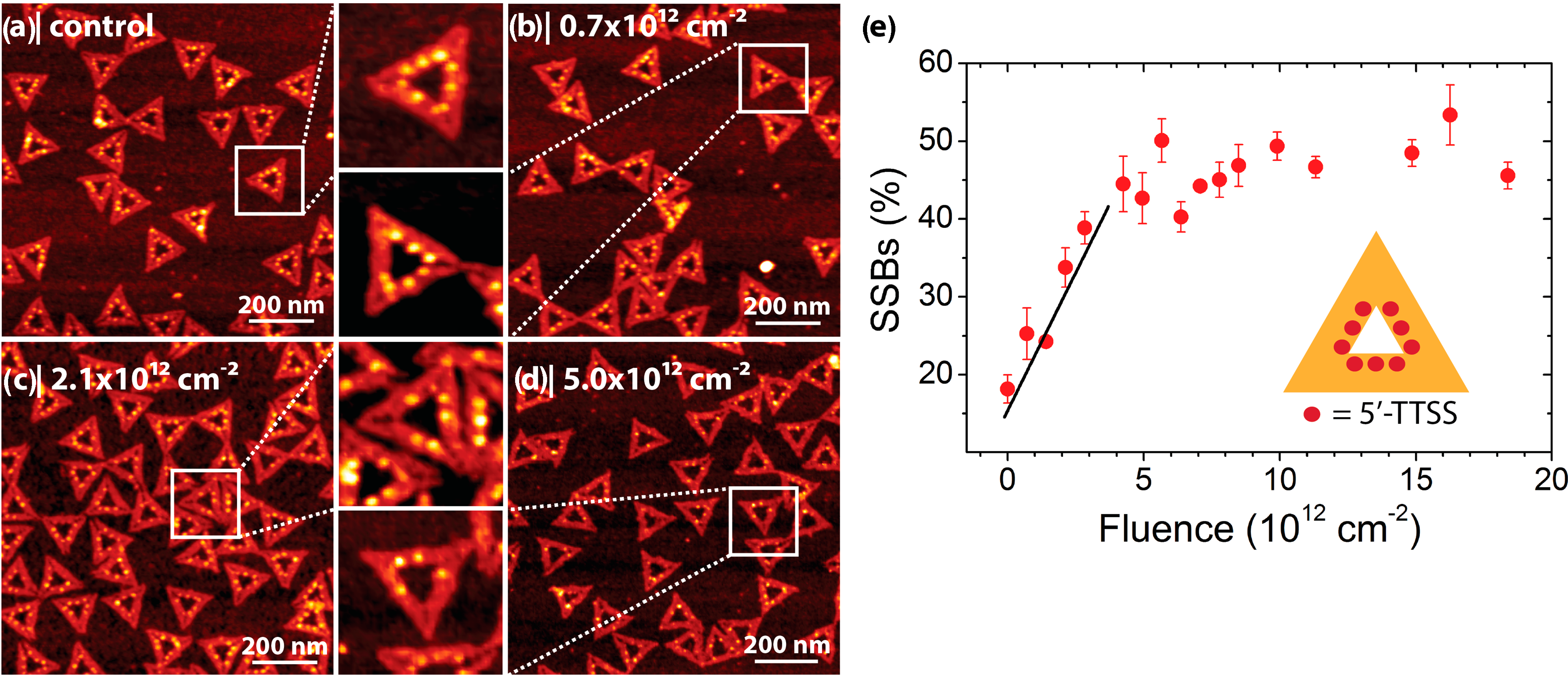
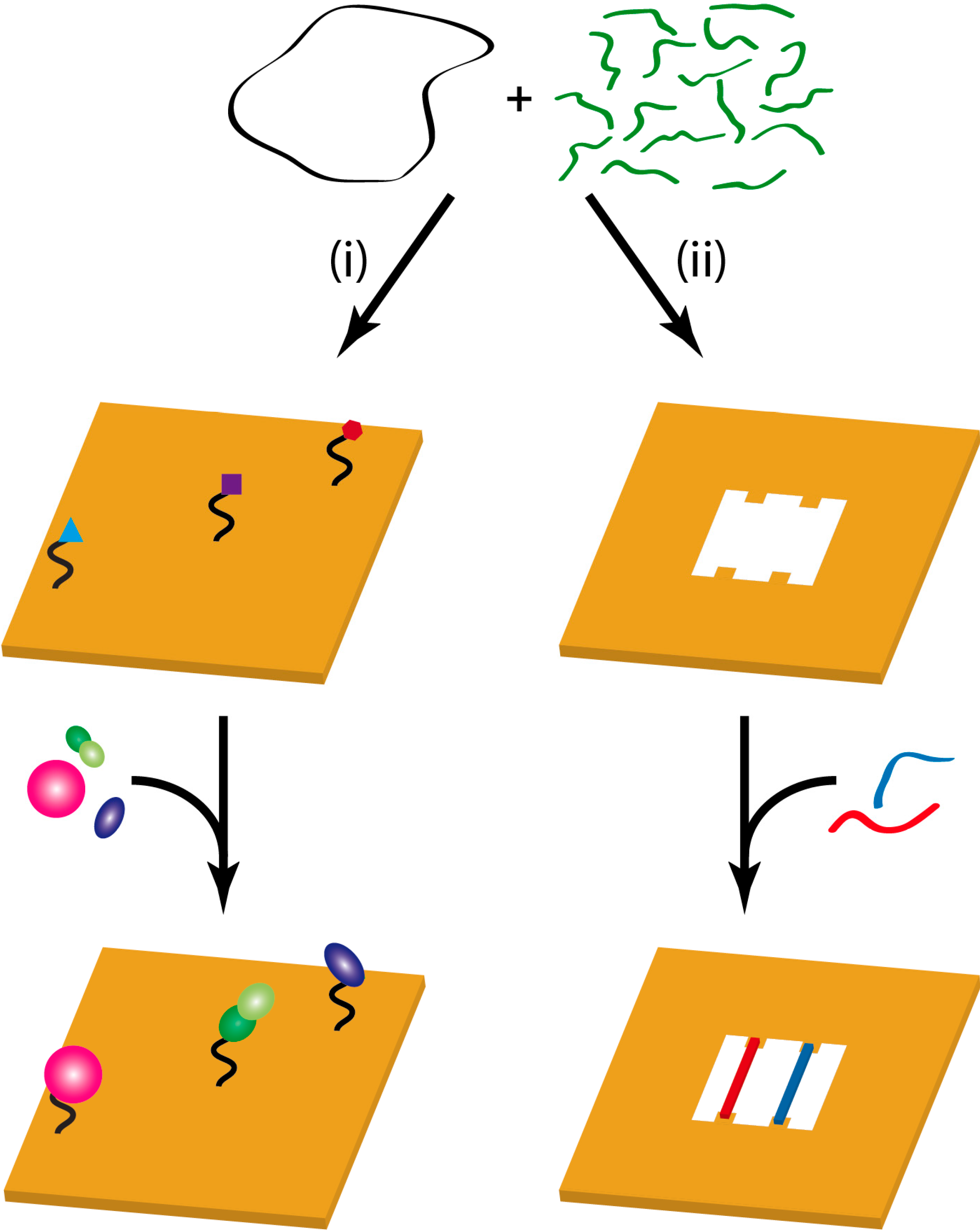
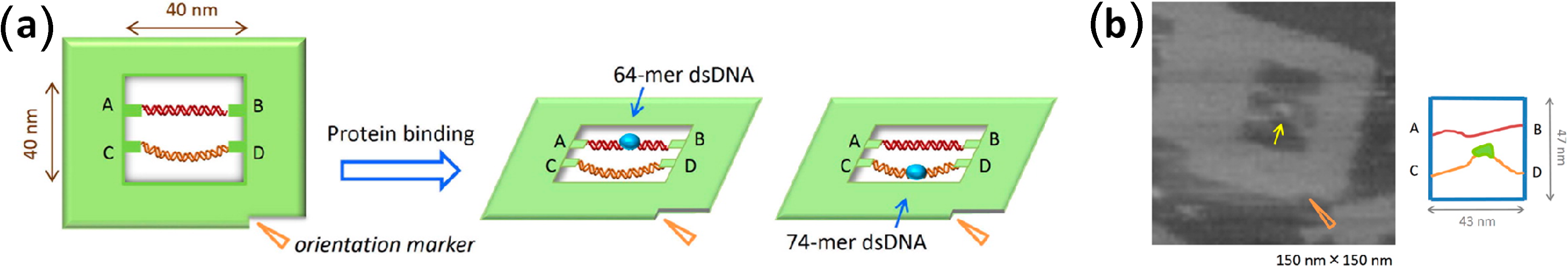
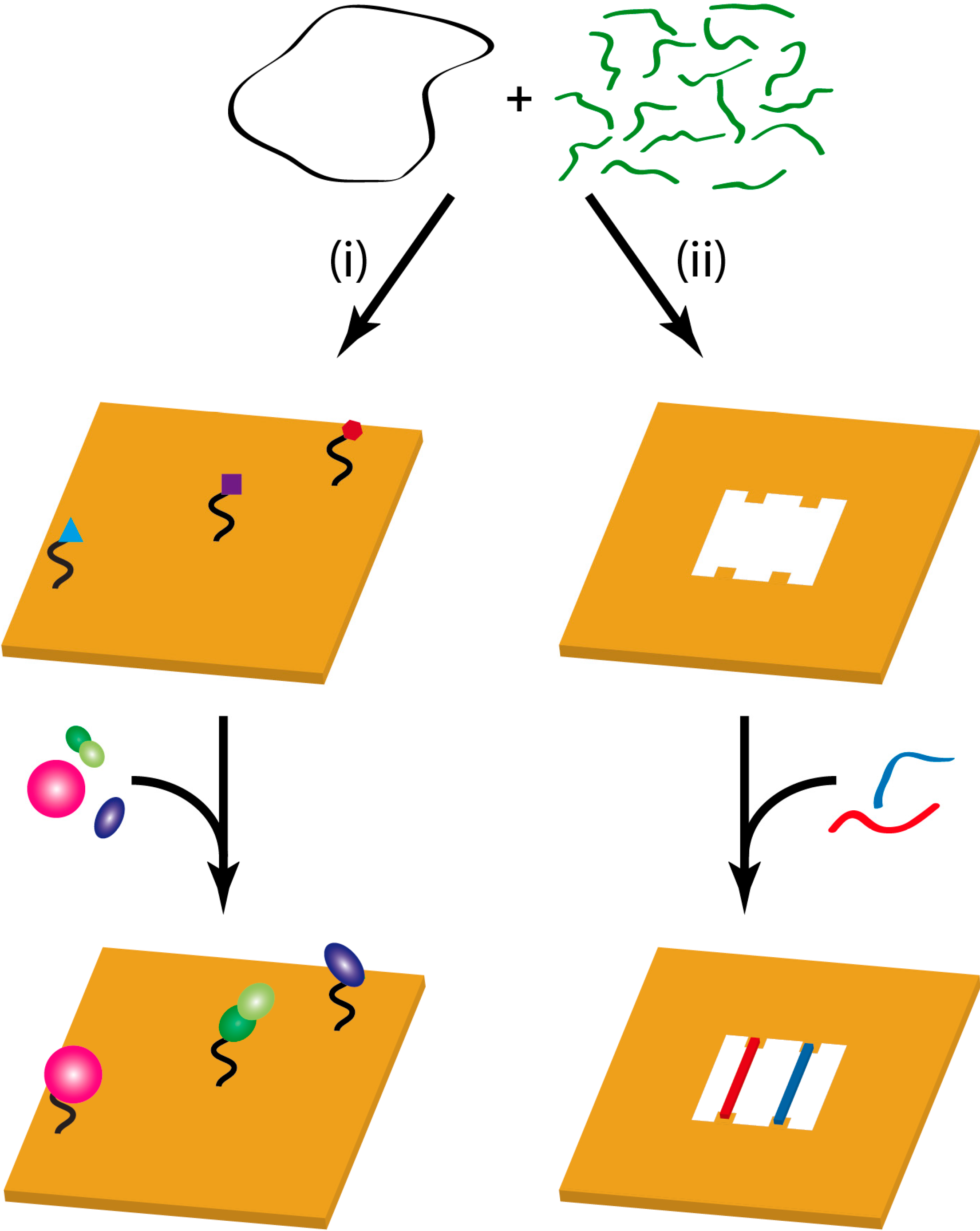
3. Protein Binding Reactions
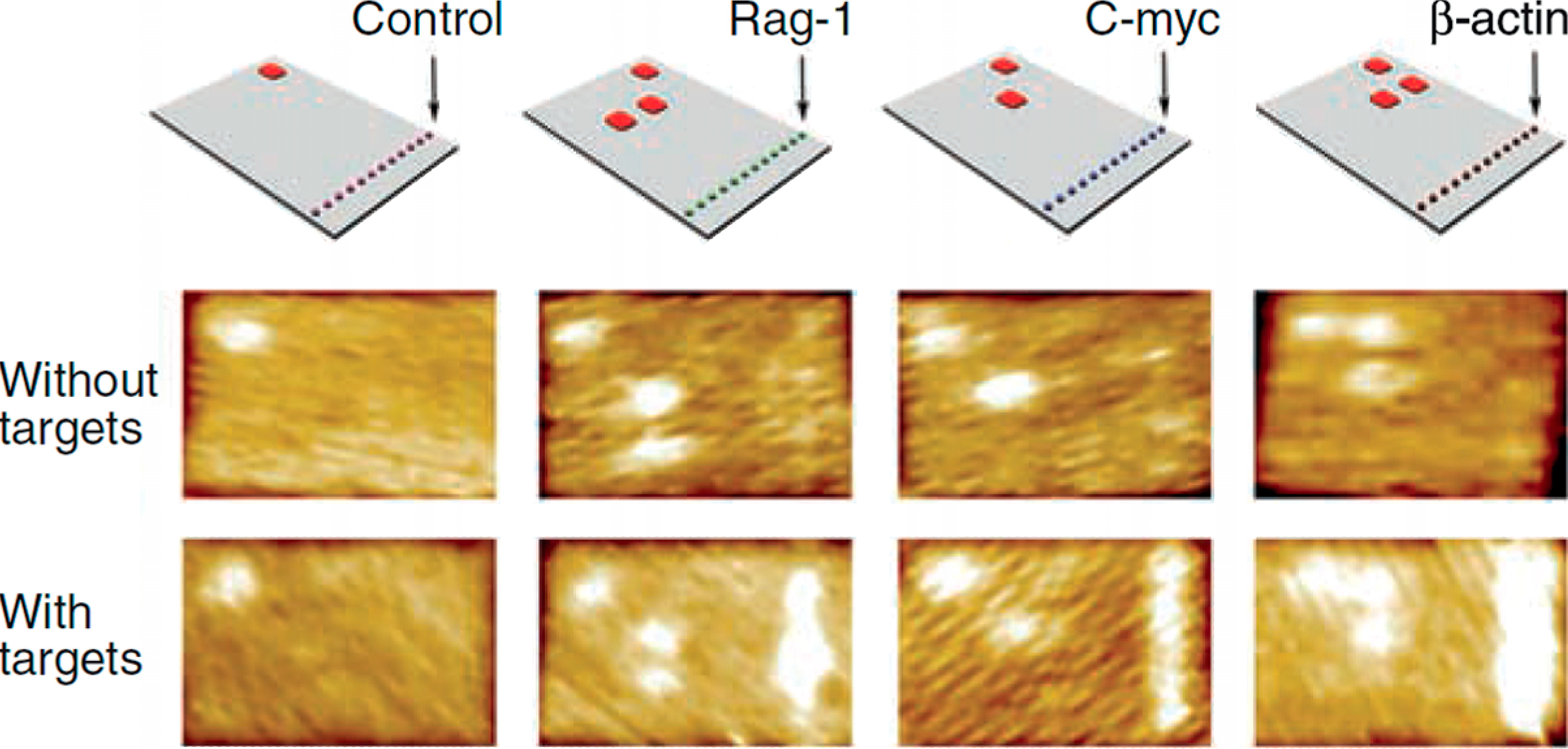
3.1. Aptamer-Protein Binding
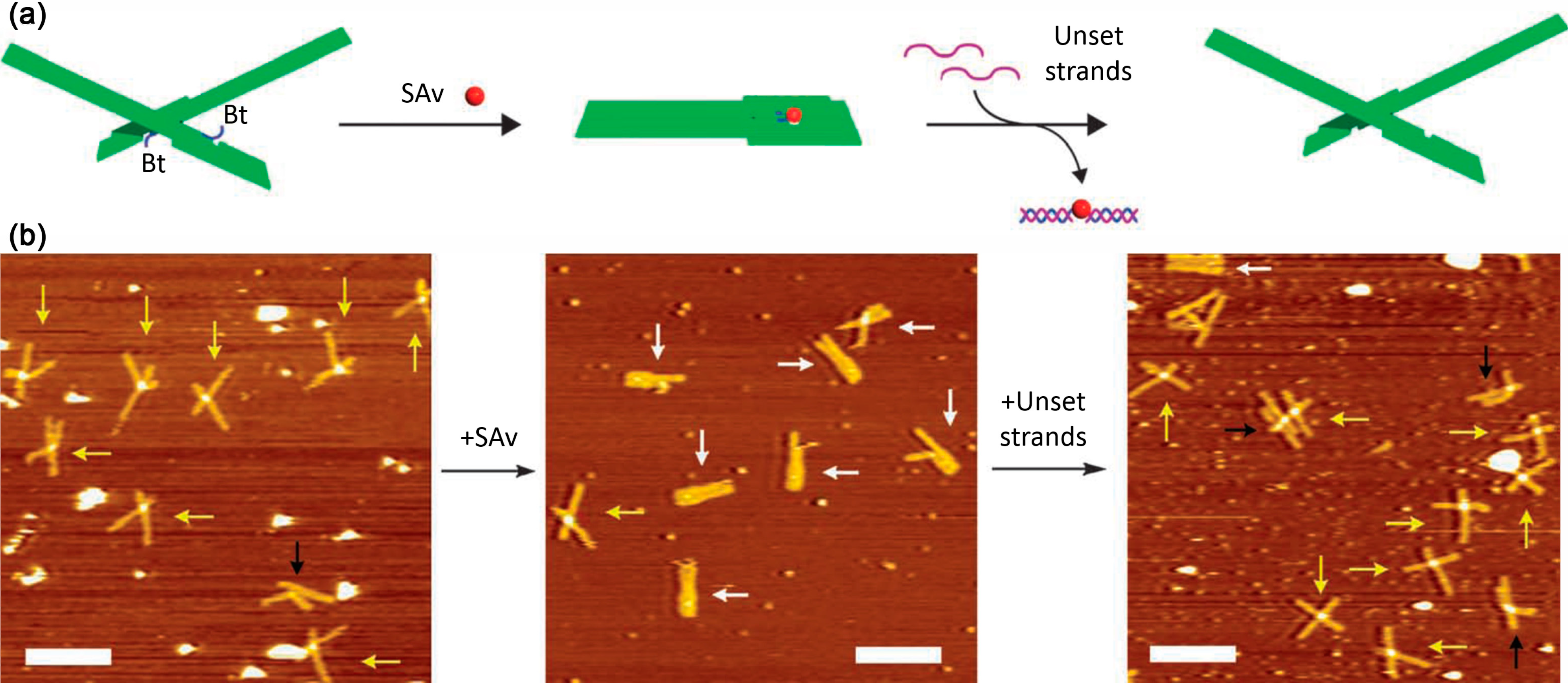
3.2. Streptavidin-Biotin Binding

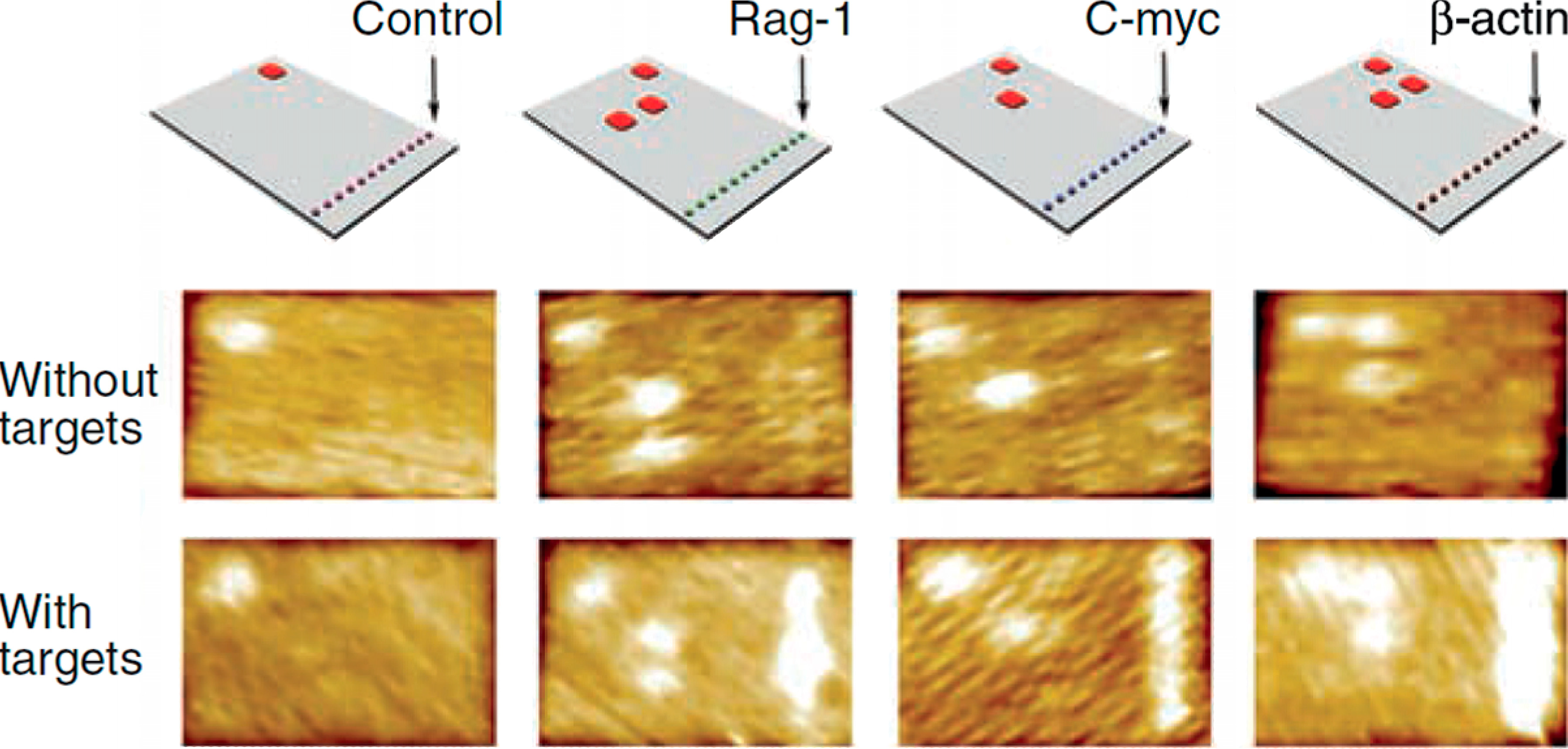
3.3. DNA-Binding Proteins
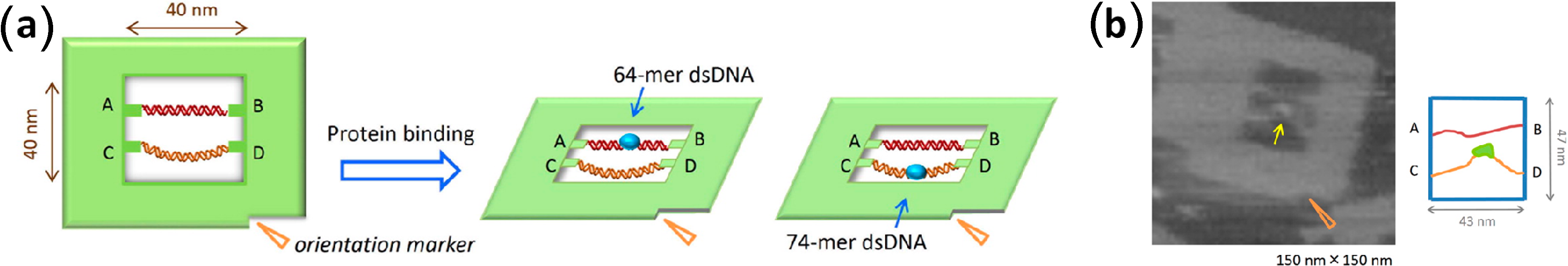
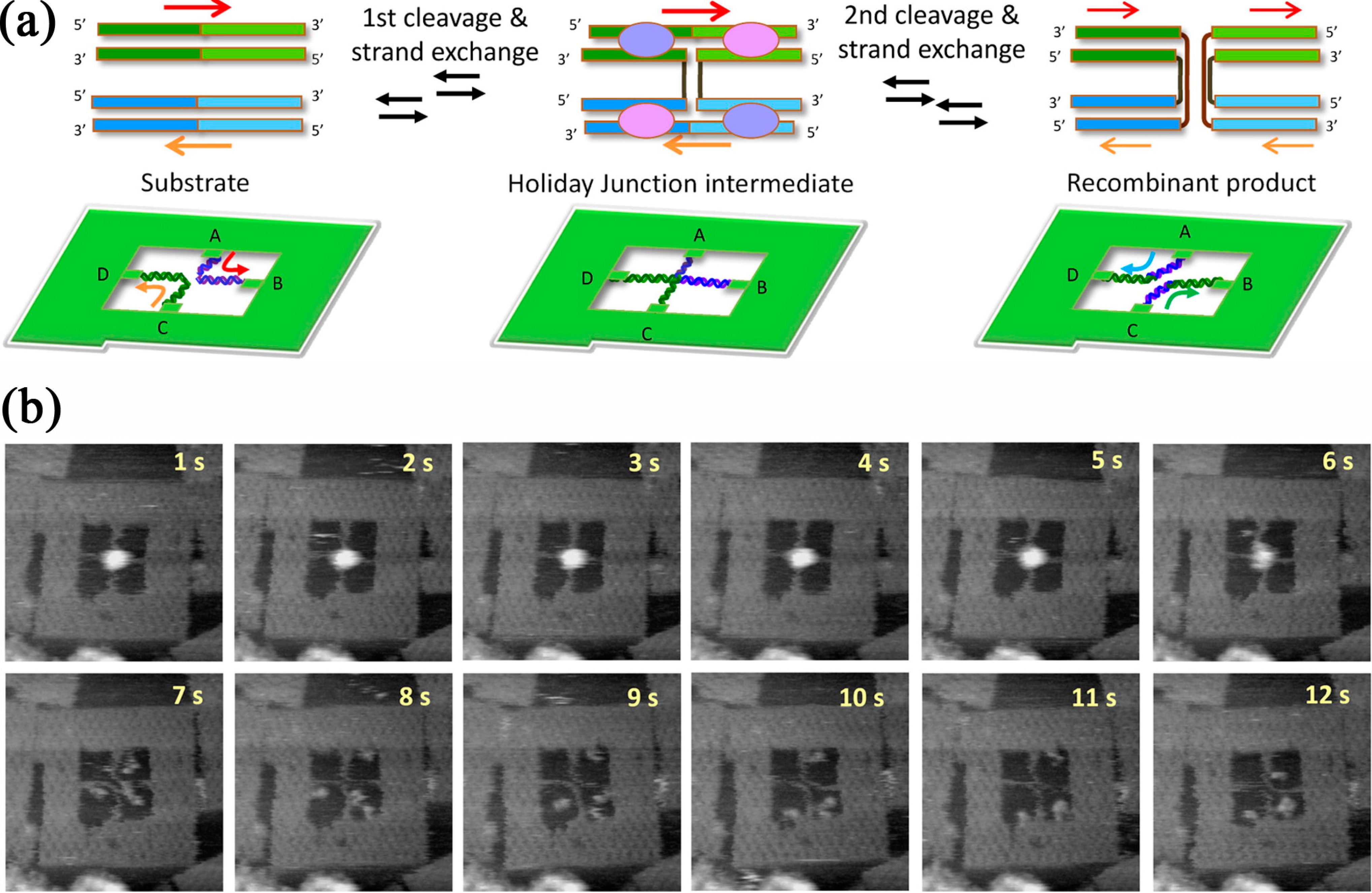
4. Enzymatic Reactions
5. Conformational Transitions of DNA
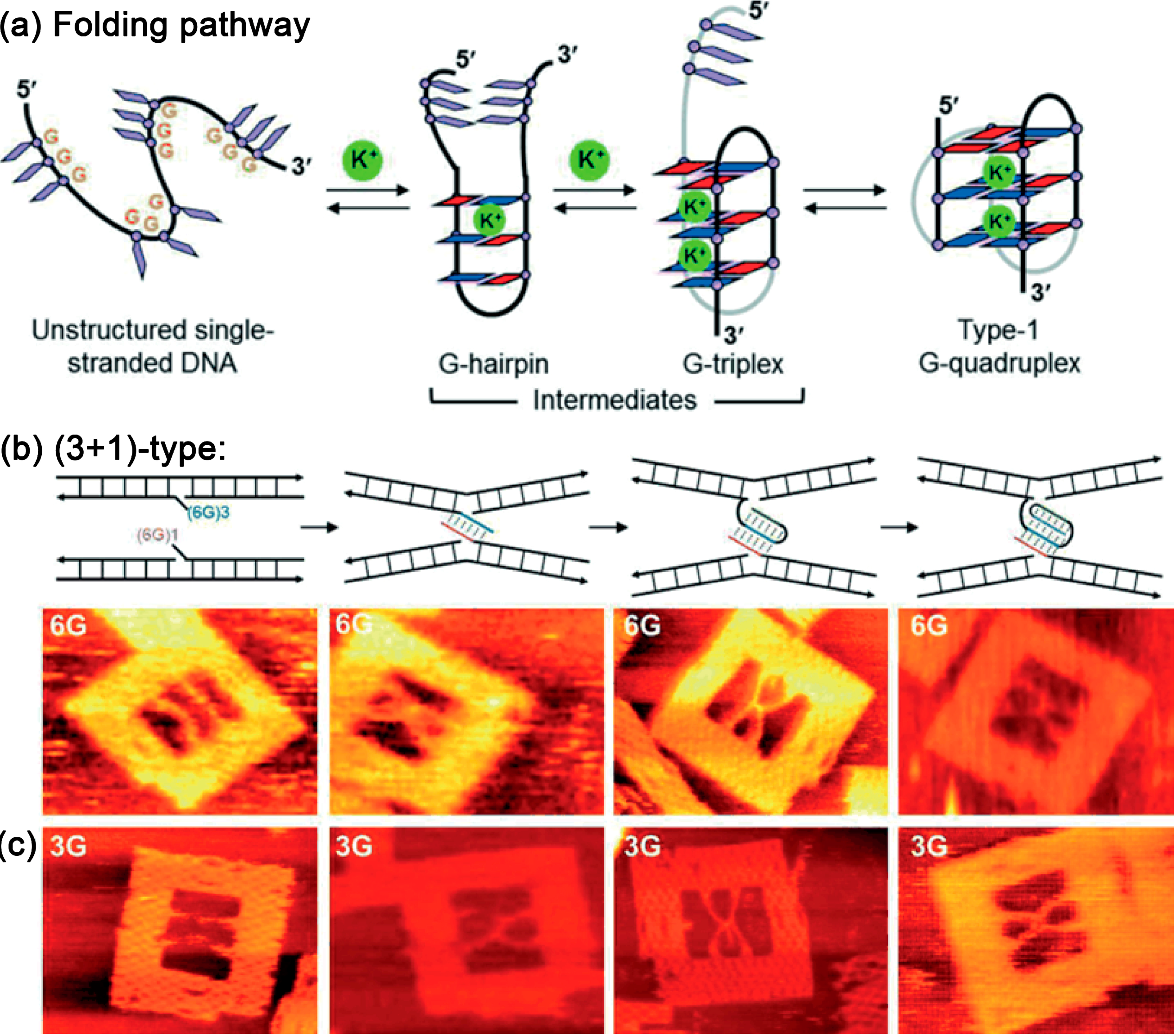
5.1. Guanine Quadruplexes
5.2. Nanomechanical DNA Origami Devices for Molecular Detection
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Seeman, N.C. Nanomaterials based on DNA. Annu. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef]
- Tørring, T.; Voigt, N.V.; Nangreave, J.; Yan, H.; Gothelf, K.V. DNA origami: A quantum leap for self-assembly of complex structures. Chem. Soc. Rev. 2011, 40, 5636–5646. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef]
- Kuzyk, A.; Schreiber, R.; Fan, Z.Y.; Pardatscher, G.; Roller, E.M.; Hogele, A.; Simmel, F.C.; Govorov, A.O.; Liedl, T. DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 2012, 483, 311–314. [Google Scholar] [CrossRef]
- Ding, B.Q.; Deng, Z.T.; Yan, H.; Cabrini, S.; Zuckermann, R.N.; Bokor, J. Gold nanoparticle self-similar chain structure organized by DNA origami. J. Am. Chem. Soc. 2010, 132, 3248–3249. [Google Scholar] [CrossRef]
- Bui, H.; Onodera, C.; Kidwell, C.; Tan, Y.; Graugnard, E.; Kuang, W.; Lee, J.; Knowlton, W.B.; Yurke, B.; Hughes, W.L. Programmable periodicity of quantum dot arrays with DNA origami nanotubes. Nano Lett. 2010, 10, 3367–3372. [Google Scholar] [CrossRef]
- Ko, S.H.; Gallatin, G.M.; Liddle, J.A. Nanomanufacturing with DNA origami: Factors affecting the kinetics and yield of quantum dot binding. Adv. Funct. Mater. 2012, 22, 1015–1023. [Google Scholar] [CrossRef]
- Steinhauer, C.; Jungmann, R.; Sobey, T.; Simmel, F.; Tinnefeld, P. DNA origami as a nanoscopic ruler for super-resolution microscopy. Angew. Chem. Int. Ed. 2009, 48, 8870–8873. [Google Scholar] [CrossRef]
- Stein, I.H.; Steinhauer, C.; Tinnefeld, P. Single-molecule four-color FRET visualizes energy-transfer paths on DNA origami. J. Am. Chem. Soc. 2011, 133, 4193–4195. [Google Scholar] [CrossRef]
- Kuzyk, A.; Laitinen, K.T.; Törmä, P. DNA origami as a nanoscale template for protein assembly. Nanotechnology 2009, 20, 235305. [Google Scholar] [CrossRef]
- Nakata, E.; Liew, F.F.; Uwatoko, C.; Kiyonaka, S.; Mori, Y.; Katsuda, Y.; Endo, M.; Sugiyama, H.; Morii, T. Zinc-finger proteins for site-specific protein positioning on DNA-origami structures. Angew. Chem. Int. Ed. 2012, 51, 2421–2424. [Google Scholar] [CrossRef]
- Birkedal, V.; Dong, M.; Golas, M.M.; Sander, B.; Andersen, E.S.; Gothelf, K.V.; Besenbacher, F.; Kjems, J. Single molecule microscopy methods for the study of DNA origami structures. Microsc. Res. Tech. 2011, 74, 688–698. [Google Scholar]
- De Oteyza, D.G.; Gorman, P.; Chen, Y.-C.; Wickenburg, S.; Riss, A.; Mowbray, D.J.; Etkin, G.; Pedramrazi, Z.; Tsai, H.-Z.; Rubio, A.; et al. Direct imaging of covalent bond structure in single-molecule chemical reactions. Science 2013, 340, 1434–1437. [Google Scholar] [CrossRef]
- Rajendran, A.; Endo, M.; Sugiyama, H. State-of-the-art high-speed atomic force microscopy for investigation of single-molecular dynamics of proteins. Chem. Rev. 2014, 1493–1520. [Google Scholar] [CrossRef]
- Acuna, G.P.; Moller, F.M.; Holzmeister, P.; Beater, S.; Lalkens, B.; Tinnefeld, P. Fluorescence enhancement at docking sites of DNA-directed self-assembled nanoantennas. Science 2012, 338, 506–510. [Google Scholar] [CrossRef]
- Acuna, G.P.; Bucher, M.; Stein, I.H.; Steinhauer, C.; Kuzyk, A.; Holzmeister, P.; Schreiber, R.; Moroz, A.; Stefani, F.D.; Liedl, T.; et al. Distance dependence of single-fluorophore quenching by gold nanoparticles studied on DNA origami. ACS Nano 2012, 6, 3189–3195. [Google Scholar] [CrossRef]
- Prinz, J.; Schreiber, B.; Olejko, L.; Oertel, J.; Rackwitz, J.; Keller, A.; Bald, I. DNA origami substrates for highly sensitive surface-enhanced raman scattering. J. Phys. Chem. Lett. 2013, 4, 4140–4145. [Google Scholar] [CrossRef]
- Thacker, V.V.; Herrmann, L.O.; Sigle, D.O.; Zhang, T.; Liedl, T.; Baumberg, J.J.; Keyser, U.F. DNA origami based assembly of gold nanoparticle dimers for surface-enhanced Raman scattering. Nat. Commum. 2014, 5. [Google Scholar] [CrossRef]
- Pilo-Pais, M.; Watson, A.; Demers, S.; LaBean, T.H.; Finkelstein, G. Surface-enhanced Raman scattering plasmonic enhancement using DNA origami-based complex metallic nanostructures. Nano Lett. 2014, 14, 2099–2104. [Google Scholar] [CrossRef]
- Kühler, P.; Roller, E.-M.; Schreiber, R.; Liedl, T.; Lohmüller, T.; Feldmann, J. Plasmonic DNA-origami nanoantennas for surface-enhanced Raman spectroscopy. Nano Lett. 2014, 14, 2914–2919. [Google Scholar] [CrossRef]
- Koirala, D.; Shrestha, P.; Emura, T.; Hidaka, K.; Mandal, S.; Endo, M.; Sugiyama, H.; Mao, H. Single-molecule mechanochemical sensing using DNA origami nanostructures. Angew. Chem. Int. Ed. 2014, 53, 8137–8141. [Google Scholar] [CrossRef]
- Ke, Y.G.; Lindsay, S.; Chang, Y.; Liu, Y.; Yan, H. Self-assembled water-soluble nucleic acid probe tiles for label-free RNA hybridization assays. Science 2008, 319, 180–183. [Google Scholar] [CrossRef]
- Zhu, J.; Feng, X.; Lou, J.; Li, W.; Li, S.; Zhu, H.; Yang, L.; Zhang, A.; He, L.; Li, C.; et al. Accurate quantification of microRNA via single strand displacement reaction on DNA origami motif. PLoS One 2013, 8, e69856. [Google Scholar]
- Voigt, N.V.; Torring, T.; Rotaru, A.; Jacobsen, M.F.; Ravnsbaek, J.B.; Subramani, R.; Mamdouh, W.; Kjems, J.; Mokhir, A.; Besenbacher, F.; et al. Single-molecule chemical reactions on DNA origami. Nat. Nanotechnol. 2010, 5, 200–203. [Google Scholar] [CrossRef]
- Helmig, S.; Rotaru, A.; Arian, D.; Kovbasyuk, L.; Arnbjerg, J.; Ogilby, P.R.; Kjems, J.; Mokhir, A.; Besenbacher, F.; Gothelf, K.V. Single molecule atomic force microscopy studies of photosensitizedsinglet oxygen behavior on a DNA origami template. ACS Nano 2010, 4, 7475–7480. [Google Scholar] [CrossRef]
- Keller, A.; Bald, I.; Rotaru, A.; Cauet, E.; Gothelf, K.V.; Besenbacher, F. Probing electron-induced bond cleavage at the single-molecule level using DNA origami templates. ACS Nano 2012, 6, 4392–4399. [Google Scholar] [CrossRef]
- Keller, A.; Kopyra, J.; Gothelf, K.V.; Bald, I. Electron-induced damage of biotin studied in the gas phase and in the condensed phase at a single-molecule level. New J. Phys. 2013, 15, 083045. [Google Scholar] [CrossRef]
- Keller, A.; Rackwitz, J.; Cauet, E.; Lievin, J.; Körzdörfer, T.; Rotaru, A.; Gothelf, K.V.; Besenbacher, F.; Bald, I. Sequence dependence of electron-induced DNA strand breakage revealed by DNA nanoarrays. Unpublished work. 2014. [Google Scholar]
- Baccarelli, I.; Bald, I.; Gianturco, F.A.; Illenberger, E.; Kopyra, J. Electron-induced damage of DNA and its components: Experiments and theoretical models. Phys. Rep.-Rev. Sec. Phys. Lett. 2011, 508, 1–44. [Google Scholar]
- Alizadeh, E.; Sanche, L. Precursors of solvated electrons in radiobiological physics and chemistry. Chem. Rev. 2012, 112, 5578–5602. [Google Scholar] [CrossRef]
- Pimblott, S.M.; LaVerne, J.A. Production of low-energy electrons by ionizing radiation. Radiat. Phys. Chem. 2007, 76, 1244–1247. [Google Scholar] [CrossRef]
- Boudaiffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science 2000, 287, 1658–1660. [Google Scholar] [CrossRef]
- Martin, F.; Burrow, P.D.; Cai, Z.L.; Cloutier, P.; Hunting, D.; Sanche, L. DNA strand breaks induced by 0–4 eV electrons: The role of shape resonances. Phys. Rev. Lett. 2004, 93, 068101. [Google Scholar] [CrossRef]
- Li, Z.J.; Cloutier, P.; Sanche, L.; Wagner, J.R. Low-energy electron-induced DNA damage: Effect of base sequence in oligonucleotide trimers. J. Am. Chem. Soc. 2010, 132, 5422–5427. [Google Scholar] [CrossRef]
- Chhabra, R.; Sharma, J.; Ke, Y.; Liu, Y.; Rinker, S.; Lindsay, S.; Yan, H. Spatially addressable multiprotein nanoarrays templated by aptamer-tagged DNA nanoarchitectures. J. Am. Chem. Soc. 2007, 129, 10304–10305. [Google Scholar] [CrossRef]
- Rinker, S.; Ke, Y.; Liu, Y.; Chhabra, R.; Yan, H. Self-assembled DNA nanostructures for distance-dependent multivalent ligand-protein binding. Nat. Nanotechnol. 2008, 3, 418–422. [Google Scholar] [CrossRef]
- Shen, W.; Zhong, H.; Neff, D.; Norton, M.L. NTA directed protein nanopatterning on DNA origami nanoconstructs. J. Am. Chem. Soc. 2009, 131, 6660–6661. [Google Scholar] [CrossRef]
- Kuzuya, A.; Sakai, Y.; Yamazaki, T.; Xu, Y.; Komiyama, M. Nanomechanical DNA origami ‘single-molecule beacons’ directly imaged by atomic force microscopy. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Numajiri, K.; Kimura, M.; Kuzuya, A.; Komiyama, M. Stepwise and reversible nanopatterning of proteins on a DNA origami scaffold. Chem. Commun. 2010, 46, 5127–5129. [Google Scholar] [CrossRef]
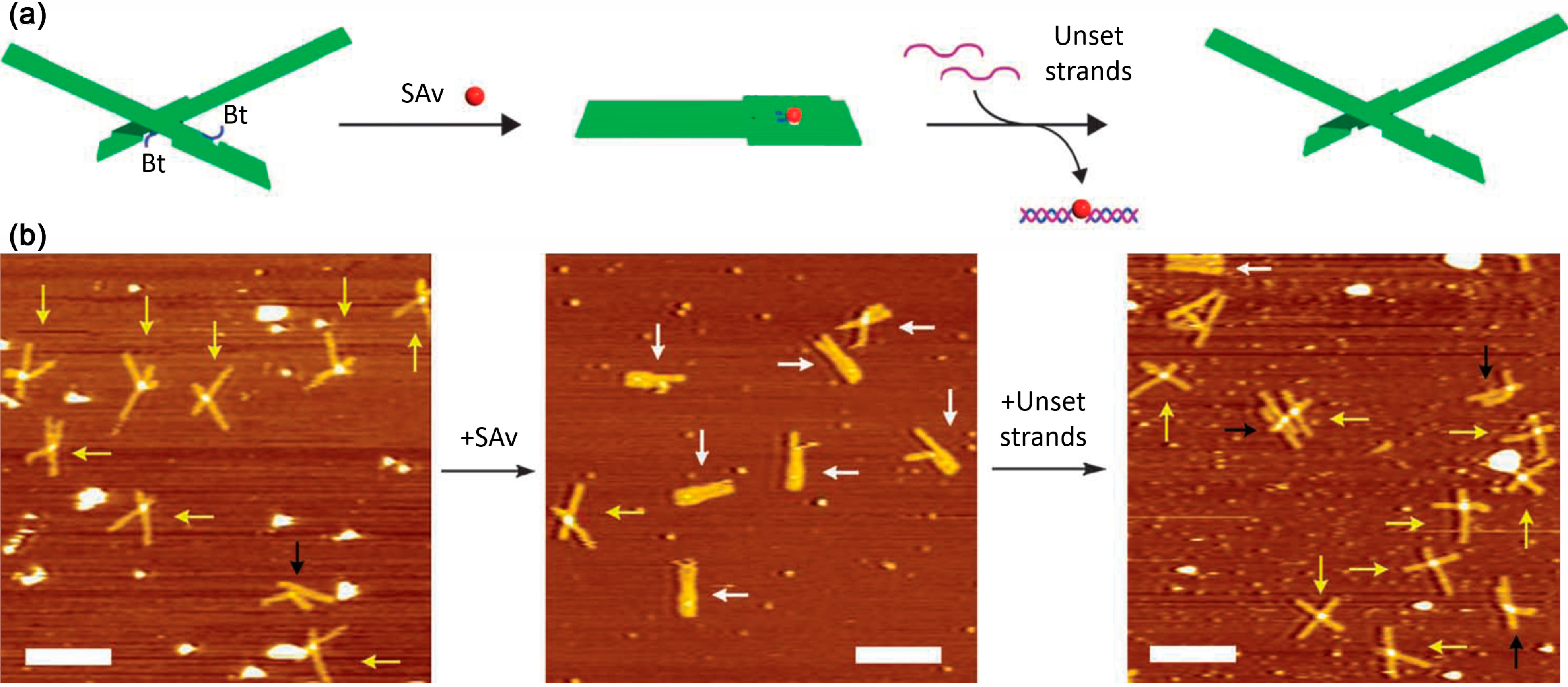
- Wu, N.; Zhou, X.; Czajkowsky, D.M.; Ye, M.; Zeng, D.; Fu, Y.; Fan, C.; Hu, J.; Li, B. In situ monitoring of single molecule binding reactions with time-lapse atomic force microscopy on functionalized DNA origami. Nanoscale 2011, 3, 2481–2484. [Google Scholar] [CrossRef]
- Wong, N.Y.; Xing, H.; Tan, L.H.; Lu, Y. Nano-encrypted morse code: A versatile approach to programmable and reversible nanoscale assembly and disassembly. J. Am. Chem. Soc. 2013, 135, 2931–2934. [Google Scholar] [CrossRef]
- Udomprasert, A.; Bongiovanni, M.N.; Sha, R.; Sherman, W.B.; Wang, T.; Arora, P.S.; Canary, J.W.; Gras, S.L.; Seeman, N.C. Amyloid fibrils nucleated and organized by DNA origami constructions. Nat. Nanotechnol. 2014, 9, 537–541. [Google Scholar] [CrossRef]
- Hermann, T. Adaptive recognition by nucleic acid aptamers. Science 2000, 287, 820–825. [Google Scholar] [CrossRef]
- Tintoré, M.; Gállego, I.; Manning, B.; Eritja, R.; Fàbrega, C. DNA origami as a DNA repair nanosensor at the single-molecule level. Angew. Chem. Int. Ed. 2013, 52, 7747–7750. [Google Scholar] [CrossRef]
- Green, N.M. Avidin. Adv. Protein Chem. 1975, 29, 85–133. [Google Scholar] [CrossRef]
- Wu, N.; Czajkowsky, D.M.; Zhang, J.; Qu, J.; Ye, M.; Zeng, D.; Zhou, X.; Hu, J.; Shao, Z.; Li, B.; et al. Molecular threading and tunable molecular recognition on DNA origami nanostructures. J. Am. Chem. Soc. 2013, 135, 12172–12175. [Google Scholar] [CrossRef]
- Sun, X.J.; Tolbert, L.P.; Hildebrand, J.G. Using laser scanning confocal microscopy as a guide for electron microscopic study: A simple method for correlation of light and electron microscopy. J. Histochem. Cytochem. 1995, 43, 329–335. [Google Scholar] [CrossRef]
- Leduc, C.; Ruhnow, F.; Howard, J.; Diez, S. Detection of fractional steps in cargo movement by the collective operation of kinesin-1 motors. Proc. Nat. Acad. Sci. USA 2007, 104, 10847–10852. [Google Scholar] [CrossRef]
- Yamamoto, S.; De, D.; Hidaka, K.; Kim, K.K.; Endo, M.; Sugiyama, H. Single molecule visualization and characterization of Sox2–Pax6 complex formation on a regulatory DNA element using a DNA origami frame. Nano Lett. 2014, 14, 2286–2292. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef]
- Fu, J.; Liu, M.; Liu, Y.; Woodbury, N.W.; Yan, H. Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 2012, 134, 5516–5519. [Google Scholar] [CrossRef]
- Endo, M.; Tatsumi, K.; Terushima, K.; Katsuda, Y.; Hidaka, K.; Harada, Y.; Sugiyama, H. Direct visualization of the movement of a single T7 RNA polymerase and transcription on a DNA nanostructure. Angew. Chem. Int. Ed. 2012, 51, 8778–8782. [Google Scholar] [CrossRef]
- Endo, M.; Katsuda, Y.; Hidaka, K.; Sugiyama, H. Regulation of DNA methylation using different tensions of double strands constructed in a defined DNA nanostructure. J. Am. Chem. Soc. 2010, 132, 1592–1597. [Google Scholar] [CrossRef]
- Endo, M.; Katsuda, Y.; Hidaka, K.; Sugiyama, H. A versatile DNA nanochip for direct analysis of DNA base-excision repair. Angew. Chem. Int. Ed. 2010, 122, 9602–9606. [Google Scholar] [CrossRef]
- Suzuki, Y.; Endo, M.; Katsuda, Y.; Ou, K.; Hidaka, K.; Sugiyama, H. DNA origami based visualization system for studying site-specific recombination events. J. Am. Chem. Soc. 2014, 136, 211–218. [Google Scholar] [CrossRef]
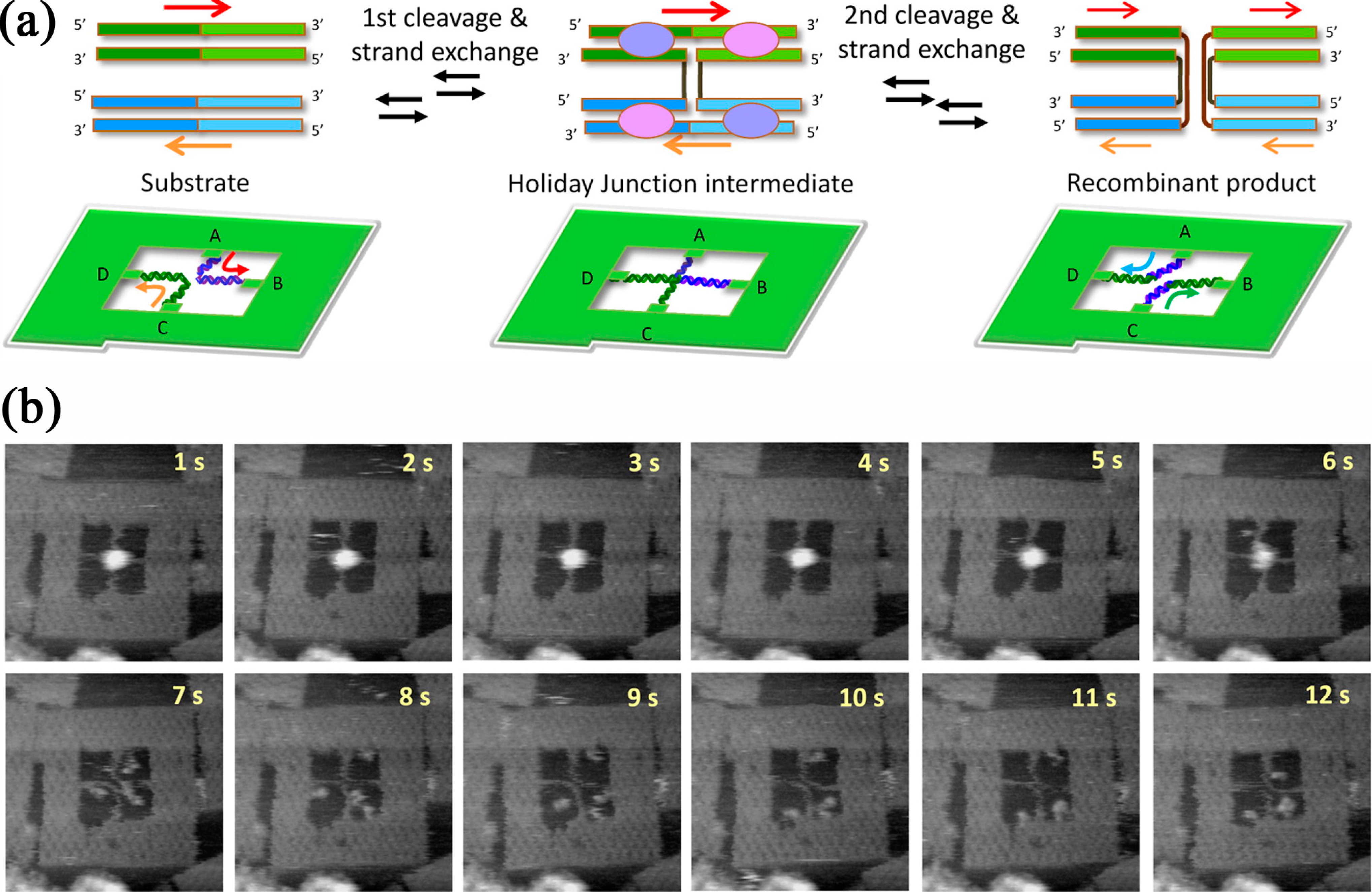
- Suzuki, Y.; Endo, M.; Cañas, C.; Ayora, S.; Alonso, J.C.; Sugiyama, H.; Takeyasu, K. Direct analysis of Holliday junction resolving enzyme in a DNA origami nanostructure. Nucleic Acids Res. 2014, 42, 7421–7428. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G.N. The structure of telomeric DNA. Curr. Opin. Struct. Biol. 2003, 13, 275–283. [Google Scholar] [CrossRef]
- Cech, T.R. Life at the end of the chromosome: Telomeres and telomerase. Angew. Chem. Int. Ed. 2000, 39, 34–43. [Google Scholar] [CrossRef]
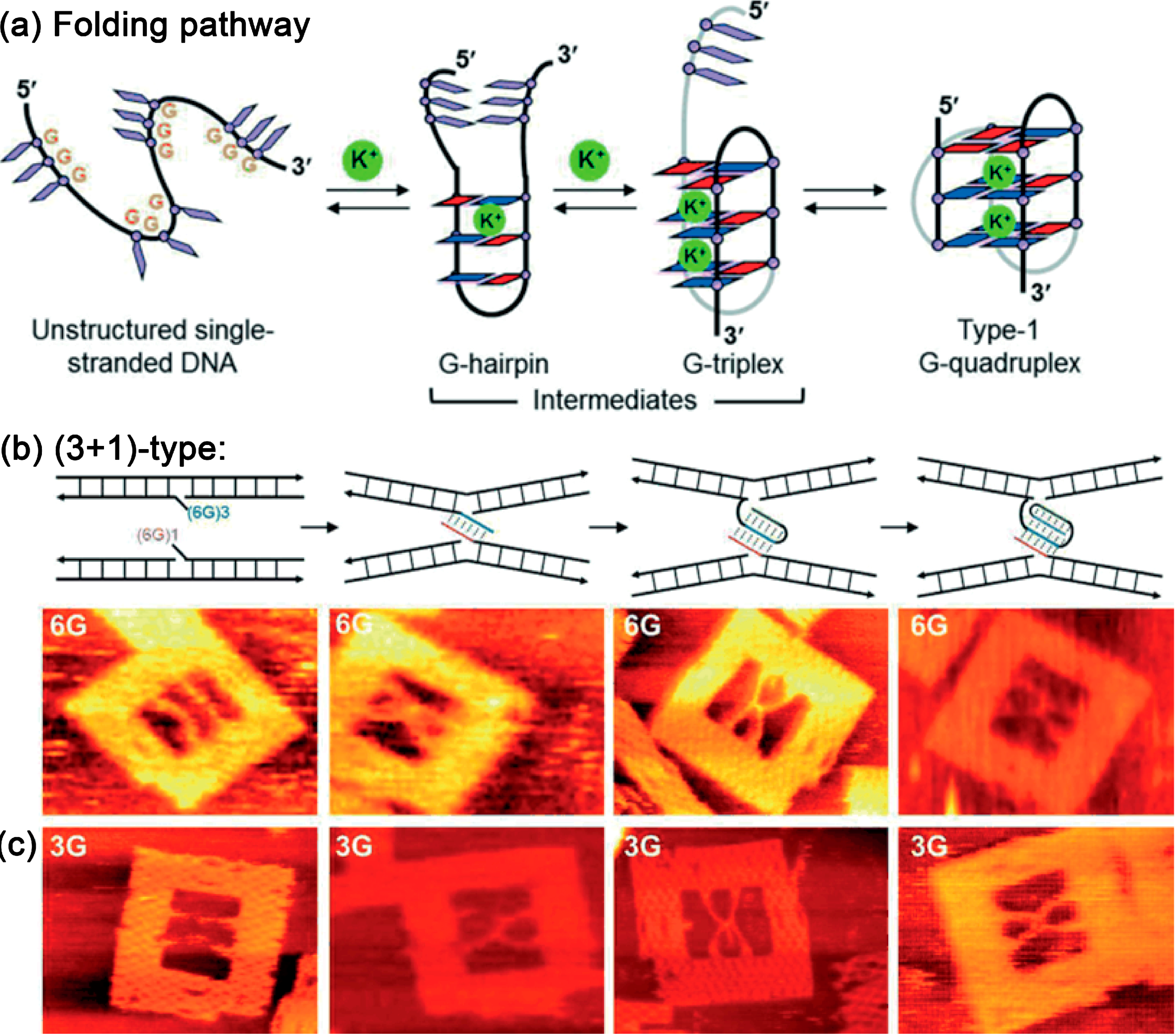
- Sannohe, Y.; Endo, M.; Katsuda, Y.; Hidaka, K.; Sugiyama, H. Visualization of dynamic conformational switching of the G-quadruplex in a DNA nanostructure. J. Am. Chem. Soc. 2010, 132, 16311–16313. [Google Scholar] [CrossRef]
- Rajendran, A.; Endo, M.; Hidaka, K.; Sugiyama, H. Direct and single-molecule visualization of the solution-state Structures of G-hairpin and G-triplex intermediates. Angew. Chem. Int. Ed. 2014, 126, 4191–4196. [Google Scholar] [CrossRef]
- Müller, S.; Sanders, D.A.; di Antonio, M.; Matsis, S.; Riou, J.-F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org. Biomol. Chem. 2012, 10, 6537–6546. [Google Scholar] [CrossRef]
- Rajendran, A.; Endo, M.; Hidaka, K.; Thao Tran, P.L.; Teulade-Fichou, M.-P.; Mergny, J.-L.; Sugiyama, H. G-quadruplex-binding ligand-induced DNA synapsis inside a DNA origami frame. RSC Adv. 2014, 4, 6346–6355. [Google Scholar] [CrossRef]
- Rajendran, A.; Endo, M.; Hidaka, K.; Tran, P.L.T.; Mergny, J.-L.; Gorelick, R.J.; Sugiyama, H. HIV-1 nucleocapsid proteins as molecular chaperones for tetramolecular antiparallel G-quadruplex formation. J. Am. Chem. Soc. 2013, 135, 18575–18585. [Google Scholar] [CrossRef]
- Esté, J.A.; Cabrera, C.; Schols, D.; Cherepanov, P.; Gutierrez, A.; Witvrouw, M.; Pannecouque, C.; Debyser, Z.; Rando, R.F.; Clotet, B.; et al. Human immunodeficiency virus glycoprotein gp120 as the primary target for the antiviral action of AR177 (Zintevir). Mol. Pharmacol. 1998, 53, 340–345. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bald, I.; Keller, A. Molecular Processes Studied at a Single-Molecule Level Using DNA Origami Nanostructures and Atomic Force Microscopy. Molecules 2014, 19, 13803-13823. https://doi.org/10.3390/molecules190913803
Bald I, Keller A. Molecular Processes Studied at a Single-Molecule Level Using DNA Origami Nanostructures and Atomic Force Microscopy. Molecules. 2014; 19(9):13803-13823. https://doi.org/10.3390/molecules190913803
Chicago/Turabian StyleBald, Ilko, and Adrian Keller. 2014. "Molecular Processes Studied at a Single-Molecule Level Using DNA Origami Nanostructures and Atomic Force Microscopy" Molecules 19, no. 9: 13803-13823. https://doi.org/10.3390/molecules190913803
APA StyleBald, I., & Keller, A. (2014). Molecular Processes Studied at a Single-Molecule Level Using DNA Origami Nanostructures and Atomic Force Microscopy. Molecules, 19(9), 13803-13823. https://doi.org/10.3390/molecules190913803