Synthesis of [13C4]-labeled ∆9-Tetrahydrocannabinol and 11-nor-9-Carboxy-∆9-tetrahydrocannabinol as Internal Standards for Reducing Ion Suppressing/Alteration Effects in LC/MS-MS Quantification


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
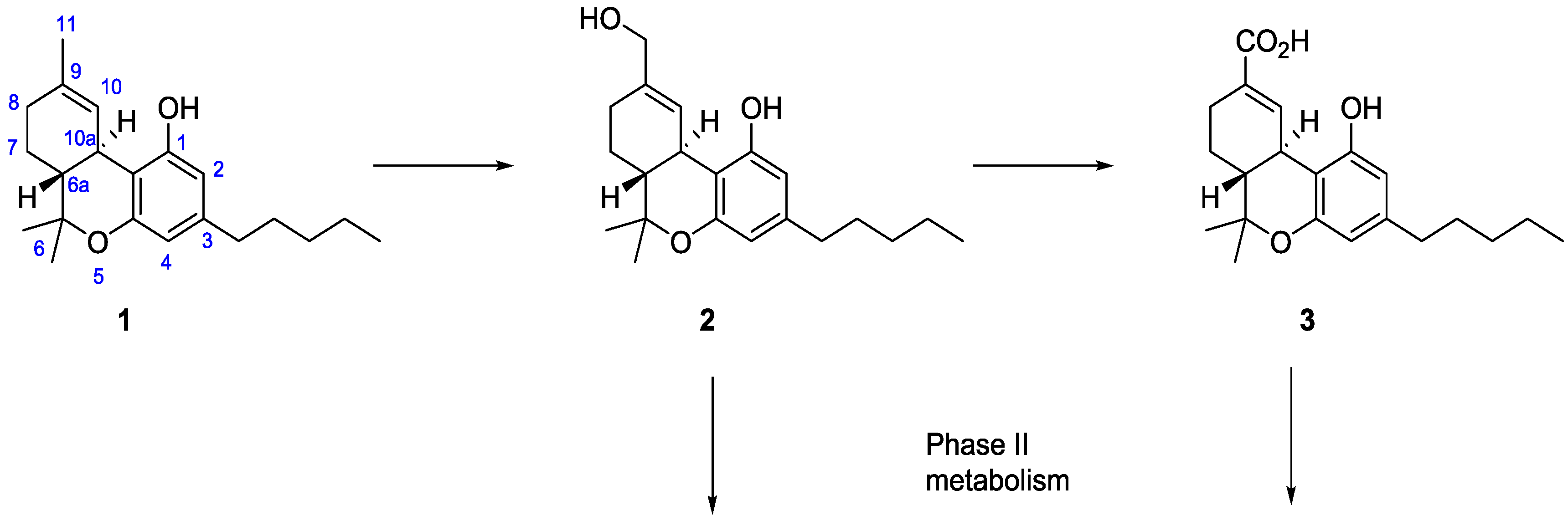{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
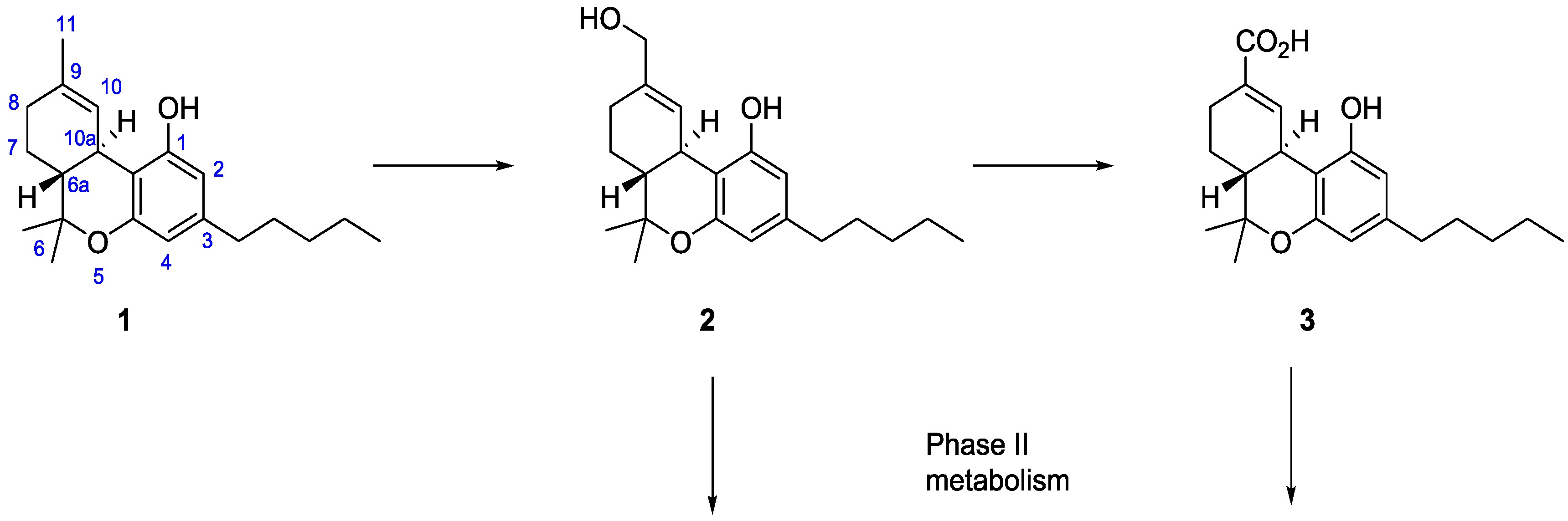
2. Results and Discussion
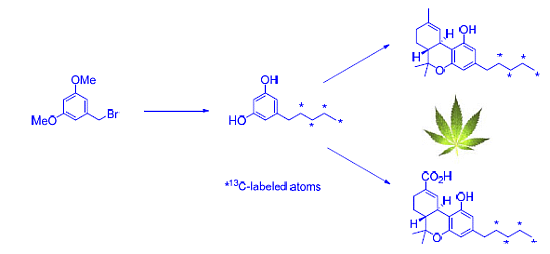
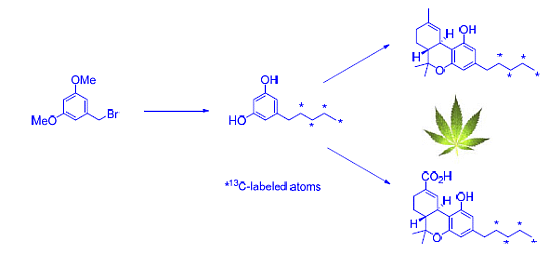
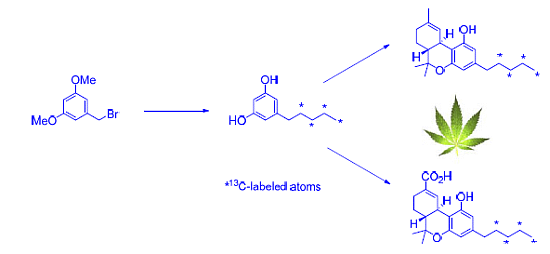
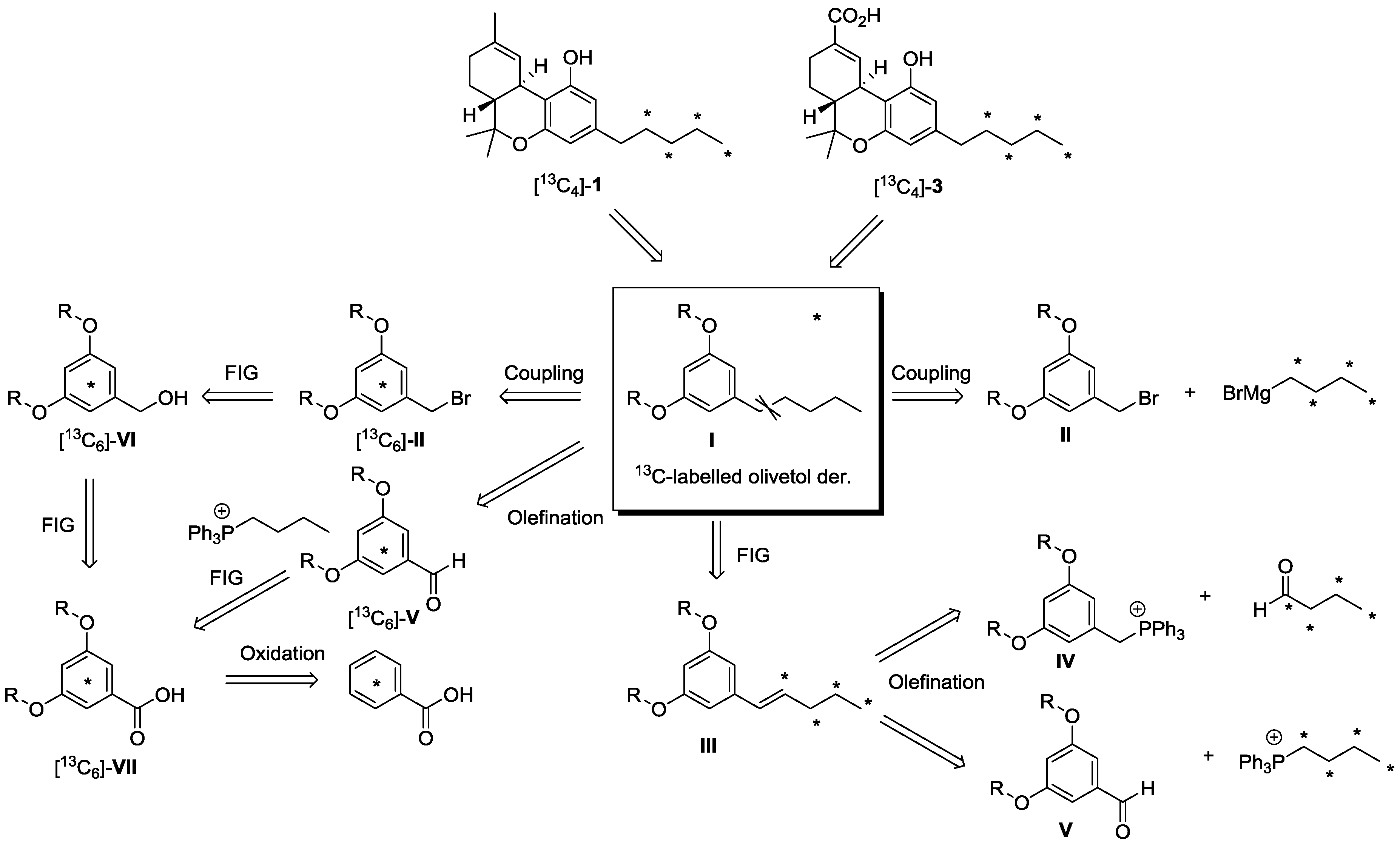
2.1. Strategy Selection and Synthesis of [13C4]-Olivetol
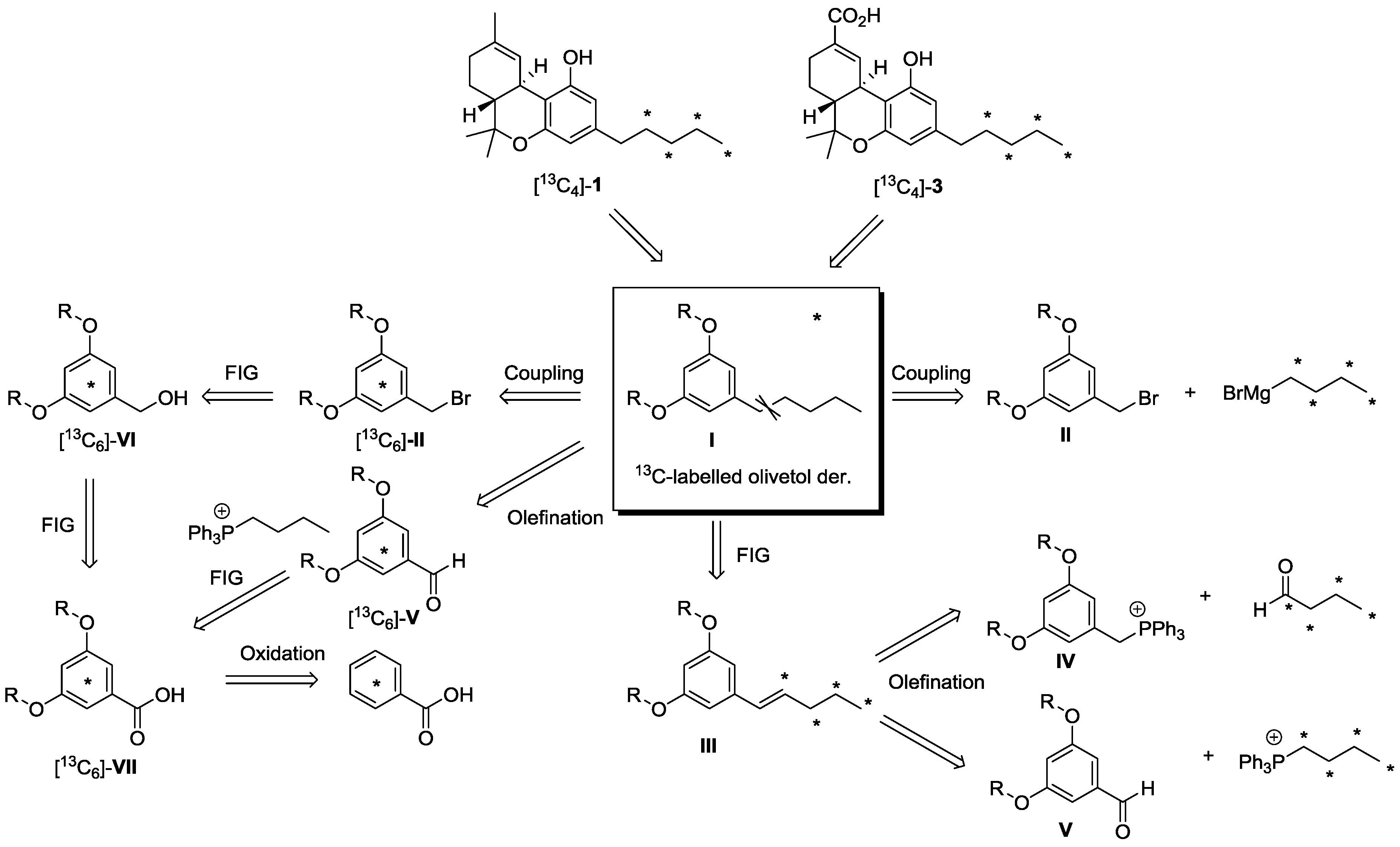
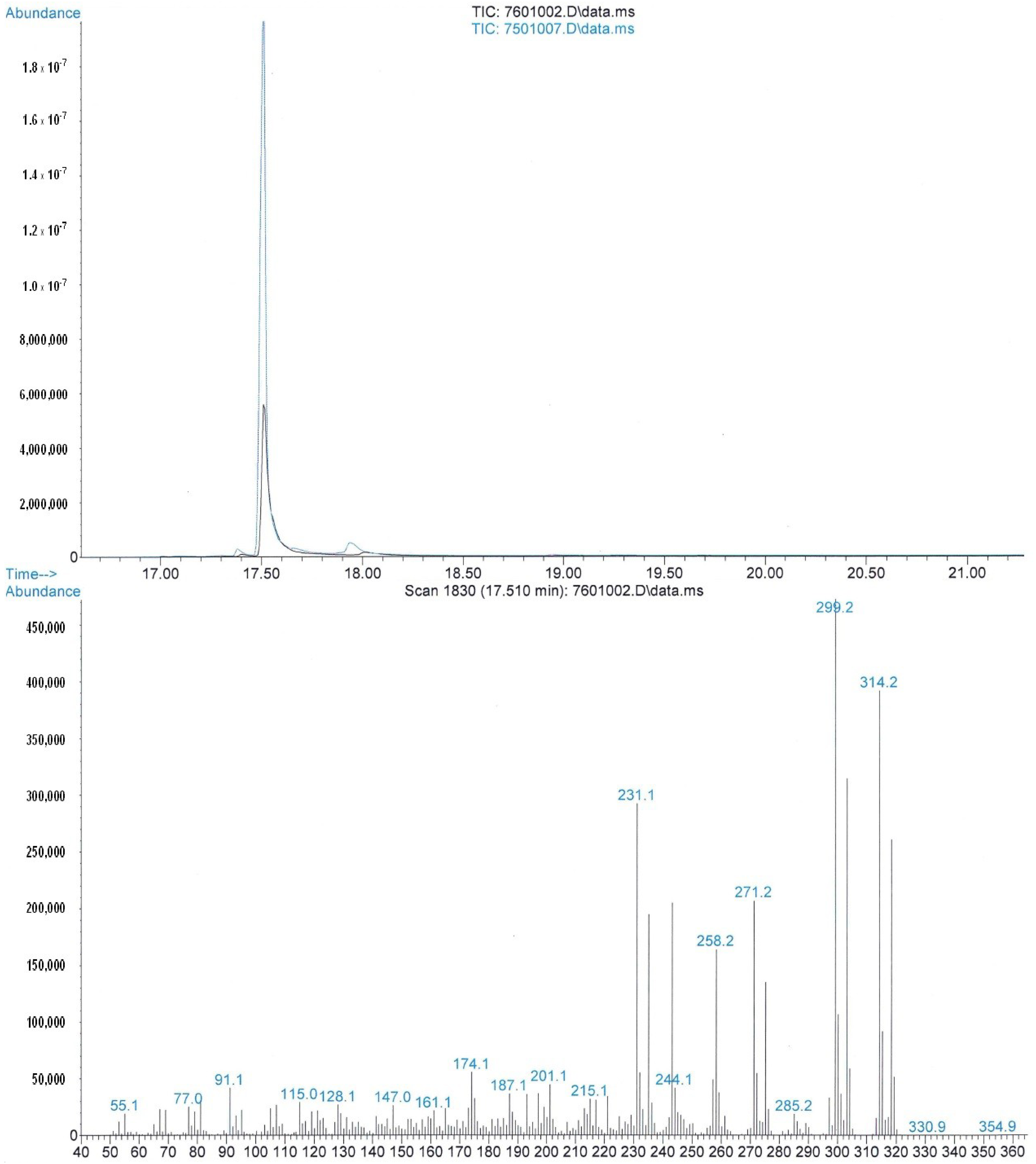
2.2. [13C4]-Labeled Δ9-THC and Δ9-THC-COOH
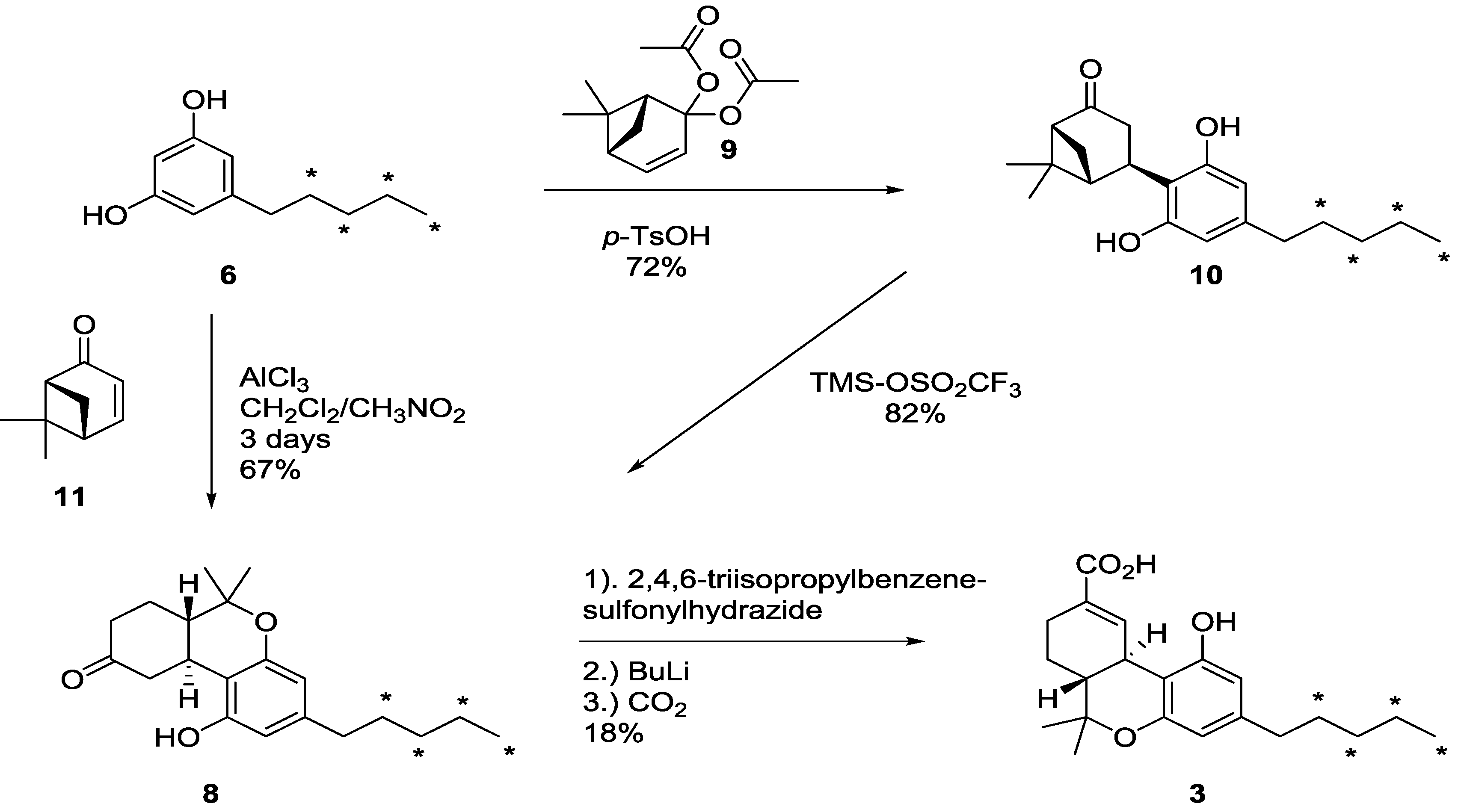
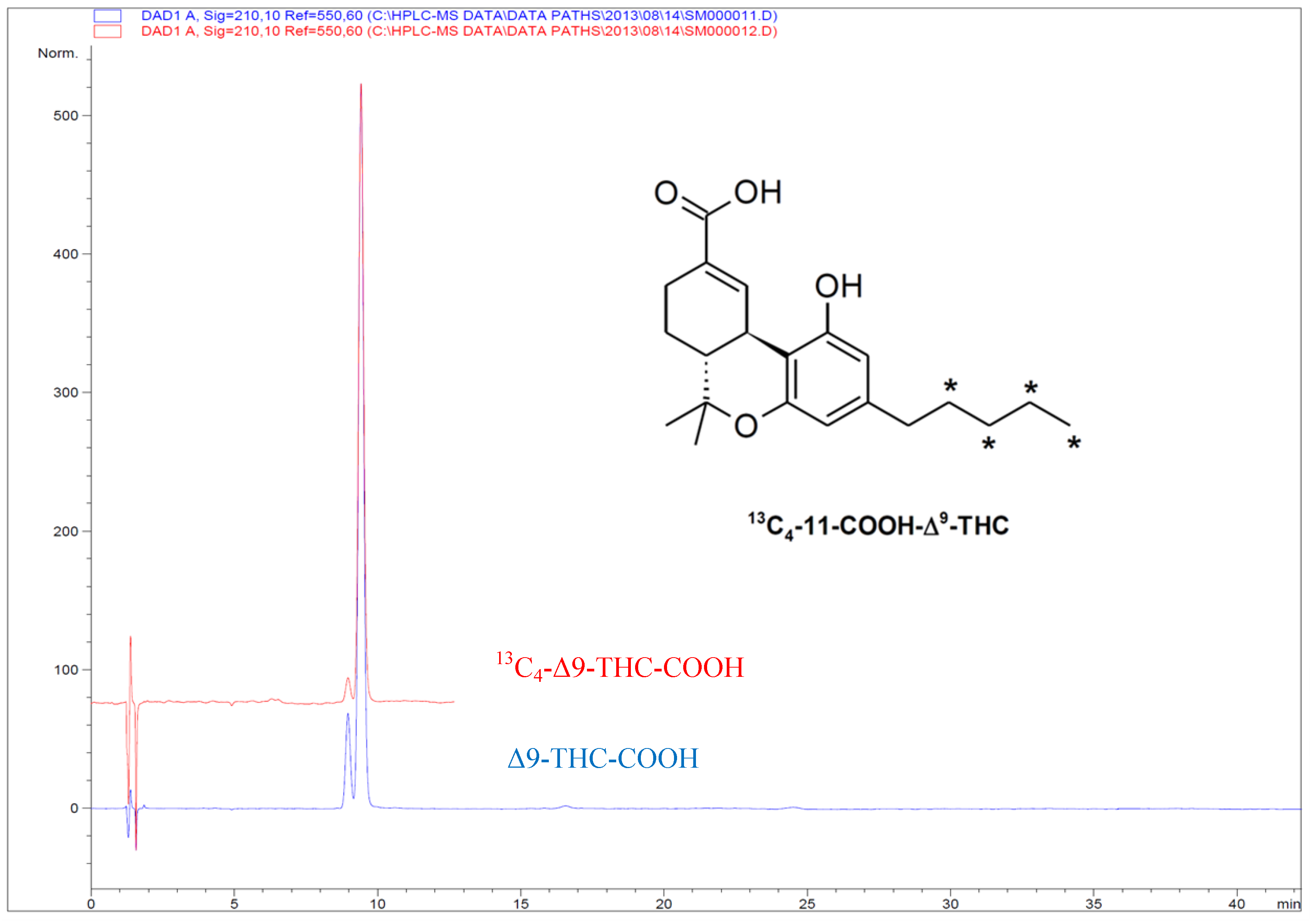
3. Experimental Section
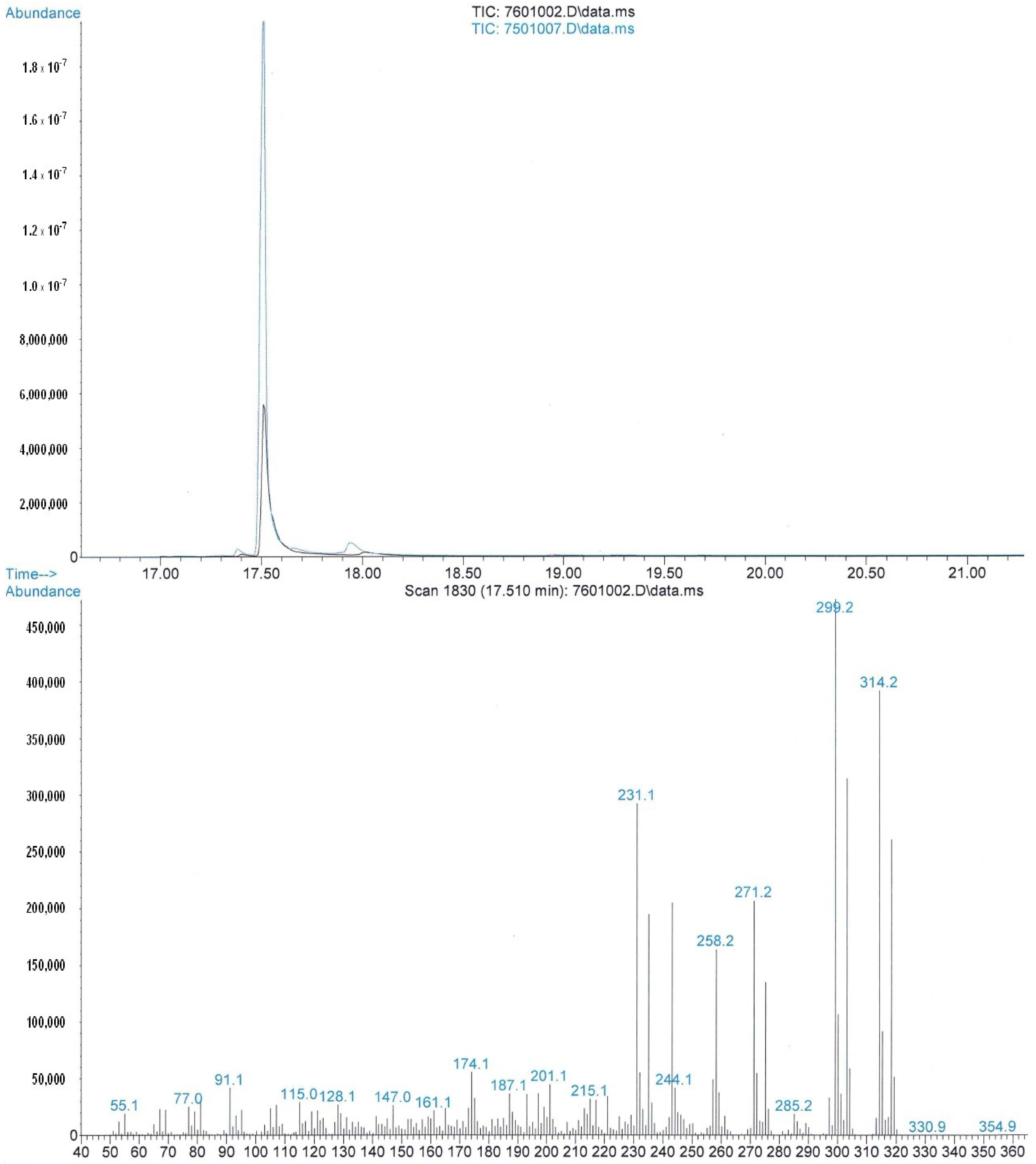
3.1. Chemicals and Analysis
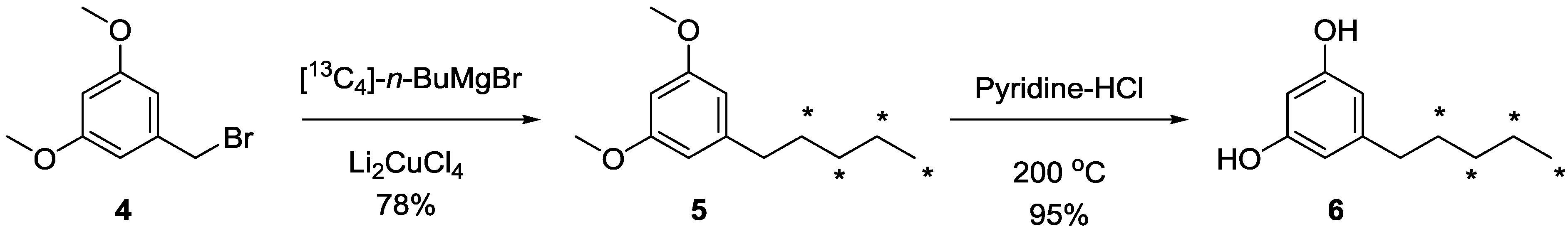
3.2. Synthesis of [13C4]-1-(3,5-Dimethoxyphenyl)pentane (5)
3.3. Synthesis of [13C4]-olivetol (6)
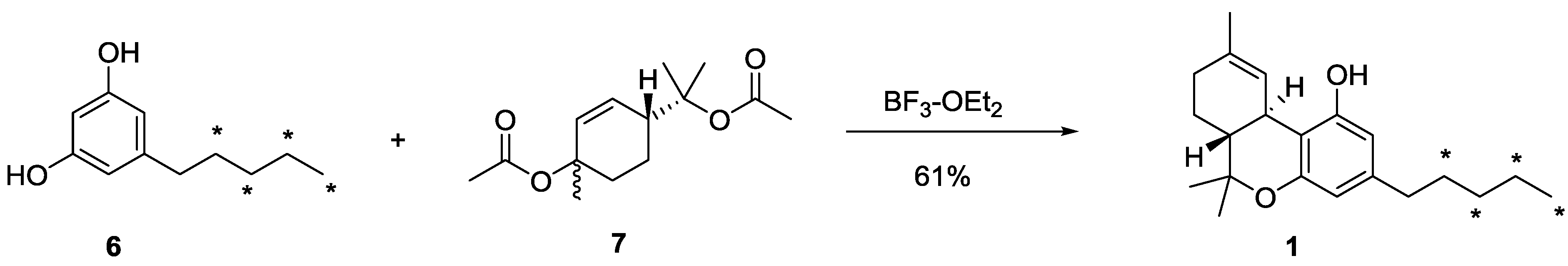
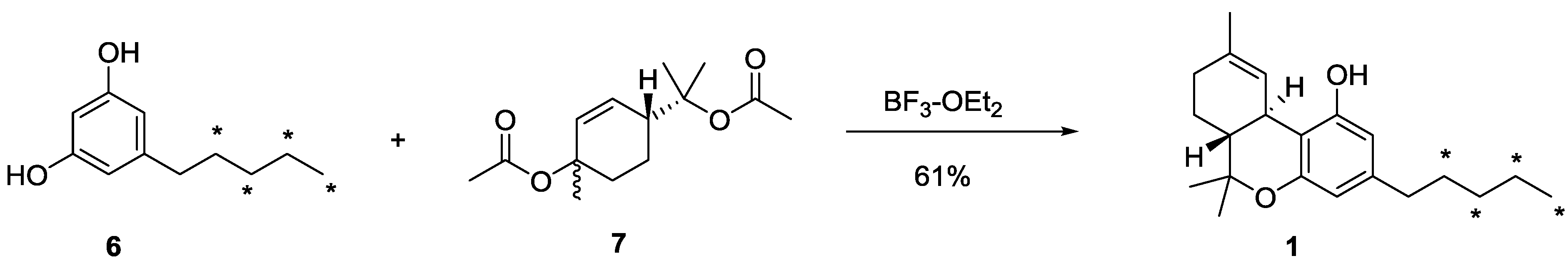
3.4. Synthesis of [13C4]-(−)-∆9-THC (1)
3.5. Synthesis of [13C4]-(4R)-4-(4-pentyl-2,6-dihydroxy-[13C6]-phenyl)-6,6-di-methyl-2-norpinanone (10)
3.6. Synthesis of [13C4]-(6aR,10aR)-6,6a,7,8,10,10a-Hexahydro-1-hydroxy-6,6-dimethyl-3-pentyl-9H-dibenzo[b,d]pyran-9-one (8)
3.7. Synthesis of [13C4]-(6aR,10aR)-6,6a,7,8,10,10a-Hexahydro-1-hydroxy-6,6-dimethyl-3-pentyl-9H-dibenzo[b,d]pyran-9-one (8) Using 11
3.8. Synthesis of [13C4]-(−)-∆9-THC Acid (3)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flachenecker, P.; Henze, T.; Zettl, U.K. Long-term effectiveness and safety of nabiximols (tetrahydrocannabinol/cannabidiol oromucosal spray) in clinical practice. Eur. Neurol. 2014, 72, 95–102. [Google Scholar] [CrossRef]
- Todaro, B. Cannabinoids in the treatment of chemotherapy-induced nausea and vomiting. J. Natl. Compr. Can. Netw. 2012, 10, 487–492. [Google Scholar]
- Wissel, J.; Haydn, T.; Muller, J.; Brenneis, C.; Berger, T.; Poewe, W.; Schelosky, L.D. Low dose treatment with the synthetic cannabinoid Nabilone significantly reduces spasticity-related pain: A double-blind placebo-controlled cross-over trial. J. Neurol. 2006, 253, 1337–1341. [Google Scholar] [CrossRef]
- Huestis, M.A.; Henningfield, J.E.; Cone, E.J. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J. Anal. Toxicol. 1992, 16, 276–282. [Google Scholar] [CrossRef]
- Watanabe, K.; Yamaori, S.; Funahashi, T.; Kimura, T.; Yamamoto, I. Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci. 2007, 80, 1415–1419. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Weinmann, W.; Vogt, S.; Goerke, R.; Müller, C.; Bromberger, A. Simultaneous determination of THC-COOH and THC-COOH-glucuronide in urine samples by LC/MS/MS. Forensic Sci. Int. 2000, 113, 381–387. [Google Scholar] [CrossRef]
- Montesano, C.; Sergi, M.; Odoardi, S.; Simeoni, M.C.; Compagnone, D.; Curini, R. A μ-SPE procedure for the determination of cannabinoids and their metabolites in urine by LC–MS/MS. J. Pharm. Biomed. Anal. 2014, 91, 169–175. [Google Scholar] [CrossRef]
- Breindahl, T.; Andreasen, K. Determination of 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in urine using high-performance liquid chromatography and electrospray ionization mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 732, 155–164. [Google Scholar] [CrossRef]
- Míguez-Framil, M.; Cocho, J.Á.; Tabernero, M.J.; Bermejo, A.M.; Moreda-Piñeiro, A.; Bermejo-Barrera, P. An improved method for the determination of Δ9-tetrahydrocannabinol, cannabinol and cannabidiol in hair by liquid chromatography–tandem mass spectrometry. Microchem. J. 2014, 117, 7–17. [Google Scholar] [CrossRef]
- Lee, K.M.; Kim, H.J.; Son, J.; Park, J.H.; Kwon, O.S.; Lee, J. Simple quantitation of formoterol and 11-nor-Δ9-tetrahydrocannabinol-9-carboxylic acid in human urine by liquid chromatography-tandem mass spectrometry in doping control. J. Chromatogr. B Biomed. Sci. Appl. 2014, 967, 8–12. [Google Scholar]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef]
- Iyer, S.S.; Zhang, Z.P.; Kellogg, G.E.; Karnes, H.T. Evaluation of deuterium isotope effects in normal-phase LC-MS-MS separations using a molecular modeling approach. J. Chromatogr. Sci. 2004, 42, 383–387. [Google Scholar] [CrossRef]
- Schwope, D.M.; Scheidweiler, K.B.; Huestis, M.A. Direct quantification of cannabinoids and cannabinoid glucuronides in whole blood by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 401, 1273–1283. [Google Scholar] [CrossRef]
- Scheidweiler, K.B.; Himes, S.K.; Chen, X.; Liu, H.F.; Huestis, M.A. 11-Nor-9-carboxy-–9-tetrahydrocannabinol quantification in human oral fluid by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6019–6027. [Google Scholar] [CrossRef]
- Berg, T.; Strand, D.H. 13C labelled internal standards-A solution to minimize ion suppression effects in liquid chromatography-tandem mass spectrometry analyses of drugs in biological samples? J. Chromatogr. A 2011, 1218, 9366–9374. [Google Scholar]
- Wang, D.; Stapleton, H.M. Analysis of thyroid hormones in serum by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2010, 397, 1831–1839. [Google Scholar] [CrossRef]
- Haubl, G.; Berthiller, F.; Krska, R.; Schuhmacher, R. Suitability of a fully 13C isotope labeled internal standard for the determination of the mycotoxin deoxynivalenol by LC-MS/MS without clean up. Anal. Bioanal. Chem. 2006, 384, 692–696. [Google Scholar] [CrossRef]
- Chavez-Eng, C.M.; Constanzer, M.L.; Matuszewski, B.K. High-performance liquid chromatographic-tandem mass spectrometric evaluation and determination of stable isotope labeled analogs of rofecoxib in human plasma samples from oral bioavailability studies. J. Chromatogr. B Biomed. Sci. Appl. 2002, 767, 117–129. [Google Scholar]
- Wu, R.; Lodwig, S.N.; Schmidt, J.G.; Williams, R.F.; Silks, L.A.P. Synthesis of 13C labeled sulfur and nitrogen mustard metabolites as mass spectrometry standards for monitoring and detecting chemical warfare agents. J. Label. Compd. Radiopharm. 2012, 55, 211–222. [Google Scholar] [CrossRef]
- Kubátová, A.; Matucha, M.; Bubner, M. Application of 13C-labelled polychlorinated biphenyl congener 153 as internal standard in the gas chromatographic-mass spectrometric analysis of polychlorinated biphenyls. J. Chromatogr. A 1996, 750, 245–251. [Google Scholar] [CrossRef]
- Mechoulam, R.; Braun, P.; Gaoni, Y. A stereospecific synthesis of (−)-Δ1- and (−)-Δ1(6)-tetrahydrocannabinols. J. Am. Chem. Soc. 1967, 89, 4552–4554. [Google Scholar] [CrossRef]
- Razdan, R.K. The total synthesis of cannabinoids. In Total Synthesis Natural Products; Apsimon, J., Ed.; Wiley: New York, NY, USA, 1981; Volume 4, pp. 185–262. [Google Scholar]
- Razdan, R.K.; Dalzell, H.C.; Handrick, G.R.; Hashish, X. A simple one-step synthesis of (−)-Δ1-tetrahydrocannabinol (THC) from p-mentha-2,8-dien-1-ol and olivetol. J. Am. Chem. Soc. 1974, 96, 5860–5865. [Google Scholar]
- Silverberg, L.J. Synthesis of Cannabinoids. WO2002096899A1. 5 December 2002. [Google Scholar]
- Trost, B.M.; Dogra, K. Synthesis of (−)-Δ9-trans-tetrahydrocannabinol: Stereocontrol via Mo-catalyzed asymmetric allylic alkylation reaction. Org. Lett. 2007, 9, 861–863. [Google Scholar] [CrossRef]
- Minuti, L.; Ballerini, E. High-pressure access to the Δ9-cis- and Δ9-trans-tetrahydrocannabinols family. J. Org. Chem. 2011, 76, 5392–5403. [Google Scholar] [CrossRef]
- Cheng, L.-J.; Xie, J.-H.; Chen, Y.; Wang, L.-X.; Zhou, Q.-L. Enantioselective total synthesis of (−)-Δ8-THC and (−)-Δ9-THC via catalytic asymmetric hydrogenation and SNAr cyclization. Org. Lett. 2013, 15, 764–767. [Google Scholar]
- Girard, M.; Moir, D.B.; ApSimon, J.W. A simple and efficient synthesis of 5'-(2H3)olivetol. Can. J. Chem. 1987, 65, 189–190. [Google Scholar] [CrossRef]
- Zhu, Y.; Soroka, D.N.; Sang, S. Synthesis and inhibitory activities against colon cancer cell growth and proteasome of alkylresorcinols. J. Agric. Food Chem. 2012, 60, 8624–8631. [Google Scholar] [CrossRef]
- Weston, A.W.; Suter, C.M. 3,5-Dihydroxybenzoic acid (α-resorcylic acid). Org. Synth. 1941, 21, 27–30. [Google Scholar] [CrossRef]
- Rosati, O.; Messina, F.; Pelosi, A.; Curini, M.; Petrucci, V.; Gertsch, J.; Chicca, A. One-pot heterogeneous synthesis of Δ3-tetrahydrocannabinol analogues and xanthenes showing differential binding to CB and CB receptors. Eur. J. Med. Chem. 2014, 85, 77–86. [Google Scholar]
- Nikas, S.P.; Thakur, G.A.; Parrish, D.; Alapafuja, S.O.; Huestis, M.A.; Makriyannis, A. A concise methodology for the synthesis of (−)-Δ9-tetrahydrocannabinol and (−)-Δ9-tetrahydrocannabivarin metabolites and their regiospecifically deuterated analogs. Tetrahedron 2007, 63, 8112–8123. [Google Scholar]
- Archer, R.A.; Blanchard, W.B.; Day, W.A.; Johnson, D.W.; Lavagnino, E.R.; Ryan, C.W.; Baldwin, J.E. Cannabinoids. 3. Synthetic approaches to 9-ketocannabinoids. Total synthesis of nabilone. J. Org. Chem. 1977, 42, 2277–2284. [Google Scholar]
- Ichii, M. Friedel-Crafts aralkylation. I. Kinetics of the aluminum trichloride-nitromethane-catalyzed phenethylation of benzene and toluene in nitromethane. Bull. Inst. Chem. Res. Kyoto Univ. 1971, 49, 114–121. [Google Scholar]
- Huffman, J.W.; Zhang, X.; Wu, M.J.; Joyner, H.H.; Pennington, W.T. Synthesis of (±)-11-nor-9-carboxy-Δ9-tetrahydrocannabinol.New synthetic approaches to cannabinoids. J. Org. Chem. 1991, 56, 1481–1489. [Google Scholar]
- Kachensky, D.F.; Hui, R.A.H.F. Preparation of racemic, (−)- and (+)-11-nor-Δ9-tetrahydrocannabinol- 9-carboxylic Acid. J. Org. Chem. 1997, 62, 7065–7068. [Google Scholar] [CrossRef]
- Hui, R.A.H.F.; Salamone, S.; Williams, T.H. Self-induced nonequivalence in the 1H-NMR spectra of the (+)- and (−)-isomers of a cannabinoid ketone intermediate. Pharmacol. Biochem. Behav. 1991, 40, 491–496. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from Chiron AS.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karlsen, M.; Liu, H.; Johansen, J.E.; Hoff, B.H. Synthesis of [13C4]-labeled ∆9-Tetrahydrocannabinol and 11-nor-9-Carboxy-∆9-tetrahydrocannabinol as Internal Standards for Reducing Ion Suppressing/Alteration Effects in LC/MS-MS Quantification. Molecules 2014, 19, 13526-13540. https://doi.org/10.3390/molecules190913526
Karlsen M, Liu H, Johansen JE, Hoff BH. Synthesis of [13C4]-labeled ∆9-Tetrahydrocannabinol and 11-nor-9-Carboxy-∆9-tetrahydrocannabinol as Internal Standards for Reducing Ion Suppressing/Alteration Effects in LC/MS-MS Quantification. Molecules. 2014; 19(9):13526-13540. https://doi.org/10.3390/molecules190913526
Chicago/Turabian StyleKarlsen, Morten, Huiling Liu, Jon Eigill Johansen, and Bård Helge Hoff. 2014. "Synthesis of [13C4]-labeled ∆9-Tetrahydrocannabinol and 11-nor-9-Carboxy-∆9-tetrahydrocannabinol as Internal Standards for Reducing Ion Suppressing/Alteration Effects in LC/MS-MS Quantification" Molecules 19, no. 9: 13526-13540. https://doi.org/10.3390/molecules190913526
APA StyleKarlsen, M., Liu, H., Johansen, J. E., & Hoff, B. H. (2014). Synthesis of [13C4]-labeled ∆9-Tetrahydrocannabinol and 11-nor-9-Carboxy-∆9-tetrahydrocannabinol as Internal Standards for Reducing Ion Suppressing/Alteration Effects in LC/MS-MS Quantification. Molecules, 19(9), 13526-13540. https://doi.org/10.3390/molecules190913526