Chemistry of Phosphorylated Formaldehyde Derivatives. Part I

Abstract
: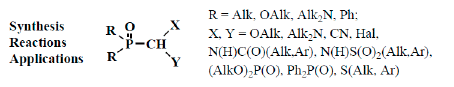
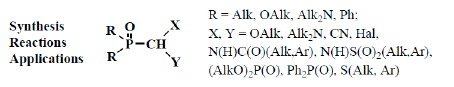{kind=link}
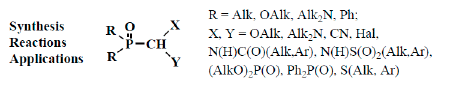{kind=link}
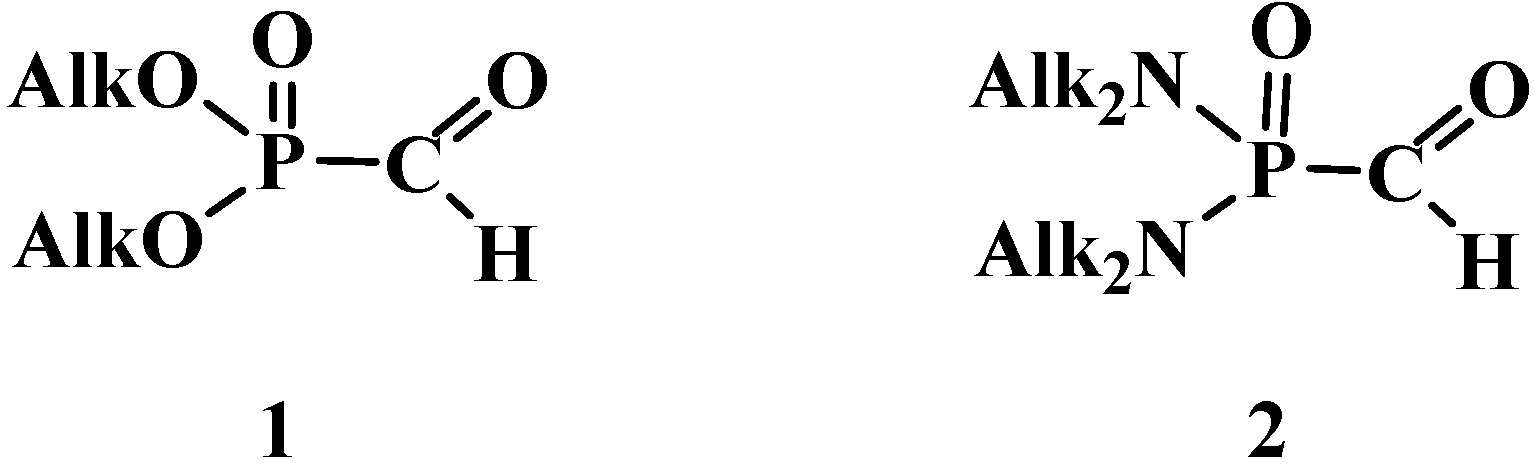{kind=link}
{kind=link}
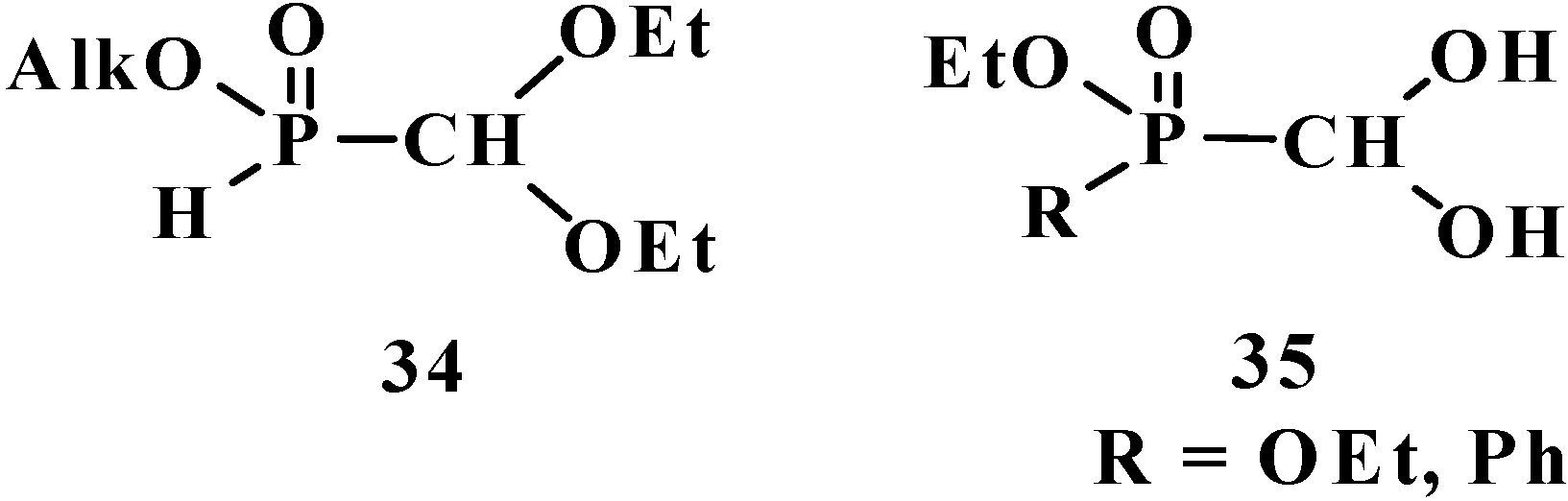{kind=link}
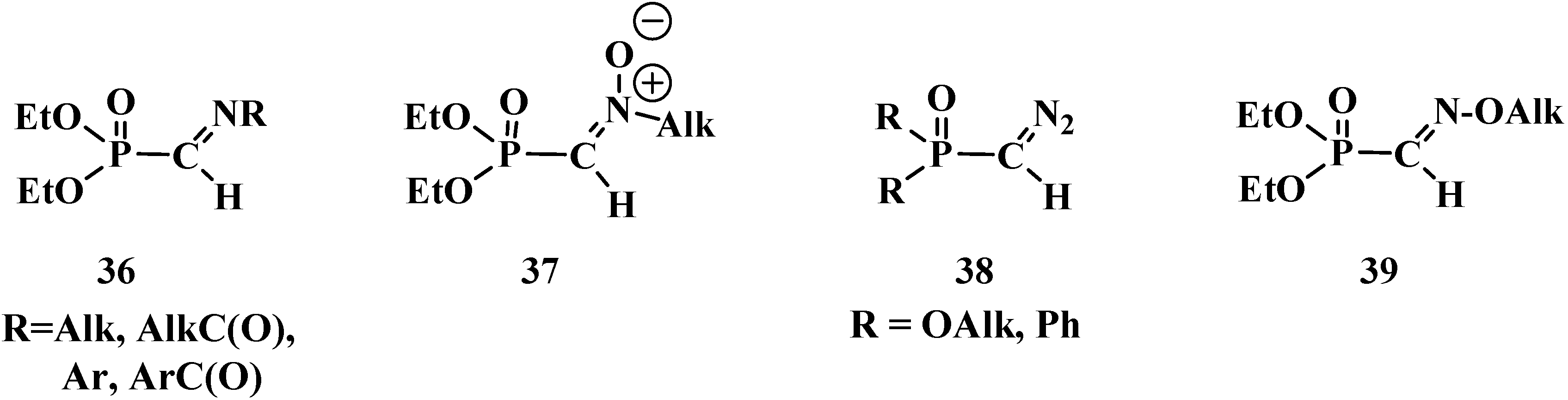{kind=link}
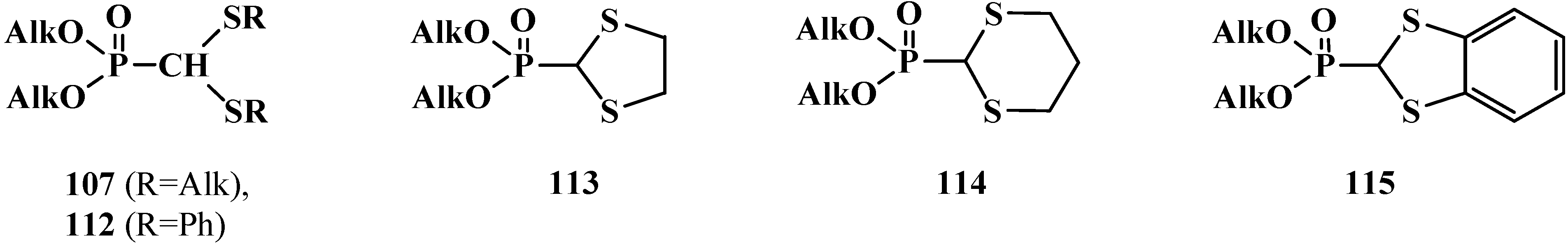{kind=link}
{kind=link}
{kind=link}
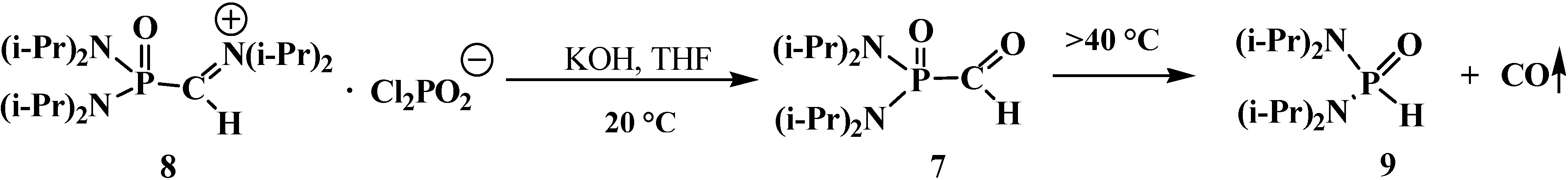{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
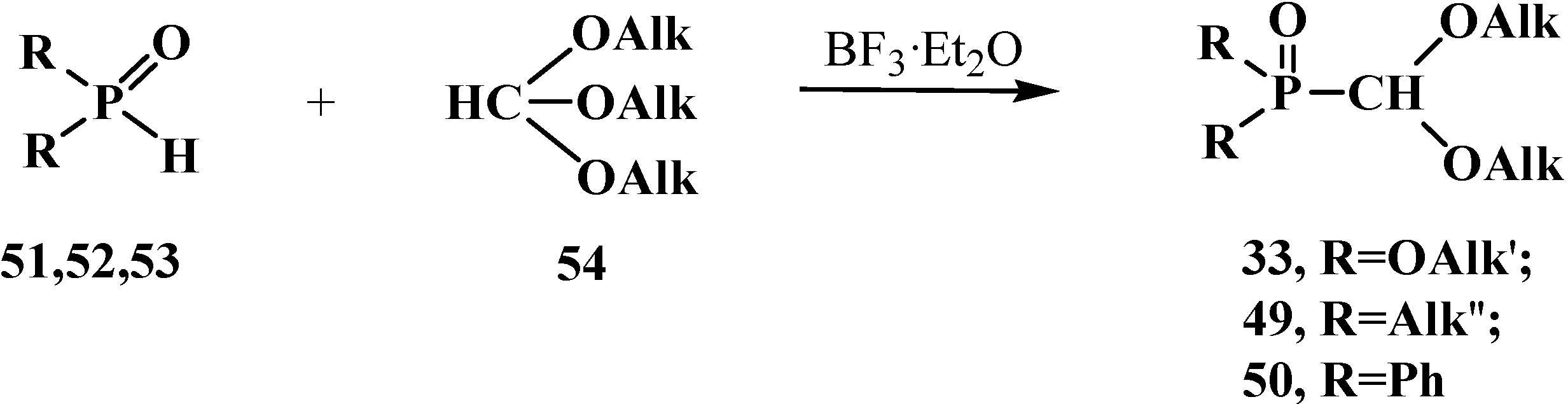{kind=link}
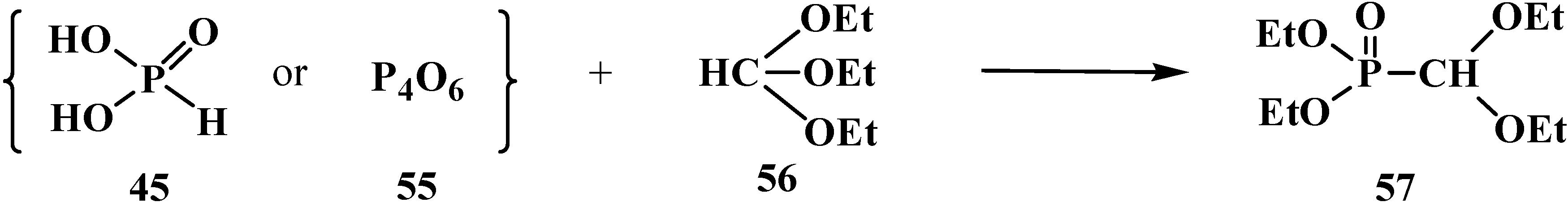{kind=link}
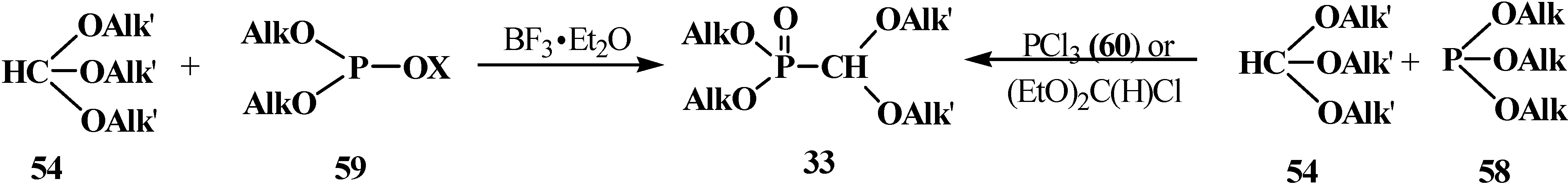{kind=link}
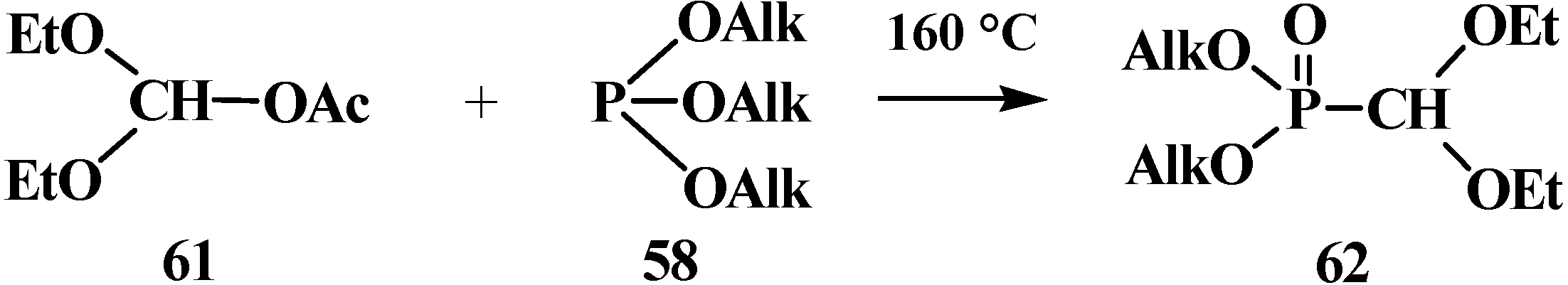{kind=link}
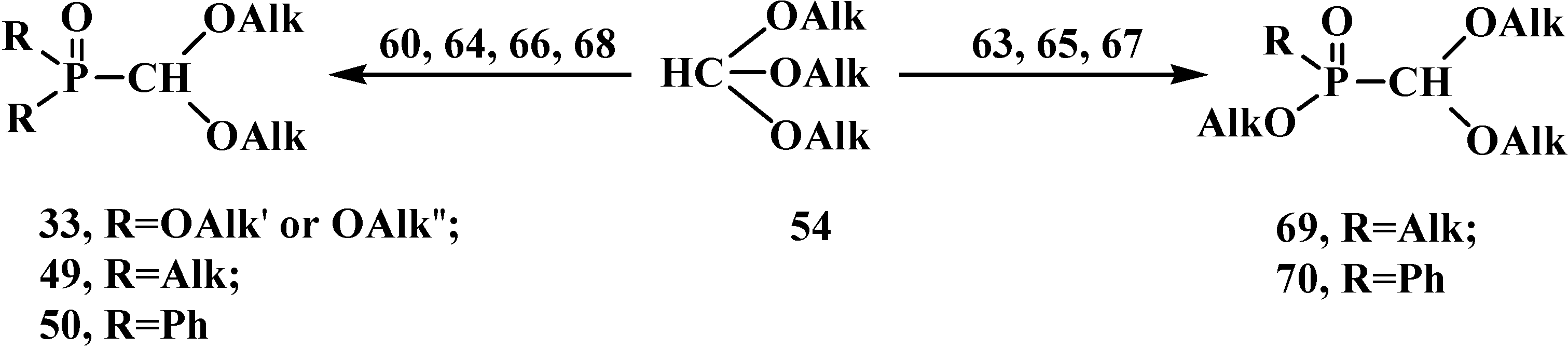{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
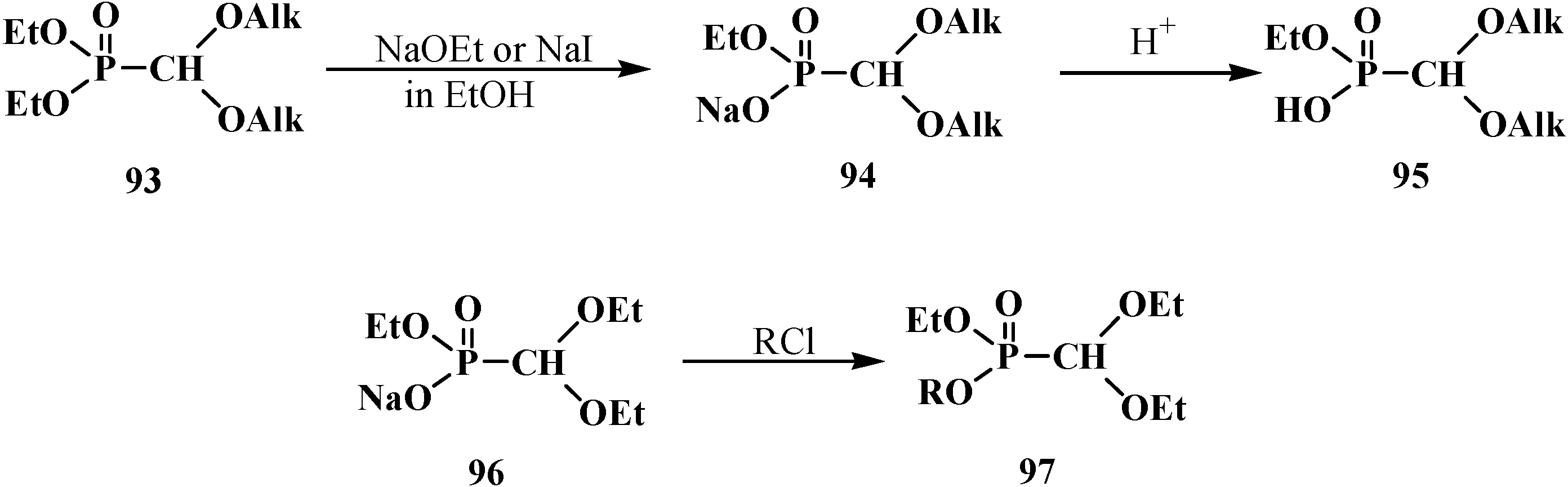{kind=link}
{kind=link}
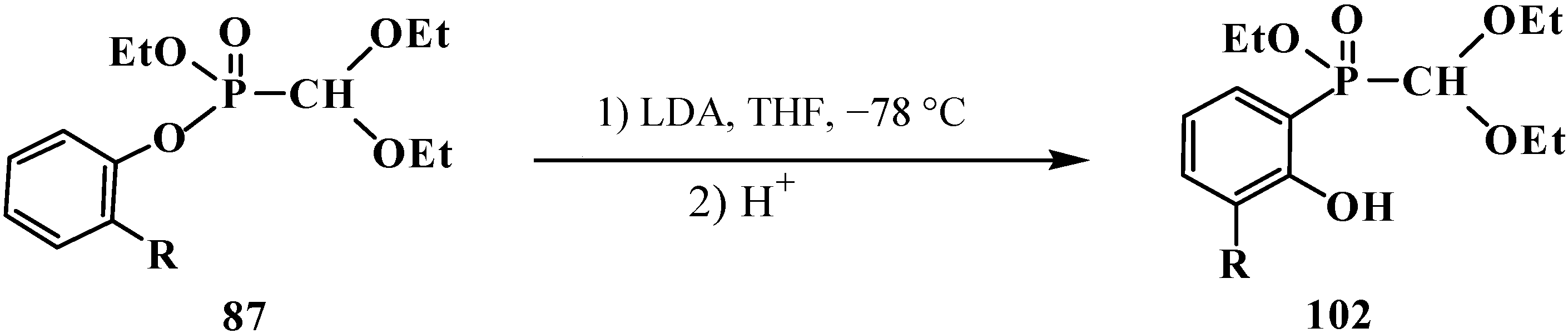{kind=link}
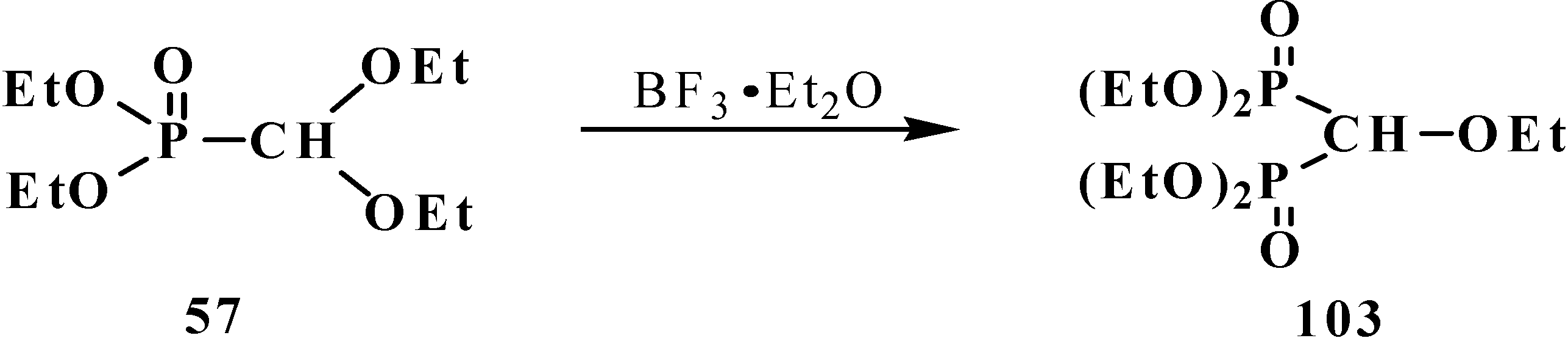{kind=link}
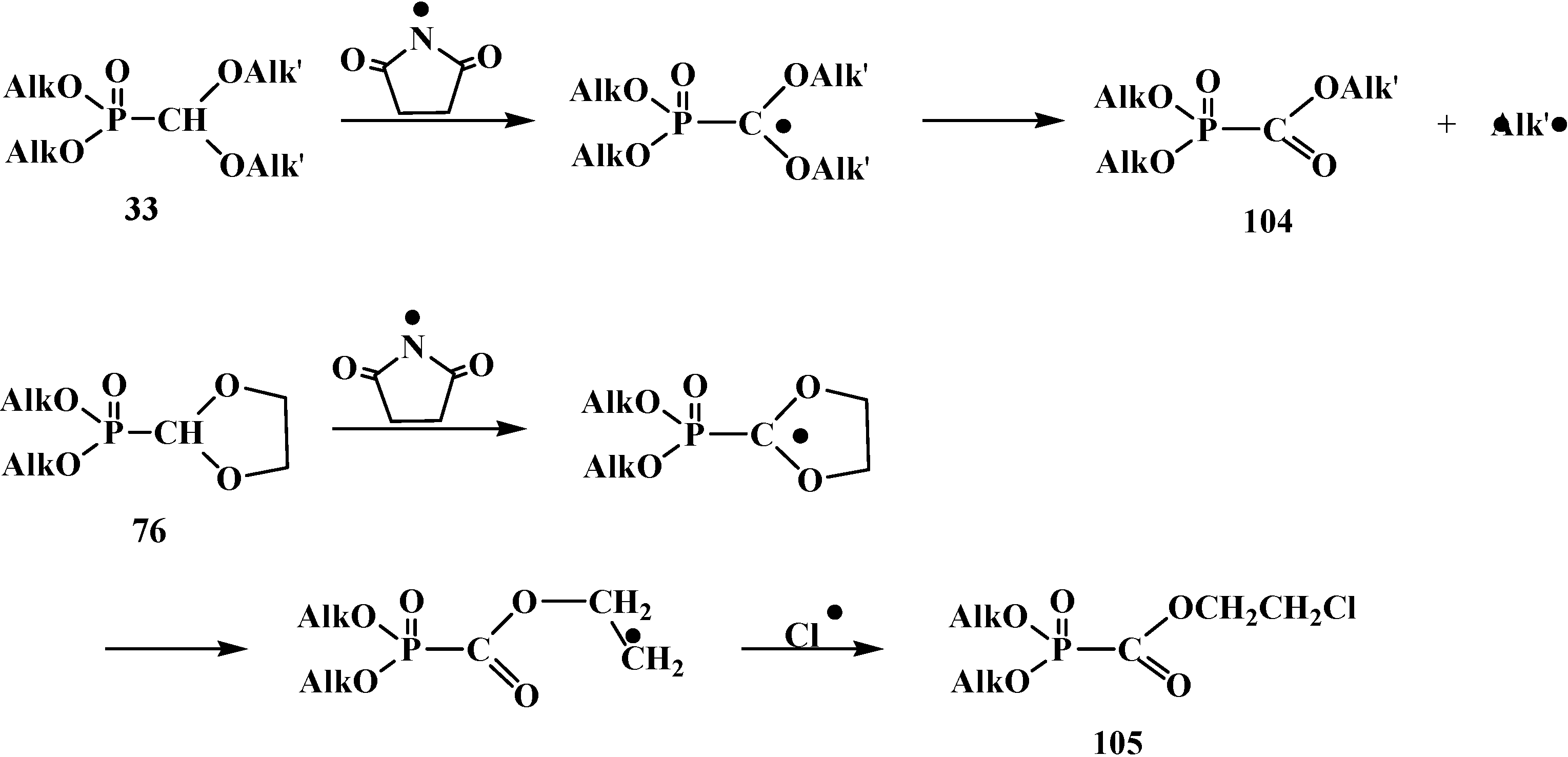{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
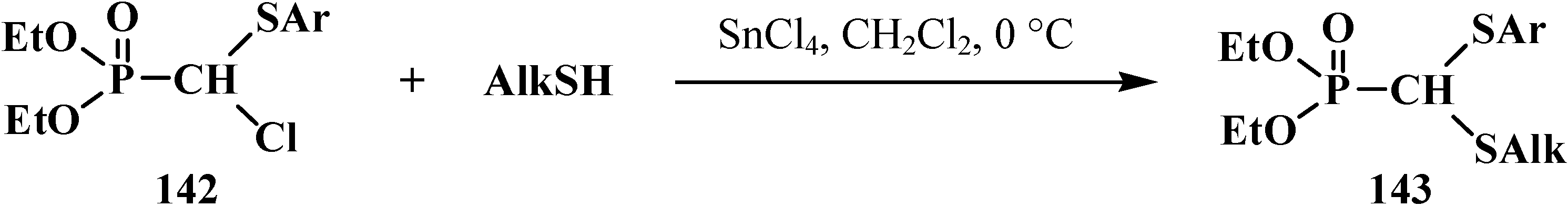{kind=link}
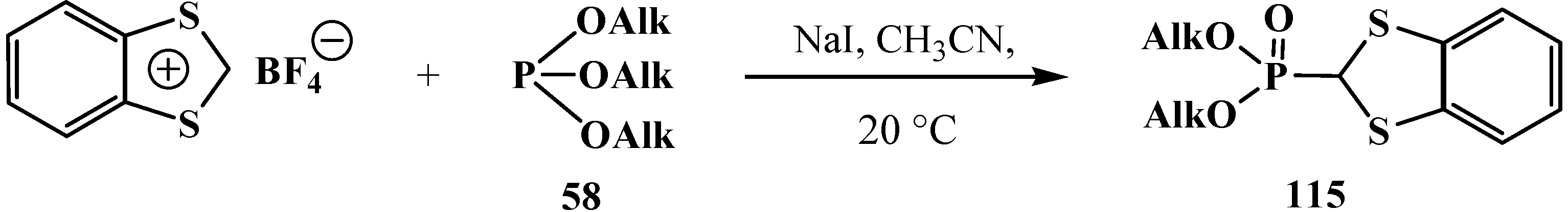{kind=link}
{kind=link}
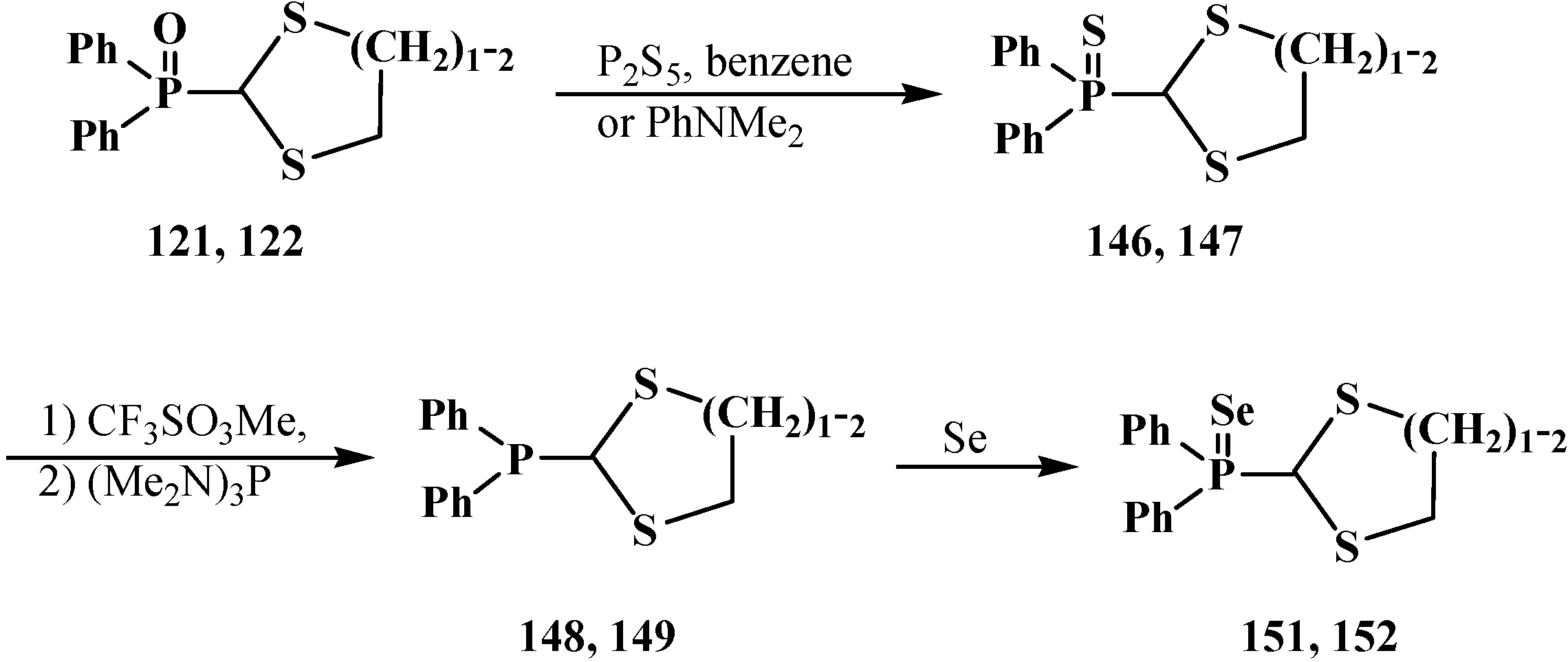{kind=link}
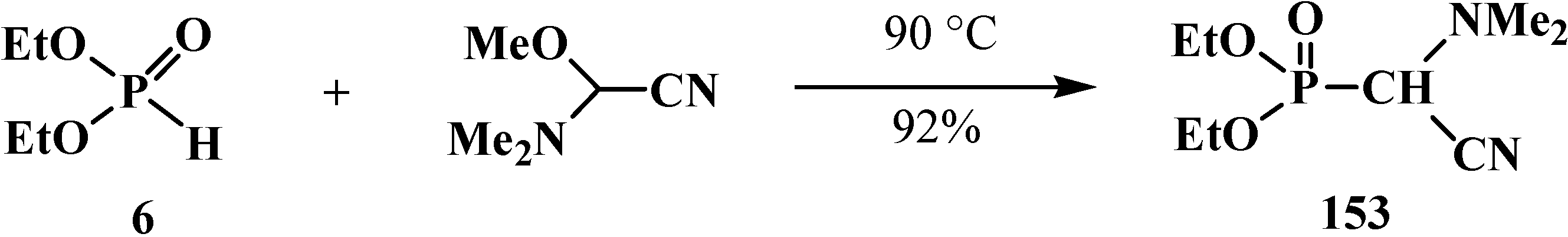{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
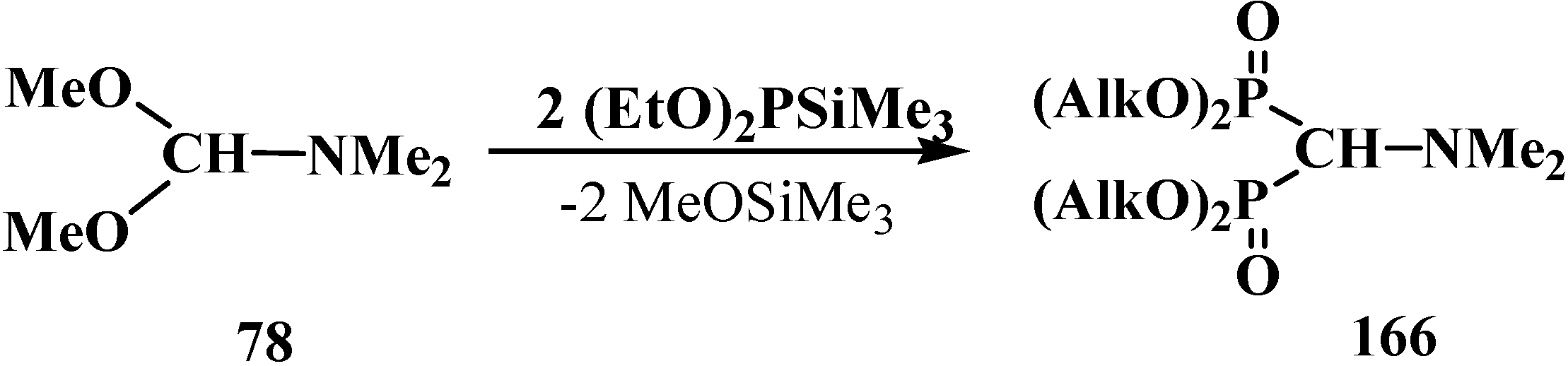{kind=link}
{kind=link}
{kind=link}
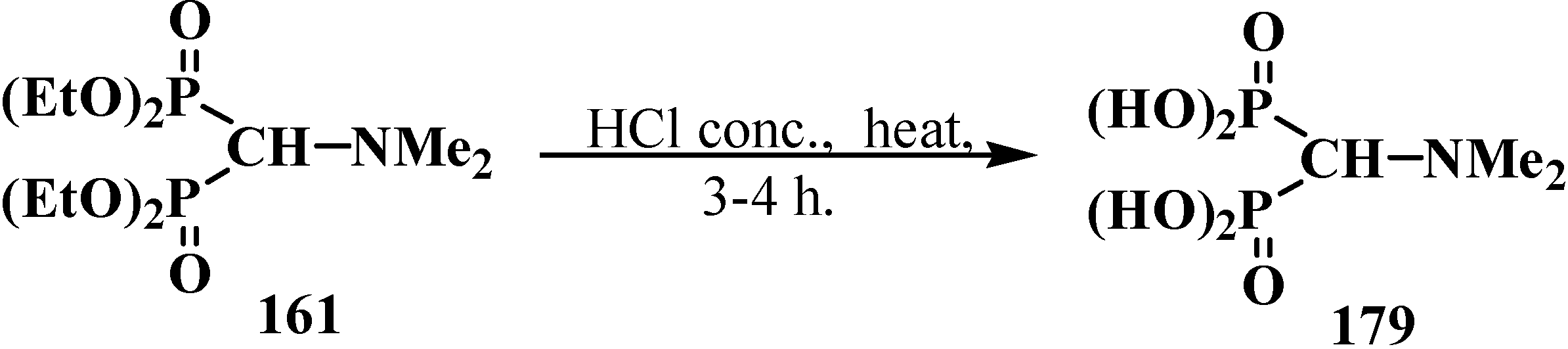{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
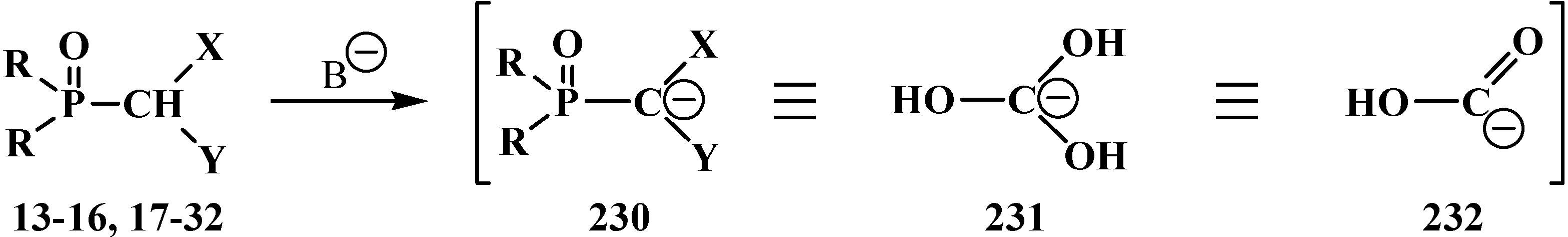{kind=link}
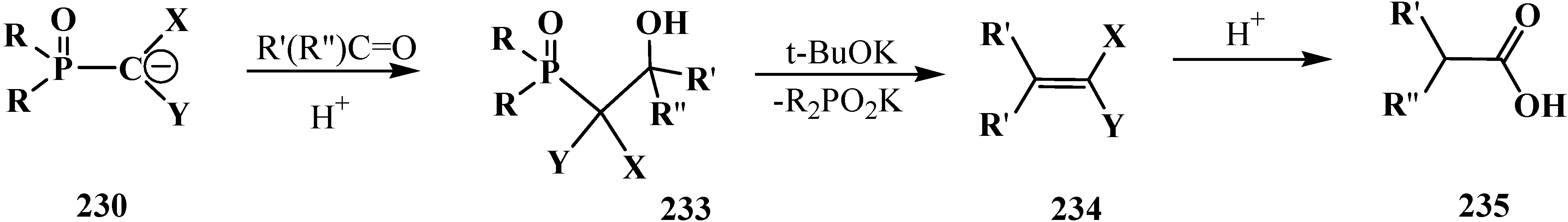{kind=link}
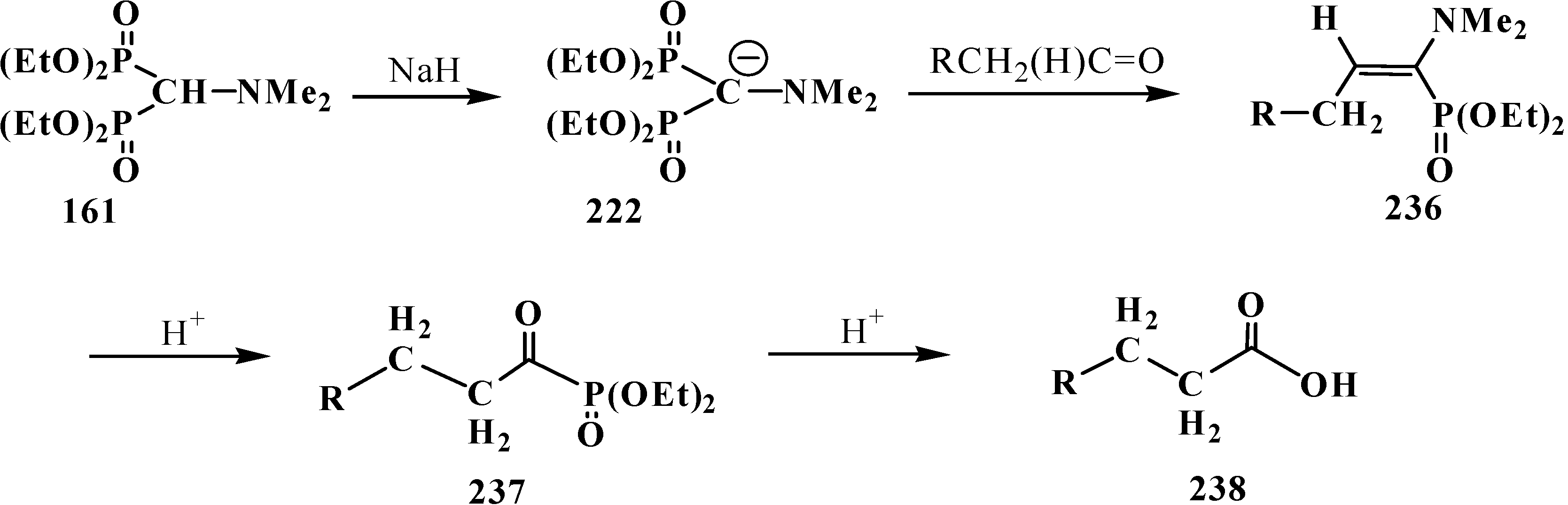{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
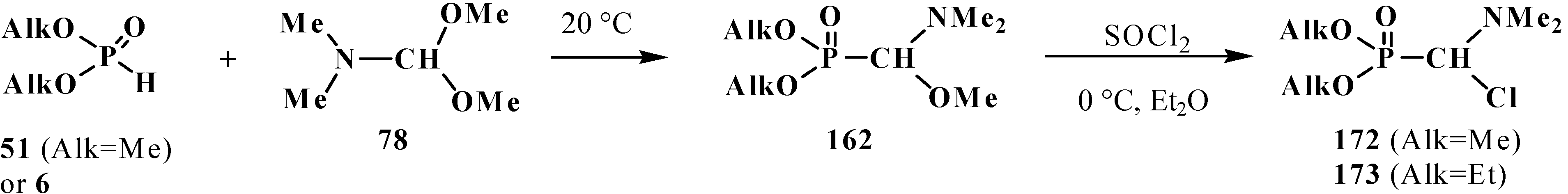{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
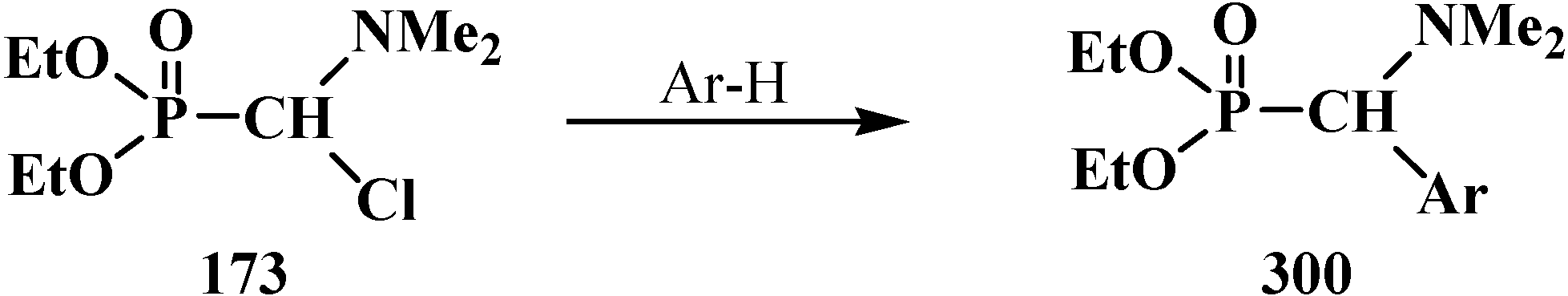{kind=link}
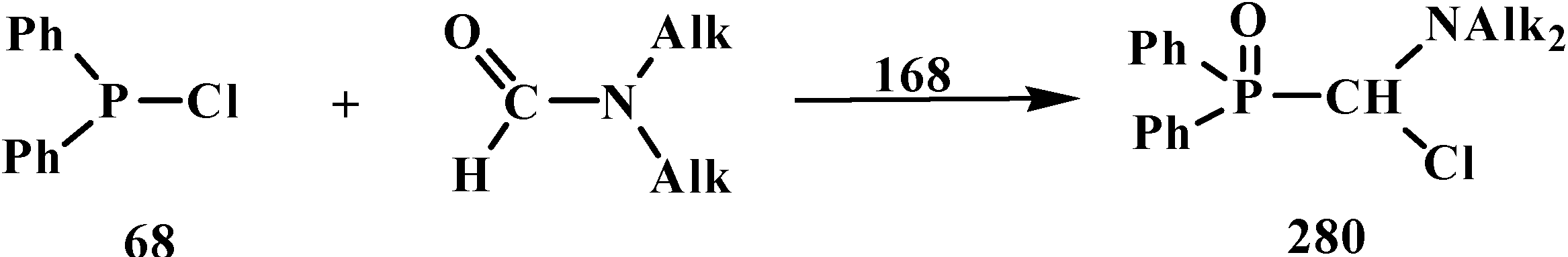{kind=link}
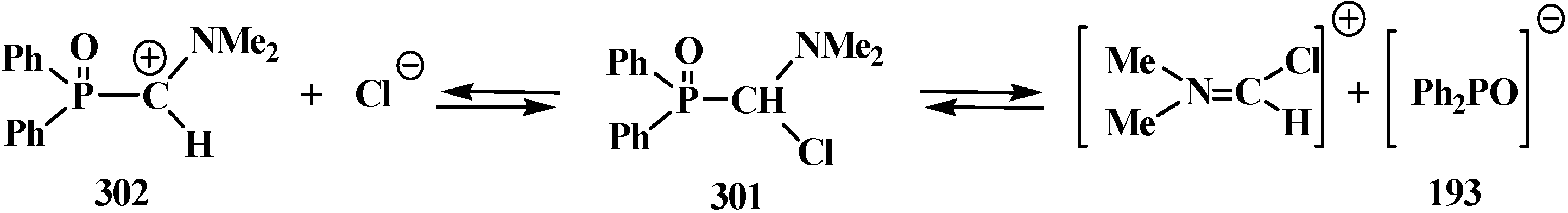{kind=link}
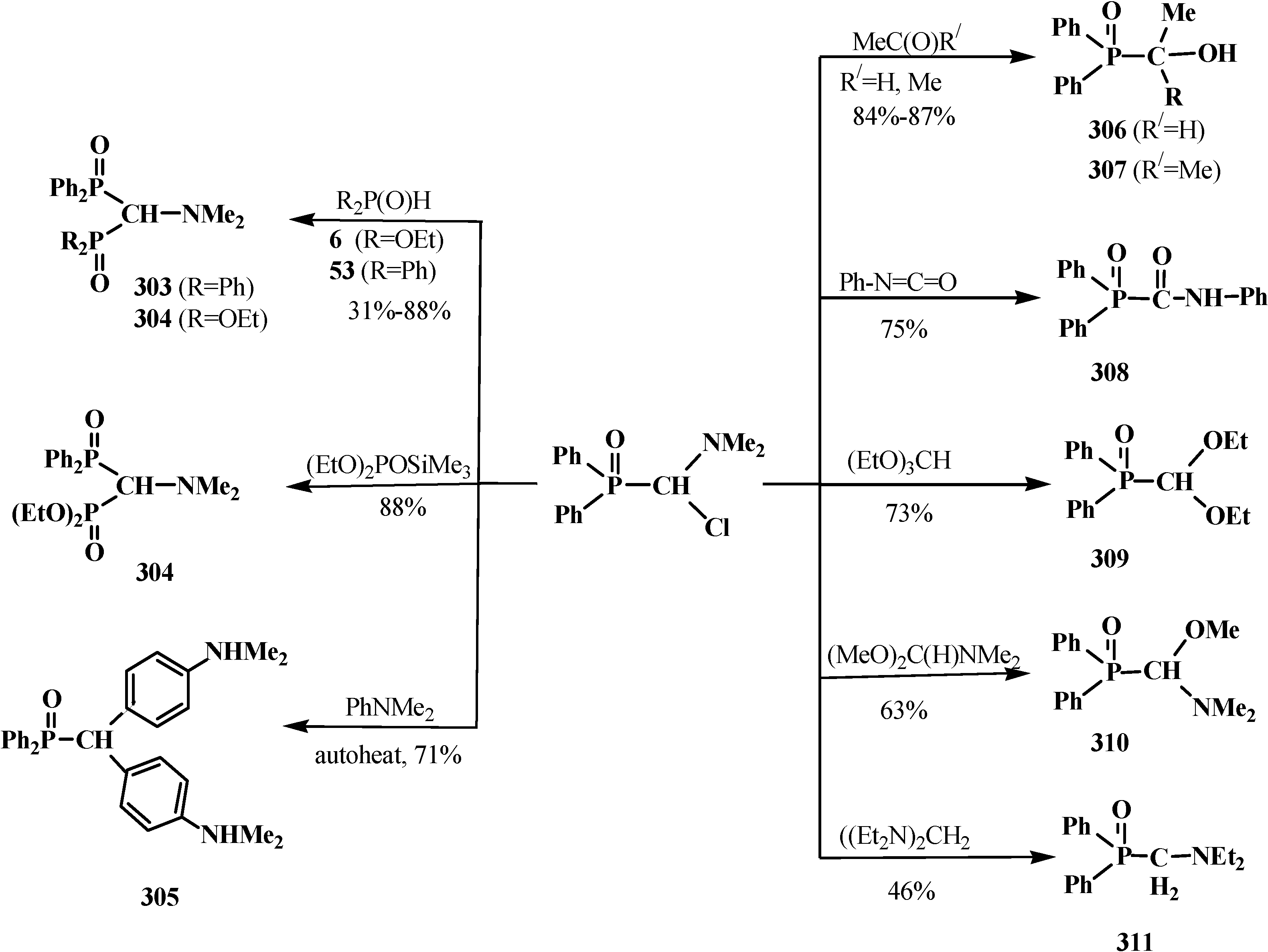{kind=link}
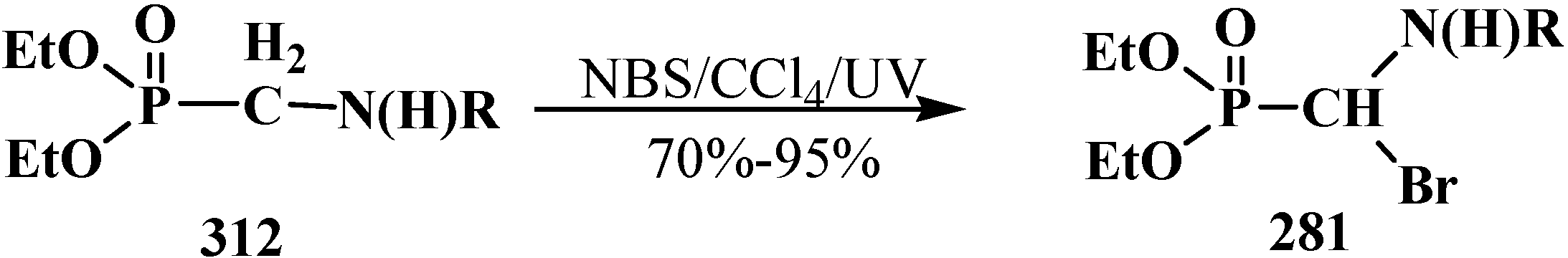{kind=link}
{kind=link}
{kind=link}
{kind=link}
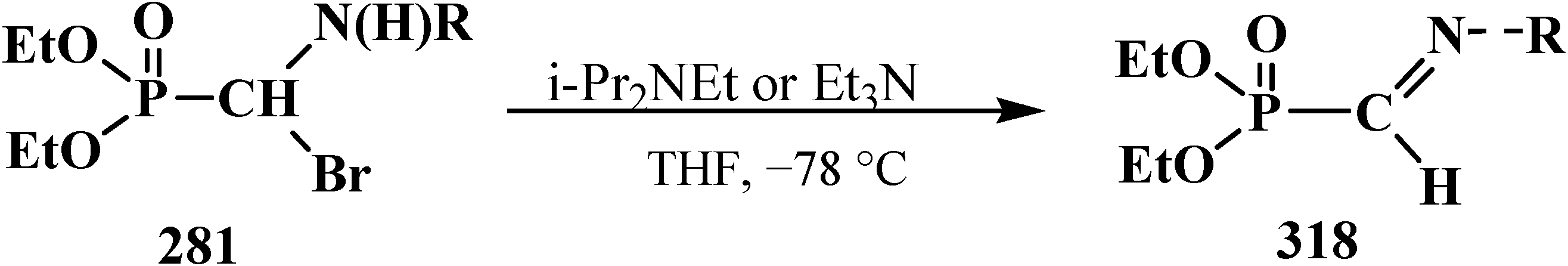{kind=link}
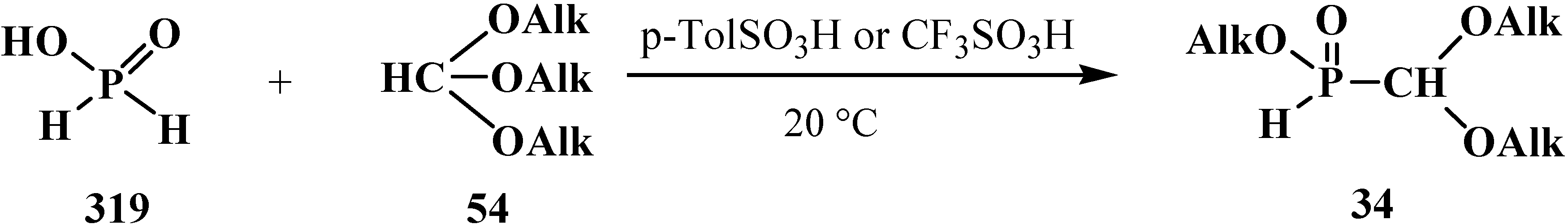{kind=link}
{kind=link}
{kind=link}
{kind=link}
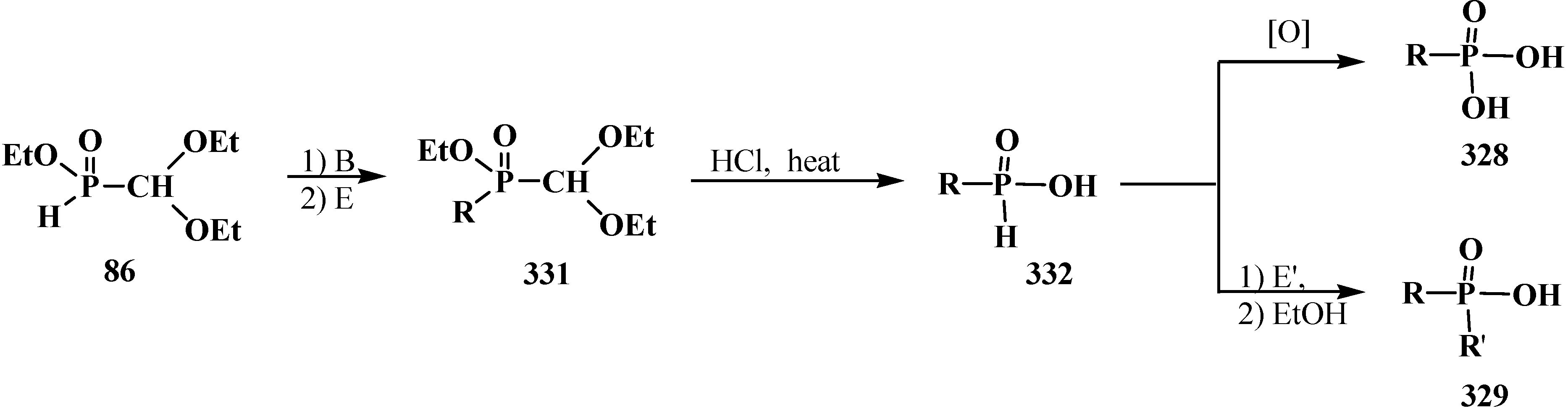{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
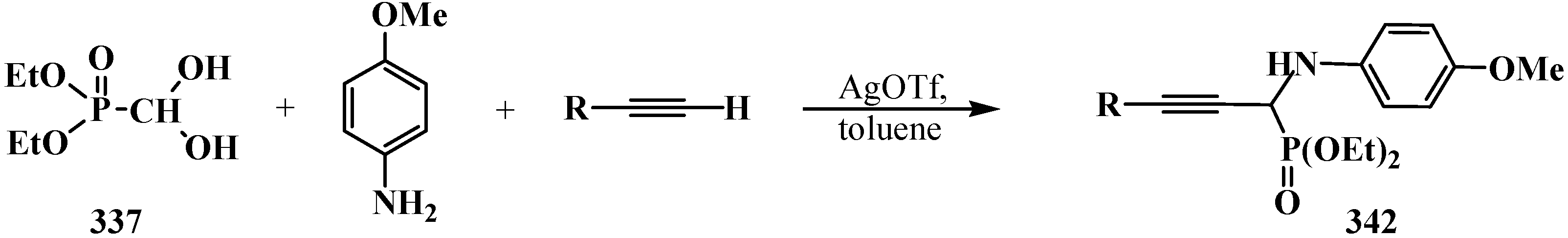{kind=link}
{kind=link}
1. Introduction
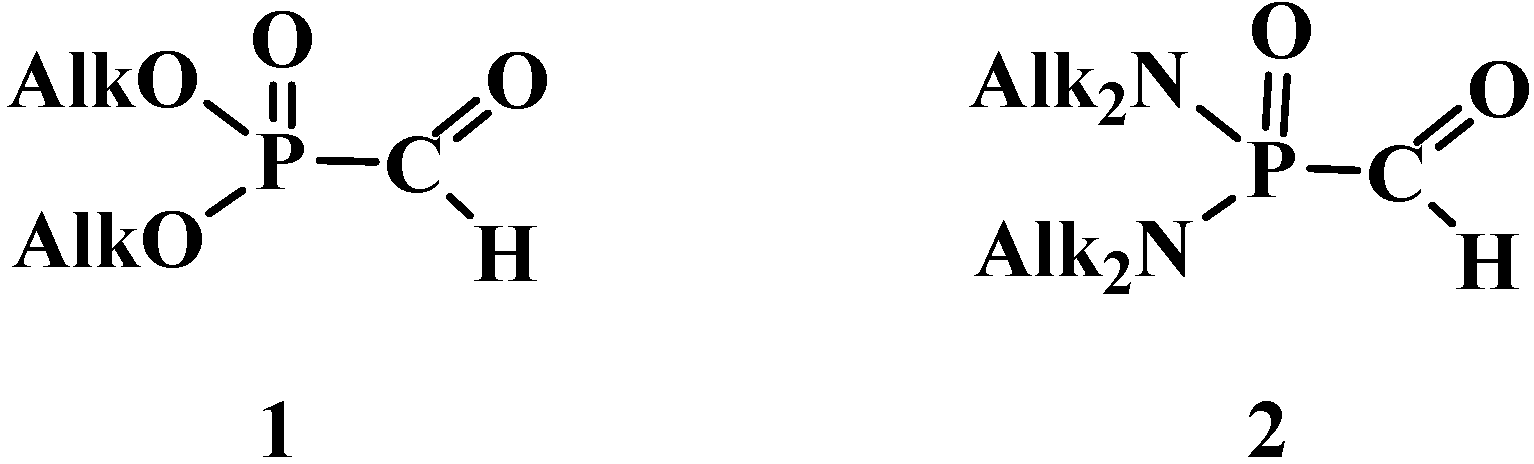
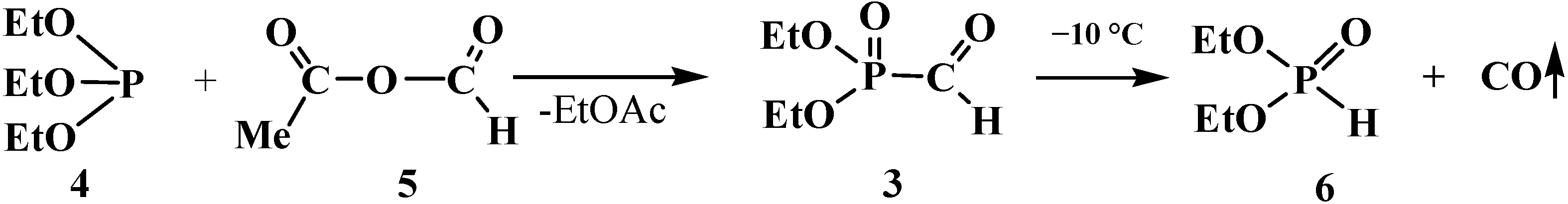
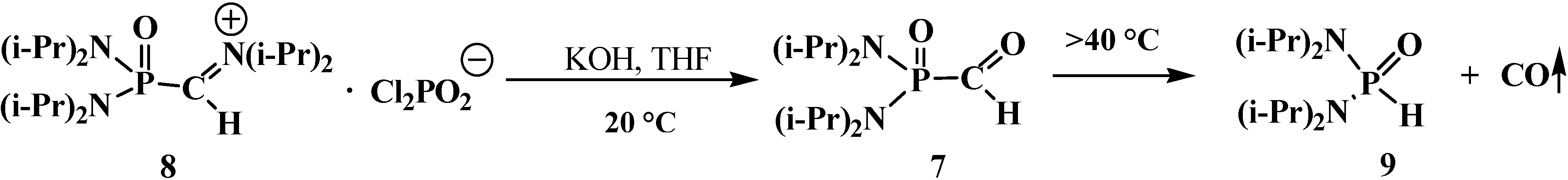

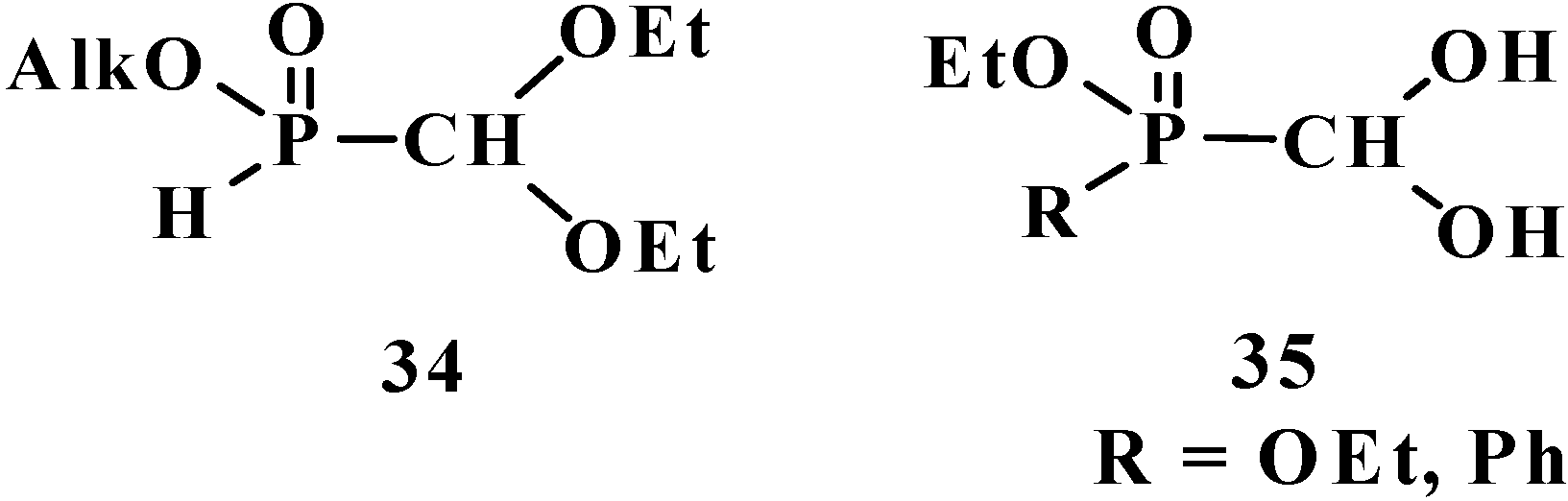
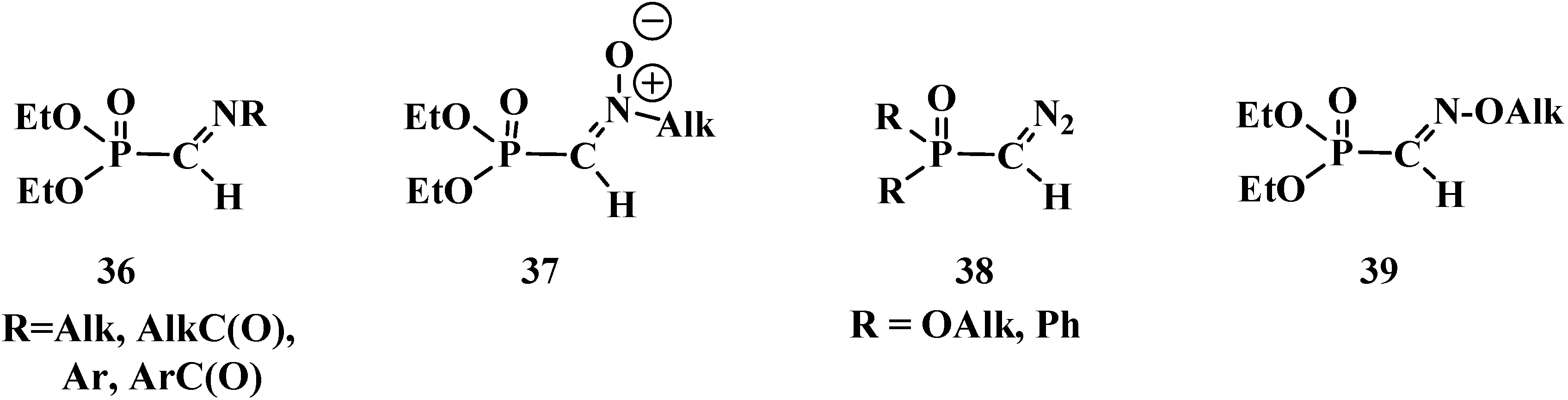
2. Chemistry of Phosphorylated Formaldehyde Derivatives
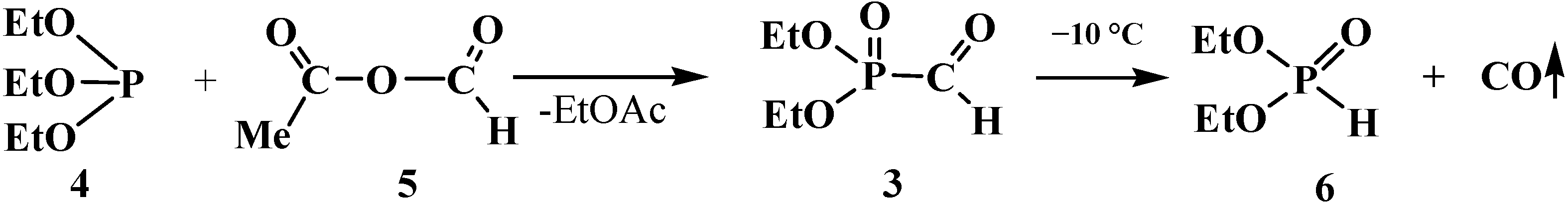
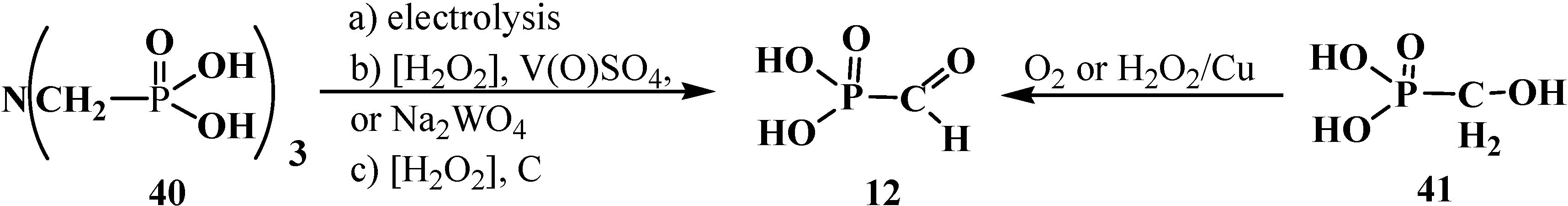
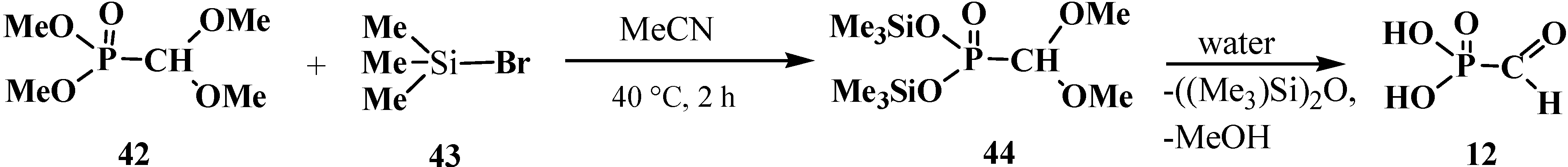
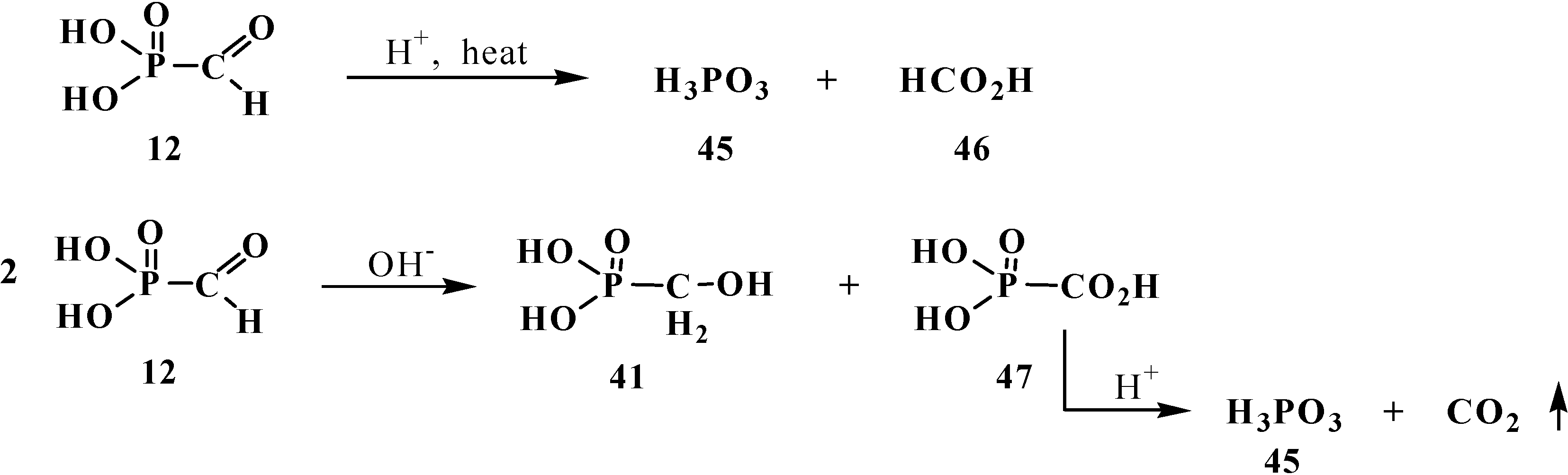
2.1. Syntheses and Chemical Properties of Formylphosphonic Acid (12)
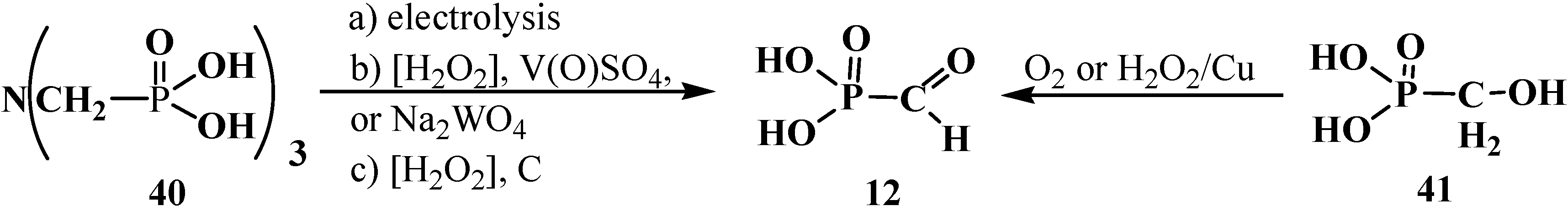
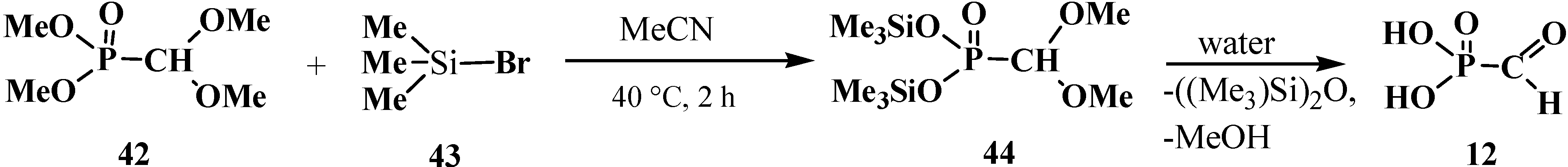
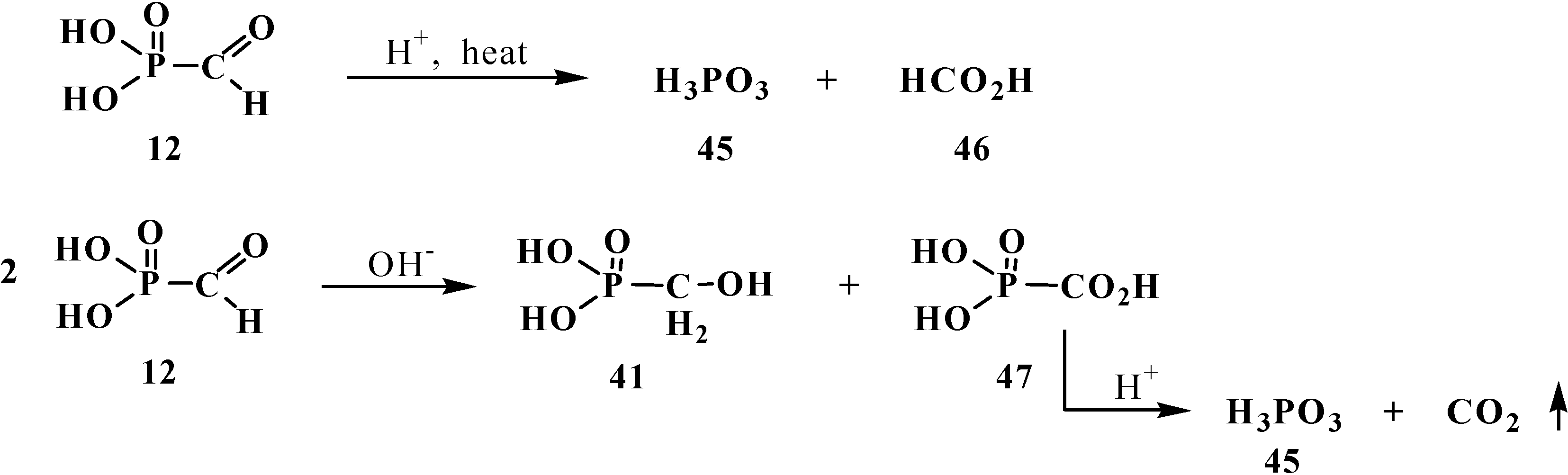
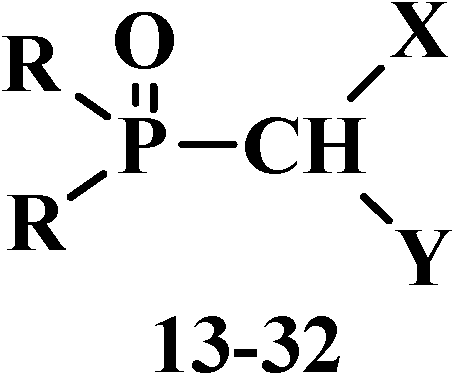
2.2. Chemistry of Phosphorylated Formaldehyde Acetals 13
2.2.1. Methods of Synthesis of Phosphorylated Formaldehyde Acetals 13
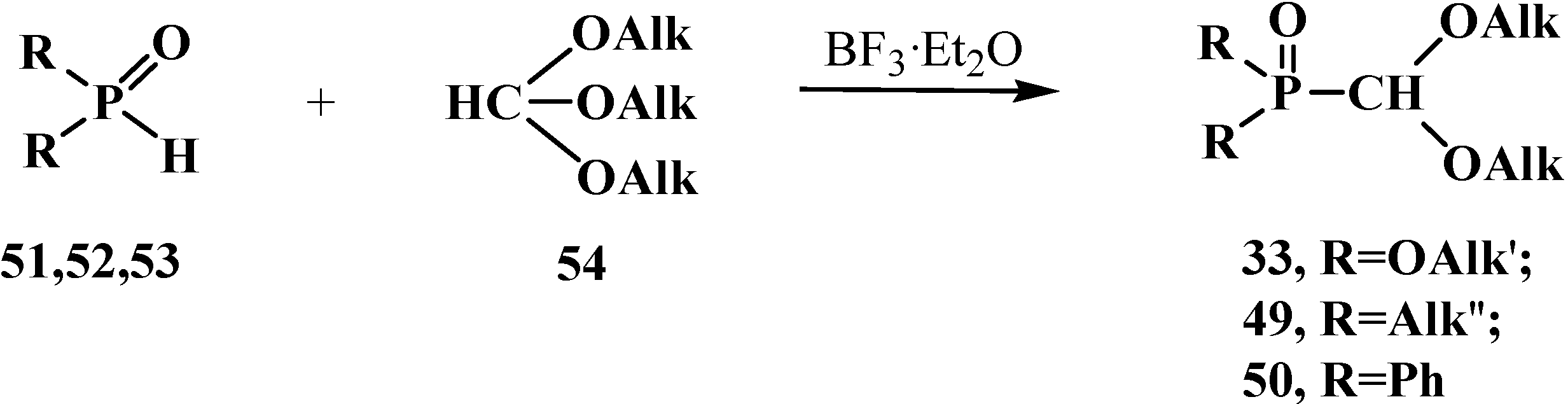
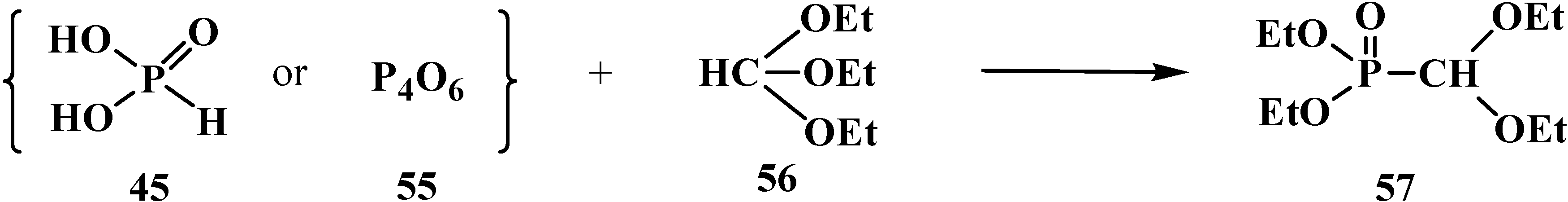
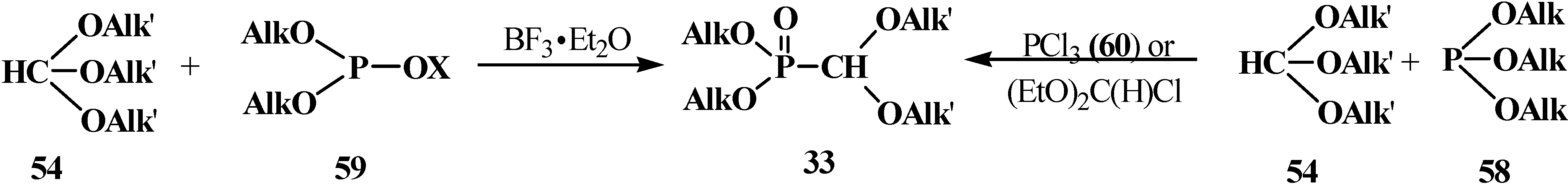
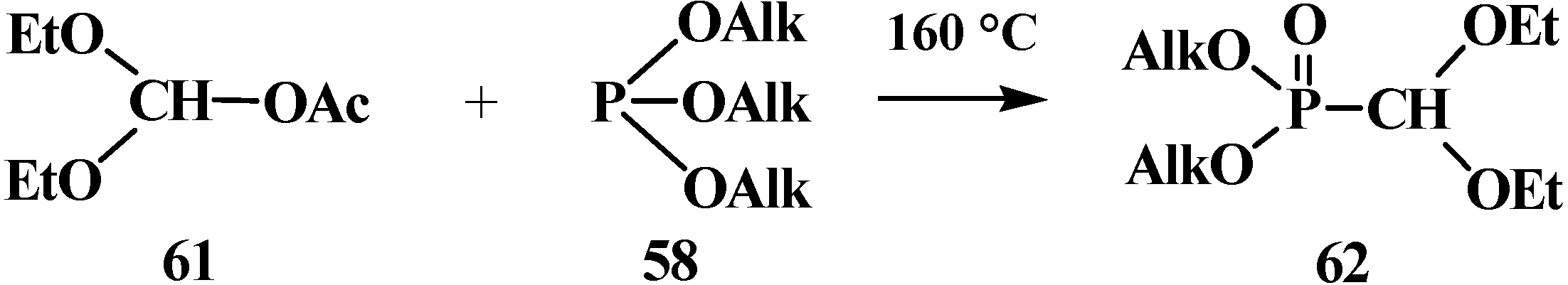
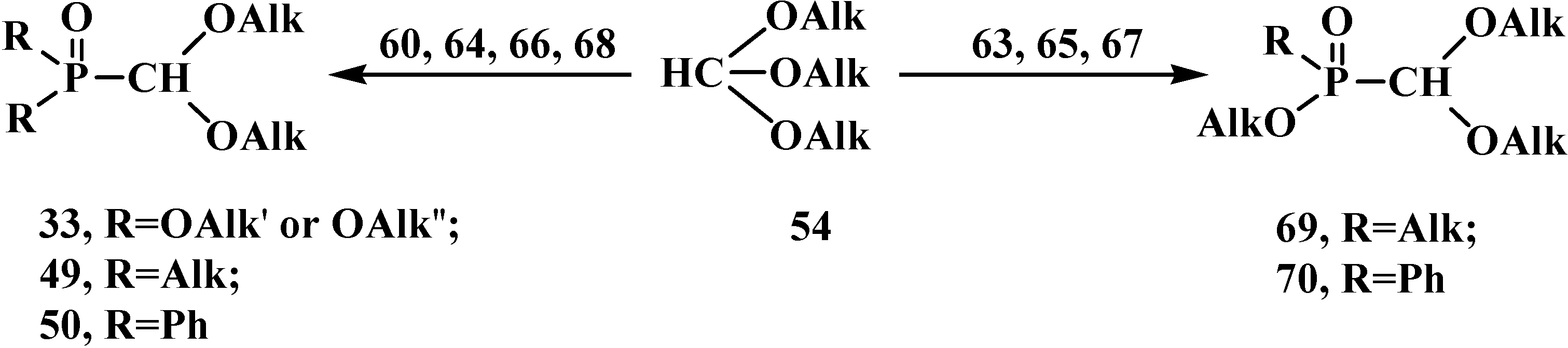
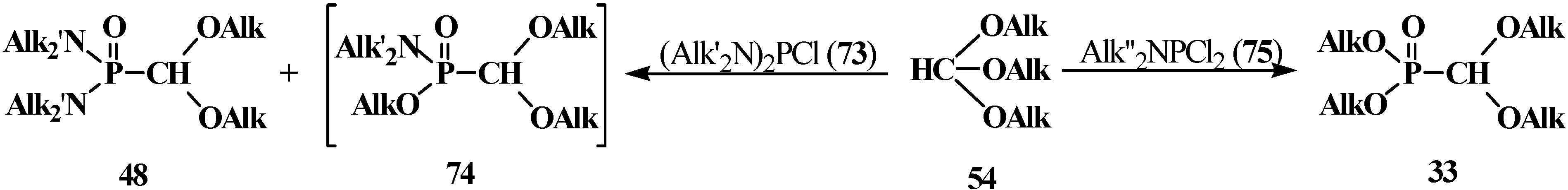
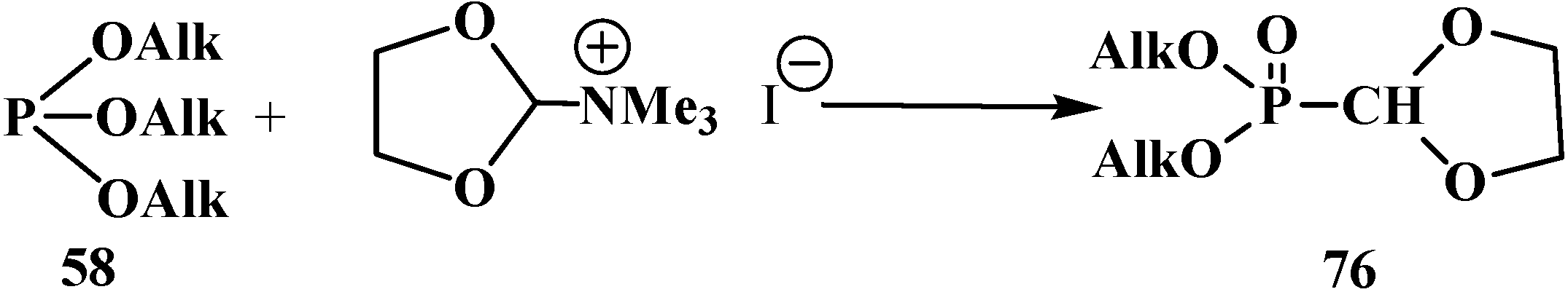
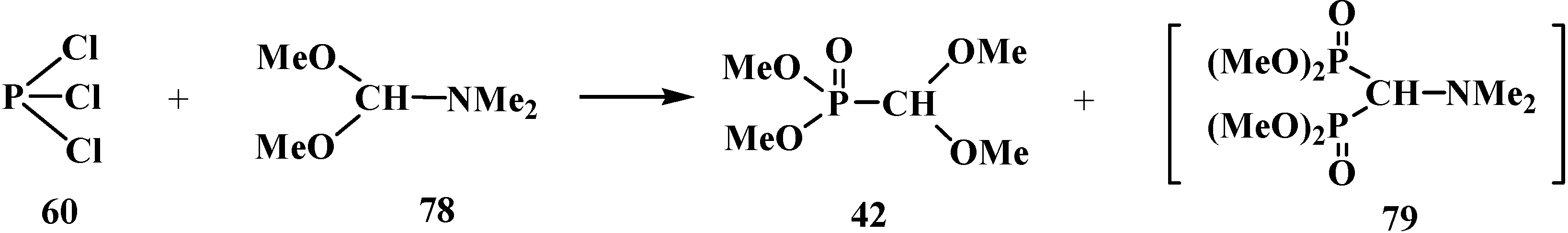
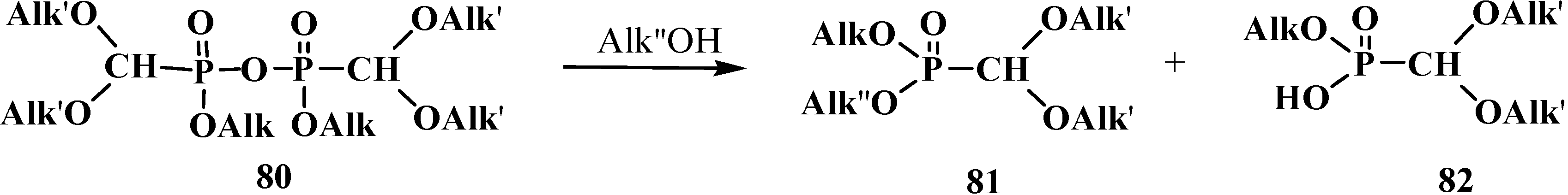
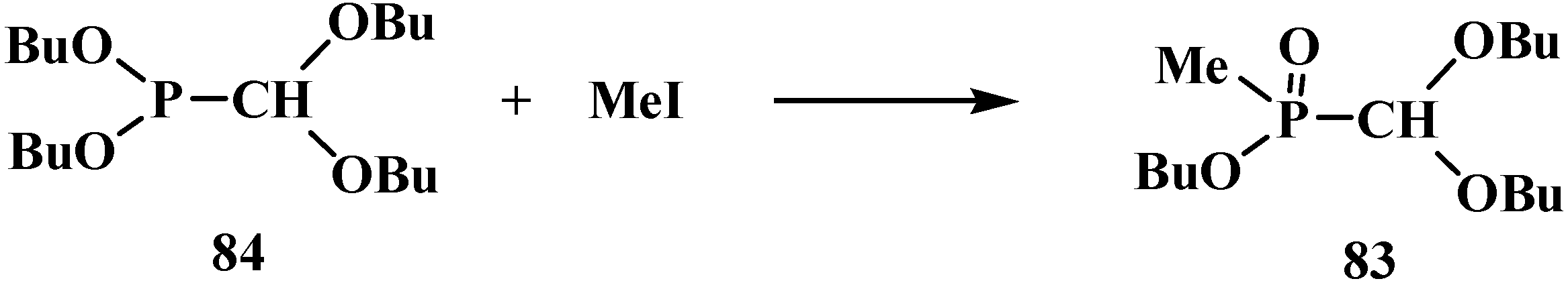

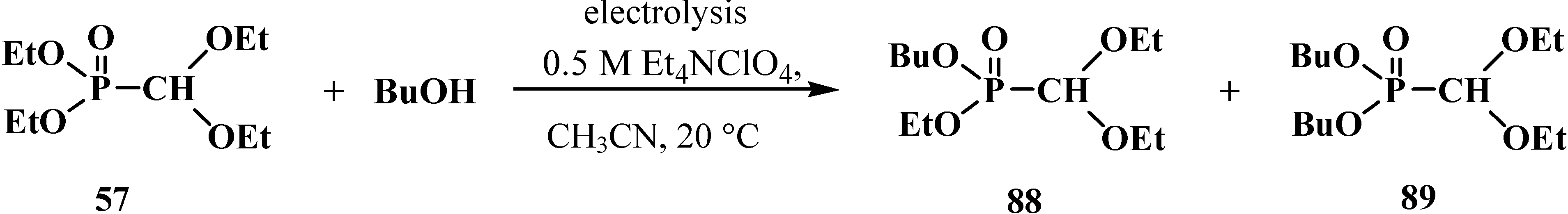
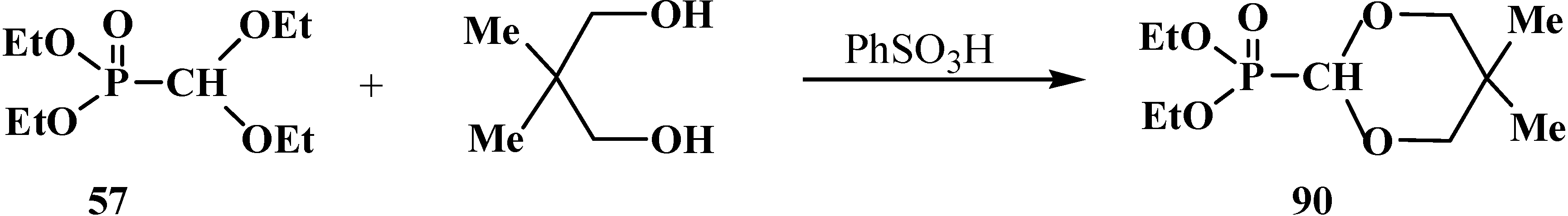
2.2.2. Chemical Properties of Phosphorylated Formaldehyde Acetals 13
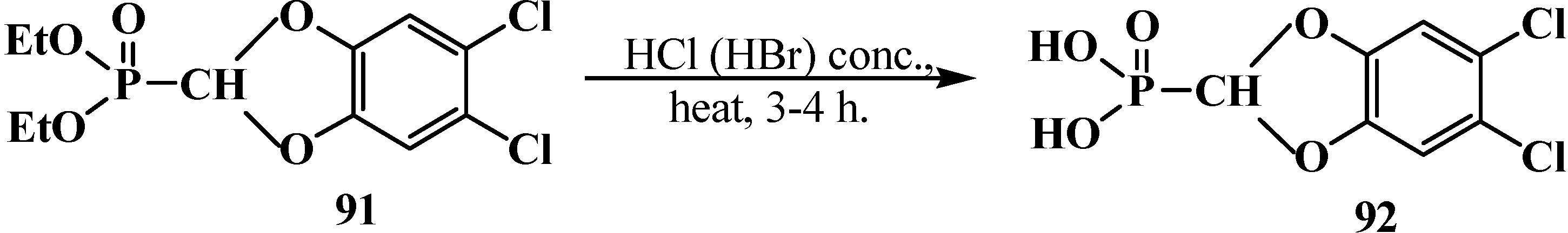
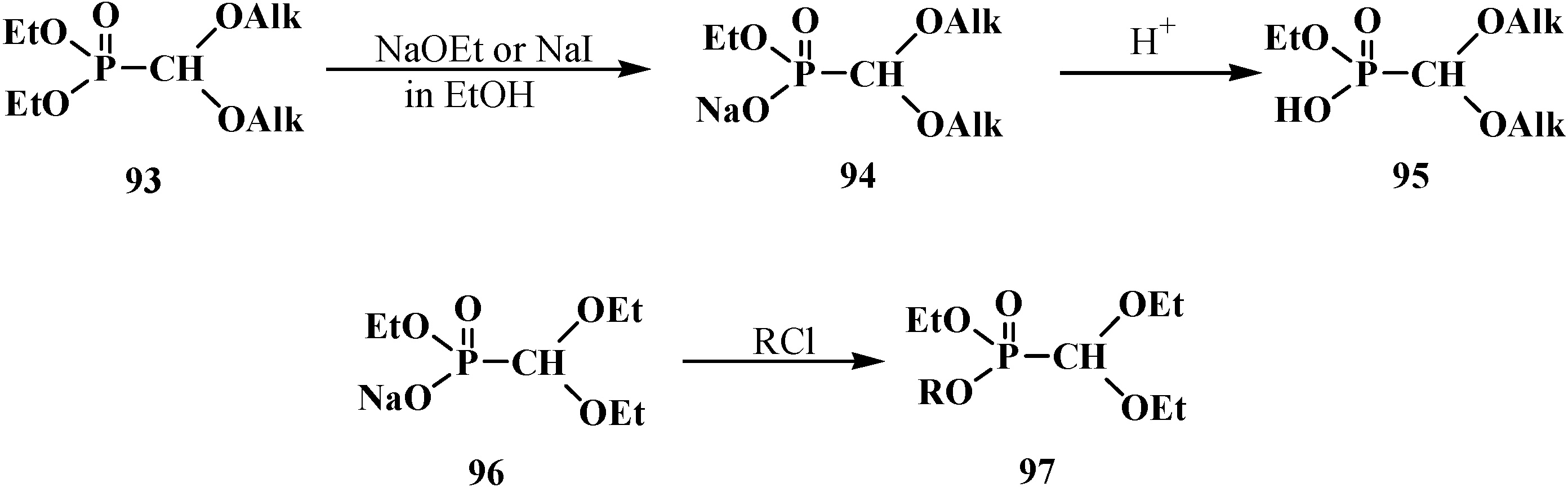
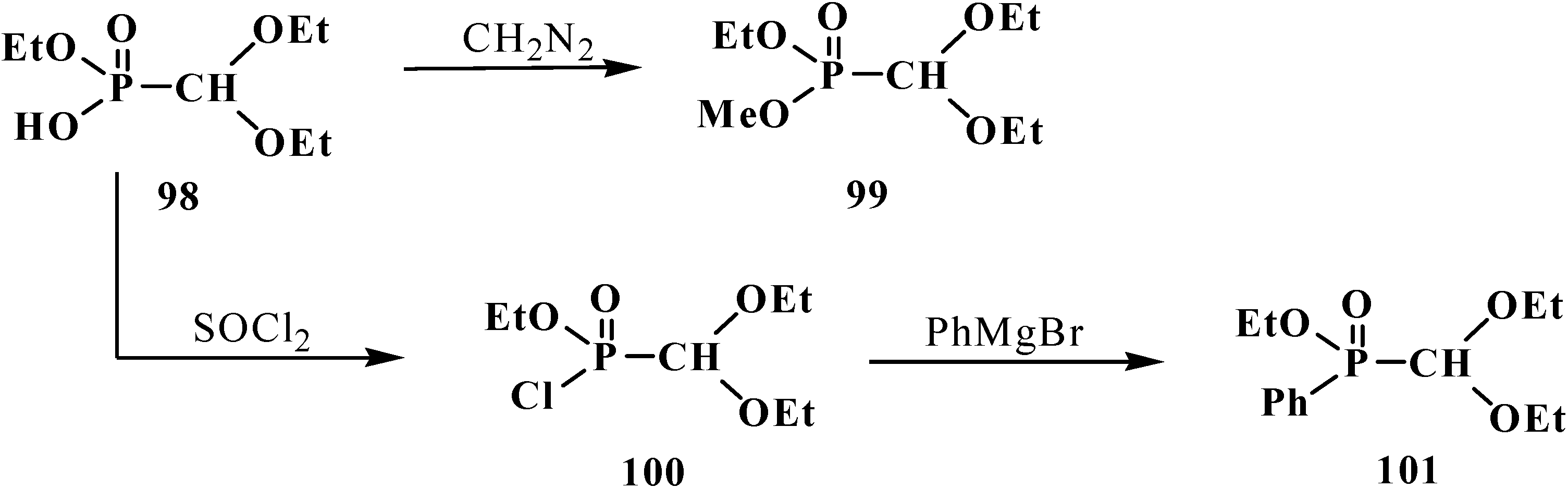
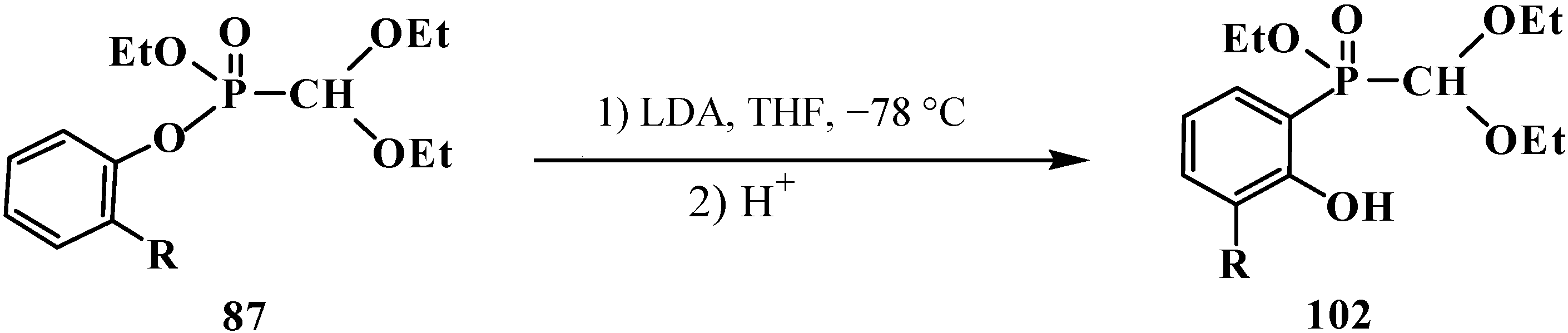
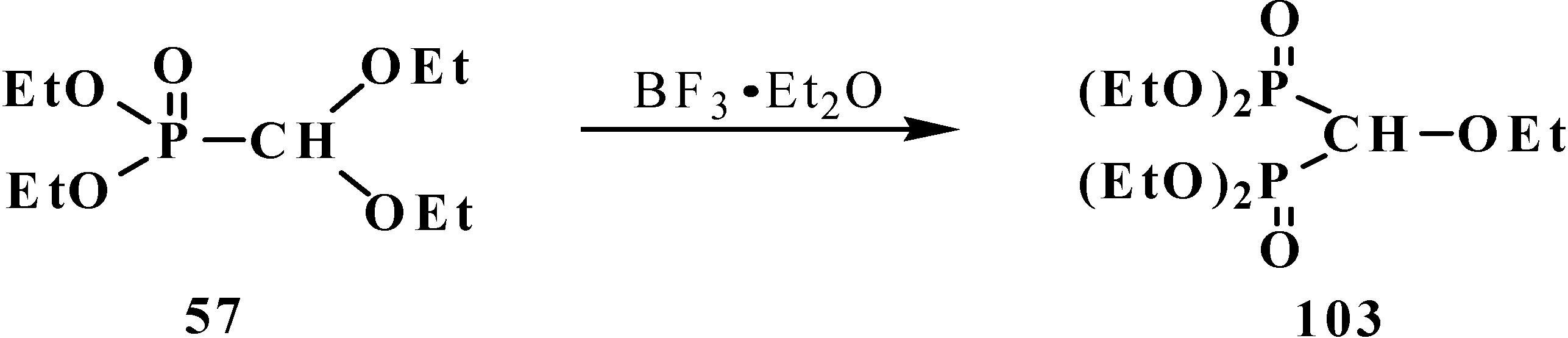
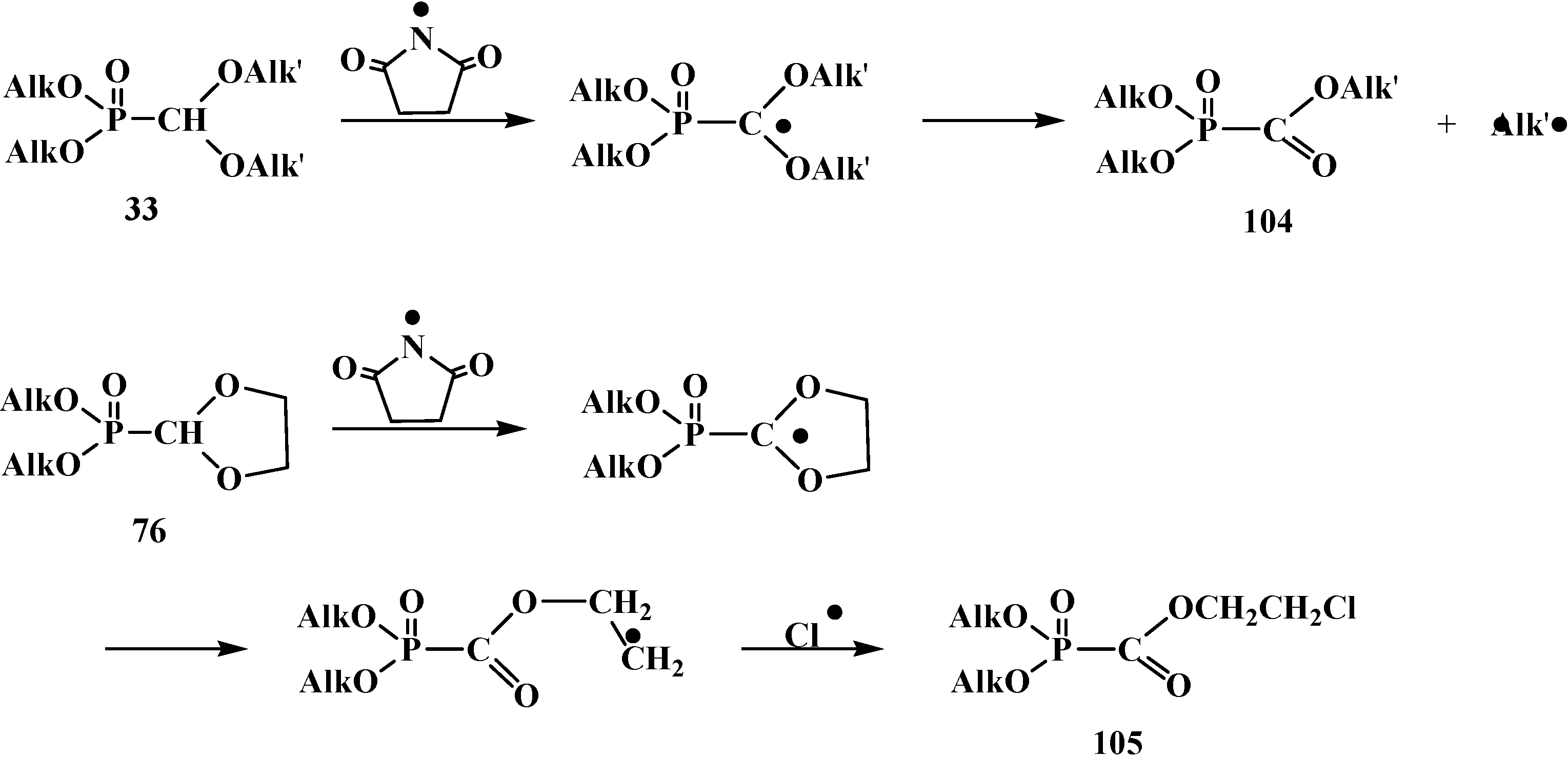
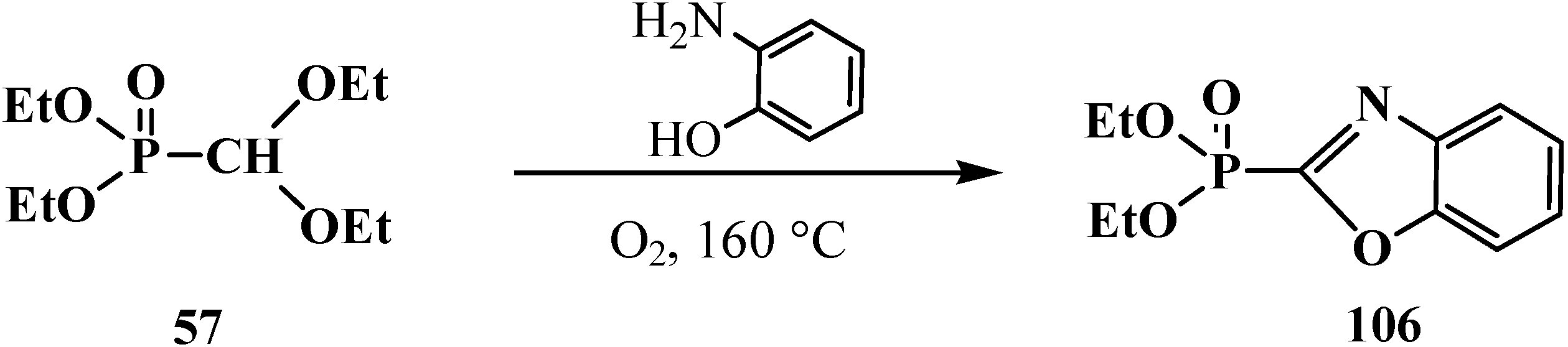
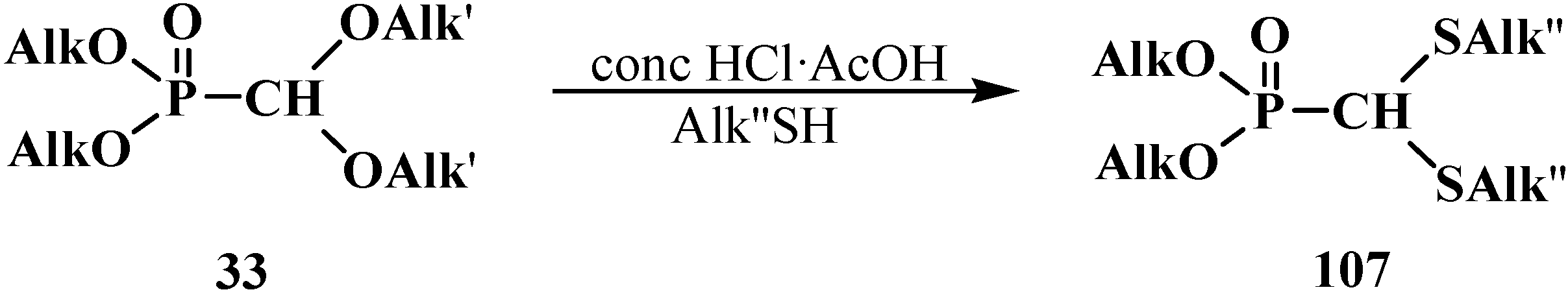

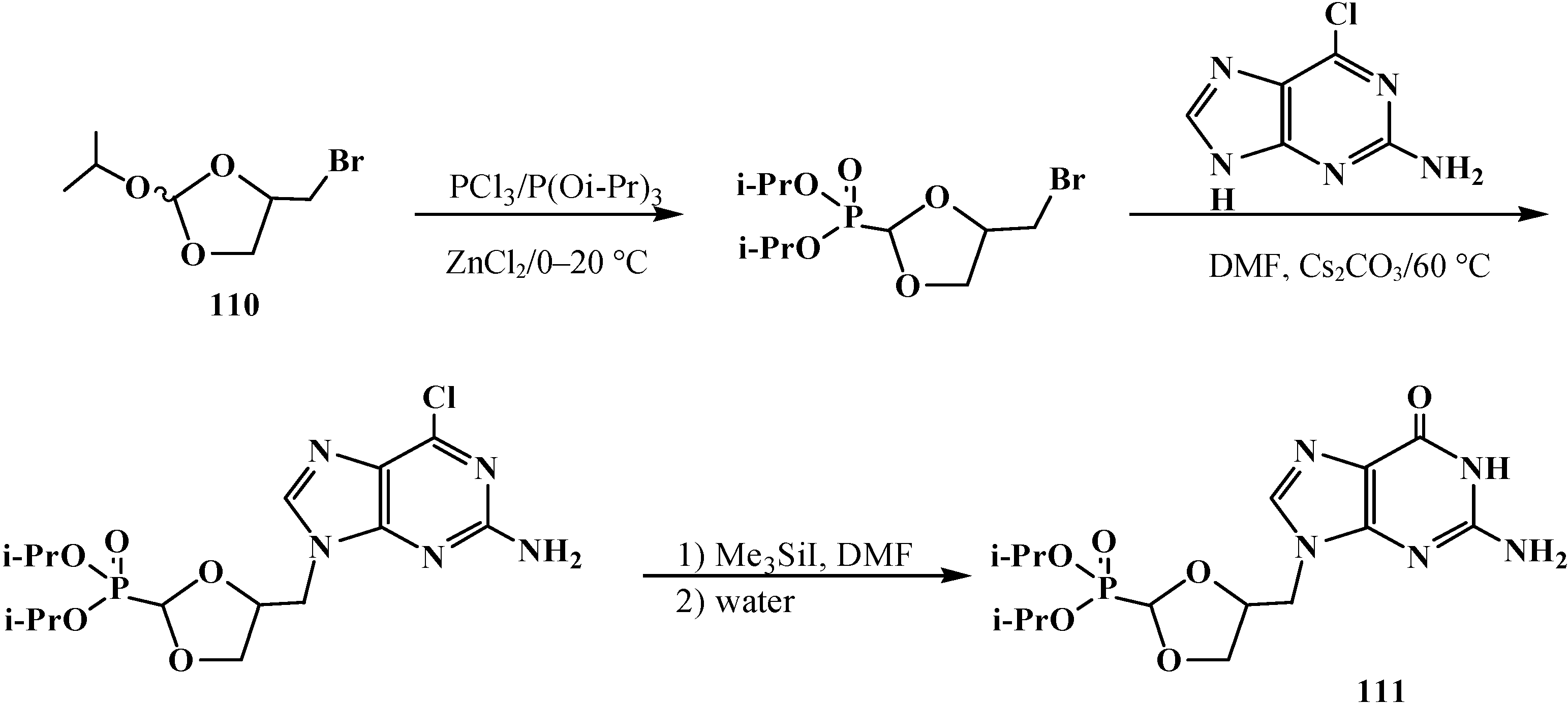
2.3. Phosphorylated Formaldehyde Thioacetals 14
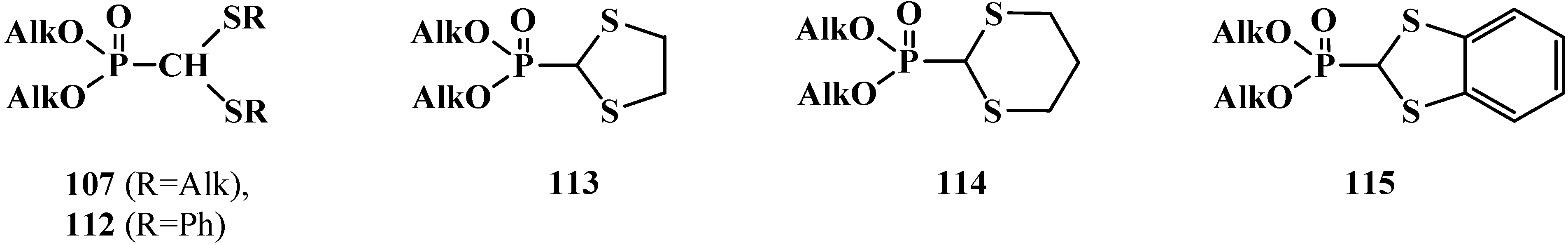
2.3.1. Methods of Synthesis of Phosphorylated Formaldehyde Thioacetals 14
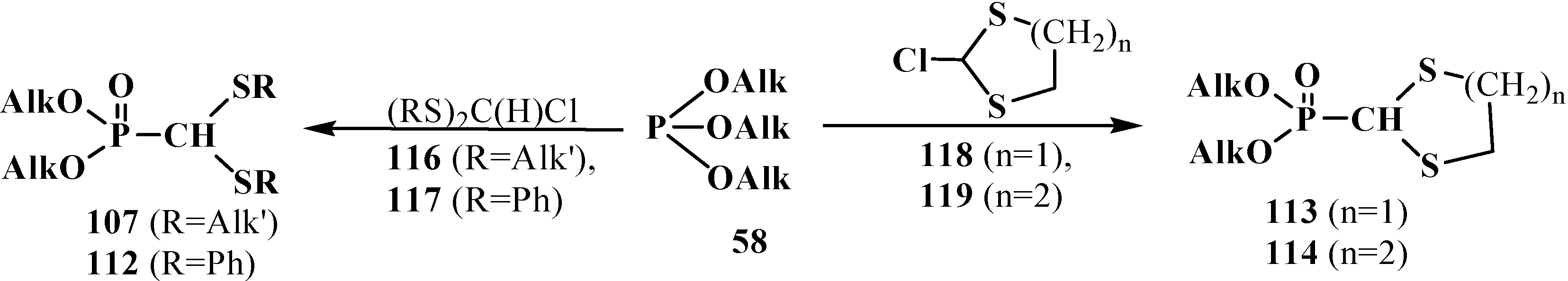
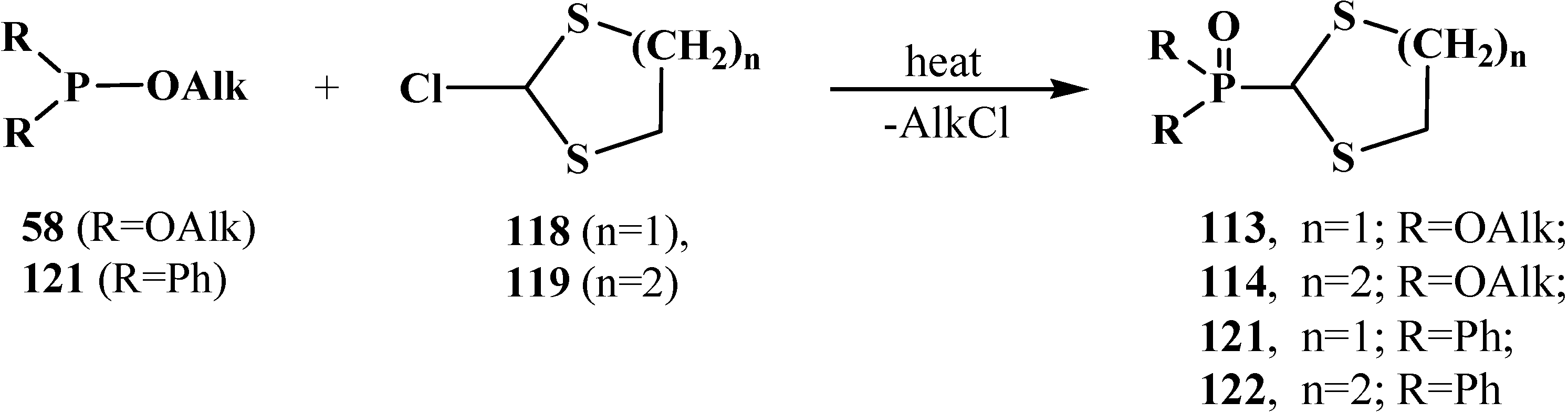
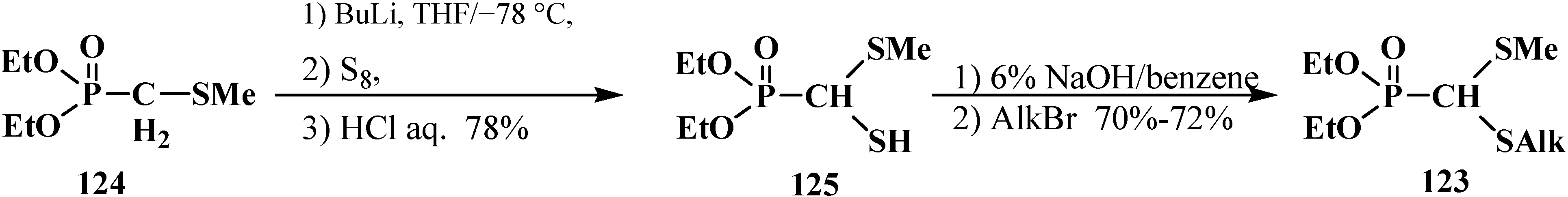
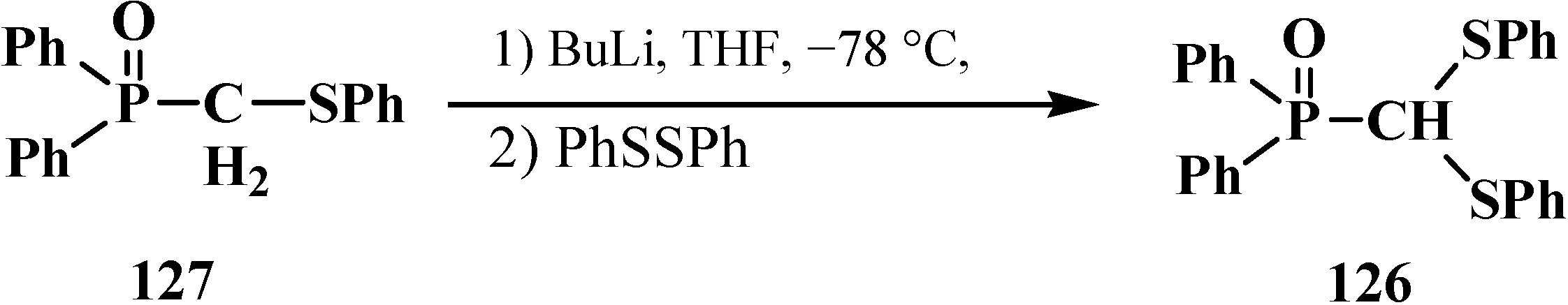
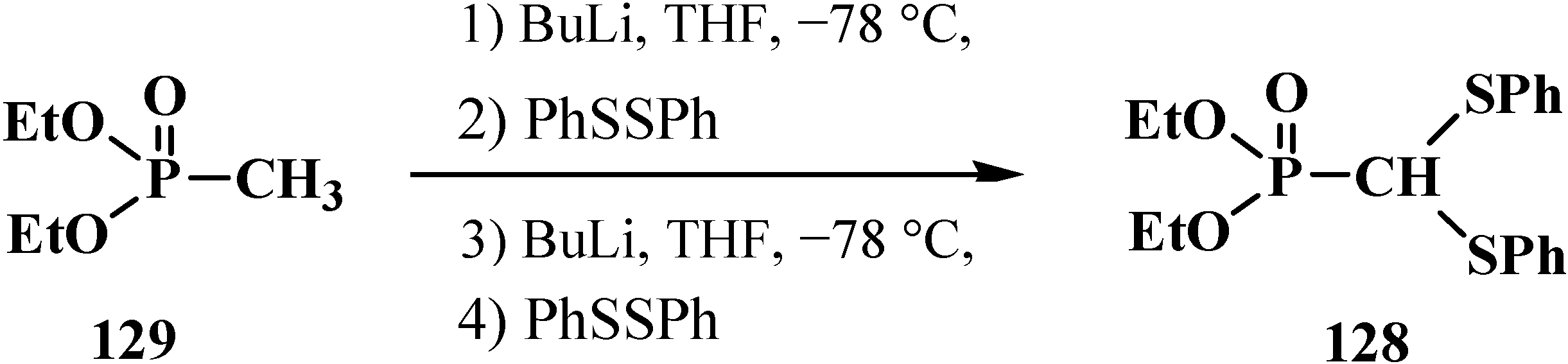

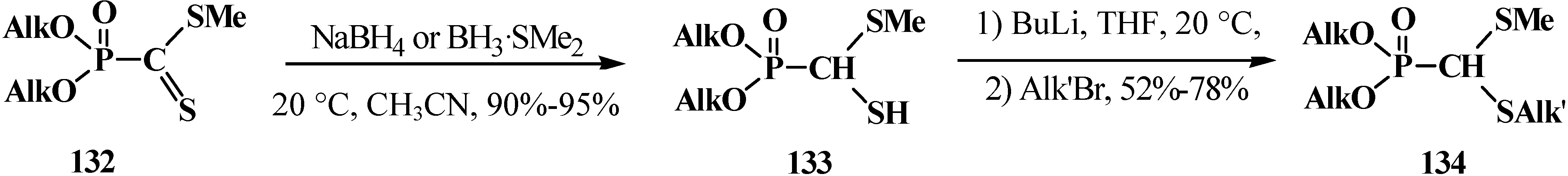
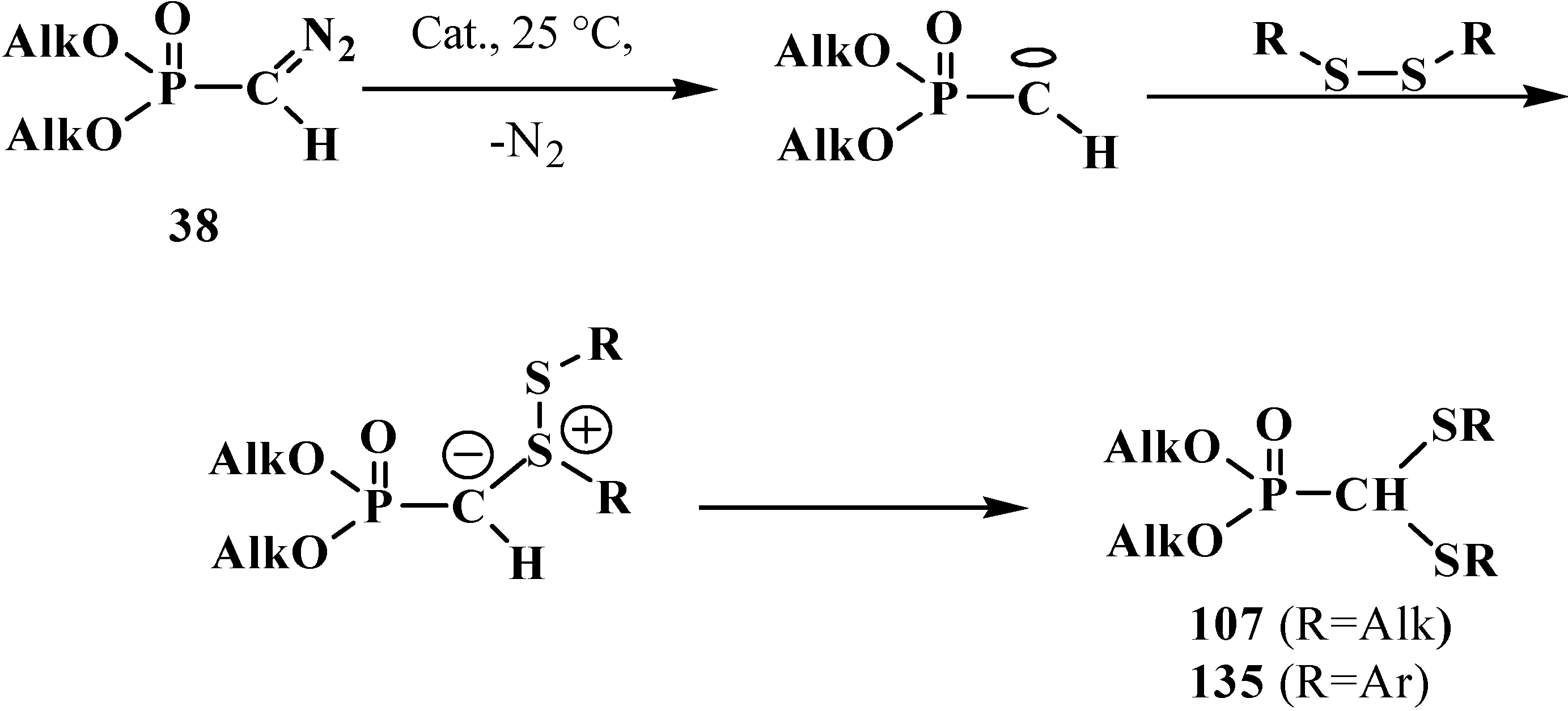
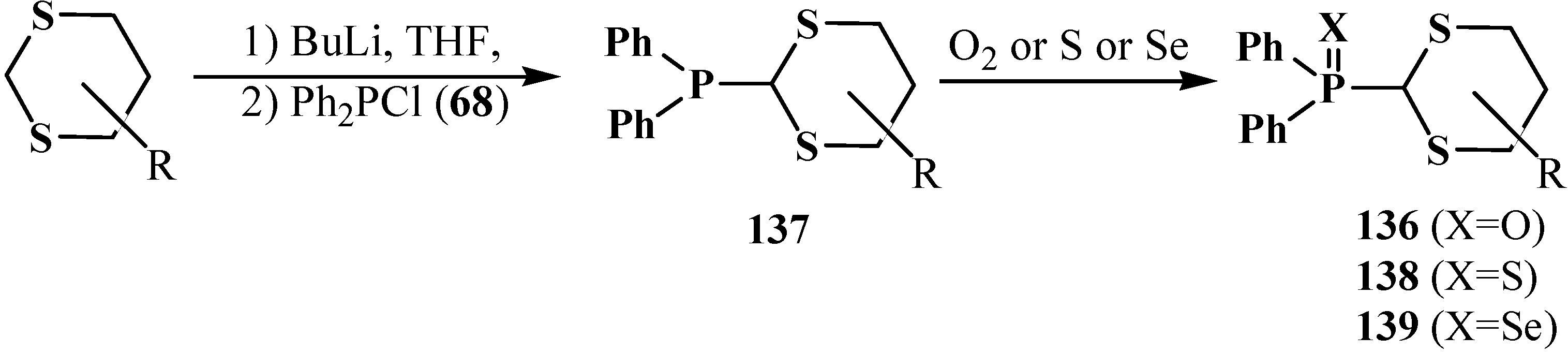
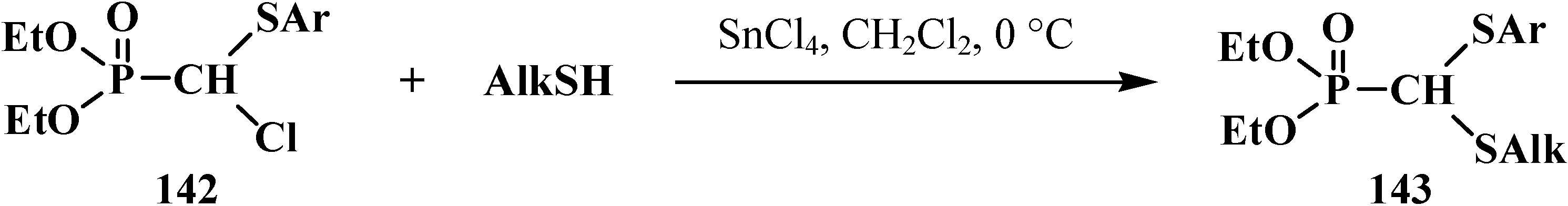
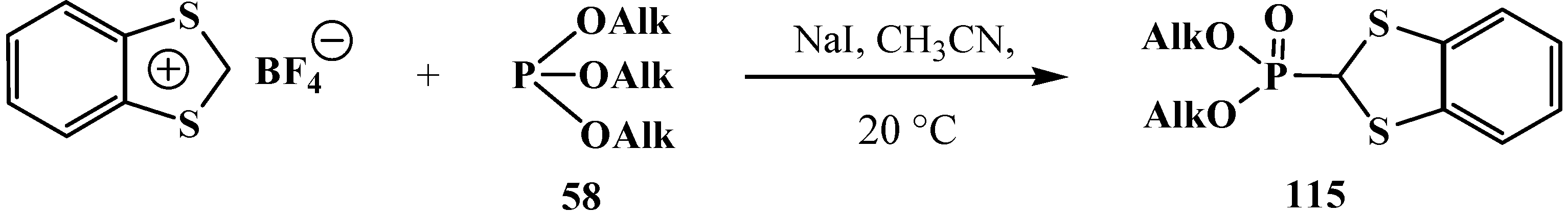
2.3.2. Chemical Properties of Phosphorylated Formaldehyde Thioacetals 14
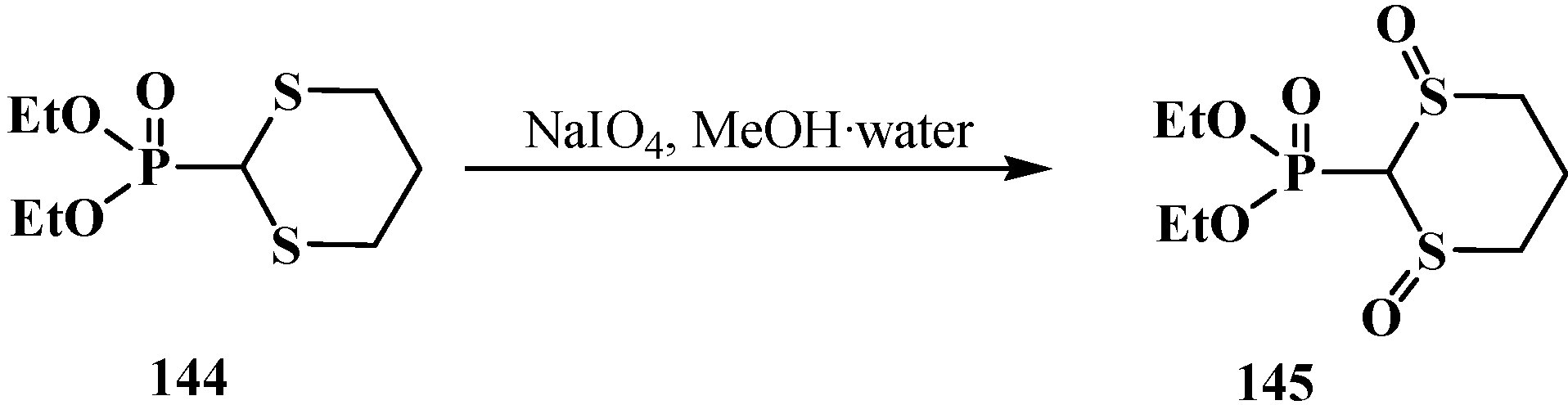
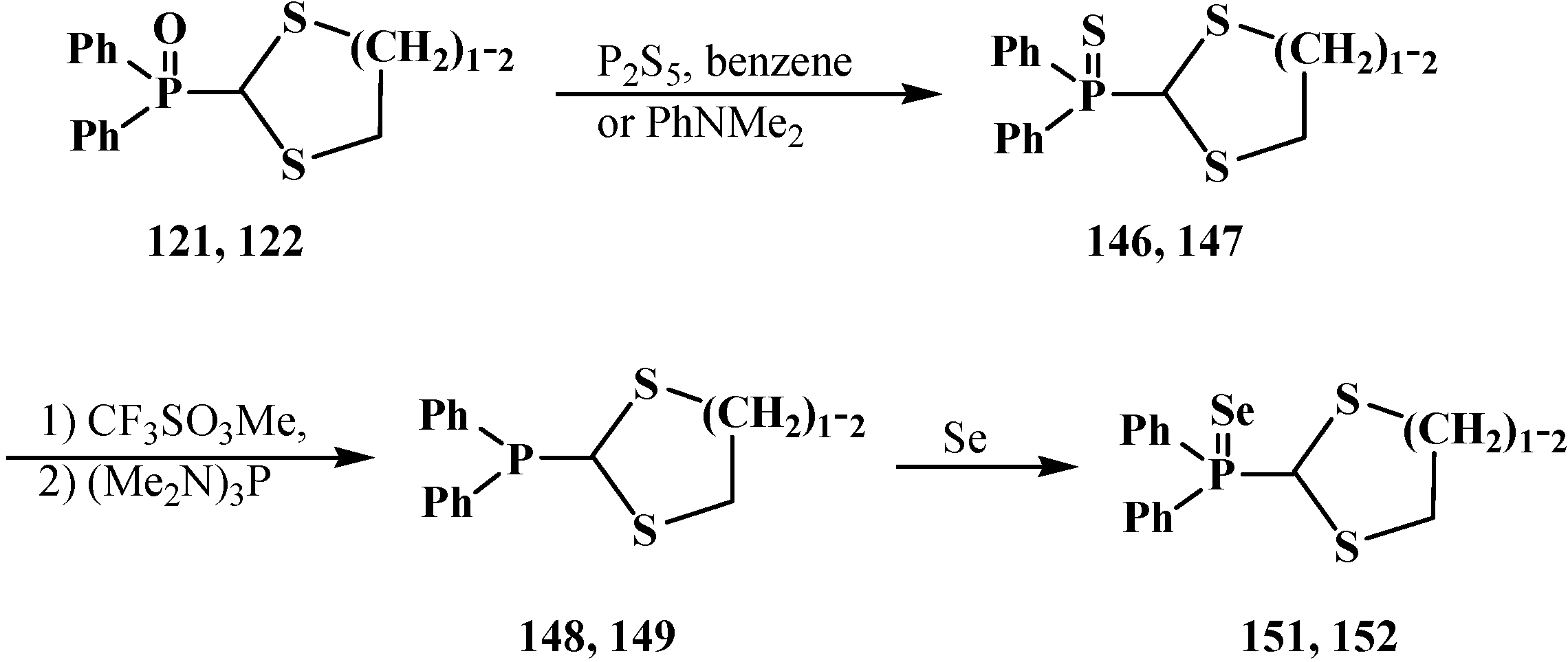
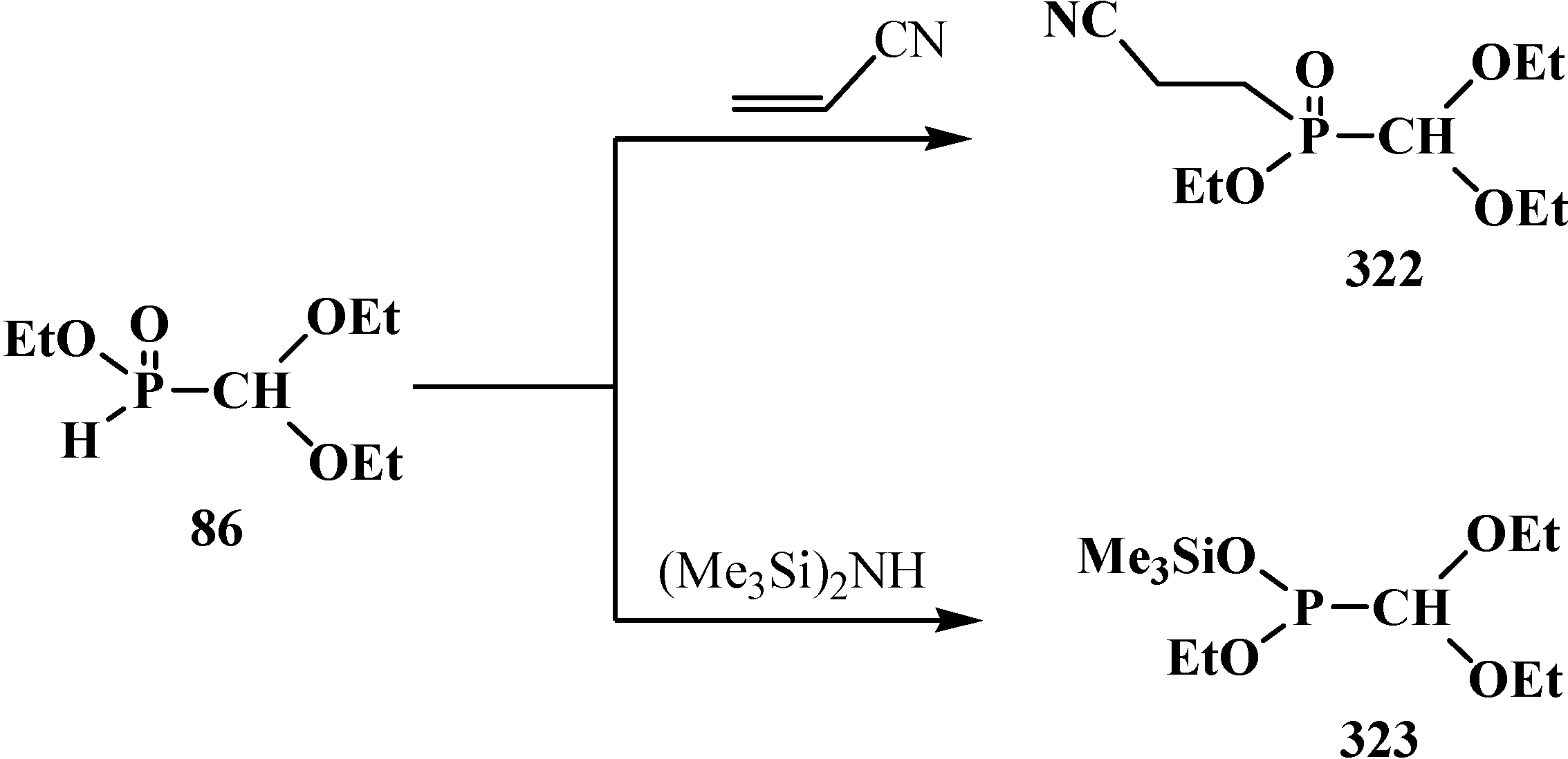
2.4. (N,N-dialkylamino)cyanomethyl Derivatives of Phosphorylated Formaldehyde (α-dialkylamino-nitriles) 15
2.4.1. Methods of Synthesis of Diethyl [(N,N-Dialkylamino)cyanomethyl]phosphonates 15
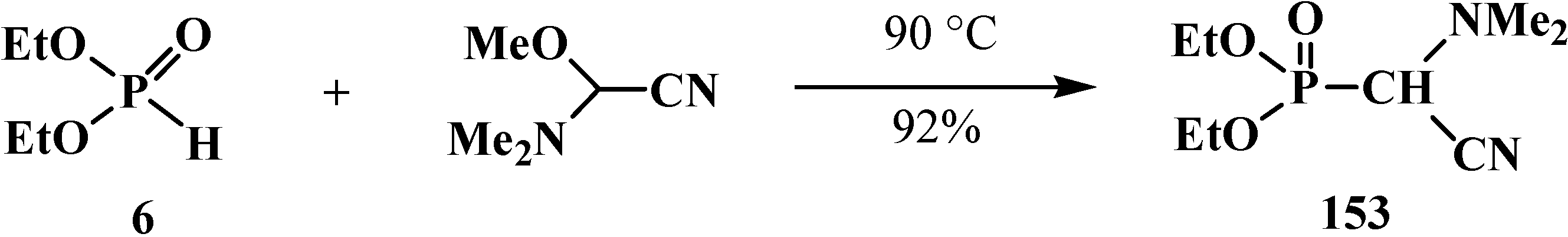

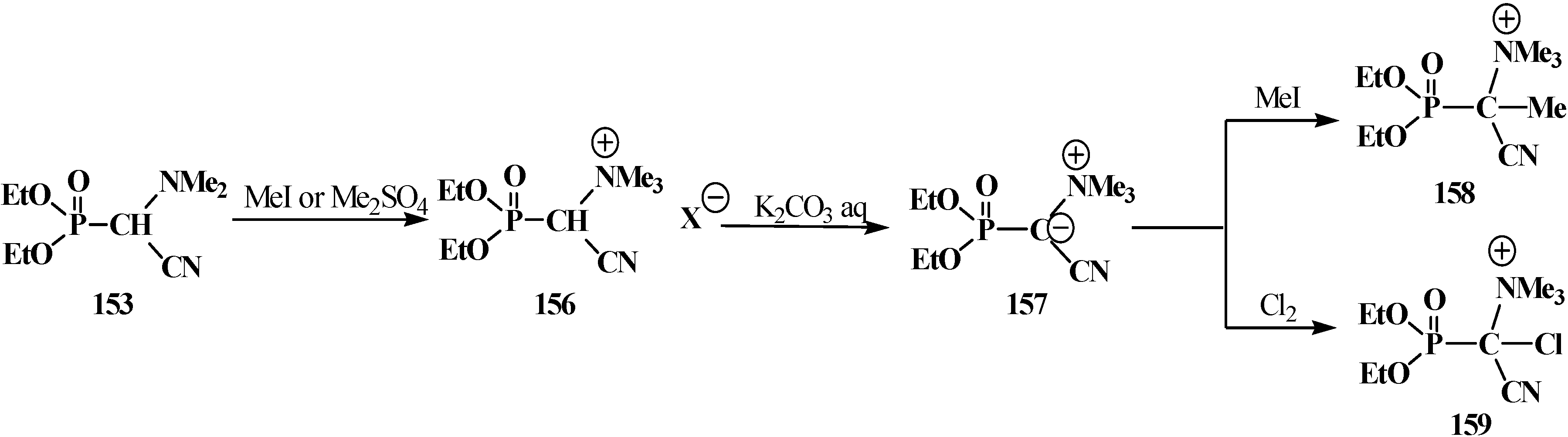
2.4.2. Chemical Properties of Diethyl [(N,N-Dialkylamino)cyanomethyl]phosphonates 15
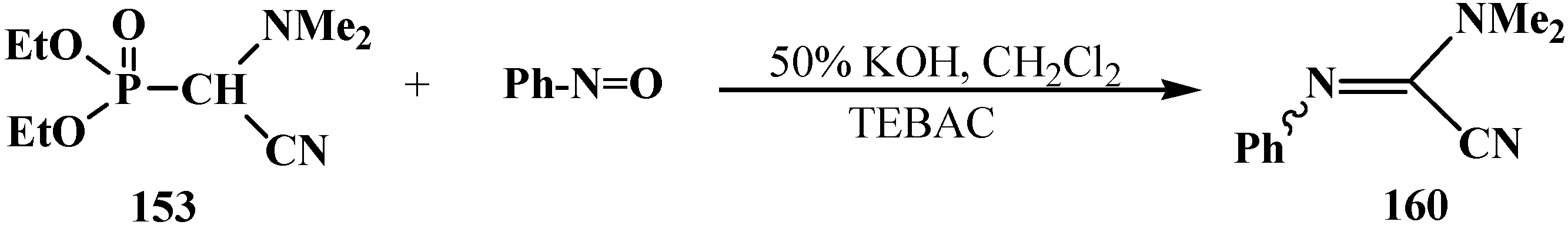
2.5. Diphosphinoyl N,N-Dialkylaminomethanes 16
2.5.1. Methods of Synthesis of Diphosphinoyl N,N-Dialkylaminomethanes 16
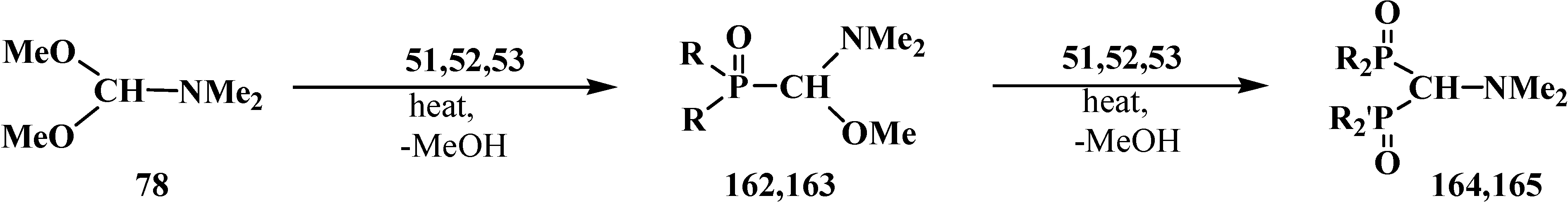
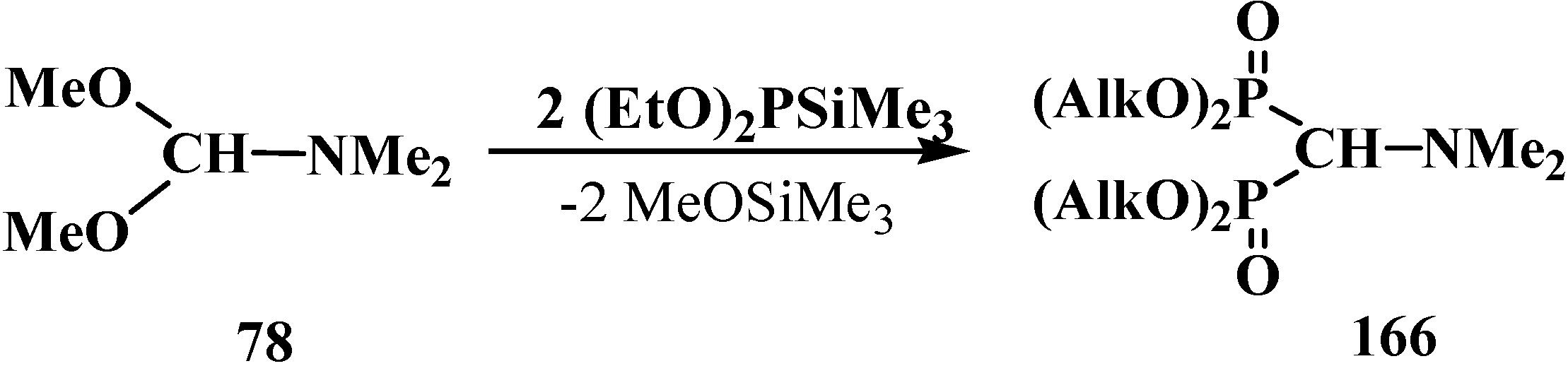
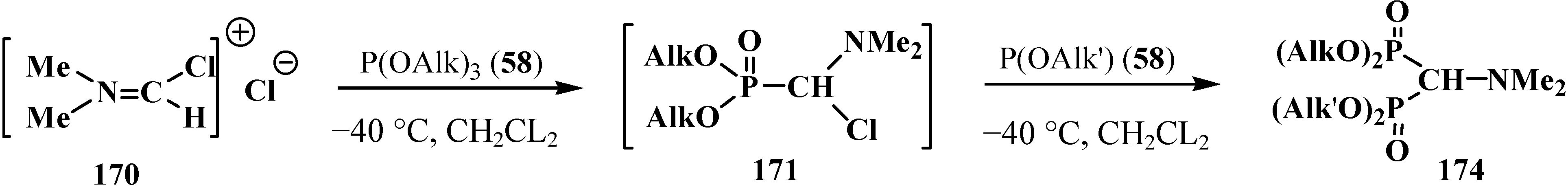
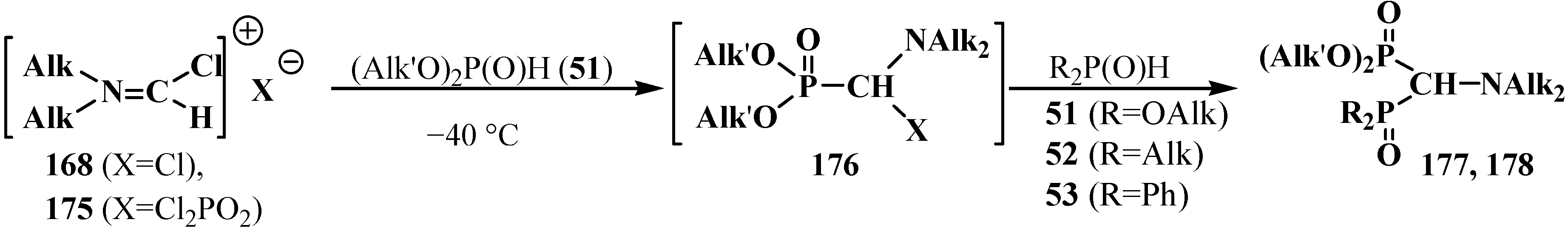
2.5.2. Chemical Properties of Diphosphinoyl N,N-Dialkylaminomethanes 16
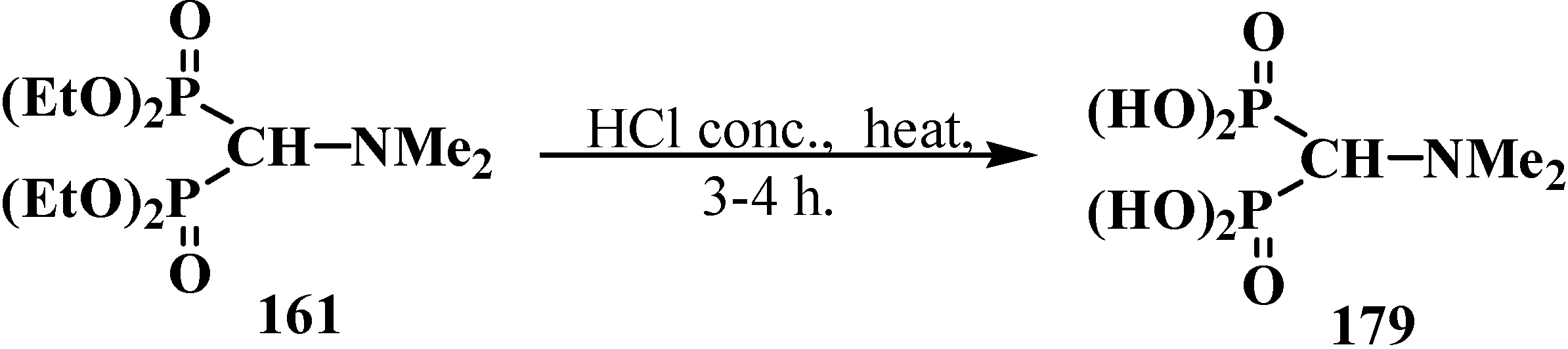
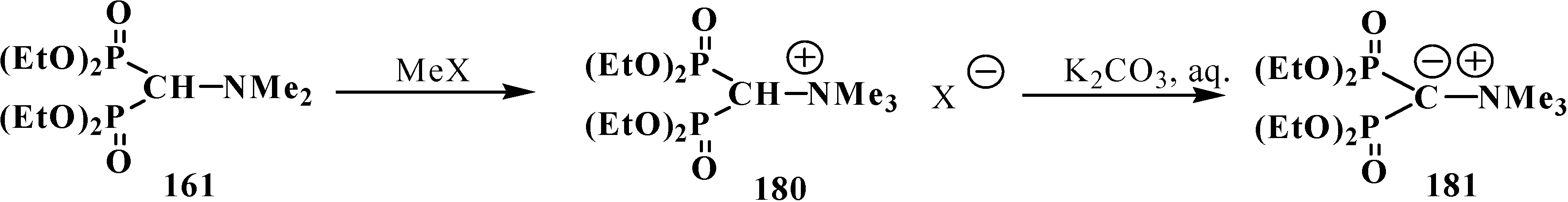
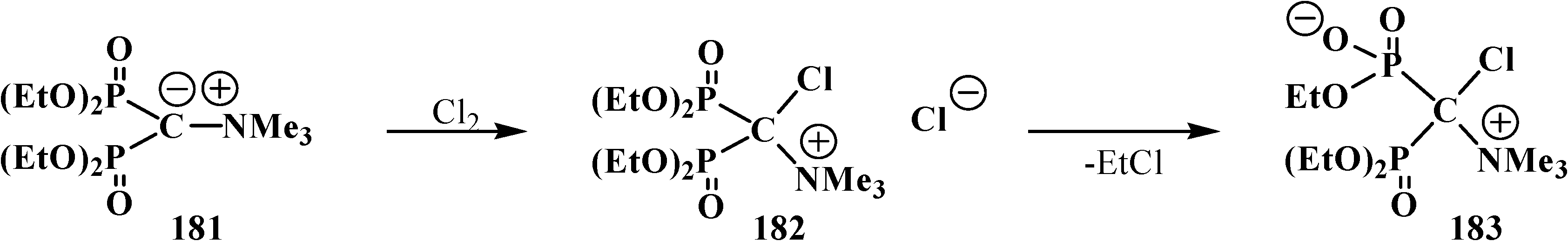
3. General Chemical Properties of Phosphorylated Formaldehyde Acetals 13 and Structurally Related Compounds 14–16
3.1. Phosphorus–Carbon Bond Cleavage
3.1.1. Cleavage of Phosphorus–Carbon Bond under the Action of Acids and Acidic Reagents
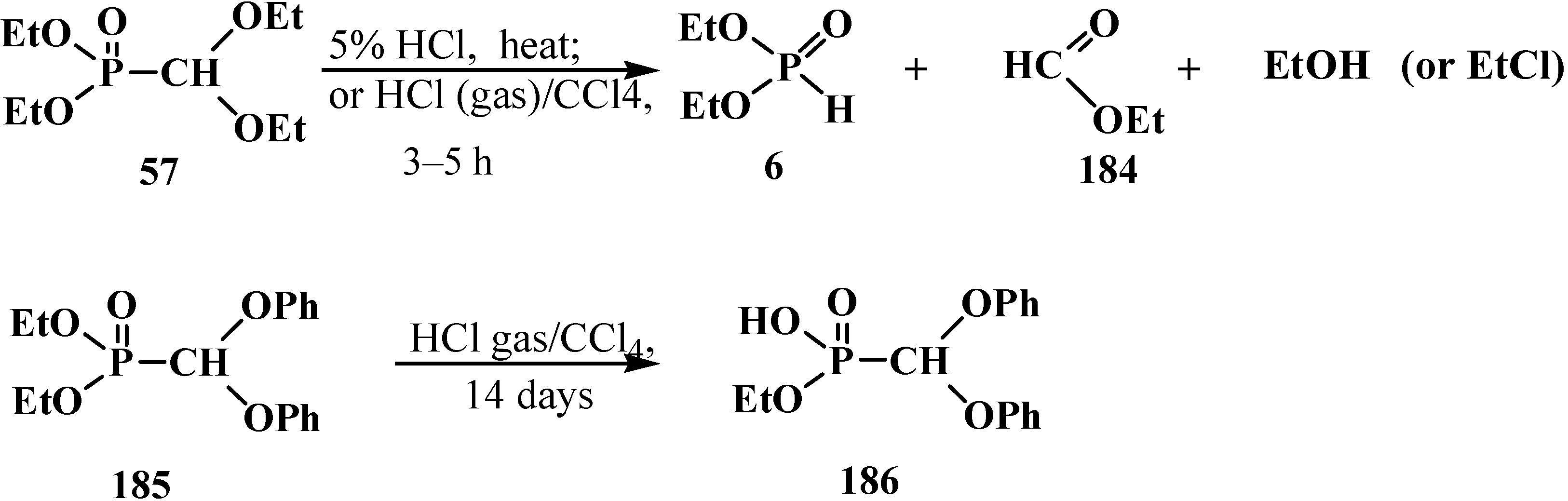
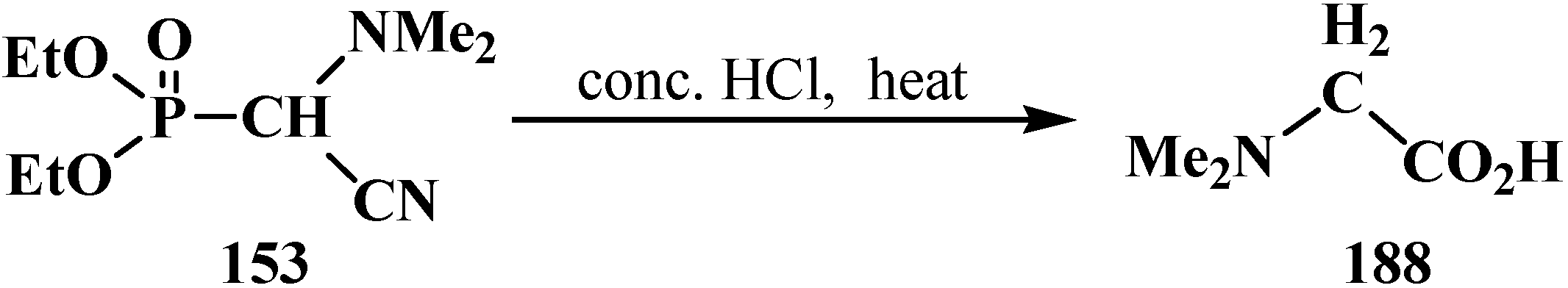
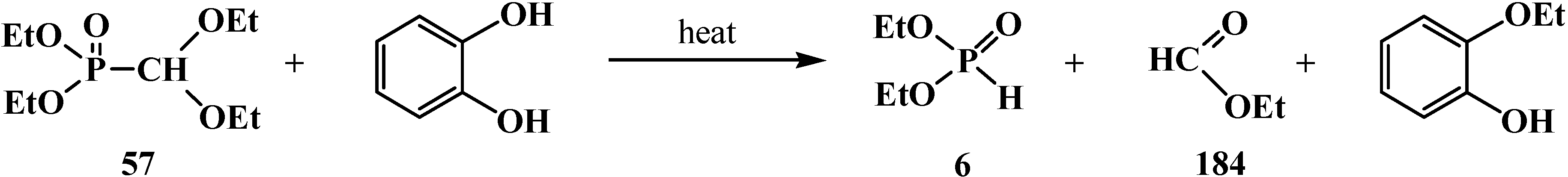
3.1.2. The Cleavage of Phosphorus–Carbon Bond in Reactions with Organic Coreactants
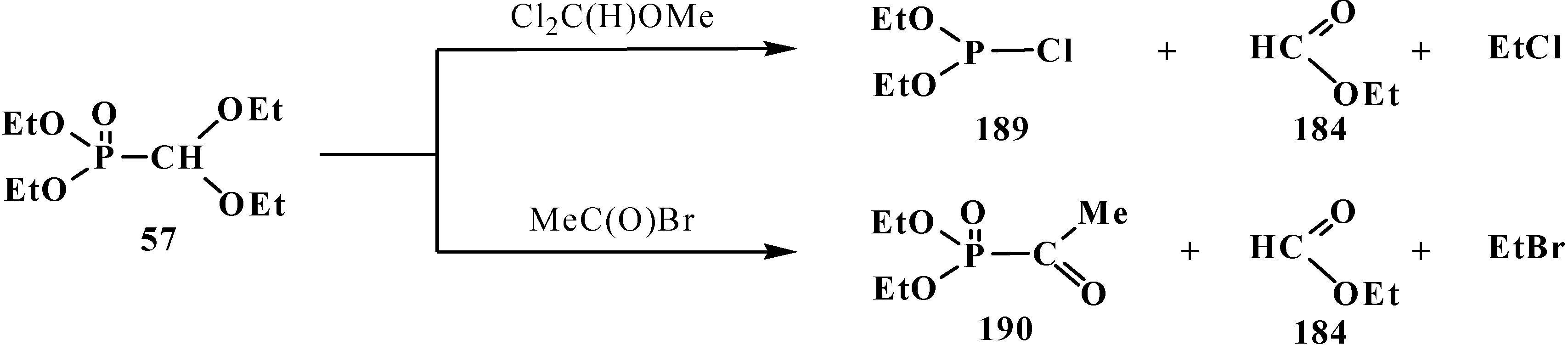
3.1.3. Phosphorus–Carbon Bond Cleavage under the Action of Bases
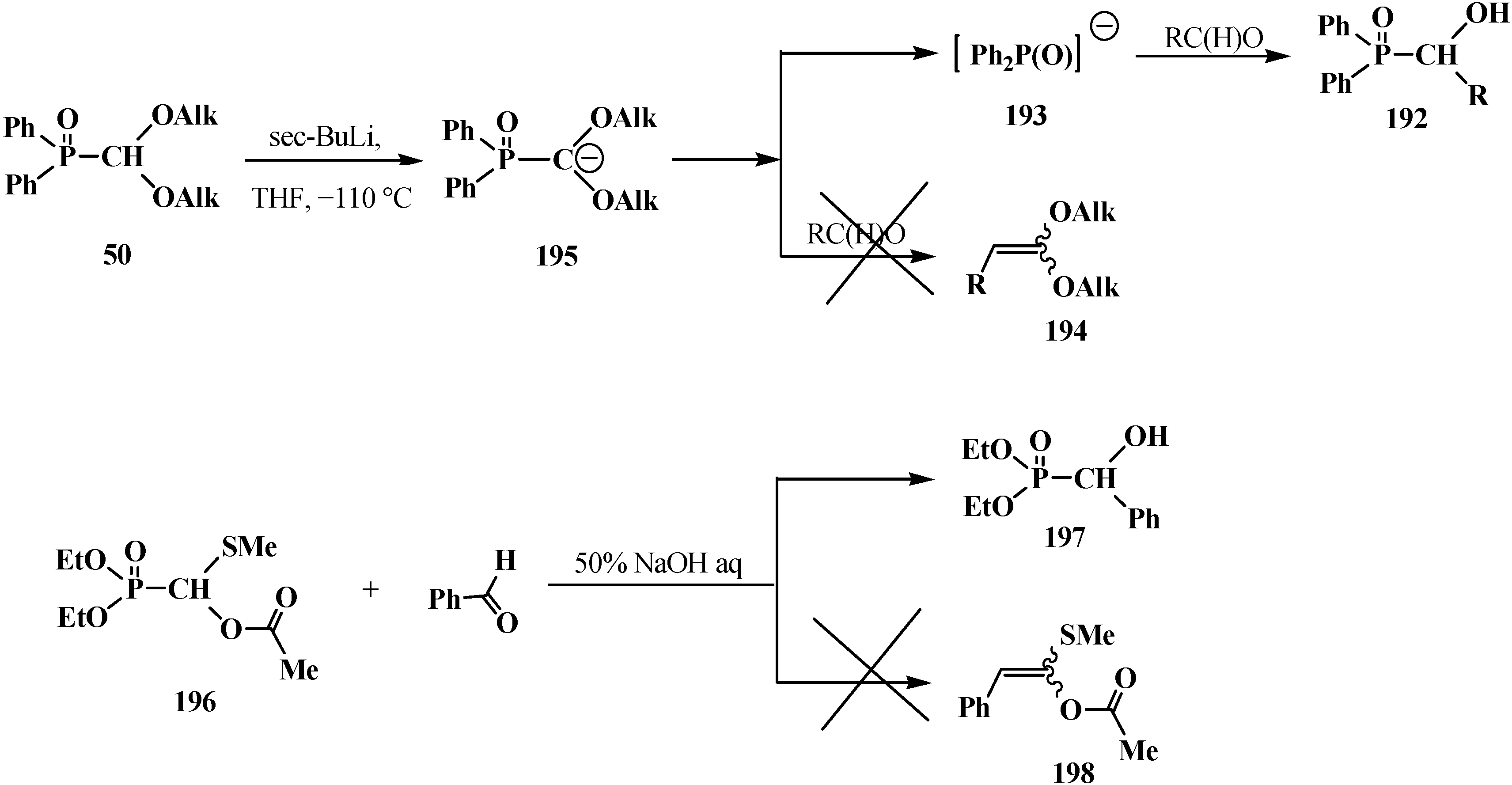
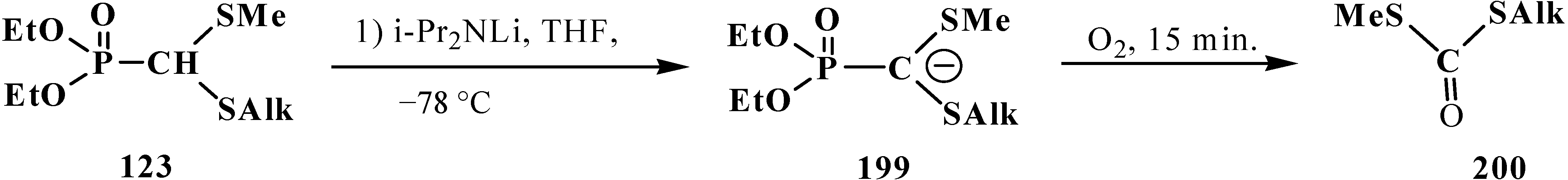
3.2. Synthesis of Formacetalphosphonic Acids
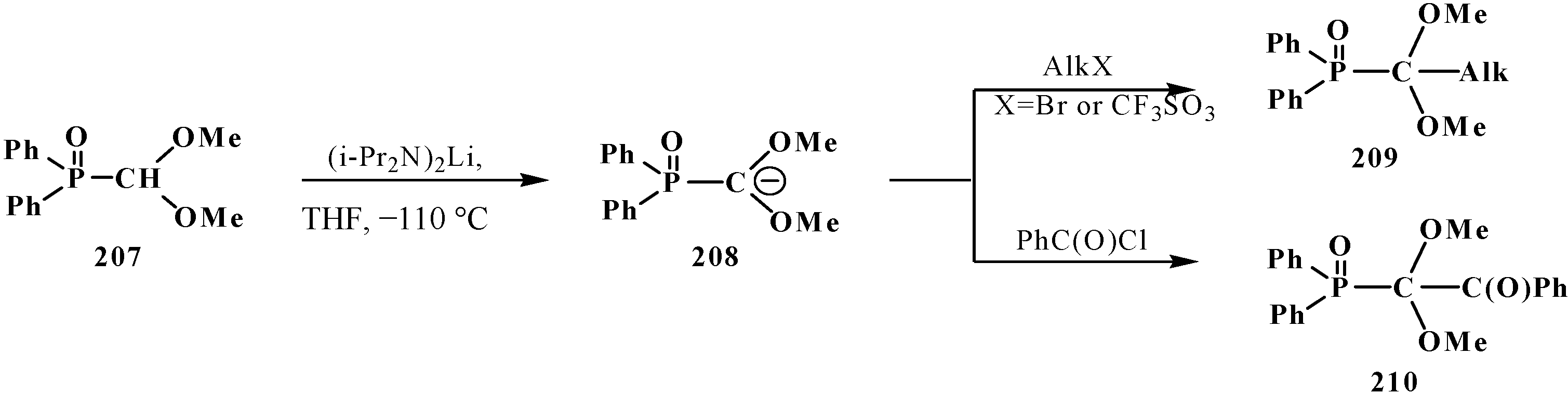
3.3. Alkylation (Acylation) of the Formacetal Carbon Atom
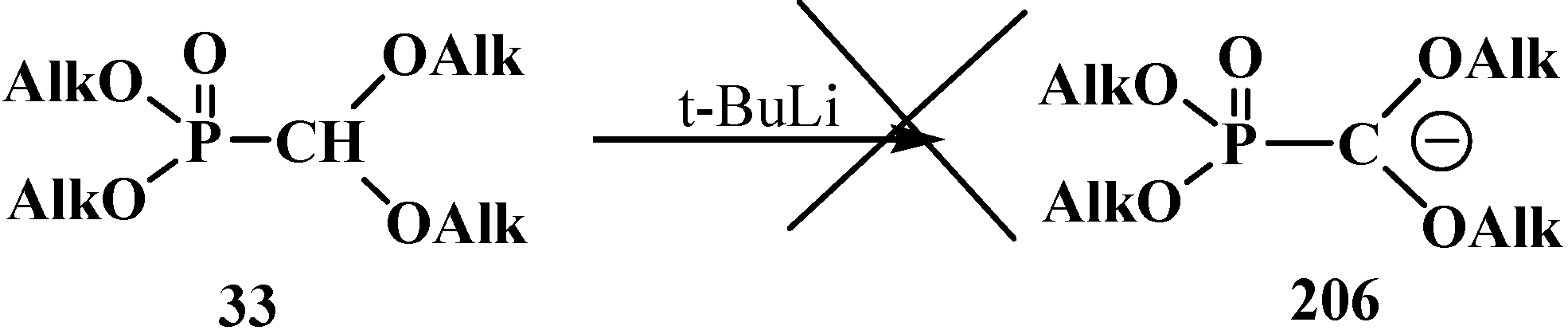
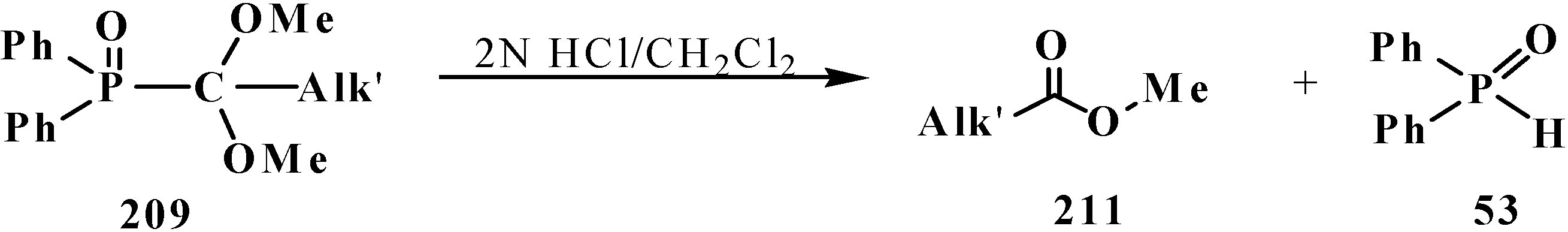
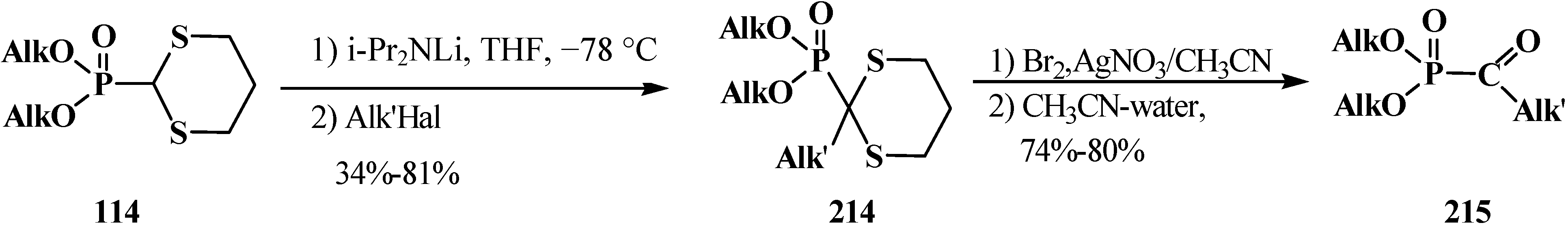
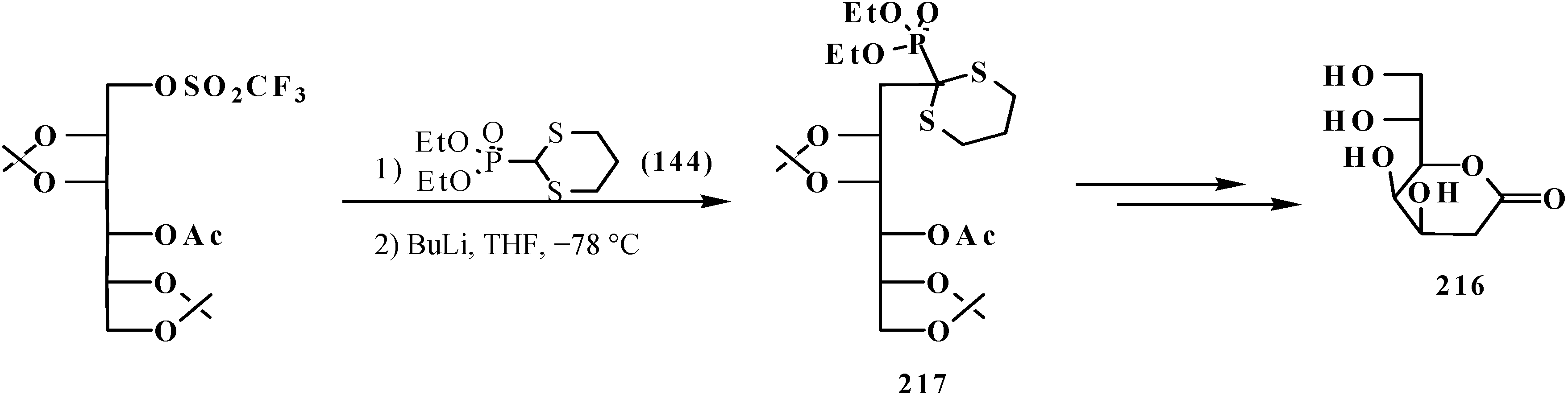
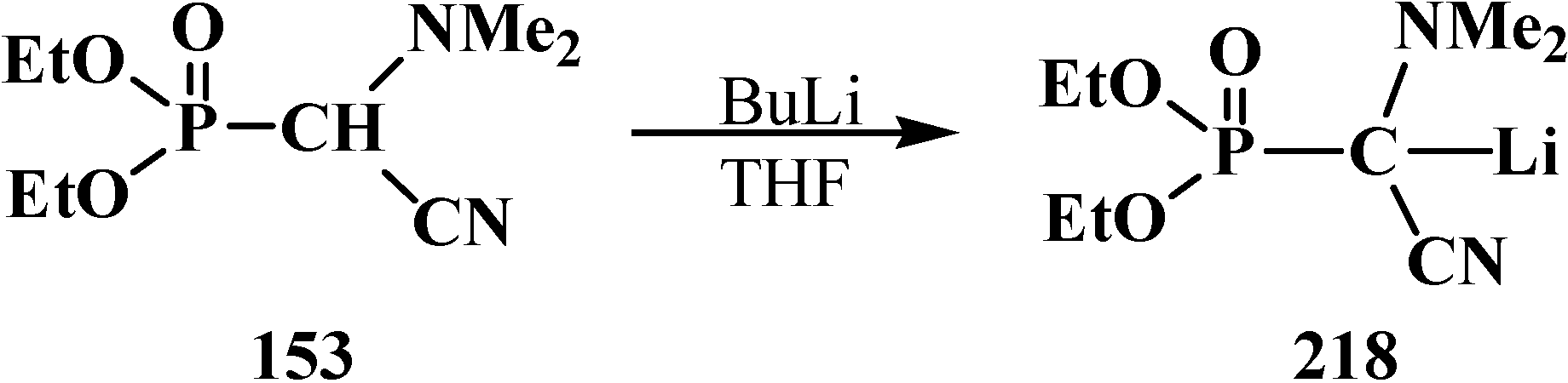
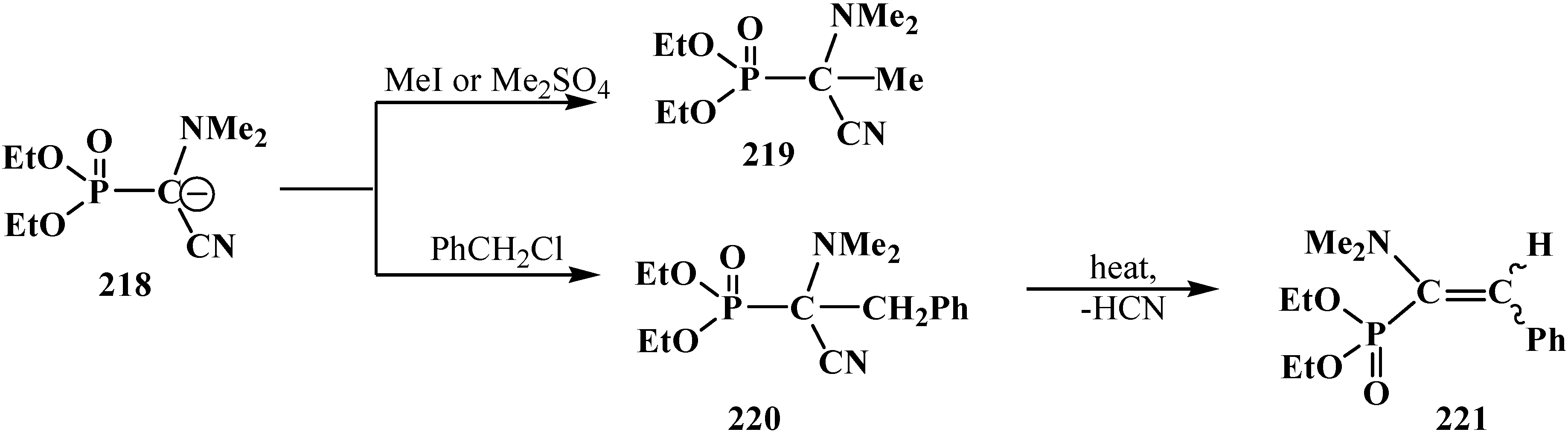
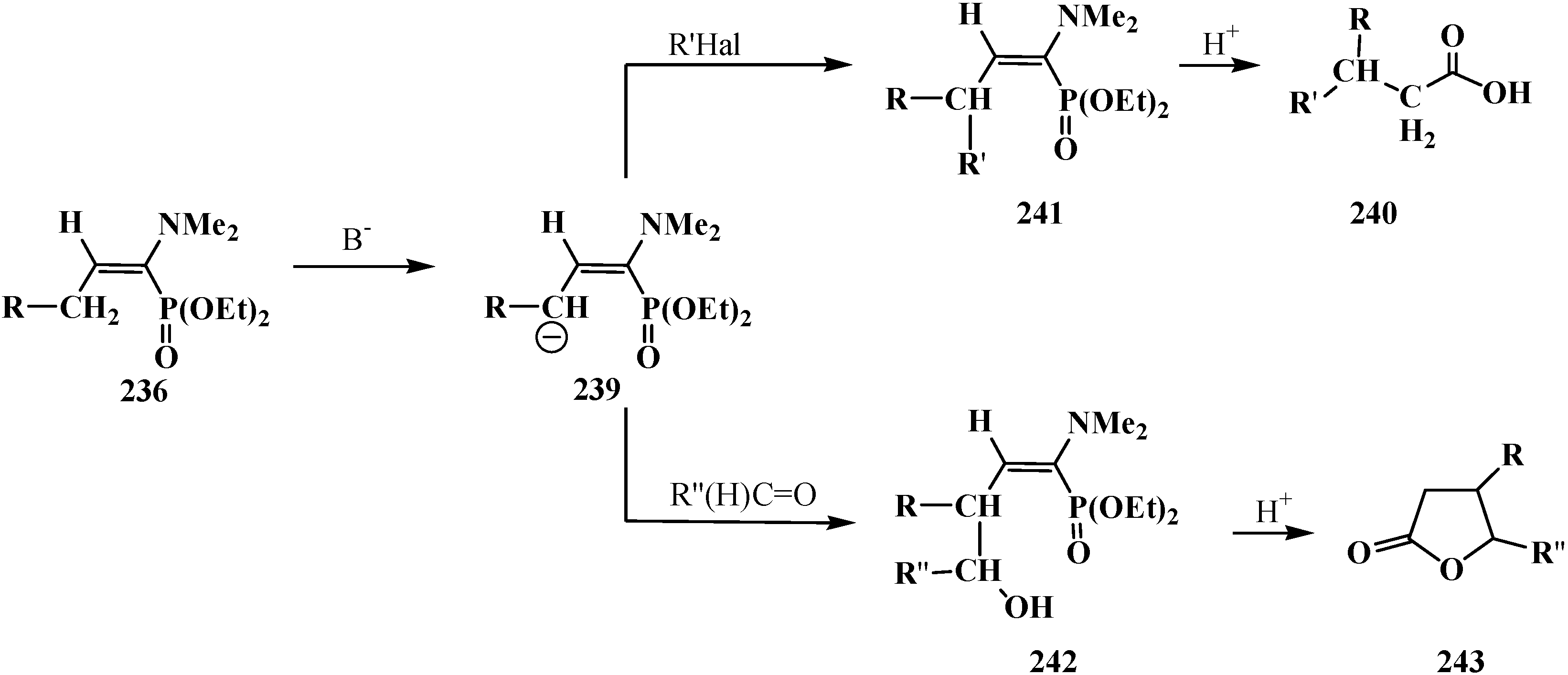
3.4. Horner Reaction
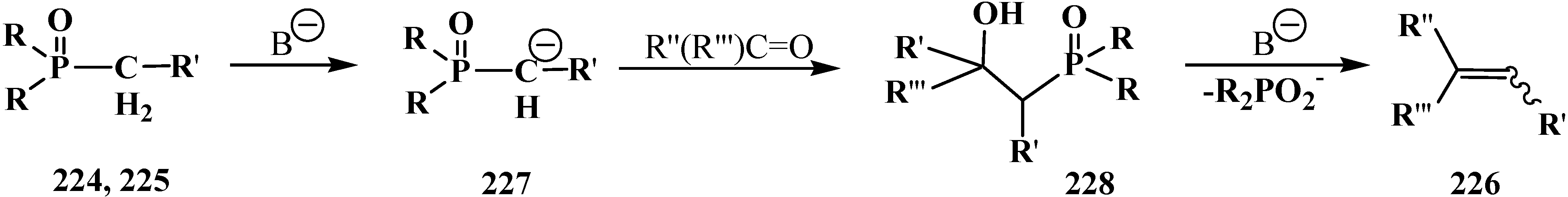
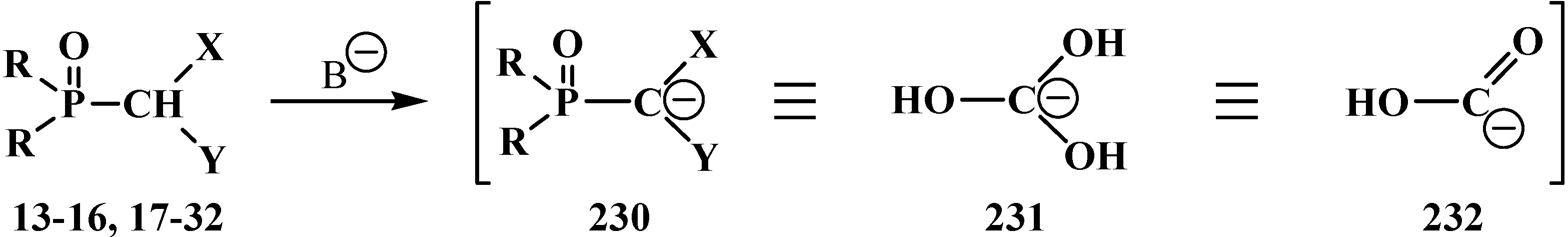
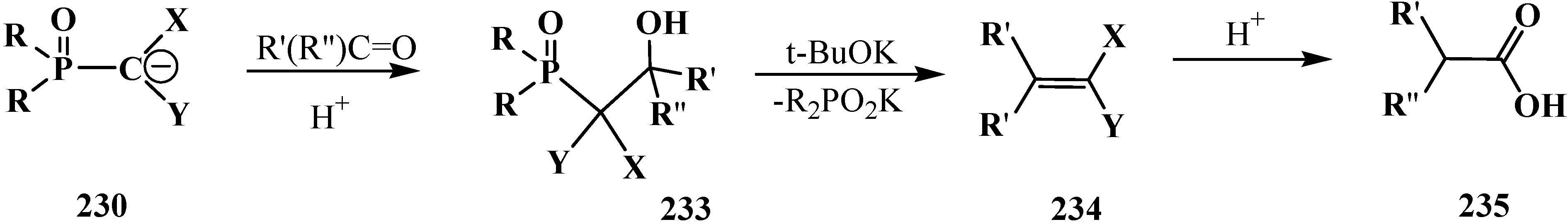
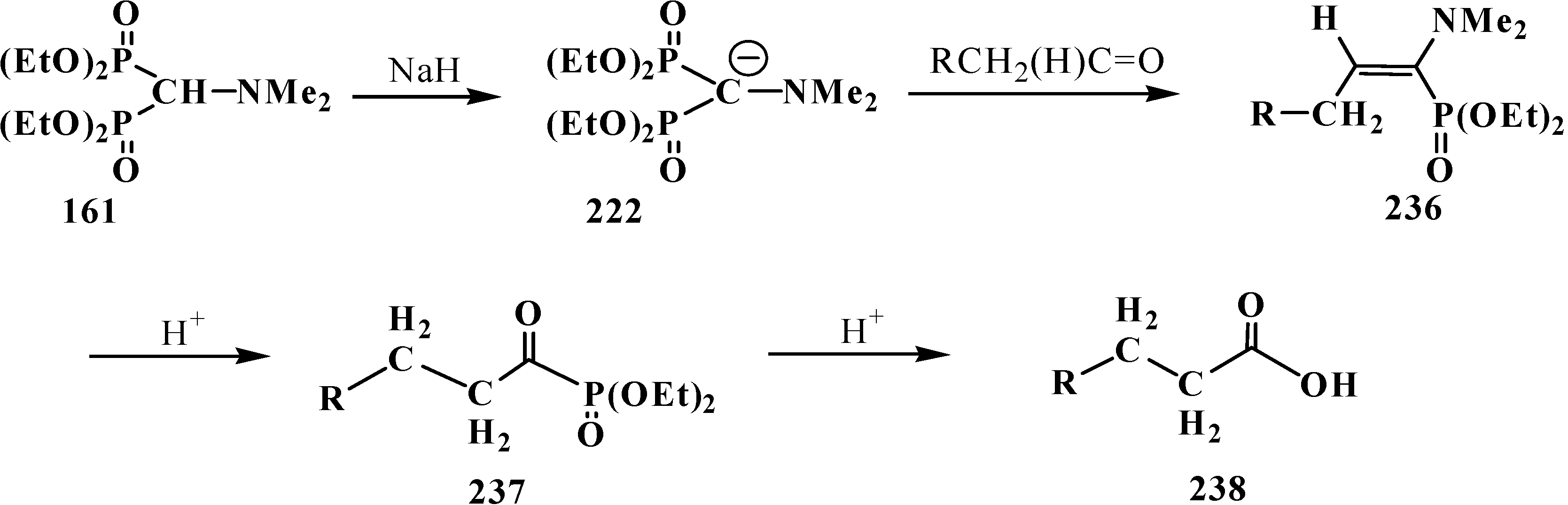
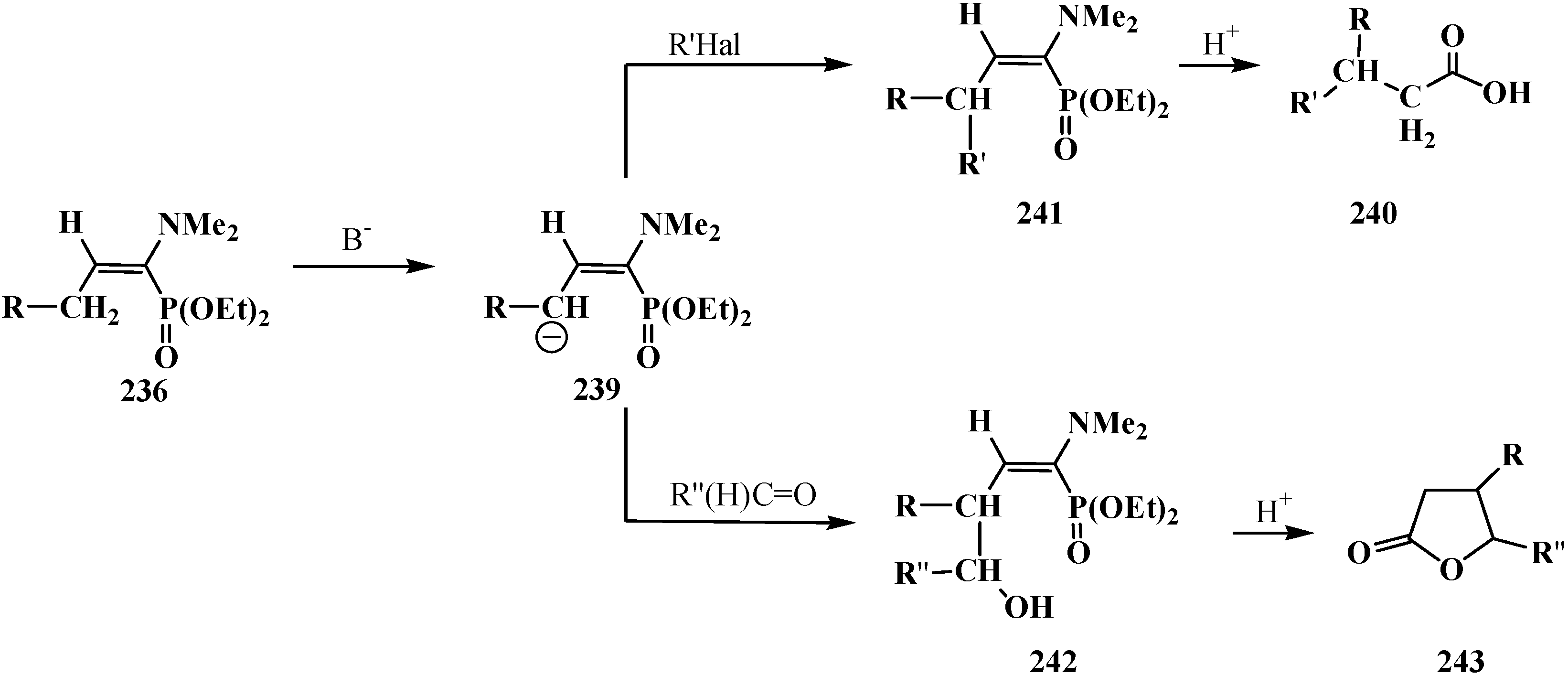
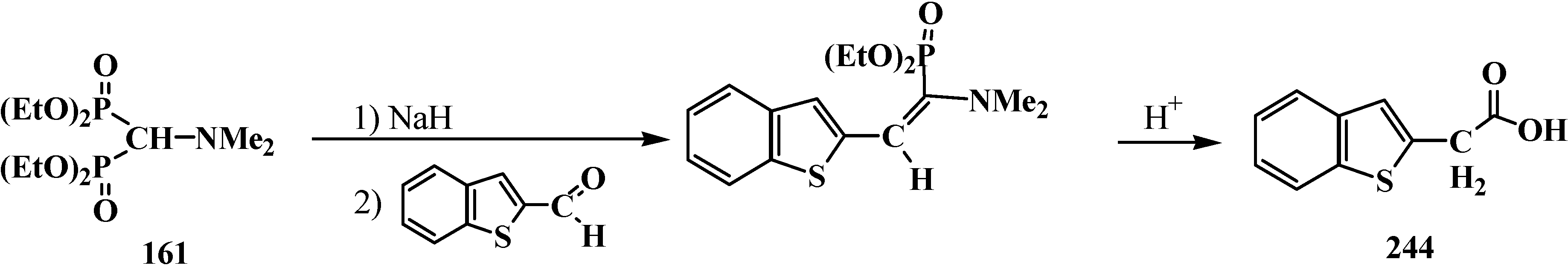
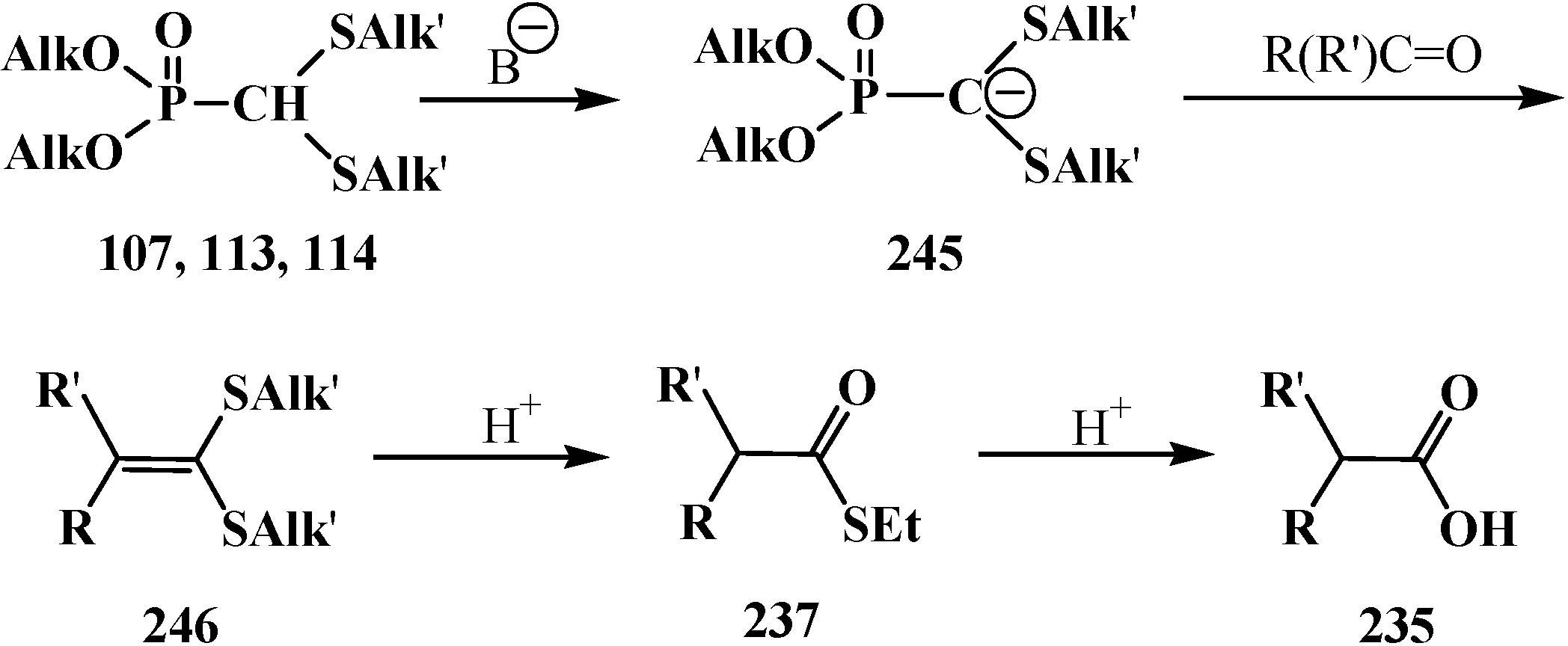
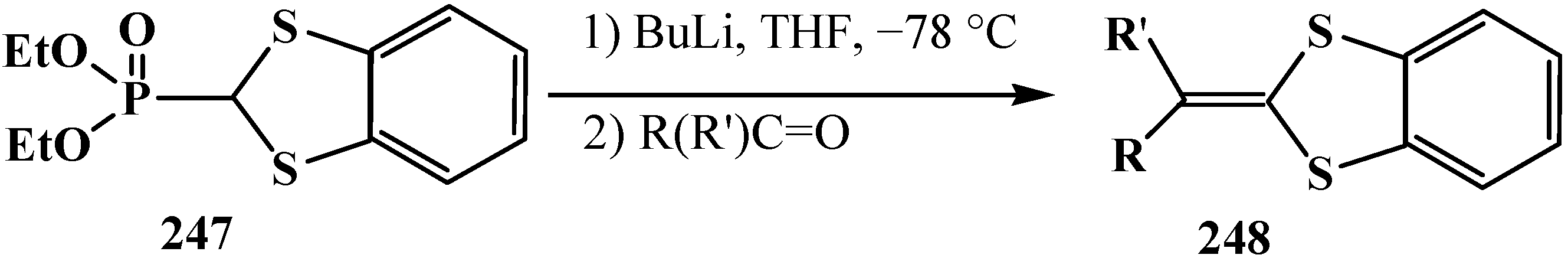
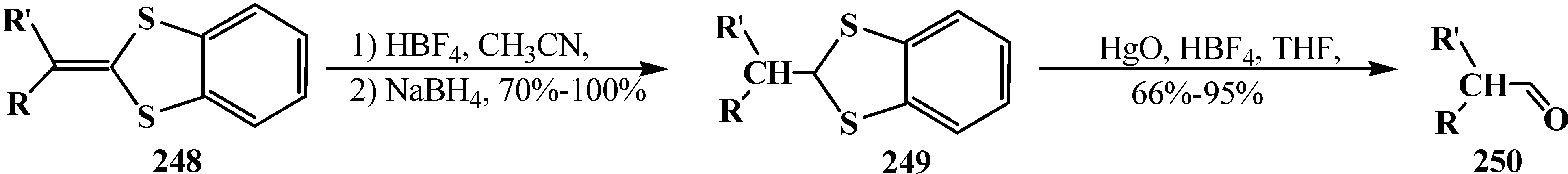

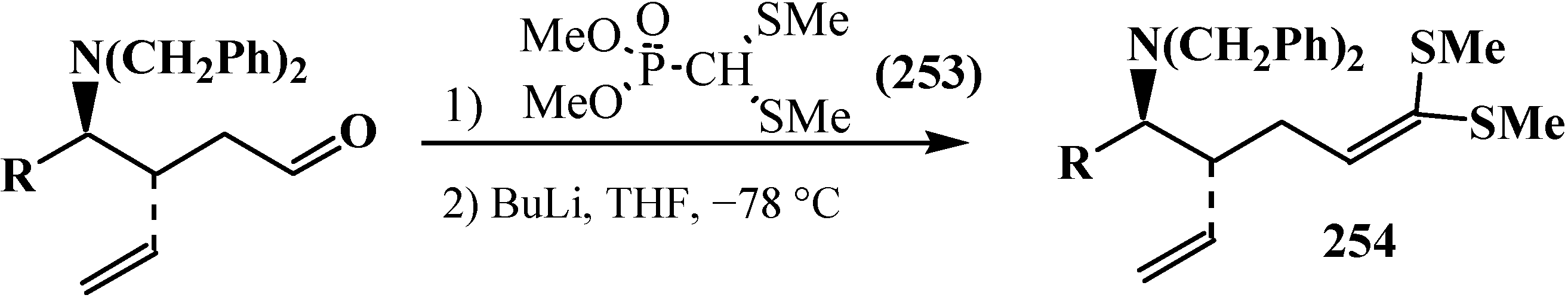
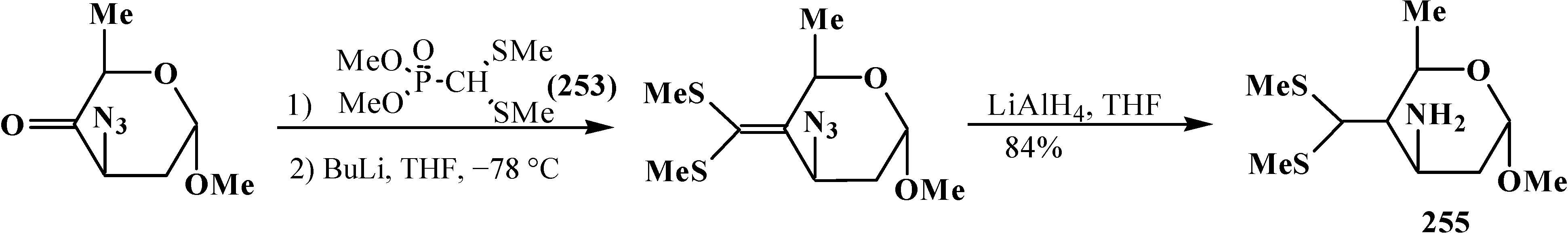
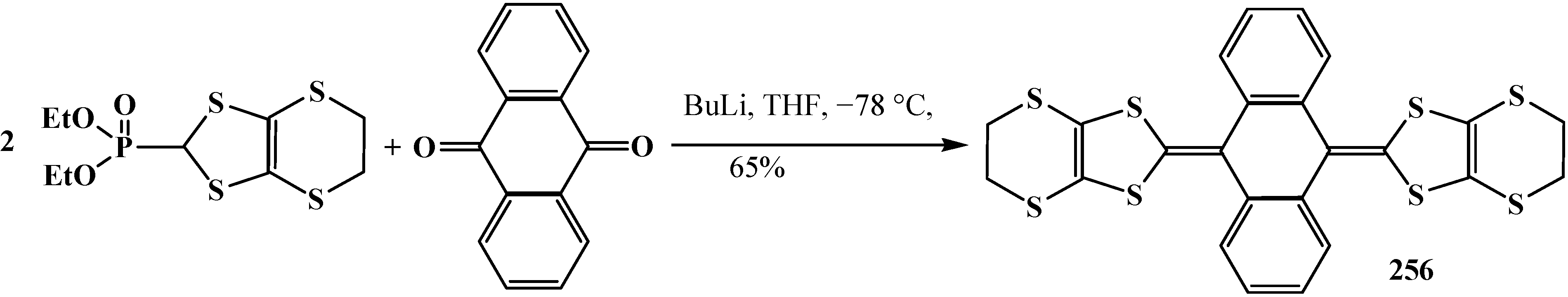

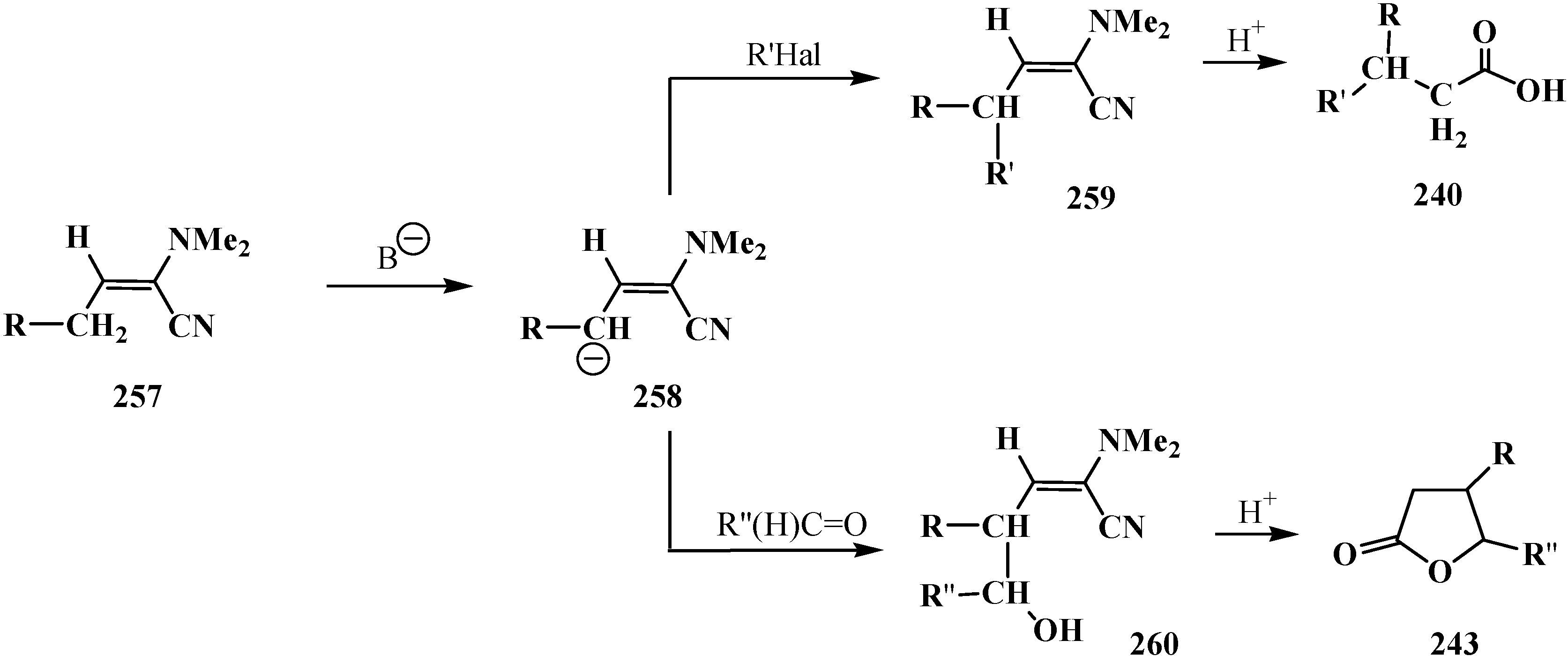

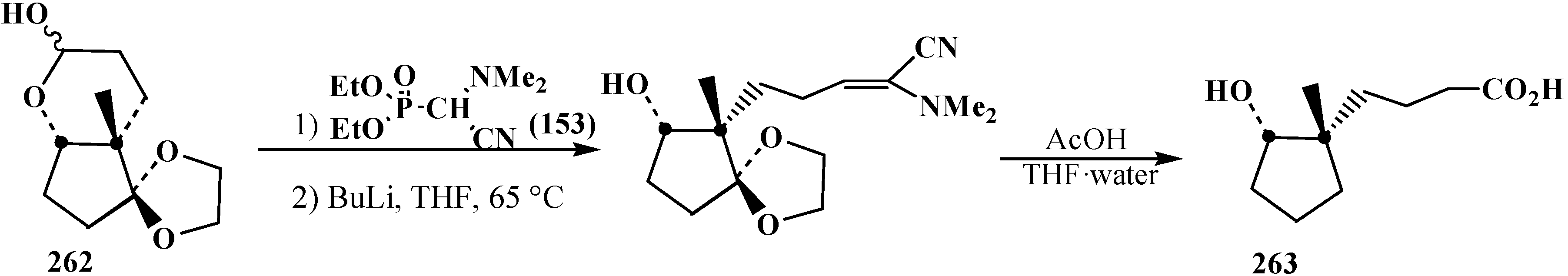
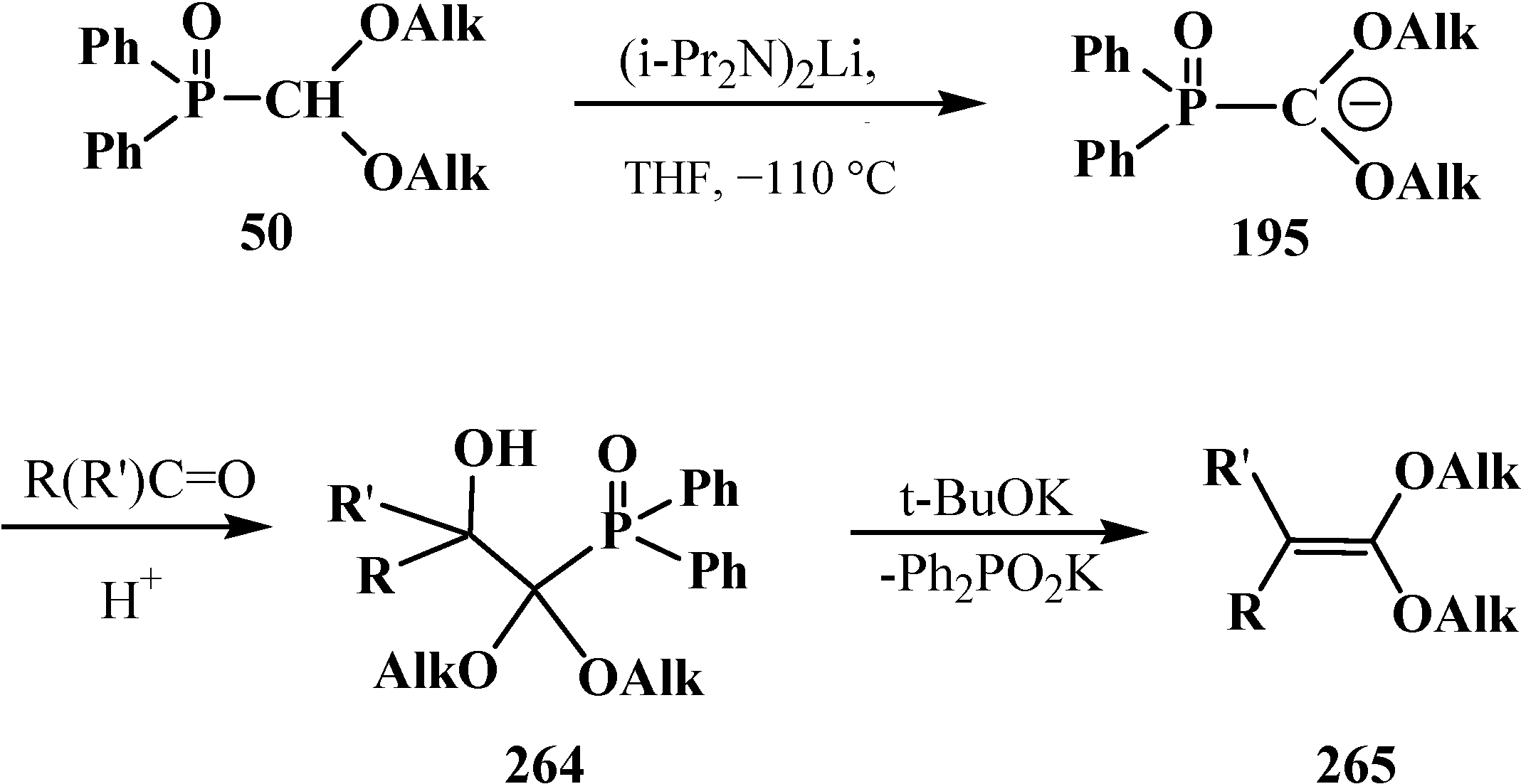
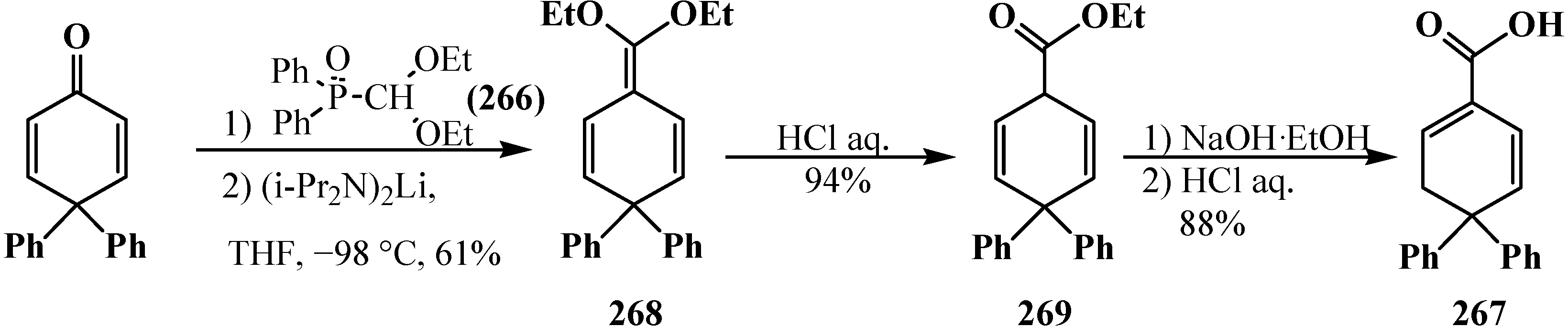
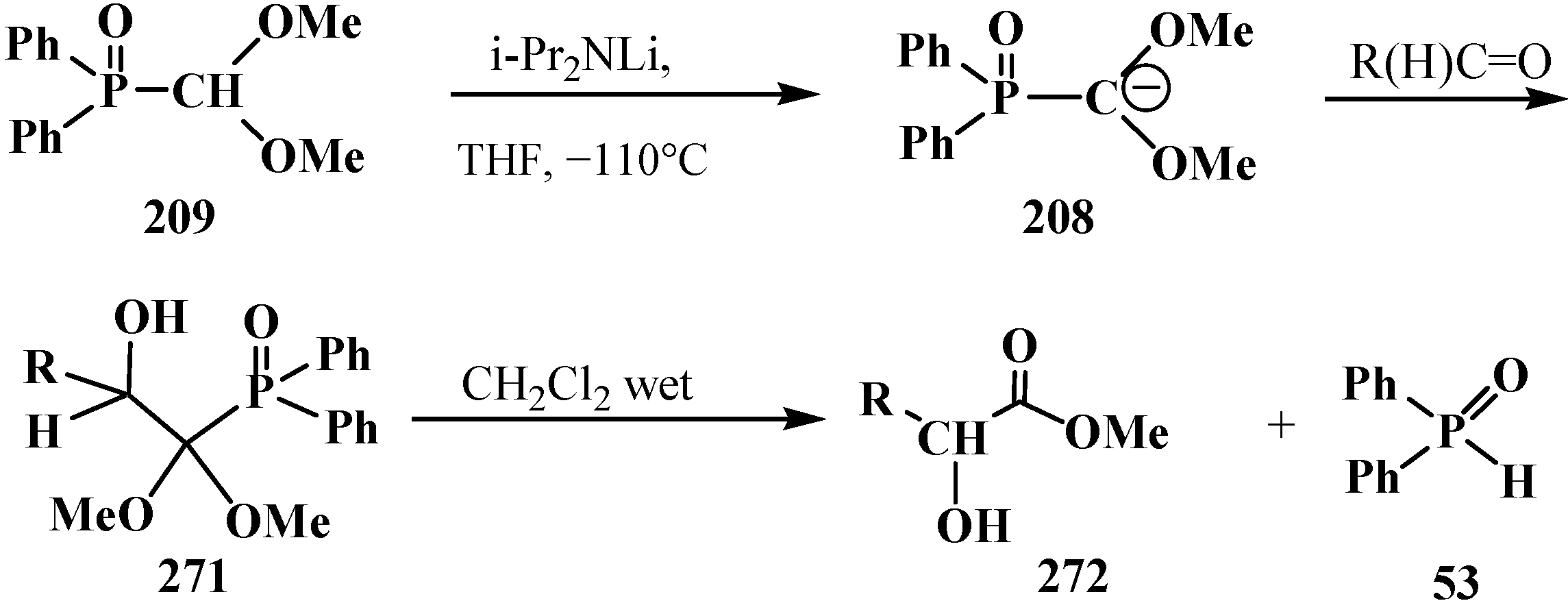
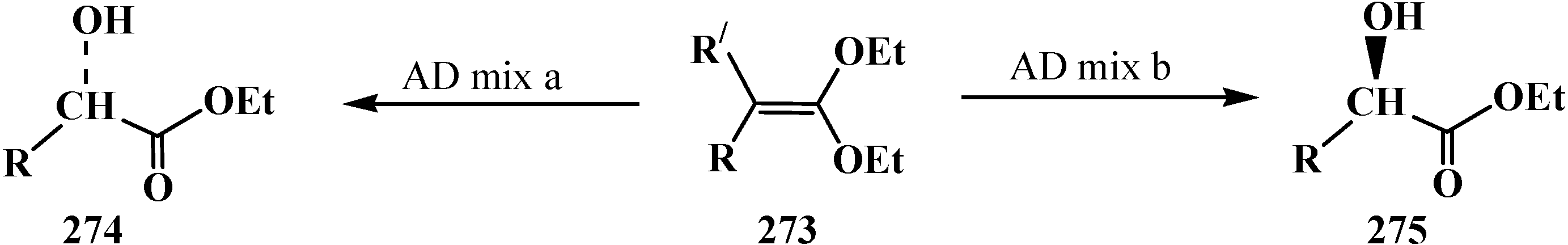
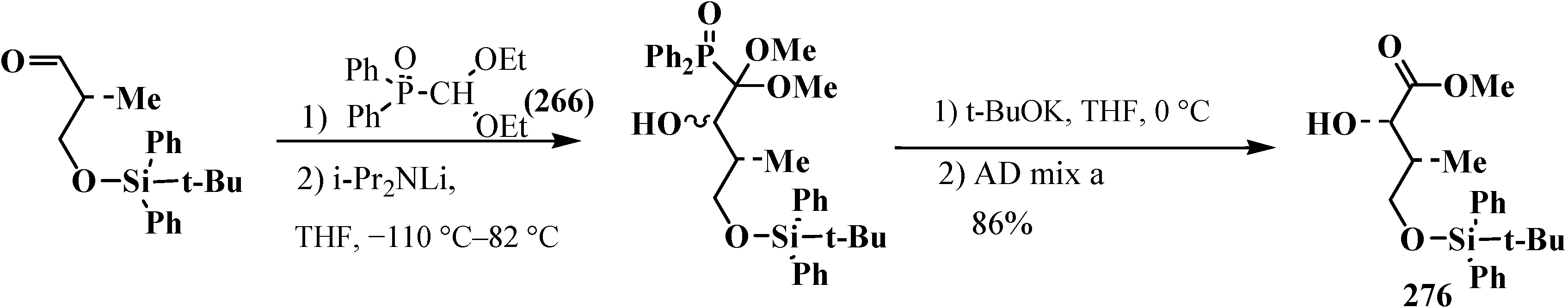

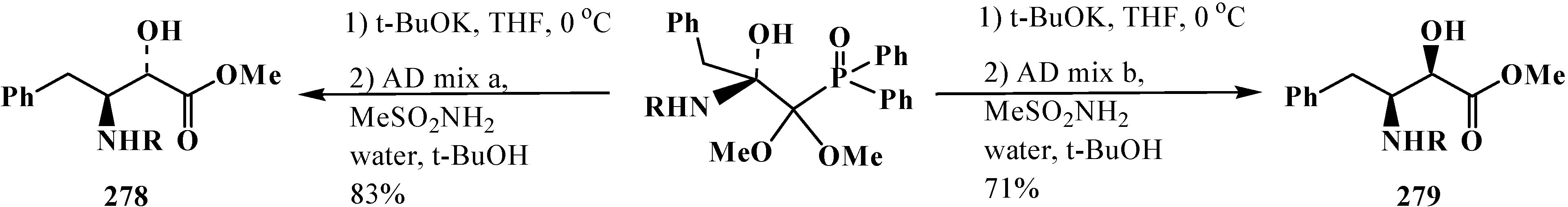
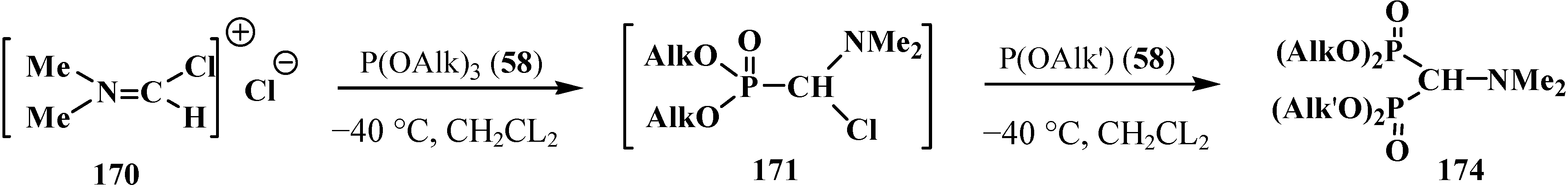
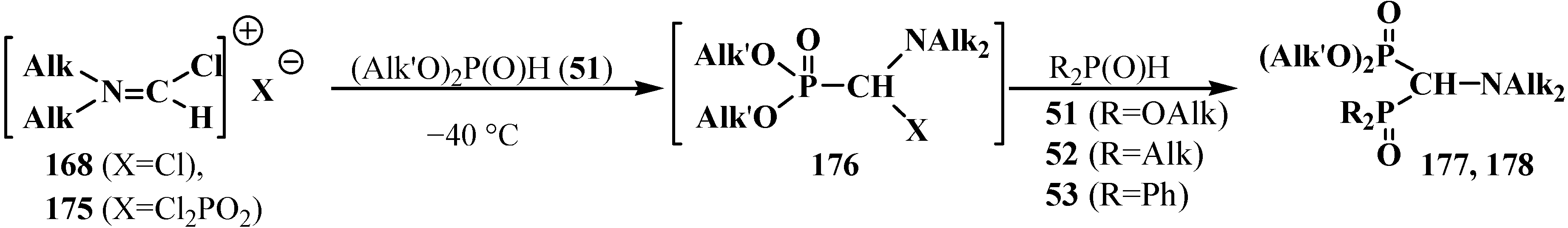
4. Phosphorylated Formaldehyde Halogenoaminals (Phosphorylated Vilsmeier–Haak Reagents) 17
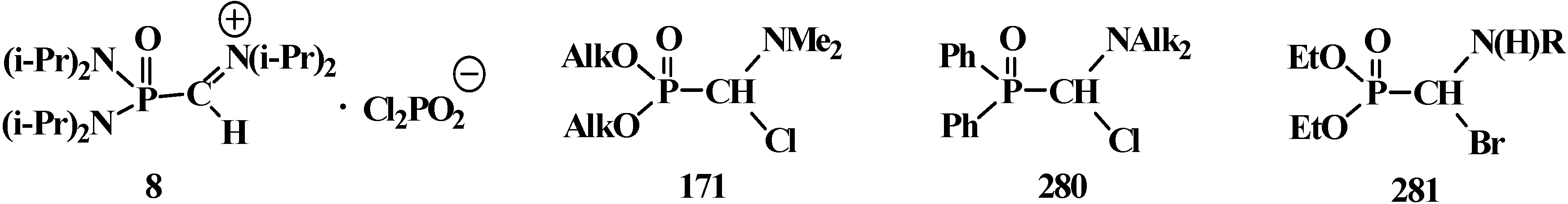
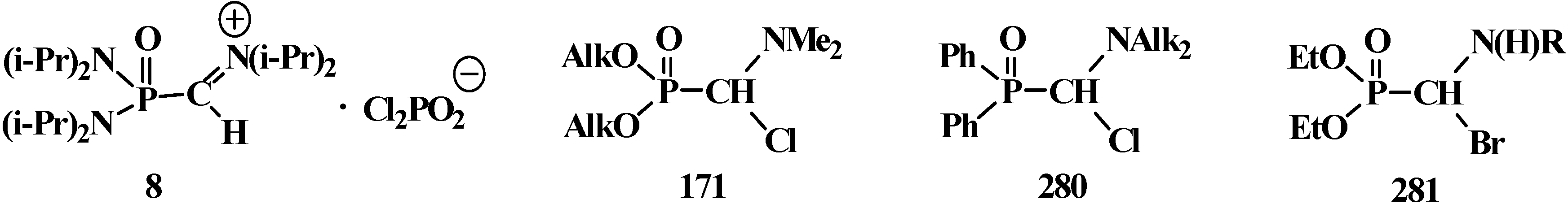
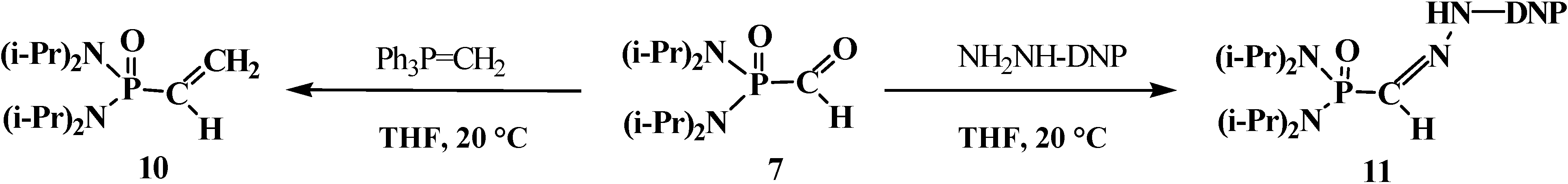
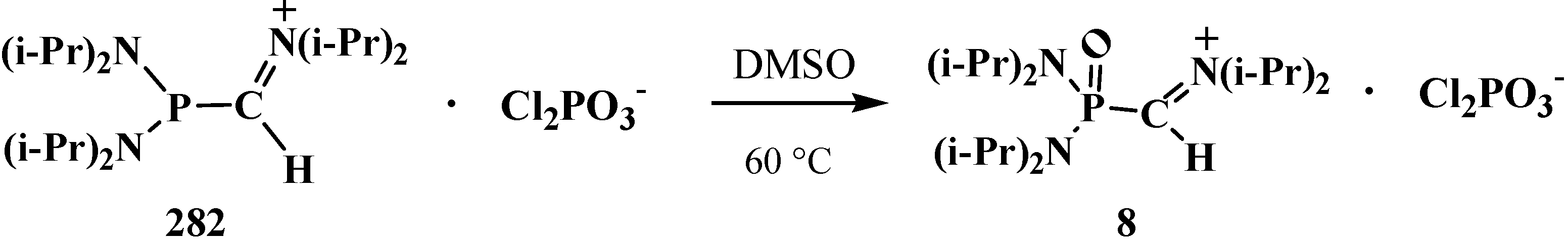
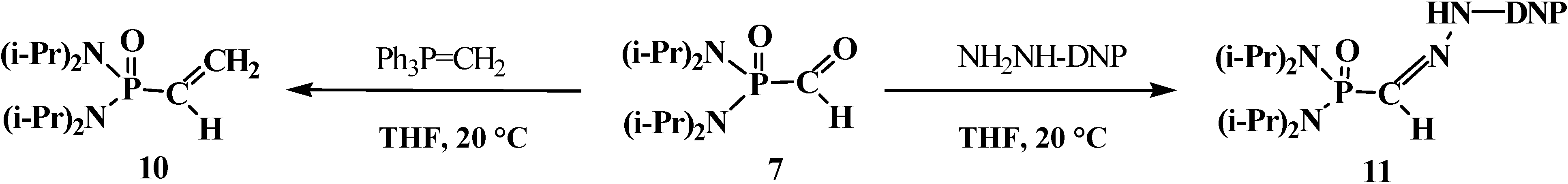
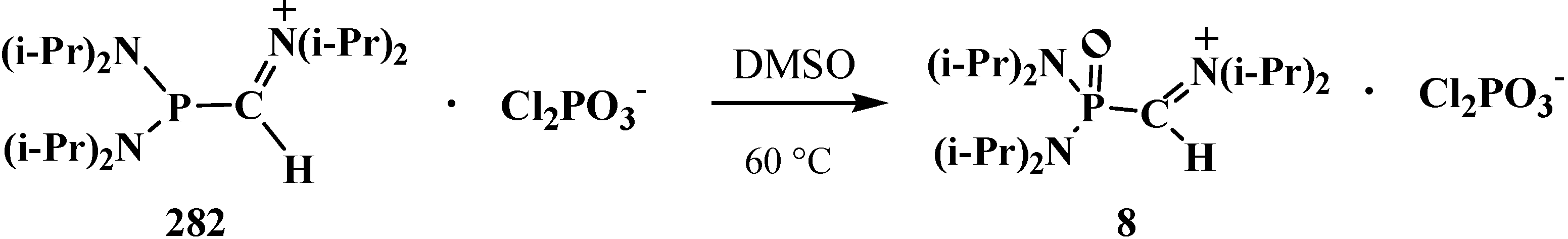
4.1. Synthesis of N,N,N',N'-Tetraisopropyl [(N'',N''-Diisopropylamino)methylydeniminium] Phosphondiamide Dichlorophosphate (8)
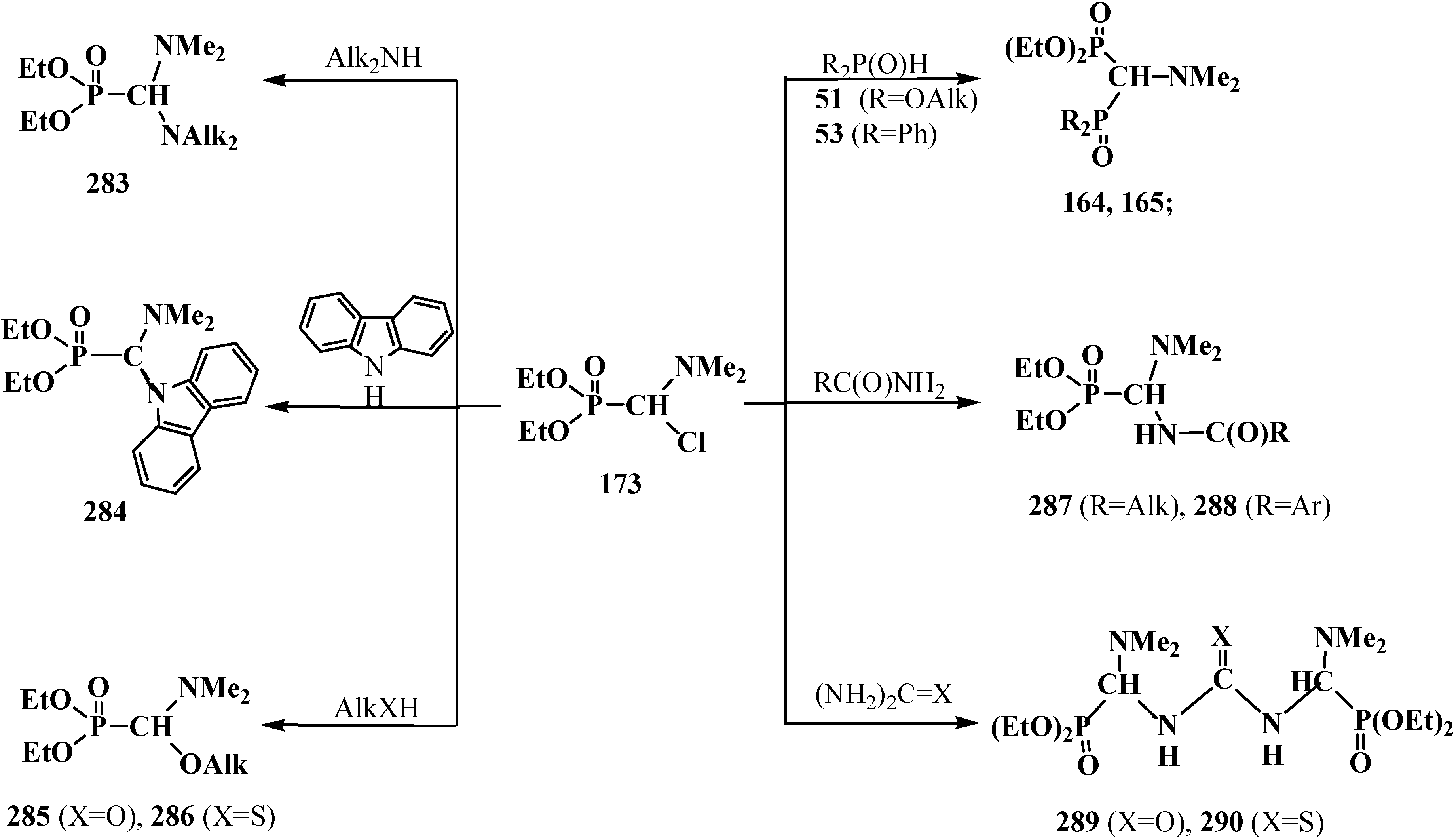
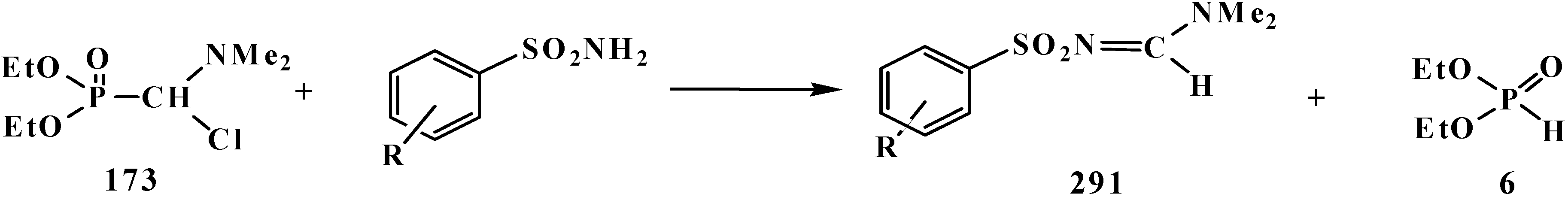
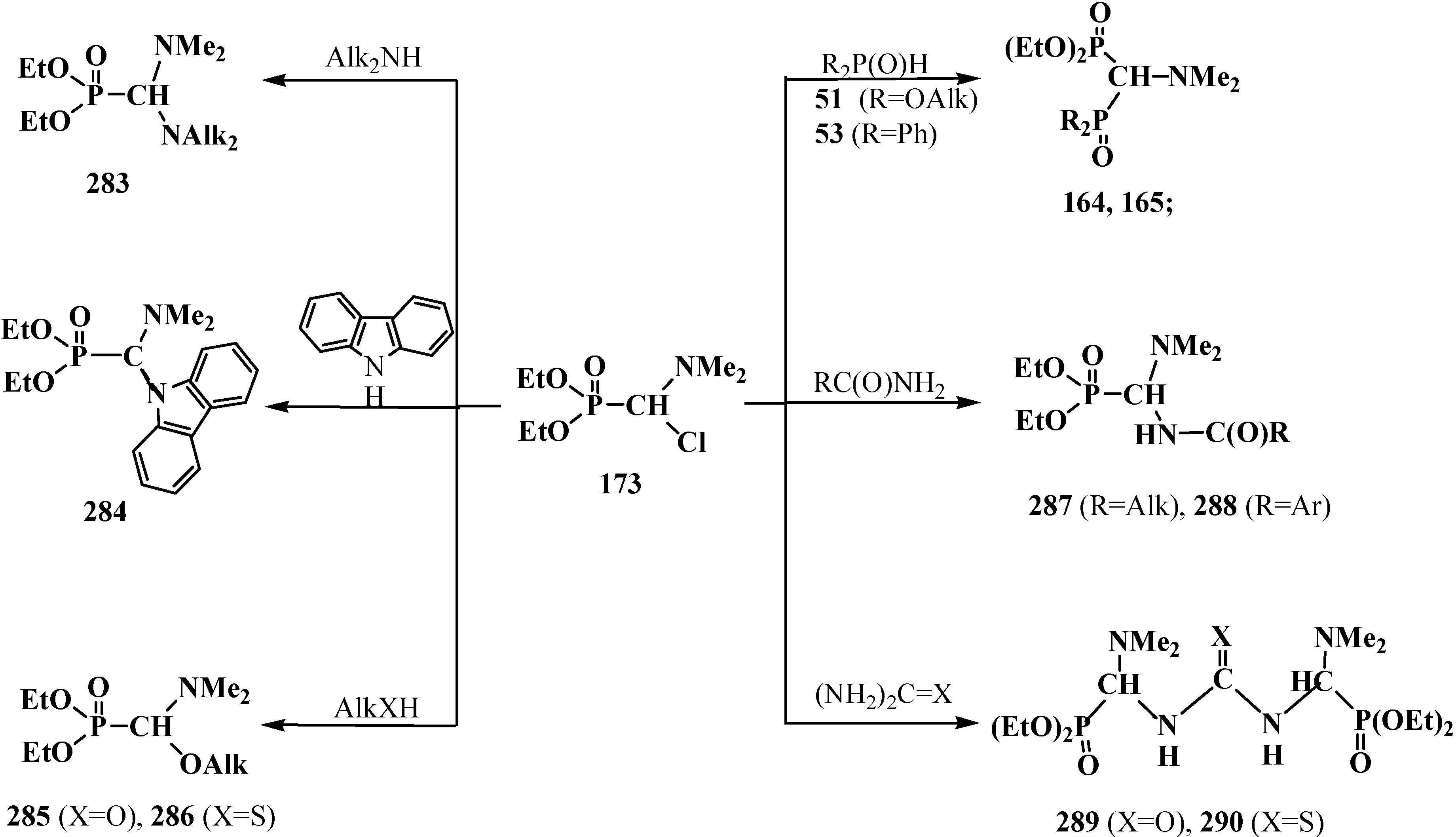
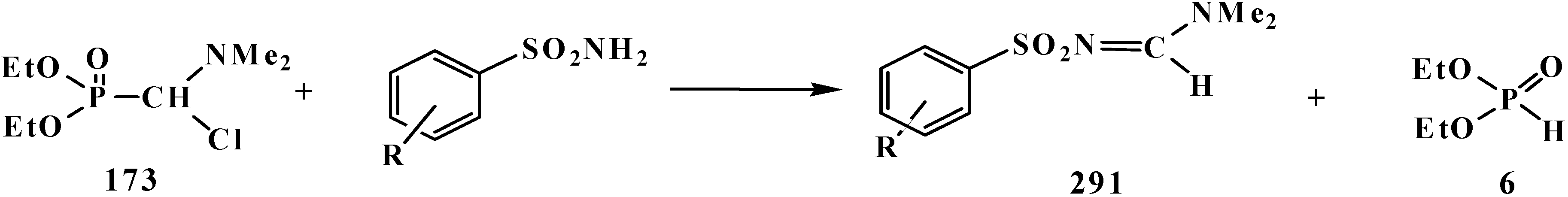
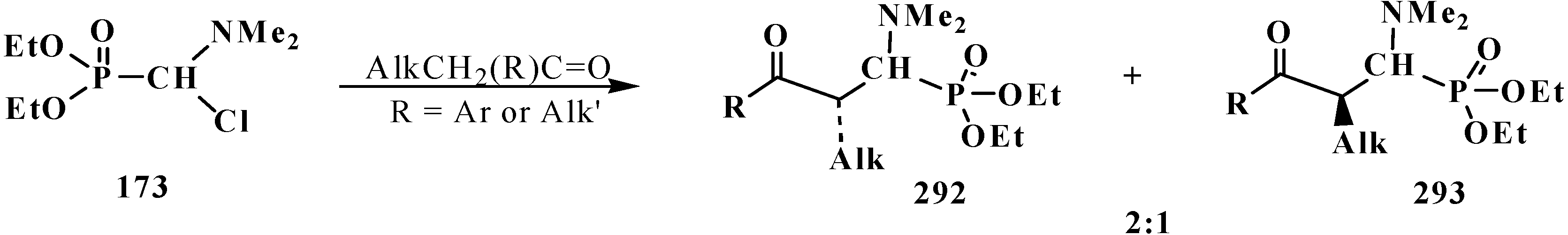
4.2. Synthesis and Chemical Properties of Dialkyl [(N,N-Dimethylamino)chloromethyl]phosphonates 171
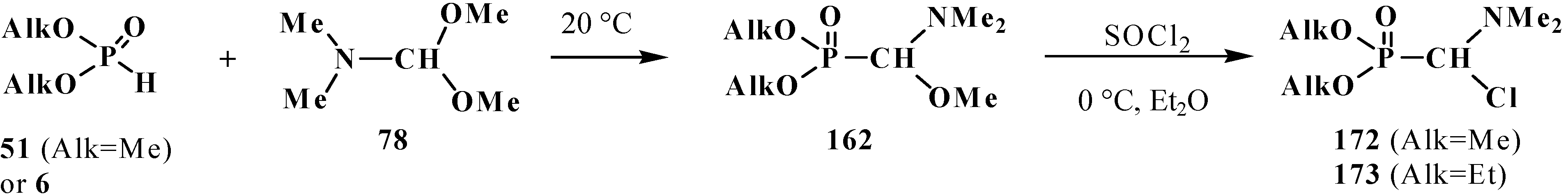
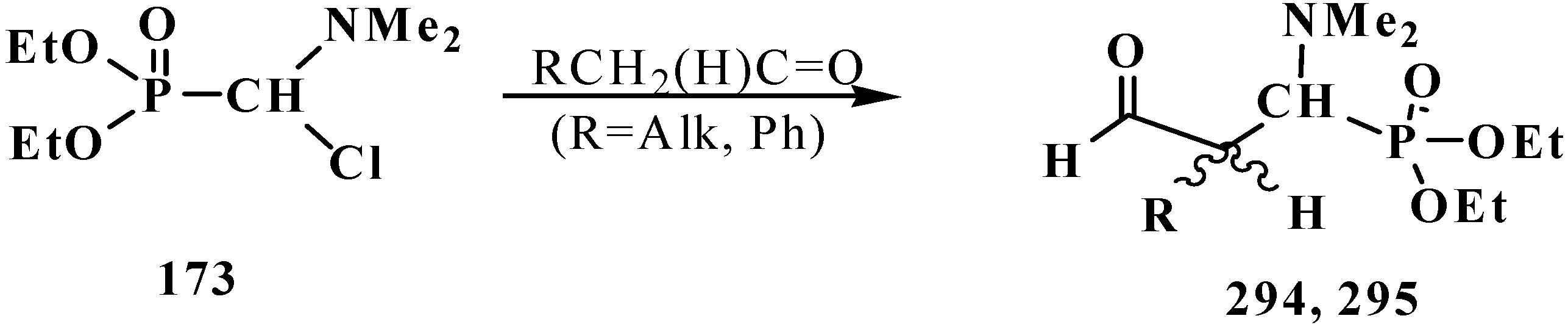
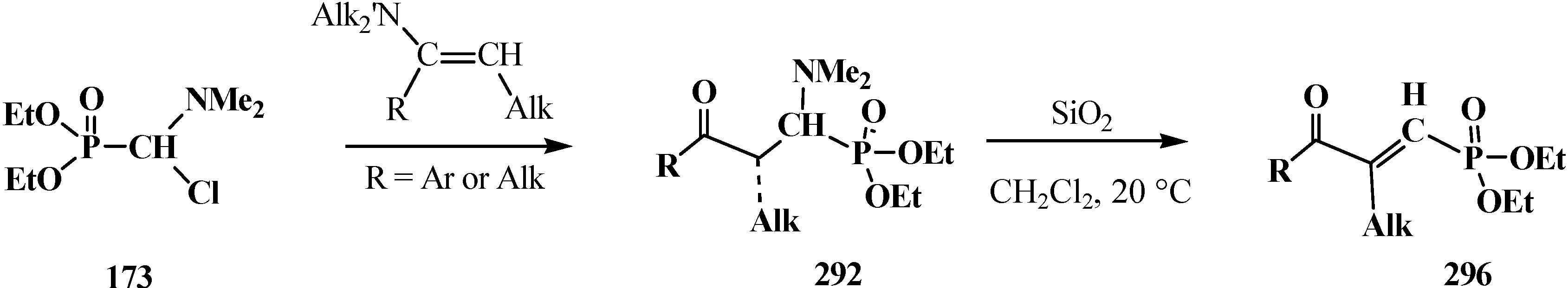
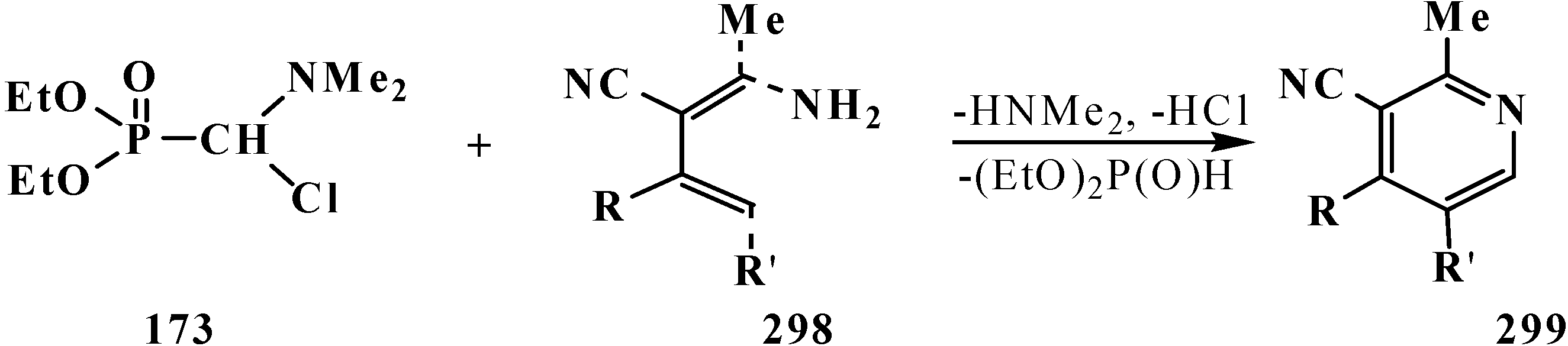

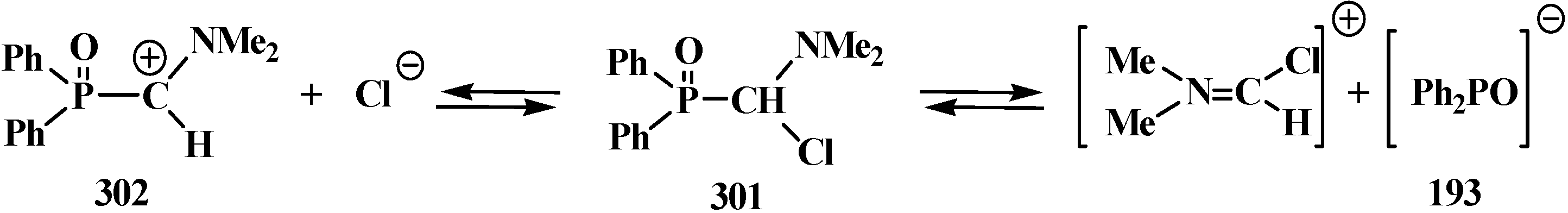
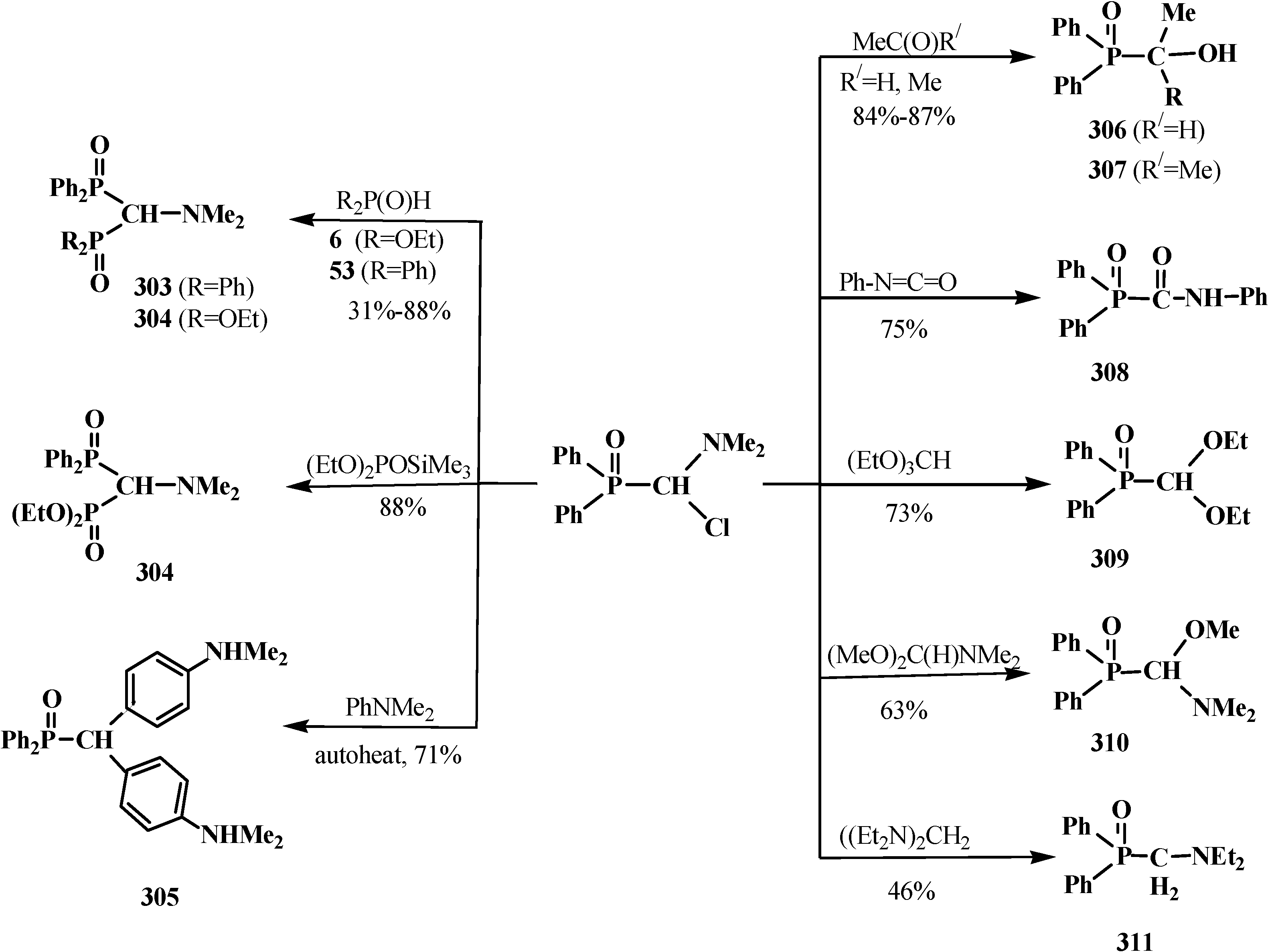
4.3. Syntheses and Chemical Properties of Diphenyl[(N,N-dialkylamino)chloromethyl]phosphine Oxides 280
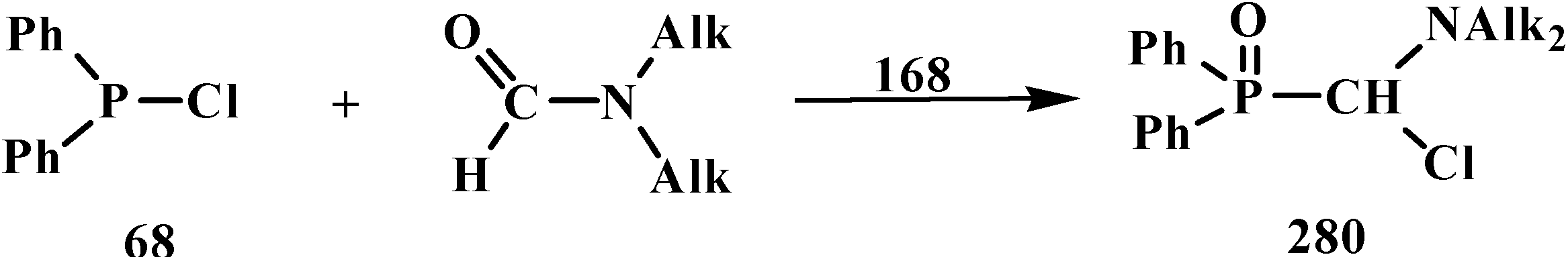
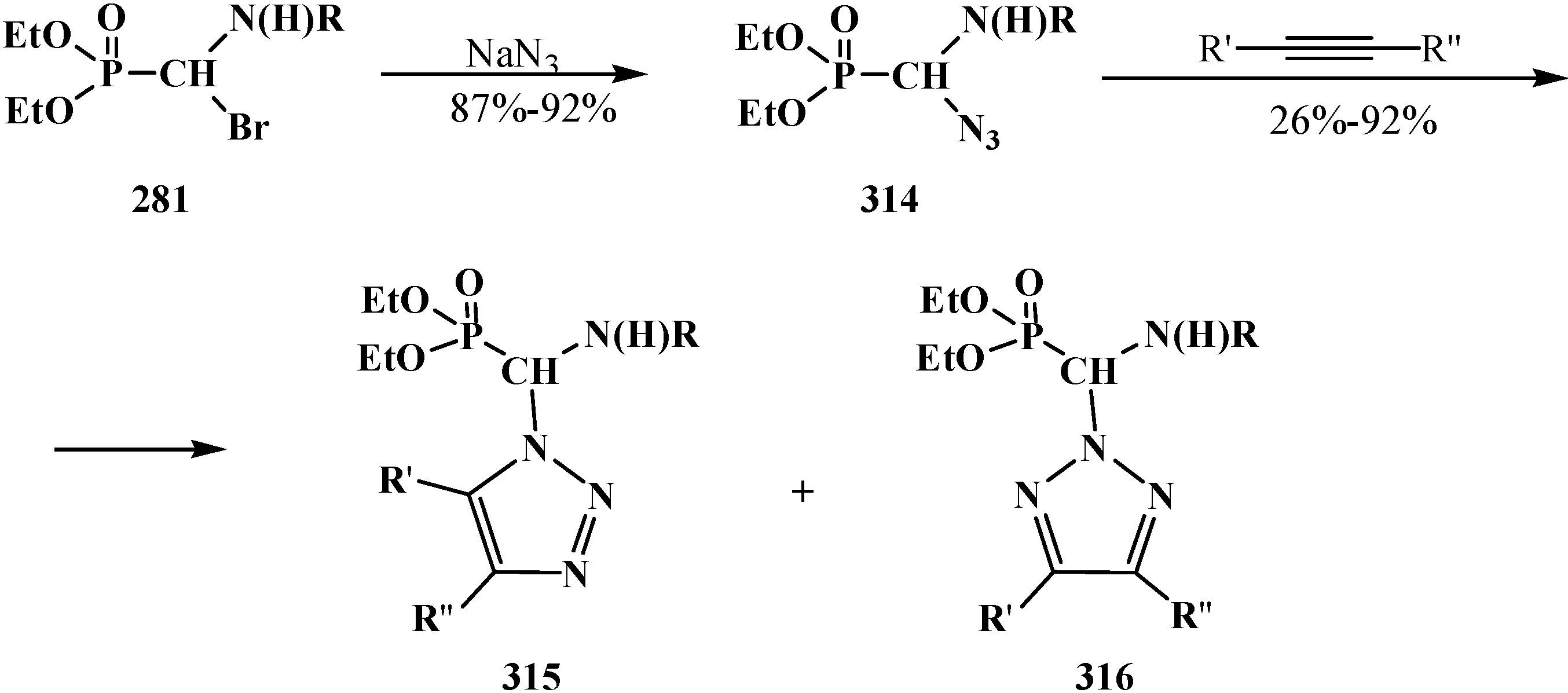
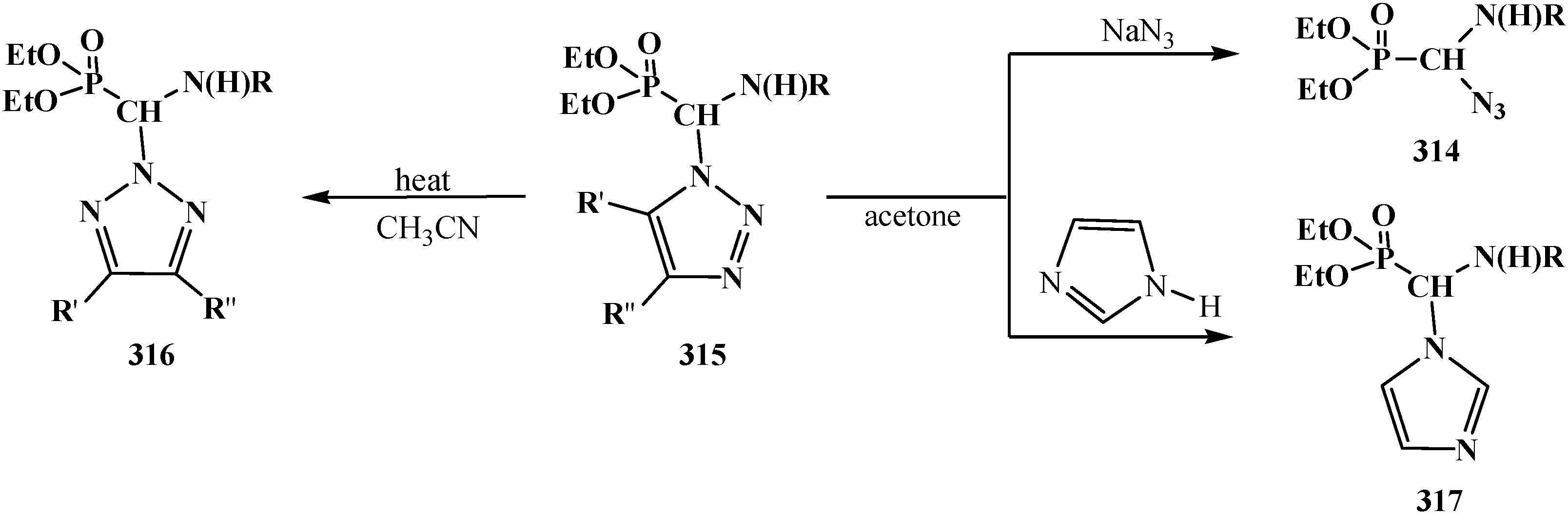
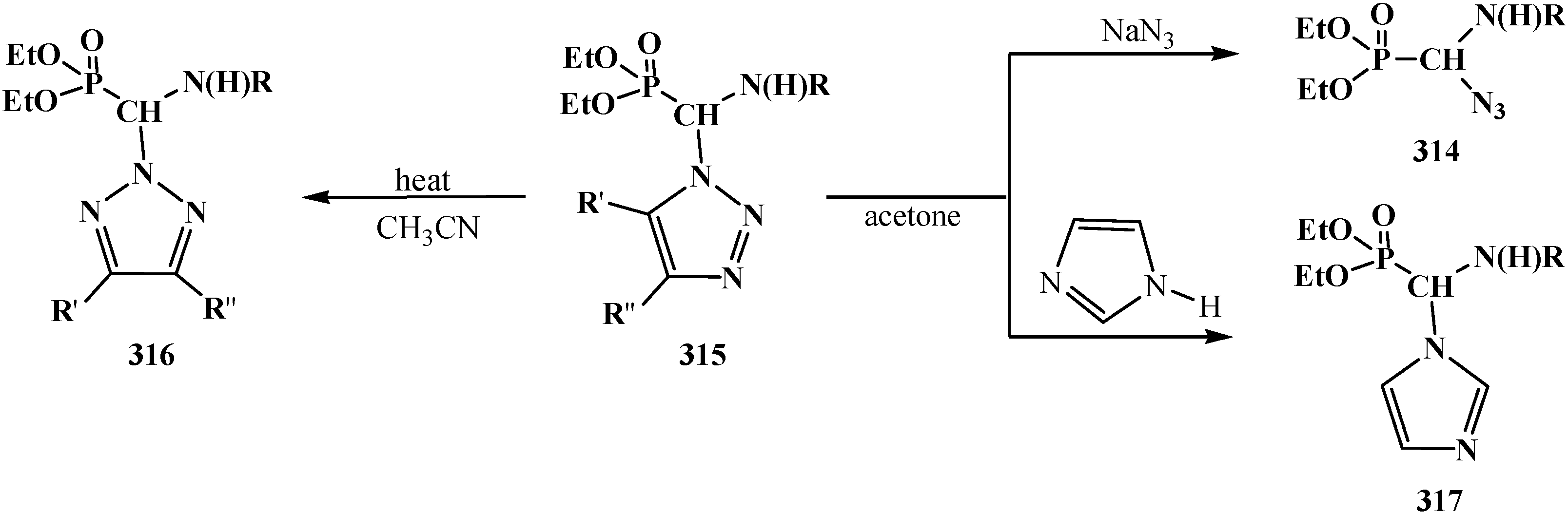
4.4. Synthesis and Chemical Properties of Diethyl [(N-Acylamino)bromomethyl]phosphonates 281
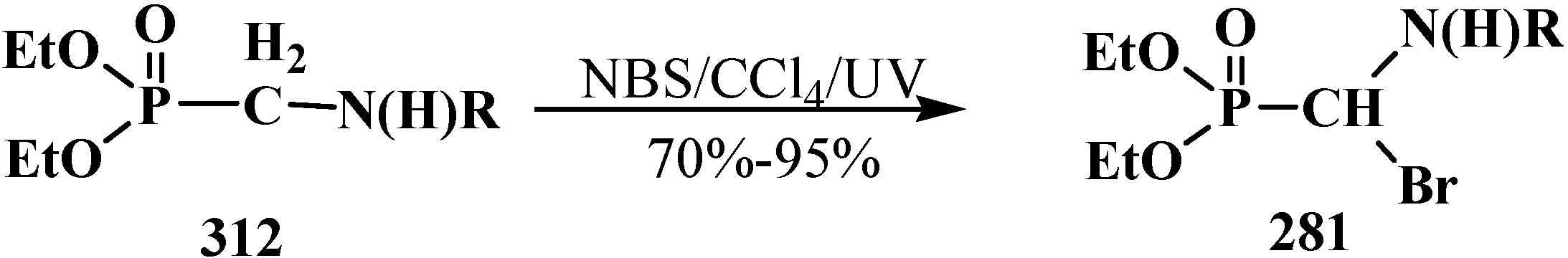

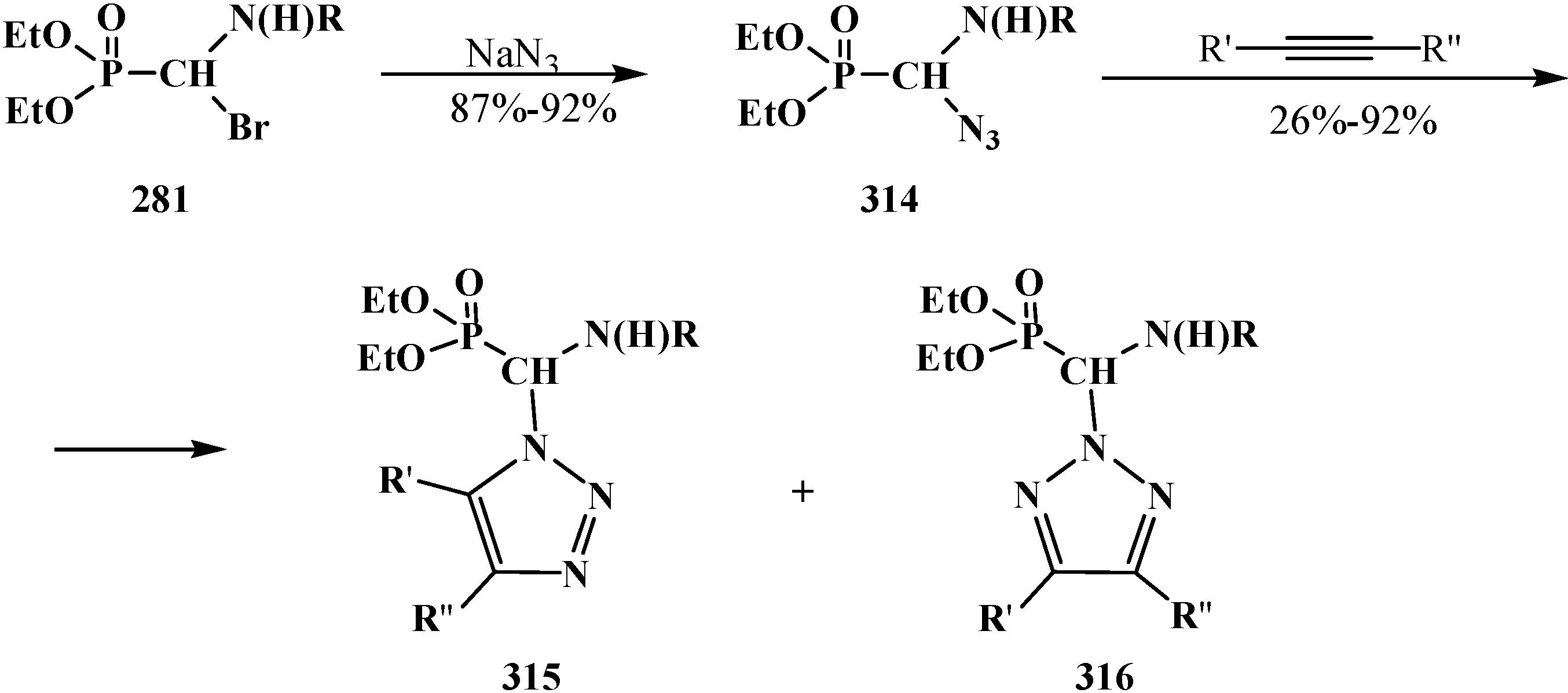
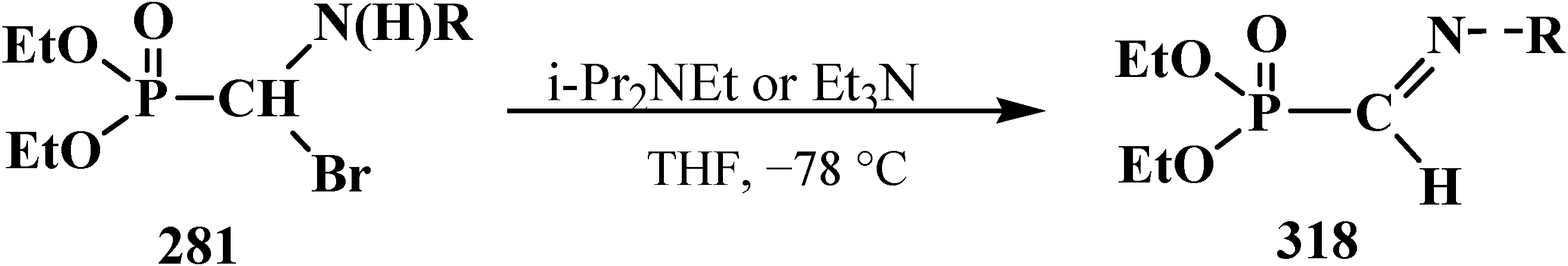
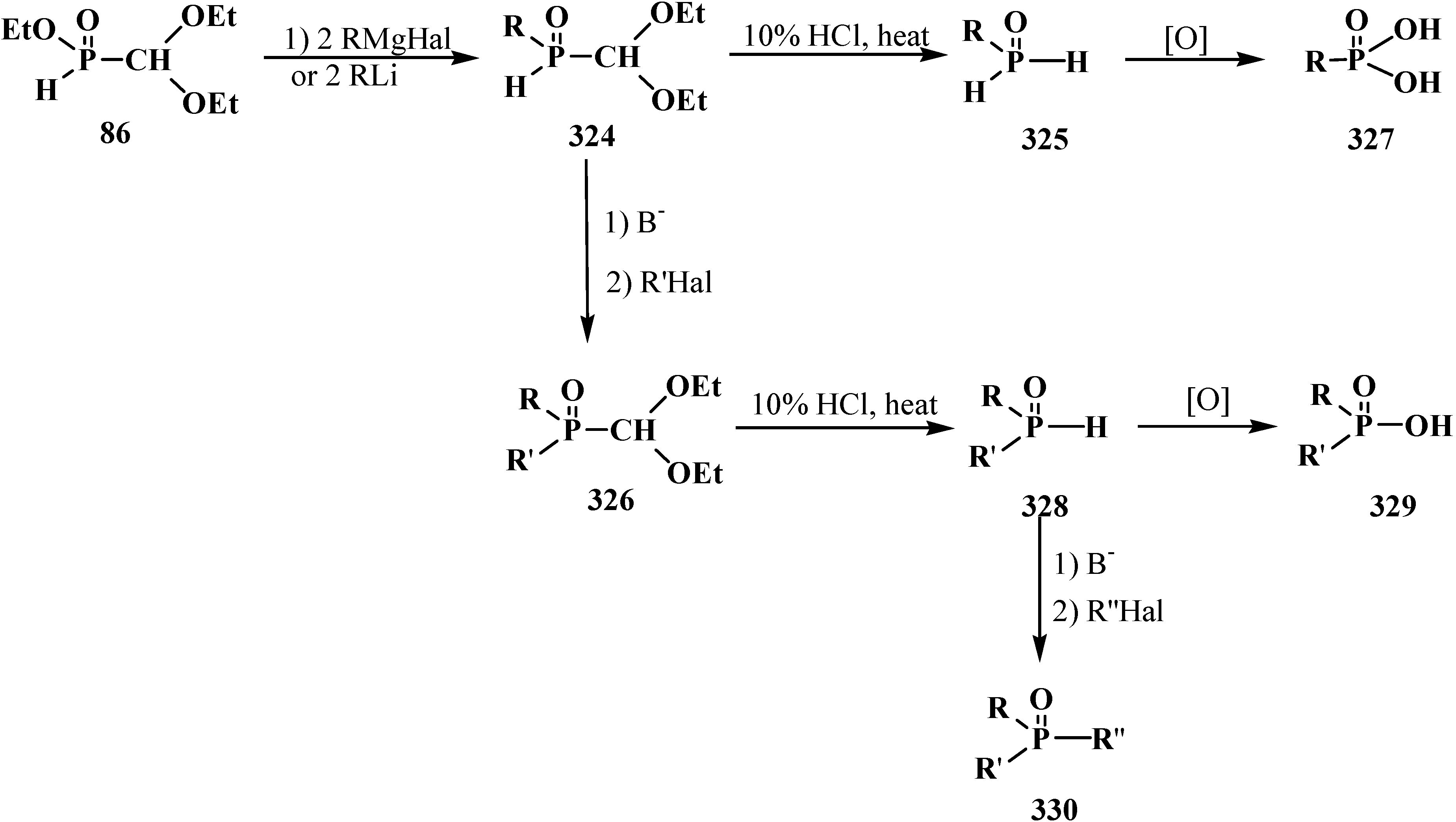
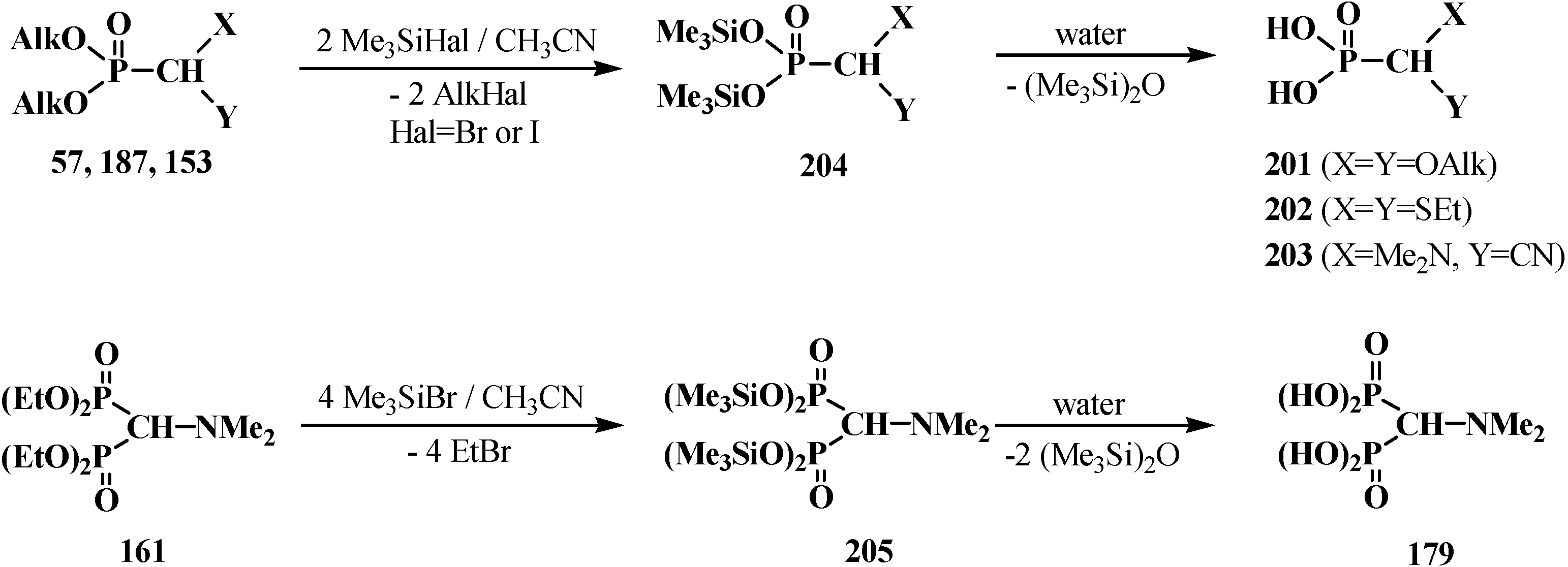
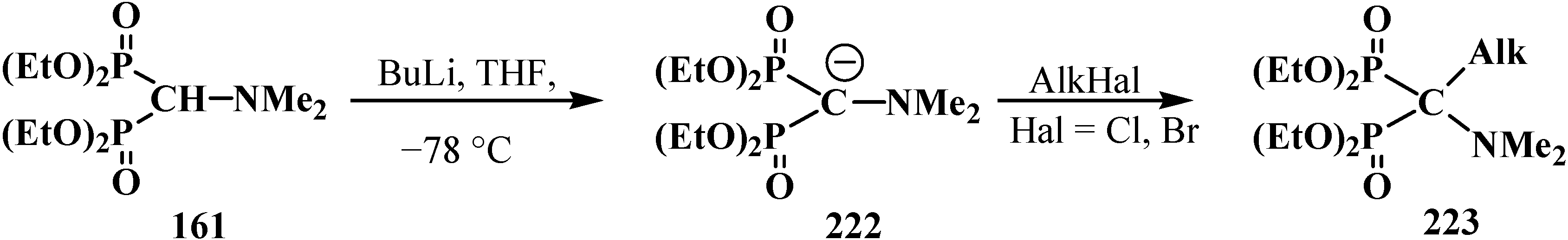
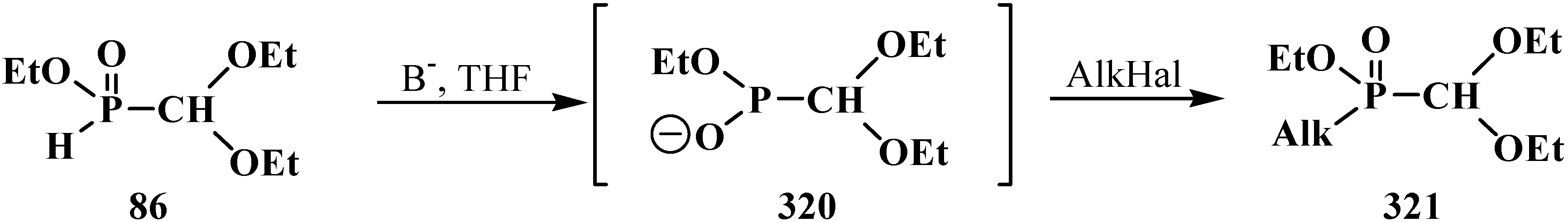
5. Alkyl (dialkoxymethyl)phosphinates—H-Phosphinates 34. Syntheses and Chemical Properties
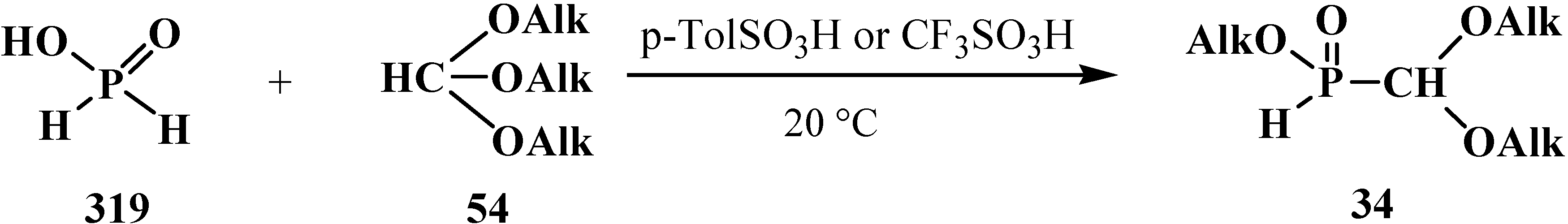

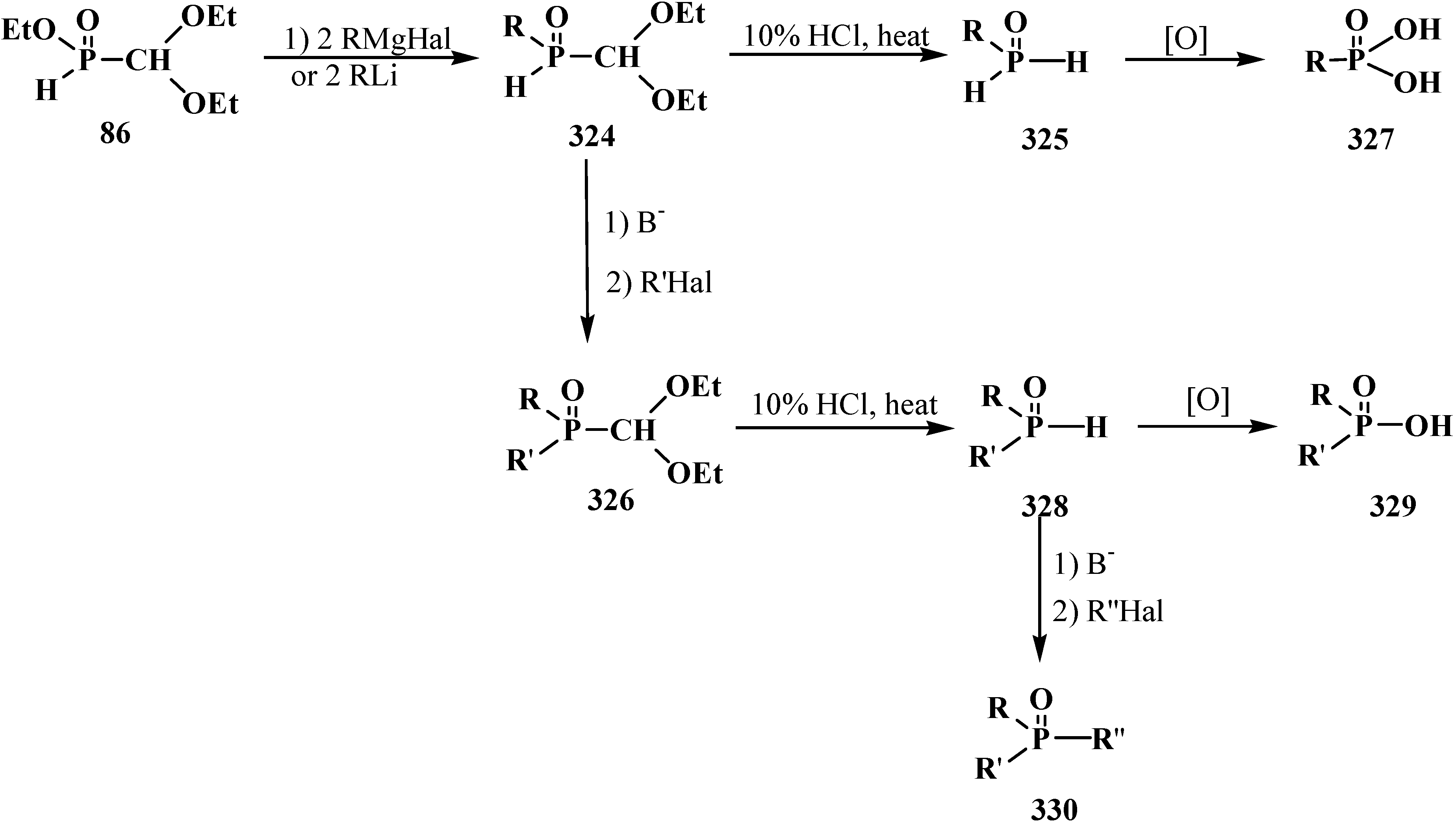
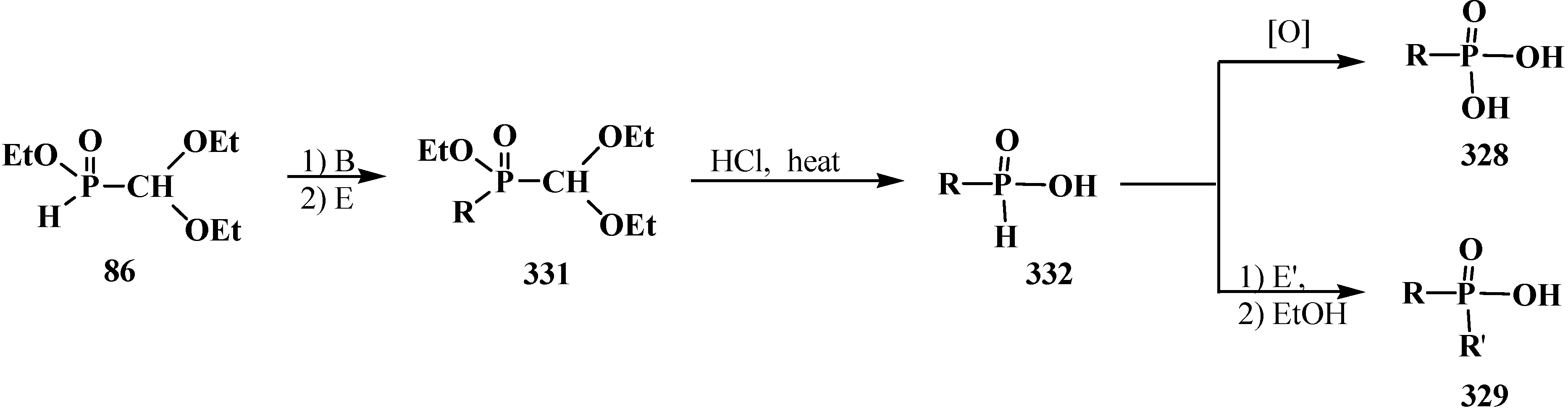
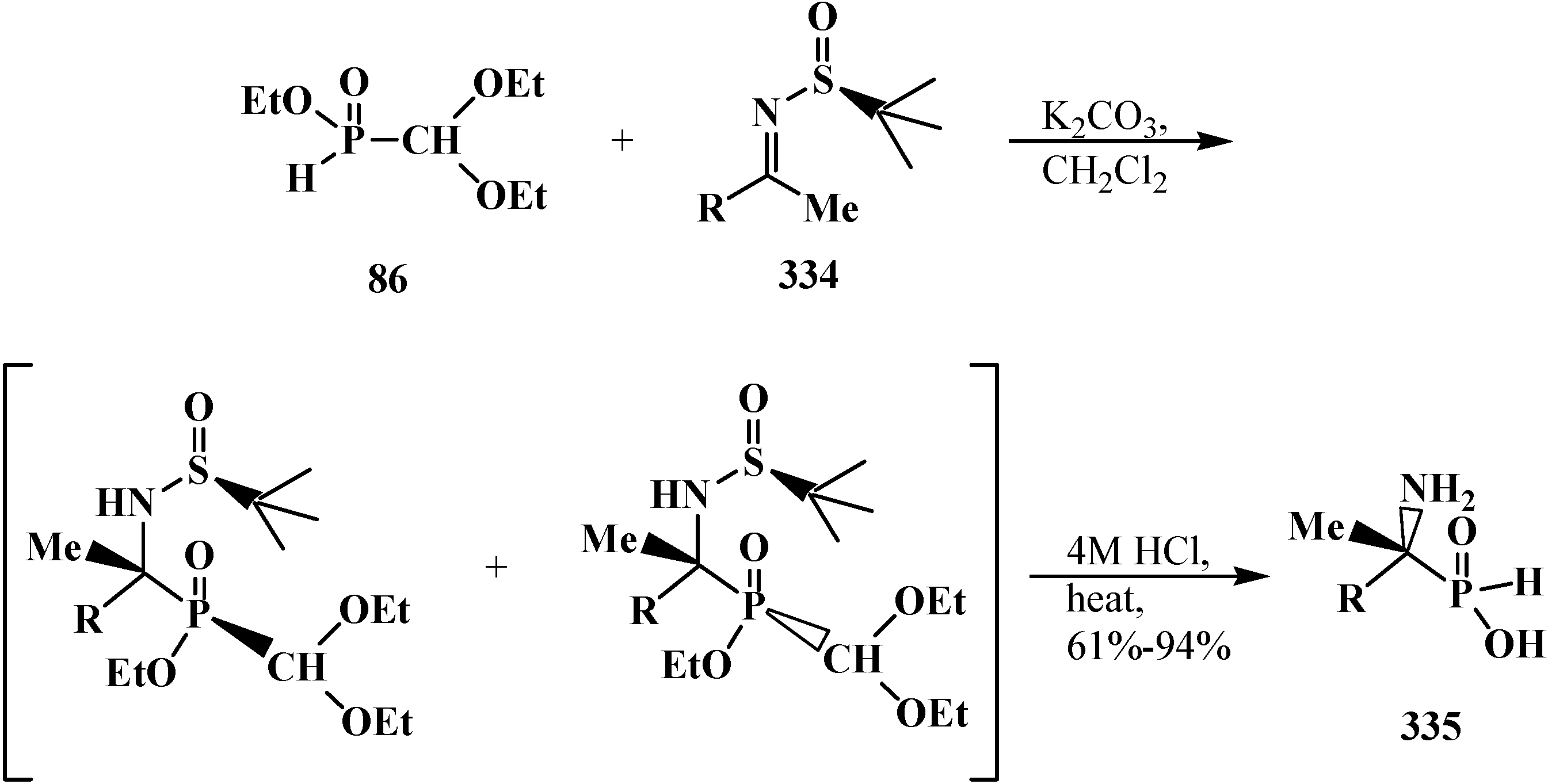
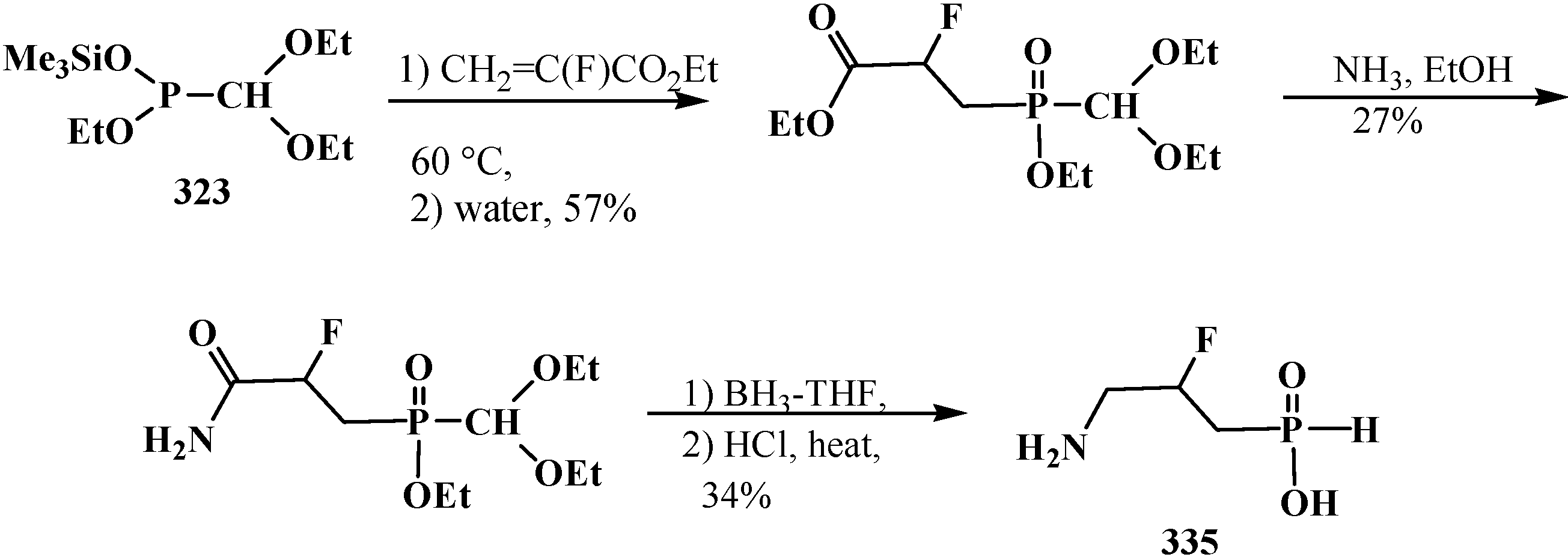
6. Phosphorylated Formaldehyde Hydrates—Geminal Diols 35. Syntheses and Chemical Properties
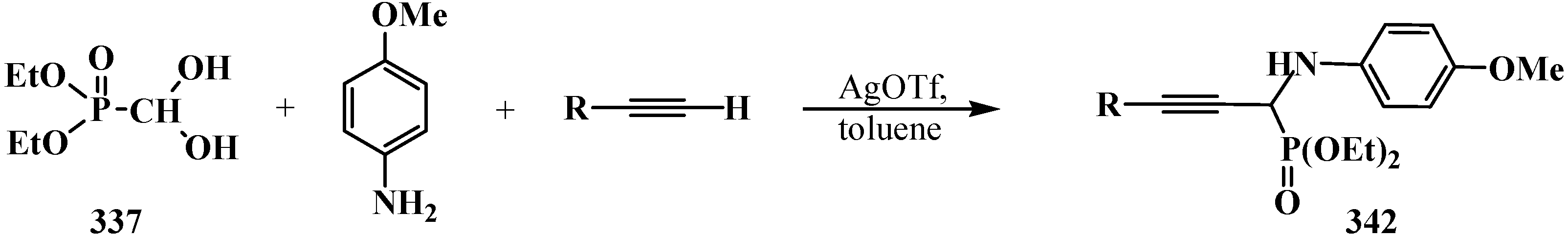
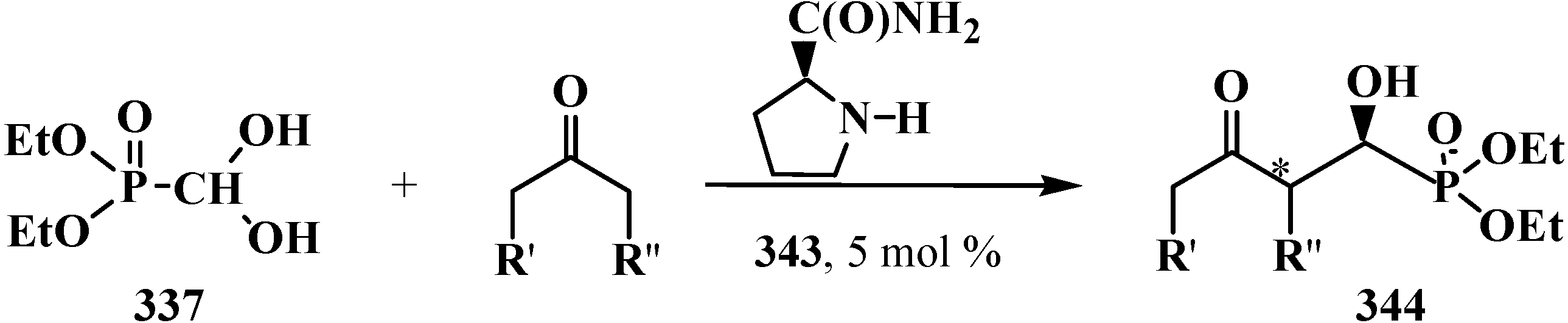
7. Conclusions
Conflicts of Interests
References
- Mitsuo, S.; Masaki, S.; Hikaru, Y.; Tsujiaki, H. Acylphosphonates: P-C Bond Cleavage of Dialkyl Acylphosphonates by Means of Amines. Substituent and Solvent Effects for Acylation of Amines. J. Org. Chem. 1980, 45, 4162–4167. [Google Scholar]
- Miller, A.; Stewart, D. Reactions of Carbonyl Compounds with Tervalent Phosporus Reagents. Part 8.l, Acetyldiphenylphosphine Oxide. J. Chem. Soc. Perkin Trans. I 1977, 17, 1898–1901. [Google Scholar]
- Frey, G.; Lesiecki, H.; Lindner, E.; Vordermaier, G. Synthese und reaktives Verhalten von Acyldiorganylphosphanoxiden. Chem. Ber. 1979, 112, 763–772. [Google Scholar] [CrossRef]
- Sekine, M.; Kume, A.; Nakajima, M.; Hata, T. A new method for acylation of enolates by means of dialkyl acylphosphonates as acylating agents. Chem. Lett. 1981, 10, 1087–1090. [Google Scholar] [CrossRef]
- Shagidullin, R.R.; Plyamovatyi, A.K.; Mukhamadeeva, R.M.; Khairullin, V.K.; Pudovik, A.N. Fourier IR—Spectroscopic study of the mechanism of anomalous Shiff base reaction with phosphorus-containing CH—Acids. Z. Obshch. Khim. 1994, 64, 926–930. [Google Scholar]
- Conti, P.; Pinto, A.; Tamborini, L.; Rizzo, V.; de Micheli, C. A regioselective route to 5-substituted pyrazole- and pyrazoline-3-phosphonic acids and esters. Tetrahedron 2007, 63, 5554–5560. [Google Scholar] [CrossRef]
- Conti, P.; Pinto, A.; Tamborini, L.; Dunkel, P.; Gambaro, V.; Visconti, G.L.; de Micheli, C. A regioselective route to 5-substituted isoxazole- and isoxazoline-3-phosphonates. Synthesis 2009, 591–596. [Google Scholar] [CrossRef]
- Vasella, A.; Voeffray, R. Asymmetric Synthesis of α- Aminophosphonic Acids by Cycloaddition of N-Glycosyl-C- dialkoxyphosphonoylnitrones. Helv. Chim. Acta 1982, 65, 1953–1964. [Google Scholar] [CrossRef]
- Moskva, V.V.; Mavrin, V.Y. O,O-Diethylformylphosphonate. J. Gen. Chem. USSR 1988, 57, 2492. [Google Scholar]
- Iorga, B.; Eumery, F.; Mouriès, V.; Savignac, P. Phosphorylated aldehydes. Tetrahedron 1998, 54, 14637–14677. [Google Scholar] [CrossRef]
- Groß, H. Zur Existenz von Estern der Formylphosphonsäure. Z. Chem. 1977, 17, 131–132. [Google Scholar]
- Amsallem, D.; Gornitzka, H.; Baceiredo, A.; Bertrand, G. New types of stable aldehydes: Formylphosphane and formylphosphane oxide. Angew. Chem. Int. Ed. Engl. 1999, 38, 2201–2203. [Google Scholar] [CrossRef] [PubMed]
- Razumov, A.I.; Liorber, B.G.; Moskva, V.V.; Sokolov, M.P. Phosphorylated aldehydes. Russ. Chem. Rev. 1973, 42, 538–550. [Google Scholar] [CrossRef]
- Wagenknecht, J. An electrochemical method for the preparation of iminodimethylenediphosphonic Acid. Synth. React. Inorg. Met. Org. Chem. 1974, 4, 567–572. [Google Scholar] [CrossRef]
- Firestone, R.A. Hydroxy and Imino Containing Phosphonic Acid Diesters. U.S. Patent 3784590, 8 January 1974. [Google Scholar]
- Mührle, H.; Vetter, W. Reaktionsbeteiligung von Phosphoester-Nachbargruppen bei Aminodehydrierungen. Z. Naturforsch. B 1988, 43, 1663–1671. [Google Scholar]
- Franczyk, T.S., II. Preparation of Formylphosphonic Acid from Tertiary Aminomethylphosphonic Acid N-oxides. U.S. Patent 6274760, 14 August 2001. [Google Scholar]
- Costisella, B.; Gross, H. Synthese und NMR-Spektren offenkettiger und cyclischer Acetale von Formylphosphonsäureestern und Formylphosphinoxid. J. Prakt. Chem. 1977, 319, 8–16. [Google Scholar] [CrossRef]
- Livantsov, M.V.; Proskurina, M.V.; Prishchenko, A.A.; Lutsenko, I.F. Synthesis and some properties of phosphorylated formals. J. Gen. Chem. USSR 1984, 54, 2504–2517. [Google Scholar]
- Brunjes, M.; Kujat, C.; Monenschein, H.; Kirschning, A. Acylation of Alkyl Halides and Amino Aldehydes with Phosphane Oxide-Based d1-Synthon. Eur. J. Org. Chem. 2004, 5, 1149–1160. [Google Scholar] [CrossRef]
- Gross, H.; Keitel, I.; Costisella, B.; Mikołajczyk, M.; Midura, W. Zur Synthese von bis-alkylmercaptomethanphosphonsäuredialkylestern. Phosphorus Sulfur Silicon Relat. Elem. 1983, 16, 257–262. [Google Scholar] [CrossRef]
- Młotkowska, B.; Gross, H.; Costisella, B.; Mikołajczyk, M.; Grejszczak, S.; Zatorski, A. Synthese offenkettiger und cyclischer S,S-Acetale von Formyl phosphonsäurestern. J. Prakt. Chem. 1977, 319, 17–22. [Google Scholar]
- Mikołajczyk, M.; Grejszczak, S.; Zatorski, A.; Młotkowska, B.; Gross, H.; Costisella, B. A new and general synthesis of ketene S,S- and S,O-Acetals based on the Horner-Wittig reaction. Tetrahedron 1978, 34, 3081–3088. [Google Scholar]
- Costisella, B.; Gross, H. 1-Dimethylamino-1-cyano methanphosphonsдurediethylester, ein neues Edukt zur Darstellung von Carbonsäuren, 1-Cyanoenaminen und Homoenolaten. Tetrahedron 1982, 38, 139–145. [Google Scholar] [CrossRef]
- Costisella, B.; Gross, H. 1-Cyano-1-dimethylamino methanphosphonsäurediethylester-Lithium—Ein “Instant-Horner-Reagent”. Z. Chem. 1987, 27, 143–144. [Google Scholar] [CrossRef]
- Marrero, Y.; Harwood, L.M. Towards the total synthesis of colletofragaranes: Constructing the macrocyclic lactone by high pressure-mediated intramolecular Diels-Alder reaction. Tetrahedron Lett. 2009, 50, 3574–3576. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Synthesis of carboxylic acids via PO-activated olefination of tetraethyl dimethilamino-methylene-diphosphonate. Angew. Chem. Int. Ed. Engl. 1968, 7, 391–392. [Google Scholar] [CrossRef]
- Qian, D.Q.; Shi, X.D.; Cao, K.Z.; Liu, L.Z. The Synthesis and Reactivity of Alkylaminosubstitutedmethylenediphosphonates. Heteroat. Chem. 1999, 10, 271–276. [Google Scholar] [CrossRef]
- McNulty, J.; Das, P. Development of one-pot method for the homologation of aldehides to carboxylic acids. Tetrahedron 2009, 65, 7794–7800. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Über α-substituerte Phosphonate, XI. Liebiegs Ann. Chem. 1971, 750, 44–52. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Derivate des Formylphosphonsäurediäthylestern. J. Prakt. Chem. 1969, 311, 925–929. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B.; Haase, L. Zur Reaction von Dimethylformamidederivaten mit Phosphorigsäurediestern und Phosphinoxiden. J. Prakt. Chem. 1969, 311, 577–585. [Google Scholar] [CrossRef]
- Risch, N.; Piper, S.; Winter, A.; Lefarth-Risse, A. An Efficient Synthesis of Novel α-Aminophosphonates Based on a Mannich-Type Reaction. Eur. J. Org. Chem. 2005, 2, 387–394. [Google Scholar] [CrossRef]
- Schrader, T.; Steglich, W. Phosphorus analogs of amino acids. IV. Syntheses of unusual 1-aminophosphonic acids via Diels-Alder reactions of diethyl (N-acyliminomethyl)phosphonates. Synthesis 1990, 1153–1156. [Google Scholar]
- Mikołajczyk, M.; Costisella, B.; Grejszczak, S.; Zatorski, A. Synthesis of O,S-thioacetals of formylphosphonates. Tetrahedron Lett. 1976, 17, 477–480. [Google Scholar]
- Kim, T.H.; Uh, D.Y. New synthetic method of O,S-thioacetals of formylphosphonates. Tetrahedron Lett. 1985, 26, 3479–3482. [Google Scholar] [CrossRef]
- Costisella, B.; Keitel, I. Synthese und Reaktionen von 1-Acetoxy-1-methylthio- methanphosphorylverbindungen. Phosphorus Sulfur Silicon Relat. Elem. 1988, 40, 161–165. [Google Scholar] [CrossRef]
- Mavrin, V.Y.; Moskva, V.V. Reaction of trihetero-substituted carbonium salts with diethyl phosphite. J. Gen. Chem. USSR 1988, 58, 1930–1931. [Google Scholar]
- Mavrin, V.Y.; Moskva, V.V. Condensation of diethyl phosphite with mixed derivatives of orthocarboxylic acid. J. Gen. Chem. USSR 1988, 58, 1490. [Google Scholar]
- Gross, H.; Seibt, H. Synthese von α-chlor-α-methoxy-methanphosphonsäureestern und α-chlor-α-methylthio- methanphosphonsäureestern. J. Prakt. Chem. 1970, 312, 475–482. [Google Scholar] [CrossRef]
- Petrov, K.A.; Chauzov, V.A.; Agafonov, S.V.; Pazhitkova, I.V. Methoxymethanediphosphonic acid esters. J. Gen. Chem. USSR 1980, 50, 1525–1526. [Google Scholar]
- Galli, R.; Scaglioni, L.; Palla, O.; Gozzo, F. Synthesis of 2-alkylthyo(or trifluoromethylthio)-2-halogenothenyl deryvatives by of Wittig (under phase transfer conditions)or Wittig-Horner reactions. Applications in the field of pyretroids. Applications in the field of pyretroids. Tetrahedron 1984, 40, 1523–1532. [Google Scholar]
- Kim, T.H.; Uh, D.Y. Synthesis of S,S-thioacetals of formylphosphonate from chloro(arylthio)methanephosphonate. Synth. Commun. 1988, 18, 1611–1614. [Google Scholar] [CrossRef]
- Otten, P.A.; Davies, H.M.; van der Gen, A. A Horner-Wittig Synthesis of 1-Chlorovinyl Sulfoxides. Tetrahedron Lett. 1995, 36, 781–784. [Google Scholar] [CrossRef]
- Otten, P.A.; Davies, H.M.; van der Gen, A. A Horner-Wittig Synthesis of 1-Chlorovinyl Sulfoxides. Phosphorus Sulfur Silicon Relat. Elem. 1996, 109, 449–452. [Google Scholar]
- Dinizo, S.E.; Freerksen, R.W.; Rabst, W.E.; Watt, D.S. Synthesis of α-Alkoxyacrylonitriles Using Substituted Diethyl Cyanomethylphoaphonates. J. Org. Chem. 1976, 41, 2846–2849. [Google Scholar] [CrossRef]
- Dinizo, S.E.; Freerksen, R.W.; Rabst, W.E.; Watt, D.S. A One-Carbon Homologation of Carbonyl Compounds to Carboxylyc Acids, Esters and Amides. J. Am. Chem. Soc. 1977, 99, 182–186. [Google Scholar] [CrossRef]
- Diamond, P.M.; Dinizo, S.E.; Freerksen, R.W.; Curtis, R.; Haltiwanger, R.C.; Watt, D.S. Ferric Chloride-catalysed Conversion of α-t-butoxy- or α-acetoxy-acrylonitryles into Imides. J. Chem. Soc. Chem. Commun. 1977, 298–299. [Google Scholar] [CrossRef]
- Koidan, G.N.; Marchenko, A.P.; Oleinik, V.A.; Pinchuk, A.M. 2-Methyl-2-tribromophosphazopropane. J. Gen. Chem. USSR 1988, 58, 1304–1309. [Google Scholar]
- Savignac, P.; Coutrot, P. Preparation of 1,1-dibromoalkanes by Halogen Exchange. Synthesis 1976, 197–199. [Google Scholar] [CrossRef]
- Majewski, P.; Koszuk, J.F. Synthesis of Dichloromethylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2009, 184, 956–962. [Google Scholar] [CrossRef]
- Bulpin, A.; Masson, S.; Sene, A. Reaction of Phosphonodithioformates with Nucleophilic Reagents; Potential synthetic Uses. Phosphorus Sulfur Silicon Relat. Elem. 1990, 49/50, 135–138. [Google Scholar]
- Costisella, B.; Oczegowski, S.; Gross, H. α-Substituierte phosphonate. 70 Untersuchungen über Methylthiomethanbisphosphorylderivate. Phosphorus Sulfur Silicon Relat. Elem. 1994, 86, 169–175. [Google Scholar]
- Binder, J.; Zbiral, E. A new procedure for homologation of carbonyl compounds to α-hydroxy-carboxylic esters by means of diethyl-[trimethylsilylethoxymethyl]phosphonate. Tetrahedron Lett. 1986, 27, 5829–5832. [Google Scholar] [CrossRef]
- Dufrechon, S.; Combert, J.-C.; Malhiac, C.; Collignon, N. Efficient synthesis of α-enamino phosphonates in the series of piperidine and morpholine. Phosphorus Sulfur Silicon Relat. Elem. 1997, 127, 1–14. [Google Scholar] [CrossRef]
- Mikołajczyk, M. α-Heterosubstituted phosphonates and phosphineoxides. Pure Appl. Chem. 1987, 59, 983–988. [Google Scholar]
- Gallagher, M.J.; Honegger, H. Dialkoxymethylation of phosphorus with trialkyl orthoformates: Reactions of phosphonic and phosphinic acids via their trivalent tautomers. Tetrahedron Lett. 1977, 18, 2987–2990. [Google Scholar] [CrossRef]
- Coudray, L.; Montchamp, J.-L. Temporary Protection of H-Phosphinic Acids as a Synthetic Strategy. Eur. J. Org. Chem. 2009, 27, 4646–4654. [Google Scholar] [CrossRef]
- Dingwall, Y.G.; Ehrenfreund, I.; Hall, R.G. Diethoxymethylphosphonites and phosphinates. Intermediates for the synthesis of α, β and γ-aminoalkylphosphinous acids. Tetrahedron 1989, 45, 3787–37808. [Google Scholar]
- Hamilton, R.; McKervey, M.A.; Rafferty, M.D.; Walker, B.J. The Reaction of Dimethyl Dioxirane with Diazomethylphosphonates; the First Synthesis of a Formylphosphonate Hydrate. J. Chem. Soc. Chem. Commun. 1994, 37–38. [Google Scholar] [CrossRef]
- Cairns, J.; Dunne, C.; Franczyk, T.S.; Hamilton, R.; Hardacre, C.; Stern, M.K.; Treacy, A.; Walker, B.J. The Synthesis and Chemistry of Formylphosphonate. Phosphorus Sulfur Silicon Relat. Elem. 1999, 144, 385–388. [Google Scholar] [CrossRef]
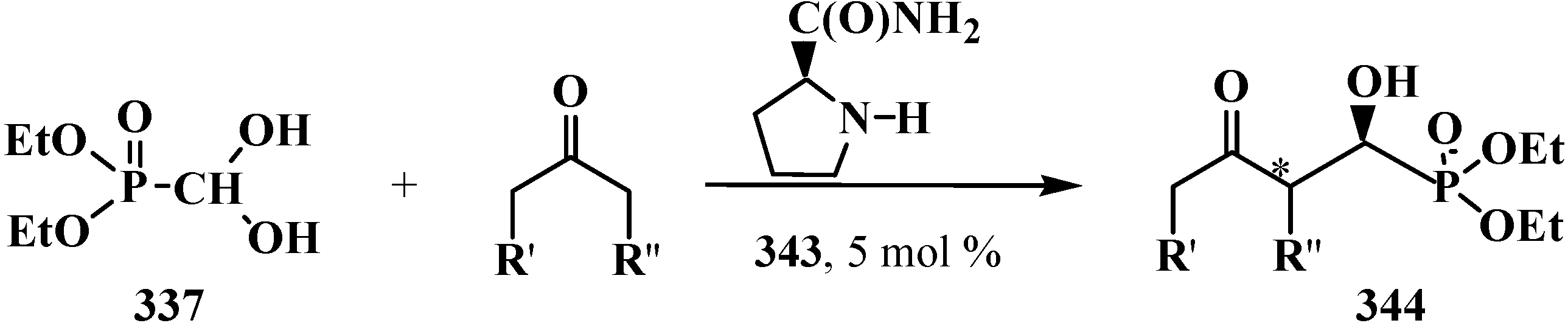
- Samanta, S.; Perera, S.; Zhao, C.-G. Organocatalytic Enantioselective Synthesis of Both Diastereomers of α-Hydroxyphosphinates. J. Org. Chem. 2010, 75, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Dodda, R.; Zhao, C.-G. Silver(I) triflate-catalyzed direct synthesis of N-PMP protected α-aminopropargylphosphonates from terminal alkynes. Org. Lett. 2007, 9, 165–167. [Google Scholar]
- Hirai, T.; Han, L.-B. Palladium-Catalyzed Ibsertion of Isocyanides into P(O)-H Bonds: Selective Formation of phosphinoyl Imines and Bisphosphinoylaminomethanes. J. Am. Chem. Soc. 2006, 128, 4722–4723. [Google Scholar] [CrossRef]
- Piotrowska, D.G. N-Substituted C-diethoxyphosphorylated nitrones as useful synthons for the synthesis of α-aminophosphonates. Tetrahedron Lett. 2006, 47, 5363–5366. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Balzarini, J.; Glowacka, I.E. Design, synthesis antiviral and cytostatic evaluation of novel isoxalidine nucleotide analogues with a 1,2,3-triazole linker. Eur. J. Med. Chem. 2012, 47, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Seufert, D.; Marmor, R.S.; Hilbert, P. Some reactions of dimethilphosphono-substituted diazoalkanes. (MeO)2P(O)CR transfer to olefins and 1,3-dipolar additions of (MeO)2P(O)CR'. J. Org. Chem. 1971, 36, 1379–1386. [Google Scholar]
- Maehr, H.; Uskokovic, M.R.; Schaffer, C.P. Concise synthesis of dimethyl(2-Oxopropyl)phosphonate and homologation of aldehydes and to alkynes in a tandem process. Synth. Commun. 2009, 39, 299–310. [Google Scholar] [CrossRef]
- Kosobokov, M.D.; Titnyuk, I.D.; Beletskaya, I.P. An expedient synthesis of diethyl diazomethylphosphonate. Mendeleev Commun. 2011, 21, 142–143. [Google Scholar] [CrossRef]
- Neidlein, R.; Keller, H. Syntheses of 1-Benzyloxyaminoalkylphosphonates. Heterocycles 1993, 36, 1925–1932. [Google Scholar] [CrossRef]
- Carter, W.A. Treatment of Human Viral Infection by Doublestranged RNA Combined with Viral Infections. Eur. Patent 286224, 12 October 1988. [Google Scholar]
- Franczyk, T.S., II. Method for Preparing Formylphosphonic Acid. U.S. Patent 7294733, 13 November 2007. [Google Scholar]
- Franczyk, T.S., II. Method of Making Phosphorus-containing Compounds and Products Thereof. U.S. Patent 6864218, 8 March 2005. [Google Scholar]
- Oediger, H.; Lieb, F.; Disselnkotter, H. Phosphonoformaldehyde,a process for its preparation and its use as an intermediate product for the preparation of medicaments. U.S. Patent 4,348,332, 7 September 1982. [Google Scholar]
- Livantsov, M.V.; Boiko, V.I.; Proskurina, M.V.; Lutsenko, I.F. Phosphorylation of orthoformates. J. Gen. Chem. USSR 1982, 52, 811. [Google Scholar]
- Razumov, A.I.; Moskva, V.V. Reaction of dialkylphosphorous acids with orthoformic esters. Z. Obshch. Khim. 1964, 34, 3125–3126. [Google Scholar]
- Razumov, A.I.; Moskva, V.V. Reaction of orthoformates with hydrogen phosphited and hydrogen phosphinites. J. Gen. Chem. USSR 1965, 35, 1599. [Google Scholar]
- Beznosko, B.K.; Usanova, V.M.; Zhuravleva, L.V.; Kharitonov, A.V.; Bondarenko, N.A.; Yarkevich, A.N.; Antoshin, A.E.; Tsvetkov, E.N. Antiinflammatory and analgesic activity of quaternary phosphine oxides. Pharm. Chem. J. 1990, 24, 244–247. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Reaktion von phosphoriger Säure bzw. deren Anhydrid mit Orthoameisensäureestern. J. Prakt. Chem. 1974, 311, 550–556. [Google Scholar]
- Gross, H.; Costisella, B. O,O-and S,S-Acetals of formylphosphoniumsalts. J. Prakt. Chem. 1978, 320, 128–132. [Google Scholar] [CrossRef]
- Gross, H.; Freiberg, J.; Costisella, B. Zur existanz von halogendialkoxyalkanen, eine einfache synthese von dialkoxymethanphosphonaten. Chem. Ber. 1968, 101, 1250–1256. [Google Scholar] [CrossRef]
- Malenko, D.M.; Gololobov, Y.G. New method for synthesis of phosphorylated formals. J. Gen. Chem. USSR 1981, 51, 1214–1215. [Google Scholar]
- Groß, H.; Costisella, B. Zur Umsetzung von Dimethylformamid-dimethyl-acetal mit Phosphortrichlorid. Z. Chem. 1970, 10, 404–405. [Google Scholar]
- Costisella, B.; Gross, H. Zur Reaktion von Orthoameisensäureestern mit Phosphor-III-Verbindungen. J. Prakt. Chem. 1969, 311, 571–576. [Google Scholar] [CrossRef]
- Krokhina, S.S.; Pyrkin, R.I.; Levin, Y.A.; Ivanov, B.E. Effect of triethyl othoformate on some trivalent phosphorus derivatives. Russ. Chem. Bull. 1968, 17, 1349. [Google Scholar] [CrossRef]
- Prishchenko, A.A.; Livantsov, M.V.; Novikova, O.P.; Livantsova, L.I.; Petrosyan, V.S. Synthesis and reactivity of substituted α-carbonylphosphonites and their derivatives. Heteroat. Chem. 2012, 23, 352–372. [Google Scholar] [CrossRef]
- Dietsche, W. Darstellung von C-phosphorylierten Formaldehydacetalen. Liebigs Ann. Chem. 1968, 712, 21–27. [Google Scholar] [CrossRef]
- Moskva, V.V.; Maikova, A.I.; Razumov, A.I. Reaction of orthocarboxylates and acetals with trivalent phosphorus acid chlorides. J. Gen. Chem. USSR 1969, 39, 563–566. [Google Scholar]
- Costisella, B.; Gross, H. Notiz zur Synthese unsymmetrisch substituirten derivate der diätoxymethanphosphonsäure. J. Prakt. Chem. 1977, 319, 343–346. [Google Scholar] [CrossRef]
- Krutskaya, L.V.; Safiulina, O.Z.; Voronina, S.G.; Bryukhovetskaya, L.V.; Tsivunin, V.S. On the reaction of 2-chloro-1,3,2-dioxaphospholanes with 2-ethoxy-1,3-dioxolanes. J. Gen. Chem. USSR 1989, 59, 2408–2411. [Google Scholar]
- Tsivunin, V.S.; Krutskii, L.N.; Ernazarov, M.; Kamai, G.K. Reaction of the diethylamido chloride of ethylphosphinous acid and ethyldichlorophosphine with orthoformic esters. J. Gen. Chem. USSR 1970, 40, 2551–2553. [Google Scholar]
- Costisella, B.; Gross, H. Zur reaktion von P(III) amiden mit orthoameisensäureesternchloriden. Phosphorus Sulfur Silicon Relat. Elem. 1980, 8, 99–103. [Google Scholar] [CrossRef]
- Młоtkowska, B.; Costisella, B.; Gross, H. Eine einfache Methode zur Synthese cyclischer Acetale des Formylphosphonsäreesters. J. Prakt. Chem. 1974, 316, 913–916. [Google Scholar]
- Bennett, S.N.L.; Hall, R.G. New syntheses of arylphosphinic acids from the reaction of ethyl diethoxymethylphosphinate with aryl bromides and phenols. J. Chem. Soc. Perkin Trans. I. 1995, 1145–1151. [Google Scholar] [CrossRef]
- Costisella, B.; Gross, H. Zur reaktivität von Acetalen des Formylphosphonsäureesters. J. Prakt. Chem. 1971, 313, 265–276. [Google Scholar] [CrossRef]
- Niyazymbetov, M.S.; Costisella, B.; Kaitel, I.; Shvatrz, K.K. Cathode-catalyzed transesterification of alkylphosphonates. Russ. Chem. Bull. 1991, 40, 182–186. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Graczyk, P.P.; Wiechorek, M.W.; Bujacz, G. A solution and solid state conformation of 2-diphenylphosphinoyl-1,3-dioxanes. The nature of O-C-P anomeric interactions. Tetrahedron 1992, 48, 4209–4230. [Google Scholar]
- Gross, H.; Böck, C.; Costisella, B.; Gloede, J. Entalkylierung von Phosphonoesthers mit labilen funktionellen Gruppen mittels Trimethylsilyl-Bromid. J. Prakt. Chem. 1978, 320, 344–350. [Google Scholar] [CrossRef]
- Rol’nik, L.Z.; Pastushenko, E.V.; Livantsov, M.V.; Proskurnina, M.V.; Zlotskii, S.S.; Rakhmankulov, D.L. Free-radical reactions of dialkyl formylphosphonate phosphonacetals. J. Gen. Chem. USSR 1983, 53, 1104–1109. [Google Scholar]
- Razumov, A.I.; Gurevich, P.A.; Liorber, B.G.; Borisova, T.B. A study in the series of phosphinic and phosphinous acid derivatives. J. Gen. Chem. USSR 1969, 39, 369–372. [Google Scholar]
- Nguyen-Ba, P.; Lee, N.; Mitchell, H.; Chan, L.; Quimpere, M. Design and synthesis of a novel class nucleotide analogs with anti-HCMV activity. Bioorg. Med. Chem. Lett. 1998, 8, 3555–3560. [Google Scholar]
- Kruse, C.G.; Broekhof, N.L.Y.M.; Wijsman, A.; van der Gen, A. Synthetic application of 2-chloro-1,3-dithiane preparation of ketene dithioacetals. Tetrahedron Lett. 1977, 18, 885–888. [Google Scholar] [CrossRef]
- Ishikawa, K.; Akiba, K.; Inamoto, N. Synthesis of 1, 4-benzodithiofulvenes via Wittig reaction. Tetrahedron Lett. 1976, 17, 3695–3698. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Graczyk, P.P.; Wieczorek, M.W. Conformational preference in 1,3-dithianes containing 2-phosphoryl, -(thiophosphoryl), and (selenophosphoryl)groups. Chemical and crystallographic implications of the nature of the anomeric effect. J. Org. Chem. 1994, 59, 1672–1679. [Google Scholar]
- Mikołajczyk, M.; Łuczak, T.; Graczyk, P.P.; Wieczorek, M.W.; Błaszczyk, Y.; Bujacz, G.D.; Majzner, W.R. Solid state conformation of the anomeric effect in 2-phosphoryl, 2- thiophosphoryl and 2-selenophosphoryl-substituted 1,3-dithiolans. J. Organomet. Chem. 1997, 536–537, 355–360. [Google Scholar]
- Aggarwal, V.K.; Barnell, J.K.; Worrall, J.M.; Alexander, R. Highly diastereoselective epoxidation of ketene dithioacetal dioxides: A new approach to asymmetric synthesis of α-amino amides. J. Org. Chem. 1998, 63, 7128–7129. [Google Scholar] [CrossRef] [PubMed]
- Mikołajczyk, M.; Grejszczak, S.; Chefchyńska, A.; Zatorski, A. Addition of elemental sulfur to phosphonate carbanions and its application for synthesis of α-phosphoryl organosulfur compounds, synthesis of oaromatic ketones. J. Org. Chem. 1979, 44, 2967–2972. [Google Scholar]
- Mikołajczyk, M.; Bałczewski, P.; Grejszczak, S. Sulphenylation of phosphonates. A facile synthesis of α-phosphoryl sulfides and S,S-acetals of oxomethanephosphonates. Synthesis 1980, 127–129. [Google Scholar]
- Grayson, J.I.; Warren, S. Acyl Anion Equivalents: Synthesis of Ketones and Enones from α-Phenylthioalkylphosphine Oxides. J. Chem. Soc. Perkin Trans. I 1977, 2263–2272. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Bałczewski, P. Synthesis and reactivity of diethyl(methylthio)(trimethylsilyl)methylphosphonate. Synthesis 1989, 101–106. [Google Scholar]
- Makomo, H.; Masson, S.; Saquet, M. Reduction of phosphonodithioformates: Syntheses of α-phosphoryl thiols and hemidithioacetals. Tetrahedron Lett. 1994, 50, 10277–10288. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Mikina, M.; Graczyk, P.P.; Bałczewski, P. Synthesis of dithio- and diselenoacetals of formylphosphonates. Synthesis 1996, 1232–1238. [Google Scholar]
- Yuaristi, E.; Cordillo, B.; Valle, L. Relative reactivity of 2-diphenylphosphinoil- and 2-diphenylthiophosphinoil-2-[1,3]-dithianyllithium as reagents Wittig-Horner. Tetrahedron 1986, 42, 1963–1970. [Google Scholar] [CrossRef]
- Yuaristi, E.; Valle, L.; Valenzuela, B.A.; Aguilar, M.A. S-C-P anomeric interactions. 4. Conformational analys of 2-(diphenylphosphinoil)-1,3-dithyane. J. Am. Chem. Soc. 1996, 108, 2000–2005. [Google Scholar]
- Iorga, B.; Mouriès, V.; Savigniac, P. Carbanionic displacement reactions of phosphorus. Synthesis and reactivity of 5,5-dimethyl-2-oxo-2-(1,1-dithian-2-yl)-1, 3, 2-dioxaphosphorinane. Bull. Chem. Soc. Fr. 1997, 134, 891–985. [Google Scholar]
- El-Wareth, A.; Sarhan, A.O.; Murakami, M.; Izumi, T. Synthesis of functionalized compounds related to π-extended tetrathiafulvenes with quinonoidal bearing a ferrocene moiety. Monatsh. Chem. 2002, 133, 1055–1066. [Google Scholar]
- Costisella, B.; Gross, H. 1-Trimethylammonium-1-diethylphosphono-1-cyanomethylid, ein stabilеs N-Ylid. J. Prakt. Chem. 1987, 324, 545–549. [Google Scholar] [CrossRef]
- Costisella, B. 1H, 13C, 31P, 15N, 17O-NMR investigations of α-substituted aminophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 1990, 51/52, 226. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Petrosyan, V.S. Reaction of some CH acids with dimethylformamide dimethylacetal. J. Gen. Chem. USSR 1993, 63, 1326–1327. [Google Scholar]
- Gross, H.; Costisella, B.; Gnauk, T.; Brenneke, L. Derivate der Aminomethan-bis-phosphonsäure. J. Prakt. Chem. 1976, 318, 116–126. [Google Scholar] [CrossRef]
- Nesterov, L.V.; Krepysheva, N.E.; Aleksandrova, N.A. Reaction of dimethylformamide dimethylacetal with silyl phosphites. J. Gen. Chem. USSR 1989, 59, 641. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Boganova, N.V.; Zhutskii, P.V.; Lutsenko, I.F. Synthesis of tetraalkyl dimethylaminomethylenediphosphorus-containing acids. J. Gen. Chem. USSR 1989, 59, 2132–2133. [Google Scholar]
- Prishchenko, A.A.; Livantsov, M.V.; Petrosyan, V.S. New types of aminomethyl organophosphorus compounds. J. Gen. Chem. USSR 1994, 63, 1181–1193. [Google Scholar]
- Degenhardt, C.R. Use of tetraethyl dimethylaminomethylene diphosphonate in the synthesis of benzothiophene-2-acetic acid and other carboxylic acids. Synth. Commun. 1982, 12, 415–421. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B. Bis(diethoxyphosphinyl)(trimethylammonio)-methylide, a Stable N-Ylide. Angew. Chem. Int. Ed. Engl. 1968, 7, 463. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B.; Bürger, W. Synthese von Alkylphosphonbetainen und stabilen N-Ylyden aus α-Trimethylammoniummethylphosphonaten. J. Prakt. Chem. 1969, 311, 563–570. [Google Scholar] [CrossRef]
- Monenschein, H.; Dräger, G.; Alexander, Jung; Kirschning, A. Asymmetric Nucleophilic Acylation of Aldehydes via 1, 1-Heterodisubstituted Alkenes. Chem. A Eur. J. 1999, 5, 2270–2280. [Google Scholar]
- Kirschning, A.; Kujat, C.; Luiken, S.; Schaumann, E. Small and Versatile—Formyl Anion and Dianion Equivalents. Eur. J. Org. Chem. 2007, 2387–2400. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Midura, W.; Grejszczak, S. Synthesis of mono- and 1, 4-dicarbonyl compounds based on the oxygenation of phosphonate carbanions. Synthesis of dihydrojasmone, allethone and methylenomycin B. Tetrahedron Lett. 1984, 25, 2489–2492. [Google Scholar]
- Morita, T.; Okamoto, Y.; Sackurai, H. Dealkylation reaction of acetals, phosphonate, and phosphate esters with chlorotrimethylsilane/metal halide reagent in acetonitrile, and its application to the synthesis of phosphonic acids and vinyl phosphonates. Bull. Chem. Soc. Jpn. 1981, 54, 267–273. [Google Scholar] [CrossRef]
- Morita, T.; Okamoto, Y.; Sackurai, H. A convenient dealkylation of dialkyl phosphonates by chlorotrimethylsilane in the presence of sodium iodide. Tetrahedron Lett. 1978, 28, 2523–2526. [Google Scholar] [CrossRef]
- Gross, H.; Keitel, I. Heterosubstituierte olefine durch horner-reaktion mit α-substituierten Phosphonaten. Z. Chem. 1982, 22, 117–126. [Google Scholar]
- Van Schaik, T.A.M.; Henzen, A.V.; van der Gen, A. A Horner-Wittig solution to the synthesis of ketene, O, O-acetals. Tetrahedron Lett. 1983, 24, 1303–1306. [Google Scholar]
- Streitwieser, A., Jr.; Juaristi, E. Carbon acidity. Equilibrium ion pair acidies of some phosphorus-substituted carbon acids. J. Org. Chem. 1982, 47, 768–770. [Google Scholar]
- Afarinkia, K.; Faller, A.; Twist, A.J. A new synthesis of α-ketophosphonates. Synthesis 2003, 357–360. [Google Scholar] [CrossRef]
- Coutrot, P.; Grison, C.; Lecouvey, M. Preparation of the phosphonic acid analogue of 3-deoxy-d-manno-2-octulosonic acid (KDO). Tetrahedron Lett. 1996, 37, 1595–1598. [Google Scholar] [CrossRef]
- Horner, L.; Hoffmann, H.; Wippel, H.S. Phosphinoxide als Olefinierungsreagenzien. Chem. Ber. 1958, 91, 61–63. [Google Scholar] [CrossRef]
- Horner, L.; Hoffmann, H.; Wippel, H.S.; Klahre, G. Phosphinoxide als olefinierungsreagenzien. Chem. Ber. 1959, 92, 2499–2505. [Google Scholar] [CrossRef]
- Wittig, G.; Schöllkopf, U. Über Triphenyl-phosphin-methylene als olefinbildende Reagenzien (I Mitteilung). Chem. Ber. 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Wardsworth, W.S.; Emmons, W.D. The Utility of Phosphonate Carbanions in Olefins Synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar] [CrossRef]
- Martin, S.F. Synthesis of Aldehydes, Ketones, and Carboxylic Acids from Lower Carbonyl Compounds by C-C Coupling Reactions. Synthesis 1979, 633–665. [Google Scholar] [CrossRef]
- Korotchenko, V.N.; Nenaidenko, V.G.; Balenkova, E.S.; Shastin, A.V. Olefination of carbonyl compounds. The newest and classical methods. Russ. Chem. Rev. 2004, 73, 957–989. [Google Scholar]
- Costisella, B.; Keitel, I.; Gross, H. α-Phosphononoenamine und Acyl phosphonate durch Horner-Olefinierung. Tetrahedron 1981, 37, 1227–1232. [Google Scholar] [CrossRef]
- Gross, H.; Costisella, B.; Schick, H. γ-Butyrolactone aus metallierten Enamine. Tetrahedron 1984, 40, 733–736. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Greene, L.M.; Bergin, O.; Fichet, J.-B.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis, evaluation and structural studies of antiproliferative tubulin-targeting azetidin-2-ones. Bioorg. Med. Chem. 2011, 19, 2306–2325. [Google Scholar]
- Mikołajczyk, M.; Grzejszczak, S.; Zatorski, A.; Młotkowska, B. A new general synthesis of ketene thioacetals. Tetrahedron Lett. 1976, 31, 2731–2734. [Google Scholar]
- Mikołajczyk, M.; Bałczewski, P. Diverse reactivity of α-carbanions derived from α-phosphoryl dithioacetals and α-phosphoryl sulphides towards α, β-unsaturated carbonyl compounds. A general synthesis of conjugated ketene dithioacetals. Tetrahedron 1992, 8, 8697–8710. [Google Scholar]
- Ceruti, M.; Degani, I.; Fochi, R. A new synthetic application of 1, 2-benzodithiolium cations: synthesis of aldehydes by 1-carbon homologation of carbonyl compounds. Synthesis 1987, 79–83. [Google Scholar]
- Hanessian, S.; Maji, D.K.; Govindan, S.; Matera, R.; Tintelnot-Blomley, M. Substrate-Controlled and Organocatalytic Asymmetric Synthesis of Carbocyclic Amino Acid Dipeptide Mimetics. J. Org. Chem. 2010, 75, 2861–2876. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Desilets, D.; Rancourt, G.; Fortin, R. The total stereocontrolled synthesis of a chemical precursor to (+)-thienamycin. A formal synthesis of the antibiotic. Can. J. Chem. 1982, 60, 2292–2294. [Google Scholar]
- Moore, A.J.; Bryce, M.R. Generation and trapping of phosphorus stabilized 4,5-ethylenedithio-1,3-dithiol-2-ide carbanions: Synthesis of ethylenedithio-1,3-dithiafulvalenes. Synthesis 1991, 26–28. [Google Scholar] [CrossRef]
- Misaki, Y.; Nishikawa, H.; Kawakami, K.; Uehara, T.; Yamabe, T. Bis(2-methylidene-1,3-dithiolo[4,5-d])tetrathiafulvalene (BDT-TTF): A tetrathiafulvalene condensed with 1,3-dithiol-2-ylidene moieties. Tetrahedron Lett. 1992, 33, 4321–4324. [Google Scholar] [CrossRef]
- Heynderick, A.; Kaou, A.M.; Moustrou, C.; Samat, A.; Guglielmetti, R. Synthesis and photochromic behaviour of new Dipyrrolylperfluorocyclopentenes. New J. Chem. 2003, 27, 1425–1432. [Google Scholar] [CrossRef]
- Seebach, D.; Bürstinghaus, R. S-methyl thiocarboxylates from aldehydes and ketones through ketene thioacetals. Reductive nucleophile thiocarboxylation. Synthesis 1975, 461–462. [Google Scholar]
- Theil, F.; Costisella, B.; Mahrwald, R.; Gross, H.; Schick, H.; Schwarz, S. Prostaglandins and Prostaglandin Intermediates. XVII. Synthesis of the Main Metabolite of the PGF2α Analogue Cloprostenol. J. Prakt. Chem. 1986, 325, 435–440. [Google Scholar]
- Theil, F.; Costisella, B.; Groß, H.; Schick, H.; Schwarz, S. A Three-step Procedure for the Conversion of γ-Lactones into δ-Lactones. J. Chem. Soc. Perkin Trans. I 1987, 2469–2472. [Google Scholar] [CrossRef]
- Zimmerman, H.E.; Baker, M.R.; Bottner, R.C.; Morrissey, M.M.; Murphy, S. Photochemistry of some allenic counterparts of cyclohexenones and 2, 5-cyclohexadienones. J. Am. Chem. Soc. 1993, 115, 459–466. [Google Scholar] [CrossRef]
- Monenschein, H.; Brünjes, M.; Kirschning, A. Lithiated dimethoxymethyl diphenyl phosphine oxide. A versatile formiate carbanion equivalent. Synlett 2002, 525–527. [Google Scholar]
- Okada, H.; Mori, T.; Saikawa, Y.; Nakata, M. Formation of α-hydroxyketones via irregular Wittig reaction. Tetrahedron Lett. 2009, 50, 1276–1278. [Google Scholar] [CrossRef]
- Kirschning, A.; Dräger, G.; Yung, A. A new asymmetric formylation of aldehydes. Angew. Chem. Int. Ed. Engl. 1997, 36, 253–255. [Google Scholar] [CrossRef]
- Becker, H.; Sharpless, B. A new ligand class for the asymmetric dihydroxylation of olefins. Angew. Chem. Int. Ed. Engl. 1996, 35, 448–451. [Google Scholar] [CrossRef]
- Wittenberg, R.; Beier, C.; Dräger, G.; Jas, G.; Jasper, C.; Monenschein, H.; Kirschning, A. Towards the total synthesis of tonantzitlolone––Preparation of key fragments and the complete carbon backbone. Tetrahedron Lett. 2004, 45, 4457–4460. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.V.; Nifant’ev, E.E. Synthesis of polyfunctionalized methylphosphine oxides. Russ. Chem. Bull. 2012, 61, 380–385. [Google Scholar] [CrossRef]
- Schrader, T.; Kober, R.; Steglich, W. Synthese von 1-aminophosphonsäure-derivaten über acyliminophosphonsäure-ester. Synthesis 1986, 372–375. [Google Scholar] [CrossRef]
- Costisella, B.; Keitel, I.; Ozegowski, S. α-Substituierte Phosphonate. 69. Diastereoselektivität bei der Knüpfung der Phosphor-kohlenstoffbindung. Phosphorus Sulfur Silicon Relat. Elem. 1993, 84, 115–120. [Google Scholar]
- Marson, C.M. Reactions of carbonyl compounds with (monohalo) methyleniminium salts (Vilsmeier reagents). Tetrahedron 1992, 48, 3659–3726. [Google Scholar] [CrossRef]
- Winter, A.; Risch, N. The vinylogous mannich reaction: An efficient access to substituted nicotinonitriles. Synlett 2003, 13, 1959–1964. [Google Scholar]
- Morgalyuk, V.P.; Petrovskii, P.V.; Lysenko, K.A.; Nifant’ev, E.E. Synthesis of polyfunctionalyzed methylphosphine oxides. Russ. Chem. Bull. 2009, 58, 248–250. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.V. Stepwise scheme for the reaction of chlorodiphenylphosphine with N,N-dialkylformamides in the presence of NaI. Russ. J. Gen. Chem. 2011, 81, 2096–2101. [Google Scholar] [CrossRef]
- Morgalyuk, V.P.; Strelkova, T.V.; Nifant’ev, E.E. Reactivity of Chloro(dialkylamino)(diphenylphosphinoyl)methanes. Bull. Chem. Soc. Jpn. 2012, 85, 93–100. [Google Scholar] [CrossRef]
- Schrader, T.; Steglich, W. Phosphoranaloge von Aminosäuren IV. Synthesen ungevönlicher 1-Aminophosphonsäuren über Diels-Alder-Reaktionen von (N-Acyliminomethyl)phosphonsäurendiethylestern. Synthesis 1990, 1153–1156. [Google Scholar]
- Boukallaba, K.; Elachqar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Labriti, B.; Atmani, A. Synthesis of α-Heterocyclic α-Aminophosphonates, Part II: Morpholine, Piperidine, Pyrrolidine, Tetrahydrofurylmethylamine, N-Benzyl-N-Methylamine, and Aniline Derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 1045–1052. [Google Scholar] [CrossRef]
- Elachqar, A.; El Hallaouiq, A.; Roumestant, M.L.; Viallefont, P. Synthesis of Heterocyclic α-Aminophosphonic Acids. Synth. Commun. 1994, 24, 1279–1286. [Google Scholar] [CrossRef]
- Achamlale, S.; Mabrouk, H.; Elachqar, A.; El Hallaoui, A.; El Hajji, S.; Alami, A.; Bellan, J.; Mazières, M.R.; Wolf, J.G.; Pierrot, M. Synthesis and Thermal Isomerization of Carboxylic and Phosphonic α-Aminoesters Substituted With a Triazole Ring. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 357–367. [Google Scholar] [CrossRef]
- Gallagher, M.J.; Honegger, H. Organophosphorus intermediates. VI. The acid-catalysed reaction of trialkyl orthoformates with phosphinic acid. Aust. J. Chem. 1980, 33, 287–294. [Google Scholar]
- Fougère, C.; Guénin, E.; Hardouin, J.; Lecouve, M. Rapid and Efficient Synthesis of Unsymmetrical Phosphinic Acids R'P(O)OHR''. Eur. J. Org. Chem. 2009, 6048–6054. [Google Scholar]
- Baylis, E.K. 1,1-Diethoxyethylphosphinates and Phosphinites. Intermediates for the Synthesis Functional Phosphorus Acids. Tetrahedron Lett. 1995, 36, 9385–9388. [Google Scholar]
- Hall, R.G.; Riebli, P. Preparation of New Phosphine Oxides: Synthesis of an Analogue of Muscarinic Antagonists. Synlett 1999, 1633–1635. [Google Scholar] [CrossRef]
- Buckler, S.A.; Epstein, M. The preparation and reactions of primary phosphine oxides. Tetrahedron 1962, 3, 1221–1230. [Google Scholar] [CrossRef]
- Zhang, D.; Yuan, C. A concise and first synthesis of α-aminophosphinates with two stereogenic atoms leading to optically pure α-amino-H-phosphinic Acids. Chem. A Eur. J. 2008, 14, 6049–6052. [Google Scholar] [CrossRef]
- Alstermark, C.; Amin, K.; Dinn, S.R.; Elebring, T.; Fjellström, O.; Fitzpatrick, K.; Geiss, W.B.; Gottfries, J.; Guzzo, P.R.; Harding, J.P.; et al. Synthesis and pharmacological evaluation of novel γ-aminobutyric acid type B (GABAB) receptor agonists as gastroesophageal reflux inhibitors. J. Med. Chem. 2008, 51, 4315–4320. [Google Scholar]
- Froestl, W.; Mickel, S.J.; von Sprecher, G.; Diel, P.J.; Hall, R.G.; Maier, L.; Strub, D.; Melillo, V.; Baumann, P.A.; Bernasconi, R.; et al. Phosphinic acid analogues of GABA. 2. Selective, orally active GABAB antagonists. J. Med. Chem. 1995, 38, 3313–3331. [Google Scholar]
- Seebach, D. Methoden und möglichkeiten der nucleophilen acylierung. Angew. Chem. Int. Ed. Engl. 1969, 81, 690–700. [Google Scholar] [CrossRef]
- Seebach, D. Methods of reactivity umpolung. Angew. Chem. Int. Ed. Engl. 1979, 18, 39–336. [Google Scholar]
- Dodda, R.; Zhao, C.-G. Organocatalytic highly enantioselective synthesis of secondary α-hydroxyphosphonates. Org. Lett. 2006, 8, 4911–4914. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Morgalyuk, V.P. Chemistry of Phosphorylated Formaldehyde Derivatives. Part I. Molecules 2014, 19, 12949-13009. https://doi.org/10.3390/molecules190912949
Morgalyuk VP. Chemistry of Phosphorylated Formaldehyde Derivatives. Part I. Molecules. 2014; 19(9):12949-13009. https://doi.org/10.3390/molecules190912949
Chicago/Turabian StyleMorgalyuk, Vasily P. 2014. "Chemistry of Phosphorylated Formaldehyde Derivatives. Part I" Molecules 19, no. 9: 12949-13009. https://doi.org/10.3390/molecules190912949
APA StyleMorgalyuk, V. P. (2014). Chemistry of Phosphorylated Formaldehyde Derivatives. Part I. Molecules, 19(9), 12949-13009. https://doi.org/10.3390/molecules190912949