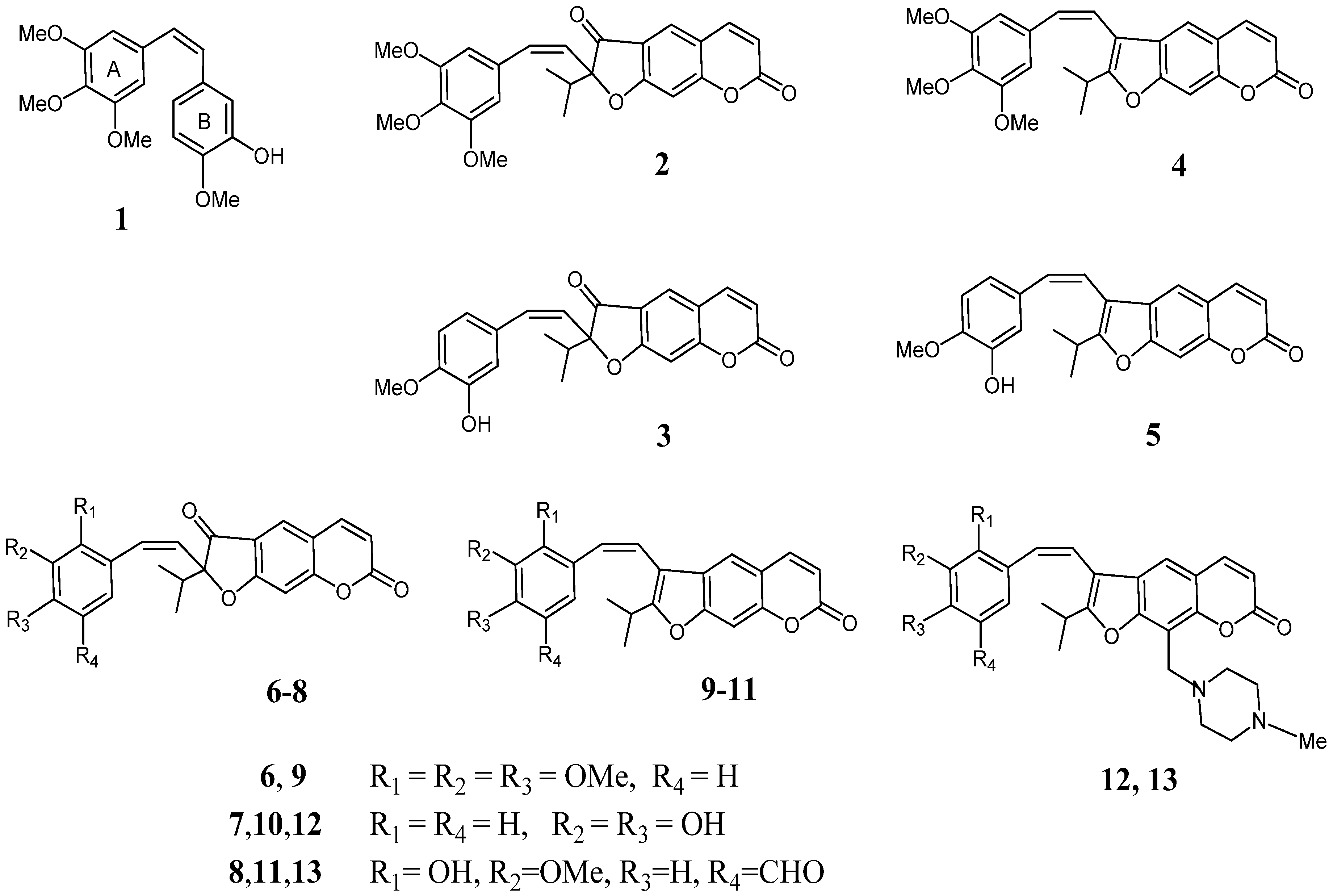
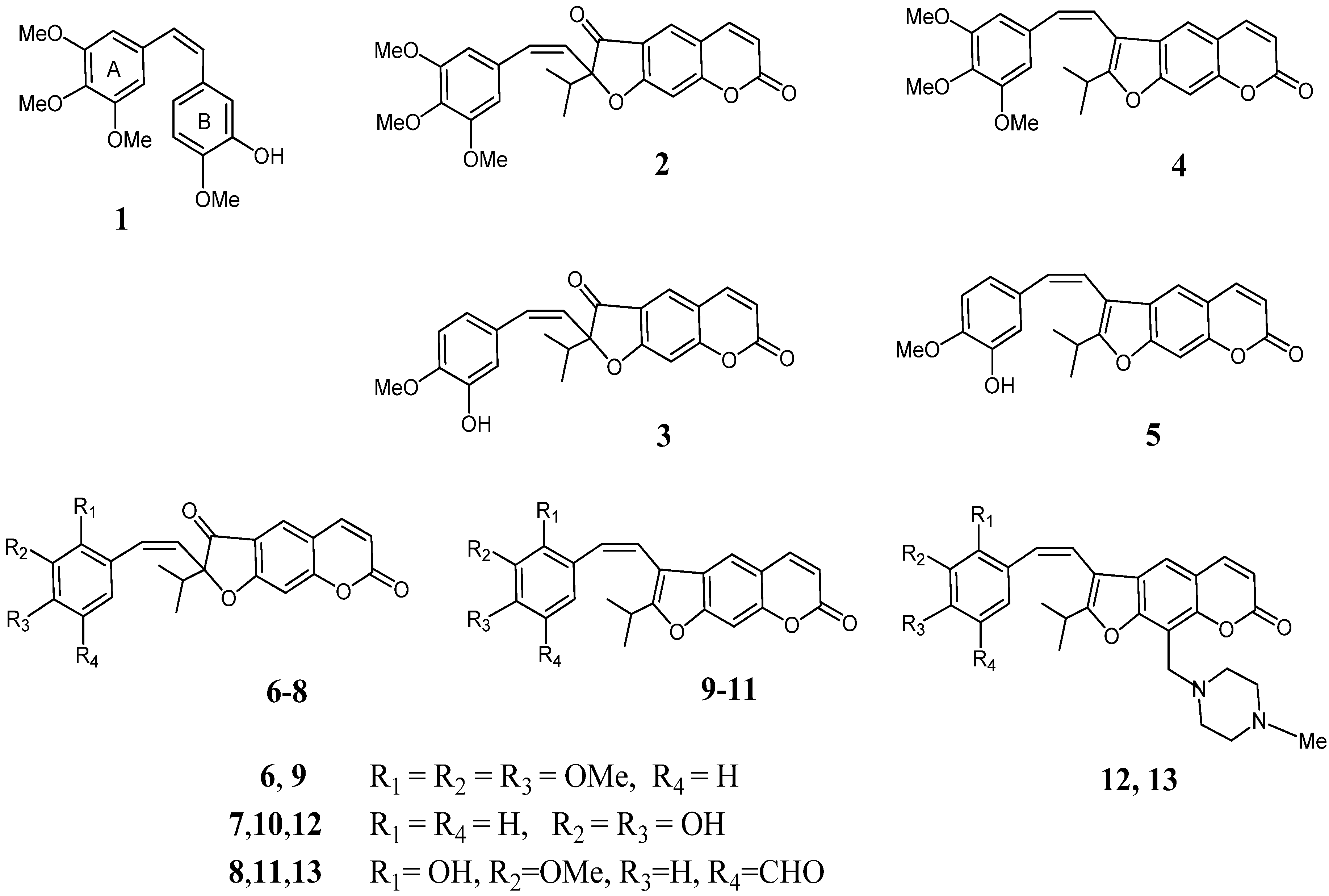
3.2. Synthesis and Spectral Data
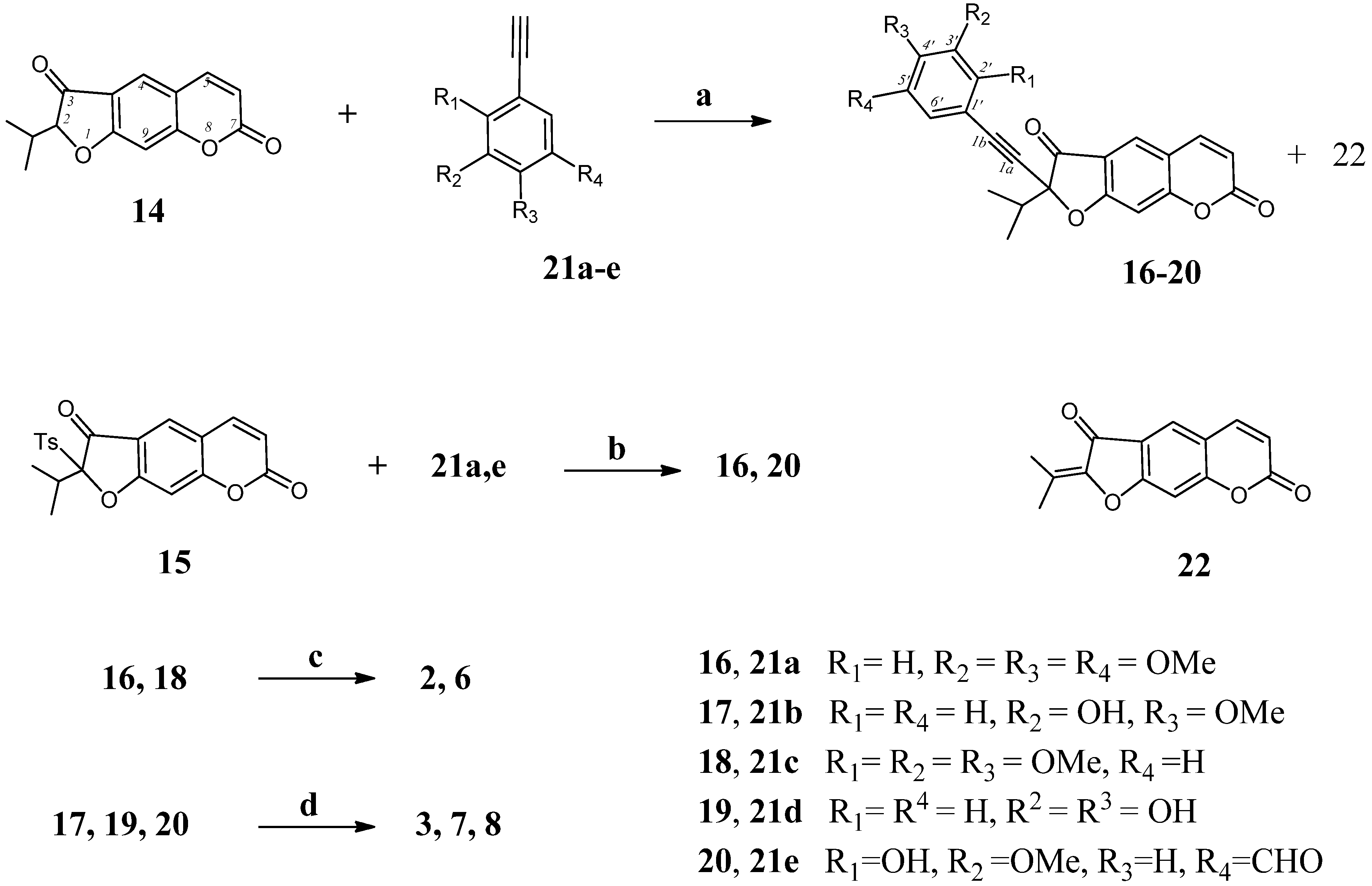
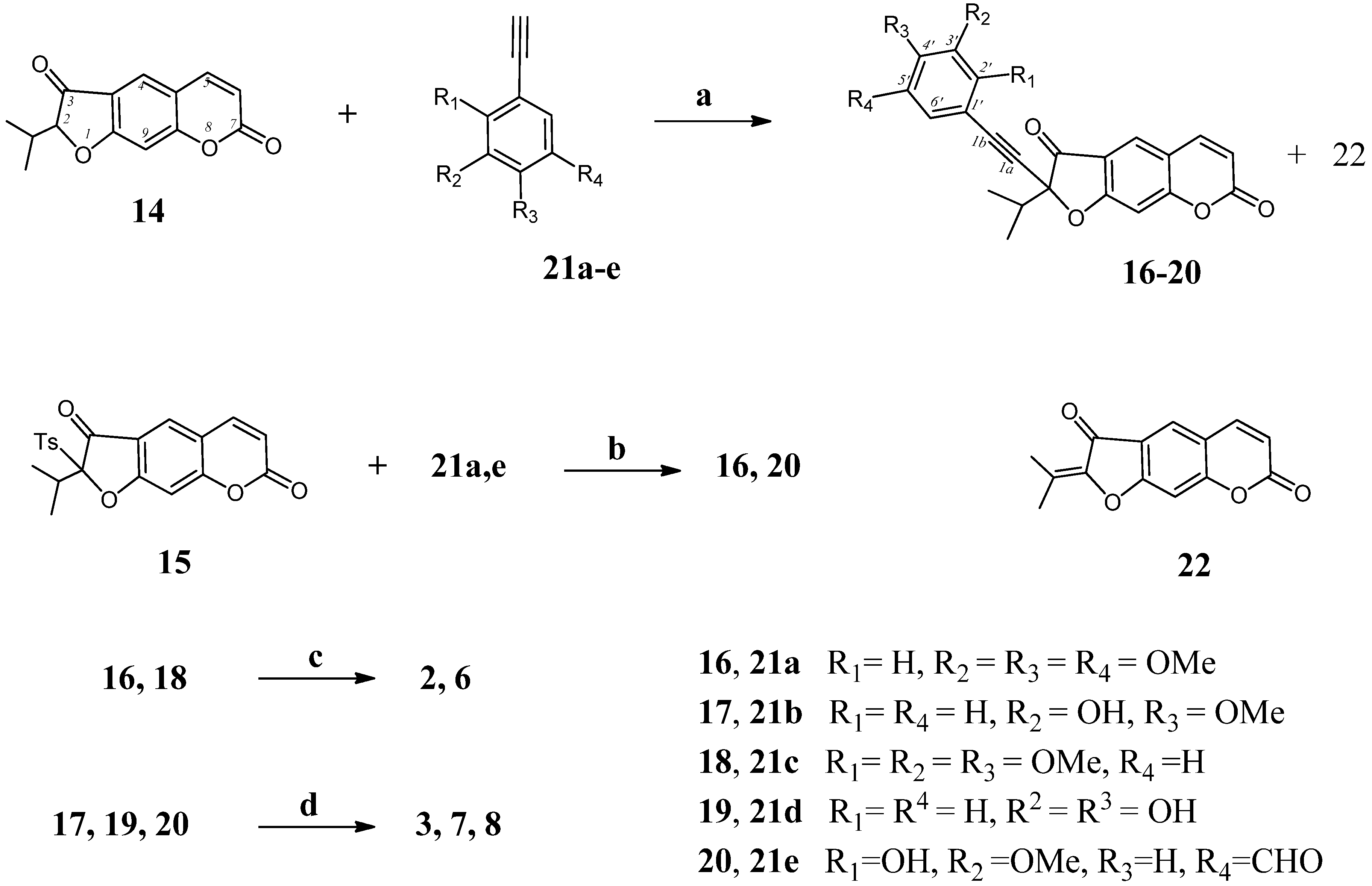
2-Isopropyl-2-[(3,4,5-trimethoxyphenyl)ethynyl]-2H-furo[3,2-g]chromene-3,7-dione (16). (a) To a solution of oreoselone (14, 488 mg, 2 mmol) in THF (7 mL) under argon was added TsCl (470 mg, 2.5 mmol) and Pd(PPh3)2Cl2 (7 mg, 0.1 mmol). The mixture was heated at 60 °C for 6 h (TLC), then 1-ethynyl-3,4,5-trimethoxybenzene (21a, 770 mg, 4 mmol) and Et3N (0.84 mL, 6 mmol) was added. The reaction mixture was heated under stirring another 7 h. After cooling, 3 mL of water was added and the mixture was extracted with methylene chloride (5 × 4 mL). The combined extract was washed with water, dried over MgSO4 and filtered. The solvent was removed under reduced pressure, and the residue was subjected to column chromatography to isolate 538 mg (62%) of 16 and 40 mg (12%) of 22. Compound 16 was recrystallized from diethyl ether, m.p. 101–102 °С. IR (KBr, ν, cm−1): 2974, 2927, 2879, 2852, 2150, 1732, 1664, 1626, 1531, 1481, 1411, 1390, 1353, 1286, 1253, 1126, 1095, 1039, 970, 948, 916, 866, 827, 734, 661. 1H-NMR (600 MHz, CDCl3, δH): 0.98 (d, J = 6.9 Hz, 3Н, СН3), 1.28 (d, J = 6.9 Hz, 3Н, СН3), 2.46 [m, 1Н, СН-(СН3)2], 3.84 (s, 9Н, 3 × OСН3), 6.34 (d, J = 9.6 Hz, 1Н, Н-6), 6.96 (s, 1Н, Н-9), 7.04 (s, 2Н, Н-2',6'), 7.68 (d, J = 9.6 Hz, 1Н, Н-5), 7.87 (s, 1Н, Н-4). 13C-NMR (150 MHz, CDCl3, δC): 16.5 (CH3), 17.4 (CH3), 36.4 (CH), 56.0 (3C-CH3), 72.1 (C-1a), 82.6 (C-1b), 99 (C-2), 101.4 (C-9), 106.5 (C-2' and C-6'), 115.2 (C-6), 115.6, 115.5 (C-3a,4a),119.4 (C-1'), 125.8 (C-4), 131.5 (C-4'), 143.0 (C-5), 153.4 (C-8a), 155.8 (C-3' and C-5'), 158.6 (C-7), 169.4 (C-9a), 193.8 (C-3). UV (EtOH) λmax, (lgε): 257 (4.56), 297 (4.3), 307 (4.27), 348 (4.11) nm. Anal. Calcd for C25H22O7: С, 69.12; Н, 5.10; found С, 69.29; Н, 5.02. (b) A mixture of 2-(p-toluenesulfonyl)oreoselone (15, 398 mg, 1 mmol), 1-ethynyl-3,4,5-trimethoxybenzene (21a, 385 mg, 2 mmol), Et3N (0.42 mL, 3 mmol), and Pd(PPh3)2Cl2 (7 mg, 0.1 mmol) in 5 mL of anhydrous THF was heated for 7 h under reflux in argon. The mixture was evaporated, the residue was treated with 10 mL of water and extracted with methylene chloride (4 × 5 mL). The combined extract was dried over MgSO4, filtered and evaporated. The residue was subjected to column chromatography on silica gel to isolate 287 mg (66%) of compound 16 and 12 mg (5%) of compound 22.
2-[(3-Hydroxy-4-methoxyphenyl)ethynyl]-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (17). This compound (374 mg, 48%) was prepared as a yellow oil from oreoselone (14, 488 mg, 2 mmol) and 1-ethynyl-3-hydroxy-4-methoxybenzene (21b (592 mg, 4 mmol) using the procedure (a) described for 16. IR (KBr, ν, cm−1): 3467, 3086, 3062, 3030, 2976, 2923, 2850, 2191, 1751, 1660, 1625, 1600, 1494, 1419, 1353, 1267, 1207, 1149, 1110, 1066, 1028, 977, 935, 806, 752, 696. 1H-NMR (400 MHz, CDCl3, δH): 1.09 (d, J = 6.9 Hz, 3Н, СН3), 1.20 (d, J = 6.9 Hz, 3Н, СН3), 3.03 [m, 1Н, СН-(СН3)2], 3.79 (s, 3Н, OСН3), 6.05 (s, 1H, OH), 6.40 (d, J = 9.6 Hz, 1Н, Н-6), 6.92 (d, J = 7.8 Hz, 1Н, Н-5'), 6.97 (dd, J = 7.8 and 1.8 Hz, 1Н, Н-6'), 6.98 (s, 1Н, Н-9), 6.99 (d, J = 1.8 Hz, 1Н, Н-2'), 7.66 (d, J = 9.6 Hz, 1Н, Н-5), 7.85 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 16.2 (CH3), 17.4 (CH3), 30.1 (CH), 54.1 (CH3), 73.5 (C-1a), 82.4 (C-1b), 96.1 (C-2), 100.1 (C-9), 112.4 (C-6), 113.1 (C-5'), 113.4 (C-3a), 115.2 (C-4a), 118.1 (C-1'), 120.5 (C-2'), 123.1 (C-6'), 125.7 (C-4), 142.3 (C-5), 147.9 (C-3'), 149.1 (C-4'), 156.6 (C-8a), 158.1 (C-7), 172.6 (C-9a), 195.5 (C-3). UV (EtOH) λmax (lgε): 258 (4.52), 295 (3.96), 307 (3.88), 351 (3.93) nm. Anal. Calcd for C23H18O6: С, 70.76; Н, 4.65; found С, 70.39; Н, 4.92.
2-Isopropyl-2-[(2,3,4-trimethoxyphenyl)ethynyl]-2H-furo[3,2-g]chromene-3,7-dione (18). Compound 18 (560 mg, 65%) was prepared from oreoselone (14, 488 mg, 2 mmol) and 1-ethynyl-2,3,4-trimethoxybenzene (21c, 770 mg, 4 mmol) using the procedure (a) described for 16. Compound 22 (30 mg, 10%) was also isolated. Compound 18, m.p. 101–102 °С (ether). IR (KBr, ν, cm−1): 3066, 2976, 2939, 2861, 2189, 1737, 1679, 1625, 1487, 1423, 1388, 1352, 1286, 1224, 1141, 1122, 1093, 1022, 983, 943, 866, 825, 790, 734, 691. 1H-NMR (400 MHz, CDCl3, δH): 0.91 (d, J = 6.9 Hz, 3Н, СН3), 1.31 (d, J = 6.9 Hz, 3Н, СН3), 2.50 [m, 1Н, СН-(СН3)2], 3.82 (s, 3Н, OСН3), 3.88 (s, 3Н, OСН3), 3.97 (s, 3Н, OСН3), 6.34 (d, J = 9.6 Hz, 1Н, Н-6), 6.70 (d, J = 8.2 Hz, 1Н, Н-5'), 6.99 (s, 1Н, Н-9), 7.52 (d, 1Н, J = 8.2 Hz, Н-6'), 7.72 (d, 1Н, J = 9.6 Hz, Н-5), 7.90 (1Н, s, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 16.6 (CH3), 17.5 (CH3), 36.4 (CH), 56.0 (CH3), 60.8 (CH3), 62.2 (CH3), 71.6 (C-1a), 82.3 (C-1b), 98.9 (C-2), 101.5 (C-9), 107.2 (C-5'), 115.4 (C-3a,4a), 115.3 (C-6), 115.6, 123.1 (C-1'), 124.0 (C-6'), 125.9 (C-4), 141.4 (C-3'), 143.1 (C-5), 156.7 (C-2'), 158.8 (C-4'), 159.1 (C-8a),161.1 (C-7), 169.5 (C-9a), 192.6 (C-3). UV (EtOH) λmax (lgε): 257 (4.42), 297 (4.17), 307 (4.14), 348 (3.97) nm. Anal. Calcd for C25H22O7: С, 69.12; Н, 5.10; found С, 69.49; Н, 5.12.
2-[(3,4-Dihydroxyphenyl)ethynyl]-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (19). Compound 19 (330 mg, 44%) was prepared from oreoselone (14, 488 mg, 2 mmol) and 1-ethynyl-2,3,4-trimethoxybenzene (21d, 540 mg, 4 mmol) using the procedure (a) described for 16. Compound 22 (82 mg, 25%) was also isolated. Compound 19, m.p. 84–86 °С (ether). IR (KBr, ν, cm−1): 3340, 3320, 3087, 3071, 2974, 2929, 2879, 2852, 2114, 1732, 1664, 1625, 1595, 1531, 1390, 1353, 1286, 1253, 1126, 1095, 1039, 948, 866, 827, 735. 1H-NMR (400 MHz, CDCl3, δH): 0.96 (d, J = 7.0 Hz, 3Н, СН3), 1.37 (d, J = 7.0 Hz, 3Н, СН3), 2.55 [m, 1Н, СН-(СН3)2], 6.40 (d, J = 9.6 Hz, 1Н, Н-6), 7.05 (s, 1Н, Н-9), 7.46 (d, J 1.8 Hz, 1Н, Н-2'), 7.52 (dd, J = 8.0 and 1.8 Hz, 1Н, Н-6'), 7.66 (d, J = 8.0 Hz, 1Н, Н-5'), 7.73 (d, J = 9.6 Hz, 1Н, Н-5), 7.93 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3): 15.6 (CH3), 16.0 (CH3), 31.1 (CH), 73.9 (C-1a), 81.9 (C-1b), 98.6 (C-2), 101.4 (C-9), 115.7 (C-3a), 116.0 (C-6), 116.7 (C-4a), 117.8 (C-1'), 120.2 (C-5'), 122.7 (C-2'), 124.1 (C-4), 126.1 (C-6'), 143.3 (C-5), 158.5 (C-8a), 159.3 (C-3'), 160.4 (C-4'), 161.9 (C-7), 171.8 (C-9a), 191.9 (C-3). UV (EtOH) λmax (lgε): 256 (4.4), 303 (3.92), 348 (3.87) nm. Anal. Calcd for C22H16O6: С, 70.21; Н, 4.29; found С, 70.49; Н, 4.20.
2-[(5-Formyl-3-hydroxy-3-methoxyphenyl)ethynyl]-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (20). Compound 20 (493 mg, 59%) was prepared from oreoselone (14, 488 mg, 2 mmol) and 1-ethynyl-5-formyl-2-hydroxy-3-methoxybenzene (21e, 705 mg, 4 mmol) using the procedure (a) described for 16, or from 2-(p-toluenesulfonyl)oreoselone (15, 398 mg, 1 mmol) and 1-ethynyl-5-formyl-2-hydroxy-3-methoxybenzene (21e, 353 mg, 2 mmol) using the procedure (b) described for 16. Yield 60%. Compound 22 was also isolated [62 mg, procedure (a) and 23 mg, procedure (b)]. Compound 20, m.p. 91–93 °С (ether). IR (KBr, ν, cm−1): 3456, 3155, 3065, 2978, 2939, 2881, 2839, 2112, 1738, 1680, 1626, 1582, 1554, 1487, 1423, 1389, 1352, 1286, 1260, 1250, 1225, 1201, 1141, 1122, 1094, 1043, 1022, 984, 970, 943, 916, 866, 826, 790, 760, 735, 690, 6800. 1H-NMR (400 MHz, CDCl3, δH): 0.91 (d, J = 6.9 Hz, 3Н, СН3), 1.29 (d, J =6.9 Hz, 3Н, СН3), 2.48 [m, 1Н, СН-(СН3)2], 3.77 (s, 3Н, OСН3), 6.32 (d, J = 9.8 Hz, 1Н, Н-6), 6.96 (s, 1Н, Н-9), 7.43 (br s, 1Н, Н-4'), 7.50 (br s, 1H, OH), 7.60 (br s, 1Н, Н-6'), 7.69 (d, J = 9.8Hz, 1Н, Н-5), 7.88 (s, 1Н, Н-4), 9.91 (s, 1H, CHO). 13C-NMR (100 MHz, CDCl3, δC): 15.2 (CH3), 15.6 (CH3), 33.7 (CH), 55.4 (OCH3), 71.3 (C-1a), 80.2 (C-1b), 98.3 (C-2), 101.1 (C-9), 115.4 (C-3a), 115.7 (C-6), 116.4 (C-4'), 118.3 (C-4a), 119.5 (C-1'), 121.5 (C-6'), 125.7 (C-4), 133.6 (C-5'), 143.1 (C-5), 156.6 (C-3'), 158.9 (C-2'), 161.6 (C-7), 165.1 (C-8a), 171.5 (C-9a), 191.6 (CHO), 192.3 (C-3). UV (EtOH) λmax (lgε): 253 (4.21), 328 (3.45), 350 (3.15) nm. Anal. Calcd for C24H18O7: С, 68.90; Н, 4.34; found С, 68.69; Н, 4.56.
(Z)-2-Isopropyl-2-(3,4,5-trimethoxystyryl)-2H-furo[3,2-g]chromene-3,7-dione (2). A solution of n-BuLi (0.08 mL, 1.04 mmol) was added dropwise to a solution of 2-isopropyl-2-[(3,4,5-trimethoxyphenyl)ethynyl]-2H-furo[3,2-g]chromene-3,7-dione (16, 100 mg, 0.23 mmol) and tetraisopropoxytitanium (140 mg, 0.52 mmol) in anhydrous THF (3 mL) at −78 °C. The stirring was continued for 10 min at the same temperature. The reaction mixture was warmed to room temperature and heated at 50 °С for 2 h. After cooling, the reaction was quenched with a saturated solution of NH4Cl (3 mL), water (3 mL), and extracted with dichloromethane (3 × 4 mL), the combined organic layers was washed with water, dried over anhydrous MgSO4, and filtered. The solvent was evaporated, and the residue was subjected to column chromatography on silica gel (chloroform and chloroform–ethanol 100:1 as eluent) to afford compound 2 (42 mg, 42% yield) as a yellow powder, m.p. 91–92 °С (ether). IR (KBr, ν, cm−1): 3047, 2974, 2935, 1736, 1701, 1628, 1585, 1474, 1390, 1356, 1286, 1226, 1195, 1120, 1034, 1011, 972, 900, 866, 840, 827, 756, 740, 700, 681. 1H-NMR (400 MHz, CDCl3, δH): 0.88 (d, J = 6.9 Hz, 3Н, СН3], 1.11 (d, J =6.9 Hz, 3Н, СН3), 3.20 [m, 1Н,СН-(СН3)2], 3.83 (s, 9Н, 3 × OСН3), 6.23 (d, J = 9.2 Hz, 1Н, Н-1a), 6.36 (d, J = 9.7 Hz, 1Н, Н-6), 6.74 (d, J = 9.2 Hz, 1Н, Н-1b), 6.94 (s, 1Н, Н-9), 7.10 (br s, 2Н, Н-2',6'), 7.67 (d, J = 9.7 Hz, 1Н, Н-5), 7.90 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 16.1 (CH3), 17.0 (CH3), 29.2 (CH), 55.6 (3×OCH3), 98.3 (C-2), 101.0 (C-9), 106.1 (C-2',6'), 114.8 (C-3a,4a), 115.1, 115.2 (C-6), 125.4 (C-4), 119.0 (C-1'), 126.8 (C-1a), 131.1 (C-1b), 131.8 (C-4'), 142.6 (C-5), 153.0 (C-8a), 155.3 (C-3',5'), 158.2 (C-7), 168.9 (C-9a), 192.4 (C-3). UV (EtOH) λmax (lgε): 255 (4.44), 296 (4.07), 310 (4.01), 344 (sh), 354 (3.9) nm. Anal. Calcd for C25H24O7: С, 68.80; Н, 5.54; found С, 68.69; Н, 5.17.
(Z)-2-(3-Hydroxy-4-methoxystyryl)-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (3). Lindlar’s catalyst (5 mg, 2 mol %) was added to a solution of 2-isopropyl-2-[(3-hydroxy-4-methoxy-phenyl)ethynyl]-2H-furo[3,2-g]chromene-3,7-dione (17, 100 mg, 0.26 mmol) in dry ethanol (6 mL). The system was filled with hydrogen, and the reaction mixture was stirred in a hydrogen flow for 20 h and then concentrated under reduced pressure. Column chromatography on silica gel afforded compound 3 (31 mg, 32% yield) as a yellow oil. IR (KBr, ν, cm−1): 3520, 3488, 3398, 2960, 2925, 2852, 1750, 1726, 1724, 1630, 1576, 1510, 1480, 1463, 1356, 1335, 1300, 1250, 1176, 1157, 1143, 1105, 1032, 978, 935, 901, 885, 831, 813, 760, 744, 700, 671. 1H-NMR δH (600 MHz, CDCl3, δH): 0.87 [d, J = 6.9 Hz, 3Н, СН3), 1.15 [d, J = 6.9 Hz, 3Н, СН3), 2.37 [m, 1Н, СН-(СН3)2], 3.76 (s, 3Н, OCH3), 6.26 (d, J = 9.5 Нz, 1Н, Н-1b), 6.33 (d, J = 9.7 Нz, 1Н, Н-6), 6.92 (d, J = 7.8 Нz, 1Н, Н-5'), 6.79 (d, J 9.5 Нz, 1Н, Н-1b), 6.89 (dd, J = 8.0 and 1.8 Нz, 1Н, Н-6'), 7.00 (s, 1Н,Н-9), 7.06 (d, J = 8.0 Нz, 1Н, Н-5'), 7.25 (d, J = 1.8 Нz, 1Н, Н-2'), 7.68 (d, J = 9.7 Нz, 1Н, Н-5), 7.78 (s, 1Н, Н-4). 13C-NMR (150 MHz, CDCl3, δC): 15.6 (CH3), 18.6 (CH3), 31.2 (CH), 55.3 (OCH3), 92.9 (C-2), 100.9 (C-9), 113.6 (C-6), 114.4 (C-5'), 114.8 (C-3a), 116.4 (C-4a), 119.3 (C-2'), 121.7 (C-6'), 124.3 (C-4), 125.2 (C-1a), 129.2 (C-1b), 134.9 (C-1'), 143.5 (C-5), 149.2 (C-3'), 150.3 (C-4'), 159.4 (C-8a), 160.1 (C-7), 173.9 (C-9a),199.4 (C-3). UV (EtOH) λmax (lgε): 253 (4.41), 296 (4.08), 308 (4.06), 341 (sh), 353 (4.1) nm, Anal. Calcd for C23H20O6: С, 70.40; Н, 5.14; found С, 70.78; Н, 4.92.
(Z)-2-Isopropyl-2-(2,3,4-trimethoxystyryl)-2H-furo[3,2-g]chromene-3,7-dione (6). Compound 6 (40 mg) was prepared from 2-isopropyl-2-[(2,3,4-trimethoxyphenyl)ethynyl]-2H-furo[3,2-g]chromene-3,7-dione (18, 100 mg, 0.23 mmol) using the procedure described for 2. Yield 40%, m.p. 112–114 °С (ether). IR (KBr, ν, cm−1): 3047, 2974, 1736, 1701, 1628, 1585, 1473, 1391, 1355, 1286, 1220, 1196, 1121, 1034, 1011, 972, 900, 866, 850, 827, 800, 756, 740, 680. 1H-NMR (400 MHz, CDCl3, δH): 0.98 (d, J = 6.9 Нz, 3Н, СН3), 1.19 (3Н, J = 6.9 Нz, d, СН3), 3.28 [m, 1Н, СН-(СН3)2], 3.91 (s, 3Н, OСН3), 3.92 (s, 3Н,OСН3), 3.94 (s, 3Н,OСН3), 6.23 (d, J = 9.3 Нz, 1Н, Н-1a), 6.45 (d, J = 9.6 Нz, 1Н, Н-6), 6.67 (d, J = 8.3 Нz, 1Н, Н-5'), 6.81 (d, J = 9.3 Нz, 1Н, Н-1b), 7.18 (d, J = 8.3 Нz, 1Н, Н-6'), 7.97 (d, J = 9.6 Нz, 1Н, Н-5), 8.19 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 16.5 (CH3), 17.4 (CH3), 31.6 (CH), 55.9 (OCH3), 62.1 (OCH3), 60.7 (OCH3), 99.5 (C-2), 101.4 (C-9), 107.2 (C-5'), 115.2 (C-6), 115.5, 115.6 (C-3a, 4a), 123.9 (C-6'), 123.0 (C-1'), 125.9 (C-4), 126.9 (C-1a), 133.1 (C-1b), 141.3 (C-5), 144.2 (C-3'), 156.6 (C-2'), 158.7 (C-4'), 159.0 (C-8a), 160.9 (C-7), 170.7 (C-9a), 190.5 (C-3). UV (EtOH) λmax (lgε): 255 (4.32), 296 (3.94), 310 (3.88), 344 (sh), 354 (3.78) nm. Anal. Calcd for C25H24O7: С, 68.80; Н, 5.54; found С, 69.09; Н, 5.42.
(Z)-2-[(3,4-Dihydroxystyryl)-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (7). Compound 7 (24 mg) was prepared by partial reduction of 2-[(3,4-dihydroxyphenyl)ethynyl]-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (19, 100 mg, 0.26 mmol) using the procedure described for 3 (reaction time 30 h). Yield 24%, m.p. 68–71 °С (ether). IR (KBr, ν, cm−1): 3470, 2979, 2922, 2851, 1751, 1661, 1626, 1601, 1598, 1495, 1420, 1354, 1327, 1267, 1207, 1150, 1111, 1086, 1028, 1002, 978, 935, 914, 823, 800, 752, 696. 1H-NMR (400 MHz, CDCl3, δH): 0.85 (d, J = 6.9 Нz, 3Н, СН3), 0.91 [d, J = 6.9 Нz, 3Н, СН3), 2.99 [m, 1Н, СН-(СН3)2], 6.21 (d, J = 9.5 Нz, 1Н, Н-1a), 6.37 (d, J = 9.7 Нz, 1Н, Н-6), 6.37 (d, J = 8.3 Нz, 1Н, Н-5'), 6.81 (d, J = 9.5 Нz, 1Н, Н-1b), 6.89 (s, 1Н, Н-9), 7.36 (d, J = 1.8 Нz, 1Н, Н-2'), 7.49 (dd, J = 8.0 and 1.8 Нz, 1Н, Н-6'), 7.65 (d, J = 8.0 Нz, 1Н, Н-5'), 7.91 (d, J = 9.6 Нz, 1Н, Н-5), 8.03 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 15.1 (CH3), 15.5 (CH3), 30.5 (CH), 98.5 (C-2), 100.9 (C-9), 115.2 (C-3a), 115.5 (C-6), 116.2 (C-5'), 117.2 (C-4a), 119.7 (C-1'), 122.2 (C-2'), 123.6 (C-6'), 125.5 (C-4), 127.1 (C-1a), 132.4 (C-1b), 142.8 (C-5), 157.9 (C-3'), 158.8 (C-4'), 159.9 (C-8a), 161.4 (C-7), 171.3 (C-9a), 190.3 (C-3). UV (EtOH) λmax (lgε): 256 (4.76), 292 (4.33), 349 (4.13) nm. Anal. Calcd for C22H18O6: С, 69.83; Н, 4.79; found С, 70.10; Н, 4.31.
(Z)-2-(5-Formyl-3-hydroxy-3-methoxystyryl)-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (8). Compound 8 (37 mg) was prepared from 2-[(5-formyl-3-hydroxy-3-methoxyphenyl)ethynyl]-2-isopropyl-2H-furo[3,2-g] chromene-3,7-dione (20, 100 mg, 0.24 mmol) using the procedure described for 3. Yield 37%, a yellow oil. IR (KBr, ν, cm−1): 3435, 3063, 3042, 2960, 2925, 2853, 2808, 1739, 1726, 1714, 1629, 1600, 1576, 1510, 1500, 1464, 1440, 1394, 1355, 1334, 1300, 1250, 1230, 1176, 1157, 1144, 1105, 1032, 978, 935, 915, 893, 831, 814, 761, 744, 720, 668. 1H-NMR (400 MHz, CDCl3, δH): 0.97 (d, J = 6.9 Нz, 3Н, СН3], 1.07 [d, J = 6.9 Нz, 3Н, СН3), 3.15 [m, 1Н, СН-(СН3)2], 3.92 (s, 3Н, OСН3), 6.23 (d, J = 9.3 Нz, 1Н, Н-1a), 6.37 (1Н, J = 9.8 Нz, d, Н-6), 6.41 (d, J = 1.8 Нz, 1Н, Н-6'), 6.83 (d, J = 9.3 Нz, 1Н, Н-1b), 7.05 (s, 1Н, Н-9), 7.69 (d, 1Н, J = 9.6 Нz, Н-5), 7.78 (1Н, J = 1.8 Нz, d, Н-4'), 7.85 (s, 1Н, Н-4), 8.07 (1Н, J = 8.8 Нz, d, Н-1b), 9.98 (s, 1Н, CНO). 13C-NMR (100 MHz, CDCl3, δC): 15.6 (CH3), 15.9 (CH3), 34.0 (CH), 55.7 (OCH3), 99.5 (C-2), 100.6 (C-9), 115.6 (C-6), 116.0 (C-3a), 116.7 (C-4a), 118.7 (C-4'), 121.8 (C-6'), 119.8 (C-1'), 126.0 (C-4), 127.5 (C-1a), 132.6 (C-1b), 133.9 (C-5'), 144.8 (C-5), 156.9 (C-3'), 158.9 (C-2'), 161.9 (C-8a), 165.3 (C-7), 171.8 (C-9a), 191.9 (СНО), 194.6 (C-3). UV (EtOH) λmax (lgε): 256 (4.41), 291 (3.97), 349 (3.78) nm. Anal. Calcd for C24H20O7: С, 68.57; Н, 4.80; found С, 68.24; Н, 4.68.
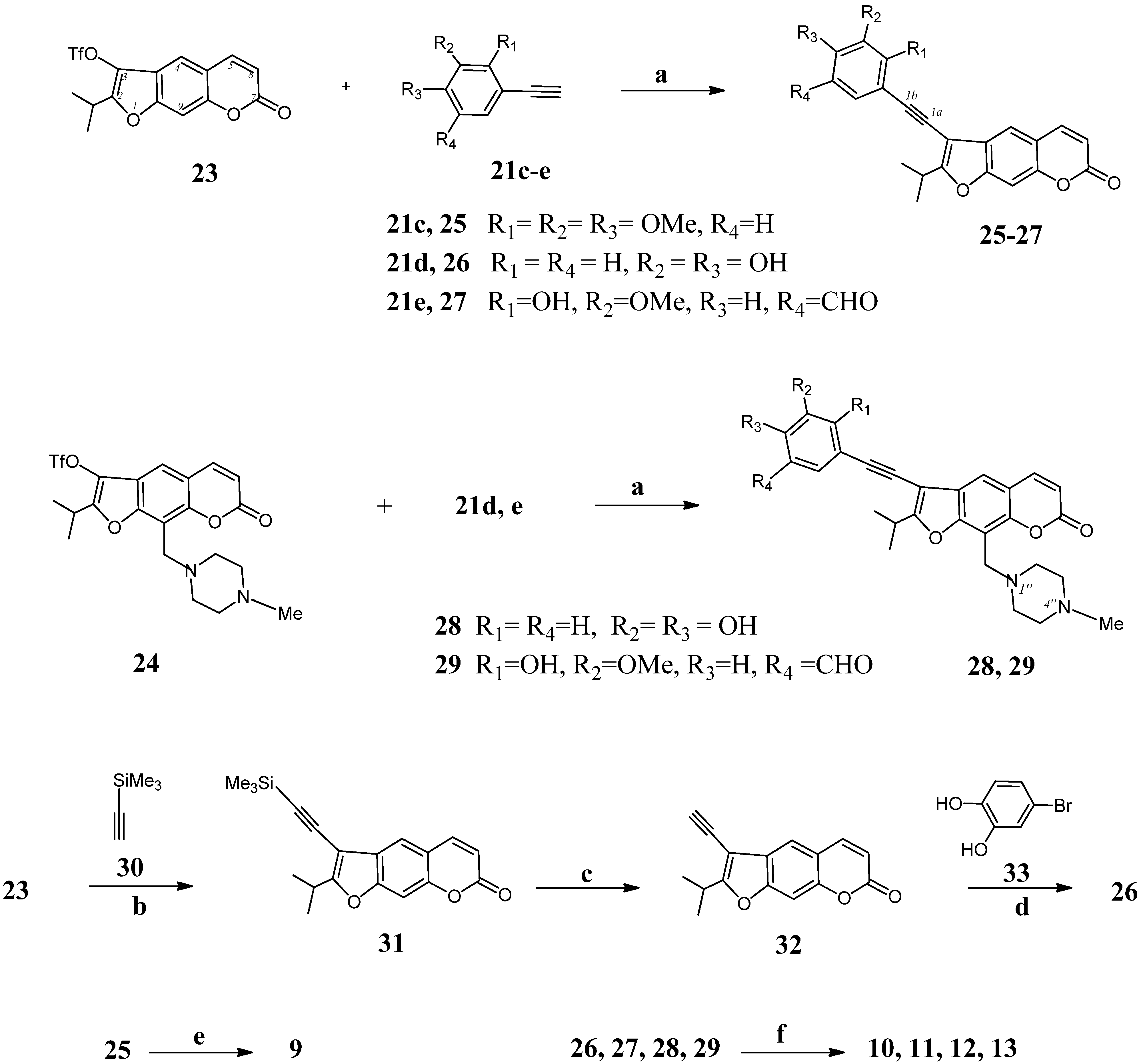
2-Isopropyl-3-[(2,3,4-trimethoxyphenyl)ethynyl]-7H-furo[3,2-g]chromene-7-one (25). To a solution of 2-isopropyl-3-(trifluoromethanesulfonyloxy)psoralene (23, 150 mg, 0.4 mmol) and 1-ethynyl-2,3,4-trimethoxybenzene (21c, 85 mg, 0.44 mmol) in benzene (5 mL) was added CuI (1.5 mg, 2 mol %), Pd(PPh3)2Cl2 (11 mg, 4 mol %), and Et3N (0.076 mL, 0.55 mmol; 1.4 equiv) under argon. The reaction mixture was stirred at 80 °C for 8 h (TLC). The mixture was cooled and 5 mL of water was added. The separated water layer was extracted with methylene chloride (5 × 4 mL). The combined organic extracts was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel. Eluting with chloroform and crystallization from diethyl ether gave 122 mg (73%) of compound 25. M.p. 121–122 °С (ether). IR (KBr, ν, cm−1): 3051, 2979, 2927, 2854, 2472, 2118, 1732, 1622, 1603, 1580, 1514, 1470, 1386, 1363, 1321, 1277, 1252, 1213, 1194, 1167, 1140, 1115, 1096, 1068, 1043, 980, 959, 935, 916, 870, 850, 820, 770, 750, 741, 702, 677. 1H-NMR (400 MHz, CDCl3, δH): 1.35 (d, J = 7.0 Hz, 3Н, СН3), 1.37 [d, J = 7.0 Hz, 3Н, СН3), 3.25 [m, 1Н, СН-(СН3)2], 3.86 (s, 3Н, OСН3), 3.92 (s, 3Н, OСН3), 4.01 (s, 3Н, OСН3), 6.40 (d, J = 9.8 Hz, 1Н, Н-6), 6.72 (d, J = 8.2 Hz, 1Н, Н-5'), 6.98 (s, 1Н, Н-9), 7.59 (d, J = 8.2 Hz, 1Н, Н-6'), 7.79 (s, 1Н, Н-4), 7.80 (d, 1Н, J = 9.8 Hz, Н-5). 13C-NMR (100 MHz, CDCl3, δC): 20.3 (CH3), 20.5 (CH3), 25.9 (CH), 56.1 (OCH3), 60.9 (OCH3), 62.3 (OCH3), 86.5 (C-1a), 94.0 (C-1b), 95.5 (C-3), 97.5 (C-9), 107.3 (C-5'), 100.5 (C-4a), 111.1 (C-6), 116.6 (C-3a), 118.9 (C-1'), 124.2 (C-6'), 126.5 (C-4), 141.6 (C-3'), 143.5 (C-5), 152.0 (C-9a), 153.1 (C-8a), 154.1, 154.7 (C-2',4'), 156.9 (C-2), 159.2 (C-7). UV (EtOH) λmax (lgε): 249 (3.65), 306 (3.75), 327 (3.73), 351 (sh), 355(2.85) nm. Anal. Calcd for C25H22O6: С, 71.76; Н, 5.30; found С, 71.49; Н, 5.12.
3-[(3,4-Dihydroxyphenyl)ethynyl]-2-isopropyl-7H-furo[3,2-g]chromene-7-one (26). Compound 26 was prepared by two methods. Reaction of psoralene derivative 23 (150 mg, 0.4 mmol) with 1-ethynyl-3,4-dihydroxybenzene (21d, 60 mg, 0.44 mmol) by the procedure described for 25 (method a) gave 98 mg (68%) of compound 26. (b) To a solution of 3-ethynyl-2-isopropylpsoralene (32, 100 mg, 0.4 mmol) in benzene (5 mL) was added 3,4-dihydroxybromobenzene (33, 83 mg, 0.44 mmol), 1.5 mg (2 mol %) of CuI, 11 mg (4 mol %) of Pd(PPh3)2Cl2, and 0.076 mL (1.4 equiv, 0.55 mmol) of Et3N under argon. The mixture was stirred at 80 °C for 10 h (TLC). Then the solution was cooled, 3 mL of water was added and reaction mixture was extracted with methylene chloride (5 × 4 mL). The combined extract was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel. Eluting with chloroform and crystallization from diethyl ether gave 60 mg (42%) of compound 26. M.p. 104–105 °С. IR (KBr, ν, cm−1): 3450, 3327, 3051, 2980, 2880, 2556, 2472, 2096, 1732, 1716, 1697, 1620, 1590, 1550, 1514, 1431, 1364, 1278, 1253, 1213, 1191, 1160, 1140, 1095, 1041, 980, 950, 876, 860, 849, 825, 770, 754, 740, 685, 650. 1H-NMR (400 MHz, CDCl3, δH): 1.38 (d, J = 7.0 Hz, 3Н, СН3), 1.40 (d, J = 7.0 Hz, 3Н, СН3), 3.30 [m, 1Н, СН-(СН3)2], 4.09 (br.s, 2Н, OH), 6.32 (d, J = 9.7 Hz, 1Н, Н-6), 7.07 (s, 1Н, Н-9), 7.26 (d, J = 2.0 Hz, 1Н, Н-2'), 7.32 (dd, J = 8.0 and 2.0 Hz, 1Н, Н-6'), 7.46 (d, J = 8.0 Hz, 1Н, Н-5'), 7.77 (d, J = 9.7 Hz, 1Н, Н-5), 7.80 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 20.0 (CH3), 20.1 (CH3), 25.8 (CH), 86.4 (C-1a), 94.0 (C-1b), 100.3 (C-3), 102.8 (C-9), 112.3 (C-4a), 115.2 (C-6), 115.3 (C-3a), 116.5 (C-1'), 120.2 (C-5'), 121.2 (C-4), 124.5 (C-2'), 126.2 (C-6'), 144.2 (C-5), 145.9 (C-3'), 147.4 (C-4'), 152.1 (C-9a), 155.3 (C-8a), 160.4 (C-2), 161.3 (C-7). UV (EtOH) λmax (lgε): 252 (3.64), 309 (3.94), 339 (3.43), 355 (sh) nm. Anal. Calcd for C22H16O5: С, 73.33; Н, 4.48; found С, 73.30; Н, 4.55.
3-[(5-Formyl-3-hydroxy-3-methoxyphenyl)ethynyl]-2-isopropyl-7H-furo[3,2-g] chromene-7-one (27). Compound 27 (112 mg) was prepared from psoralene derivative 23 (150 mg, 0.4 mmol) and 1-ethynyl-5-formyl-2-hydroxy-3-methoxybenzene (21e, 75 mg, 0.44 mmol) using the procedure described for 25. Yield 70%, m.p. 110–112 °С (ether). IR (KBr, ν, cm−1): 3430, 3325, 2989, 2880, 2713, 2472, 2046, 1944, 1782, 1732, 1716, 1697, 1620, 1602, 1514, 1469, 1431, 1363, 1315, 1278, 1253, 1220, 1191, 1139, 1095, 1041, 1020, 980, 904, 860, 848, 769, 754, 695, 650, 600. 1H-NMR (400 MHz, CDCl3, δH): 1.39 (d, J = 7.0 Hz, 3Н, СН3), 1.42 (d, J = 7.0 Hz, 3Н, СН3), 3.29 [m, 1Н, СН-(СН3)2], 3.98 (s, 3Н, OСН3), 6.36 (d, J = 9.8 Hz, 1Н, Н-6), 7.09 (s, 1Н, Н-9), 7.44 (br.s, 1Н, Н-4'), 7.50 (br.s, 1H, OH), 7.60 (br.s, 1Н, Н-6'), 7.75 (d, J = 9.8 Hz, 1Н, Н-5), 7.82 (s, 1Н, Н-4), 10.01 (s, 1H, CHO). 13C-NMR (100 MHz, CDCl3, δC): 20.5 (CH3), 20.6 (CH3), 26.2 (CH), 60.6 (OCH3), 85.6 (C-1a), 91.0 (C-1b), 99.5 (C-3), 104.4 (C-9), 115.8 (C-3a), 116.3 (C-6), 116.9 (C-4'), 118.9 (C-4a), 119.8 (C-1'), 120.6 (C-6'), 128.7 (C-4), 132.2 (C-5'), 143.9 (C-5), 150.7 (C-3'), 152.5 (C-8a), 153.1 (C-9a), 154.6 (C-2'), 159.2 (C-2), 160.7 (C-7), 191.8 (CHO). UV (EtOH) λmax (lgε): 246 (3.92), 305 (sh), 329 (3.63), 355 (2.81) nm. Anal. Calcd for C24H18O6: С, 71.64; Н, 4.51; found С, 71.30; Н, 4.58.
3-[(3,4-Dihydroxyphenyl)ethynyl]-2-isopropyl-9-[(4-methylpiperazin-1-yl)methyl]-7H-furo[3,2-g]-chromene-7-one (28). Compound 28 (79 mg) was prepared from 2-isopropyl-9-((4-methylpiperazin-1-yl)methyl)-7-oxo-7H-furo[3,2-g]chromen-3-yl trifluoromethanesulfonate (24, 195 mg, 0.4 mmol) and 1-ethynyl-3,4-dihydroxybenzene (21d, 60 mg, 0.44 mmol) using the procedure described for 25. Yield 42%, yellow powder, m.p. 118–120 °С (ether). IR (KBr, ν, cm−1): 3437, 2967, 2922, 2851, 2111, 1732, 1635, 1628, 1585, 1511, 1496, 1465, 1431, 1388, 1348, 1319, 1250, 1213, 1198, 1140, 1114, 1095, 1047, 957, 870, 820, 737, 700, 626, 602. 1H-NMR (400 MHz, CDCl3, δH): 1.33 (d, J = 7.0 Hz, 3Н, СН3), 1.36 (3Н, d, J = 7.0 Hz, СН3), 2.32 (s, 3Н, NСН3), 2.38 (m, 4H, H-3'',5''), 2.68 (m, 4H, H-2'',6''), 3.25 [m, 1Н, СН-(СН3)2], 4.46 (d, 1H, J = 9.8 Hz, 1H, CH2), 4.58 (d, 1H, J = 9.8 Hz, 1H, CH2), 6.27 (d, J = 9.8 Hz, 1Н, Н-6), 6.52 (dd, J = 8.0 and 2.0 Hz, 1Н, Н-6'), 6.64 (d, J = 2.0 Hz, 1Н, Н-2'), 6.79 (d, J = 8.0 Hz, 1Н, Н-5'), 7.59 (d, J = 9.8 Hz, 1Н, Н-5), 7.74 (s, 1Н, Н-4), 8.00 (br.s, 2Н, OН). 13C-NMR (100 MHz, CDCl3, δC): 20.1 (CH3), 20.3 (CH3), 25.9 (CH), 42.7 (NСН3), 48.5 (СН2N), 51.6 (C-2'',6''), 53.4 (C-3'',5''), 85.3 (C-1a), 90.7 (C-1b), 99.2 (C-3), 104.4 (C-9), 115.0 (C-1'), 115.6 (C-3a), 116.0 (C-6), 116.6 (C-4a), 117.6 (C-5'), 122.0 (C-2'), 128.4 (C-4), 130.8 (C-6'), 143.5 (C-5), 147.1 (C-2'), 148.7 (C-4'), 152.2 (C-8a), 153.0 (C-9a), 156.9 (C-2), 160.4 (C-7). UV (EtOH) λmax (lgε): 250 (3.76), 284 (2.65), 327 (3.20), 346 (2.78) nm. Anal. Calcd for C28H28N2O5: С, 71.17; Н, 5.97; N, 5.93; found С, 70.91; Н, 6.02; N, 5.63.
3-[(5-Formyl-3-hydroxy-3-methoxyphenyl)ethynyl]-2-isopropyl-9-[(4-methylpiperazin-1-yl)methyl]-7H-furo[3,2-g]chromen-7-one (29). Compound 29 (119 mg) was prepared from 2-isopropyl-9-((4-methylpiperazin-1-yl)methyl)-7-oxo-7H-furo[3,2-g]chromen-3-yl trifluoromethanesulfonate (24, 195 mg, 0.4 mmol) and 1-ethynyl-5-formyl-2-hydroxy-3-methoxybenzene (21e, 5 mg, 0.44 mmol) using the procedure described for 25. Yield 58%, m.p. 104–105 °С (ether). IR (KBr, ν, cm−1): 3402, 3117, 3080, 2958, 2850, 2783, 2711, 2611, 2172, 2133, 1726, 1693, 1610, 1593, 1537, 1464, 1427, 1402, 1367, 1331, 1304, 1252, 1229, 1213, 1180, 1142, 1107, 1065, 1030, 993, 968, 908, 876, 845, 752, 741, 715, 667, 633, 621. 1H-NMR (400 MHz, CDCl3, δH): 1.36 (d, J = 7.0 Hz, 3Н, СН3), 1.39 (d, J = 7.0 Hz, 3Н, СН3), 2.38 (s, 3Н, NСН3), 2.42 (m, 4H, H-3'',5''), 2.65 (m, 4H, H-2'',6''), 3.25 [m, 1Н, СН-(СН3)2], 4.03 (s, 3Н, OСН3), 4.41 (d, J = 9.8 Hz, 1H, CH2), 4.48 (d, J = 9.8 Hz, 1H, CH2), 6.41 (d, J = 9.8 Hz, 1Н, Н-6), 6.89 (br.s, 1H, Н-4'), 7.41 (br.s, 1Н, OH), 7.44 (br.s, 1Н, Н-6'), 7.57 (s, 1Н, Н-4), 7.79 (d, J = 9.8 Hz, 1Н, Н-5), 9.99 (s, 1H, CHO). 13C-NMR (100 MHz, CDCl3, δC): 20.2 (CH3), 20.5 (CH3), 25.9 (CH), 42.5 (NСН3), 48.2 (СН2N), 51.5 (C-2'',6''), 52.9 (C-3'',5''), 58.3 (OCH3), 85.2 (C-1a), 93.5 (C-1b), 100.5 (C-9), 104.2 (C-3), 115.5 (C-6), 115.7 (C-3a), 116.6 (C-4'), 118.9 (C-4a), 119.8 (C-1'), 120.5 (C-6'), 128.4 (C-4), 132.1 (C-5'), 143.5 (C-5), 150.6 (C-8a), 152.2 (C-9a), 153.4 (C-3'), 154.7 (C-2'), 156.9 (C-2), 160.4 (C-7), 189.3 (CHO). UV (EtOH) λmax (lgε): 253 (3.81), 289 (2.52), 328 (3.11), 350 (3.08) nm. Anal. Calcd for C30H30N2O6: С, 70.02; Н, 5.88; N, 5.44; found С, 69.79; Н, 5.85; N, 5.16.
(Z)-2-Isopropyl-3-(2,3,4-trimethoxystyryl)-2H-furo[3,2-g]chromene-3,7-dione (9). Compound 9 (60 mg) was prepared from 2-isopropyl-3-[(2,3,4-trimethoxyphenyl)ethynyl]-7H-furo[3,2-g]chromene-7-one (25, 100 mg, 0.24 mmol) using the procedure described for 2. Yield 60%, m.p. 94–97 °С (ether). IR (KBr, ν, cm−1): 3062, 3049, 2980, 2950, 1732, 1700, 1636, 1589, 1495, 1465, 1433, 1389, 1346, 1288, 1259, 1203, 1169, 1140, 1094, 1047, 1029, 1009, 962, 937, 903, 870, 831, 820, 781, 754, 700, 678, 650, 623, 602. 1H-NMR (600 MHz, CDCl3, δH): 1.26 (d, J 6.9 Hz, 3Н, СН3), 1.28 [d, J = 6.9 Hz, 3Н, СН3), 3.16 [m, 1Н,СН-(СН3)2], 3.87 (s, 3Н, OСН3), 3.88 (s, 3Н, OСН3), 3.90 (s, 3Н, OСН3), 6.23 (d, J = 9.8 Hz, 1Н, Н-6), 6.40 (d, J = 8.8 Hz, 1Н, Н-1a), 6.71 (d, J = 8.8 Hz, 1Н, Н-1b), 6.83 (d, 1Н, J = 8.2 Hz, Н-5'), 6.90 (s, 1Н, Н-9), 7.19 (d, J = 8.2 Hz, 1Н, Н-6'), 7.77 (d, J = 9.8 Hz, 1Н, Н-5), 7.94 (s, 1Н, Н-4). 13C-NMR (150 MHz, CDCl3, δC): 20.1 (CH3), 20.3 (CH3), 25.9 (CH), 55.3 (OCH3), 58.7 (OCH3), 60.6 (OCH3), 95.9 (C-3), 98.4 (C-9), 108.5 (C-3a), 114.0 (C-6), 115.5, 115.6 (C-5',4a), 123.0 (C-1'), 124.2 (C-6'), 125.9 (C-4), 130.0 (C-1a), 133.4 (C-1b), 141.5 (C-3'), 143.5 (C-5), 152.2 (C-8a), 153.0 (C-2'), 153.8 (C-4'), 154.7 (C-9a), 157.5 (C-2), 160.4 (C-7). UV (EtOH) λmax (lgε): 240 (4.25), 252 (4.14), 296 (3.82), 352 (3.49) nm. Anal. Calcd for C25H24O6: С, 71.41; Н, 5.75; found С, 71.09; Н, 5.68.
(Z)-3-[(3,4-Dihydroxystyryl)-2-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (10). Compound 10 (42 mg) was prepared from 3-[(3,4-dihydroxyphenyl)ethynyl]-2-isopropyl-7H-furo[3,2-g]chromene-7-one (26, 100 mg, 0.27 mmol) using the procedure described for 3. Yield 42%, a yellow oil. IR (KBr, ν, cm−1): 3450, 3050, 2980, 1732, 1717, 1670, 1610, 1603, 1514, 1470, 1431, 1412, 1364, 1279, 1254, 1192, 1140, 1110, 1086, 1041, 990, 975, 928, 910, 860, 849, 820, 770, 754, 740, 648, 627, 601. 1H-NMR (400 MHz, CDCl3, δH): 1.36 (d, J = 6.9 Hz, 3Н, (СН3), 1.37 (d, J = 6.9 Hz, 3Н, (СН3), 3.25 [m, 1Н, СН-(СН3)2], 3.78 (br.s, 2Н, OH), 6.30 (d, J = 9.8 Hz, 1Н, Н-6), 6.39 (d, J = 9.0 Hz, 1Н, Н-1a), 6.65 (d, J = 9.0 Hz, 1Н, Н-1b), 6.80 (d, J = 8.0 Hz, 1Н, Н-5'), 6.89 (s, 1Н, Н-9), 7.36 (d, J = 1.6 Hz, 1Н, Н-2'), 7.42 (dd, J = 8.0 and 1.6 Hz, 1Н, Н-6'), 7.79 (d, J = 9.8 Hz, 1Н, Н-5), 7.80 (s, 1Н, Н-4). 13C-NMR (100 MHz, CDCl3, δC): 20.1 (CH3), 20.3 (CH3), 24.4 (CH), 96.0 (C-3), 98.4 (C-9), 106.6 (C-3a), 114.0 (C-6), 114.9, 115.1 (C-1',5'), 115.5 (C-2'), 117.2 (C-4a), 117.6 (C-6'), 125.6 (C-4), 130.9 (C-1a), 133.8 (C-1b), 143.5 (C-5), 146.5 (C-3'), 147.9 (C-4'), 152.2 (C-8a), 153.0 (C-9a), 156.4 (C-2), 160.3 (C-7). UV (EtOH) λmax (lgε): 252 (4.47), 287 (4.02), 350 (3.62) nm. Anal. Calcd for C22H18O5: С, 72.92; Н, 5.01; found С, 72.78; Н, 4.88.
(Z)-3-(5-Formyl-3-hydroxy-3-methoxystyryl)-3-isopropyl-2H-furo[3,2-g]chromene-3,7-dione (11). Compound 11 (61 mg) was prepared from 3-[(5-formyl-3-hydroxy-3-methoxyphenyl)ethynyl]-2-isopropyl-7H-furo[3,2-g]chromene-7-one (27, 100 mg, 0.24 mmol) using the procedure described for 3. Yield 60%, m.p. 104–105 °С (ether). IR (KBr, ν, cm−1): 3435, 3063, 3042, 2960, 2925, 2853, 2808, 1739, 1726, 1714, 1629, 1600, 1576, 1510, 1500, 1464, 1440, 1394, 1355, 1334, 1300, 1250, 1230, 1176, 1157, 1144, 1105, 1032, 978, 935, 915, 893, 831, 814, 761, 744, 720, 668. 1H-NMR (400 MHz, CDCl3, δH): 1.43 (d, J = 6.9 Hz, 3Н, СН3), 1.46 (d, J = 6.9 Hz, 3Н, СН3), 3.34 [m, 1Н, СН-(СН3)2], 3.98 (s, 3Н, OСН3), 6.39 (d, J = 9.8 Hz, 1Н, Н-6), 6.41 (d, J = 1.8 Hz, 1Н, Н-6'), 6.48 (d, J = 8.8 Hz, 1Н, Н-1a), 6.82 (d, J = 8.8 Hz, 1Н, Н-1b), 7.05 (s, 1Н, Н-9), 7.10 (br.s, 1Н, OН), 7.59 (d, J = 1.8 Hz, 1Н, Н-4'), 7.64 (s, 1Н, Н-4), 7.86 (d, J = 9.8 Hz, 1Н, Н-5), 10.03 (1Н, s, CНO). 13C-NMR (100 MHz, CDCl3, δ): 20.1 (CH3), 20.3 (CH3), 25.8 (CH), 55.9 (OCH3), 99.1 (C-3), 104.1 (C-9), 115.4 (C-3a), 115.9 (C-6), 116.5 (C-4'), 117.1 (C-4a), 118.6 (C-1'), 121.4 (C-6'), 126.8 (C-4), 128.3 (C-1b), 132.1 (C-1a), 132.6 (C-5'), 143.4 (C-5), 149.9 (C-3'), 152.1 (C-8a), 152.9 (C-9a), 154.4 (C-2'), 156.8 (C-2), 160.3 (C-7), 188.2 (СНО). UV (EtOH) λmax (lgε): 252 (4.26), 284 (3.65), 346 (3.53) nm. Anal. Calcd for C24H20O6: С, 70.92; Н, 4.88; found С, 71.28; Н, 4.98.
(Z)-3-[(3,4-Dihydroxystyryl)-2-isopropyl-9-[(4-methylpiperazin-1-yl)methyl]-2H-furo[3,2-g]-chrom-ene-3,7-dione (12). The compound 12 (49 mg) was prepared from 3-[(3,4-dihydroxyphenyl)ethynyl]-2-isopropyl-9-[(4-methylpiperazin-1-yl)methyl]-7H-furo[3,2-g]-chromene-7-one (28, 123 mg, 0.26 mmol) using the procedure described for 3. Yield 40%, a yellow oil. IR (KBr, ν, cm−1): 3433, 3060, 3049, 2955, 2922, 2851, 1732, 1660, 1628, 1580, 1496, 1467, 1431, 1389, 1349, 1310, 1286, 1250, 1213, 1198, 1140, 1115, 1071, 1047, 988, 957, 905, 870, 820, 790, 768, 752, 736, 725, 708, 690, 675, 650. 1H-NMR (600 MHz, CDCl3, δH): 1.35 (d, J= 7.0 Hz, 3Н, СН3), 1.37 (d, J = 7.0 Hz, 3Н, СН3), 2.28 (s, 3Н, СН3), 2.48 (m, 4H, H-3'',5''), 2.89 (m, 4H, H-2'',6''), 3.25 [m, 1Н, СН-(СН3)2], 4.57 (d, J = 9.8 Hz, 1H, CH2), 4.62 (d, J = 9.8 Hz, 1H, CH2), 6.28 (d, J = 9.7 Hz, 1Н, Н-6), 6.37 (d, J = 9.0 Hz, 1Н, Н-1a), 6.77 (d, J = 8.3 Hz, 1Н, Н-5'), 6.95 (d, J = 9.0 Hz, 1Н, Н-1b), 7.38 (d, J = 1.8 Hz, 1Н, Н-2'), 7.49 (dd, J = 8.0 and 1.8 Hz, 1Н, Н-6'), 7.65 (d, J = 8.0 Hz, 1Н, Н-5'), 7.78 (s, 1Н, Н-4), 7.91 (d, 1Н, J = 9.7 Hz, Н-5), 8.17 (br.s, 2Н, OH). 13C-NMR (100 MHz, CDCl3, δC): 19.9 (CH3), 20.2 (CH3), 25.6 (CH), 42.4 (СН3), 48.2 (СН2), 52.8 (C-3'',5''), 51.3 (C-2'',6''), 98.9 (C-3), 103.9 (C-9), 115.2 (C-3a), 116.3 (C-6), 117.0 (C-5'), 117.4 (C-4a), 120.0 (C-1'), 126.1 (C-2'), 127.8 (C-6'), 128.1 (C-4), 130.6, 131.6 (C-1a,1b), 143.2 (C-5), 147.1 (C-3'), 148.1 (C-4'), 151.9 (C-9a), 152.7 (C-8a), 157.7 (C-2), 160.0 (C-7). UV (EtOH) λmax (lgε): 252 (4.42), 275(sh), 288 (3.91), 306 (3.84), 322 (sh), 354 (3.28) nm. Anal. Calcd for C28H30N2O5: С, 70.87; Н, 6.37; N, 5.90; found С, 71.02; Н, 6.33; N, 5.81.
(Z)-2-(5-Formyl-3-hydroxy-3-methoxystyryl)-3-isopropyl-9-[(4-methylpiperazin-1-yl)methyl]-2H-furo-[3,2-g]chromene-3,7-dione (13). Compound 13 (56 mg) was prepared from 3-[(5-formyl-3-hydroxy-3-methoxyphenyl)ethynyl]-2-isopropyl-9-[(4-methylpiperazin-1-yl)methyl]-7H-furo[3,2-g]chromen-7-one (29, 133 mg, 0.26 mmol) using the procedure described for 3. Yield 42%, m.p. 100–102 °С (ether). IR (KBr, ν, cm−1): 3402, 3200, 3061, 3049, 2968, 2954, 2922, 2874, 2818, 1732, 1705, 1628, 1580, 1510, 1497, 1466, 1431, 1410, 1389, 1348, 1300, 1287, 1250, 1214, 1198, 1140, 1115, 1070, 1047, 1020, 975, 956, 910, 870, 820, 780, 752, 737, 720, 700, 660. 1H-NMR (400 MHz, CDCl3, δH): 1.34 (d, J = 6.9 Hz, 3Н, (СН3), 1.39 (d, J = 6.9 Hz, 3Н, (СН3), 2.28 (s, 3Н, СН3), 2.41 (m, 4H, H-3'',5''), 2.65 (m, 4H, H-2'',6''), 3.24 [m, 1Н, СН-(СН3)2], 4.03 (s, 3Н, OСН3), 4.48 (d, J = 9.8 Hz, 1H, CH2), 4.52 (d, J = 9.8 Hz, 1H, CH2), 6.30 (d, J = 9.8 Hz, 1Н, Н-6), 6.38 (d, J = 9.1 Hz, 1Н, Н-1a), 6.45 (d, J = 1.8 Hz, 1Н, Н-6'), 6.95 (d, J = 9.1 Hz, 1Н, Н-1b), 7.48 (d, J = 1.8 Hz, 1Н, Н-4'), 7.68 (s, 1Н, Н-4), 7.77 (d, J = 9.8 Hz, 1Н, Н-5), 9.95 (br.s, 1Н, CНO). 13C-NMR (100 MHz, CDCl3, δC): 19.9 (CH3), 20.1 (CH3), 25.8 (CH), 42.5 (СН3), 48.3 (СН2), 51.4 (C-2'',6''), 52.8 (C-3'',5''), 58.3 (OCH3), 102.6 (C-3), 104.0 (C-9), 115.4 (C-3a), 115.8 (C-6), 116.4 (C-4a), 118.8 (C-4'), 119.9 (C-1'), 120.0 (C-6'),125.2 (C-4), 128.6 (C-1a),131.9 (C-1b), 133.1 (C-5'), 145.1 (C-5), 145.6 (C-3'), 151.3 (C-8a), 152.0 (C-9a), 154.6 (C-2'), 156.8 (C-2), 158.9 (C-7), 189.2 (СНО). UV (EtOH) λmax (lgε): 252 (4.59), 290 (3.82), 336 (sh), 353(3.42) nm. Anal. Calcd for C30H32N2O6: С, 69.75; Н, 6.24; N, 5.42; found С, 69.52; Н, 6.38; N, 5.35.
2-Isopropyl-3-[(trimethylsilyl)ethynyl]-7Н-furo[3,2-g]chromen-7-оne (31). To a solution of oreoselone triflate 23 (200 mg, 0.5 mmol) and (trimethylsilyl)acetylene (30, 73 mg, 0.75 mmol) in benzene (5 mL) was added CuI (1.5 mg, 2 mol %), Pd(PPh3)2Cl2 (11 mg, 4 mol %), and Et3N (0.076 mL, 0.55 mmol; 1.1 equiv) under argon. The reaction mixture was stirred at 80 °C for 10 h (TLC). The mixture was cooled, and 5 mL of water was added. The separated water layer was extracted with methylene chloride (5 × 4 mL). The combined organic extracts was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel. Eluting with chloroform and crystallization from diethyl ether gave 90 mg (58%) of compound 31, m.p. 94–96 °С. IR (KBr, ν, cm−1): 3435, 2962, 2925, 2152, 1730, 1625, 1597, 1577, 1485, 1431, 1388, 1355, 1284, 1249, 1211, 1197, 1139, 1101, 1068, 1047, 914, 869, 754, 688. 1H-NMR (600 MHz, CDCl3, δH): 0.16 [9Н, s, (СН3)3Si], 1.35 (d, J = 7 Hz, 3Н, СН3), 1.38 (d, J = 7 Hz, 3Н, СН3), 3.25 [1H, m, СH(СН3)2], 6.40 (d, J = 9.6 Hz, 1Н, Н-6), 7.40 (s, 1Н, Н-9), 7.57 (s, 1Н, Н-4), 7.79 (d, J = 9.6 Hz, 1Н, Н-5). 13C-NMR (150 MHz, CDCl3, δC): 9.5 (3×СН3), 20.1 (СН3), 20.3 (СН3), 27.3 (СН), 89.2 (С-1a), 94.8 (С-1b), 96.2 (С-3), 98.6 (С-9), 106.9 (С-3а), 114.3 (С-6), 115.7 (С-4а), 115.8 (С-4), 116.8 (С-3), 142.7 (С-5), 152.4 (С-8а), 153.2 (С-9a), 159.0 (С-2), 160.8 (С-7). UV (EtOH) λmax (lgε): 222 (4.36), 251 (4.07), 294 (2.74), 338 (2.64) nm. Anal. Calcd for C19H20O3Si: С, 70.34; Н, 6.21; Si 8.66; found С, 69.98; Н, 5.99; Si, 8.35.
3-Ethynyl-2-isopropyl-7H-furo[3,2-g]chromen-7-one (32). To a solution of compound 31 (100 mg, 0.3 mmol) in methanol (5 mL) were added CsF (230 mg, 1.5 mmol) and benzyltrimethylammonium chloride (28 mg, 0.15 mmol). The mixture was stirred at rt for 10 h in under argon (TLC). Then 10 mL of water was added and the mixture was extracted with methylene chloride (5 × 4 mL). The combined extract was washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel. Eluting with chloroform and crystallization from diethyl ether gave compound 32 (50 mg, 66%). M.p. 82–83 °С (ether). IR (KBr, ν, cm−1): 3435, 3059, 2979, 2935, 2877, 2185, 1732, 1685, 1625, 1577, 1471, 1433, 1388, 1321, 1286, 1249, 1211, 1197, 1137, 1116, 1047, 869, 819, 721, 694. 1H-NMR (400 MHz, CDCl3, δH): 1.25 (d, J = 7 Hz, 3Н, СН3), 1.29 (d,3Н, J = 7 Hz, СН3), 2.37 (s, 1H, ≡CH), 3.19 [m, 1H, СH(СН3)2], 6.32 (d, J = 9.6 Hz, 1Н, Н-6), 7.33 (s, 1Н, Н-9), 7.50 (s, 1Н, Н-4), 7.71 (d, J = 9.6 Hz, 1Н, Н-5). 13C-NMR (100 MHz, CDCl3, δC): 20.2 (СН3), 20.4 (СН3), 25.8 (СН), 80.5 (С-1a), 87.6 (С-1b), 96.1 (С-3), 100.4 (С-9), 107.3 (С-3а), 115.6 (С-6), 116.0 (С-4а), 116.4 (С-4), 116.6 (С-3), 143.5 (С-5), 152.2 (С-8а), 153.1 (С-9a), 156.9 (С-2), 160.2 (С-7). UV (EtOH) λmax (lgε): 224 (3.91), 250 (3.94), 285 (sh), 335 (3.34) nm. Anal. Calcd for C16H12O3: С, 76.18; Н, 4.79; found С, 76.31; Н, 5.09.



{kind=link}
{kind=link}
{kind=link}