Pharmacological Importance of Optically Active Tetrahydro-β-carbolines and Synthetic Approaches to Create the C1 Stereocenter


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
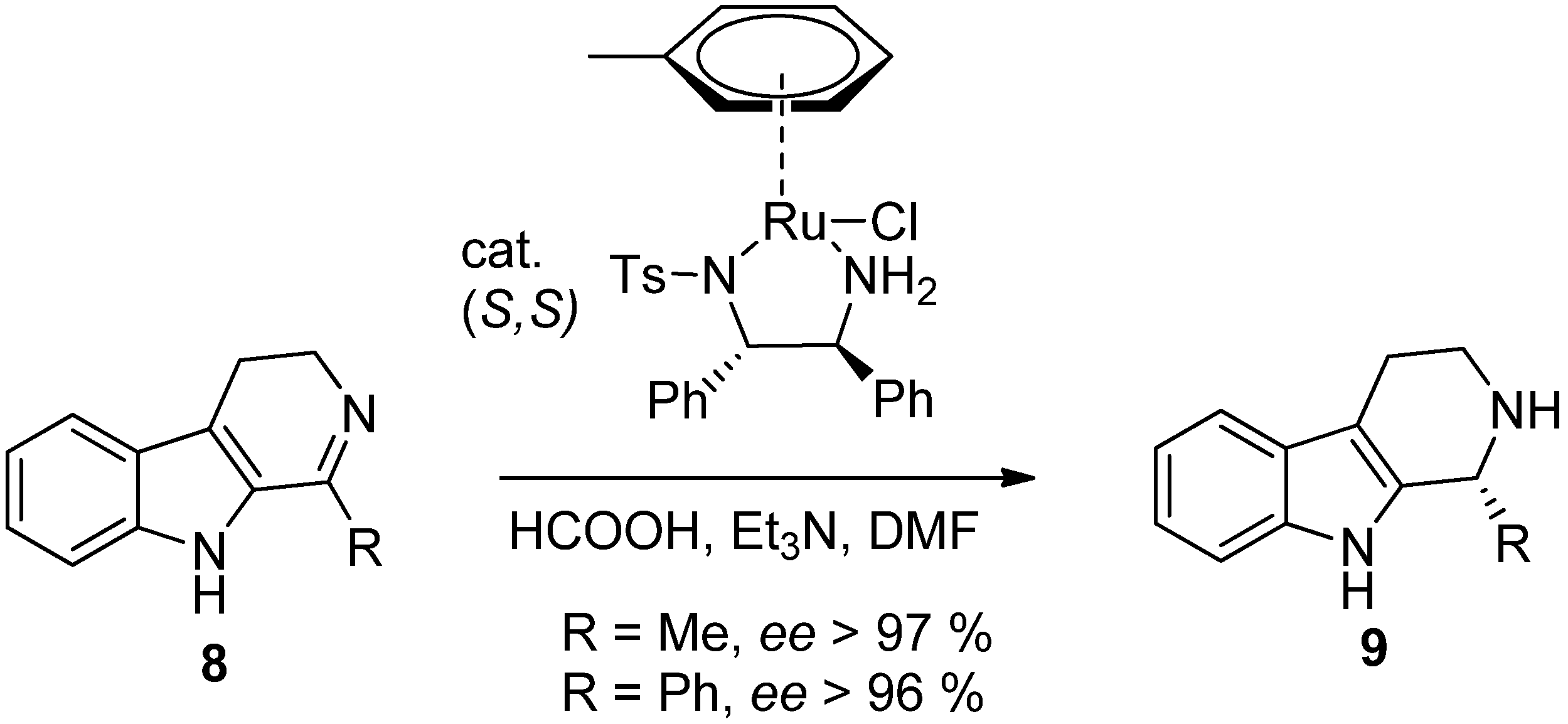{kind=link}
{kind=link}
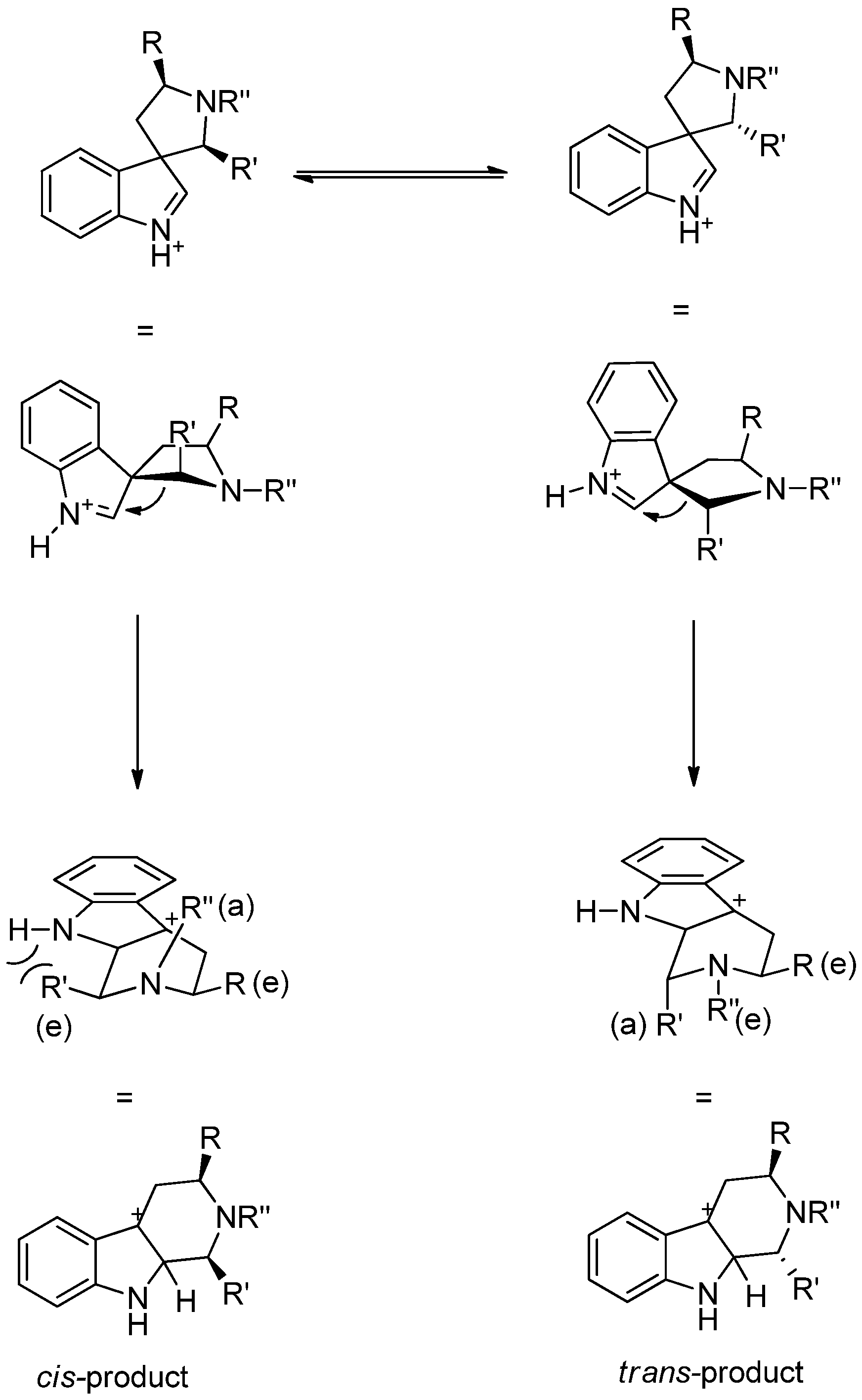{kind=link}
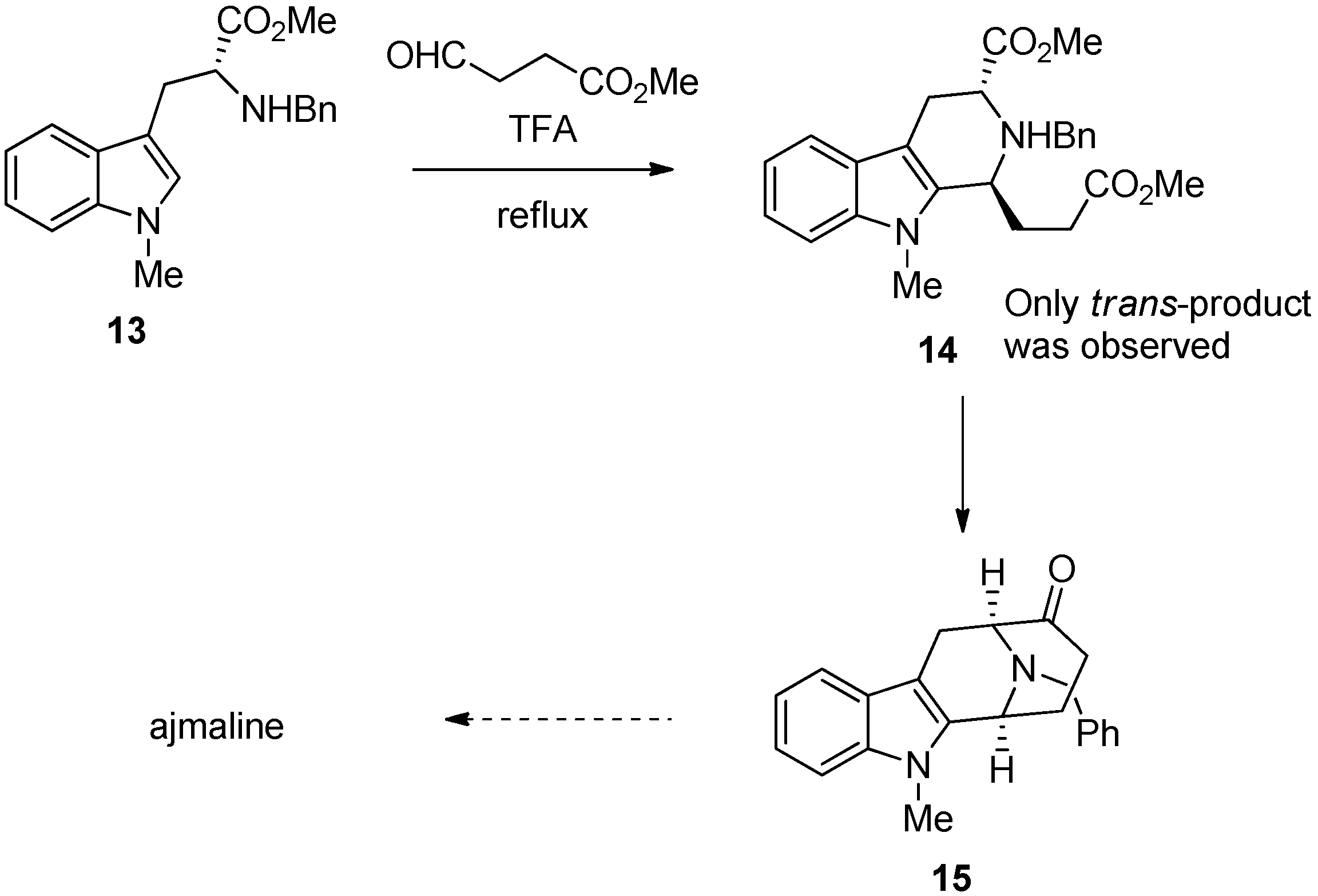{kind=link}
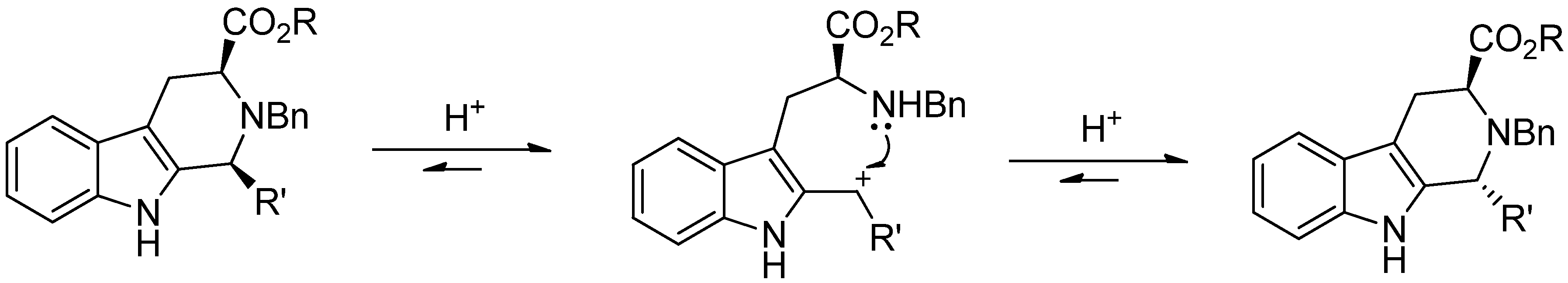{kind=link}
{kind=link}
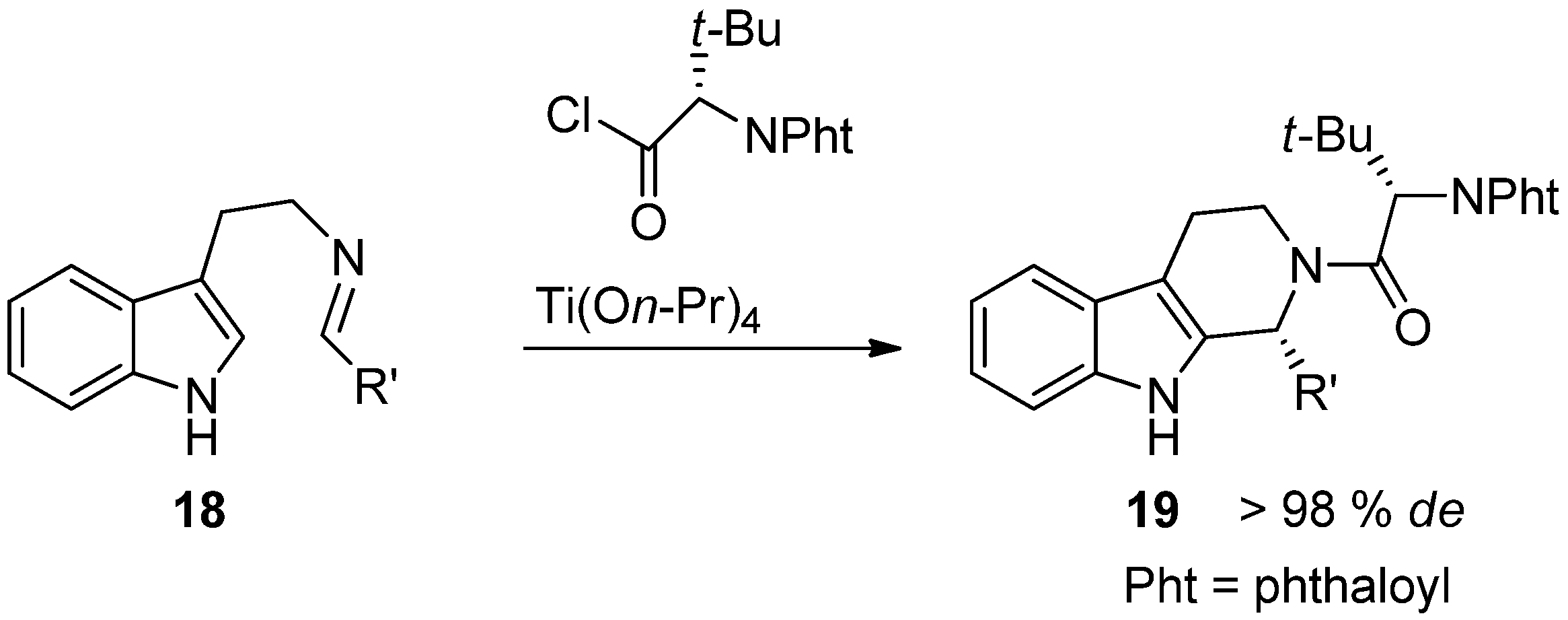{kind=link}
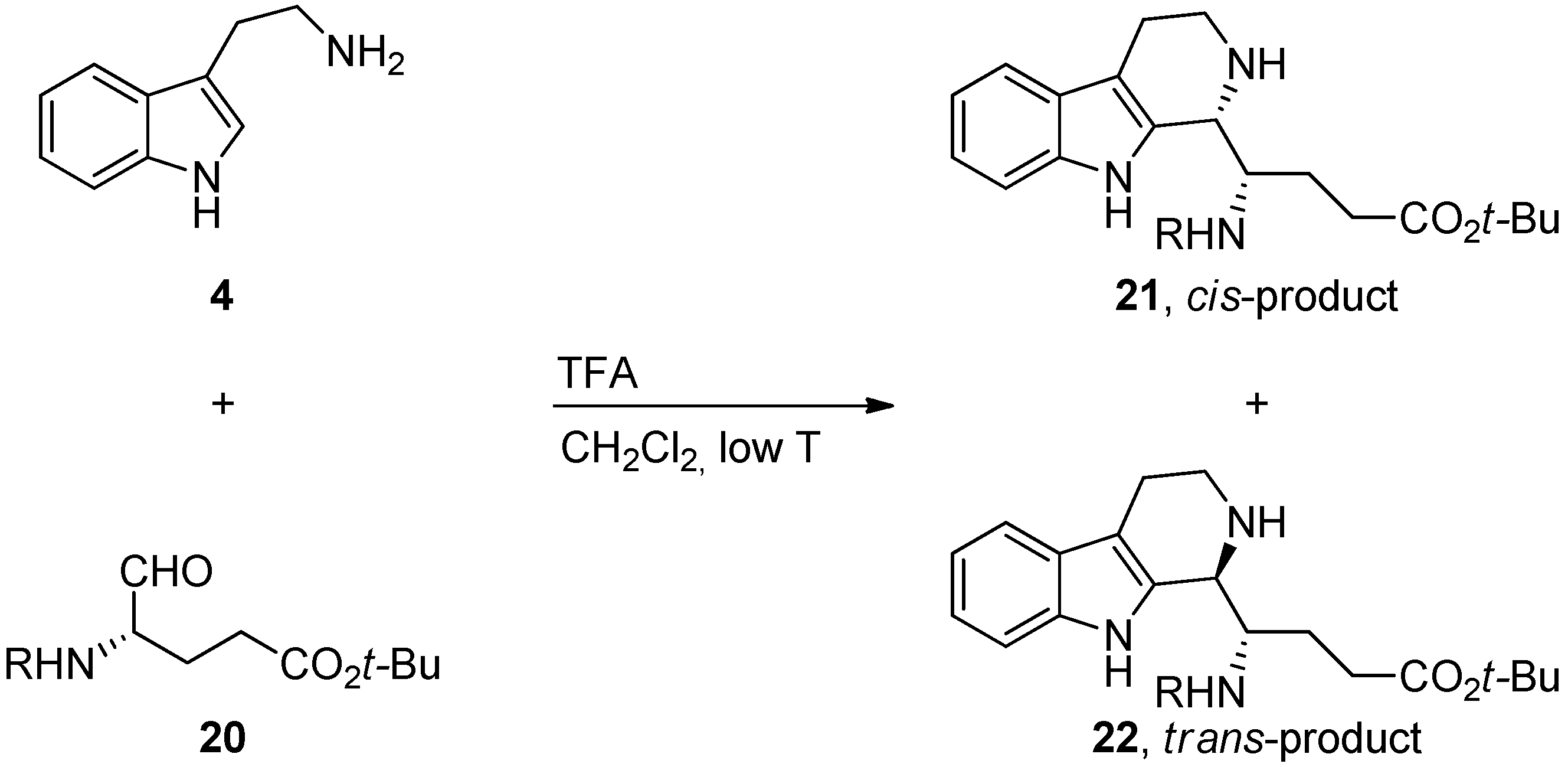{kind=link}
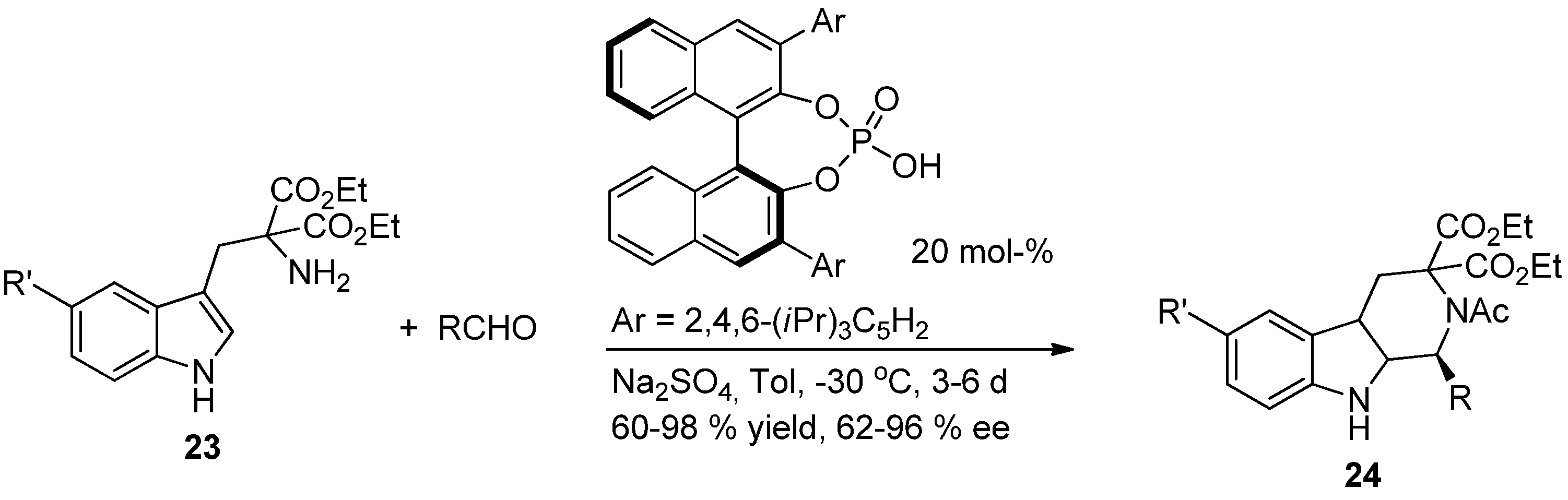{kind=link}
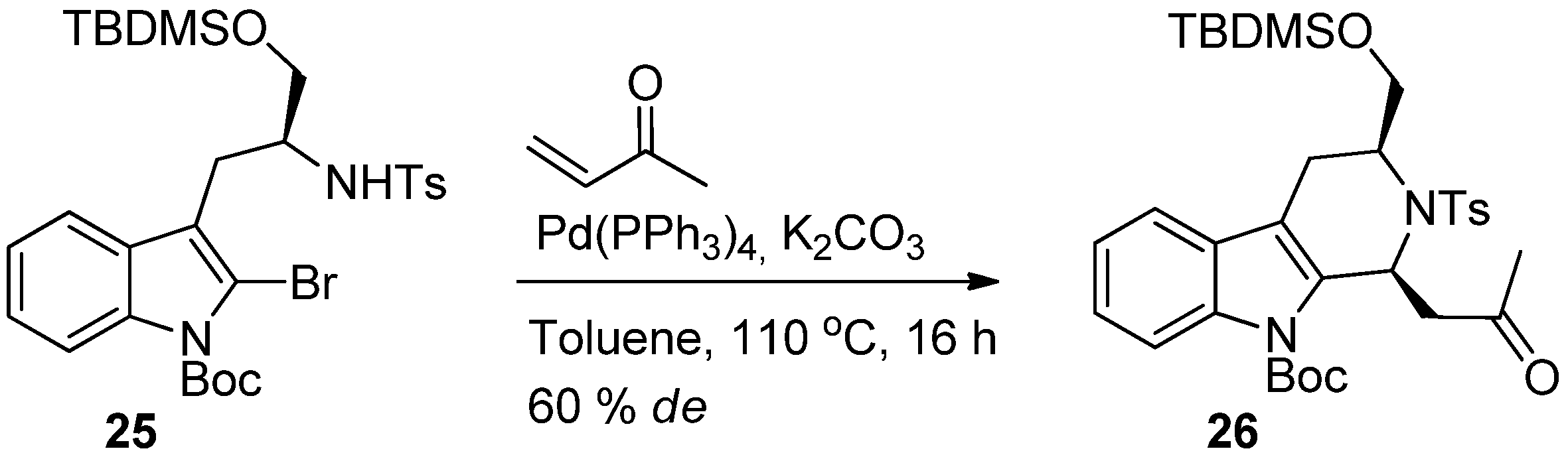{kind=link}
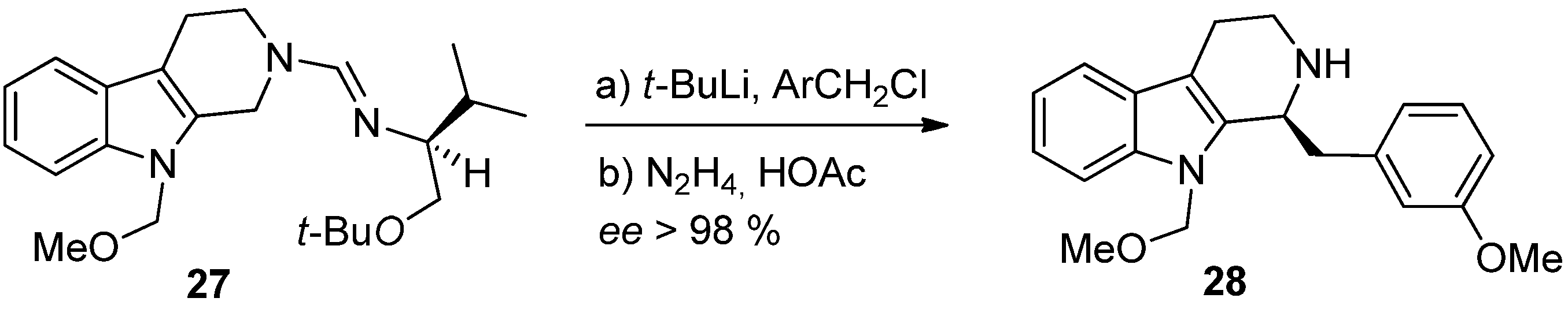{kind=link}
1. Introduction
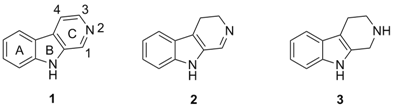
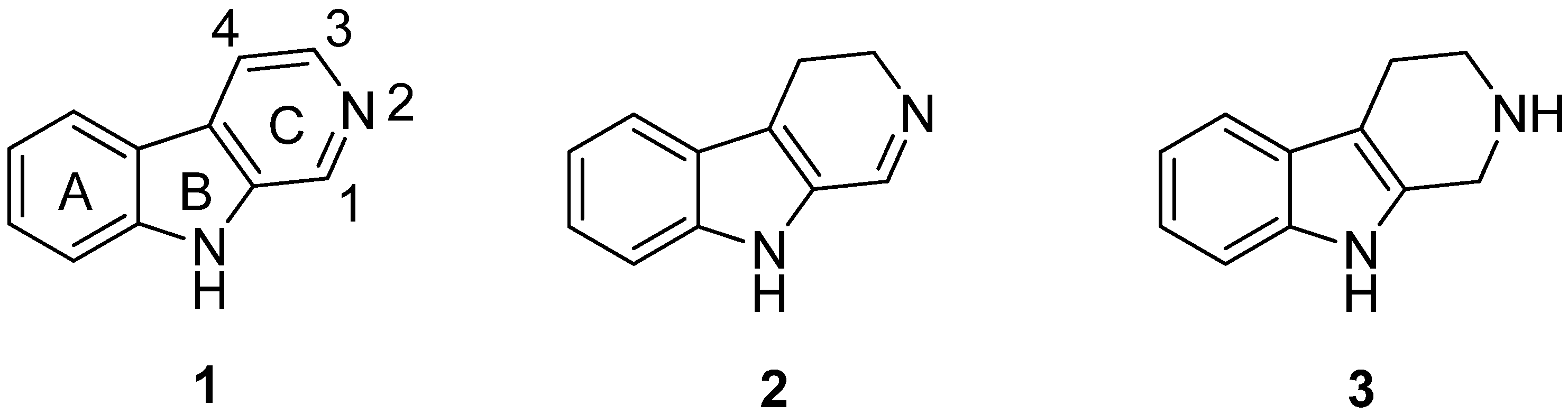
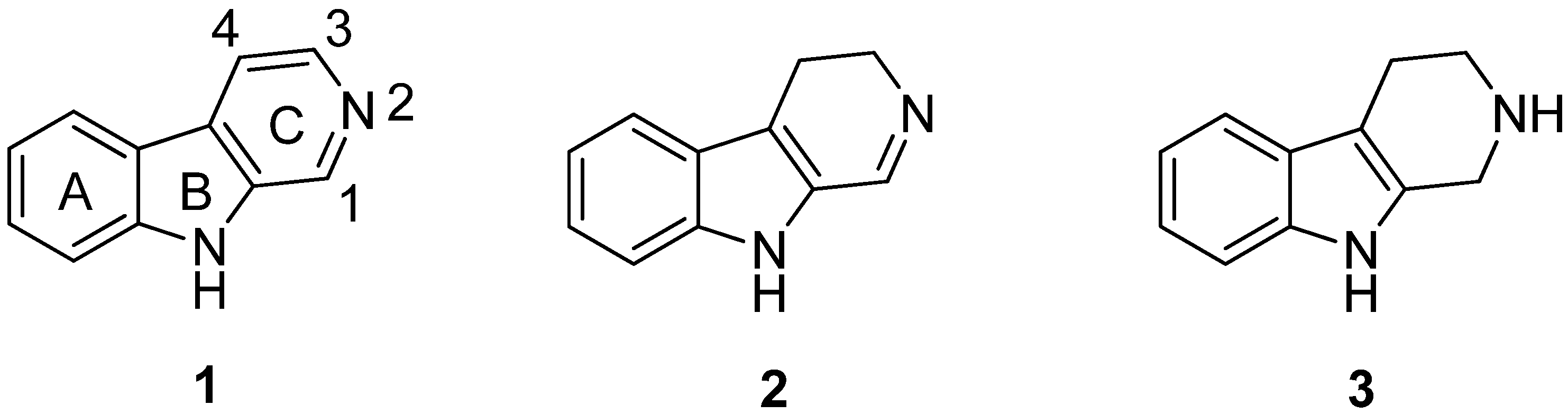
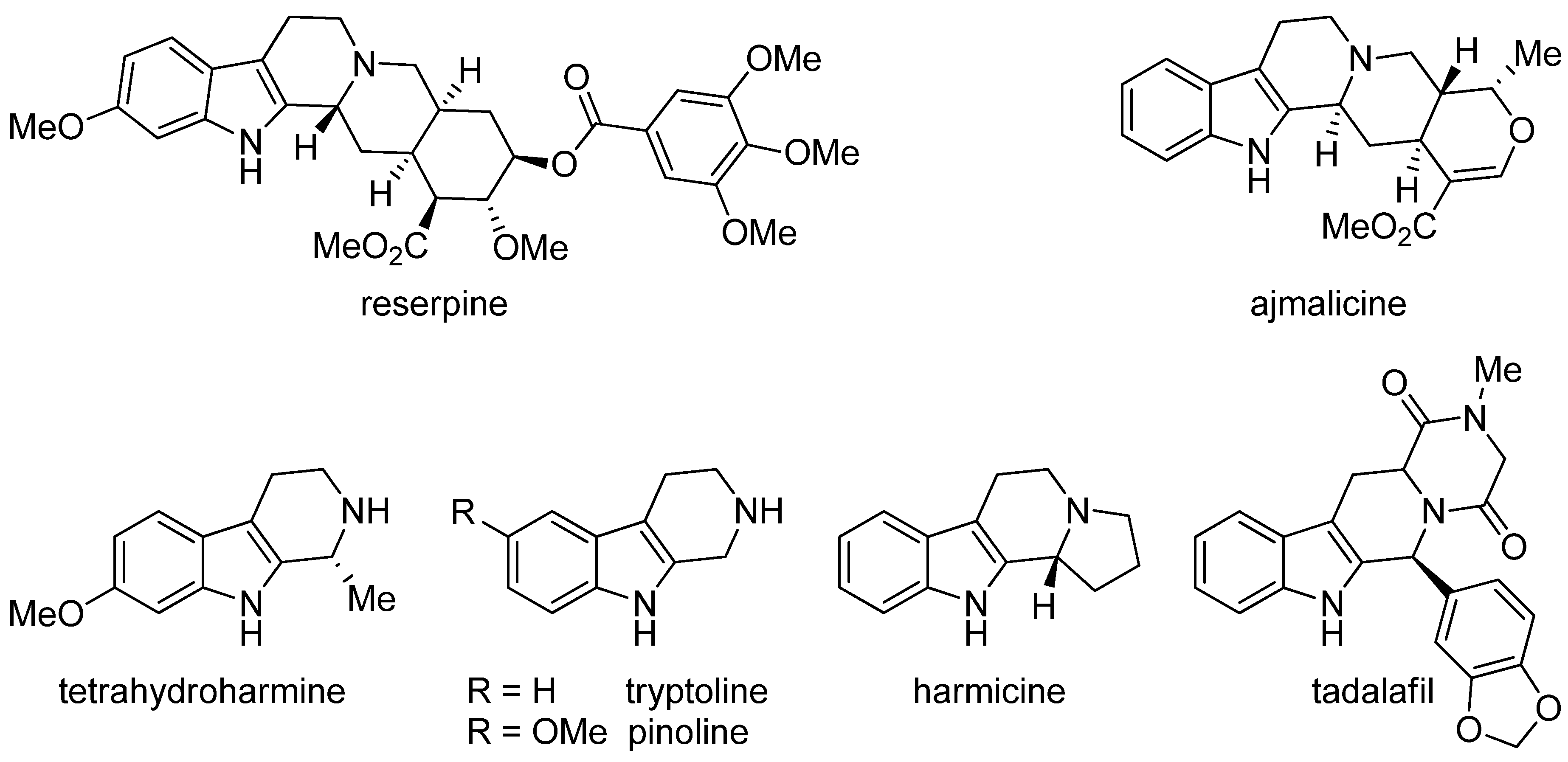
2. Structure and Occurrence
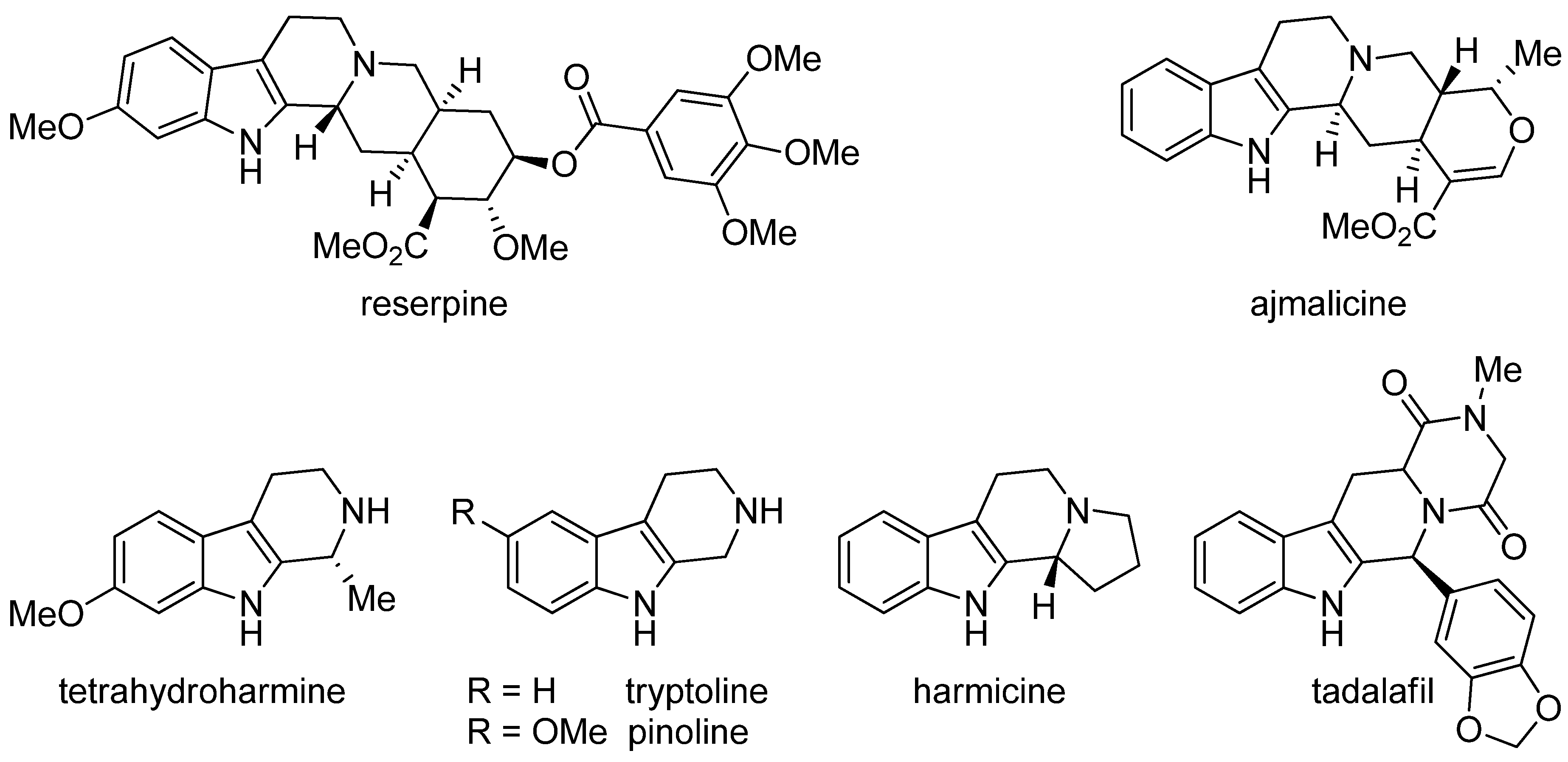
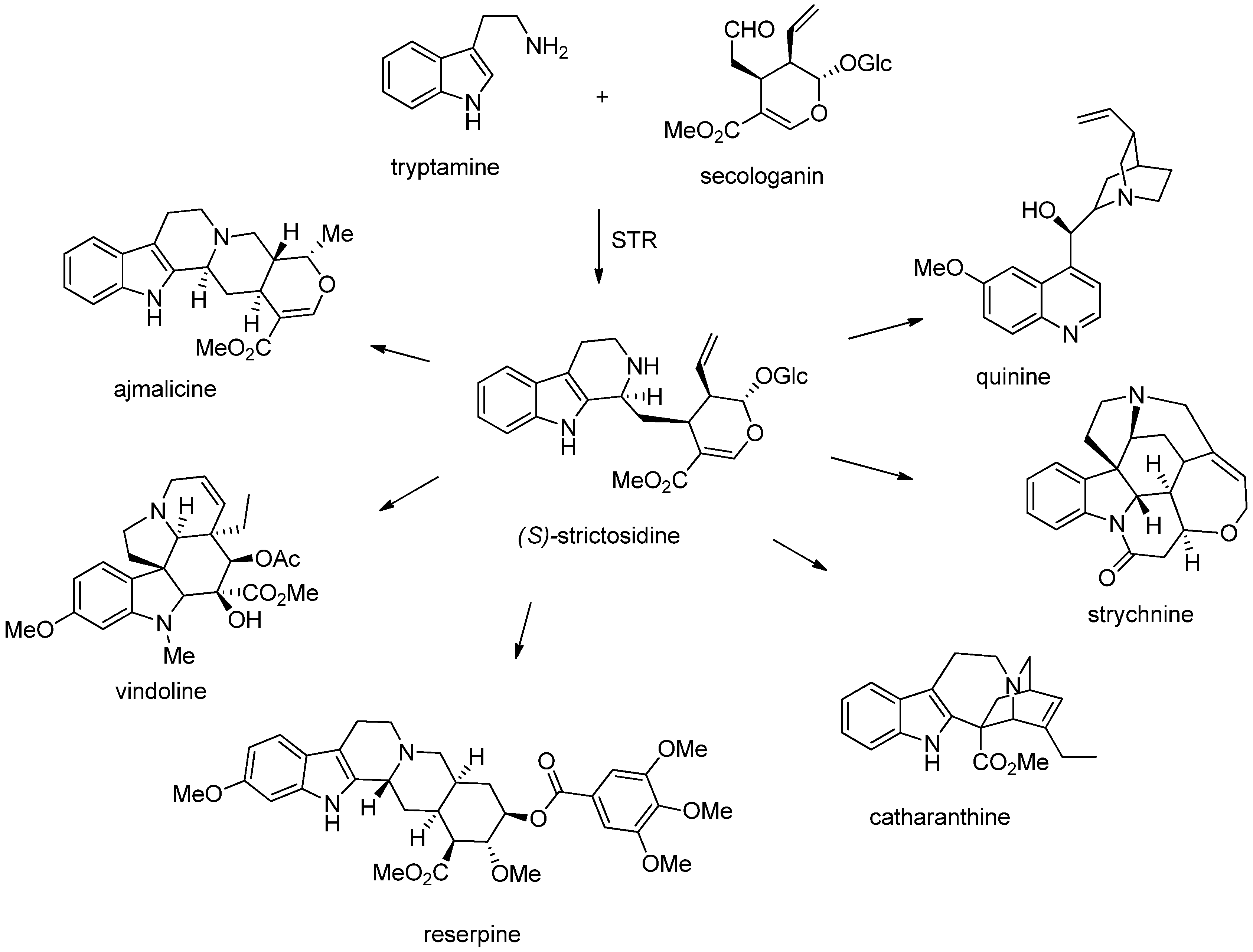
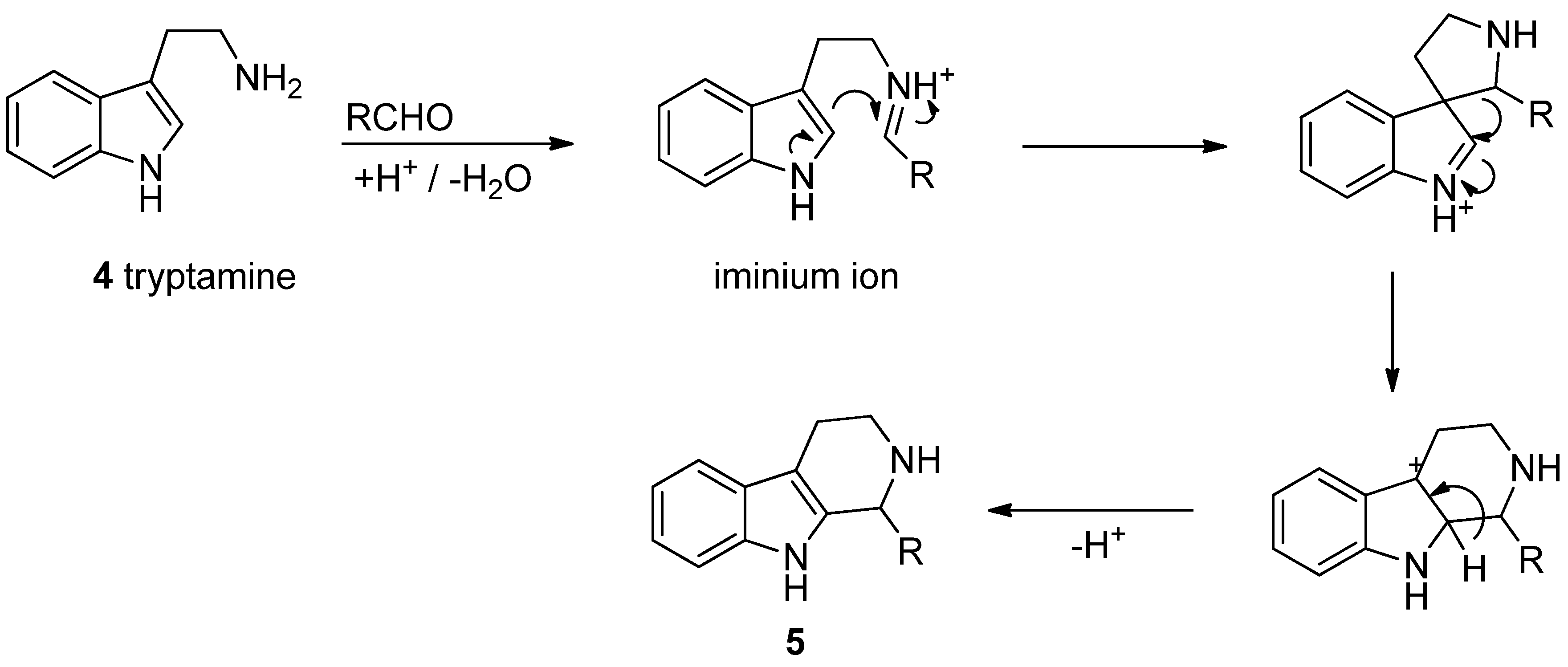
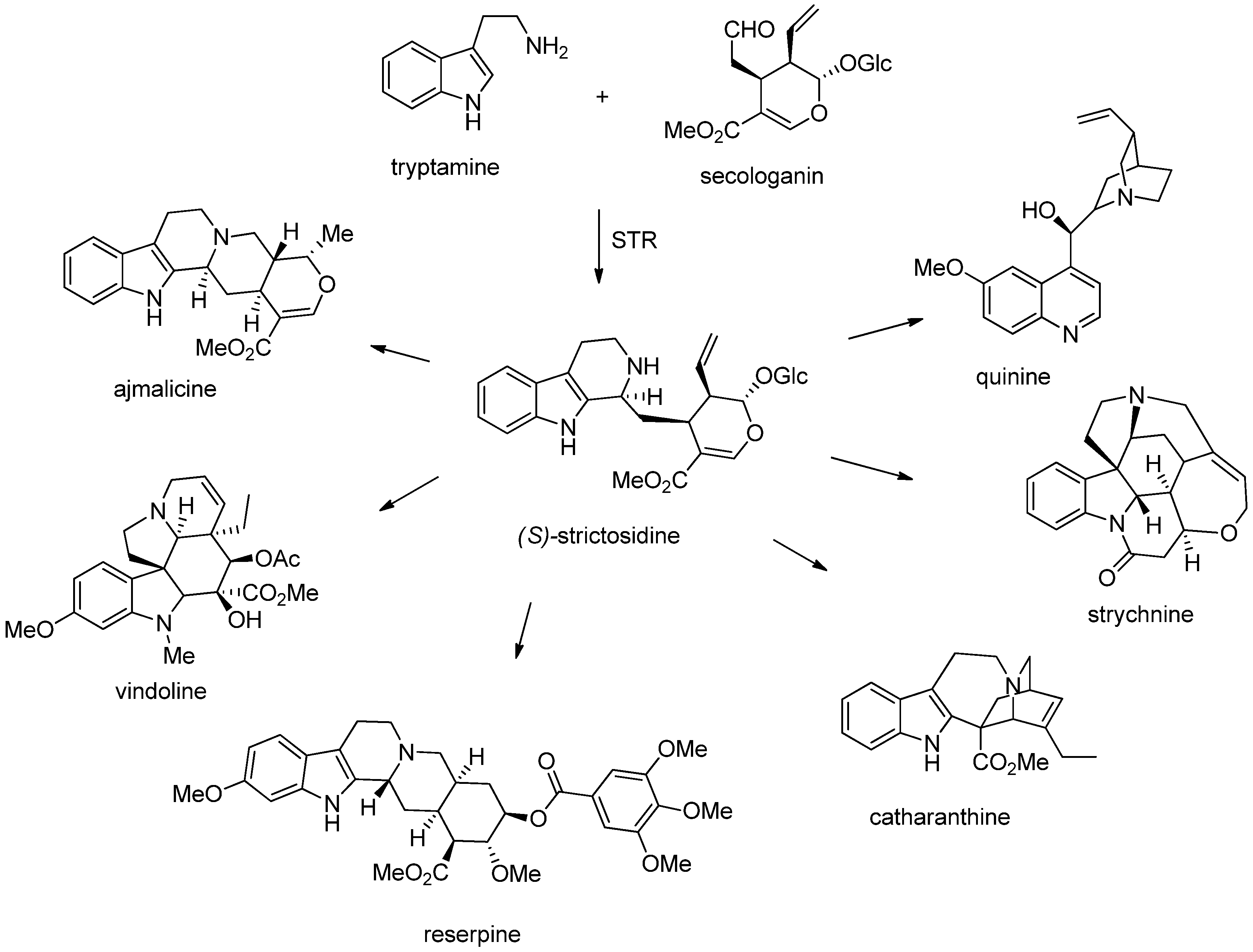
3. Biosynthesis

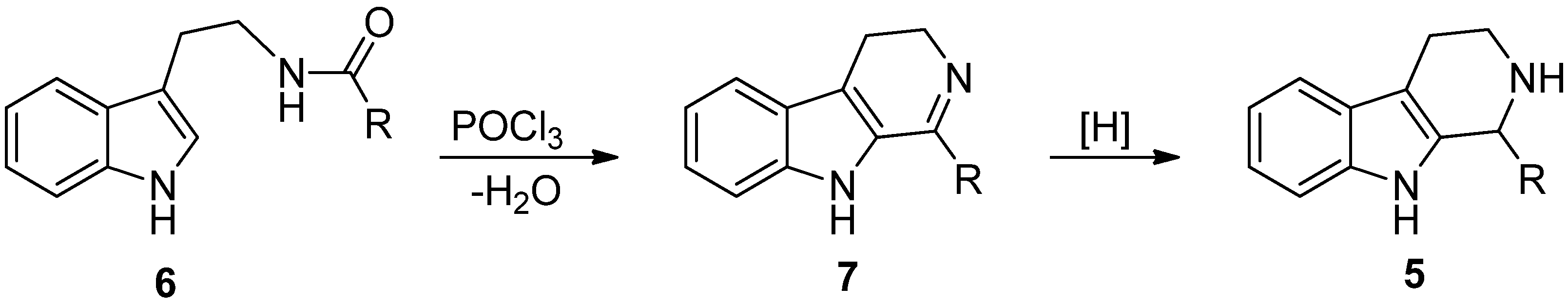
4. Synthetic Methods to Create the C1 Stereocenter
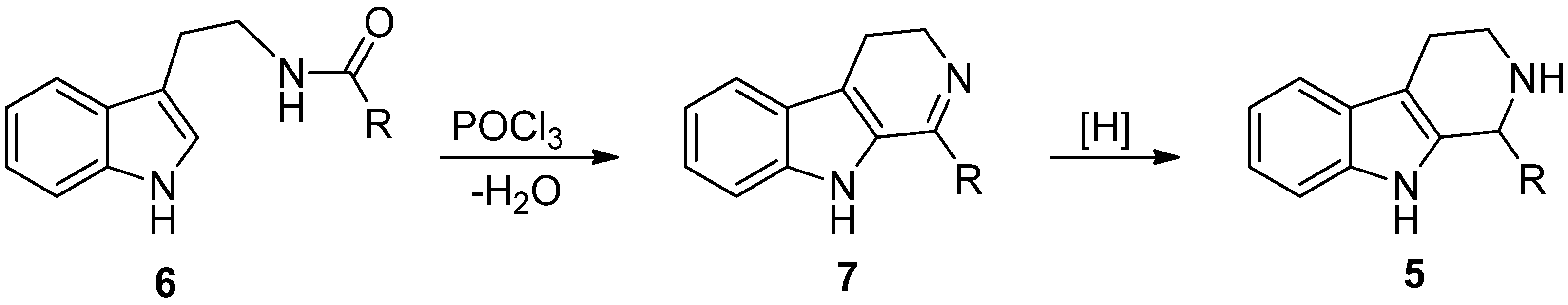
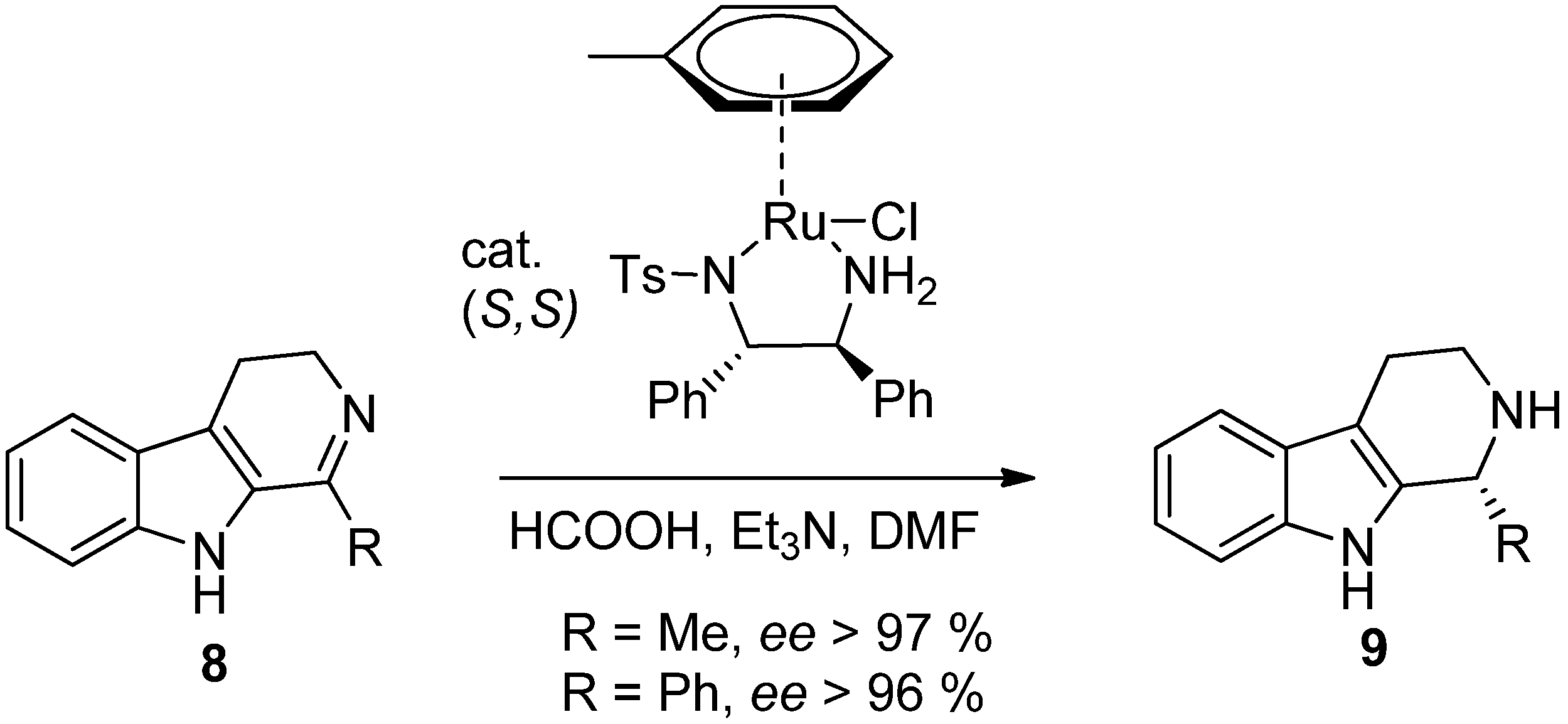


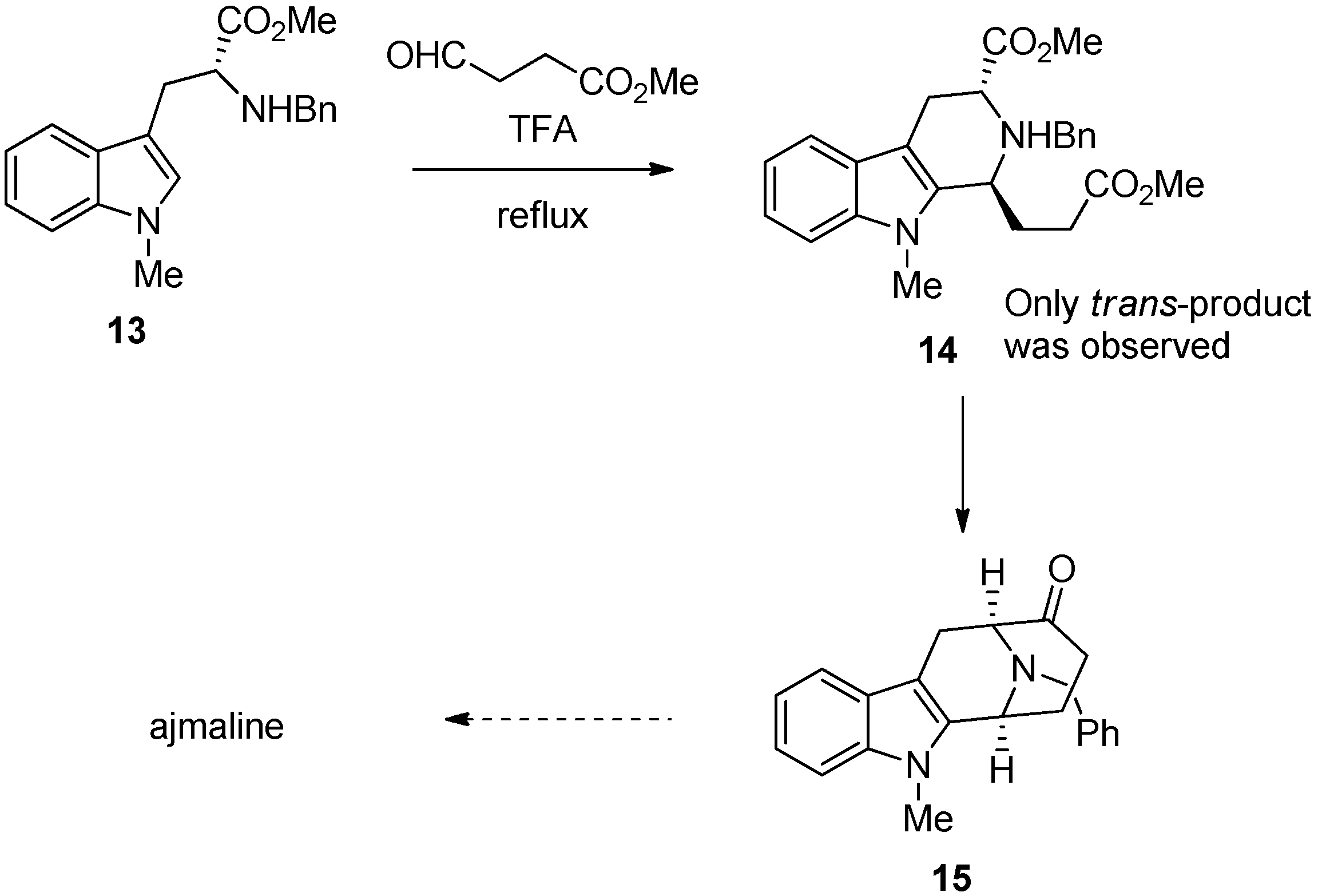
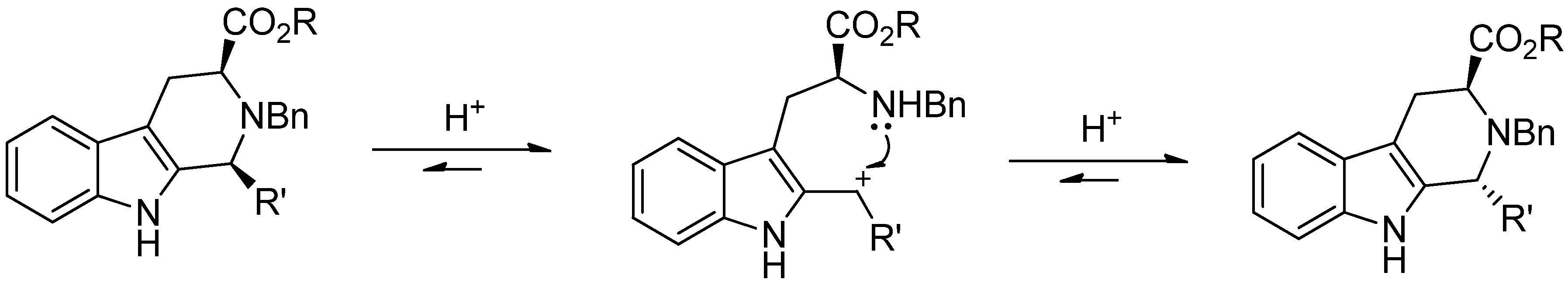
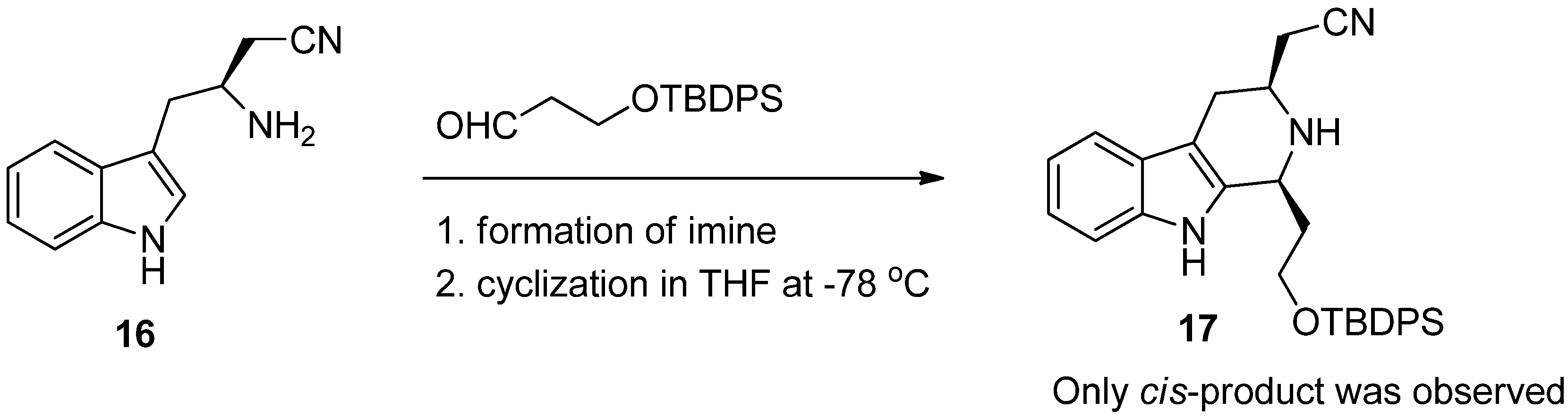

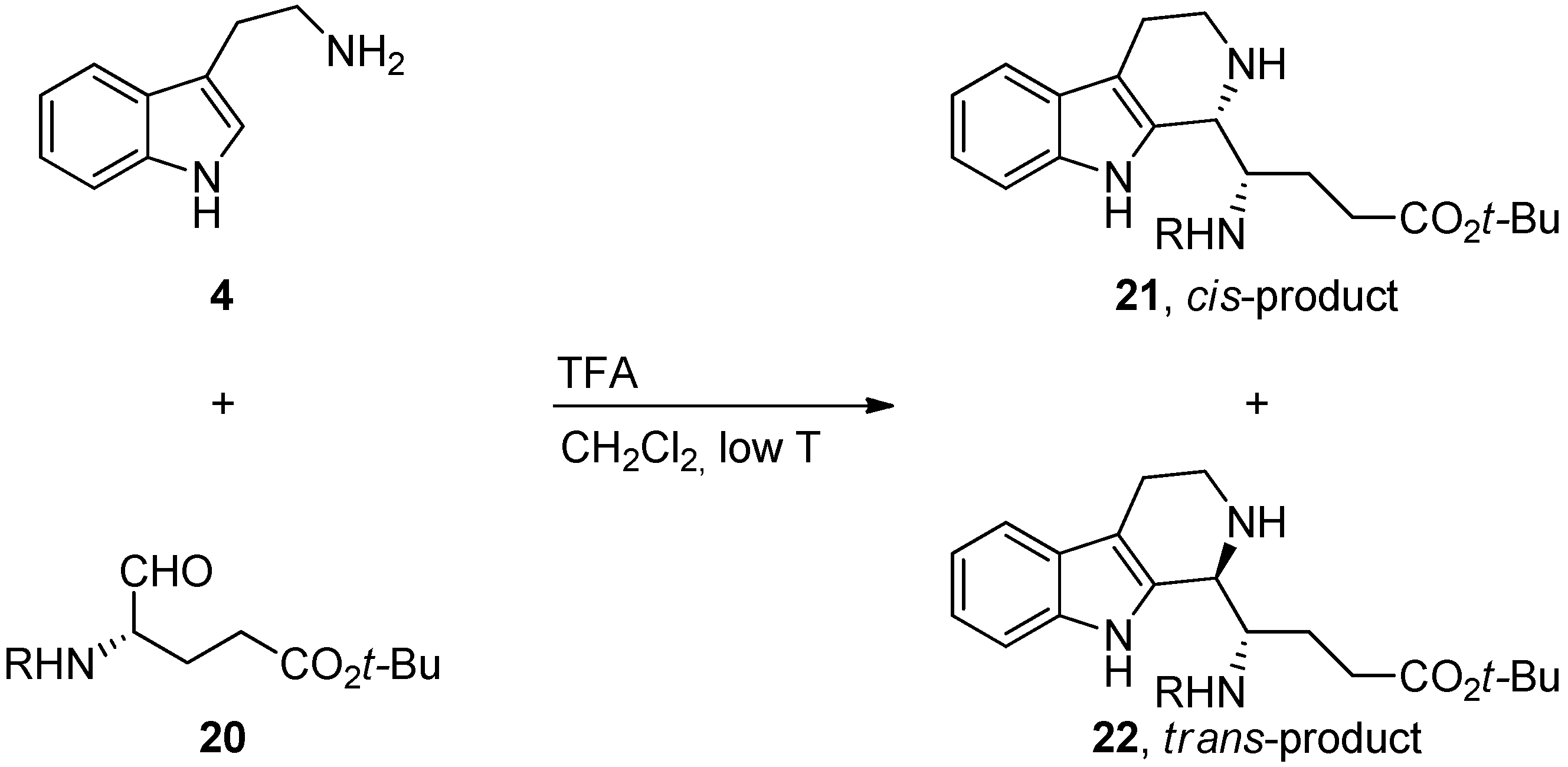
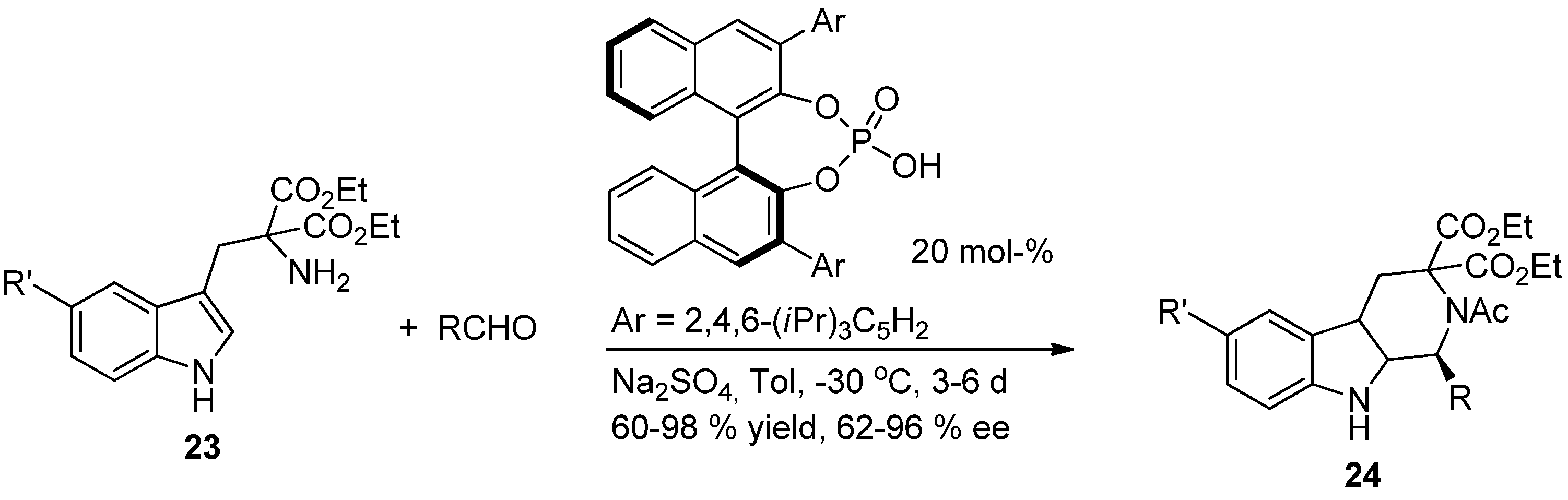
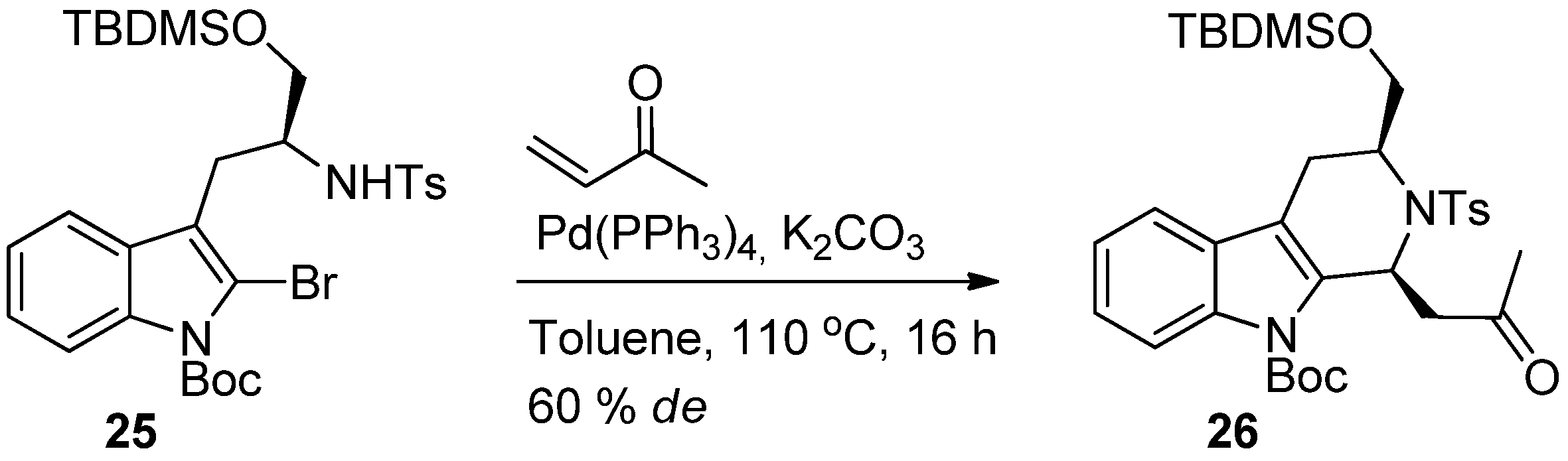
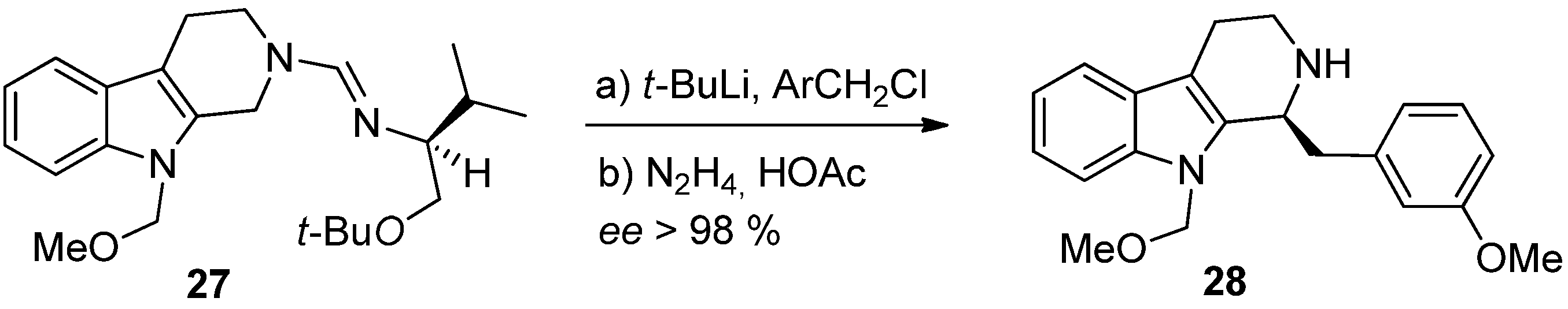
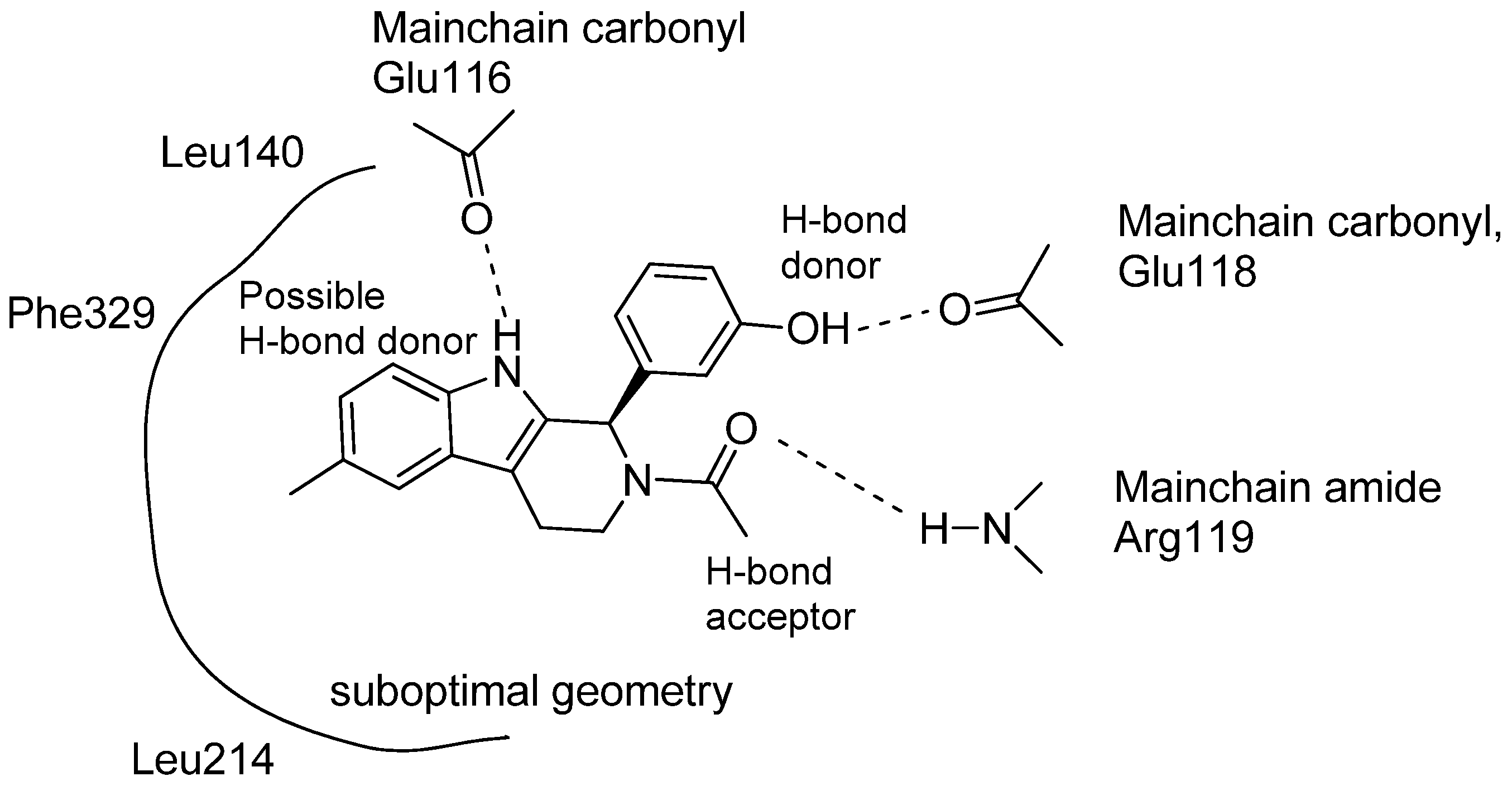
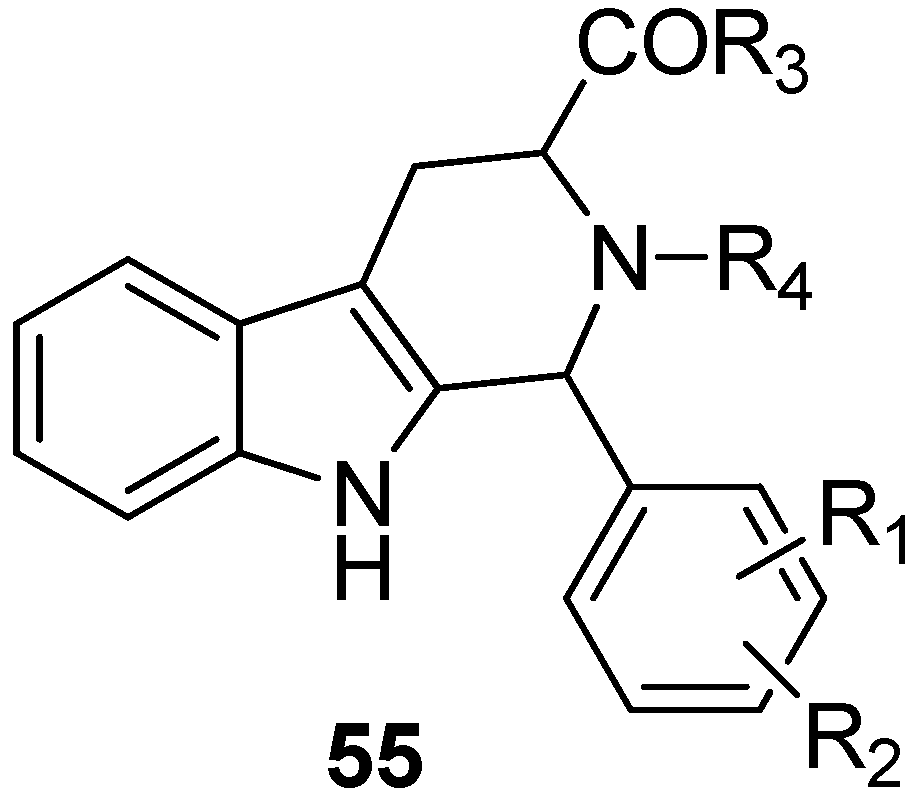
5. Pharmacological Importance
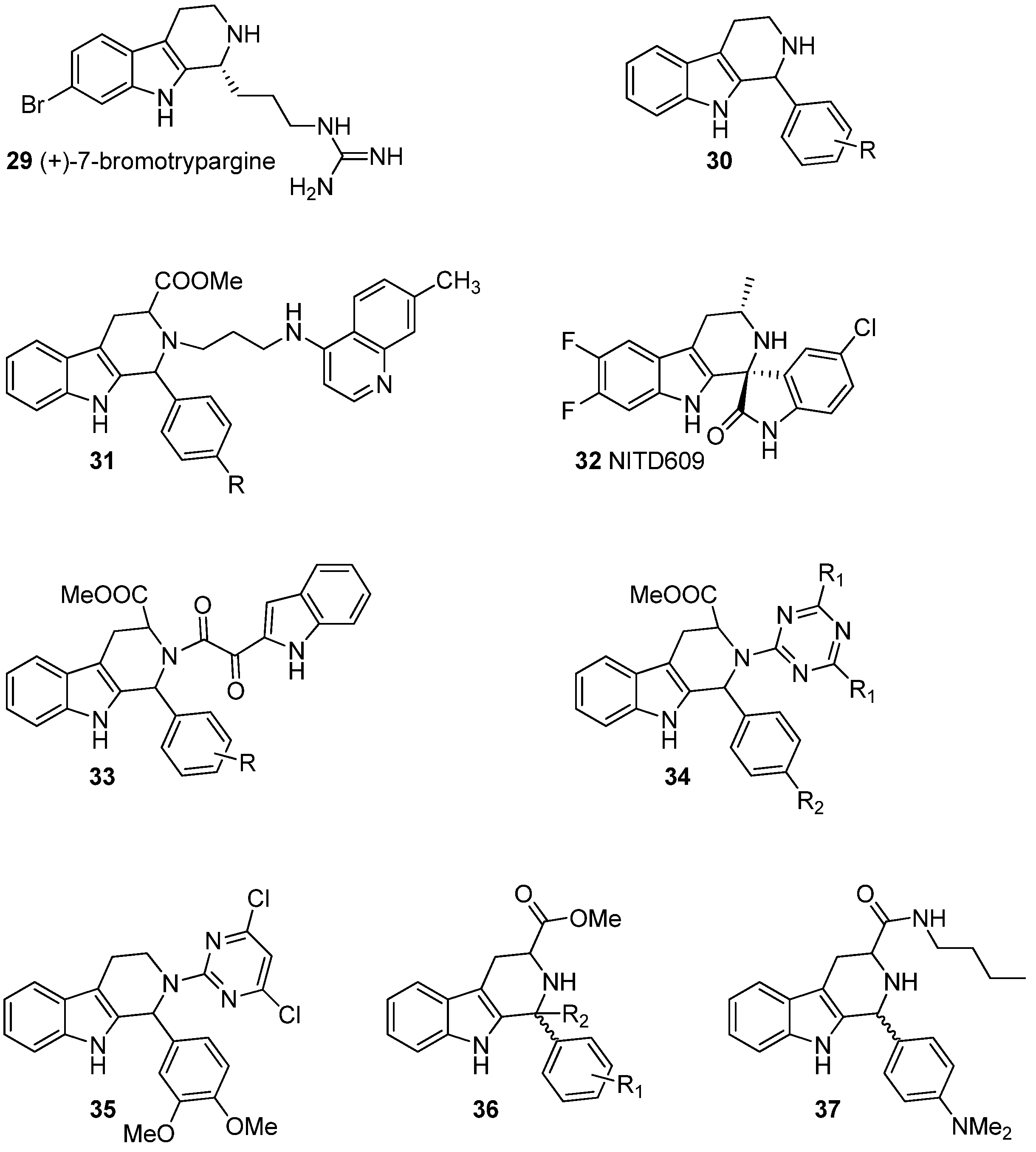
5.1. Antiprotozoal Activity
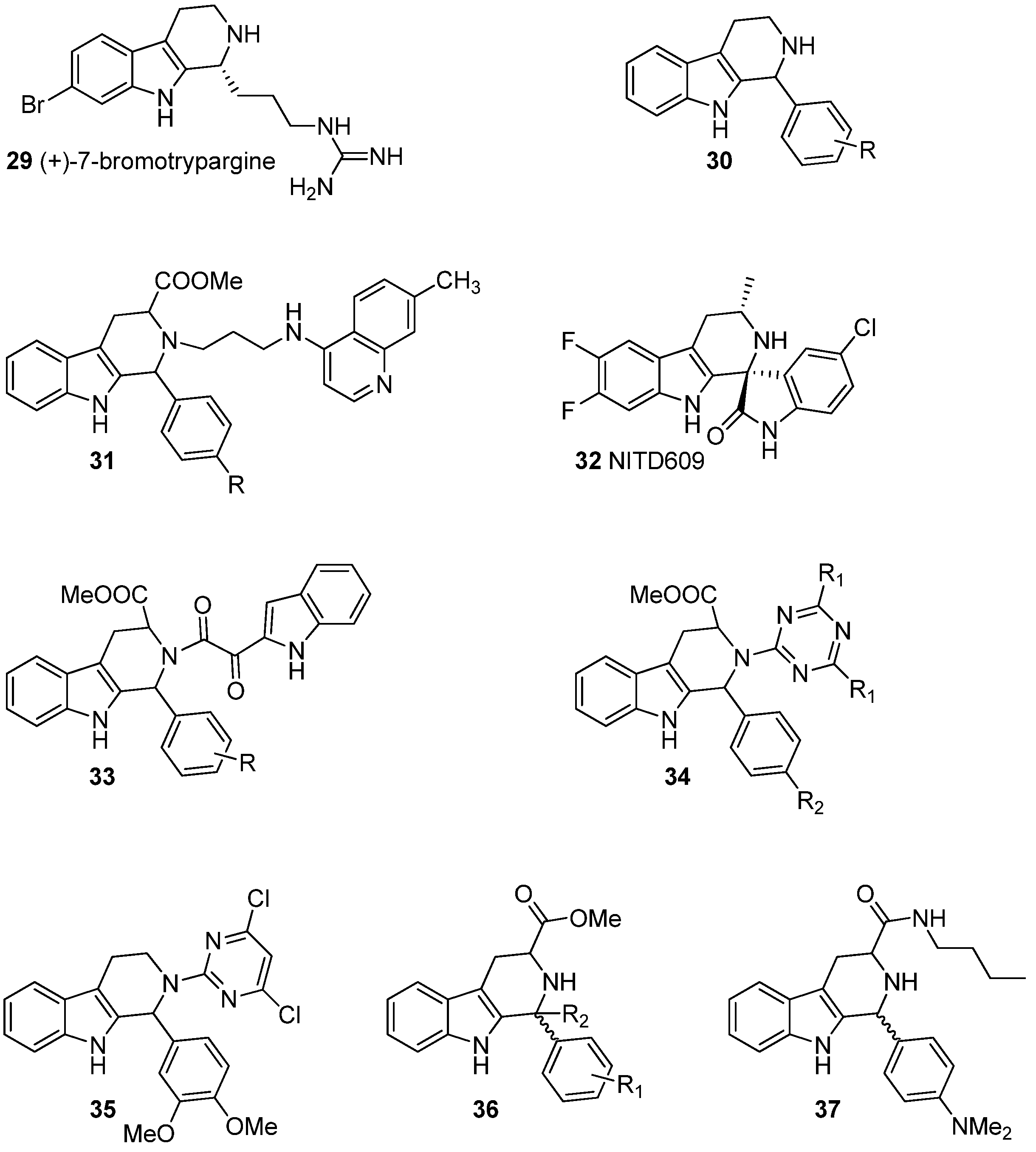
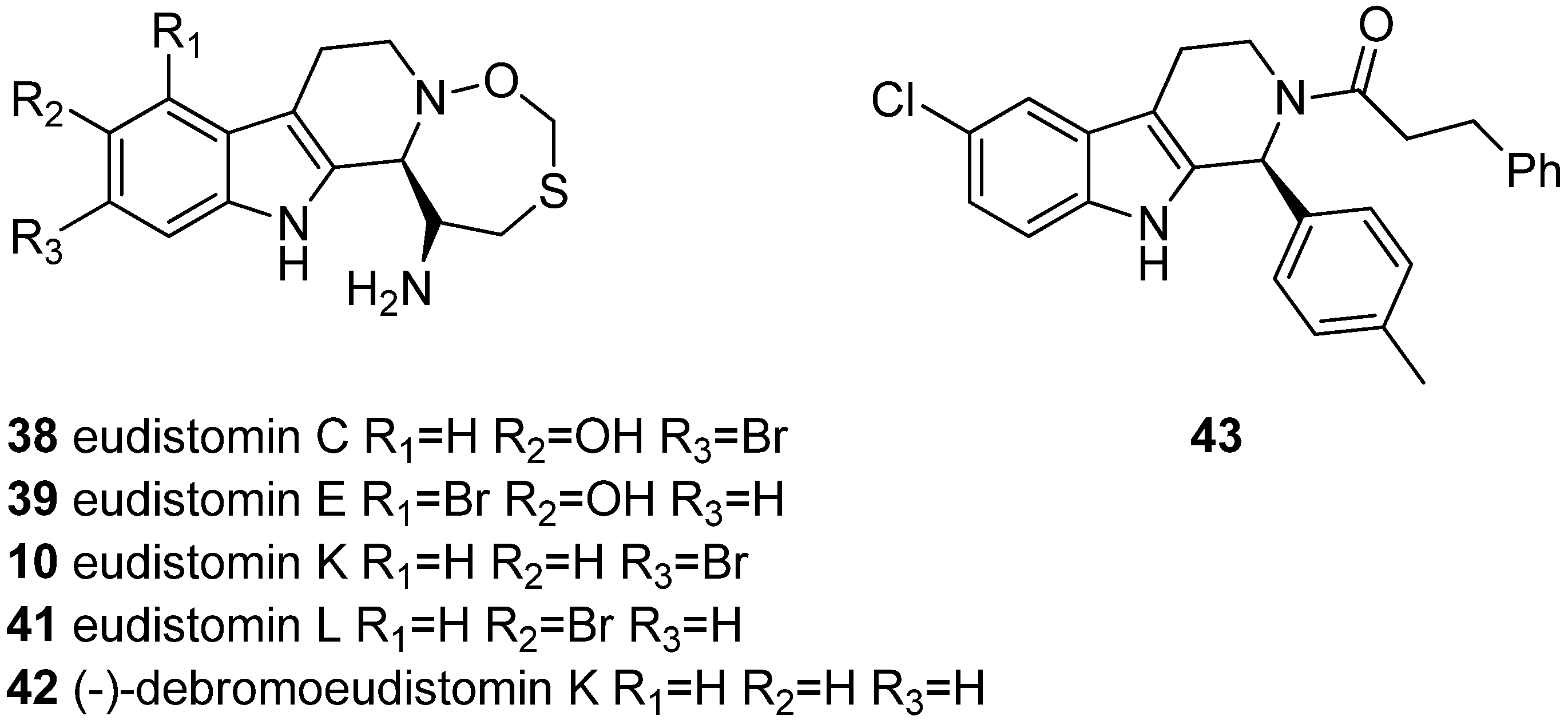
5.2. Antiviral Activity
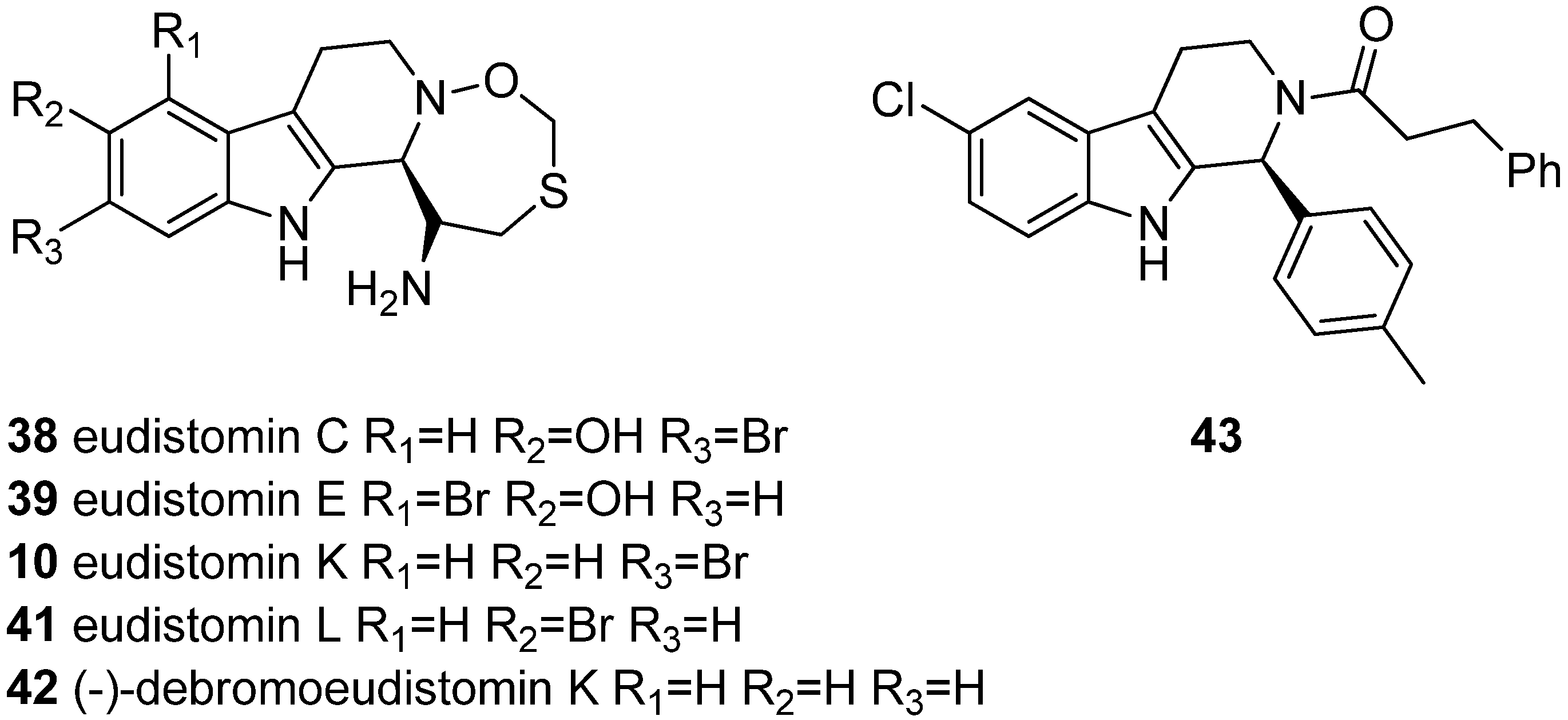
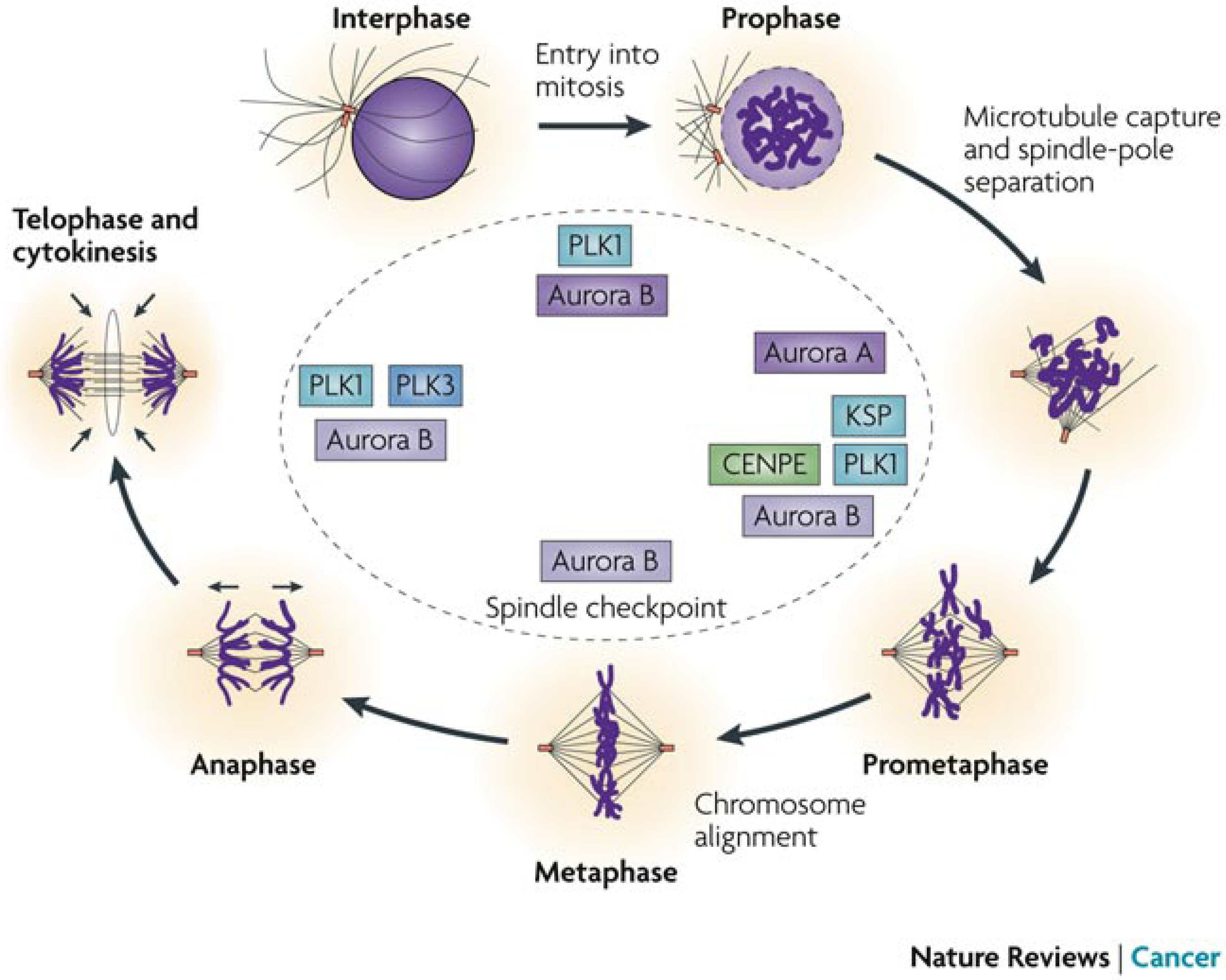
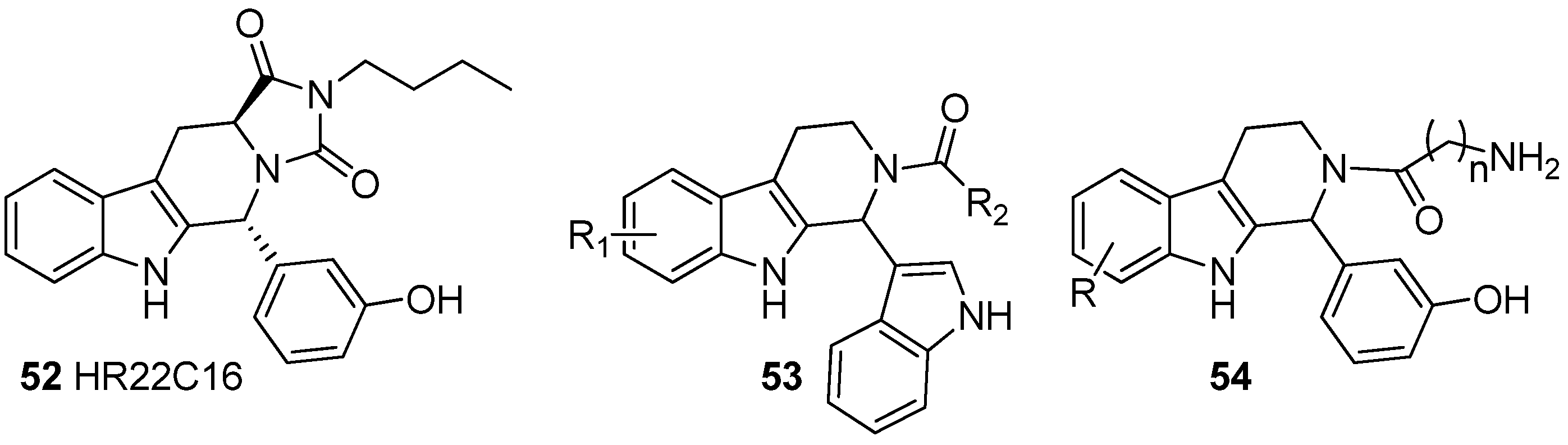
5.3. Anticancer
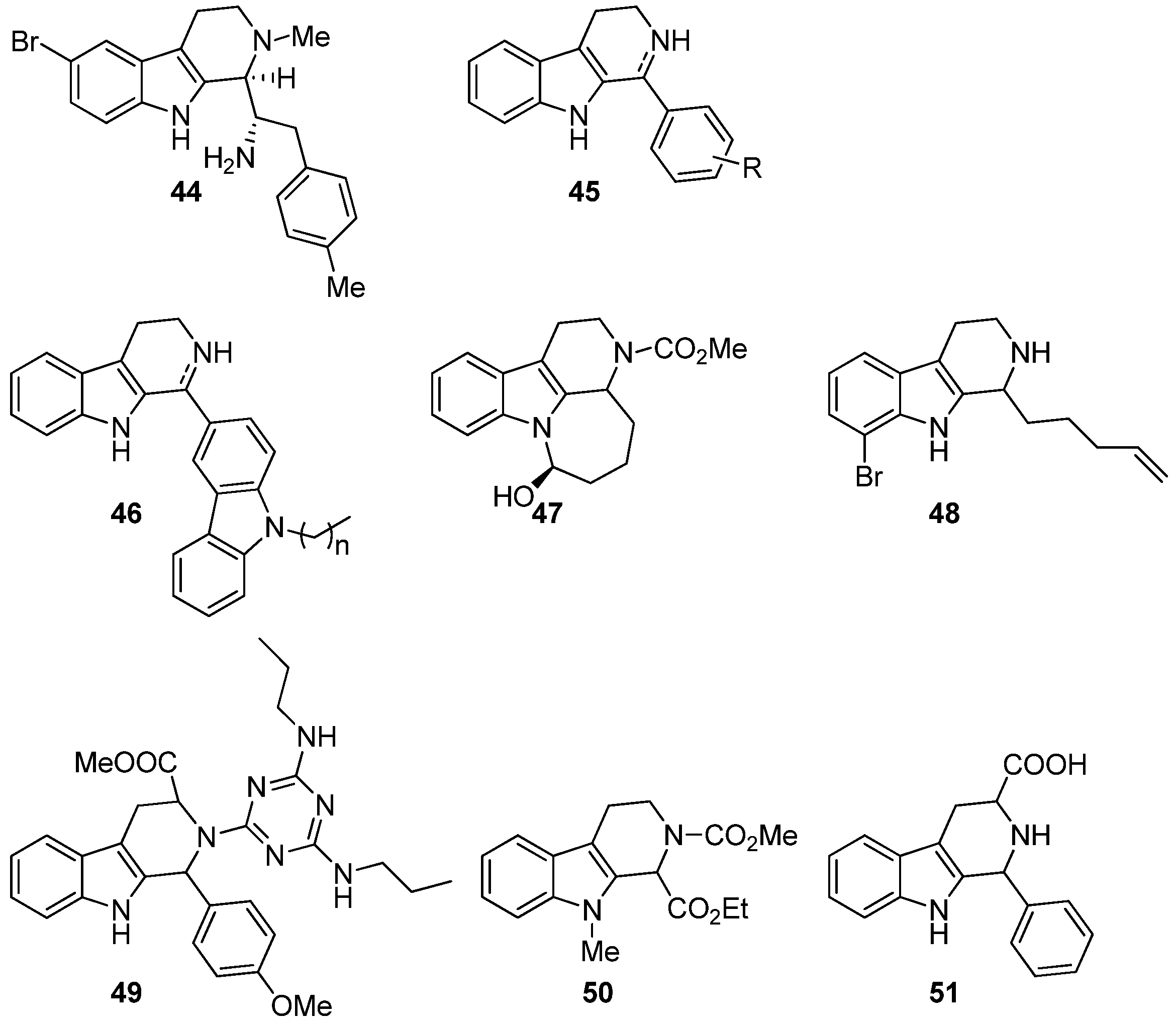
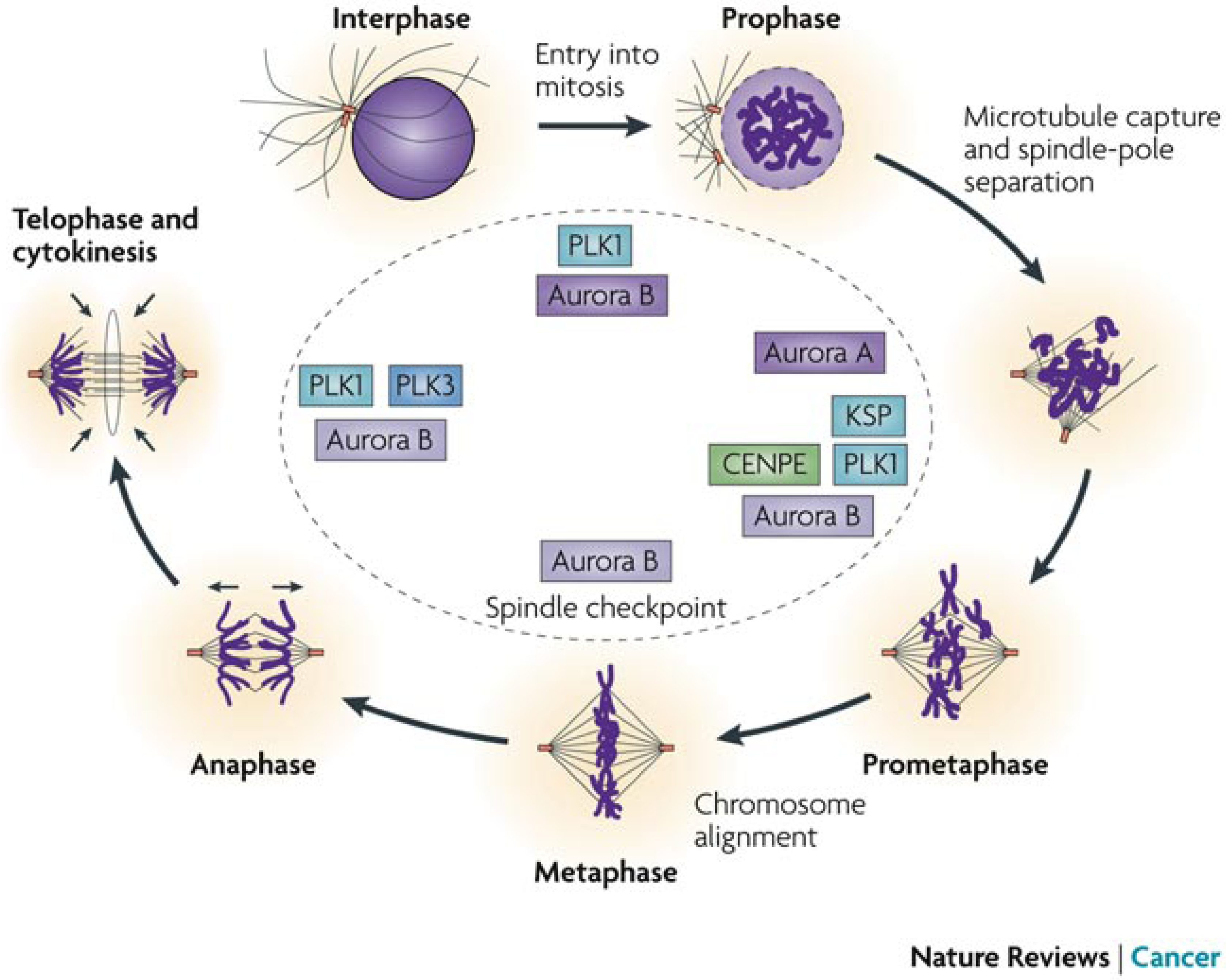
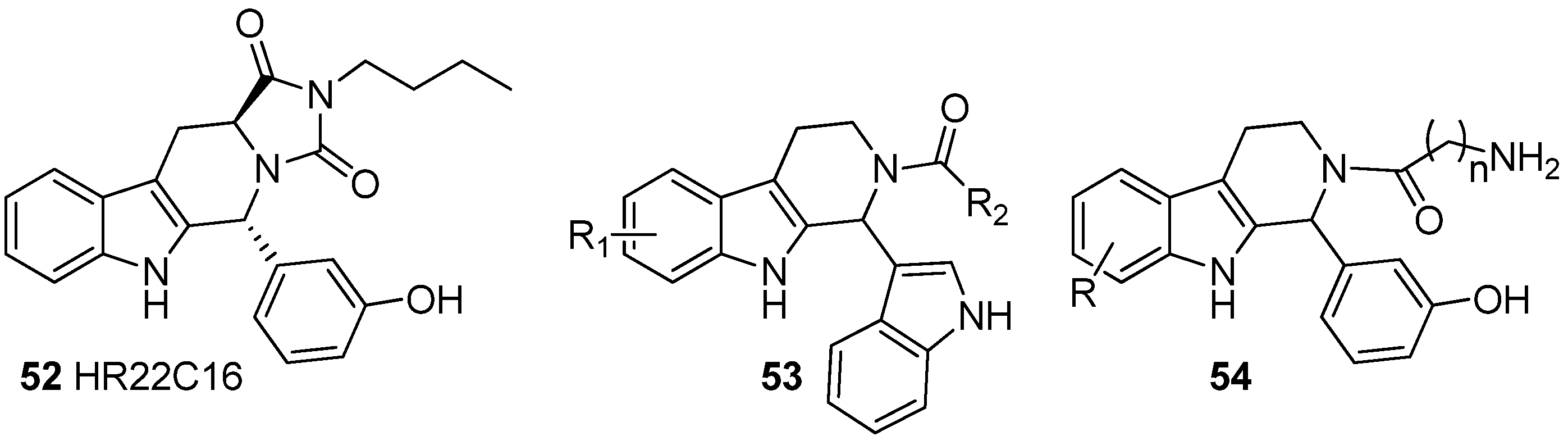
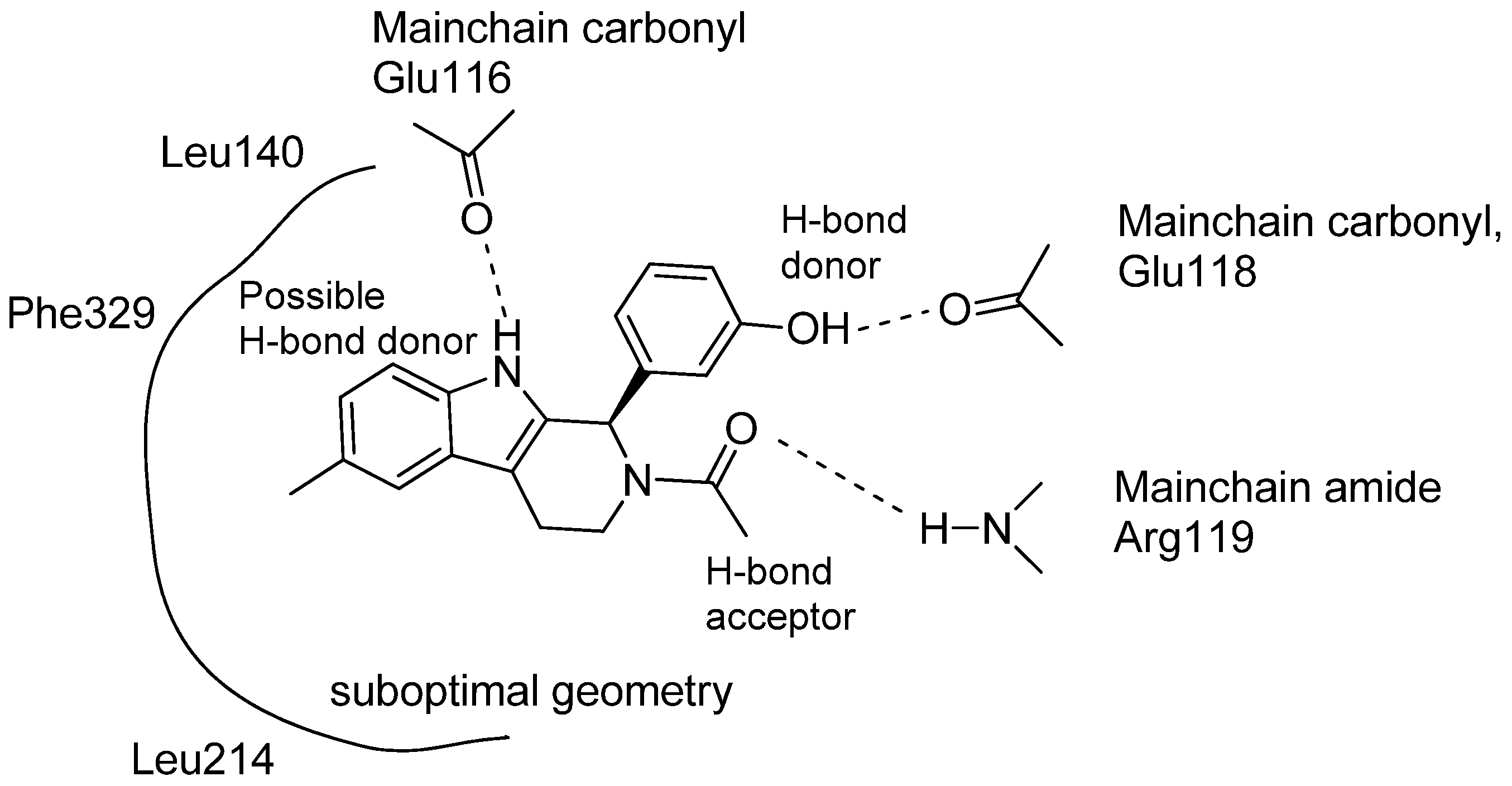
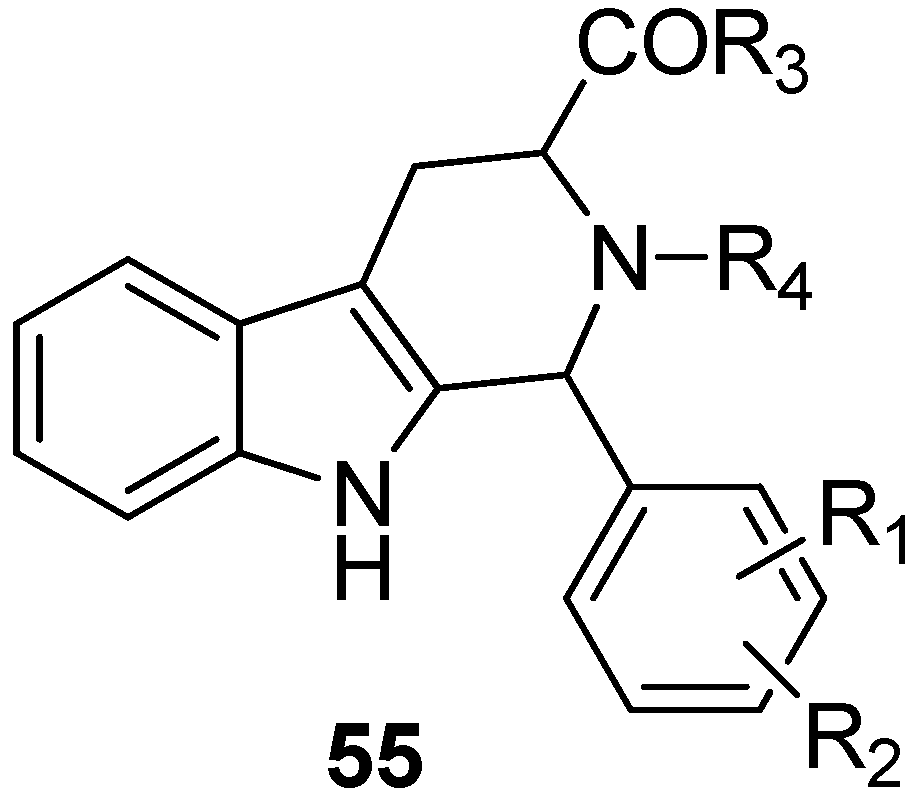
5.4. Other Pharmacological Uses
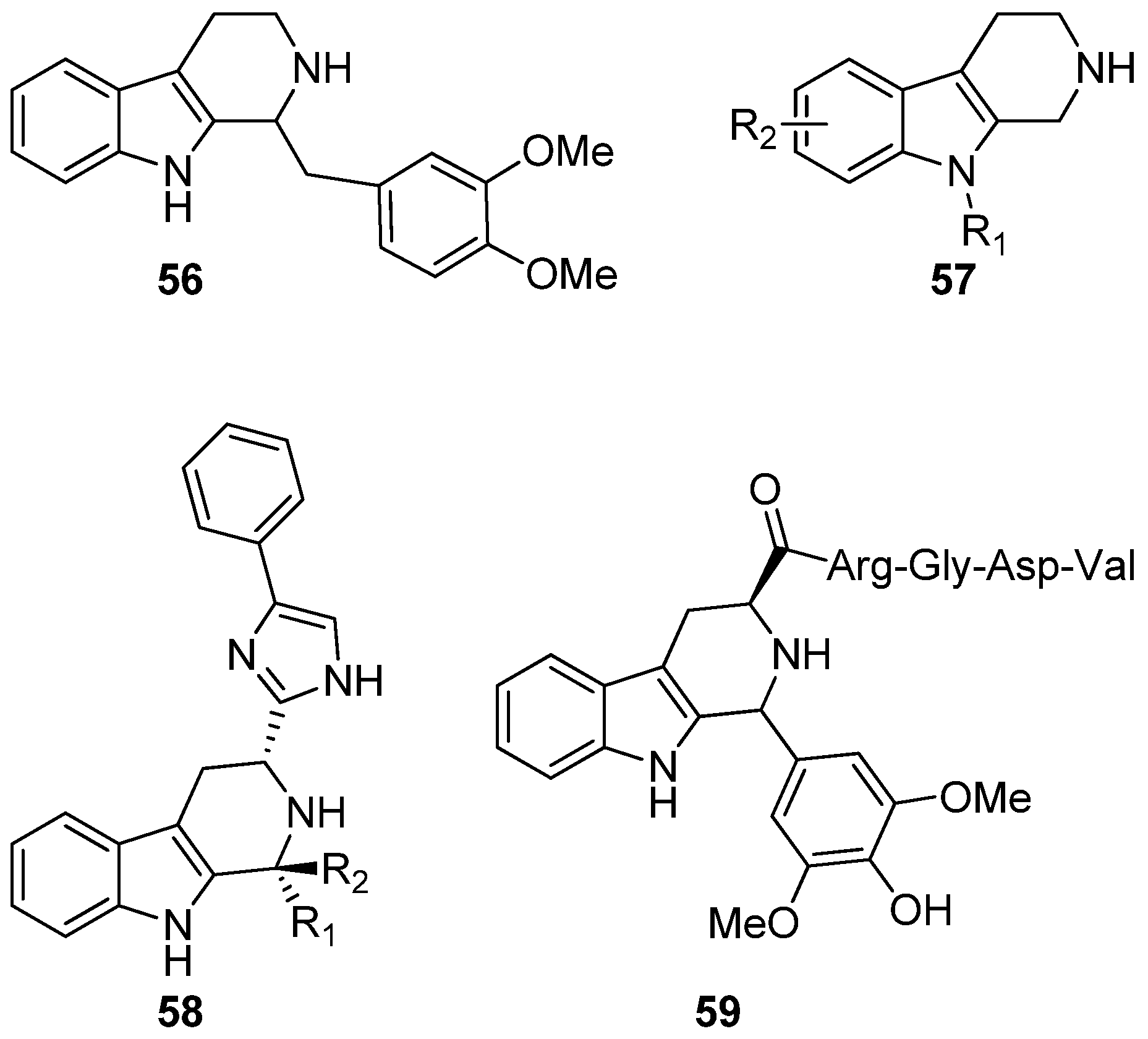
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Hesse, M. Alkaloids: Nature’s Curse or Blessing; Wiley-VCH: Zürich, Switzerland, 2002; pp. 14–27. [Google Scholar]
- Daugan, A.; Grondin, P.; Ruault, C.; le Monnier, D.G.; Coste, H.; Kirilovsky, J.; Hyafil, F.; Labaudiniere, R. The discovery of tadalafil: A novel and highly selective PDE5 inhibitor. 1: 5,6,11,11a-Tetrahydro-1H-imidazo[1',5':1,6]pyrido[3,4-B]indole-1,3(2H)-dione analogues. J. Med. Chem. 2003, 46, 4525–4532. [Google Scholar] [CrossRef]
- Davis, R.A.; Duffy, S.; Avery, V.M.; Camp, D.; Hooper, J.N.A.; Quinn, R.J. (+)-7-Bromotrypargine: An Antimalarial β-Carboline from the Australian Marine Sponge Ancorina Sp. Tetrahedron Lett. 2010, 51, 583–585. [Google Scholar] [CrossRef]
- Chan, S.T.S.; Pearce, A.N.; Page, M.J.; Kaiser, M.; Copp, B.R. Antimalarial β-Carbolines from the New Zealand Ascidian Pseudodistoma opacum. J. Nat. Prod. 2011, 74, 1972–1979. [Google Scholar] [CrossRef]
- Gellis, A.; Dumètre, A.; Lanzada, G.; Hutter, S.; Ollivier, E.; Vanelle, P.; Azas, N. Preparation and antiprotozoal evaluation of promising β-carboline alkaloids. Biomed. Pharmacother. 2012, 66, 339–347. [Google Scholar]
- Gupta, L.; Srivastava, K.; Singh, S.; Puri, S.K.; Chauhan, P.M.S. Synthesis of 2-[3-(7-Chloro-Quinolin-4-ylamino)-Alkyl]-1-(Substituted Phenyl)-2,3,4,9-Tetrahydro-1H-β-Carbolines as a New Class of Antimalarial Agents. Bioorg. Med. Chem. Lett. 2008, 18, 3306–3309. [Google Scholar] [CrossRef]
- Rottmann, M.; McNamara, C.; Yeung, B.K.S.; Lee, M.C.S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. [Google Scholar] [CrossRef]
- Yeung, B.K.S.; Zou, B.; Rottmann, M.; Lakshminarayana, S.B.; Ang, S.H.; Leong, S.Y.; Tan, J.; Wong, J.; Keller-Maerki, S.; Fischli, C.; et al. Spirotetrahydro β-carbolines (spiroindolones): A new class of potent and orally efficacious compounds for the treatment of Malaria. J. Med. Chem. 2010, 53, 5155–5164. [Google Scholar] [CrossRef]
- IFPMA Developing World Health Partnerships: Novartis R&D for Malaria. Available online: http://partnerships.ifpma.org/partnership/novartis-r-d-for-malaria (accessed on 23 July 2013).
- Rinehart, K.L.; Kobayashi, J.; Harbour, G.C.; Hughes, R.G.; Mizsak, S.A.; Scahill, T.A. Eudistomins C, E, K, and L, potent antiviral compounds containing a novel oxathiazepine ring from the caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1524–1526. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Mascal, M.; Holt, T.G.; Shield, L.S.; Lafargue, F. Eudistomins A-Q, β-Carbolines from the Antiviral Caribbean Tunicate Eudistoma. olivaceum. J. Am. Chem. Soc. 1987, 109, 3378–3387. [Google Scholar] [CrossRef]
- Van Maarseveen, J.H.; Hermkens, P.H.H.; de Clercq, E.; Balzarini, J.; Scheeren, H.W.; Kruse, C.G. Antiviral and antitumor structure-activity relationship studies on tetracyclic eudistomines. J. Med. Chem. 1992, 35, 3223–3230. [Google Scholar] [CrossRef]
- Miller, J.F.; Turner, E.M.; Sherrill, R.G.; Gudmundsson, K.; Spaltenstein, A.; Sethna, P.; Brown, K.W.; Harvey, R.; Romines, K.R.; Golden, P. Substituted tetrahydro-β-carbolines as potential agents for the treatment of human papillomavirus infection. Bioorg. Med. Chem. Lett. 2010, 20, 256–259. [Google Scholar] [CrossRef]
- Shen, Y.; Chang, Y.; Lin, C.; Liaw, C.; Kuo, Y.H.; Tu, L.; Yeh, S.F.; Chern, J. Synthesis of 1-substituted carbazolyl-1,2,3,4-tetrahydro- and carbazolyl-3,4-dihydro-î²-carboline analogs as potential antitumor agents. Mar. Drugs 2011, 9, 256–277. [Google Scholar] [CrossRef]
- Skouta, R.; Hayano, M.; Shimada, K.; Stockwell, B.R. Design and synthesis of pictet–spengler condensation products that exhibit oncogenic-ras synthetic lethality and induce non-apoptotic cell death. Bioorg. Med. Chem. Lett. 2012, 22, 5707–5713. [Google Scholar] [CrossRef]
- Hotha, S.; Yarrow, J.C.; Yang, J.G.; Garrett, S.; Renduchintala, K.V.; Mayer, T.U.; Kapoor, T.M. HR22C16: A potent small-molecule probe for the dynamics of cell division. Angew. Chem. 2003, 115, 2481–2484. [Google Scholar] [CrossRef]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef]
- Allen, J.R.F.; Holmstedt, B.R. The simple β-carboline alkaloids. Phytochemistry 1980, 19, 1573–1582. [Google Scholar] [CrossRef]
- Goebel, F. Über das Harmalin. Justus Liebigs. Ann. Chem. 1841, 38, 363–366. [Google Scholar] [CrossRef]
- Sumpter, W.C.; Miller, F.M. Heterocyclic Compounds with Indole and Carbazole Systems; Wiley Interscience: New York, NY, USA, 1954. [Google Scholar]
- Maresh, J.J.; Giddings, L.; Friedrich, A.; Loris, E.A.; Panjikar, S.; Trout, B.L.; Stöckigt, J.; Peters, B.; O’Connor, S.E. Strictosidine synthase: Mechanism of a pictet-spengler catalyzing enzyme. J. Am. Chem. Soc. 2008, 130, 710–723. [Google Scholar] [CrossRef]
- Stöckigt, J.; Antonchick, A.P.; Wu, F.; Waldmann, H. The pictet-spengler reaction in nature and in organic chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef]
- Pictet, A.; Spengler, T. Über die bildung von isochinolin-derivaten durch einwirkung von methylal auf phenyl-äthylamin, phenyl-alanin und tyrosin. Ber. Deutsch. Chem. Gesell. 1911, 44, 2030–2036. (in German). [Google Scholar] [CrossRef]
- Cox, E.D.; Cook, J.M. The pictet-spengler condensation: A new direction for an old reaction. Chem. Rev. 1995, 95, 1797–1842. [Google Scholar] [CrossRef]
- Bischler, A.; Napieralski, B. Zur kenntniss einer neuen isochinolinsynthese. Ber. Deutsch. Chem. Gesell. 1893, 26, 1903–1908. (in German). [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Woodward, R.B.; Bader, F.E.; Bickel, H.; Frey, A.J.; Kierstead, R.W. The total synthesis of reserpine. Tetrahedron 1958, 2, 1–57. [Google Scholar] [CrossRef]
- Bailey, P.D.; Cochrane, P.J.; Lorenz, K.; Collier, I.D.; Pearson, D.P.J.; Rosair, G.M. A concise, efficient route to fumitremorgins. Tetrahedron Lett. 2001, 42, 113–115. [Google Scholar] [CrossRef]
- Koskinen, A.M.P. Asymmetric Synthesis of Natural Products, 2nd ed.; John Wiley & Sons: Chichester, UK, 2012; pp. 259–261. [Google Scholar]
- Bailey, P.D.; Hollinshead, S.P.; McLay, N.R.; Morgan, K.; Palmer, S.J.; Prince, S.N.; Reynolds, C.D.; Wood, S.D. Diastereo- and enantio-selectivity in the pictet-spengler reaction. J. Chem. Soc. Perkin Trans. 1 1993, 4, 431–439. [Google Scholar]
- Ardeo, A.; Garcı́a, E.; Arrasate, S.; Lete, E.; Sotomayor, N. A practical approach to the fused β-carboline system. asymmetric synthesis of indolo[2,3-a]indolizidinones via a diastereoselective intramolecular α-amidoalkylation reaction. Tetrahedron Lett. 2003, 44, 8445–8448. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Yu, P.; Peterson, A.; Weber, R.; Soerens, D.; Grubisha, D.; Bennett, D.; Cook, J.M. General approach for the synthesis of ajmaline/sarpagine indole alkaloids: Enantiospecific total synthesis of (+)-ajmaline, alkaloid g, and norsuaveoline via the asymmetric pictet-spengler reaction. J. Am. Chem. Soc. 1999, 121, 6998–7010. [Google Scholar] [CrossRef]
- Liu, X.; Deschamp, J.R.; Cook, J.M. Regiospecific, enantiospecific total synthesis of the alkoxy-substituted indole bases, 16-epi-na-methylgardneral, 11-methoxyaffinisine, and 11-methoxymacroline as well as the indole alkaloids alstophylline and macralstonine. Org. Lett. 2002, 4, 3339–3342. [Google Scholar] [CrossRef]
- Bailey, P.D.; Clingan, P.D.; Mills, T.J.; Price, R.A.; Pritchard, R.G. Total synthesis of (−)-raumacline. Chem. Commun. 2003, 22, 2800–2801. [Google Scholar]
- Alberch, L.; Bailey, P.D.; Clingan, P.D.; Mills, T.J.; Price, R.A.; Pritchard, R.G. The cis-specific pictet-spengler reaction. Eur. J. Org. Chem. 2004, 2004, 1887–1890. [Google Scholar]
- Bailey, P.D.; Beard, M.A.; Phillips, T.R. Unexpected cis selectivity in the pictet–spengler reaction. Tetrahedron Lett. 2009, 50, 3645–3647. [Google Scholar] [CrossRef]
- Yamada, H.; Kawate, T.; Nishida, A.; Nakagawa, M. Asymmetric addition of alkyllithium to chiral imines: α-naphthylethyl group as a chiral auxiliary. J. Org. Chem. 1999, 64, 8821–8828. [Google Scholar] [CrossRef]
- Soe, T.; Kawate, T.; Fukui, N.; Nakagawa, M. Asymmetric pictet-spengler reaction with chiral-n-(β-3-indolyl)ethyl-1-methylbenzylamine. Tetrahedron Lett. 1995, 36, 1857–1860. [Google Scholar] [CrossRef]
- Waldmann, H.; Schmidt, G.; Henke, H.; Burkard, M. Asymmetric pictet-spengler reactions employing n,n-phthaloyl amino acids as chiral auxiliary groups. Angew. Chem. Int. Ed. Engl. 1995, 34, 2402–2403. [Google Scholar] [CrossRef]
- Ducrot, P.; Rabhi, C.; Thal, C. Synthesis of tetrahydro-β-carbolines and studies of the pictet–spengler reaction. Tetrahedron 2000, 56, 2683–2692. [Google Scholar] [CrossRef]
- Kawate, T.; Yamada, H.; Soe, T.; Nakagawa, M. Enantioselective asymmetric pictet-spengler reaction catalyzed by diisopinocampheylchloroborane. Tetrahedron Asymmetry 1996, 7, 1249–1252. [Google Scholar] [CrossRef]
- Raheem, I.T.; Thiara, P.S.; Peterson, E.A.; Jacobsen, E.N. Enantioselective pictet-spengler-type cyclizations of hydroxylactams: H-bond donor catalysis by anion binding. J. Am. Chem. Soc. 2007, 129, 13404–13405. [Google Scholar] [CrossRef]
- Klausen, R.S.; Jacobsen, E.N. Weak bronsted acid thiourea co-catalysis: Enantioselective, catalytic protio-pictet-spengler reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef]
- Seayad, J.; Seayad, A.M.; List, B. Catalytic asymmetric pictet-spengler reaction. J. Am. Chem. Soc. 2006, 128, 1086–1087. [Google Scholar] [CrossRef]
- Wanner, M.J.; van der Haas, R.N.S.; de Cuba, K.R.; van Maarseveen, J.H.; Hiemstra, H. Catalytic asymmetric pictet-spengler reactions via sulfenyliminium ions. Angew. Chem. 2007, 119, 7629–7631. [Google Scholar] [CrossRef]
- Priebbenow, D.L.; Henderson, L.C.; Pfeffer, F.M.; Stewart, S.G. Domino Heck-aza-Michael reactions: Efficient access to 1-substituted tetrahydro-β-carbolines. J. Org. Chem. 2010, 75, 1787–1790. [Google Scholar] [CrossRef]
- Rixson, J.E.; Chaloner, T.; Heath, C.H.; Tietze, L.F.; Stewart, S.G. The development of Domino reactions incorporating the Heck reaction: The formation of N-heterocycles. Eur. J. Org. Chem. 2012, 2012, 544–558. [Google Scholar]
- Priebbenow, D.L.; Stewart, S.G.; Pfeffer, F.M. Asymmetric induction in domino heck-aza-michael reactions. Tetrahedron Lett. 2012, 53, 1468–1471. [Google Scholar] [CrossRef]
- Meyers, A.I.; Sohda, T.; Loewe, M.F. An asymmetric synthesis and absolute configuration of (S)(−)-deplancheine. J. Org. Chem. 1986, 51, 3108–3112. [Google Scholar] [CrossRef]
- Meyers, A.I.; Miller, D.B.; White, F.H. Chiral and achiral formamidines in synthesis. The first asymmetric route to (−)-yohimbine and an efficient total synthesis of (±)-yohimbine. J. Am. Chem. Soc. 1988, 110, 4778–4787. [Google Scholar] [CrossRef]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef]
- World Malaria Report 2012. Available online: http://www.who.int/malaria/publications/world_malaria_report_2012/report/en/index.html (accessed on 23 July 2013).
- Kaufman, T.S.; Rúveda, E.A. The quest for quinine: Those who won the battles and those won the war. Angew. Chem. Int. Ed. 2005, 44, 854–885. [Google Scholar] [CrossRef]
- Sidhu, A.B.S.; Verdier-Pinard, D.; Fidock, D.A. Chloroquine resistance in Plasmodium falciparum malaria parasites conferred by Pfcrt mutations. Science 2002, 298, 210–213. [Google Scholar] [CrossRef]
- Staerk, D.; Lemmich, E.; Christensen, J.; Kharazmi, A.; Olsen, C.E.; Jaroszewski, J.W. Leishmanicidal, Antiplasmodial and cytotoxic activity of indole alkaloids from Corynanthe. pachyceras. Planta Med. 2000, 66, 531–536. [Google Scholar] [CrossRef]
- Kam, T.; Sim, K.; Koyano, T.; Komiyama, K. Leishmanicidal alkaloids from Kopsia. griffithii. Phytochemistry 1999, 50, 75–79. [Google Scholar]
- Chauhan, S.S.; Gupta, L.; Mittal, M.; Vishwakarma, P.; Gupta, S.; Chauhan, P.M.S. Synthesis and Biological evaluation of indolyl glyoxylamides as a new class of antileishmanial agents. Bioorg. Med. Chem. Lett. 2010, 20, 6191–6194. [Google Scholar] [CrossRef]
- Kumar, A.; Katiyar, S.B.; Gupta, S.; Chauhan, P.M.S. Syntheses of new substituted triazino tetrahydroisoquinolines and β-carbolines as novel antileishmanial agents. Eur. J. Med. Chem. 2006, 41, 106–113. [Google Scholar]
- Kumar, R.; Khan, S.; Verma, A.; Srivastava, S.; Viswakarma, P.; Gupta, S.; Meena, S.; Singh, N.; Sarkar, J.; Chauhan, P.M.S. Synthesis of 2-(Pyrimidin-2-yl)-1-phenyl-2,3,4,9-tetrahydro-1H-β-carbolines as antileishmanial agents. Eur. J. Med. Chem. 2010, 45, 3274–3280. [Google Scholar] [CrossRef]
- Düsman Tonin, L.T.; Barbosa, V.A.; Bocca, C.C.; Ramos, É.R.F.; Nakamura, C.V.; Ferreira da Costa, W.; Basso, E.A.; Nakamura, T.U.; Sarragiotto, M.H. Comparative study of the trypanocidal activity of the methyl 1-nitrophenyl-1,2,3,4-9H-tetrahydro-β-carboline-3-carboxylate derivatives and benznidazole using theoretical calculations and cyclic voltammetry. Eur. J. Med. Chem. 2009, 44, 1745–1750. [Google Scholar]
- Valdez, R.H.; Tonin, L.T.D.; Ueda-Nakamura, T.; Silva, S.O.; Dias Filho, B.P.; Kaneshima, E.N.; Yamada-Ogatta, S.F.; Yamauchi, L.M.; Sarragiotto, M.H.; Nakamura, C.V. In Vitro and in vivo Trypanocidal synergistic activity of n-butyl-1-(4-dimethylamino)phenyl-1,2,3,4-tetrahydro-β-carboline-3-carboxamide associated with benznidazole. Antimicrob. Agent. Chemother. 2012, 56, 507–512. [Google Scholar] [CrossRef]
- Lake, R.J.; Blunt, J.W.; Munro, M. Eudistomins from the New Zealand ascidian Ritterella. sigillinoides. Aust. J. Chem. 1989, 42, 1201–1206. [Google Scholar] [CrossRef]
- Gudmundsson, K.; Miller, J.F.; Sherrill, R.G.; Turner, E.M. Useful Compounds for HPV Infection. WO2006015035A1, 9 February 2006. [Google Scholar]
- Kobayashi, J.; Cheng, J.F.; Ohta, T.; Nozoe, S.; Ohizumi, Y.; Sasaki, T. Eudistomidins B, C, and D: Novel antileukemic alkaloids from the okinawan marine tunicate Eudistoma glaucus. J. Org. Chem. 1990, 55, 3666–3670. [Google Scholar] [CrossRef]
- Adesanya, S.A.; Chbani, M.; Paîs, M.; Debitus, C. Brominated beta-carbolines from the marine tunicate Eudistoma album. J. Nat. Prod. 1992, 55, 525–527. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, Y.; Hsieh, P.; Duh, C.; Lin, Y.; Ko, C. The preparation and evaluation of 1-substituted 1,2,3,4-tetrahydro- and 3,4-dihydro-β-carboline derivatives as potential antitumor agents. Chem. Pharm. Bull. 2005, 53, 32–36. [Google Scholar] [CrossRef]
- Santos, L.S.; Theoduloz, C.; Pilli, R.A.; Rodriguez, J. Antiproliferative activity of arborescidine alkaloids and derivatives. Eur. J. Med. Chem. 2009, 44, 3810–3815. [Google Scholar] [CrossRef]
- Kumar, R.; Gupta, L.; Pal, P.; Khan, S.; Singh, N.; Katiyar, S.B.; Meena, S.; Sarkar, J.; Sinha, S.; Kanaujiya, J.K.; et al. Synthesis and cytotoxicity evaluation of (tetrahydro-β-carboline)-1,3,5-triazine hybrids as anticancer agents. Eur. J. Med. Chem. 2010, 45, 2265–2276. [Google Scholar]
- Zeng, Y.; Zhang, Y.; Weng, Q.; Hu, M.; Zhong, G. Cytotoxic and insecticidal activities of derivatives of harmine, a natural insecticidal component isolated from Peganum harmala. Molecules 2010, 15, 7775–7791. [Google Scholar] [CrossRef]
- Sarli, V.; Giannis, A. Targeting the kinesin spindle protein: Basic principles and clinical implications. Clin. Cancer Res. 2008, 14, 7583–7587. [Google Scholar] [CrossRef]
- Jackson, J.R.; Patrick, D.R.; Dar, M.M.; Huang, P.S. Targeted anti-mitotic therapies: Can we improve on tubulin agents? Nat. Rev. Cancer 2007, 7, 107–117. [Google Scholar] [CrossRef]
- Sunder-Plassmann, N.; Sarli, V.; Gartner, M.; Utz, M.; Seiler, J.; Huemmer, S.; Mayer, T.U.; Surrey, T.; Giannis, A. Synthesis and biological evaluation of new tetrahydro-β-carbolines as inhibitors of the mitotic kinesin eg5. Bioorg. Med. Chem. 2005, 13, 6094–6111. [Google Scholar] [CrossRef]
- Vennemann, M.; Braunger, J.; Gimmnich, P.; Baer, T. Indolopyridines as Inhibitors of the Kinesin Spindle Protein (Eg5). WO2009024190 (A1), 25 February 2009. [Google Scholar]
- 4SC: Product pipeline/SC4–205. Available online: http://www.4sc.de/product-pipeline/clinical/4SC-205 (accessed on 24 July 2013).
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef]
- Liu, F.; Yu, L.; Jiang, C.; Yang, L.; Wu, W.; You, Q. Discovery of Tetrahydro-Β-Carbolines as Inhibitors of the Mitotic Kinesin KSP. Bioorg. Med. Chem. 2010, 18, 4167–4177. [Google Scholar] [CrossRef]
- Barsanti, P.A.; Wang, W.; Ni, Z.; Duhl, D.; Brammeier, N.; Martin, E.; Bussiere, D.; Walter, A.O. The discovery of tetrahydro-β-carbolines as inhibitors of the kinesin Eg5. Bioorg. Med. Chem. Lett. 2010, 20, 157–160. [Google Scholar]
- Schmidt, M.; Bastians, H. Mitotic drug targets and the development of novel anti-mitotic anticancer drugs. Drug Resist. Updat. 2007, 10, 162–181. [Google Scholar] [CrossRef]
- Chan, K.; Koh, C.; Li, H. Mitosis-targeted anti-cancer therapies: Where they stand. Cell. Death Dis. 2012, 3, e411. [Google Scholar] [CrossRef]
- Sandeep, G.; Bhasker, S.; Ranganath, Y.S. Phosphodiesterase as a novel target in cancer chemotherapy. Internet J. Pharmacol. 2009, 7, 1. [Google Scholar]
- El-Gamil, D.S.; Ahmed, N.S.; Gary, B.D.; Piazza, G.A.; Engel, M.; Hartmann, R.W.; Abadi, A.H. Design of novel β-carboline derivatives with pendant 5-bromothienyl and their evaluation as phosphodiesterase-5 inhibitors. Arch. Pharm. 2013, 346, 23–33. [Google Scholar]
- Abadi, A.H.; Lehmann, J.; Piazza, G.A.; Abdel-Halim, M.; Ali, M.S.M. Synthesis, molecular modeling, and biological evaluation of novel tetrahydro-β-carboline hydantoin and tetrahydro-β-carboline thiohydantoin derivatives as phosphodiesterase 5 inhibitors. Int. J. Med. Chem. 2011, 2011. [Google Scholar] [CrossRef]
- Mohamed, H.A.; Girgis, N.M.R.; Wilcken, R.; Bauer, M.R.; Tinsley, H.N.; Gary, B.D.; Piazza, G.A.; Boeckler, F.M.; Abadi, A.H. Synthesis and molecular modeling of novel tetrahydro-β-carboline derivatives with phosphodiesterase 5 inhibitory and anticancer properties. J. Med. Chem. 2011, 54, 495–509. [Google Scholar] [CrossRef]
- Ahmed, N.S.; Gary, B.D.; Tinsley, H.N.; Piazza, G.A.; Laufer, S.; Abadi, A.H. Design, synthesis and structure-activity relationship of functionalized tetrahydro-β-carboline derivatives as novel PDE5 inhibitors. Arch. Pharm. 2011, 344, 149–157. [Google Scholar]
- Abadi, A.H.; Piazza, G. Tetrahydro-β-Carboline Derivatives, Synthesis and Use Thereof. WO2011063223(A1), 26 May 2011. [Google Scholar]
- Saxton, J.E. Monoterpenoid Indole Alkaloids: Supplement to Part. 4.; Wiley: Chichester, UK, 1994; pp. 162–176, 719–725. [Google Scholar]
- Audia, J.E.; Evrard, D.A.; Murdoch, G.R.; Droste, J.J.; Nissen, J.S.; Schenck, K.W.; Fludzinski, P.; Lucaites, V.L.; Nelson, D.L.; Cohen, M.L. Potent, selective tetrahydro-β-carboline antagonists of the serotonin 2B (5HT2B) contractile receptor in the rat stomach fundus. J. Med. Chem. 1996, 39, 2773–2780. [Google Scholar] [CrossRef]
- Giorgioni, G.; Accorroni, B.; Stefano, A.; Marucci, G.; Siniscalchi, A.; Claudi, F. Benzimidazole, Benzoxazole and benzothiazole derivatives as 5ht2b receptor ligands. synthesis and preliminary pharmacological evaluation. Med. Chem. Res. 2005, 14, 57–73. [Google Scholar] [CrossRef]
- Glennon, R.A.; Grella, B.; Tyacke, R.J.; Lau, A.; Westaway, J.; Hudson, A.L. Binding of β-carbolines at imidazoline I2 receptors: A structure–affinity investigation. Bioorg. Med. Chem. Lett. 2004, 14, 999–1002. [Google Scholar]
- Poitout, L.; Roubert, P.; Contour-Galcera, M.; Moinet, C.; Lannoy, J.; Pommier, J.; Plas, P.; Bigg, D.; Thurieau, C. Identification of potent non-peptide somatostatin antagonists with Sst3 selectivity. J. Med. Chem. 2001, 44, 2990–3000. [Google Scholar] [CrossRef]
- Pasternak, A.; Feng, Z.; de Jesus, R.; Ye, Z.; He, S.; Dobbelaar, P.; Bradley, S.A.; Chicchi, G.G.; Tsao, K.; Trusca, D.; et al. Stimulation of glucose-dependent insulin secretion by a potent, selective Sst3 antagonist. ACS Med. Chem. Lett. 2012, 3, 289–293. [Google Scholar] [CrossRef]
- Ganong, W. (Ed.) Review of Medical Physiology, 22nd ed.; Lange Medical Books/McGraw-Hill: New York, NY, USA, 2005; pp. 113–114.
- Zhou, Y.; Li, J.; Wu, W.; Shang, J.; Thompson, J.R.; Thornberry, N.A. Diagnosis and Treatment of Type 2 Diabetes and Other Disorders. U.S. Patent 7,879,859, 2009. [Google Scholar]
- Mjalli, A.M.M.; Andrews, R.C.; Xie, R.; Yarragunta, R.R.; Ren, T. Beta-Carboline Derivatives as PTP-Inhibitors. WO03033496A1 24 April 2003. [Google Scholar]
- Brandt, P.; Bremberg, U.; Crossley, R.; Graffner Nordberg, M.; Jenmalm Jensen, A.; Ringberg, E.; Ward, T. Novel Beta-Carbolines as Growth Hormone Secretagogue Receptor (GHSR) Antagonists. WO2005048916A2 29 December 2005. [Google Scholar]
- Hung, D.; Protter, A.; Chakravarty, S.; Jain, R.; Dugar, S. Pyrido[3,4-B]indoles and methods of use. WO2009120717 3 December 2009. [Google Scholar]
- Ortar, G.; Petrocellis, L.D.; Moriello, A.S.; Allarà, M.; Morera, E.; Nalli, M.; Marzo, V.D. Tetrahydro-β-Carboline derivatives targeting fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channels. Bioorg. Med. Chem. Lett. 2013, 23, 138–142. [Google Scholar]
- Bi, W.; Bi, L.; Cai, J.; Liu, S.; Peng, S.; Fischer, N.O.; Tok, J.B.-H.; Wang, G. Dual-acting agents that possess free radical scavenging and antithrombotic activities: Design, Synthesis, and evaluation of phenolic tetrahydro-β-carboline RGD peptide conjugates. Bioorg. Med. Chem. Lett. 2006, 16, 4523–4527. [Google Scholar] [CrossRef]
- Bi, W.; Cai, J.; Liu, S.; Baudy-Floc’h, M.; Bi, L. Design, Synthesis and cardioprotective effect of a new class of dual-acting agents: Phenolic tetrahydro-β-carboline RGD peptidomimetic conjugates. Bioorg. Med. Chem. 2007, 15, 6909–6919. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laine, A.E.; Lood, C.; Koskinen, A.M.P. Pharmacological Importance of Optically Active Tetrahydro-β-carbolines and Synthetic Approaches to Create the C1 Stereocenter. Molecules 2014, 19, 1544-1567. https://doi.org/10.3390/molecules19021544
Laine AE, Lood C, Koskinen AMP. Pharmacological Importance of Optically Active Tetrahydro-β-carbolines and Synthetic Approaches to Create the C1 Stereocenter. Molecules. 2014; 19(2):1544-1567. https://doi.org/10.3390/molecules19021544
Chicago/Turabian StyleLaine, Aino E., Christopher Lood, and Ari M. P. Koskinen. 2014. "Pharmacological Importance of Optically Active Tetrahydro-β-carbolines and Synthetic Approaches to Create the C1 Stereocenter" Molecules 19, no. 2: 1544-1567. https://doi.org/10.3390/molecules19021544
APA StyleLaine, A. E., Lood, C., & Koskinen, A. M. P. (2014). Pharmacological Importance of Optically Active Tetrahydro-β-carbolines and Synthetic Approaches to Create the C1 Stereocenter. Molecules, 19(2), 1544-1567. https://doi.org/10.3390/molecules19021544