Synthesis, Characterisation and Reactions of Phosphine-Substituted Alkynylboronates and Alkynyltrifluoroborate Salts

Abstract
: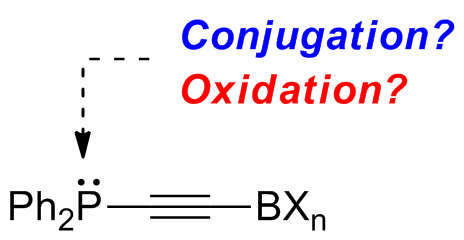
1. Introduction
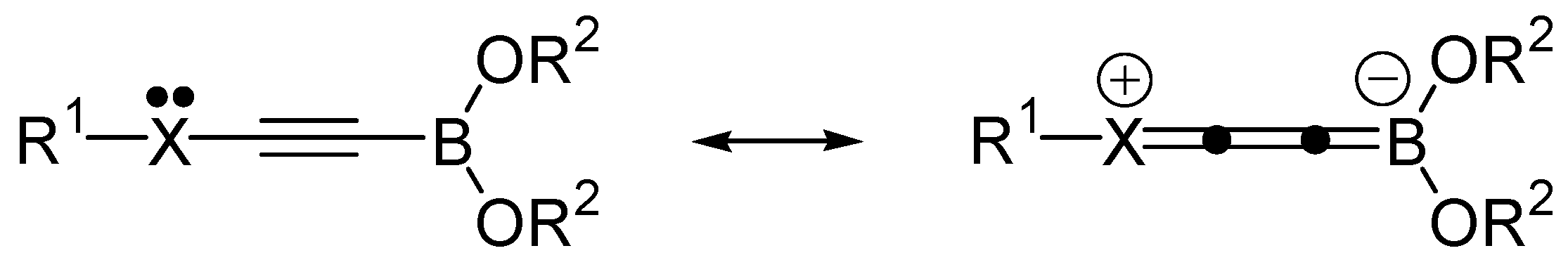
2. Results and Discussion

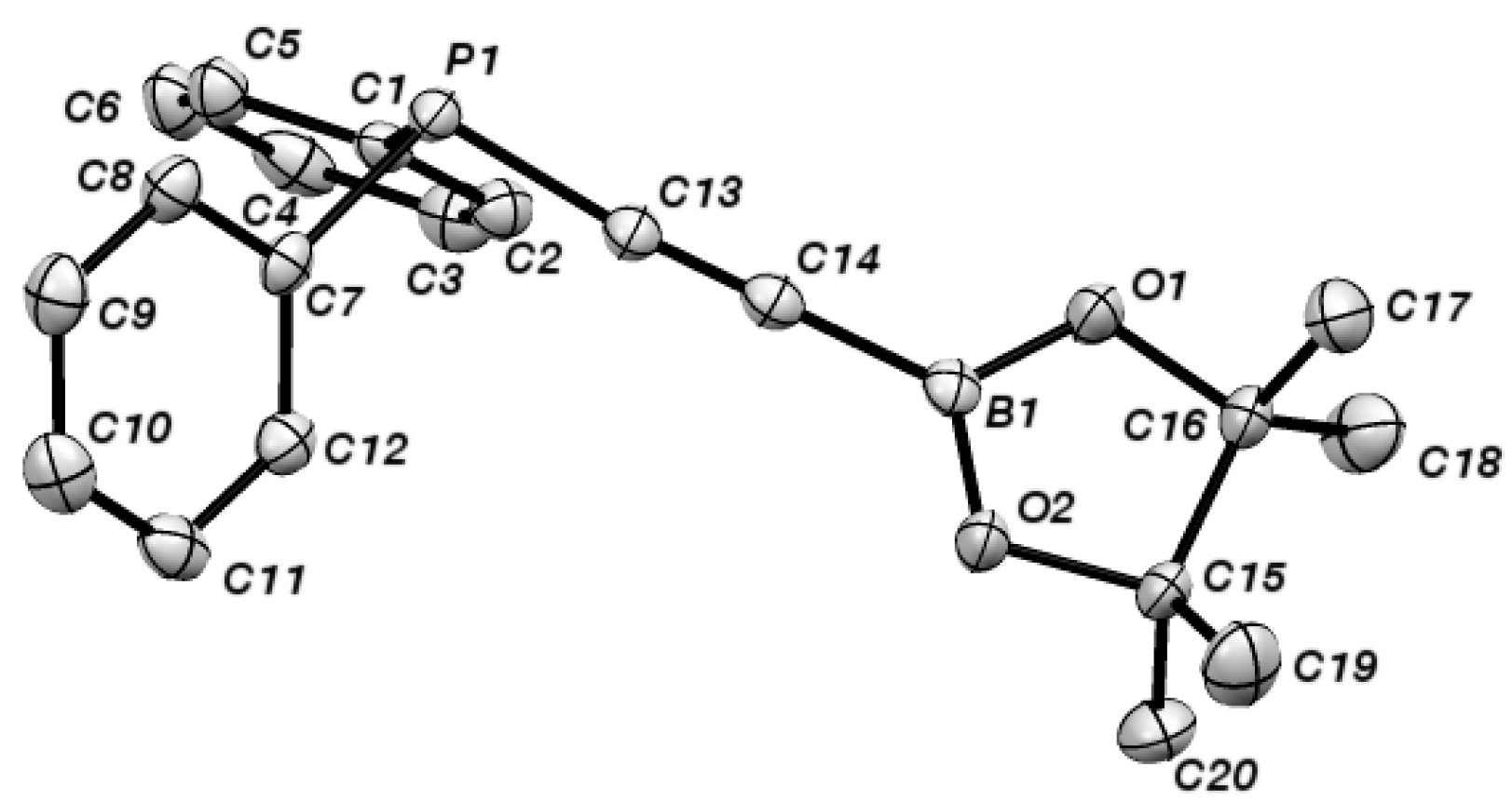

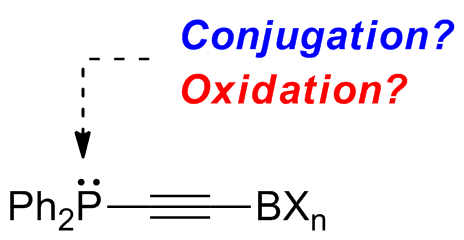{kind=link}
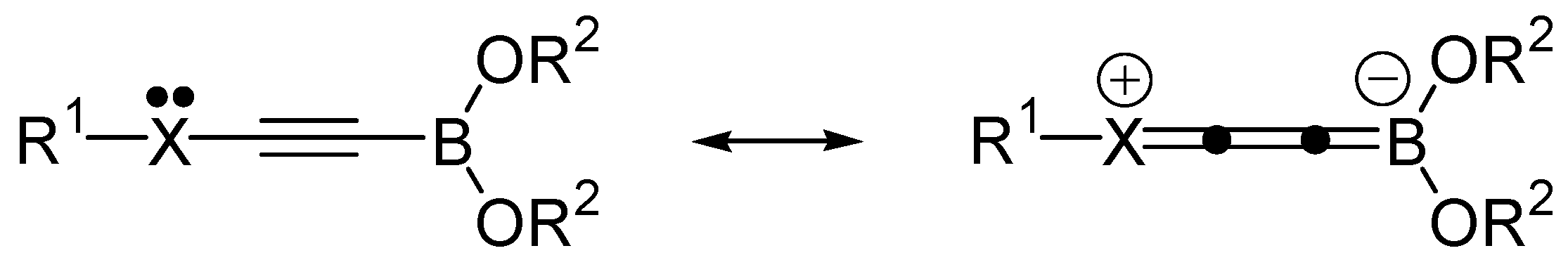{kind=link}
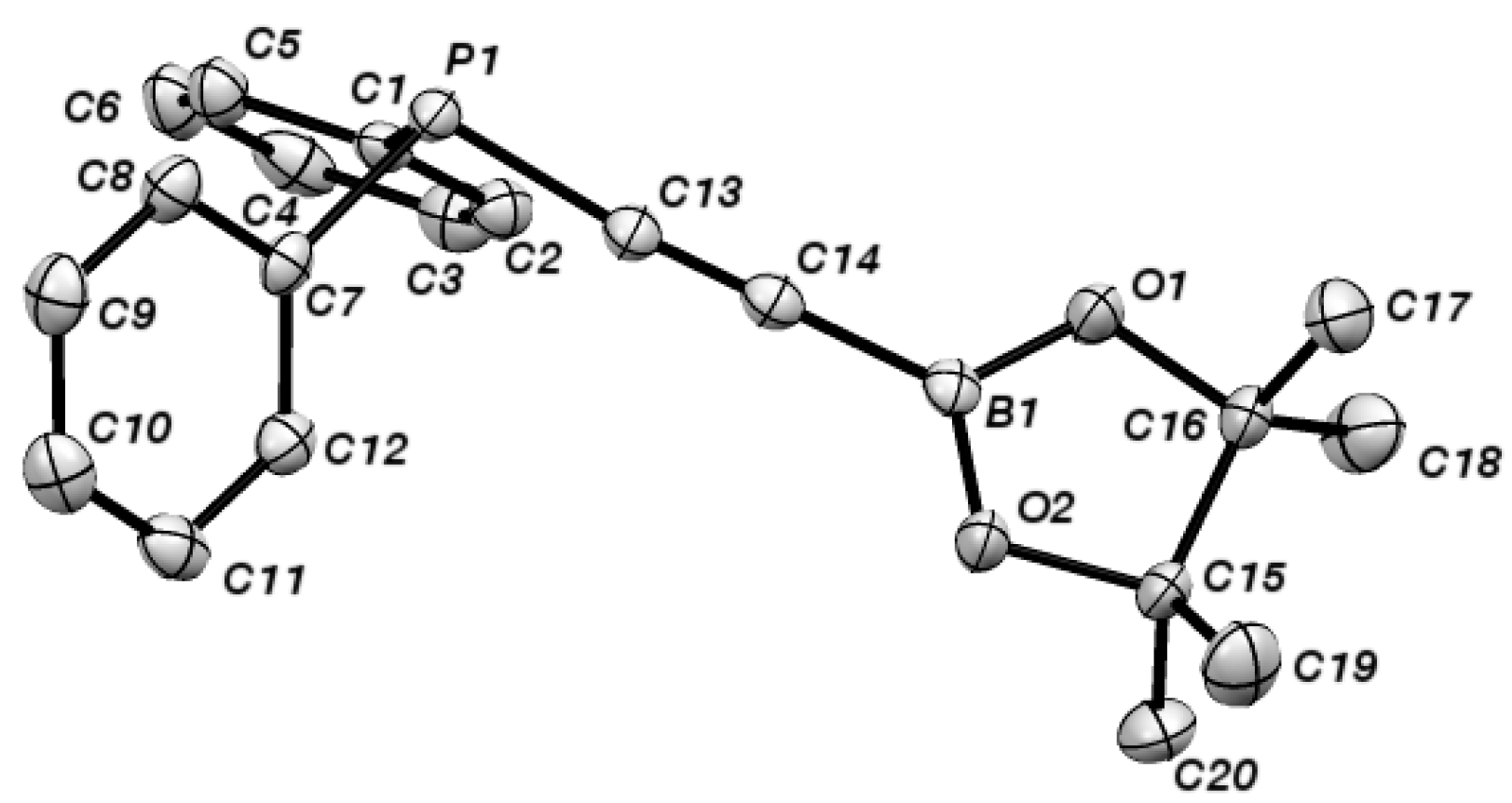{kind=link}
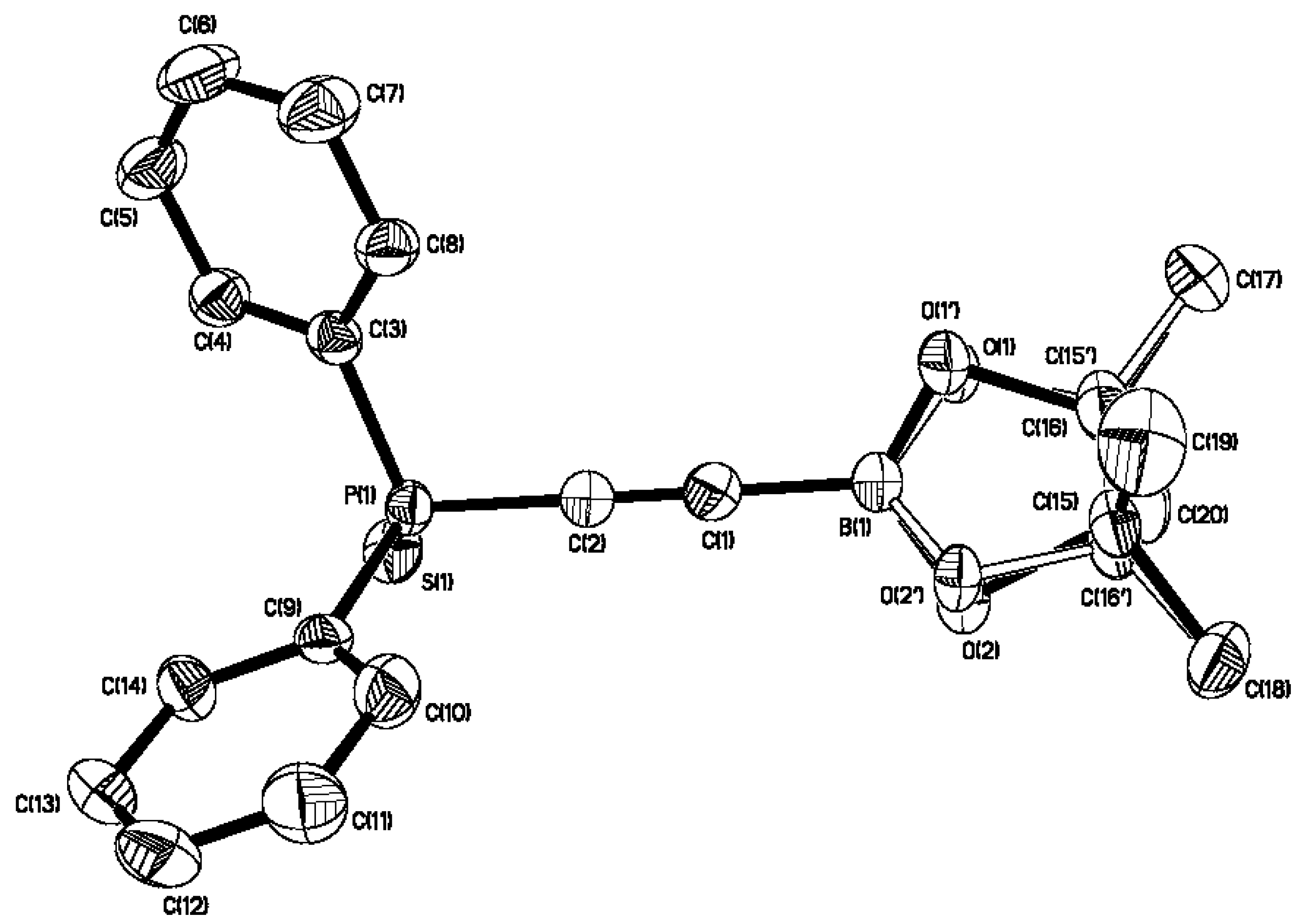{kind=link}
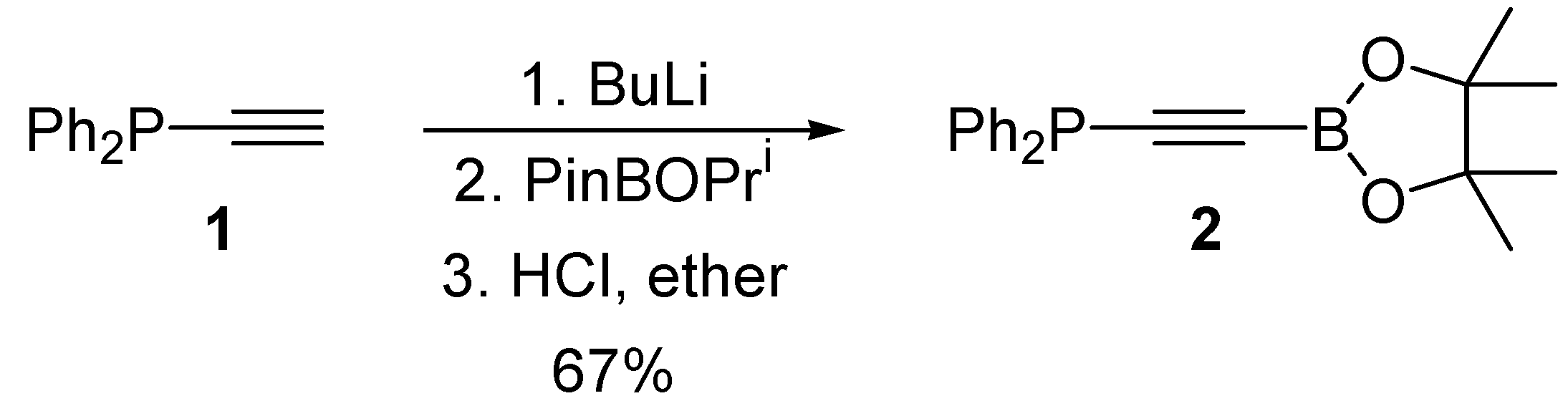{kind=link}
{kind=link}
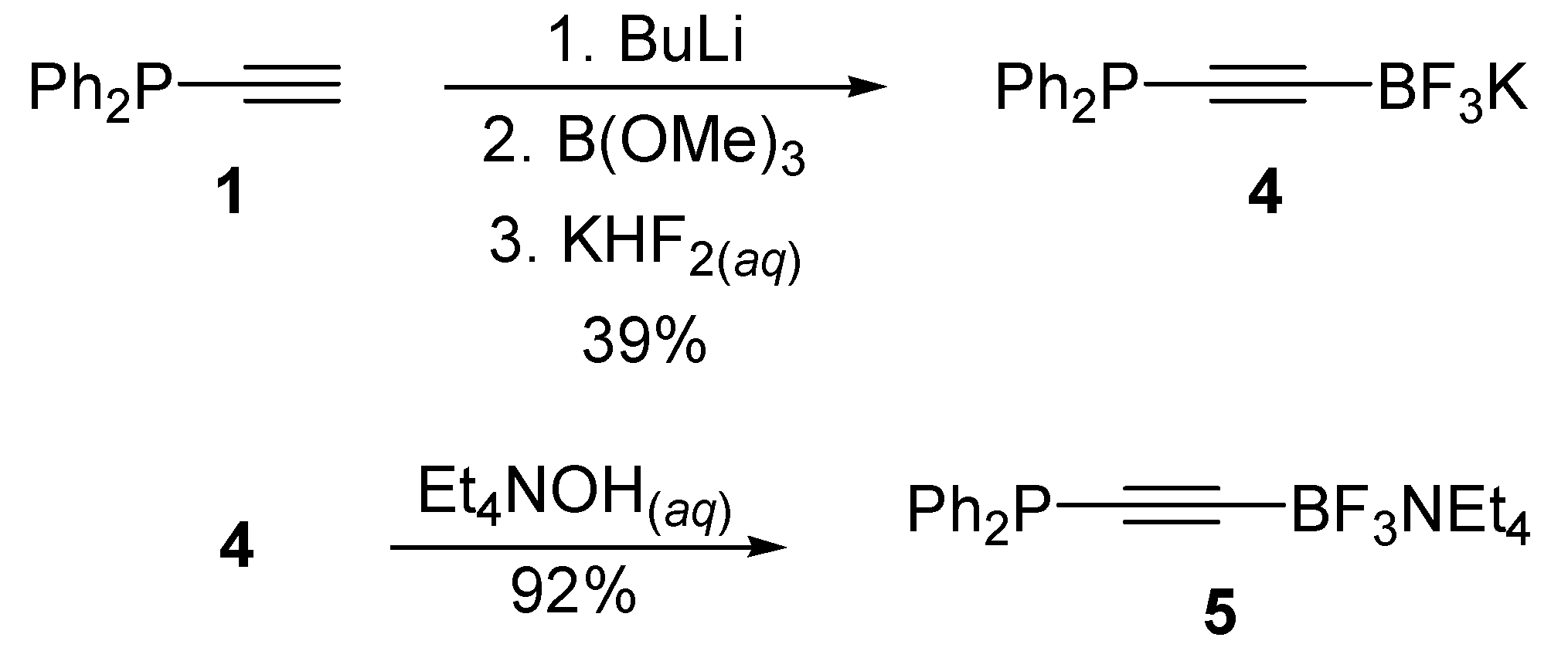{kind=link}
{kind=link}
{kind=link}
| 2 | |
|---|---|
| C(13A)-C(14A) | 1.203(4) |
| B(1A)-C(14A) | 1.531(4) |
| P(1A)-C(13A) | 1.761(3) |
| B(1A)-C(14A)-C(13A) | 177.8(3) |
| P(1A)-C(13A)-C(14A) | 175.5(2) |
| O(1A)-B(1A)-O(2A) | 114.8(2) |
| O(1A)-B(1A)-C(14A) | 122.0(2) |
| O(2A)-B(1A)-C(14A) | 123.2(2) |
| C(7A)-P(1A)-C(1A) | 102.19(12) |
| C(1A)-P(1A)-C(13A) | 100.56(13) |
| C(7A)-P(1A)-C(13A) | 100.46(12) |
| 3 | |
|---|---|
| C(1)-C(2) | 1.197(4) |
| B(1)-C(1) | 1.544(5) |
| P(1)-C(2) | 1.770(3) |
| B(1)-C(1)-C(2) | 179.3(4) |
| P(1)-C(2)-C(1) | 176.7(3) |
| O(1)-B(1)-O(2) | 114.7(4) |
| O(1)-B(1)-C(1) | 122.1(4) |
| O(2)-B(1)-C(1) | 123.0(4) |
| C(9)-P(1)-C(2) | 103.82(16) |
| C(3)-P(1)-C(2) | 104.20(16) |
| C(3)-P(1)-C(9) | 106.05(14) |


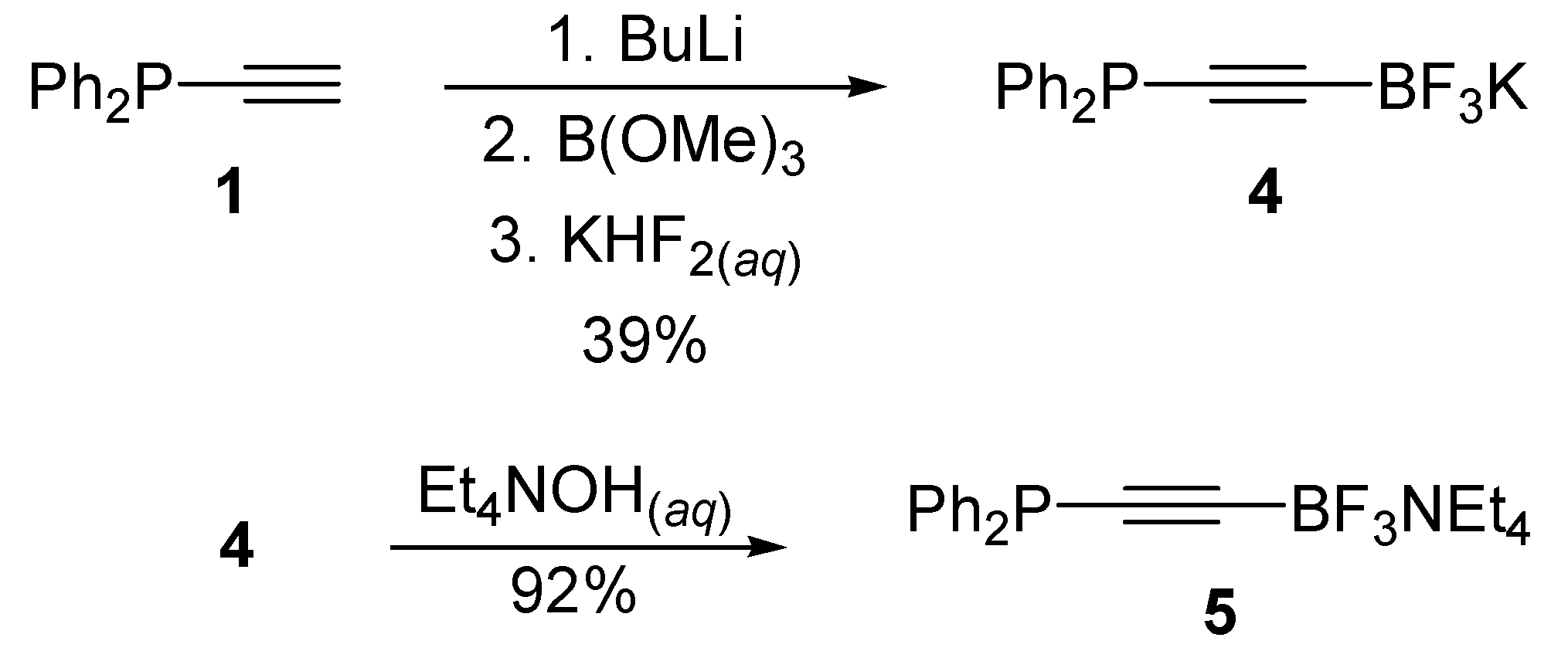
3. Experimental Section
3.1. General Remarks
3.2. Synthesis of Diphenyl((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethynyl)phosphine 2
3.3. Synthesis of Diphenyl((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethynyl)thiophosphine 3
3.4. Synthesis of Potassium ((Diphenylphosphino)ethynyl)trifluoroborate 4
3.5. Synthesis of Tetraethylammonium ((Diphenylphosphino)-ethynyl)trifluoroborate 5
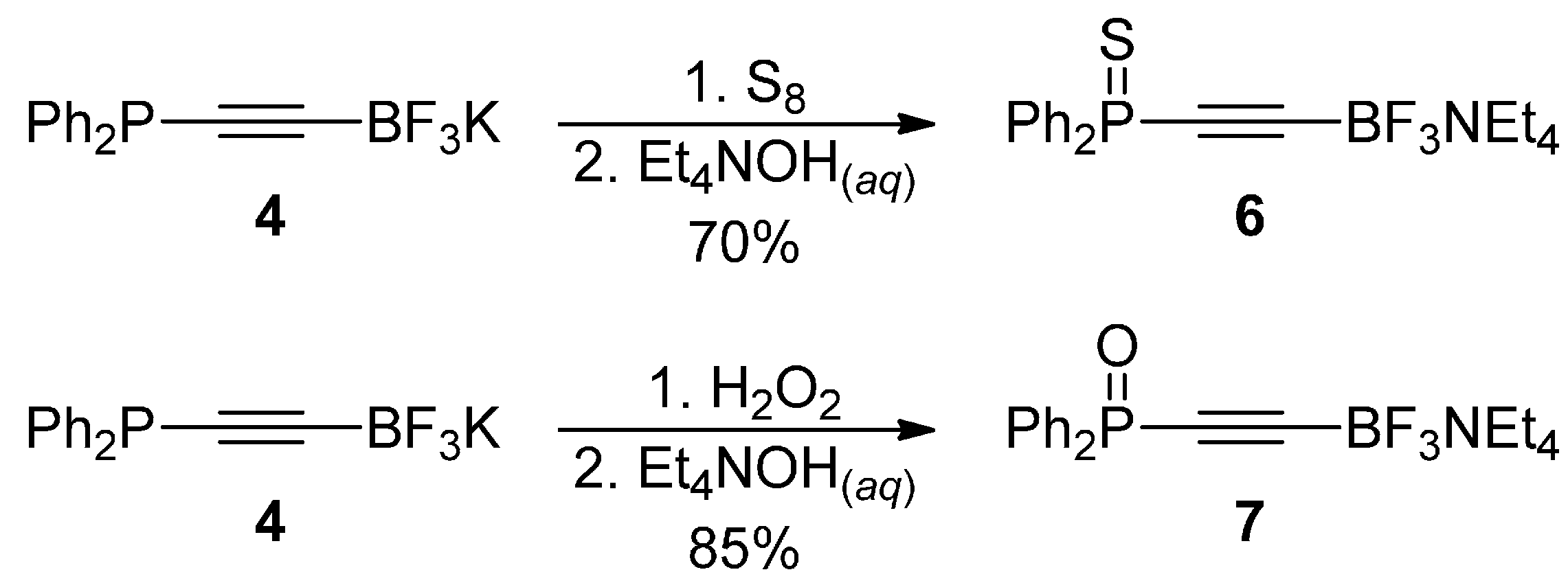
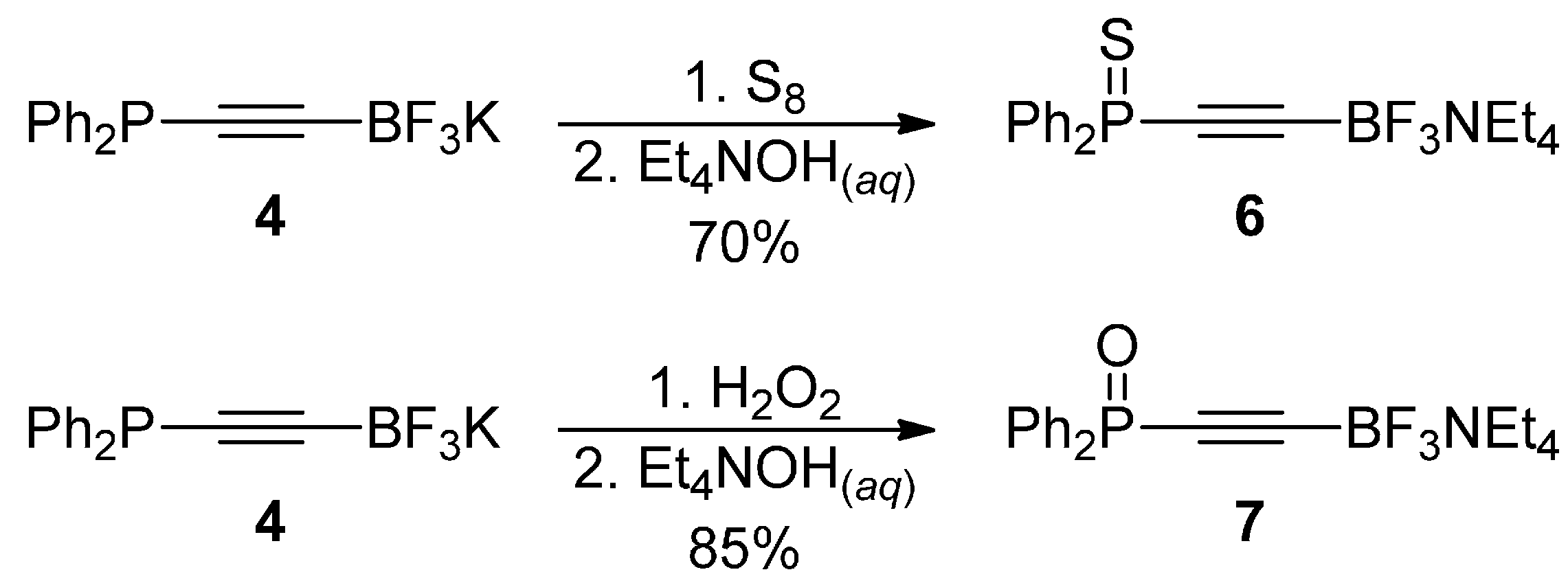
3.6. Synthesis of Tetraethylammonium ((Diphenylphosphorothioyl)-ethynyl)trifluoroborate 6
3.7. Synthesis of Tetraethylammonium ((Diphenylphosphoryl)ethynyl)-trifluoroborate 7
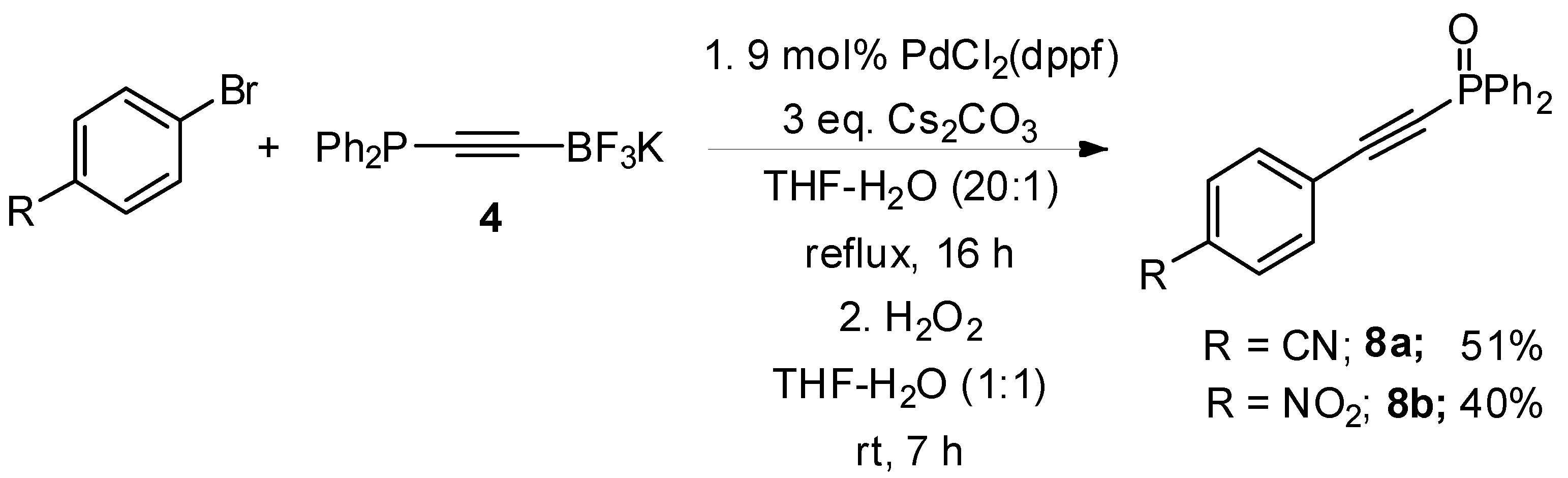
3.8. Synthesis of 4-((Diphenylphosphoryl)ethynyl)benzonitrile 8a
3.9. Synthesis of ((4-Nitrophenyl)ethynyl)diphenylphosphine Oxide 8b
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Matteson, D.S.; Peacock, K. Dibutyl acetyleneboronate. J. Am. Chem. Soc. 1960, 82, 5759–5760. [Google Scholar] [CrossRef]
- Castanet, A.-S.; Colobert, F.; Schlama, T. Suzuki-Miyaura coupling of alkynylboronic esters generated in situ from acetylenic derivatives. Org. Lett. 2000, 2, 3559–3561. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, Y.; Okamoto, M.; Inoue, Y.; Miyazaki, M.; Miyasaka, M.; Takagi, K. Synthesis of symmetrical 1,3-butadiynes by homocoupling reactions of alkynylboronates mediated by a copper salt. Tetrahedron Lett. 2005, 46, 8661–8664. [Google Scholar] [CrossRef]
- Nishihara, Y.; Saito, D.; Inoue, E.; Okada, Y.; Miyazaki, M.; Inoue, Y.; Takagi, K. Palladium- and base-free synthesis of conjugated ynones by cross-coupling reactions of alkynylboronates with acid chlorides mediated by CuCl. Tetrahedron Lett. 2010, 51, 306–308. [Google Scholar] [CrossRef]
- Hall, D.G. (Ed.) Boronic Acids; WILEY-VCH: Weinheim, Germany, 2005.
- Srebnik, M.; Bhat, N.G.; Brown, H.C. A new convenient approach to the preparation of Z-1-alkenylboronates by the cis-Hydrogenation of 1-Alkynyldiisopropoxyboranes. Tetrahedron Lett. 1988, 29, 2635–2638. [Google Scholar] [CrossRef]
- Deloux, L.; Skrzypczak-Jankun, E.; Cheesman, B.V.; Srebnik, M. First example of stable 1,1-bimetalloalkenes of boron and zirconium: Synthesis, reactivity, X-ray analysis, and NMR studies. J. Am. Chem. Soc. 1994, 116, 10302–10303. [Google Scholar] [CrossRef]
- Hyodo, K.; Suetsugu, M.; Nishihara, Y. Diborylation of alkynyl MIDA boronates and sequential chemoselective Suzuki-Miyaura couplings: A formal carboborylation of alkynes. Org. Lett. 2014, 16, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Ouellet, S.G. Novel synthesis of cyclic alkenylboronates via ring-closing metathesis. J. Am. Chem. Soc. 1998, 120, 7995–7996. [Google Scholar] [CrossRef]
- Moore, J.E.; Davies, M.W.; Goodenough, K.M.; Wybrow, R.A.J.; York, M.; Johnson, C.N.; Harrity, J.P.A. Investigation of the scope of a [3+2] cycloaddition approach to isoxazole boronic esters. Tetrahedron 2005, 61, 6707–6714. [Google Scholar] [CrossRef]
- Helm, M.D.; Moore, J.E.; Plant, A.; Harrity, J.P.A. Synthesis of highly substituted pyridazines via alkynylboronic ester cycloaddition reactions. Angew. Chem. Int. Ed. 2005, 44, 3889–3892. [Google Scholar] [CrossRef]
- Moore, J.E.; York, M.; Harrity, J.P.A. A metal-free cycloaddition approach to highly substituted aromatic boronic esters. Synlett 2005, 860–862. [Google Scholar]
- Delaney, P.M.; Moore, J.E.; Harrity, J.P.A. An alkynylboronic ester cycloaddition route to functionalised aromatic boronic esters. Chem. Commun. 2006, 42, 3323–3325. [Google Scholar] [CrossRef]
- Browne, D.L.; Vivat, J.F.; Plant, A.; Gomez-Bengoa, E.; Harrity, J.P.A. An investigation of the scope and regiochemistry of alkynylboronate cycloadditions with sydnones. J. Am. Chem. Soc. 2009, 131, 7762–7769. [Google Scholar] [CrossRef]
- Huang, J.; Macdonald, S.J.F.; Harrity, J.P.A. A cycloaddition route to novel triazole boronic esters. Chem. Commun. 2009, 45, 436–438. [Google Scholar] [CrossRef]
- Gandon, V.; Leboeuf, D.; Amslinger, S.; Vollhardt, K.P.C.; Malacria, M.; Aubert, C. Chemo-, regio-, and stereoselective cobalt-mediated [2+2+2] cycloaddition of alkynyl boronates to alkenes: 1,3- and 1,4-diboryl-1,3-cyclohexadienes. Angew. Chem. Int. Ed. 2005, 44, 7114–7118. [Google Scholar] [CrossRef]
- Hilt, G.; Smolko, K.I. Alkynylboronic esters as efficient dienophiles in cobalt-catalyzed Diels-Alder reactions. Angew. Chem. Int. Ed. 2003, 42, 2795–2797. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ishii, J.-I.; Nishiyama, H.; Itoh, K. Ru(II)-catalyzed chemo- and regioselective cyclotrimerization of three unsymmetrical alkynes through boron temporary tether. One-pot four-component coupling via cyclotrimerization/Suzuki−Miyaura coupling. J. Am. Chem. Soc. 2004, 126, 3712–3713. [Google Scholar] [CrossRef] [PubMed]
- Auvinet, A.-L.; Harrity, J.P.A.; Hilt, G. Ambient-temperature cobalt-catalyzed cycloaddition strategies to aromatic boronic esters. J. Org. Chem. 2010, 75, 3893–3896. [Google Scholar] [CrossRef] [PubMed]
- Auvinet, A.-L.; Harrity, J.P.A. A nickel catalyzed benzannulation approach to aromatic boronic esters. Angew. Chem. Int. Ed. 2011, 50, 2769–2772. [Google Scholar] [CrossRef]
- Matteson, D.S.; Peacock, K. Use of the bromoethyl leaving group for the synthesis of an ethoxyacetyleneboronic ester. J. Organomet. Chem. 1964, 2, 190–192. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Moritani, I. Carbon-13 nuclear magnetic resonance studies of organoboranes. Relative importance of mesomeric B-C π-bonding forms in alkenyl- and alkynylboranes. J. Org. Chem. 1975, 40, 3434–3437. [Google Scholar] [CrossRef]
- Brown, H.C.; Srebnik, M.; Bhat, N.G. A simple, general synthesis of 1-alkynyldiisopropoxyboranes. Tetrahedron Lett. 1988, 29, 2631–2634. [Google Scholar] [CrossRef]
- Delaney, P.M.; Browne, D.L.; Adams, H.; Plant, A.; Harrity, J.P.A. A 2-pyrone cycloaddition route to functionalised aromatic boronic esters. Tetrahedron 2008, 64, 866–873. [Google Scholar] [CrossRef]
- Bestmann, H.J.; Behl, H.; Bremer, M. Phosphonioboratoacetylenes: C2 stabilized by donor and acceptor molecules. Angew. Chem. Int. Ed. Engl. 1989, 28, 1219–1221. [Google Scholar] [CrossRef]
- Yuan, Z.; Taylor, N.J.; Sun, Y.; Marder, T.B.; Williams, I.D.; Cheng, L.-T. Synthesis and second-order nonlinear optical properties of three coordinate organoboranes with diphenylphosphino and ferrocenyl groups as electron donors: Crystal and molecular structures of (E)-DCHCHB(mes)2 and DCCB(mes)2 [D = P(C6H5)2, (η-C5H5)Fe(η-C5H4); mes = 2,4,6-(CH3)3C6H2]. J. Organomet. Chem. 1993, 449, 27–37. [Google Scholar] [CrossRef]
- Darses, S.; Genêt, J.-P. Potassium organotrifluoroborates: new perspectives in organic synthesis. Chem. Rev. 2008, 108, 288–325. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Ellis, N. Organotrifluoroborates: Protected boronic acids that expand the versatility of the Suzuki coupling reaction. Acc. Chem. Res. 2007, 40, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, J.D.; Butlin, R.J.; Harrity, J.P.A. A mild benzannulation via directed cycloaddition reactions. Angew. Chem. Int. Ed. 2012, 51, 6402–6405. [Google Scholar] [CrossRef]
- Crépin, D.F.P.; Harrity, J.P.A.; Jiang, J.; Meijer, A.J.H.M.; Nassoy, A.-C.M.A.; Raubo, P. A mechanistic study of the Lewis base directed cycloaddition of 2-pyrones and alkynylboranes. J. Am. Chem. Soc. 2014, 136, 8642–8653. [Google Scholar] [CrossRef] [PubMed]
- Bachollet, S.P.J.T.; Vivat, J.F.; Cocker, D.C.; Adams, H.; Harrity, J.P.A. Development of a mild and versatile directed cycloaddition approach to pyridines. Chem. Eur. J. 2014, 20, 12889–12893. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Katona, B.W.; Machrouhi, F. Development of the Suzuki-Miyaura cross-coupling reaction: Use of air-stable potassium alkynyltrifluoroborates in aryl alkynylations. J. Org. Chem. 2002, 67, 8416–8423. [Google Scholar] [CrossRef] [PubMed]
- The cross-coupling reactions provide a mixture of coupled phosphine and phosphine oxide compounds. The oxidative workup provided a convenient method to isolate the cross-coupled products.
- Huc, V.; Balueva, A.; Sebastian, R.-M.; Caminade, A.-M.; Majoral, J.-P. Synthesis of functionalized mono-, di-, tri-, and tetraphosphines: Attempted application to prepare hyperbranched polymers and dendrimers built with phosphines at each branching point. Synthesis 2000, 726–730. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vivat, J.F.; Bachollet, S.P.J.T.; Adams, H.; Harrity, J.P.A. Synthesis, Characterisation and Reactions of Phosphine-Substituted Alkynylboronates and Alkynyltrifluoroborate Salts. Molecules 2014, 19, 21324-21334. https://doi.org/10.3390/molecules191221324
Vivat JF, Bachollet SPJT, Adams H, Harrity JPA. Synthesis, Characterisation and Reactions of Phosphine-Substituted Alkynylboronates and Alkynyltrifluoroborate Salts. Molecules. 2014; 19(12):21324-21334. https://doi.org/10.3390/molecules191221324
Chicago/Turabian StyleVivat, Jérôme F., Sylvestre P. J. T. Bachollet, Harry Adams, and Joseph P. A. Harrity. 2014. "Synthesis, Characterisation and Reactions of Phosphine-Substituted Alkynylboronates and Alkynyltrifluoroborate Salts" Molecules 19, no. 12: 21324-21334. https://doi.org/10.3390/molecules191221324
APA StyleVivat, J. F., Bachollet, S. P. J. T., Adams, H., & Harrity, J. P. A. (2014). Synthesis, Characterisation and Reactions of Phosphine-Substituted Alkynylboronates and Alkynyltrifluoroborate Salts. Molecules, 19(12), 21324-21334. https://doi.org/10.3390/molecules191221324