Characterization and Quantification of the Compounds of the Ethanolic Extract from Caesalpinia ferrea Stem Bark and Evaluation of Their Mutagenic Activity

 ,
,
Abstract
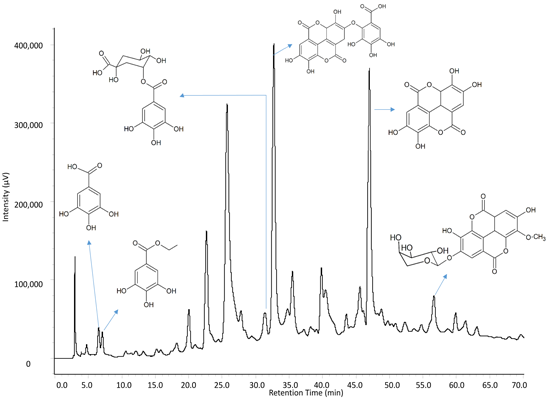
:
1. Introduction
2. Results and Discussion
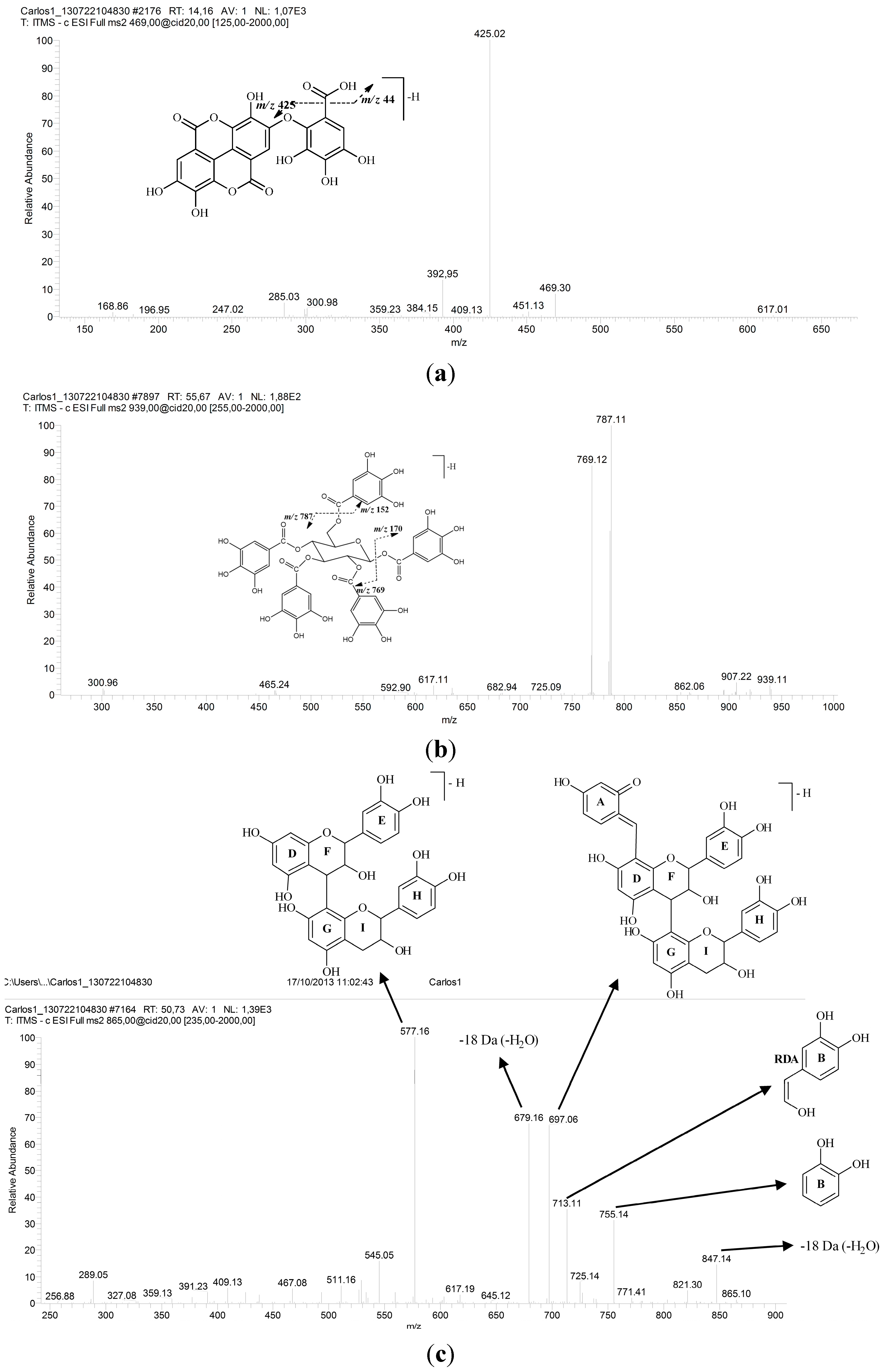
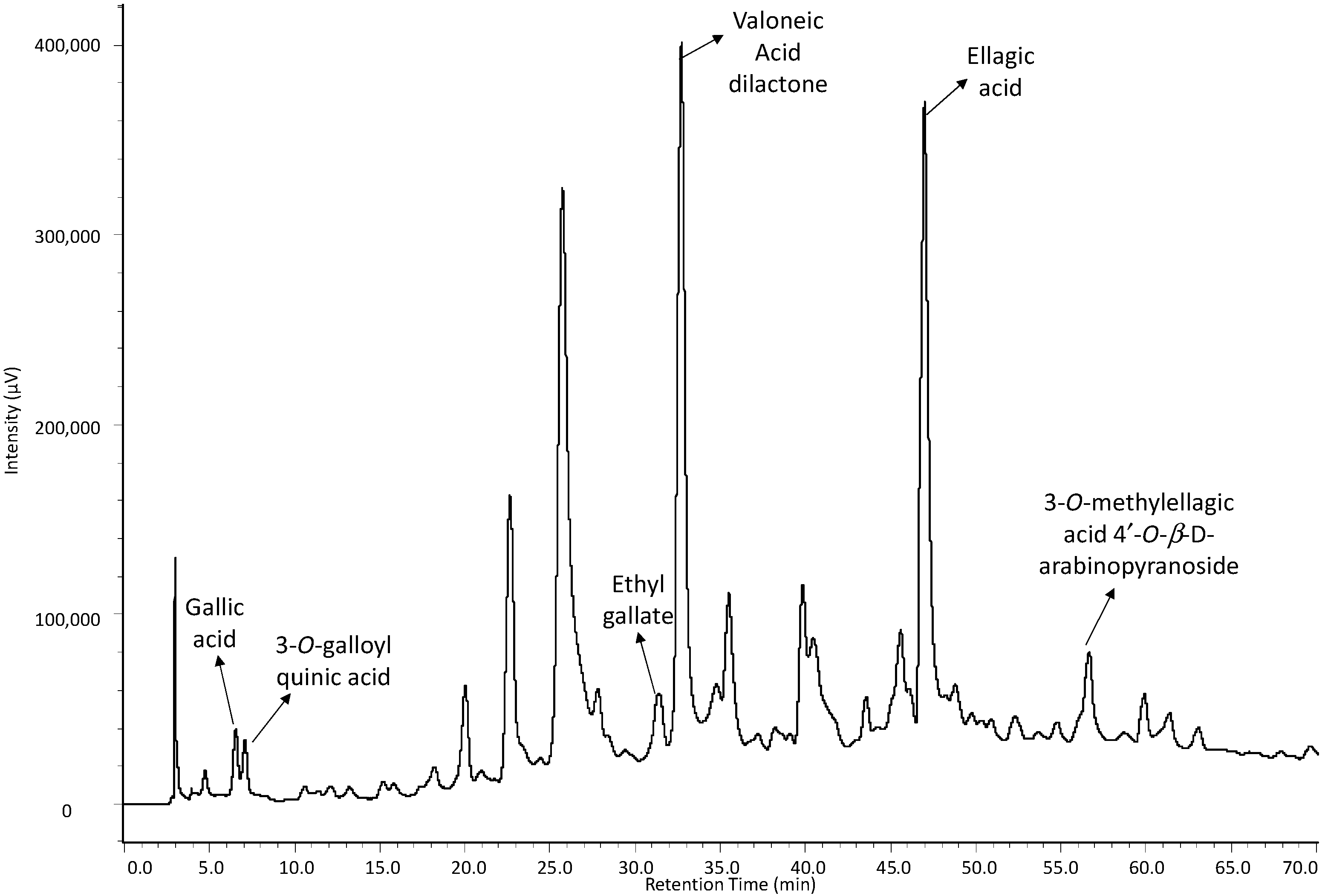
2.1. Identification of Constituents by FIA-ESI-IT-MS/MSn
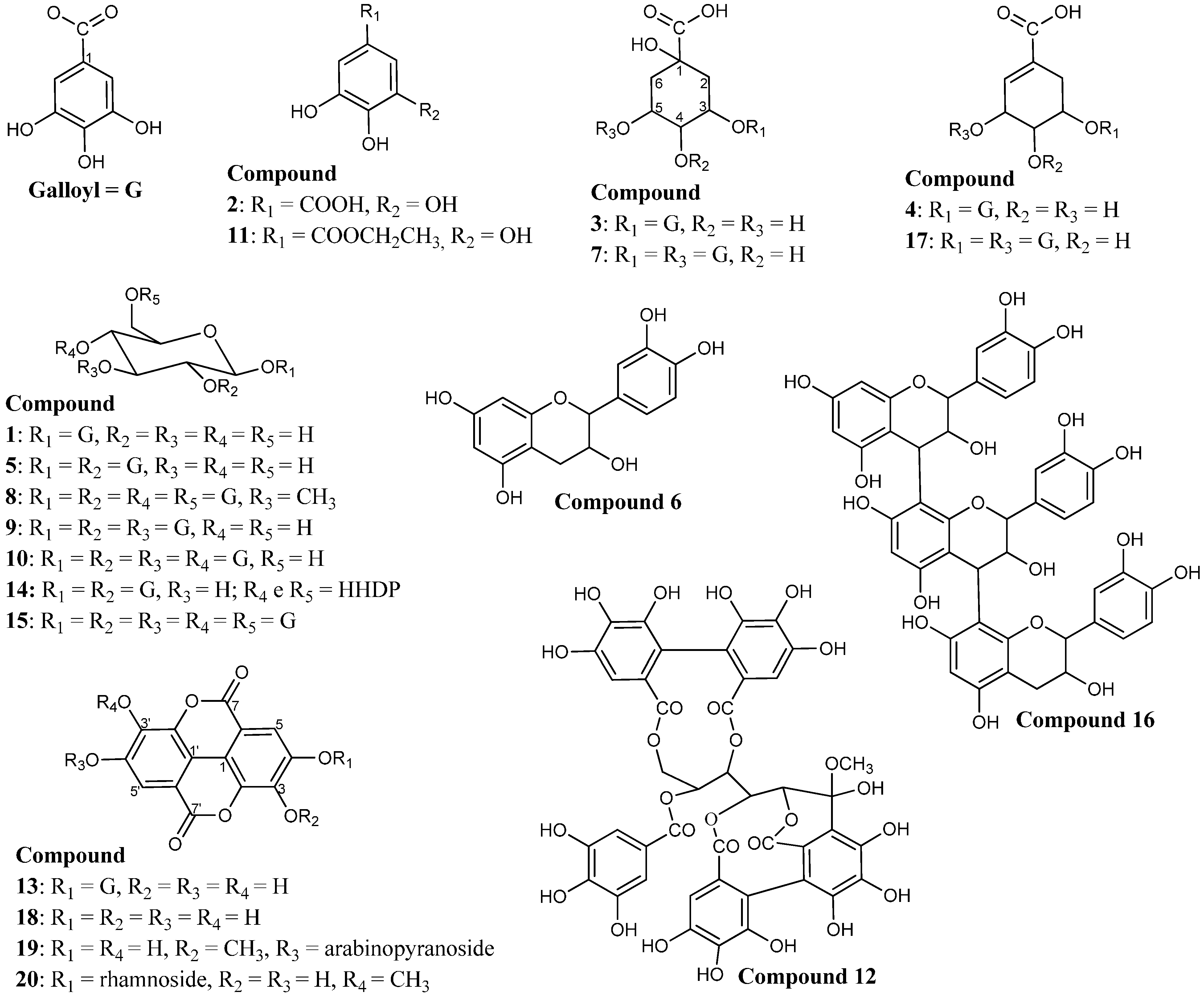

{kind=link}
{kind=link}
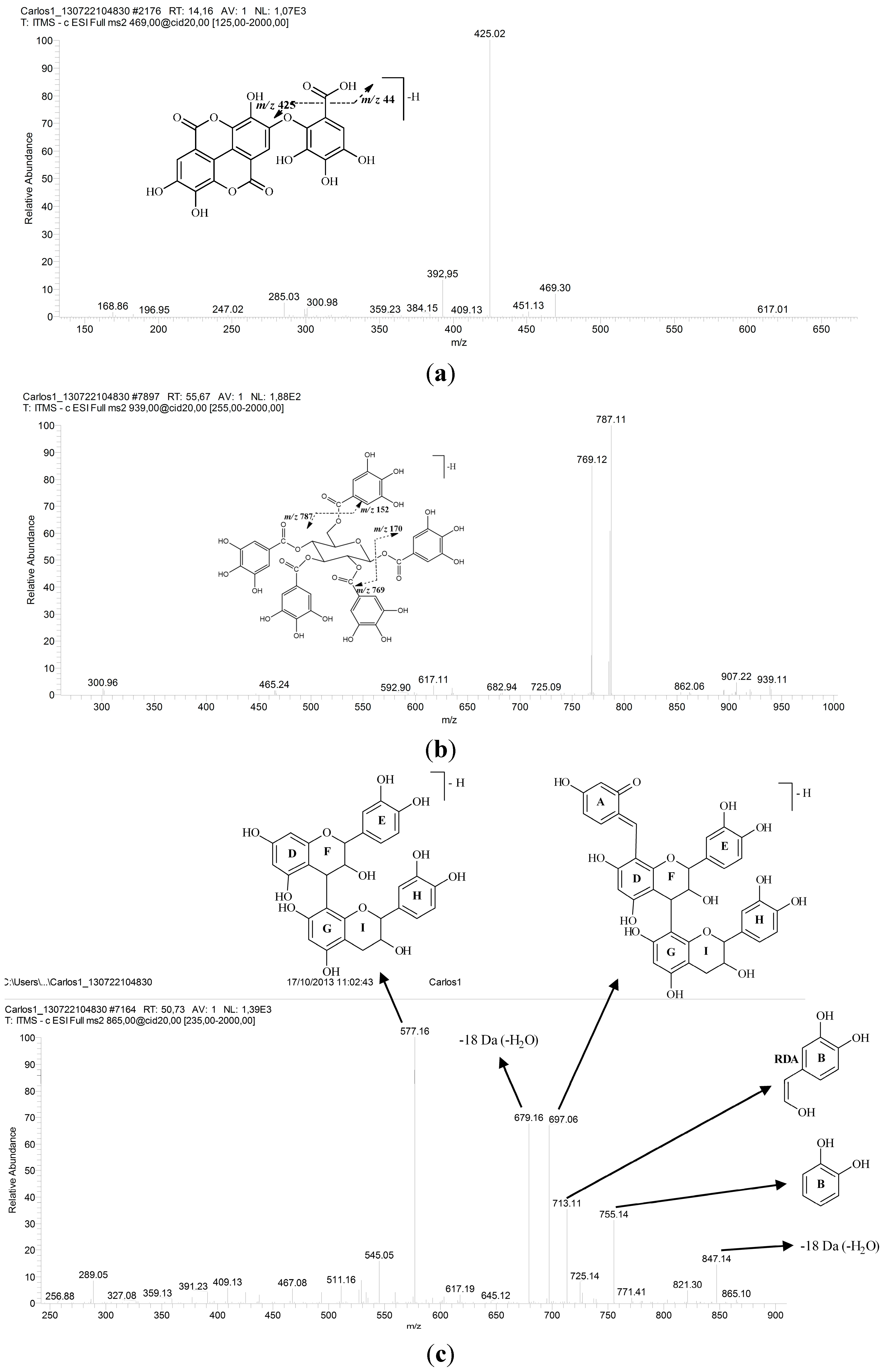{kind=link}
{kind=link}
| Peak (Compound) | Rt (min) | UV-Vis (λmax) | LC-MS [M − H]− | ESI-IT-MS/MSn Ions | Identification | Reference |
|---|---|---|---|---|---|---|
| 1 | 9.58 | 279 | 331 | 313, 271, 211, 193, 169, 125 | Monogalloylglucose | [19,20,21,22] |
| 2 | 11.72 | 271 | 169 | 125 | Gallic acid | [23,24] |
| 3 | 12.75 | 274 | 343 | 191, 169, 125 | 3-O-galloyl quinic acid | [25] |
| 4 | 17.48 | 274 | 325 | 169, 125 | Galloylshikimic acid | [23,26,27,28] |
| 5 | 19.63 | 280 | 483 | 271, 211, 193, 169, 125 | Digalloylglucose | [19,20,21,22] |
| 6 | 20.62 | 279 | 289 | 271, 245, 205, 139 | (epi) catechin | [20,29,30] |
| 7 | 21.52 | 278 | 495 | 343 | Digalloylquinic acid | [25] |
| 8 | 23.30 | 273 | 801 | 757, 713, 633, 631, 613 | Galloyltannin | [31] |
| 9 | 25.68 | 278 | 635 | 483, 465, 423, 301 | Trigalloylglucose | [19,20,21,22] |
| 10 | 26.61 | 279 | 787 | 635, 617, 301 | Tetragalloylglucose | [19,20,21,22] |
| 11 | 29.01 | 277 | 197 | 169, 125 | Ethyl gallate | [32] |
| 12 | 29.78 | 279 | 965 | 933, 301 | Castalagin derivative | [33] |
| 13 | 31.06 | 257, 365 | 469 | 425 | Valoneic acid dilactone | [34,35] |
| 14 | 31.94 | 278 | 785 | 633, 301 | hexahydroxydiphenyl-digalloylglucose acid | [20,36,37] |
| 15 | 32.60 | 279 | 939 | 787, 769, 635, 617 | Pentagalloylglucose | [19,20,21,22] |
| 16 | 34.01 | 279 | 865 | 847, 755, 713, 697, 679, 577, 529, 289 | Procyanidin trimer | [30,38] |
| 17 | 36.86 | 276 | 477 | 325, 315, 169 | Digalloylshikimic acid | [23,26,27,28] |
| 18 | 40.71 | 254, 365 | 301 | 257, 229, 185 | Ellagic acid | [39,40] |
| 19 | 42.27 | 254, 366 | 447 | 315, 300 | 3-O-methylellagic acid 4'-O-β-d-arabinopyranoside | [40,41] |
| 20 | 43.24 | 254, 363 | 461 | 315 | Methylellagic acid rhamnoside | [42] |
2.2. Validation Method
| Property | Ethanolic Extract | ||
|---|---|---|---|
| Gallic Acid | Ellagic Acid | ||
| Linear range (μg·mL−1) | 500–1.95 | 333–2.60 | |
| Calibration equation | y = 34178x − 105370 | y = 169688x − 1257.8 | |
| Correlation coefficient (r2) | 0.9999 | 1.0 | |
| LOQ (μg·mL−1) | 2.35 | 2.60 | |
| LOD (μg·mL−1) | 0.78 | 0.46 | |
| Interday precision (%RSD) | 4.76 | 3.66 | |
| Intraday precision (%RSD) | 0.59 | 0.76 | |
| Selectivity | Selective | Selective | |
| Compound | Concentration ± SD (µg·mL−1) | Standard |
|---|---|---|
| Gallic acid | 17.68 ± 0.10 | GA |
| 3-O-galloyl quinic acid | 13.26 ± 0.28 | GA |
| Ethyl gallate | 28.26 ± 0.81 | GA |
| Valoneic acid dilactone | 63.00 ± 0.93 | EA |
| Ellagic acid | 57.64 ± 1.22 | EA |
| Methylellagic acid-pentoside | 4.98 ± 0.06 | EA |
2.3. Mutagenic Activity
| Treatments | Number of Revertants (M ± SD)/Plate and MI | |||||||
|---|---|---|---|---|---|---|---|---|
| TA 98 | TA 100 | TA 102 | TA 97a | |||||
| mg/plate | −S9 | +S9 | −S9 | +S9 | −S9 | +S9 | −S9 | +S9 |
| 0.00 a | 57 ± 3 | 39 ± 5 | 128 ± 39 | 104 ± 3 | 401 ± 27 | 369 ± 33 | 75 ± 5 | 110 ± 14 |
| 0.26 | 68 ± 12(1.2) | 40 ± 3(1.0) | 130 ± 18(1.0) | 121 ± 8(1.2) | 434 ± 37(1.1) | 414 ± 6(1.1) | 109 ± 8(1.5) | 145 ± 9(1.3) |
| 0.52 | 70 ± 7(1.2) | 38 ± 1(1.0) | 135 ± 2(1.1) | 116 ± 1(1.1) | 425 ± 42(1.1) | 414 ± 15(1.1) | 84 ± 18(1.1) | 166 ± 5(1.5) |
| 1.04 | 65 ± 7(1.1) | 38 ± 6(1.0) | 159 ± 8(1.2) | 121 ± 13(1.2) | 394 ± 16(1.0) | 409 ± 17(1.1) | 93 ± 10(1.2) | 151 ± 7(1.4) |
| 1.56 | 66 ± 14(1.2) | 38 ± 4(1.0) | 142 ± 13(1.1) | 118 ± 10(1.1) | 393 ± 20(1.0) | 407 ± 17(1.1) | 91 ± 9(1.2) | 165 ± 10(1.5) |
| 2.08 | 61 ± 5(1.1) | 37 ± 3(1.0) | 152 ± 11(1.2) | 102 ± 2(1.0) | 392 ± 21(1.0) | 403 ± 8(1.0) | 81 ± 9(1.1) | 164 ± 29(1.5) |
| C+ | 797 ± 79 b | 2204 ± 255 e | 1193 ± 39 c | 1229 ± 94 e | 1192 ± 49 d | 1804 ± 43 e | 716 ± 74 b | 2636 ± 132 e |
3. Experimental
3.1. Chemicals and Reagents
3.2. Plant Material
3.3. Extraction
3.4. Isolation and Identification of the Compounds
3.5. HPLC/ESI-IT-MS Analyses
3.6. FIA-ESI-IT-MSn Analyses
3.7. Quantification by HPLC-DAD of the Ethanolic Extract Obtained from the Stem Bark
3.8. Preparation of Samples and Standards for Analysis by HPLC-DAD
3.9. Identification of Peaks
3.10. Quantitative Determination of Constituents
3.11. Linearity, Detection Limit, Quantification Limit and Precision
3.12. Salmonella/Microsome Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alzugaray, D. Plantas Que Curam; Hemus: São Paulo, Brazil, 1984; p. 260. [Google Scholar]
- Cavalheiro, M.G.; Farias, D.F.; Fernandes, G.S.; Nunes, E.P.; Cavalcanti, F.S.; Vasconcelos, I.M.; Melo, V.M.M.; Carvalho, A.F.U. Biological and enzymatic activities of aqueous extract of seeds from Caesalpinia ferrea Mart., Leguminosae. Rev. Bras. Farmacog. 2009, 19, 586–591. [Google Scholar] [CrossRef]
- Queiroz, L.P. Leguminosas da Caatinga, 1st ed.; State University of Feira de Santana Press: Feira de Santana, BA, Brazil, 2009; p. 443. [Google Scholar]
- Carvalho, J.C.T.; Teixeira, J.R.M.; Souza, P.J.C.; Bastos, J.K.; Santos-Filho, D.; Sarti, S.J. Preliminary studies of analgesic and anti-inflammatory propertiesof Caesalpinia ferrea crude extract. J. Ethnopharm. 1996, 53, 175–178. [Google Scholar] [CrossRef]
- Pereira, L.P.; Silva, R.O.; Bringel, P.H.S.F.; Silva, K.E.S.; Assreuy, A.M.S.; Pereira, M.G. Polysaccharide fractions of Caesalpinia ferrea pods: Potential anti-inflammatory usage. J. Ethnopharmacol. 2012, 139, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, E.M.; Sertie, J.A.A.; Villa, N.; Katz, H. Antiulcer ation and toxicity Styrax camporum and Caesalpinia ferrea. Planta Med. 1995, 61, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.S.; Kurosaki, F.; Arisawa, M.; Mukainaka, T.; Okuda, M.; Tokuda, H. Cancer chemopreventive effects of constituents of Caesalpiniaferrea and related compounds. Cancer Lett. 2002, 177, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, F.C.; Pereira, M.S.V.; Dias, C.S.; Costa, V.C.O.; Conde, N.C.O.; Buzalaf, M.A.R. In vitro antimicrobial activity of Caesalpinia ferrea Martius fruits against oral pathogens. J. Ethnopharm. 2009, 124, 289–294. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Batista, J.S.; Paiva, E.S.; Silva, A.E.; Farias, Y.J.M.D.; Damasceno, C.A.R.; Brito, P.D.; Queiroz, S.A.C.; Rodrigues, C.M.F.; Freitas, C.I.A. Avaliação da atividade cicatrizante do jucá (Caesalpinia ferrea Mart.) em lesões cutâneas de caprinos. Rev. Bras. Plantas Med. 2010, 12, 302–310. [Google Scholar] [CrossRef]
- Bragança, L.A.R. Plantas Medicinais Antidiabéticas: Uma Abordagem Multidisciplinar; Universidade Federal Fluminense: Niteroi, Brazil, 1996; p. 172. [Google Scholar]
- Vasconcelos, C.F.B.; Maranhão, H.M.L.; Batista, T.M.; Carneiro, E.M.; Ferreira, F.; Costa, J.; Soares, L.A.L.; Sá, M.D.C.; Souza, T.P.; Wanderley, A.G. Hypoglycaemic activity and molecular mechanisms of Caesalpinia ferrea Martius bark extract on streptozotocin-induced diabetes in Wistar rats. J. Ethnopharm. 2011, 137, 1533–1541. [Google Scholar] [CrossRef]
- Gonzalez, F.G.; Barros, S.B.M.; Bacchi, E.M. Atividade Antioxidante e perfil fitoquímico de Caesalpinia ferrea Mart. In Proceedings of IX Semana Farmacêutica de Ciência e Tecnologia, São Paulo, Brazil, 18 October 2004.
- Nozaki, H.; Hayashi, K.; Kido, M.; Kakumoto, K.; Ikeda, S.; Matsuura, N.; Tani, H.; Takaoka, D.; Iinuma, M.; Akao, Y. Pauferrol, “A novel chalcone trimer with a cyclobutane ring from Caesalpinia ferrea mart exhibiting DNA topoisomerase II inhibition and apoptosis-inducing activity”. Tetrahedron Lett. 2007, 48, 8290–8292. [Google Scholar] [CrossRef]
- Ohira, S.; Takaya, K.; Mitsui, T.; Kido, M.; Kakumoto, K.; Hayashi, K.; Kuboki, A.; Tani, H.; Ikeda, S.; Iinuma, M.; et al. New chalcone dimers from Caesalpiniaferrea Mart act as potent inhibitors of DNA topoisomerase II. Tetrahedron Lett. 2013, 54, 5052–5055. [Google Scholar] [CrossRef]
- Kaur, S.; Michael, H.; Arora, S.; Härkönen, P.L.; Kumar, S. The in vitro cytotoxic and apoptotic activity of Triphala—An Indian herbal drug. J. Ethnopharm. 2005, 97, 15–20. [Google Scholar] [CrossRef]
- Santos, F.V.; Tubaldini, F.R.; Colus, I.M.S.; Andréo, M.A.; Bauab, T.M.; Leite, C.Q.F.; Vilegas, W.; Varanda, E.A. Mutagenicity of Mouriripusa and Mouririelliptica Martius. Food Chem. Toxicol. 2008, 46, 2721–2727. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, C.R.P.; Cólus, I.M.S.; Bernardi, C.C.; Sannomiya, M.; Vilegas, W.; Varanda, E.A. Evaluation of the mutagenic activity promoted by amentoflavone and methanolic extract of Byrsonima crassa Niedenzu. Toxicology 2006, 225, 55–63. [Google Scholar] [CrossRef]
- Biso, I.; Rodrigues, C.M.; Rinaldo, D.; Reis, M.B.; Bernardi, C.C.; Matos, J.C.P.; Araújo, A.C.; Vilegas, W.; Colus, I.M.S.; Varanda, E.A. Assessment of DNA damage induced by extracts, fractions and isolated compounds of Davilla nitida and Davilla elliptica (Dilleniaceae). Mutat. Res. 2010, 702, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.O.; Vilela, C.; Freire, C.S.R.; PascoalNeto, C.; Silvestre, A.J.D. Ultra-high performance liquid chromatography coupled to mass spectrometry applied to the identificatíon of valuable phenoliccompounds from Eucalyptus wood. J. Chromatogr. B 2013, 938, 65–74. [Google Scholar] [CrossRef]
- Sandhu, A.K.; Gu, L. Antioxidant Capacity, Phenolic Content, and Profiling of Phenolic Compounds in the Seeds, Skin, and Pulp of Vitisrotundifolia (Muscadine Grapes) as Determined by HPLC-DAD-ESI-MSn. J. Agric. Food Chem. 2010, 58, 4681–4692. [Google Scholar] [CrossRef] [PubMed]
- Regueiro, J.; Sánchez-González, C.; Vallverdú-Queralt, A.; Simal-Gándara, J.; Lamuela-Raventós, R.; Izquierdo-Pulido, M. Comprehensive identification of walnut polyphenols by liquid chromatography coupled to linear ion trap–Orbitrap mass spectrometry. Food Chem. 2014, 152, 340–348. [Google Scholar] [CrossRef]
- Nuengchamnong, N.; Boonpathanasak, S.; Tepwitukkij, P. Rapid Screening of Antioxidant Compounds in Homemade Fruit Fermented Juice Using an On Line LC-ESI-MS/MS and DPPH Assay. Chiang Mai J. Sci. 2011, 38, 430–438. [Google Scholar]
- Mendes, L.; Freitas, V.; Baptista, P.; Carvalho, M. Comparative antihemolytic and radical scavenging activities of strawberry tree (Arbutus unedo L.) leaf and fruit. Food Chem. Toxicol. 2011, 49, 2285–2291. [Google Scholar] [CrossRef]
- Mena, P.; Calani, L.; Dall’asta, C.; Galaverna, G.; García-Viguera, C.; Bruni, R.; Crozier, A.; Del Rio, D. Rapid and Comprehensive Evaluation of (Poly)phenolic Compounds in Pomegranate (Punicagranatum L.) Juice by UHPLC-MSn. Molecules 2012, 17, 14821–14840. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N.; Stoupi, S.; Kuhnert, N. Profiling and Characterization by LC-MSn of the Galloylquinic Acids of Green Tea, Tara Tannin, and Tannic Acid. J. Agric. Food Chem. 2007, 55, 2797–2807. [Google Scholar] [CrossRef] [PubMed]
- Pawlowska, A.M.; Leo, M.; Braca, A. Phenolics of Arbutus unedoL. (Ericaceae) fruits: Identificatíon of anthocyanins and gallic acid derivatives. J. Agric. Food Chem. 2006, 54, 10234–10238. [Google Scholar]
- Tavares, L.; Fortalezas, S.; Carrilho, C.; McDougall, G.J.; Stewart, D.; Ferreira, R.B.; Santos, C.N. Antioxidant and antiproliferative properties of strawberry tree tissues. J. Berry Res. 2010, 1, 3–12. [Google Scholar]
- Guimarães, R.; Barros, L.; Dueñas, M.; Carvalho, A.M.; Queiroz, M.J.R.P.; Santos-Buelga, C.; Ferreira, I.C.F.R. Characterisatíon of phenolic compounds in wild fruits from Northeastern Portugal. Food Chem. 2013, 141, 3721–3730. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Zhang, Z.; Beecher, G.; Holden, J.; Haytowitz, D.; Prior, R.L. Liquid chromatographic/electrospray ionization mass spectrometric studies of proanthocyanidins in foods. J. Mass Spectrom. 2003, 38, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Hamed, A.I.; Al-Ayed, A.S.; Moldoch, J.; Piacente, S.; Oleszek, W.; Stochmal, A. Profiles analysis of proanthocyanidins in the argun nut (Medemiaargun—an ancient Egyptian palm) by LC–ESI–MS/MS. J. Mass Spectrom. 2014, 49, 306–315. [Google Scholar] [CrossRef]
- Romani, A.; Campo, M.; Pinelli, P. HPLC/DAD/ESI-MS analyses and anti-radical activity of hydrolyzable tannins from different vegetal species. Food Chem. 2012, 130, 214–221. [Google Scholar] [CrossRef]
- Sun, J.; Liang, F.; Bin, Y.; Li, P.; Duan, C. Screening Non-colored Phenolics in Red Wines using Liquid Chromatography/Ultraviolet and Mass Spectrometry/Mass Spectrometry Libraries. Molecules 2007, 12, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.A.; Carle, R.; Kammerer, D.R. Identificatíon and quantificatíon of phenolic compounds from pomegranate (Punicagranatum L.) peel, mesocarp, aril and differently produced juices by HPLC-DAD–ESI/MSn. Food Chem. 2011, 127, 807–821. [Google Scholar] [CrossRef]
- Silva, H.R.; Silva, C.C.M.; Neto, L.B.C.; Lopes, J.A.D.; Citó, A.M.G.L.; Chaves, M.H. Constituintes químicos das cascas do caule de Cenostigma macrophyllum: Ocorrência de colesterol. Quim. Nova 2007, 30, 1877–1881. [Google Scholar] [CrossRef]
- Barakat, H.H.; Hussein, S.A.M.; Marzouk, M.S.; Merfort, I.; Linscheid, M.; Nawwar, M.A.M. Polyphenoic metabolites of epilobium hirsutum. Phytochemistry 1997, 46, 935–941. [Google Scholar] [CrossRef]
- Lee, J.H.; Johnson, J.V.; Talcott, S.T. Identificatíon of ellagic acid conjugates and other polyphenolics in muscadine grapes by HPLCESI-MS. J. Agric. Food Chem. 2005, 53, 6003–6010. [Google Scholar] [CrossRef] [PubMed]
- Hanhineva, K.; Rogachev, I.; Kokko, H.; Mintz-Oron, S.; Venger, I.; Karenlampi, S.; Aharoni, A. Non-targeted analysis of spatial metabolite composition in strawberry (Fragariaxananassa) flowers. Phytochemistry 2008, 69, 2463–2481. [Google Scholar] [CrossRef] [PubMed]
- Sannomiya, M.; Montoro, P.; Piacente, S.; Pizza, C.; Brito, A.R.M.S.; Vilegas, W. Application of liquid chromatography/electrospray ionization tandem mass spectrometry to the analysis of polyphenolic compounds from an infusion of Byrsonima crassa Niedenzu. Rapid Commun. Mass Spectrom. 2005, 19, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Mullen, W.; Yokota, T.; Lean, M.E.J. Crozier, Analysis of ellagitannins and conjugates of ellagic acid and quercetinin raspberry fruits by LC–MSn. Phytochemistry 2003, 64, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Seeram, N.P.; Lee, R.; Scheuller, H.S.; Heber, D. Identification of phenolic compounds in strawberries by liquid chromatography electrospray ionization mass spectroscopy. Food Chem. 2006, 97, 1–11. [Google Scholar] [CrossRef]
- Fracassetti, D.; Costa, C.; Moulay, L.; Tomás-Barberán, F.A. Ellagic acid derivatives, ellagitannins, proanthocyanidins and other phenolics, vitamin C and antioxidant capacity of two powder products from camu-camu fruit (Myrciaria dubia). Food Chem. 2013, 139, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.P.; Lee, I.K.; Yun, B.S.; Chung, S.H.; Shim, G.S.; Koshino, H.; Yoo, I.D. Ellagic acid rhamnosides from the stem bark of Eucalyptus globulus. Phytochemistry 2001, 57, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Meyers, K.J.; Swiecki, T.J.; Mitchell, A.E. Understanding the Native Californian Diet: Identification of Condensed and Hydrolyzable Tannins in Tanoak Acorns (Lithocarpus densiflorus). J. Agric. Food Chem. 2006, 54, 7686–7691. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nonaka, G.I.; Nishioka, I. Seven Quinic Acids Gallates from Quercus stenophylla. Phytochemistry 1984, 23, 2621–2623. [Google Scholar] [CrossRef]
- Nono, R.N.; Barboni, L.; Teponno, R.B.; Quassinti, L.; Bramucci, M.; Vitali, L.A.; Petrelli, D.; Lupidi, G.; Tapondjou, A.L. Antimicrobial, antioxidant, anti-inflammatory ctivities and phytoconstituents of extracts from the roots of Dissotisthollonii Cogn. (Melastomataceae). S. Afr. J. Bot. 2014, 93, 19–26. [Google Scholar] [CrossRef]
- Benigni, R.; Bossa, C. Structure alerts for carcinogenicity, and the Salmonella assay system: A novel insight through the chemical relational databases technology. Mutat. Res. 2008, 659, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.; Dusinska, M.; Franklin, M.; Somorovska, M.; Petrovska, H.; Duthie, S.; Fillion, L.; Panayiotidis, M.; Raslova, K.; Vaughan, N. Comet assay in human biomonitoring studies: reliability, validation, and applications. Environ. Mol. Mutagen. 1997, 30, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, H.; Galehdari, H.; Zaree, Z.; Gesztelyi, R.; Varga, B.; Haines, D.; Bombicz, M.; Tosaki, A.; Juhasz, B. Toxicological and mutagenic analysis of Artemisia dracunculus (tarragon) extract. Food Chem. Toxicol. 2013, 51, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for Salmonella mutagenicity test. Mutat. Res. 1983, 113, 175–215. [Google Scholar]
- Myers, L.N.; Adams, N.; Kier, T.K.; Rao, B.; Shaw, B.; Williams, L. Microcomputer software for data management and statistical analyses of the Ames/Salmonella. In Statistical Methods in Toxicological Research; Krewski, D., Ed.; Gordon and Brech: New York, NY, USA, 1991; pp. 265–279. [Google Scholar]
- Bernstein, L.; Kaldor, J.; Mccann, J.; Pike, M.C. An empirical approach to the statistical analysis of mutagenesis data from the Salmonella test. Mutat. Res. 1982, 97, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Resende, F.A.; Barbosa, L.C.; Tavares, D.C.; Camargo, M.S.; Souza-Rezende, K.C.; Silva, M.; Varanda, E.A. Mutagenicity and antimutagenicity of (−)-hinokinin a trypanosomicidal compound measured by Salmonella microsome and comet assays. BMC Complement. Altern. Med. 2012, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are available from authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyrepkowski, C.C.; Gomes da Costa, D.L.M.; Sinhorin, A.P.; Vilegas, W.; De Grandis, R.A.; Resende, F.A.; Varanda, E.A.; Dos Santos, L.C. Characterization and Quantification of the Compounds of the Ethanolic Extract from Caesalpinia ferrea Stem Bark and Evaluation of Their Mutagenic Activity. Molecules 2014, 19, 16039-16057. https://doi.org/10.3390/molecules191016039
Wyrepkowski CC, Gomes da Costa DLM, Sinhorin AP, Vilegas W, De Grandis RA, Resende FA, Varanda EA, Dos Santos LC. Characterization and Quantification of the Compounds of the Ethanolic Extract from Caesalpinia ferrea Stem Bark and Evaluation of Their Mutagenic Activity. Molecules. 2014; 19(10):16039-16057. https://doi.org/10.3390/molecules191016039
Chicago/Turabian StyleWyrepkowski, Carlos César, Daryne Lu Maldonado Gomes da Costa, Adilson Paulo Sinhorin, Wagner Vilegas, Rone Aparecido De Grandis, Flavia Aparecida Resende, Eliana Aparecida Varanda, and Lourdes Campaner Dos Santos. 2014. "Characterization and Quantification of the Compounds of the Ethanolic Extract from Caesalpinia ferrea Stem Bark and Evaluation of Their Mutagenic Activity" Molecules 19, no. 10: 16039-16057. https://doi.org/10.3390/molecules191016039
APA StyleWyrepkowski, C. C., Gomes da Costa, D. L. M., Sinhorin, A. P., Vilegas, W., De Grandis, R. A., Resende, F. A., Varanda, E. A., & Dos Santos, L. C. (2014). Characterization and Quantification of the Compounds of the Ethanolic Extract from Caesalpinia ferrea Stem Bark and Evaluation of Their Mutagenic Activity. Molecules, 19(10), 16039-16057. https://doi.org/10.3390/molecules191016039