Abstract
A palladium-catalyzed synthesis of the carbazole framework is described, including the preparation of 2-, 5-, and 7-oxygenated natural and unnatural carbazole alkaloids. A series of N-arylcyclohexane enaminones, generated by condensation of cyclohexane-1,3-dione with diverse anilines, were aromatized by a Pd(0)-catalyzed thermal treatment to afford the corresponding diarylamines. The latter were submitted to a Pd(II)-catalyzed cyclization and methylation processes to provide the desired carbazoles, including clausine V. Following an inverse strategy, a new and short total synthesis of glycoborine is also reported.
1. Introduction
Biologically active carbazole alkaloids, a family of natural products with a variety of molecular structures, are isolated from higher order plants of the genera Clausena, Glycosmis, Micromelum, and Murraya (Rutaceae), among other sources [1,2,3,4,5]. Specifically, a great number of 2-, 5-, 6-, 7-mono- and bis-oxygenated tricyclic carbazoles isolated from these genera [1,2,3] exhibit a broad range of significant biological activities, including compounds with anti-tumor [6,7], antiplatelet aggregative [8], antibiotic [6,9,10], anti-viral [11,12,13], anti-plasmodial [14], anti-convulsant [15], and sigma receptor antagonist [16,17] properties. Carbazole derivatives 1a–g are examples of these natural alkaloids [8,18,19,20,21,22,23] (Figure 1).
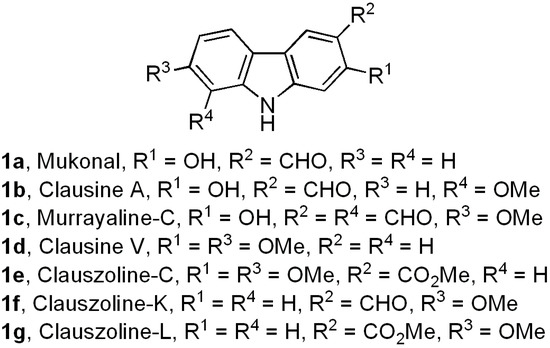
Figure 1.
Examples of naturally occurring 2-, 7-, and 2,7-oxygenated tricyclic carbazoles.
In spite of the large number of 2-, 5-, 6-, 7-mono- and bis-oxygenated tricyclic natural carbazoles that have been isolated, the wide range of functional groups and substitution patterns that exists among these compounds, and their important pharmacological activity, only recently a considerable number of synthetic approaches for their efficient preparation have been published [1,2,3,24,25,26,27,28,29,30,31,32,33,34,35,36].
We recently described a general synthetic approach for the construction of 1-methoxycarbazoles, including the naturally occurring alkaloid glycozolicine, which was accomplished with high overall yields through a three-step reaction sequence [37]. Based on this approach, we describe herein a new synthetic route for the preparation of 2-, 7-, and 2,7-oxygenated carbazoles 1. Starting from cyclohexene-1,3-dione (2) and the respective anilines 3a–e, enaminones 4a–e were prepared (Scheme 1). The latter were converted into diarylamines 5a–e and then cyclized to the desired carbazoles 1, via an efficient Pd-catalyzed aromatization and cyclization sequence of reactions.
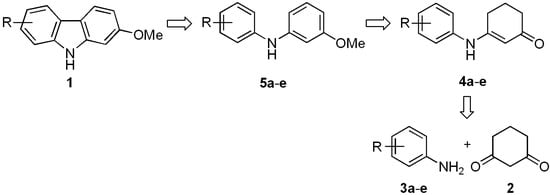
Scheme 1.
Synthetic approach for the preparation of 2-oxygenated tricyclic carbazoles 1.
2. Results and Discussion
2.1. Synthesis of Diarylamines
The catalyst-free condensation of cyclohexane-1,3-dione (2) with anilines 3a–e provided 3-anilino-2-cyclohexen-1-ones 4a–e in high yields (Table 1). However, the use of deactivated anilines, such as 3-nitro- and 4-nitroanilines, failed to provide the desired enaminones, thus limiting this procedure to anilines substituted with electron-donating groups. Applying our previous procedure for aromatization using Pd(OAc)2 (30% mol) [37], derivatives 4a–b did not lead to the desired diarylamines 6a–b, but instead furnished the carbazole frame compounds 7a–b in good yields (Scheme 2). Similar results via Pd-mediated procedures have been reported for analogous substrates [1,38,39,40,41], which in turn have been transformed into the 4-oxygenated carbazoles [42]. When using other catalysts, such as mercuric acetate [43] and 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) [44,45], diarylamines 6 were indeed produced, but in very low yields (15%–20%).

Table 1.
Scope of the reaction between cyclohexane-1,3-dione (2) and anilines 3a–e a. 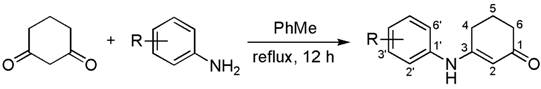
| Entry | 3 (Ar) | 4 (%) b |
|---|---|---|
| 1 | 3a (C6H4-4-Me) | 4a (92) |
| 2 | 3b (C6H4-4-OMe) | 4b (95) |
| 3 | 3c (C6H4-3-Me) | 4c (90) |
| 4 | 3d (C6H4-3-OMe) | 4d (93) |
| 5 | 3e (C6H3-3,5-(OMe)2 | 4e (96) |
a Standard conditions: 2 (3.57 mmol), 3 (3.57 mmol), toluene (150 mL), reflux, 12 h.
b Isolated yields.
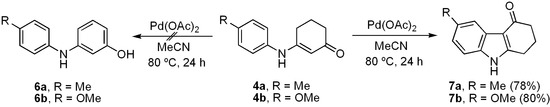
Scheme 2.
Pd(II)-catalyzed treatment of 3-anilino-2-cyclohexen-1-ones 4a–b.
Due to the fact that the insertion of the aryl and cyclohexenone rings takes place via a Pd(II)-catalyzed pathway [1,2,38,39,40,41,46], we chose a Pd(0)-mediated method for carrying out such an aromatization. Initially, when 4b was treated with Pd/C (5%) at different concentrations (1–6 mol%) with MeOH as the solvent and heating to 50–200 °C in a sealed vessel, diarylamine 6b was not obtained and the starting material was recovered. However, the desired transformation was achieved by increasing both the palladium(0) loading on charcoal (10%) (1.9%–5.7% mol) and the reaction temperature (Table 2, entries 1–3). The use of the Pd(0)-mediated aromatization method for similar substrates or carbazole derivatives in moderate to good yields has been reported [42,47,48,49,50,51,52]. Reagents such as DDQ [44,45,53] and chloranil [54] have also been successfully applied to achieve analogous conversions [43,55].
Although the preparation and purification of diarylamines 6a–b and 6d–e resulted in high yields (Table 2, entries 3–4 and 6–7), the relative instability of these compounds under the conditions of the following cyclization reaction made it necessary to protect the phenol moiety. In order to achieve this protection and taking into account that there are many naturally occurring methoxy-containing oxygenated carbazoles, we decided to obtain the methylated derivatives 5a–e. For this purpose, we employed a direct sequential procedure for the dehydrogenation and methylation of phenols 6 without purification (Table 2, entries 3–7). Thus, the series of compounds 5a–e was prepared in high yields (81%–87%).
Table 2.
Conversion of 3-anilino-2-cyclohexen-1-ones 4a–e into diarylamines 6a–b, 6d–e and 5a–e a. 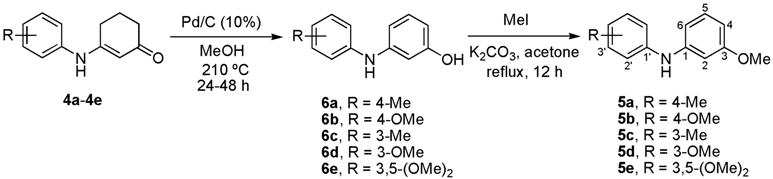
| Entry | 4 (R) | Pd/C (10%) (mol%) b | 6 (%) c | 5 (%) d |
|---|---|---|---|---|
| 1 e | 4b (4-OMe) | 1.9 | 6b (65) | ---- |
| 2 f | 4b (4-OMe) | 3.8 | 6b (75) | ---- |
| 3 | 4b (4-OMe) | 5.7 | 6b (87) | 5b (85) |
| 4 | 4a (4-Me) | 5.7 | 6a (85) | 5a (83) |
| 5 | 4c (3-Me) | 5.7 | (g) | 5c (87) |
| 6 | 4d (3-OMe) | 5.7 | 6d (84) | 5d (81) |
| 7 | 4e (3,5-(OMe)2 | 5.7 | 6e (88) | 5e (86) |
a Standard conditions: (a) Preparation of diarylamines 6: 4 (0.81–1.00 mmol), Pd/C (10%), MeOH, 210 °C, 48 h; (b) Preparation of diarylamines 5a–e: Aromatization step: 4 (0.82–1.00 mmol), Pd/C (10%), MeOH, 210 °C, 24 h; Methylation step: 6 (1.0 mol equiv.), MeI (2.0 mol equiv.), K2CO3 (1.5 mol equiv.), acetone, reflux, 12 h. b Calculated for Pd(0). c Isolated yields. d Isolated yields for the two steps. e At 180 °C for 12 h. f At 200 °C for 48 h. g Not isolated.
2.2. Synthesis of Carbazoles
The final cyclization step of diarylamines 5a–e was successfully carried out by following the protocol originally developed by Knölker and coworkers [40,56,57], later applied by others [28,29], and optimized in our syntheses of natural carbazoles [37,44]. Thus, the conversion of the series 5a–c and 5e into the carbazole derivatives 1h–k resulted in good yields (80%–92%) (Table 3). It is noteworthy that the cyclization of 5d provided clausine V (1d) in high yield (90%) [22,33].
With the aim of testing the utility of this methodology for the total synthesis of natural 7-oxygenated tricyclic carbazoles, we carried out the conversion of derivative 1h into clauszoline-K (1f) and clauszoline-L (clausine C, 1g). Thus, upon applying the well-known procedure [58,59] for the synthesis of these [32] and other natural carbazoles [44], carbazole 1h was treated with DDQ in a mixture of MeOH/H2O/acetone (1:1:1) at room temperature for 45 min to give 1f in 70% yield (Scheme 3).
Table 3.
Preparation of carbazoles 1d and 1h–k via Pd(II)-catalyzed cyclization of diarylamines 5a–e a. 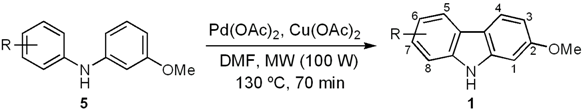
| Entry | 5 (R) | 1 | Isolated yield (%) b |
|---|---|---|---|
| 1 | 5a (4-Me) | 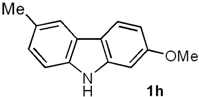 | 80 |
| 2 | 5b (4-OMe) | 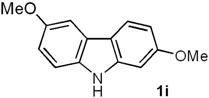 | 87 |
| 3 | 5c (3-Me) | 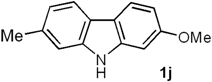 | 82 |
| 4 | 5d (3-OMe) | 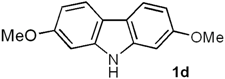 | 90 |
| 5 | 5e (3,5-(OMe)2 | 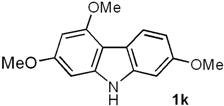 | 92 |
a Standard conditions: 5 (0.32 –0.47 mmol), Pd(OAc)2 (10 mol%), Cu(OAc)2 (2.5 mol equiv.), DMF, MW (100 W), 130 °C, 70 min. b Isolated yields.
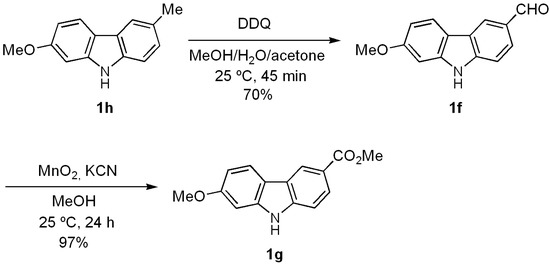
Scheme 3.
Preparation of natural carbazoles clauszoline-K (1f) and clauszoline-L (1g).
The latter was oxidized with a mixture of MnO2/KCN in MeOH [58] to furnish clauszoline-L (1g) in almost quantitative yield. The spectral data of the products obtained agree with those described for the natural [20,23] and synthetic [58] products.
2.3. Total Synthesis of Glycoborine (Glycrophylamine, 9)
Recently, 5-methoxy-3-methylcarbazole (9) was isolated from the roots and branches of Glycosmis macrophylla and named glycrophylamine. This compound showed cytotoxic activity against NC1-H187 cancerigene cells [60]. However, the same carbazole had been isolated from Glycosmis arborea a decade earlier, and named glycoborine. This was the first 5-oxygenated tricyclic natural carbazole ever isolated [61]. Nowadays, three routes of synthesis have been developed for 9 based on Fischer [61], Japp-Klingemann [62], and Cadogan cyclizations [33] as the key step. We herein describe a new total synthesis of 9 starting from the key precursor tetrahydrocarbazole 7a (Scheme 4), which was efficiently prepared from 4a (Scheme 2).
When a mixture of 7a, Pd/C (10%) (5.7% mol) and anhydrous MeOH was heated in a sealed vessel to 270 °C for 48 h, 5-hydroxy-3-methylcarbazole (8) was isolated and then purified in good yield (Scheme 4). Methylation of the latter under the usual reaction conditions provided the desired natural carbazole 9, which was synthesized in four steps with high overall yield (53%). The spectral data of 9 agree with those described for the natural [60,61] and synthetic [33,62] products.
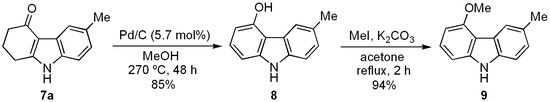
Scheme 4.
Preparation of natural carbazole glycoborine (9).
All the structures of intermediates and products described in these synthetic sequences were characterized by 1H- and 13C-NMR spectroscopy, with the help of 2D (HMQC and HMBC) experiments and mass spectrometric techniques (MS and HRMS).
3. Experimental
3.1. General
Melting points (uncorrected) were determined with an Electrothermal capillary melting point apparatus. IR spectra were recorded on a Perkin-Elmer 2000 spectrophotometer. 1H (300 or 500 MHz) and 13C-NMR (75 or 125 MHz) spectra were recorded on Varian Mercury-300 or Varian VNMR System instruments, with TMS as internal standard. Mass spectra (MS) and high-resolution mass spectra (HRMS) were obtained, in electron impact (EI) (70 eV) mode, on Thermo-Finnigan Polaris Q and Jeol JSM-GcMateII spectrometers, respectively. Microwave (MW) irradiation was performed on a CEM MW reactor. Analytical thin-layer chromatography was carried out using E. Merck silica gel 60 F254 coated 0.25 plates, visualized by a long- and short-wavelength UV lamp. Flash column chromatography was performed over Natland International Co. silica gel (230–400 mesh). All air moisture sensitive reactions were carried out under nitrogen using oven-dried glassware. Toluene, MeOH, and MeCN were freshly distilled over sodium and DMF over calcium hydride prior to use. Acetone was dried by distillation after treatment with potassium permanganate. K2CO3 was dried overnight at 200 °C prior to use. All other reagents were used without further purification.
3.2. Synthesis and Characterization
3-(p-Tolylamino)cyclohex-2-en-1-one (4a) [63]. In a 250 mL, three necked, round-bottomed flask equipped with a magnetic stirring bar, rubber septum, a water condenser and a Dean-Stark trap, under N2 atmosphere, a mixture of 2 (0.400 g, 3.57 mmol) and 3a (0.382 g, 3.37 mmol) in dry toluene (150 mL) was stirred at reflux for 12 h. The solvent was removed under vacuum, and the residue purified by column chromatography over silica gel (10 g/g of crude, hexane/EtOAc, 1:1) to give 4a (0.66 g, 92%) as a pale yellow solid. Rf 0.15 (hexane/EtOAc, 1:1); mp 248–249 °C. IR (KBr): νmax 3214, 3029, 2937, 1573, 1512, 1361, 1311, 1245, 1183, 1141, 818 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.98 (qu, J = 6.5 Hz, 2H, H-5), 2.31 (br t, J = 6.5 Hz, 2H, H-6), 2.32 (s, 3H, CH3), 2.48 (t, J = 6.5 Hz, 2H, H-4), 5.48 (s, 1H, H-2), 6.99–7.03 (m, 2H, H-2′), 7.07–7.11 (m, 2H, H-3′), 7.12 (br s, 1H, NH). 13C-NMR (125 MHz, CDCl3): δ = 20.9 (CH3), 21.8 (C-5), 29.5 (C-4), 36.4 (C-6), 99.0 (C-2), 124.0 (C-2′), 129.7 (C-3′), 135.3 (C-4′), 135.5 (C-1′), 163.1 (C-3), 198.1 (C-1). MS (70 eV): m/z (%) 201 (M+, 74), 184 (26), 173 (100), 144 (53), 130 (29), 106 (13), 91 (12), 77 (10). HRMS (EI): m/z [M+] calcd for C13H15NO: 201.1154; found: 201.1156.
3-(4-Methoxyphenylamino)cyclohex-2-en-1-one (4b). Following the procedure described for 4a, using 2 (0.400 g, 3.57 mmol) and 3b (0.439 g, 3.57 mmol), 4b (0.74 g, 95%) was obtained as a pale yellow solid. Rf 0.12 (hexane/EtOAc, 1:1); mp 166–167 °C [Lit. [64] 164–166 °C]. IR (KBr): νmax 3218, 3039, 2946, 1513, 1412, 1365, 1243, 1180, 1135, 1032, 834, 716 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.97 (qu, J = 6.5 Hz, 2H, H-5), 2.30 (t, J = 6.5 Hz, 2H, H-6), 2.47 (t, J = 6.5 Hz, 2H, H-4), 3.78 (s, 3H, CH3O), 5.34 (s, 1H, H-2), 6.80–6.84 (m, 2H, H-3′), 7.02–7.06 (m, 2H, H-2′), 7.13 (br s, 1H, NH). 13C-NMR (125 MHz, CDCl3): δ = 21.8 (C-5), 29.3 (C-4), 36.4 (C-6), 55.4 (CH3O), 98.6 (C-2), 114.4 (C-3′), 126.1 (C-2′), 130.8 (C-1′), 157.5 (C-4′), 164.0 (C-3), 198.0 (C-1). MS (70 eV): m/z (%) 217 (M+, 100), 200 (55), 189 (43), 174 (20), 160 (98), 146 (30), 130 (23), 117 (12), 77 (10). HRMS (EI): m/z [M+] calcd for C13H15NO2: 217.1103; found: 217.1110.
3-(m-Tolylamino)cyclohex-2-en-1-one (4c). Following the procedure described for 4a, 4c (0.65 g, 90%) was obtained as a pale yellow oil from 2 (0.400 g, 3.57 mmol) and 3c (0.382 g, 3.57 mmol). Rf 0.15 (hexane/EtOAc, 1:1). IR (film): νmax 3256, 3066, 2959, 1546, 1453, 1360, 1244, 1185, 1136, 829, 795, 728 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.98 (qu, J = 6.5 Hz, 2H, H-5), 2.29 (s, 3H, CH3), 2.32 (t, J = 6.5 Hz, 2H, H-6), 2.49 (t, J = 6.5 Hz, 2H, H-4), 5.55 (s, 1H, H-2), 6.91-6.95 (m, 2H, H-4′, H-6′), 6.96 (br s, 1H, H-2′), 7.17 (t, J = 8.0 Hz, 1H, H-5′), 7.18 (br s, 1H, NH). 13C-NMR (125 MHz, CDCl3): δ 21.3 (CH3), 21.8 (C-5), 29.6 (C-4), 36.4 (C-6), 99.3 (C-2), 120.9 (C-6′), 124.4 (C-2′), 126.2 (C-4′), 129.0 (C-5′), 138.0 (C-3′), 139.1 (C-1′), 162.9 (C-3), 198.3 (C-1). MS (70 eV): m/z (%) 201 (M+, 79), 184 (49), 173 (100), 158 (16), 144 (88), 130 (42), 106 (13), 91 (19), 77 (16). HRMS (EI): m/z [M+] calcd for C13H15NO: 201.1154; found: 201.1160.
3-(3-Methoxyphenylamino)cyclohex-2-en-1-one (4d). Following the procedure described for 4a, using 2 (0.400 g, 3.57 mmol) and 3d (0.439 g, 3.57 mmol), 4d (0.72 g, 93%) was obtained as a pale yellow solid. Rf 0.11 (hexane/EtOAc, 1:1); mp 126–127 °C [Lit. [64] 122.5–124 °C; [65] 126–128 °C]. IR (KBr): νmax 3278, 3196, 3133, 2937, 1540, 1425, 1357, 1318, 1245, 1191, 1143, 1051, 871, 733 cm−1. 1H-NMR (500 MHz, CDCl3): δ 1.98 (qu, J = 6.5 Hz, 2H, H-5), 2.32 (t, J = 6.5 Hz, 2H, H-6), 2.50 (t, J = 6.5 Hz, 2H, H-4), 3.75 (s, 3H, CH3O), 5.59 (s, 1H, H-2), 6.66–6.70 (m, 2H, H-2′, H-6′), 6.72 (dm, J = 8.0 Hz, 1H, H-4′), 7.16–7.21 (m, 1H, H-5′), 7.29 (br s, 1H, NH). 13C-NMR (125 MHz, CDCl3): δ 21.8 (C-5), 29.5 (C-4), 36.4 (C-6), 55.2 (CH3O), 99.6 (C-2), 109.7 (C-2′), 110.8 (C-6′), 116.0 (C-4′), 129.9 (C-5′), 139.4 (C-1′), 160.2 (C-3′), 162.7 (C-3), 198.4 (C-1). MS (70 eV): m/z (%) 217 (M+, 99), 200 (59), 189 (46), 160 (100), 146 (32), 130 (23), 117 (11), 77 (7). HRMS (EI): m/z [M+] calcd for C13H15NO2: 217.1103; found: 217.1101.
3-(3,5-Dimethoxyphenylamino)cyclohex-2-en-1-one (4e). Following the procedure described for 4a, with 2 (0.400 g, 3.57 mmol) and 3e (0.546 g, 3.57 mmol), 4e (0.85 g, 96%) was obtained as a pale yellow solid. Rf 0.12 (hexane/EtOAc, 1:1); mp 139–140 °C. IR (KBr): νmax 3272, 2940, 1598, 1582, 1538, 1462, 1423, 1361, 1253, 1186, 1153, 1055, 824 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 1.99 (qu, J = 6.5 Hz, 2H, H-5), 2.33 (t, J = 6.5 Hz, 2H, H-6), 2.49 (t, J = 6.5 Hz, 2H, H-4), 3.73 (s, 6H, 2CH3O), 5.64 (s, 1H, H-2), 6.24 (t, J = 2.0 Hz, 1H, H-4′), 6.30 (d, J = 2.0 Hz, 2H, H-2′, H-6′), 7.06 (br s, 1H, NH). 13C-NMR (125 MHz, CDCl3): δ = 21.8 (C-5), 29.6 (C-4), 36.4 (C-6), 55.3 (CH3O), 97.4 (C-4′), 100.2 (C-2), 102.1 (C-2′, C-6′), 139.9 (C-1′), 161.2 (C-3′, C-5′), 162.3 (C-3), 198.3 (C-1). MS (70 eV): m/z (%) 247 (M+, 37), 230 (100), 219 (25), 190 (83), 160 (18), 135 (30), 120 (14), 77 (7). HRMS (EI): m/z [M+] calcd for C14H17NO3: 247.1208; found: 247.1207.
3-(p-Tolylamino)phenol (6a). In a threaded ACE glass pressure tube with a sealed Teflon screw cap, under N2 atmosphere, a mixture of 4a (0.20 g, 1.0 mmol) and Pd/C (10%) (0.060 g, 0.057 mmol) in dry MeOH (2.5 mL) was stirred at 210 °C for 48 h. The solvent was removed under vacuum, and the residue purified by column chromatography over silica gel (20 g/g of crude, hexane/EtOAc, 80:20), to give 6a (0.168 g, 85%) as a pale grey solid. Rf 0.55 (hexane/EtOAc, 7:3); mp 81–82 °C [Lit. [66] 82 °C]. IR (film): νmax 3394, 1606, 1512, 1493, 1332, 1243, 1155, 969, 815, 766 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.27 (s, 3H, CH3), 5.58 (br s, 1H, NH), 6.31 (dd, J = 7.8, 2.1 Hz, 1H, H-4), 6.44 (t, J = 2.1 Hz, 1H, H-2), 6.51 (dd, J = 7.8, 2.1 Hz, 1H, H-6), 6.92–6.99 (m, 2H, H-2′), 6.99–7.07 (m, 3H, H-3′, H-5). 13C-NMR (75.4 MHz, CDCl3): δ = 20.6 (CH3), 103.2 (C-2), 107.1 (C-4), 108.9 (C-6), 119.5 (C-2′), 129.8 (C-3′), 130.2 (C-5), 131.2 (C-4′), 139.7 (C-1′), 145.6 (C-1), 156.6 (C-3). MS (70 eV): m/z (%) 199 (M+, 100), 183 (19), 170 (22), 154 (35), 128 (14), 91 (83), 65 (18). HRMS (EI): m/z [M+] calcd for C13H13NO: 199.0997; found: 199.0998.
3-(4-Methoxyphenylamino)phenol (6b). Following the procedure described for 6a, with 4b (0.200 g, 0.92 mmol) and Pd/C (10%) (0.060 g, 0.057 mmol), 6b (0.172 g, 87%) was obtained as a pale grey solid. Rf 0.50 (hexane/EtOAc, 8:2); mp 66–67 °C [Lit. [66] 67–68 °C]. IR (KBr): νmax 3379, 1601, 1526, 1504, 1459, 1291, 1239, 1174, 1110, 1027, 735 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 3.79 (s, 3H, CH3O), 6.50 (br s, 1H, NH), 6.28 (ddd, J = 8.0, 2.4, 0.6 Hz, 1H, H-4), 6.37 (t, J = 2.4 Hz, 1H, H-2), 6.44 (ddd, J = 8.0, 2.4, 0.6 Hz, 1H, H-6), 6.82–6.89 (m, 2H, H-3′), 7.04 (t, J = 8.0 Hz, 1H, H-5), 7.04–7.10 (m, 2H, H-2′). 13C-NMR (75.4 MHz, CDCl3): δ = 55.5 (CH3O), 101.9 (C-2), 106.3 (C-4), 108.0 (C-6), 114.6 (C-3′), 122.9 (C-2′), 130.3 (C-5), 135.1 (C-1′), 146.9 (C-1), 155.4 (C-4′), 156.7 (C-3). MS (70 eV): m/z (%) 215 (M+, 100), 201 (6), 185 (7), 172 (5), 146 (4), 132 (5), 91 (11). HRMS (EI): m/z [M+] calcd for C13H13NO2: 215.0946; found: 215.0952.
3-(3-Methoxyphenylamino)phenol (6d) [66]. Following the procedure described for 6a, with 4d (0.200 g, 0.92 mmol) and Pd/C (10%) (0.060 g, 0.057 mmol), 6d (0.166 g, 84%) was obtained as a purple oil. Rf 0.51 (hexane/EtOAc, 8:2). IR (film): νmax 3411, 1645, 1489, 1156, 764 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 3.71 (s, 3H, CH3O), 5.76 (br s, 1H, NH), 6.38 (ddd, J = 8.1, 2.4, 0.9 Hz, 1H, H-4), 6.46 (dm, J = 7.8 Hz, 1H, H-4′), 6.54 (t, J = 2.4 Hz, 1H, H-2), 6.56–6.65 (m, 3H, H-2′, H-6, H-6′), 7.05 (t, J = 8.1 Hz, 1H, H-5), 7.11 (t, J = 7.8 Hz, 1H, H-5′). 13C-NMR (75.4 MHz, CDCl3): δ = 55.1 (CH3O), 103.8 (C-2′), 104.6 (C-2), 106.3 (C-4′), 108.0 (C-4), 110.1 (C-6′), 110.7 (C-6), 130.0 (C-5′), 130.2 (C-5), 144.0 (C-1′), 144.3 (C-1), 156.6 (C-3), 160.3 (C-3′). MS (70 eV): m/z (%) 215 (M+, 26), 199 (21), 182 (25), 160 (31), 146 (45), 130 (25), 109 (23), 51 (100). HRMS (EI): m/z [M+] calcd for C13H13NO2: 215.0946; found: 215.0952.
3-(3,5-Dimethoxyphenylamino)phenol (6e). Following the procedure described for 6a, with 4e (0.200 g, 0.81 mmol) and Pd/C (10%) (0.060 g, 0.057 mmol), 6e (0.174 g, 88%) was obtained as a yellow oil. Rf 0.49 (hexane/EtOAc, 8:2). IR (film): νmax 3379, 2917, 1594, 1481, 1203, 1152, 1065, 821 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 3.73 (s, 6H, 2CH3O), 5.74 (br s, 1H, NH), 6.07 (t, J = 2.1 Hz, 1H, H-4′), 6.23 (d, J = 2.1 Hz, 2H, H-2′, H-6′), 6.39 (dd, J = 8.1, 2.4 Hz, 1H, H-4), 6.55 (dd, J = 2.4, 2.1 Hz, 1H, H-2), 6.62 (ddd, J = 8.1, 2.1, 0.9 Hz, 1H, H-6), 7.08 (t, J = 8.1 Hz, 1H, H-5). 13C-NMR (75.4 MHz, CDCl3): δ = 55.3 (2CH3O), 93.2 (C-4′), 96.3 (C-2′, C-6′), 105.0 (C-2), 108.2 (C-4), 110.6 (C-6), 130.3 (C-5), 144.1 (C-1′), 144.7 (C-1), 156.6 (C-3), 161.4 (C-3′, C-5′). MS (70 eV): m/z (%) 245 (M+, 3), 154 (14), 153 (96), 125 (15), 124 (100), 94 (25), 92 (22). HRMS (EI): m/z [M+] calcd for C14H15NO3: 245.1052; found: 245.1059.
3-Methoxy-N-(p-tolyl)aniline (5a) [67]. In a threaded ACE glass pressure tube with a sealed Teflon screw cap, under N2 atmosphere, a mixture of 4a (0.20 g, 1.0 mmol) and Pd/C (10%) (0.060 g, 0.057 mmol) in dry MeOH (2.5 mL) was stirred at 210 °C for 24 h. After removing the solvent under vacuum, K2CO3, (0.200 g, 1.45 mmol) and CH3I (0.281 g, 1.98 mmol) in dry acetone (20 mL) were added, and the mixture was heated to reflux for 12 h. The solvent was removed under vacuum and the residue purified by column chromatography over silica gel (20 g/g of crude, hexane/EtOAc, 90:10), to give 5a (0.176 g, 83%) as a white solid. Rf 0.60 (hexane/EtOAc, 7:3); mp 49–50 °C. IR (KBr): νmax 3367, 1598, 1493, 1462, 1256, 1157, 1032, 950, 832, 774 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.30 (s, 3H, CH3), 3.75 (s, 3H, CH3O), 6.00 (br s, 1H, NH), 6.42 (ddd, J = 8.5, 2.5, 0.5 Hz, 1H, H-4), 6.55-6.59 (m, 2H, H-2, H-6), 6.98–7.02 (m, 2H, H-2′), 7.06-7.10 (m, 2H, H-3′), 7.12 (tm, J = 8.5 Hz, 1H, H-5). 13C-NMR (125 MHz, CDCl3): δ = 20.7 (CH3), 55.1 (CH3O), 102.4 (C-2), 105.5 (C-4), 109.4 (C-6), 119.4 (C-2′), 129.8 (C-3′), 130.0 (C-5), 131.2 (C-4′), 140.0 (C-1′), 145.4 (C-1), 160.7 (C-3). MS (70 eV): m/z (%) 213 (M+, 100), 200 (23), 189 (24), 174 (21), 160 (39), 130 (11), 91 (12), 84 (9). HRMS (EI): m/z [M+] calcd for C14H15NO: 213.1154; found: 213.1153.
3-Methoxy-N-(4-methoxyphenyl)aniline (5b) [29]. Following the procedure described for 5a using 4b (0.200 g, 0.92 mmol), Pd/C (10%) (0.055 g, 0.052 mmol), K2CO3 (0.190 g, 1.38 mmol) and MeI (0.261 g, 1.84 mmol), 5b (0.179 g, 85%) was obtained as a white solid. Rf 0.55 (hexane/EtOAc, 7:3); mp 99–100 °C. IR (KBr): νmax 3400, 1597, 1509, 1460, 1239, 1157, 1035, 825, 768 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 3.75 (s, 3H, CH3O), 3.79 (s, 3H, CH3O), 6.51 (br s, 1H, NH), 6.38 (br dd, J = 7.8, 2.1 Hz, 1H, H-4), 6.44–6.51 (m, 2H, H-2, H-6), 6.83–6.89 (m, 2H, H-3′), 7.04–7.10 (m, 2H, H-2′), 7.11 (t, J = 7.8 Hz, 1H, H-5). 13C-NMR (75.4 MHz, CDCl3): δ = 55.1 (CH3O-C3), 55.5 (CH3O-C4′), 101.1 (C-2), 104.6 (C-4), 108.2 (C-6), 114.6 (C-3′), 122.7 (C-2′), 130.0 (C-5), 135.3 (C-1′), 146.6 (C-1), 155.4 (C-4′), 160.7 (C-3). MS (70 eV): m/z (%) 229 (M+, 90), 216 (47), 214 (100), 186 (19), 171 (21), 142 (15), 115 (21). HRMS (EI): m/z [M+] calcd for C14H15NO2: 229.1103; found: 229.1111.
3-Methoxy-N-(m-tolyl)aniline (5c). Following the procedure described for 5a, with 4c (0.20 g, 1.0 mmol), Pd/C (10%) (0.060 g, 0.057 mmol), K2CO3 (0.200 g, 1.45 mmol) and MeI (0.281 g, 1.98 mmol), 5c (0.18 g, 87%) was obtained as a yellow oil. Rf 0.59 (hexane/EtOAc, 7:3). IR (film): νmax 3391, 1589, 1490, 1266, 1203, 1155, 1042, 765, 687 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 2.31 (s, 3H, CH3), 3.77 (s, 3H, CH3O), 5.67 (br s, 1H, NH), 6.47 (br dd, J = 7.8, 2.4 Hz, 1H, H-4), 6.62-6.67 (m, 2H, H-2, H-6), 6.76 (br d, J = 7.2 Hz, 1H, H-4′), 6.88–6.94 (m, 2H, H-2′, H-6′), 7.12–7.20 (m, 2H, H-5, H-5′). 13C-NMR (75.4 MHz, CDCl3): δ = 21.5 (CH3), 55.2 (CH3O), 103.2 (C-2), 105.9 (C-4), 110.2 (C-6), 115.4 (C-6′), 119.0 (C-2′), 122.1 (C-4′), 129.1 (C-5′), 130.0 (C-5), 139.2 (C-3′), 142.7 (C-1′), 144.6 (C-1), 160.6 (C-3). MS (70 eV): m/z (%) 213 (M+, 100), 200 (32), 189 (35), 174 (26), 160 (44), 130 (13), 92 (11), 77 (11). HRMS (EI): m/z [M+] calcd for C14H15NO: 213.1154; found: 213.1161.
bis(3-Methoxyphenyl)amine (5d) [26]. Following the procedure described for 5a, with 4d (0.20 g, 0.92 mmol), Pd/C (10%) (0.055 g, 0.052 mmol), K2CO3 (0.190 g, 1.38 mmol) and MeI (0.261 g, 1.84 mmol), 5d (0.171 g, 81%) was obtained as a white solid. Rf 0.55 (hexane/EtOAc, 7:3); mp 154–155 °C. IR (film): νmax 3393, 1592, 1490, 1270, 1207, 1155, 1040, 832, 760, 685 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 3.74 (s, 6H, 2CH3O), 5.78 (br s, 1H, NH), 6.47 (ddm, J = 8.1, 2.4 Hz, 2H, H-4, H-4′), 6.61–6.68 (m, 4H, H-2, H-2′, H-6, H-6′), 7.14 (t, J = 8.1 Hz, 2H, H-5, H-5′). 13C-NMR (75.4 MHz, CDCl3): δ = 55.1 (2CH3O), 103.6 (C-2, C-2′), 106.3 (C-4, C-4′), 110.4 (C-6, C-6′), 130.0 (C-5, C-5′), 144.1 (C-1, C-1′), 160.5 (C-3, C-3′). MS (70 eV): m/z (%) 229 (M+, 100), 217 (10), 200 (12), 189 (6), 170 (11), 160 (9), 154 (12), 142 (9), 115 (5). HRMS (EI): m/z [M+] calcd for C14H15NO2: 229.1103; found: 229.1104.
3,5-Dimethoxy-N-(3-methoxyphenyl)aniline (5e). Following the procedure described for 5a, with 4e (0.200 g, 0.818 mmol), Pd/C (10%) (0.050 g, 0.047 mmol), K2CO3 (0.167 g, 1.21 mmol) and MeI (0.230 g, 1.62 mmol), 5e (0.18 g, 86%) was obtained as a colorless oil. Rf 0.52 (hexane/EtOAc, 7:3). IR (film): νmax 3735, 1590, 1541, 1457, 1203, 1150, 1057 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 3.74 (s, 6H, 2CH3O), 3.76 (s, 3H, CH3O-3), 5.73 (br s, 1H, NH), 6.07 (t, J = 2.0 Hz, 1H, H-4), 6.24 (d, J = 2.0 Hz, 2H, H-2, H-6), 6.49 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H, H-4′), 6.65 (t, J = 2.0 Hz, 1H, H-2′), 6.67 (ddd, J = 8.0, 2.0, 1.0 Hz, 1H, H-6′), 7.15 (t, J = 8.0 Hz, 1H, H-5′). 13C-NMR (125 MHz, CDCl3): δ = 55.1 (CH3O-3′), 55.2 (2CH3O), 93.3 (C-4), 96.2 (C-2, C-6), 104.2 (C-2′), 106.7 (C-4′), 111.0 (C-6′), 130.0 (C-5′), 144.0 (C-1), 144.9 (C-1′), 160.6 (C-3′), 161.6 (C-3, C-5). MS (70 eV): m/z (%) 259 (M+, 7), 257 (97), 242 (100), 214 (49), 199 (42), 184 (13), 156 (8), 128 (7). HRMS (EI): m/z [M+] calcd for C15H17NO3: 259.1208; found: 259.1209.
2-Methoxy-6-methyl-9H-carbazole (1h). A mixture of 5a (0.100 g, 0.47 mmol), Pd(AcO)2 (0.0105 g, 0.047 mmol) and Cu(AcO)2 (0.211 g, 1.17 mmol) in dry DMF (0.5 mL), under N2 atmosphere, was stirred and heated at 130 °C for 70 min under MW irradiation (100 W). The solvent was removed under vacuum by adding toluene, and the azeotropic distillation was continued until no solvent remained. The residue was purified by column chromatography over silica gel (10 g/g of crude, hexane/EtOAc, 95:5), to give 1h (0.079 g, 80%) as a white solid. Rf 0.60 (hexane/EtOAc, 7:3); mp 226–227 °C [Lit. [32] 227–228 °C]. IR (film): νmax 3392, 1659, 1026, 826, 764, 687 cm−1. 1H-NMR (500 MHz, DMSO-d6/acetone-d6, 3:7): δ = 2.46 (s, 3H, CH3), 3.86 (s, 3H, CH3O), 6.76 (dd, J = 8.4, 2.4 Hz, 1H, H-3), 6.99 (d, J = 2.4 Hz, 1H, H-1), 7.11 (dd, J = 8.2, 1.5 Hz, 1H, H-7), 7.33 (d, J = 8.2 Hz, 1H, H-8), 7.78 (br s, 1H, H-5), 7.91 (d, J = 8.4 Hz, 1H, H-4), 10.6 (br s, 1H, NH). 13C-NMR (125 MHz, DMSO-d6/acetone-d6, 3:7): δ = 21.2 (CH3), 55.4 (CH3O), 95.0 (C-1), 108.1 (C-3), 110.8 (C-8), 117.2 (C-4a), 119.6 (C-5), 121.1 (C-4), 124.0 (C-4a), 125.9 (C-7), 128.0 (C-6), 139.0 (C-8a), 142.4 (C-9a), 159.5 (C-2). MS (70 eV): m/z (%) 211 (M+, 100), 196 (51), 168 (76), 139 (10), 86 (6). HRMS (EI): m/z [M+] calcd for C14H13NO: 211.0997; found: 211.0994.
2,6-Dimethoxy-9H-carbazole (1i). Following the procedure described for 1h, with 5b (0.100 g, 0.44 mmol), Pd(AcO)2 (0.0099 g, 0.044 mmol) and Cu(AcO)2 (0.198 g, 1.1 mmol), 1i (0.086 g, 87%) was obtained as a white solid. Rf 0.55 (hexane/EtOAc, 7:3); mp 162–163 °C [Lit. [29] 163–164 °C]. IR (KBr): νmax 3398, 1626, 1491, 1465, 1284, 1221, 1202, 1162, 1029, 820 cm−1. 1H-NMR (300 MHz, CDCl3): δ = 3.85 (s, 3H, CH3O), 3.86 (s, 3H, CH3O), 6.75 (dd, J = 8.4, 2.4 Hz, 1H, H-3), 6.92 (dd, J = 8.7, 2.5 Hz, 1H, H-7), 6.95 (d, J = 2.4 Hz, 1H, H-1), 7.31 (d, J = 8.7 Hz, 1H, H-8), 7.51 (d, J = 2.5 Hz, 1H, H-5), 7.89 (d, J = 8.4 Hz, 1H, H-4), 9.90 (br s, 1H, NH). 13C-NMR (75.4 MHz, CDCl3): δ = 53.9 (CH3O), 54.3 (CH3O), 93.3 (C-1), 101.2 (C-5), 106.6 (C-3), 110.0 (C-8), 112.0 (C-7), 115.9 (C-4a), 119.7 (C-4), 122.7 (C-4b), 133.8 (C-8a), 141.1 (C-9a), 152.8 (C-6), 158.0 (C-2). MS (70 eV): m/z (%) 227 (M+, 100), 212 (86), 184 (69), 169 (28), 141 (19), 114 (7). HRMS (EI): m/z [M+] calcd for C14H13NO2: 227.0946; found: 227.0951.
2-Methoxy-7-methyl-9H-carbazole (1j). Following the procedure described for 1h, with 5c (0.100 g, 0.47 mmol), Pd(AcO)2 (0.0105 g, 0.047 mmol) and Cu(AcO)2 (0.211 g, 1.17 mmol), 1j (0.081 g, 82%) was obtained as a white solid. Rf 0.61 (hexane/EtOAc, 7:3); mp 162–163 °C [Lit. [68] 280 °C]. IR (film): νmax 3399, 1654, 1047, 1025, 995, 827, 766 cm−1. 1H-NMR (500 MHz, DMSO-d6/acetone-d6, 3:7): δ = 2.46 (s, 3H, CH3), 3.85 (s, 3H, CH3O), 6.75 (dd, J = 8.5, 2.5 Hz, 1H, H-3), 6.95 (dd, J = 8.3, 1.0 Hz, 1H, H-6), 6.99 (d, J = 2.5 Hz, 1H, H-1), 7.25 (br s, 1H, H-8), 7.84 (d, J = 8.3 Hz, 1H, H-5), 7.88 (d, J = 8.5 Hz, 1H, H-4), 10.7 (br s, 1H, NH). 13C-NMR (125 MHz, DMSO-d6/acetone-d6, 3:7): δ = 21.4 (CH3), 54.9 (CH3O), 94.2 (C-1), 106.9 (C-3), 110.3 (C-8), 116.1 (C-4a), 118.4 (C-5), 119.6 (C-6), 119.9 (C-4), 120.2 (C-4b), 133.1 (C-7), 140.0 (C-8a), 140.8 (C-9a), 157.8 (C-2). MS (70 eV): m/z (%) 211 (M+, 100), 196 (58), 168 (66), 139 (10), 86 (6). HRMS (EI): m/z [M+] calcd for C14H13NO: 211.0997; found: 211.1000.
2,7-Dimethoxy-9H-carbazole (Clausine V, 1d). Following the procedure described for 1h, with 5d (0.100 g, 0.44 mmol), Pd(AcO)2 (0.0099 g, 0.044 mmol) and Cu(AcO)2 (0.198 g, 1.10 mmol), 1d (0.089 g, 90%) was obtained as a white solid. Rf 0.56 (hexane/EtOAc, 7:3); mp 229–230 °C [Lit. [22] 228–230 °C]. IR (KBr): νmax 3382, 2927, 1608, 1575, 1502, 1457, 1322, 1265, 1233, 1160, 1118, 1026, 825, 805 cm−1. 1H-NMR (300 MHz, DMSO-d6/acetone-d6, 3:7): δ = 3.85 (s, 6H, 2CH3O), 6.75 (dd, J = 8.4, 2.4 Hz, 2H, H-3, H-6), 6.99 (d, J = 2.4 Hz, 2H, H-1, H-8), 7.85 (d, J = 8.4 Hz, 2H, H-4, H-5), 10.81 (br s, 1H, NH). 13C-NMR (75.4 MHz, DMSO-d6/acetone-d6, 3:7): δ = 54.9 (2CH3O), 94.6 (C-1, C8), 107.3 (C-3, C-6), 116.8 (C-4a, C-4b), 119.7 (C-4, C-5), 141.4 (C-8a, C-9a), 157.9 (C-2, C-7). MS (70 eV): m/z (%) 227 (M+, 77), 212 (100), 184 (42), 169 (54), 153 (13), 141 (27), 114 (5). HRMS (EI): m/z [M+] calcd for C14H13NO2: 227.0946; found: 227.0946.
2,4,7-Trimethoxy-9H-carbazole (1k). Following the procedure described for 1h, with 5e (0.101 g, 0.39 mmol), Pd(AcO)2 (0.0087 g, 0.039 mmol) and Cu(AcO)2 (0.175 g, 0.97 mmol), 1k (0.092 g, 92%) was obtained as a white solid. Rf 0.20 (hexane/EtOAc, 7:3); mp 167–168 °C. IR (KBr): νmax 3383, 1617, 1580, 1510, 1453, 1428, 1260, 1213, 1149, 1119, 1032, 803 cm−1. 1H-NMR (500 MHz, CDCl3/acetone-d6, 7:3): δ = 3.83 (s, 6H, 2CH3O), 3.98 (s, 3H, CH3O), 6.27 (d, J = 1.3 Hz, 1H, H-3), 6.46 (d, J = 1.3 Hz, 1H, H-1), 6.76 (dd, J = 8.5, 2.2 Hz, 1H, H-6), 6.82 (d, J = 2.2 Hz, 1H, H-8), 7.99 (d, J = 8.5 Hz, 1H, H-5), 9.32 (br s, 1H, NH). 13C-NMR (125 MHz, CDCl3/acetone-d6, 7:3): δ = 54.7 (CH3O), 54.9 (CH3O), 55.0 (CH3O), 86.7 (C-1), 90.2 (C-3), 94.0 (C-8), 106.0 (C-4a), 107.0 (C-6), 116.1 (C-4b), 121.7 (C-5), 139.7 (C-8a), 141.3 (C-9a), 155.2 (C-4), 156.8 (C-7), 158.7 (C-2). MS (70 eV): m/z (%) 257 (M+, 39), 247 (36), 230 (100), 219 (24), 214 (22), 190 (82), 176 (21), 160 (19), 117 (7). HRMS (EI): m/z [M+] calcd for C15H15NO3: 257.1052; found: 257.1052.
7-Methoxy-9H-carbazole-3-carbaldehyde (Clauszoline-K) (1f). A mixture of 1h (0.030 g, 0.14 mmol) and DDQ (0.129 g, 0.57 mmol) in acetone/MeOH/H2O (1:1:1) (10 mL) was stirred at 25 °C for 45 min. The solvent was removed under vacuum and the residue purified by column chromatography over silica gel (10 g/g of crude, hexane/EtOAc, 8:2), to give 1f (0.022 g, 70%) as a white solid. Rf 0.25 (hexane/EtOAc, 8:2); mp 184–185 °C [Lit. [32] 183–186 °C]. IR (KBr): νmax 3296, 1670, 1604, 1570, 1479, 1322, 1237, 1160, 1026, 821 cm−1. 1H-NMR (500 MHz, DMSO-d6/CDCl3, 3:7): δ = 3.88 (s, 3H, CH3O), 6.84 (dd, J = 8.5, 2.0 Hz, 1H, H-6), 7.00 (d, J = 2.0 Hz, 1H, H-8), 7.51 (d, J = 8.0, 1H, H-1), 7.83 (dd, J = 8.0, 1.0 Hz, 1H, H-2), 7.96 (d, J = 8.5 Hz, 1H, H-5), 8.45 (s, 1H, H-4), 10.05 (s, 1H, CHO), 11.40 (br s, 1H, NH). 13C-NMR (125 MHz, DMSO-d6/CDCl3, 3:7): δ = 54.0 (CH3O), 93.8 (C-8), 107.6 (C-6), 109.5 (C-1), 115.0 (C-4b), 119.7 (C-5), 121.3 (C-4), 121.8 (C-4a), 124.1 (C-2), 126.9 (C-3), 140.8 (C-8a), 142.6 (C-9a), 158.0 (C-7), 190.2 (CHO). MS (70 eV): m/z (%) 225 (M+, 40), 210 (28), 180 (72), 167 (97), 160 (44), 146 (30), 130 (32), 115 (28), 77 (29), 51 (100). HRMS (EI): m/z [M+] calcd for C14H11NO2: 225.0790; found: 225.0796.
Methyl 7-methoxy-9H-carbazole-3-carboxylate (Clauszoline-L, Clausine C) (1g). A mixture of 1f(0.200 g, 0.89 mmol), MnO2 (0.20 g, 2.3 mmol), and KCN (0.028 g, 0.43 mmol) in MeOH (10 mL) was stirred at 25 °C for 24 h. The solvent was removed under vacuum and the residue purified by column chromatography over silica gel (10 g/g of crude, hexane/EtOAc, 8:2), to give 1g (0.22 g, 97%) as a white solid. Rf 0.29 (hexane/EtOAc, 8:2); mp 194–195 °C [Lit. [20] 195–197 °C; [32] 195 °C; [33] 194–195 °C]. IR (KBr): νmax 3288, 1698, 1605, 1439, 1327, 1259, 1195, 1159, 1094, 815, 728 cm−1. 1H-NMR (500 MHz, acetone-d6): δ = 3.88 (s, 3H, CH3O), 3.91 (s, 3H, CH3O), 6.88 (dd, J = 8.5, 2.0 Hz, 1H, H-6), 7.08 (d, J = 2.0 Hz, 1H, H-8), 7.51 (d, J = 8.5, 1H, H-1), 7.99 (dd, J = 8.5, 1.5 Hz, 1H, H-2), 8.09 (d, J = 8.5 Hz, 1H, H-5), 8.69 (d, J = 1.5 Hz, 1H, H-4), 10.80 (br s, 1H, NH). 13C-NMR (125 MHz, acetone-d6): δ = 51.8 (CH3O), 55.7 (CH3O), 95.7 (C-8), 109.6 (C-6), 110.9 (C-1), 117.4 (C-4b), 121.6 (C-5), 121.9 (C-4a), 122.1 (C-4), 123.9 (C-3), 126.4 (C-2), 143.0 (C-8a), 144.0 (C-9a), 160.5 (C-2), 167.9 (CO2Me). MS (70 eV): m/z (%) 255 (M+, 100), 240 (22), 224 (44), 212 (61), 196 (38), 181 (33), 153 (67), 126 (15), 84 (20), 51 (21). HRMS (EI): m/z [M+] calcd for C15H13NO3: 255.0895; found: 255.0900.
6-Methyl-2,3-dihydro-1H-carbazol-4(9H)-one (7a). In a threaded ACE glass pressure tube with a sealed Teflon screw cap, under N2 atmosphere, a mixture of 4a (0.10 g, 0.5 mmol) and Pd(AcO)2 (0.034 g, 0.15 mmol) in dry MeCN (2.5 mL) was stirred at 80 °C for 24 h. The solvent was removed under vacuum and the residue purified by column chromatography over silica gel (20 g/g of crude, hexane/EtOAc, 80:20), to give 7a (0.077 g, 78%) as a white solid. Rf 0.15 (hexane/EtOAc, 1:1); mp 281–282 °C [Lit. [69] 280–282 °C]. IR (film): νmax 3154, 2934, 1615, 1469, 1406, 1375, 1213, 1183, 1122, 1070, 1016, 797 cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 2.09 (qu, J = 6.3 Hz, 2H, H-2), 2.38 (s, 3H, CH3), 2.41 (t, J = 6.3 Hz, 2H, H-3), 2.93 (t, J = 6.3 Hz, 2H, H-1), 6.97 (br d, J = 8.1 Hz, 1H, H-7), 7.27 (d, J = 8.1 Hz, 1H, H-8), 7.77 (br s, 1H, H-5), 11.75 (br s, 1H, NH) 13C-NMR (75.4 MHz, DMSO-d6): δ = 21.2 (CH3), 22.7 (C-1), 23.4 (C-2), 37.8 (C-3), 111.1 (C-8), 111.4 (C-4a), 120.1 (C-5), 123.7 (C-7), 124.7 (C-4b), 130.2 (C-6), 134.1 (C-8a), 152.2 (C-9a), 192.8 (C-4). MS (70 eV): m/z (%) 199 (M+, 100), 198 (45), 183 (13), 170 (11), 154 (20), 128 (8), 91 (40).
6-Methoxy-2,3-dihydro-1H-carbazol-4(9H)-one (7b). Following the procedure described for 7a, with 4b (0.100 g, 0.46 mmol) and Pd(AcO)2 (0.0309 g, 0.138 mmol), 7b (0.08 g, 80%) was obtained as a white solid. Rf 0.13 (hexane/EtOAc, 1:1); mp 252–253 °C [Lit. [70] 250–254 °C]. IR (KBr): νmax 3416, 1578, 1482, 1459, 1259, 1217, 1175, 1031, 796, 780 cm−1. 1H-NMR (300 MHz, DMSO-d6): δ = 2.09 (qu, J = 6.3 Hz, 2H, H-2), 2.41 (t, J = 6.3 Hz, 2H, H-3), 2.92 (t, J = 6.3 Hz, 2H, H-1), 3.76 (s, 3H, CH3O), 6.77 (dd, J = 8.7, 2.7 Hz, 1H, H-7), 7.28 (d, J = 8.7 Hz, 1H, H-8), 7.45 (d, J = 2.7 Hz, 1H, H-5), 11.74 (br s, 1H, NH) 13C-NMR (75.4 MHz, DMSO-d6): δ = 22.8 (C-1), 23.4 (C-2), 37.7 (C-3), 55.2 (CH3O), 102.4 (C-5), 111.6 (C-7), 111.7 (C-4a), 112.2 (C-8), 125.2 (C-4b), 130.5 (C-8a), 152.3 (C-9a), 155.1 (C-6), 192.8 (C-4). MS (70 eV): m/z (%) 215 (M+, 2), 155 (37), 153 (100), 127 (12), 125 (35), 90 (23).
6-Methyl-9H-carbazol-4-ol (8). In a threaded ACE glass pressure tube with a sealed Teflon screw cap, under N2 atmosphere, a mixture of 7a (0.20 g, 1.0 mmol) and Pd/C (10%) (0.060 g, 0.057 mmol) in dry MeOH (2.5 mL) was stirred at 270 °C for 48 h. The solvent was removed under vacuum and the residue purified by column chromatography over silica gel (20 g/g of crude, hexane/EtOAc, 80:20), to give 8 (0.168 g, 85%) as a white solid. Rf 0.30 (hexane/EtOAc, 7:3); mp 125–126 °C. IR (film): νmax 3404, 1615, 1589, 1455, 1341, 1297, 1267, 1047, 803, 752, 724 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.52 (s, 3H, CH3), 5.38 (br s, 1H, OH), 6.53 (d, J = 8.5 Hz, 1H, H-3), 6.95 (d, J = 8.5 Hz, 1H, H-1), 7.17–7.22 (m, 2H, H-2, H-7), 7.26 (t, J = 8.5 Hz, 1H, H-8), 7.89 (br s, 1H, NH), 8.06 (br s, 1H, H-5). 13C-NMR (125 MHz, CDCl3): δ = 21.4 (CH3), 103.3 (C-1), 104.9 (C-3), 109.7 (C-8), 111.6 (C-4a), 122.5 (C-4b), 122.7 (C-5), 126.3 (C-2 or C-7), 126.4 (C-7 or C-2), 129.0 (C-6), 137.0 (C-8a), 141.7 (C-9a), 151.8 (C-4). HRMS (EI): m/z [M+] calcd for C13H11NO: 197.0841; found: 197.0844.
5-Methoxy-3-methyl-9H-carbazole (Glycoborine, Glycrophylamine, 9). A mixture of 8 (0.150 g, 0.76 mmol), MeI (0.216 g, 1.52 mmol) and K2CO3 (0.157 g, 1.14 mmol) in dry acetone (10 mL) was heated to reflux for 2 h. The solvent was removed under vacuum and the residue purified by column chromatography over silica gel (10 g/g of crude, hexane/EtOAc, 95:5), to give 9 (0.151 g, 94%) as a white solid. Rf 0.35 (hexane/EtOAc, 8:2); mp 133–134 °C [Lit. [33] 154–156 °C; [60] 132–134.6 °C; [61] 155–156 °C; [62] 135 °C]. IR (KBr): νmax 3402, 1586, 1508, 1458, 1346, 1261, 1103, 804, 719 cm−1. 1H-NMR (500 MHz, CDCl3): δ = 2.52 (s, 3H, CH3), 4.06 (s, 3H, CH3O), 6.64 (d, J = 8.0 Hz, 1H, H-6), 6.97 (d, J = 8.0 Hz, 1H, H-8), 7.18 (dd, J = 8.0, 1.2 Hz, 1H, H-2), 7.24 (d, J = 8.0 Hz, 1H, H-1), 7.29 (t, J = 8.0 Hz, 1H, H-7), 8.00 (br s, 1H, NH), 8.11 (br s, 1H, H-4). 13C-NMR (125 MHz, CDCl3): δ = 21.4 (CH3), 55.3 (CH3O), 100.1 (C-6), 103.5 (C-8), 109.5 (C-1), 112.4 (C-4b), 122.8 (C-4a), 122.9 (C-4), 126.1 (C-2), 126.4 (C-7), 128.8 (C-3), 136.9 (C-9a), 141.2 (C-8a), 156.2 (C-5). HRMS (EI): m/z [M+] calcd for C14H13NO: 211.0997; found: 211.0995.
4. Conclusions
In this work, a short and efficient synthetic route for the construction of 2-, 5-, and 7-oxygenated carbazole alkaloids including natural clausine V (1d) is described. As the key steps, this approach includes a palladium(0)-catalyzed aromatization and a palladium(II)-catalyzed cyclization to provide the 2- and 7-oxygenated tricyclic carbazole framework. In the case of the natural 5-oxygenated carbazole glycoborine (glycrophylamine, 9), the palladium-catalyzed sequence was inverted, with cyclization performed before aromatization. The preparation of natural carbazoles clauszoline-K (1f) and clauszoline-L (1g) was also carried out by transformation of carbazole 1h. This methodology is currently being applied to the synthesis of diverse carbazoles, and the results will be reported in due course.
Acknowledgments
We thank Professors Francisco Delgado and Gerardo Zepeda for their help in spectrometric analyses, and Bruce A. Larsen for reviewing the use of English in the manuscript. J.T. acknowledges SIP/IPN (Grants 20110172, 20120830, and 20130686) and CONACYT (Grants 83446 and 178319) for financial support. R.B., P.A.M., and A.R. thank CONACYT for awarding them graduate scholarships, and SIP/IPN (PIFI) for scholarship complements. J.T. and E.B. are fellows of the EDI-IPN and COFAA-IPN programs.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Knölker, H.J.; Reddy, K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002, 102, 4303–4427. [Google Scholar] [CrossRef]
- Knölker, H.J.; Reddy, K.R. Chemistry and Biology of Carbazole Alkaloids. In The Alkaloids Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2008; Volume 65. [Google Scholar]
- Schmidt, A.W.; Reddy, K.R.; Knölker, H.J. Occurrence, biogenesis, and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2012, 112, 3193–3328. [Google Scholar] [CrossRef]
- Knölker, H.J. Occurrence, biological activity, and convergent organometallic synthesis of carbazole alkaloids. Top. Curr. Chem. 2005, 244, 115–148. [Google Scholar]
- Knölker, H.J. Synthesis of biologically active carbazole alkaloids using selective transition-metal-catalyzed coupling reactions. Chem. Lett. 2009, 38, 8–13. [Google Scholar] [CrossRef]
- Chakraborty, D.P. Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G.W., Eds.; Springer-Verlag: Wien, Austria, 1977; Volume 34. [Google Scholar]
- Chaichantipyuth, C.; Pummangura, S.; Naowsaran, K.; Thanyavuthi, D. Two new bioactive carbazole alkaloids from the rootbark of clausena harmandiana. J. Nat. Prod. 1988, 51, 1285–1288. [Google Scholar] [CrossRef]
- Wu, T.S.; Huang, S.C.; Wu, P.L.; Teng, C.M. Carbazole alkaloids from Clausena excavata and their biological activity. Phytochemistry 1996, 43, 133–140. [Google Scholar]
- Sunthitikawinsakul, A.; Kongkathip, N.; Kongkathip, B.; Phonnakhu, S.; Daly, J.W.; Spande, T.F.; Nimit, Y.; Rochanaruangrai, S. Coumarins and carbazoles from Clausena excavata exhibited antimycobacterial and antifungal activities. Planta Med. 2003, 69, 155–157. [Google Scholar] [CrossRef]
- Ma, C.; Case, R.J.; Wang, Y.; Zhang, H.J.; Tan, G.T.; Hung, N.V.; Cuong, N.M.; Franzblau, S.G.; Soejarto, D.D.; Fong, H.H.S.; Pauli, G.F. Anti-tuberculosis constituents from the stem bark of Micromelum hirsutum. Planta Med. 2005, 71, 261–267. [Google Scholar] [CrossRef]
- Meragelman, K.M.; McKee, T.C.; Boyd, M.R. Siamenol, a new carbazole alkaloid from Murraya siamensis. J. Nat. Prod. 2000, 63, 427–428. [Google Scholar] [CrossRef]
- Kongkathip, B.; Kongkathip, N.; Sunthitikawinsakul, A.; Napaswat, C.; Yoosook, C. Anti-HIV-1 constituents from Clausena excavata: Part II. Carbazoles and a pyranocoumarin. Phytother. Res. 2005, 19, 557–731. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, Y.; Efferth, T.; Wang, R.; Shen, Y.; Hao, X. Indole and carbazole alkaloids from Glycosmis montana with weak anti-HIV and cytotoxic activities. Phytochemistry 2005, 66, 697–701. [Google Scholar] [CrossRef]
- Yenjai, C.; Sripontan, S.; Sriprajun, P.; Kittakoop, P.; Jintasirikul, A.; Tanticharoen, M.; Thebtaranonth, Y. Coumarins and carbazoles with antiplasmodial activity from Clausena harmandiana. Planta Med. 2000, 66, 277–279. [Google Scholar] [CrossRef]
- Shoeb, A.; Anwer, F.; Kapil, R.S.; Popli, S.P. N-Alkylaminocarbazoles as potential anticonvulsant and diuretic agents. J. Med. Chem. 1973, 16, 425–427. [Google Scholar] [CrossRef]
- Ferris, R.M.; White, H.L.; Tang, F.L.M.; Russell, A.; Harfenist, M. Rimcazole (BW 234U), a novel antipsychotic agent whose mechanism of action cannot be explained by a direct blockade of postsynaptic dopaminergic receptors in brain. Drug Dev. Res. 1986, 9, 171–188. [Google Scholar] [CrossRef]
- Gilmore, D.L.; Liu, Y.; Matsumoto, R.R. Review of the pharmacological and clinical profile of rimcazole. CNS Drug Rev. 2004, 10, 1–22. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Chakraborty, A. Mukonal, a probable biogenetic intermediate of pyranocarbazole alkaloids from Murraya koenigii. Phytochemistry 1984, 23, 471–472. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Bhattacharyya, P.; Islam, A.; Roy, M.S. Structure of murrayacinine: A new carbazole alkaloid from Murraya koenigii spreng. Chem. Ind. 1974, 1974, 165–166. [Google Scholar]
- Wu, T.S.; Huang, S.C.; Wu, P.L. Carbazole alkaloids from stem bark of Clausena excavata. Phytochemistry 1996, 43, 1427–1429. [Google Scholar] [CrossRef]
- Ito, C.; Nakagawa, M.; Wu, T.-S.; Furukawa, H. New carbazole alkaloids from Murraya euchrestifolia. Chem. Pharm. Bull. 1991, 39, 2525–2528. [Google Scholar] [CrossRef]
- Wu, T.S.; Huang, S.C.; Wu, P.L.; Kuoh, C.S. Alkaloidal and other constituents from the root bark of Clausena excavata. Phytochemistry 1999, 52, 523–527. [Google Scholar]
- Ito, C.; Katsuno, S.; Ohta, H.; Omura, M.; Kajiura, I.; Furukawa, H. Constituents of Clausena excavate. Isolation and structural elucidation of new carbazole alkaloids. Chem. Pharm. Bull. 1997, 45, 48–52. [Google Scholar] [CrossRef]
- Knölker, H.J.; Wolpert, M. Transition metal complexes in organic synthesis. Part 68: Iron-mediated total synthesis of mukonine and mukonidine by oxidative cyclization with air as the oxidizing agent. Tetrahedron 2003, 59, 5317–5322. [Google Scholar] [CrossRef]
- St. Jean, D.J., Jr.; Poon, S.F.; Schwarzbach, J.L. A tandem cross-coupling/SNAr approach to functionalized carbazoles. Org. Lett. 2007, 9, 4893–4896. [Google Scholar] [CrossRef]
- Forke, R.; Krahl, M.P.; Däbritz, F.; Jäger, A.; Knölker, H.J. Transition metals in organic synthesis, Part 87: An efficient palladium-catalyzed route to 2-oxygenated and 2,7-dioxygenated carbazole alkaloids—Total synthesis of 2-methoxy-3-methylcarbazole, glycosinine, clausine L, mukonidine, and clausine V. Synlett 2008, 2008, 1870–1876. [Google Scholar] [CrossRef]
- Forke, R.; Jäger, A.; Knölker, H.J. First total synthesis of clausine L and pityriazole, a metabolite of the human pathogenic yeast Malassezia furfur. Org. Biomol. Chem. 2008, 6, 2481–2483. [Google Scholar] [CrossRef]
- Watanabe, T.; Oishi, S.; Fujii, N.; Ohno, H. Palladium-catalyzed direct synthesis of carbazoles via one-pot N-arylation and oxidative biaryl coupling: Synthesis and mechanistic study. J. Org. Chem. 2009, 74, 4720–4726. [Google Scholar] [CrossRef]
- Sridharan, V.; Martín, M.A.; Menéndez, J.C. Acid-free synthesis of carbazoles and carbazolequinones by intramolecular Pd-catalyzed, microwave-assisted oxidative biaryl coupling reactions—Efficient syntheses of murrayafoline A, 2-methoxy-3-methylcarbazole, and glycozolidine. Eur. J. Org. Chem. 2009, 4614–4621. [Google Scholar] [CrossRef]
- Tsang, W.C.P.; Munday, R.H.; Brasche, G.; Zheng, N.; Buchwald, S.L. Palladium-catalyzed method for the synthesis of carbazoles via tandem C-H functionalization and C-N bond formation. J. Org. Chem. 2008, 73, 7603–7610. [Google Scholar] [CrossRef]
- Witulski, B.; Alayrac, C. A highly efficient and flexible synthesis of substituted carbazoles by rhodium-catalyzed inter- and intramolecular alkyne cyclotrimerizations. Angew. Chem. Int. Ed. 2002, 41, 3281–3284. [Google Scholar] [CrossRef]
- Krahl, M.P.; Jäger, A.; Krause, T.; Knölker, H.J. First total synthesis of the 7-oxygenated carbazole alkaloid clauszoline-K, 3-formyl-7-hydroxycarbazole, clausine M, clausine N and the anti-HIV active siamenol using a highly efficient palladium-catalyzed approach. Org. Biomol. Chem. 2006, 4, 3215–3219. [Google Scholar] [CrossRef]
- Kuethe, J.T.; Childers, K.G. Suzuki-miyaura cross-coupling of 2-nitroarenediazonium tetrafluoroborates: Synthesis of unsymmetrical 2-nitrobiphenyls and highly functionalized carbazoles. Adv. Synth. Catal. 2008, 350, 1577–1586. [Google Scholar] [CrossRef]
- Kataeva, O.; Krahl, M.P.; Knölker, H.J. First total synthesis of the biologically active 2,7-dioxygenated tricyclic carbazole alkaloids 7-methoxy-O-methylmukonal, clausine H (clauszoline-C), clausine K (clauszoline-J) and clausine O. Org. Biomol. Chem. 2005, 3, 3099–3101. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Alonso, J.M.; Fernández, I. Carbocyclization versus oxycyclization on the metal-catalyzed reactions of oxyallenyl C3-linked índoles. J. Org. Chem. 2013, 78, 6688–6701. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Alonso, J.M.; Cembellín, S.; Fernández, I.; Martínez del Campo, T.; Torres, M.R. Iodine recycling via 1,3-migration in iodoindoles under metal catalysis. Chem. Commun. 2013, 49, 7779–7781. [Google Scholar] [CrossRef]
- Bautista, R.; Jerezano, A.V.; Tamariz, J. Synthetic approach for constructing the 1-oxygenated carbazole core and its application to the preparation of natural alkaloids. Synthesis 2012, 44, 3327–3336. [Google Scholar] [CrossRef]
- Knölker, H.J.; Fröhner, W. Palladium-catalyzed total synthesis of the antibiotic carbazole alkaloids carbazomycin G and H. J. Chem. Soc. Perkin Trans. 1 1998, 1998, 173–175. [Google Scholar]
- Knölker, H.J.; Reddy, K.R. Indoloquinones, Part 6. First palladium-mediated oxidative cyclization of arylamino-1,2-benzoquinones to carbazole-3,4-quinones—Application to the total synthesis of carbazoquinocin C and (±)-carquinostatin A. Synlett 1999, 1999, 596–598. [Google Scholar] [CrossRef]
- Knölker, H.J.; O’Sullivan, N. Palladium-promoted synthesis of hydroxy-substituted 5-cyano-5H-benzo[b]carbazole-6,11-diones. Tetrahedron 1994, 50, 10893–10908. [Google Scholar] [CrossRef]
- Hagelin, H.; Oslob, J.D.; Åkermark, B. Oxygen as oxidant in palladium-catalyzed inter- and intramolecular coupling reactions. Chem. Eur. J. 1999, 5, 2413–2416. [Google Scholar] [CrossRef]
- Sissouma, D.; Maingot, L.; Collet, S.; Guingant, A. Concise and efficient synthesis of calothrixin B. J. Org. Chem. 2006, 71, 8384–8389. [Google Scholar] [CrossRef]
- Iida, H.; Yuasa, Y.; Kibayashi, C. A convenient synthesis of m-aminophenols by mercury(II) acetate oxidation of 3-amino-2-cyclohexenones. Synthesis 1982, 1982, 471–472. [Google Scholar] [CrossRef]
- Bautista, R.; Bernal, P.; Montiel, L.E.; Delgado, F.; Tamariz, J. Total synthesis of the natural carbazoles glycozolicine, mukoline, and mukolidine, starting from 4,5-dimethyleneoxazolidin-2-ones. Synthesis 2011, 2011, 929–933. [Google Scholar] [CrossRef]
- Mithani, S.; Weeratunga, G.; Taylor, N.J.; Dmitrienko, G.I. The kinamycins are diazofluorenes and not cyanocarbazoles. J. Am. Chem. Soc. 1994, 116, 2209–2210. [Google Scholar] [CrossRef]
- Yogo, M.; Ito, C.; Furukawa, H. Synthesis of some carbazolequinone alkaloids and their analogues. Facile palladium-assisted intramolecular ring closure of arylamino-1,4-benzoquinones to carbazole-1,4-quinones. Chem. Pharm. Bull. 1991, 39, 328–334. [Google Scholar] [CrossRef]
- Saha, C.; Chowdhury, B.K. Carbazoloquinones from Murraya koenigii. Phytochemistry 1998, 48, 363–366. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, A. The first synthesis of optically pure biscarbazoles and determination of their absolute configurations. Tetrahedron Lett. 1999, 40, 341–344. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.M.; Almendros, P. Fused carbazoles by tandem aza Wittig/electrocyclic ring closure, preparation of 6H-pyrido[4,3-b]carbazole, 11H-pyrido[4,3-a]carbazole and 11H-pyrido[3,4-a]carbazole derivatives. Tetrahedron 1993, 49, 1223–1236. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Chowdhury, B.K. Synthesis of murrayanine. J. Org. Chem. 1968, 33, 1265–1268. [Google Scholar] [CrossRef]
- Murakami, Y.; Yokoo, H.; Watanabe, T. New syntheses of murrayaquinone—A via Fischer indolization of 2-sulfonyloxyphenylhydrazone (Fischer indolization and its related compounds. Part 29). Heterocycles 1998, 49, 127–132. [Google Scholar] [CrossRef]
- Chakraborty, D.P.; Islam, A.; Bhattacharyya, P. Synthesis of murrayanine. J. Org. Chem. 1973, 38, 2728–2729. [Google Scholar] [CrossRef]
- Murphy, W.S.; Bertrand, M. Bromoquinone-enaminone annulations: Syntheses of murrayaquinone-A and (±)-bismurrayaquinone-A. J. Chem. Soc. Perkin Trans. 1 1998, 1998, 4115–4119. [Google Scholar] [CrossRef]
- Crum, J.D.; Sprague, P.W. The synthesis of murrayanine. Chem. Commun. 1966, 1966, 417–418. [Google Scholar]
- Sissouma, D.; Collet, S.C.; Guingant, A.Y. A synthesis of calothrixin B. Synlett 2004, 2004, 2612–2614. [Google Scholar]
- Forke, R.; Krahl, M.P.; Krause, T.; Schlechtingen, G.; Knölker, H.J. Transition metals in organic synthesis, Part 82. First total synthesis of methyl 6-methoxycarbazole-3-carboxylate, glycomaurrol, the anti-TB active micromeline, and the furo[2,3-c]carbazole alkaloid eustifoline-D. Synlett 2007, 2007, 268–272. [Google Scholar] [CrossRef]
- Schmidt, M.; Knölker, H.J. Transition metals in organic synthesis, Part 91: Palladium-catalyzed approach to 2,6-dioxygenated carbazole alkaloids—First total synthesis of the phytoalexin carbalexin C. Synlett 2009, 2009, 2421–2424. [Google Scholar] [CrossRef]
- Knölker, H.J.; Goesmann, H.; Hofmann, C. Transition metal complexes in organic synthesis, Part 31. A novel molybdenum-mediated synthesis of carbazole derivatives: Application to the total synthesis of mukonal and 1,1′-bis(2-hydroxy-3-methylcarbazole). Synlett 1996, 1996, 737–740. [Google Scholar] [CrossRef]
- Roy, S.; Bhattacharyya, L.; Chakraborty, D.P. Structure and synthesis of mukoline and mukolidine, two new carbazole alkaloids from Murraya koenigii spreng. J. Indian Chem. Soc. 1982, 59, 1369–1371. [Google Scholar]
- Cheenpracha, S.; Laphookhieo, S. Alkaloids and amides from Glycosmis macrophylla. Phytochem. Lett. 2011, 4, 187–189. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Sarkar, T.; Masuda, K.; Takey, T.; Doi, H.; Kotani, E.; Shiojima, K. Structure and synthesis of glycoborine, a new carbazole alkaloid from the roots of Glycosmis arborea: A note on the structure of glycozolicine. Indian J. Chem. 2001, 40B, 484–489. [Google Scholar]
- Jash, S.S.; Biswas, G.K.; Bhattacharyya, S.K.; Bhattacharyya, P.; Chakraborty, A.; Chowdhury, B.K. Carbazole alkaloids from Glycosmis pentaphylla. Phytochemistry 1992, 31, 2503–2505. [Google Scholar] [CrossRef]
- Makawana, J.A.; Patel, M.P.; Patel, R.G. Synthesis and in vitro antimicrobial activity of N-arylquinoline derivatives bearing 2-morpholinoquinoline moiety. Chinese Chem. Lett. 2012, 23, 427–430. [Google Scholar] [CrossRef]
- Iida, H.; Yuasa, Y.; Kibayashi, C. Intramolecular cyclization of enaminones involving arylpalladium complexes. Synthesis of carbazoles. J. Org. Chem. 1980, 45, 2938–2942. [Google Scholar] [CrossRef]
- Andrade, C.K.Z.; Barreto, A.F.S.; Wender, A.S. Microwave assisted solvent-, support- and catalyst-free synthesis of enaminones. Arkivoc 2008, xii, 226–232. [Google Scholar]
- Maiti, D.; Buchwald, S.L. Orthogonal Cu- and Pd-based catalyst systems for the O- and N-arylation of aminophenols. J. Am. Chem. Soc. 2009, 131, 17423–17429. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Xu, J.H.; Verkade, J.G. Application of a new bicyclic triaminophosphine ligand in Pd-catalyzed Buchwald-Hartwig amination reactions of aryl chlorides, bromides, and iodines. J. Org. Chem. 2003, 68, 8416–8423. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Kapil, R.S. Synthesis of O-methylcycloclausanitin. Indian J. Chem. Soc. 1984, 23B, 296–299. [Google Scholar]
- Oikawa, Y.; Yonemitsu, O. Selective oxidation of the side chain at C-3 of indoles. J. Org. Chem. 1977, 42, 1213–1216. [Google Scholar] [CrossRef]
- Weng, B.; Liu, R.; Li, J.-H. An improved method for the synthesis of carbazolones by palladium/copper-catalyzed intramolecular annulation of N-arylenaminones. Synthesis 2010, 2010, 2926–2930. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4a–e, 5a, 5d, 5e, 1d, 1f–g, 1h–i, and 1k are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).