1. Introduction
Glycosylated peptides play a fundamental role in biological systems: in fact, more than half of all proteins carry carbohydrate moieties, generating different glycoforms whose exact composition often controls protein function and distribution in biological systems [
1,
2,
3,
4]. The details of glycan regulation of protein activity and stability are still under intense scrutiny and the synthesis of well-defined glycopeptides is therefore an important target [
5,
6], which still presents many challenges to organic chemistry. One of the processes that is currently being elucidated is adenosine diphosphate ribosylation (ADP-ribosylation), a wide-occurring post-translational modification effected by enzymes that transfer ADP-ribose from NAD
+ to Asn, Glu, Asp, Arg or Cys residues of proteins, so altering their function [
7]. An important contribution for the clarification of the role of ADP-ribosylation events came in 2010, when Filippov and coworkers [
8] reported the synthesis of ribosylated oligopeptides using α-
N-ribosyl-asparagine (α-
N-ribosyl-Asn) and α-
N-ribosyl-glutamine (α-
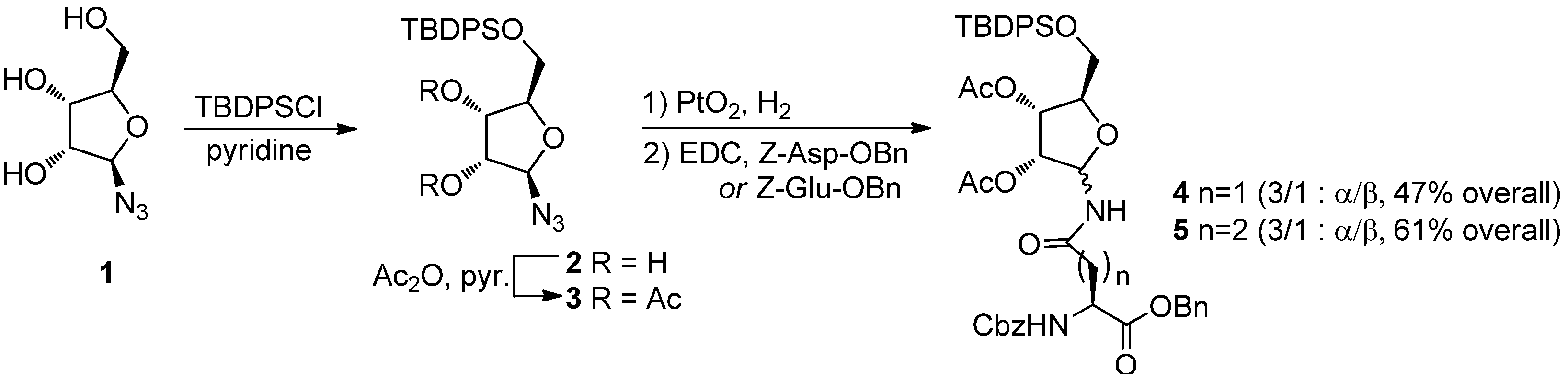
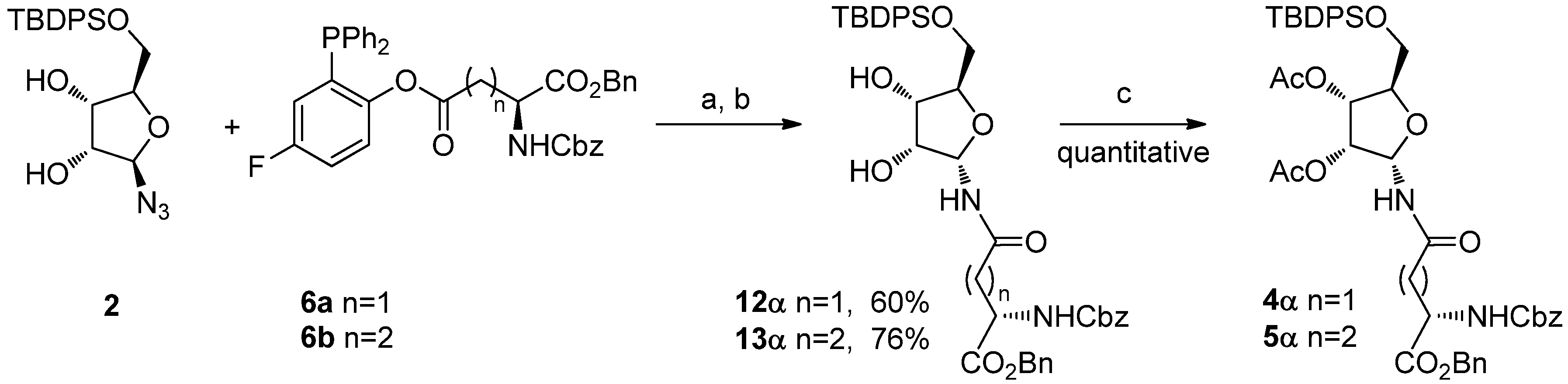
N-ribosyl-Gln) building blocks. Despite the relevance of this work, the approach suffers from a poorly selective synthesis of the required ribosylated building blocks
4α and
5α (
Scheme 1), which were prepared by PtO
2 reduction of azide
3, followed by EDC-mediated coupling of the resulting mixture of epimeric amines with Z-Asp-OBn or Z-Glu-OBn, respectively. The
N-ribosyl-aminoacids were obtained as a 3:1 α: β anomeric mixtures, from which the desired α-anomers were to be chromatographically isolated. Acetyl transfer to the anomeric nitrogen from the 2-
O-acetate also occurs during the reaction, further reducing the process yields, particularly for the asparagine derivative
4α.
Scheme 1.
Filippov’s synthesis of ribosylated amino acids (from ref. [
8]).
Scheme 1.
Filippov’s synthesis of ribosylated amino acids (from ref. [
8]).
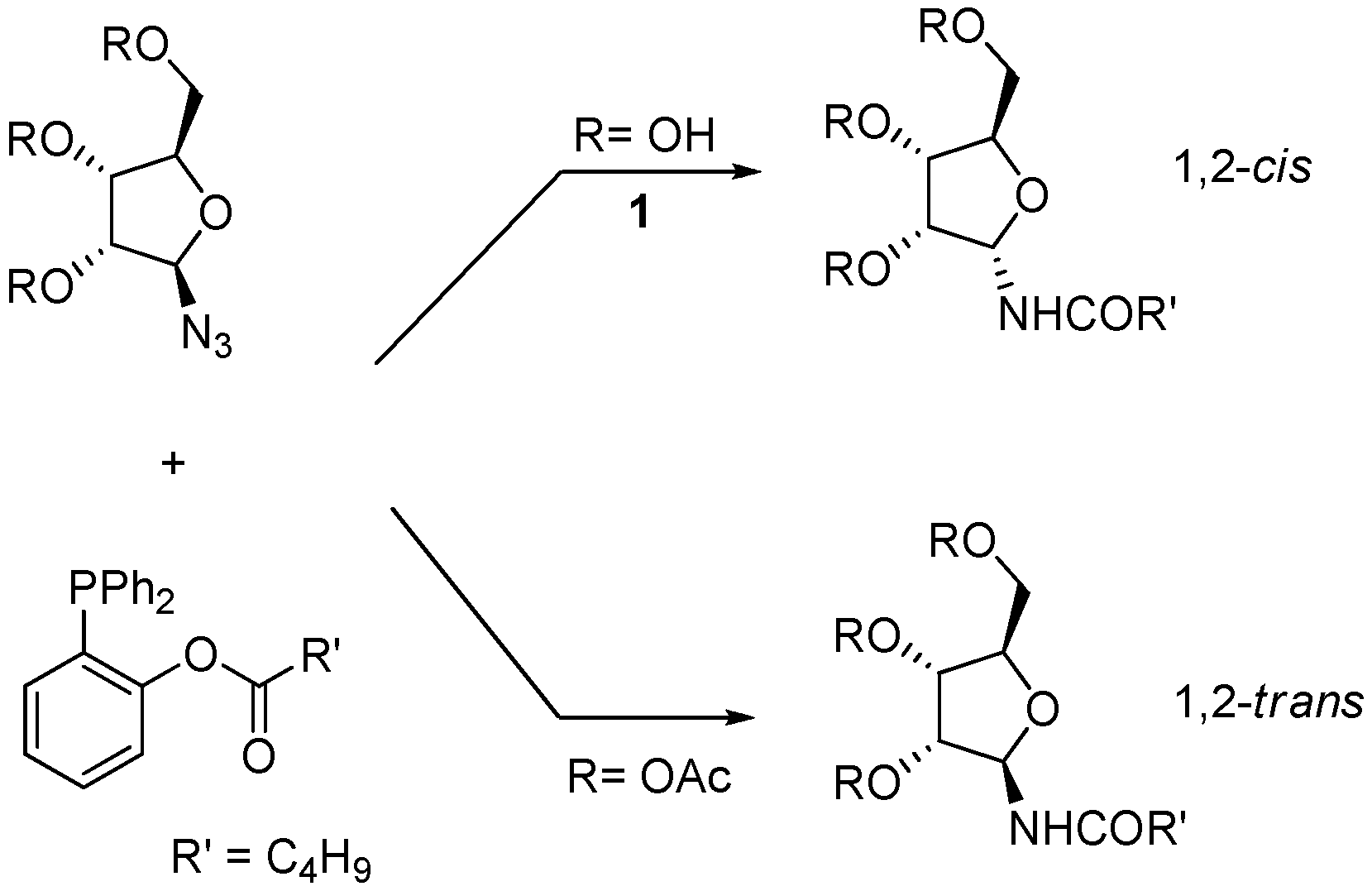
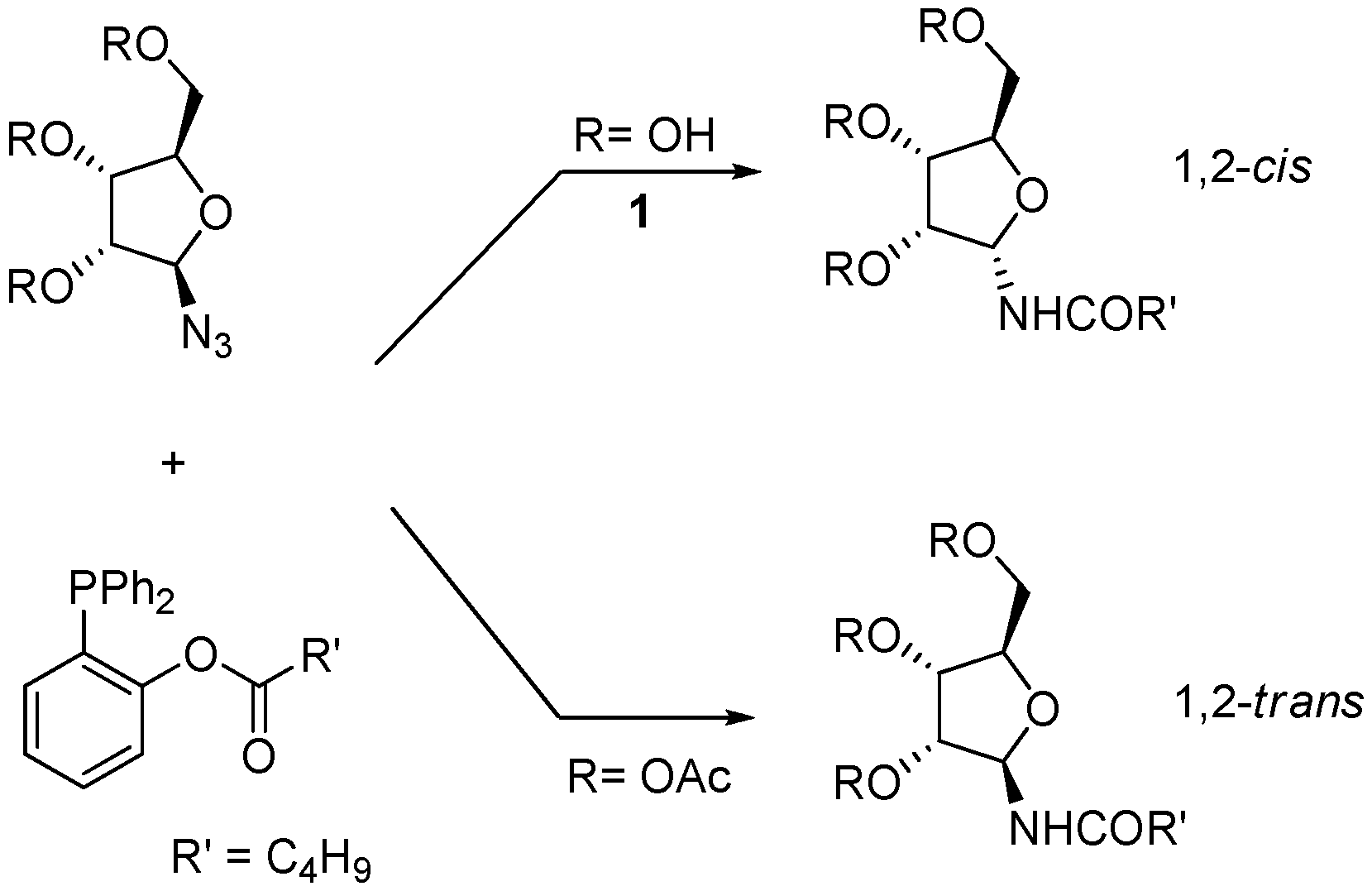
We have recently developed a protocol for the stereoselective synthesis of either α- or β-
N-glycofuranosyl amides using the Staudinger traceless ligation [
9,
10]. In this reaction, a starting anomeric glycosyl azide is partially reduced by an appropriate phosphine and then intramolecularly acylated in order to afford the amide (
Scheme 2) [
11]. We have shown that for furanosyl azides the outcome of the process is controlled by the configuration and the protection state of the contiguous hydroxyl group, so that 1,2-
cis amides (α-, in the
ribo series) are obtained when this group is unprotected and 1,2-
trans amides (β-, in the
ribo series) when it is acetylated. For unprotected furanoses, the anomeric configuration of the product amides appears to be dictated by O-P coordination, as supported by
31P-NMR studies [
10].
Scheme 2.
Traceless Staudinger ligation of furanosyl azides with functionalized phosphines (from ref [
9]).
Scheme 2.
Traceless Staudinger ligation of furanosyl azides with functionalized phosphines (from ref [
9]).
As an application of this methodology, we here report the facile stereoselective synthesis of 4α and 5α that could be obtained in good yields and with full stereoselectivity with a single synthetic operation starting from β-5-O-tert-butyldiphenylsilyl-ribosylazide 2.
2. Results and Discussion
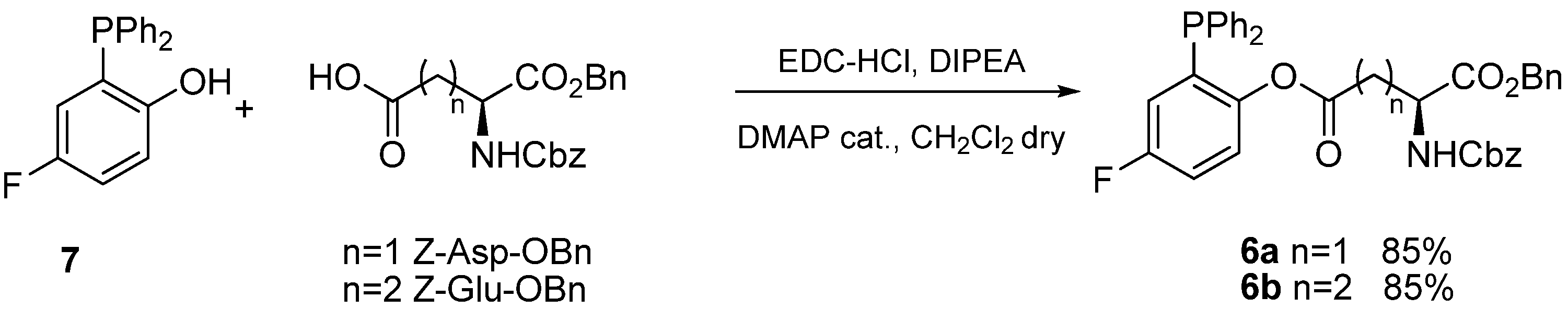
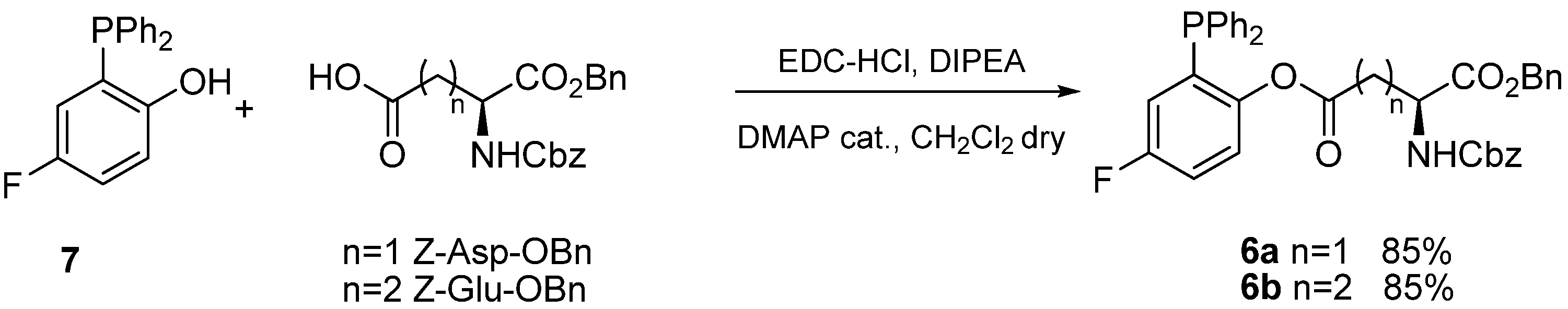
Previous research in our laboratories had shown that transfer of aminoacid chains in Staudinger ligations of glycosyl azides was particularly effective using fluorinated phosphines
6 [
12]. Thus we envisaged that phosphines
6a and
6b (
Scheme 3), functionalized with Z-Asp-OBn and Z-Glu-OBn acyl chains, respectively, would be the reagent of choice to prepare the target α-
N-ribosyl-Asn and α-
N-ribosyl-Gln building blocks. Phosphines
6a and
6b were synthesized in excellent yields by EDC-mediated acylation of 2-diphenylphosphanyl-5-fluorophenol (
7) [
13] and purified by filtration on short pads of silica.
Scheme 3.
Synthesis of functionalized phosphines 6a and 6b.
Scheme 3.
Synthesis of functionalized phosphines 6a and 6b.
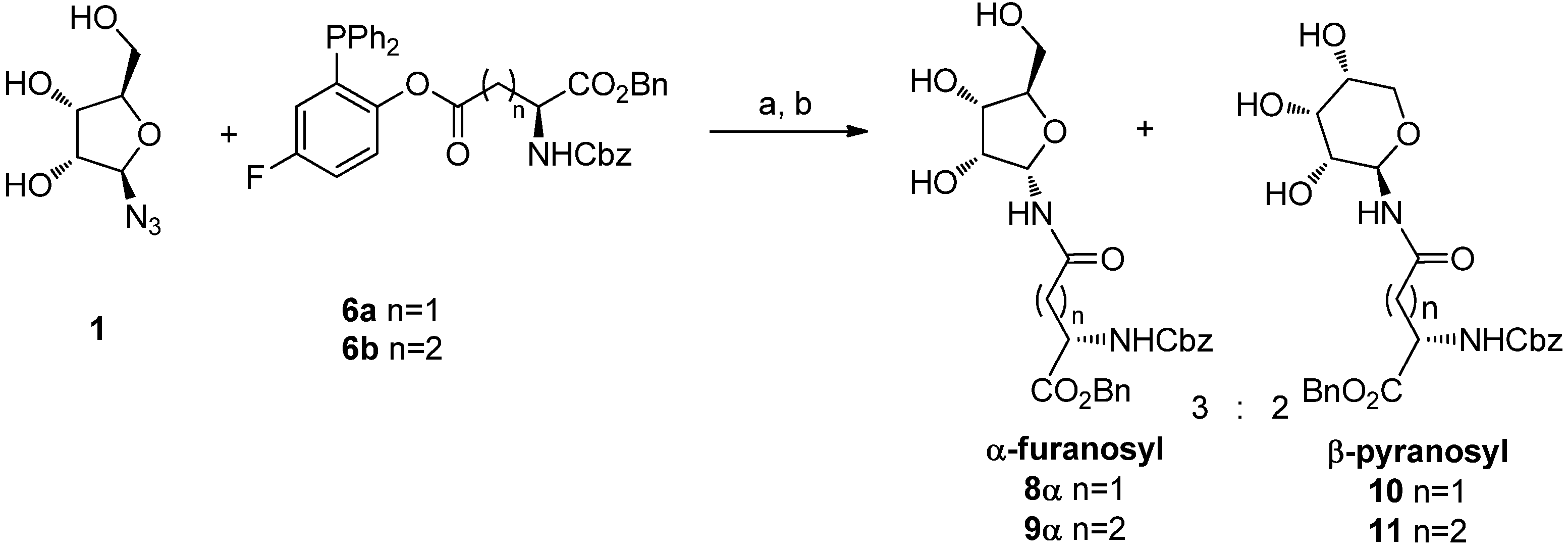
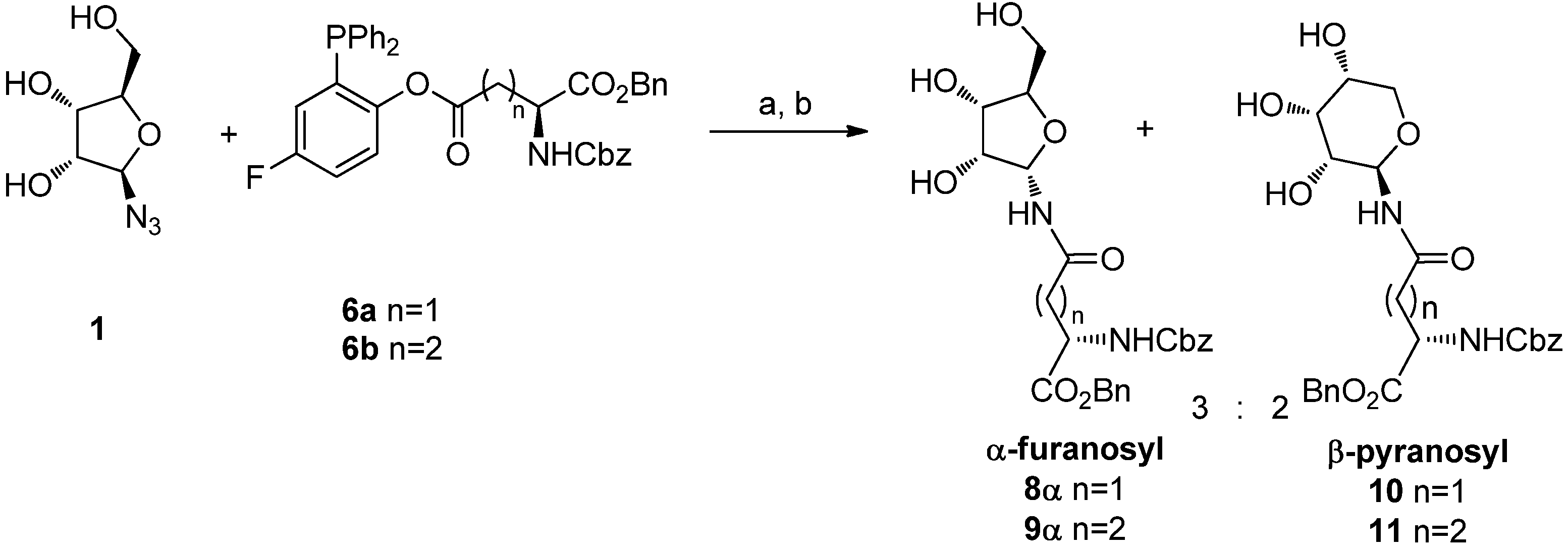
An early trial of ligation was conducted using the unprotected β-
d-ribofuranosyl azide
1 [
14] with phosphines
6a and
6b for 20 h at 70 °C in a 98:2 DMA:DMPU [
15] solution, followed by water quenching (
Scheme 4). In both ligation reactions 45% of a 3:2 mixture of α-ribofuranosyl amide (
8α or
9α) and β-ribopyranosyl amide (
10 or
11) was obtained. The isomer distribution was determined by
1H-NMR analysis of the crude reaction mixtures: the α-ribofuranosyl amide (
8α or
9α) was characterized by the presence of the H1 proton signal at 5.65 ppm and by the high chemical shift of C4 (84.3 ppm), which is diagnostic for the furanose form. The structures of
10 and
11 were assigned on the basis of the vicinal coupling constant of the anomeric protons (
J1,2 = 8 Hz, H1 = 5.12 ppm) and of the low chemical shift value of C4 (68.8 ppm), that confirms the pyranose structure. The β-glycopyranosyl isomers
10 and
11 obtained in these reactions must derive from ring-expansion occurring after a ring-opening process.
Scheme 4.
Ligation of 1 with phosphines 6a and 6b.
Scheme 4.
Ligation of 1 with phosphines 6a and 6b.
Reagents and Conditions: DMA/DMPU 98:2, 70 °C, 20 h; (b) H2O, 2 h, 70 °C.
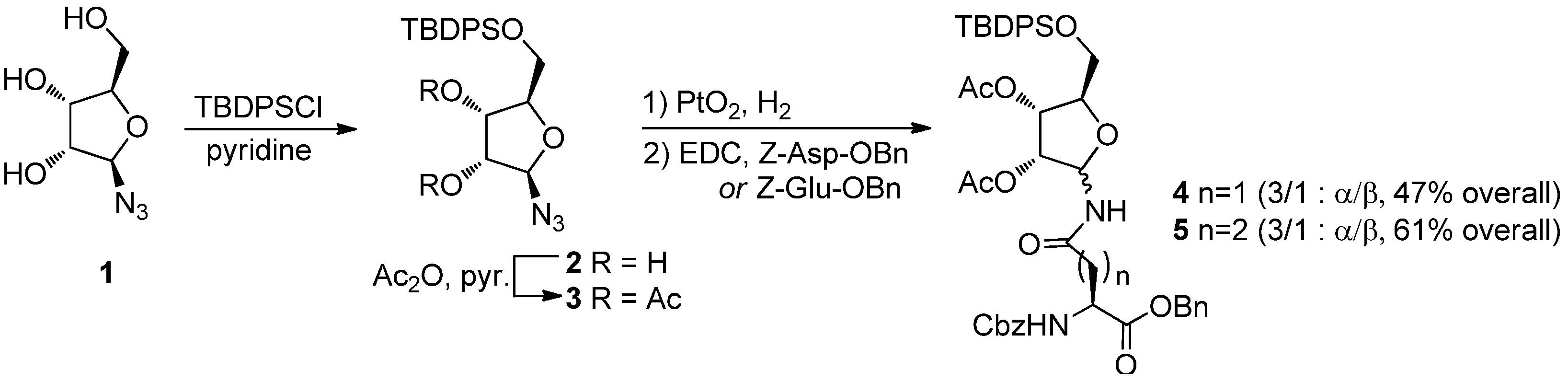
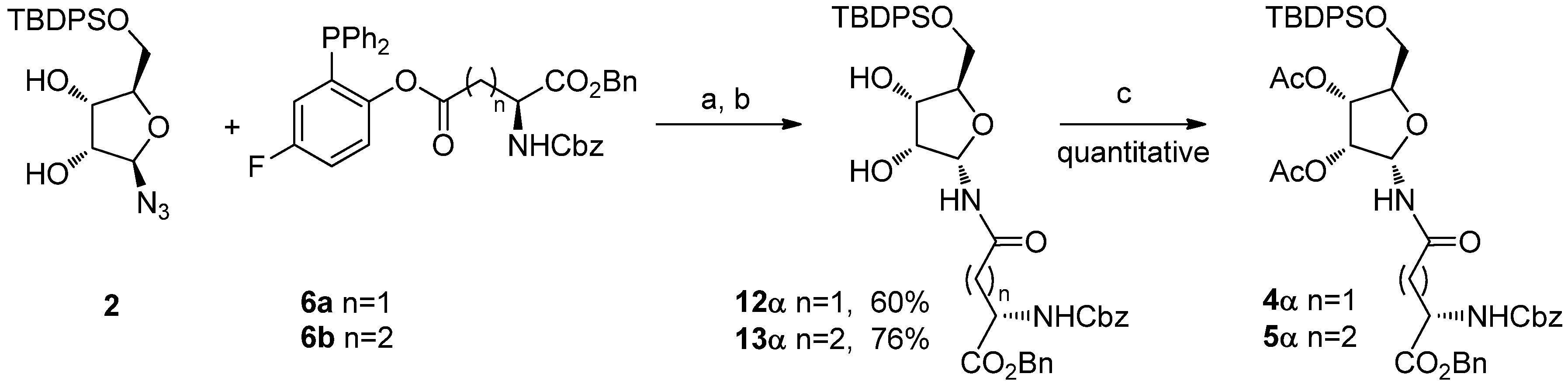
To avoid ring expansion, the 5-hydroxy group of
1 was protected as a
tert-butyldiphenylsilyl ether and the 5-
tert-butyldiphenylsilyl-β-D-ribofuranosyl azide
2 was synthesized in good yields as described by Filippov [
8]. Ligation of
2 with
6a or
6b was carried out as above (20 h at 70 °C in a 98:2 DMA: DMPU mixture) (
Scheme 5). In both cases, 400 MHz
1H-NMR analysis of the crude showed only one signal in the furanose anomeric region, implying that no pyranose was formed, as expected, and that the α/β ratio of the resulting ribofuranosyl amides
12α and
13α was ≥ 99:1. The two compounds were isolated by flash chromatography, which afforded the desired building blocks in 60% and 76% yields, respectively, as single α isomers. After acetylation (Ac
2O, cat. DMAP, CH
2Cl
2, quant.),
4α and
5α were obtained. Their spectroscopic data were fully consistent with the already published characterization [
8].
Scheme 5.
Synthesis of ribofuranosyl amides 4α and 5α.
Scheme 5.
Synthesis of ribofuranosyl amides 4α and 5α.
Reagents and Conditions: DMA/DMPU 98:2, 70 °C, 20 h; (b) H2O, 2 h, 70 °C; (c) Ac2O, cat. DMAP, CH2Cl2.
3. Experimental
Solvents were dried by standard procedures: dichloromethane and N,N-diisopropylethylamine were dried over calcium hydride; N,N-dimethylacetamide (DMA), 1,3-dimethyltetrahydro-2(1H)-pyrimidinone (DMPU), chloroform and pyridine were dried over activated molecular sieves. Reactions requiring anhydrous conditions were performed under nitrogen. 1H-, 13C- and 31P-NMR spectra were recorded at 400 MHz on a Bruker AVANCE-400 instrument. Chemical shifts (δ) for 1H and 13C spectra are expressed in ppm relative to internal Me4Si as standard. Chemical shifts (δ) for 31P are expressed in ppm relative to internal H3PO4 as standard. Signals were abbreviated as s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Mass spectra were obtained with a Bruker ion-trap Esquire 3000 apparatus (ESI ionization) or FT-ICR Mass Spectrometer APEX II & Xmass software (Bruker Daltonics)—4.7 Magnet and Autospec Fission Spectrometer (FAB ionization). Thin layer chromatography (TLC) was carried out with pre-coated Merck F254 silica gel plates. Flash chromatography (FC) was carried out with Macherey-Nagel silica gel 60 (230–400 mesh). The typical scale used for the ligation reactions in this paper was 20 mg (0.1 mmol) of unprotected glycosyl azide.
General Procedure for the Synthesis of 2-(diphenylphosphanyl)-4-fluorophenyl Esters 6
A solution of the o-diphenylphosphinophenol (7, 1 equiv.), the commercially available N-carbobenzyloxy-L-aminoacid-1-benzyl ester (Z-Asp-OBn or Z-Glu-OBn, 1.2 equiv.) and N,N-dimethylaminopyridine (0.1 equiv.) in dry CH2Cl2(0.1 M) were added, at room temperature and under nitrogen, to a suspension of N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC, 1.4 equiv.) and dry N,N-diisopropylethylamine (1.4 equiv.) in dry CH2Cl2. The mixture was stirred at RT for 2 h, monitoring by TLC (60:40 hexane/AcOEt). The reaction mixture was diluted with CH2Cl2 and washed with 5% aqueous HCl and water: the organic layer was dried over Na2SO4 and concentrated. The crude product obtained was purified by flash chromatography, as indicated in each case.
1-Benzyl 5-[2-(Diphenylphosphanyl)-4-fluorophenyl] N-(Benzyloxycarbonyl)-l-aspartate (6a). The crude product was purified by flash chromatography (hexane/AcOEt 60:40). yield = 85%. 1H-NMR (CDCl3, 25 °C): δ = 7.37–7.23 (m, 20H, Ph), 7.02–6.84 (m, 2H, H-2, H-3), 6.49 (m, 1H, H-1), 5.69 (d, JNH-CH = 8.6 Hz, 1H, NH), 5.17 (s, 2H, CH2O), 5.13 (s, 2H, CH2O), 4.62 (m, JCH-CH2 = 4.4 Hz, 1H, CH), 3.00 (dt, J = 4.9 Hz, J = 17.4 Hz, 1H, Ha, CH2), 2.67 (dd, J = 4.3 Hz, J = 17.4 Hz, 1H, Hb, CH2). 13C-NMR (CDCl3, 25 °C): δ = 170.4, 169.2, 161.9, 159.4 (CO(Cbz)), 134.6, 134.0, 132.0, 131.7, 129.0, 128.9, 128.7, 128.6, 128.4, 128.3, 128.2 (CAr), 124.0, 116.9, 116.6, 67.8 (CH2O), 67.3 (CH2O), 50.4 (CH), 36.5 (CH2). 31P-NMR (161 MHz, CDCl3, 25 °C): δ = −14.3 ppm. FAB-MS: m/z 636 (M+1).
1-Benzyl 5-[2-(Diphenylphosphanyl)-4-fluorophenyl] N-(Benzyloxycarbonyl)-l-glutamate (6b). The crude product was purified by flash chromatography (hexane/AcOEt 70:30). yield = 85%. 1H-NMR (CDCl3, 25 °C): δ = 7.37–7.24 (m, 20 H, Ph), 7.05 (m, 1H, H-2), 6.99 (m, 1H, H-3), 6.45 (m, 1H, H-1), 5.37 (d, JNH-CH = 8.1 Hz, 1H, NH), 5.16 (s, 2H, CH2-O), 5.07 (s, 2H, CH2-O), 4.38 (m, 1H, CH), 2.26 (m, 2H, CH2-CO), 2.05 (m, 1H, Ha, CH2-CH), 1.80 (m, 1H, Hb, CH2-CH). 13C-NMR (CDCl3, 25 °C): δ = 170.7, 161.7, 159.3, 156.1 (CO(Cbz)), 134.4, 134.3, 134.2, 134.0, 129.1, 129.0, 128.8, 128.5, 128.4 (CAr), 124.2, 124.1, 120.0, 119.8, 116.8, 116.5, 67.6 (CH2-O), 67.4 (CH2-O), 53.5 (CH), 30.1 (CH2-CO), 27.4 (CH2). 31P-NMR (161 MHz, CDCl3, 25 °C): δ = −14.3 ppm. FAB-MS: m/z 650 (M+1).
General Procedure for the Staudinger Traceless Ligation of Ribofuranosyl Azides
Phosphine 6a or 6b (2 equiv.) was added, at room temperature, to a 0.1 M solution of ribofuranosyl azide 2 (1 equiv.) in 98: 2 N,N-dimethylacetamide and DMPU. The solution was stirred for 20 h at 70 °C, then 10% water was added and the mixture was stirred for an additional 2 h at the same temperature. The solvent was evaporated under reduced pressure, and the residue was purified by flash chromatography as indicated in each case.
General Procedure for the Acetylation of the Ribofuranosyl Amides
Ac
2O (6 equiv.), pyridine (6 equiv.) and a catalytic amount of
N,N-dimethylaminopyridine were added, at room temperature, to a solution of substrate (1 equiv.) in dry CH
2Cl
2 (0.1 M). The solution was stirred for 24 h and then was concentrated in vacuo. The residue was dissolved in AcOEt and washed with aqueous 5% HCl, aqueous 5% NaHCO
3 and water. The organic layer was dried over Na
2SO
4 and concentrated. The crude was then purified by flash chromatography (hexane:AcOEt, 80:20 to 50:50). Purification afforded products with spectroscopic data coherent with the fully characterized compounds published by Filippov and coworkers [
8].
Nδ-(5-tert-Butyldiphenylsilyl-α-D-ribosyl)-Nα-benzyloxycarbonyl asparagine benzyl ester (12α). The crude product was purified by flash chromatography (CHCl3/MeOH 95:5). yield = 60%. 1H-NMR (CD3OD, 25 °C): δ = 7.71–7.64, 7.42–7.24 (3 × m, 20H, Arom.), 5.73 (d, J1,2 = 4.8 Hz, 1H, H-1), 5.15, 5.07 (2 × s, 2H, CH2Bn, CH2Cbz), 4.65 (m, 1H, CH α-Asn), 4.25 (t, J = 5 Hz, 1H, H-3), 4.15 (t, J = 4.8 Hz, 1H, H-2), 3.97 (m, 1H, H-4), 3.72 (dd, J4,5 = 3.2 Hz, Jgem = 11.3 Hz, 2H, H-5 H-5’), 2.84 (m, 2H, CH2 β-Asn), 1.04 (s, 9H, t-Bu). 13C-NMR (CD3OD, 25 °C): 172.9, 172.4 (CO α-Asn, CO γ-Asn), 158.6 (CO Cbz), 138.2 (Cq Arom.), 137.3 (Cq Arom.), 136.9, 136.8 (Arom.), 134.6, 134.4 (Cq Arom.), 131.1, 131.0, 129.7, 129.6, 129.5, 129.5, 129.3, 129.3, 129.0, 128.8, 128.4, 128.1 (Arom.), 84.7 (C-4), 81.9 (C-1), 72.8 (C-3), 72.1 (C-2), 68.4, 67.9 (CH2 Bn, CH2 Cbz), 65.3 (C-5), 52.4 (CH, α-Asn), 38.9 (CH2, β-Asn), 27.4 (CH3, t-Bu), 20.2 (Cq, t-Bu). FT-ICR (ESI) calcd. for C40H46N2O9Si [M+Na]+ 749.29726; found 749.29721.
Nδ-(5-tert-Butyldiphenylsilyl-α-D-ribosyl)-Nα-benzyloxycarbonyl glutamine benzyl ester (13α). The crude product was purified by flash chromatography (CHCl3/MeOH 95:5). yield = 76%. 1H-NMR (CD3OD, 25 °C): δ = 7.71–7.64, 7.43–7.23 (3 x m, 20H, Arom.), 5.72 (d, J1,2 = 4.6 Hz, 1H, H-1), 5.15, 5.06 (2 × s, 2H, CH2Bn, CH2Cbz), 4.25 (m, 1H, CH α-Gln), 4.23 (t, J = 4.9 Hz, 1H, H-3), 4.15 (t, J = 4.8 Hz, 1H, H-2), 3.95 (m, 1H, H-4), 3.70 (dd, J4,5 = 3.2 Hz, Jgem= 10.2 Hz, 2H, H-5 H-5’), 2.35 (m, 2H, CH2 γ-Asn), 2.13 (m, 1H, CH2 β-Asn), 1.95 (m, 1H, CH2 β-Asn), 1.02 (s, 9H, t-Bu). 13C-NMR (CD3OD, 25 °C): 136.7, 136.6 (Arom.), 134.6, 134.4 (Cq Arom.), 130.8, 130.8, 129.7, 129.5, 129.4, 129.1, 128.9, 128.8, 128.7 (Arom.),84.5 (C-4), 81.6 (C-1), 72.7 (C-3), 71.9 (C-2), 67.9, 67.6 (CH2 Bn, CH2 Cbz), 65.0 (C-5), 55.1 (CH α-Gln), 33.3 (CH2 β-Gln), 28.0 (CH2 γ-Gln), 27.3 (t-Bu), 22.9 (Cq t-Bu). FT-ICR (ESI) calcd. for C41H48N2O9Si [M+Na]+ 763.31291; found 763.31298.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}