Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks

Abstract
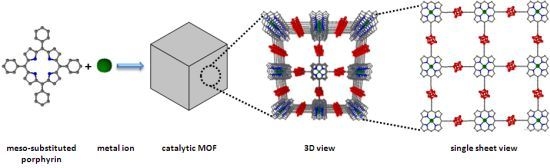
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
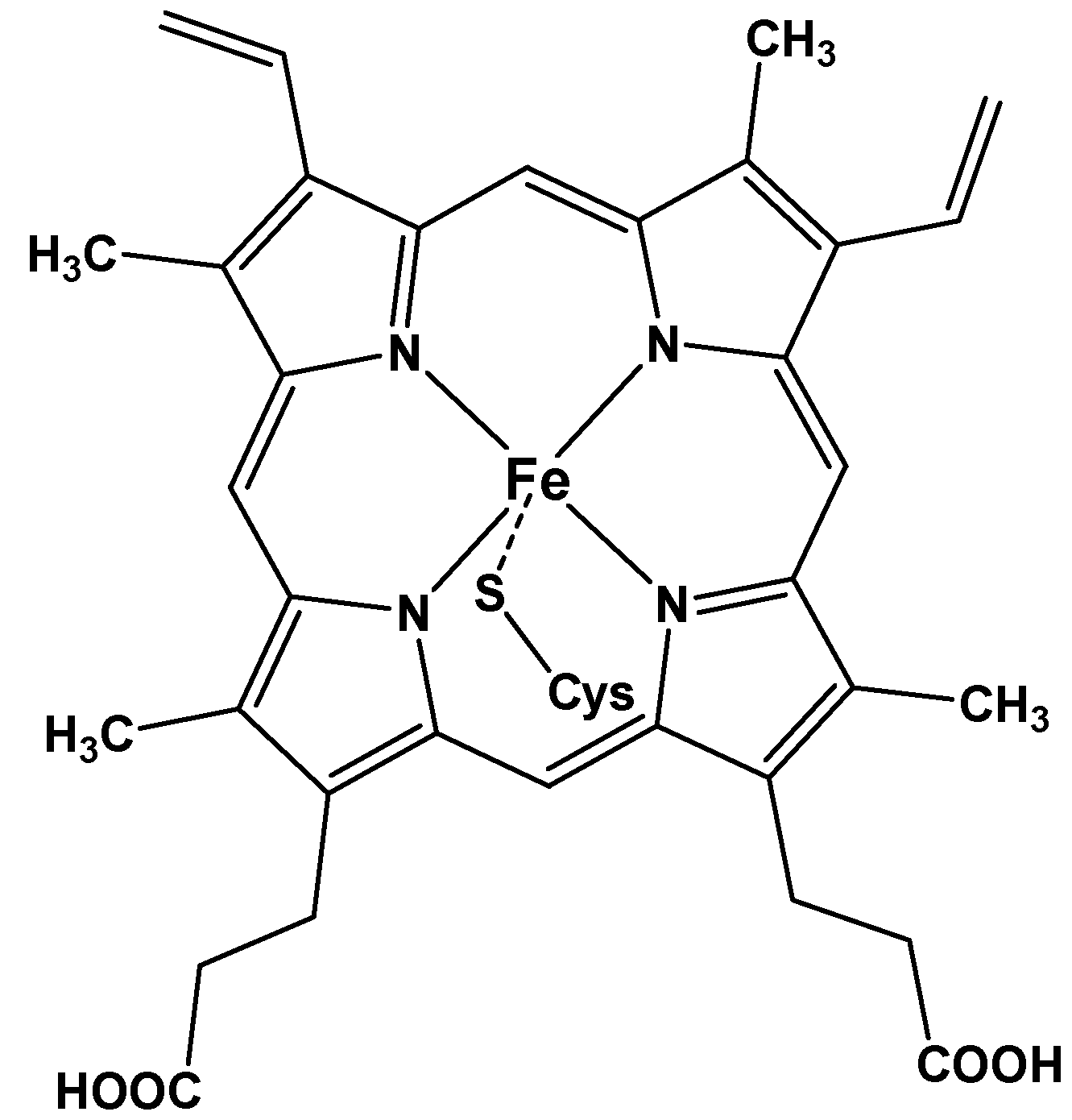
1. Metalloporphyrins as Bioinspired Systems
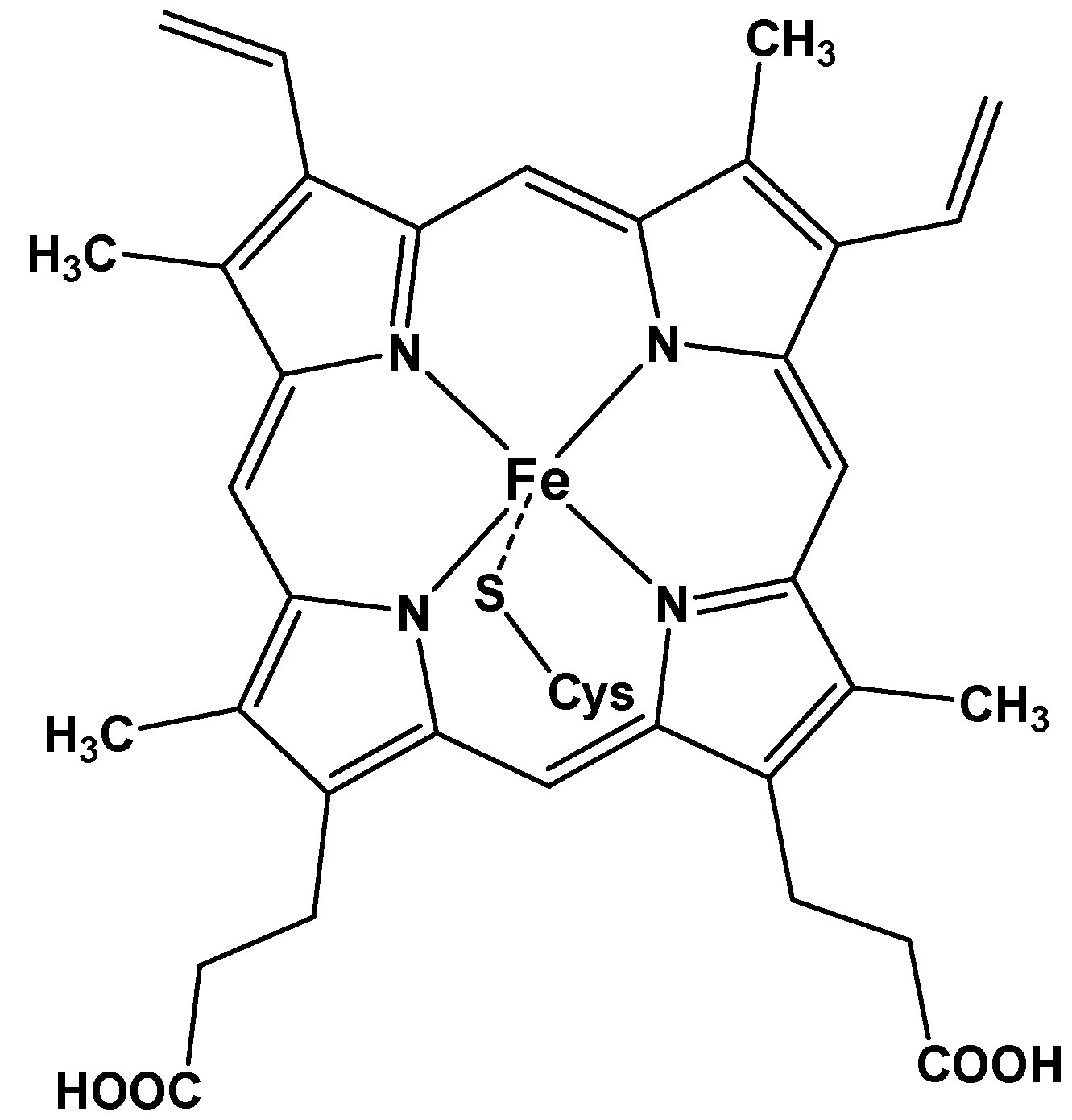
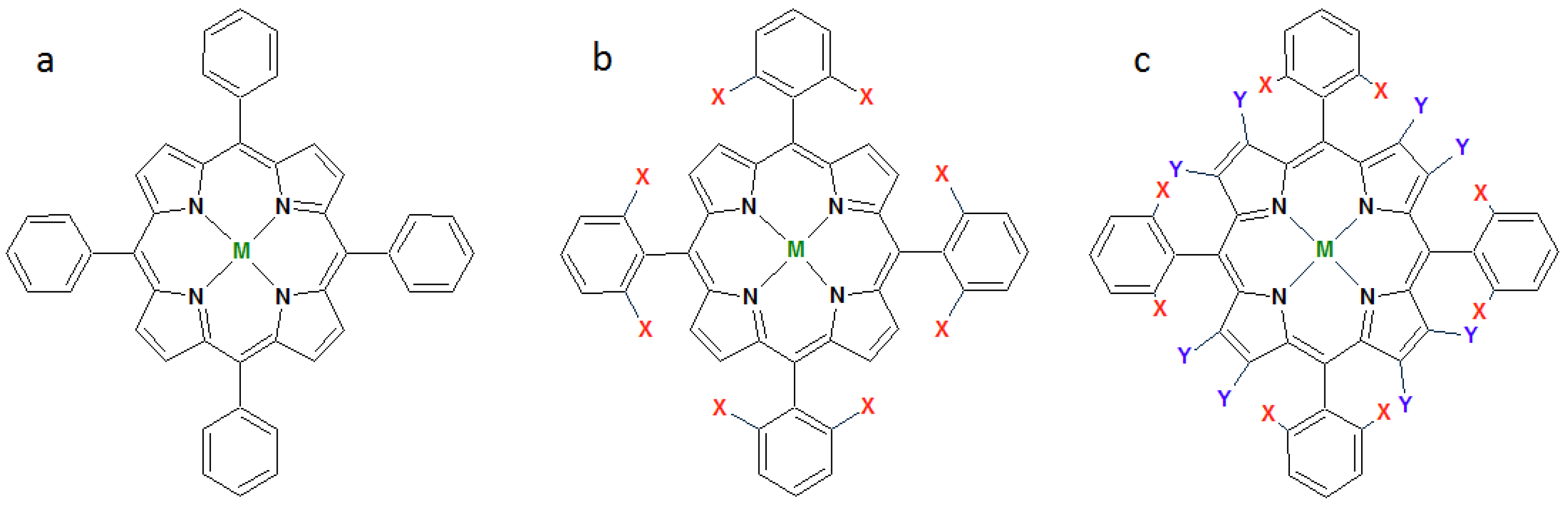
1.1. Synthetic Porphyrins
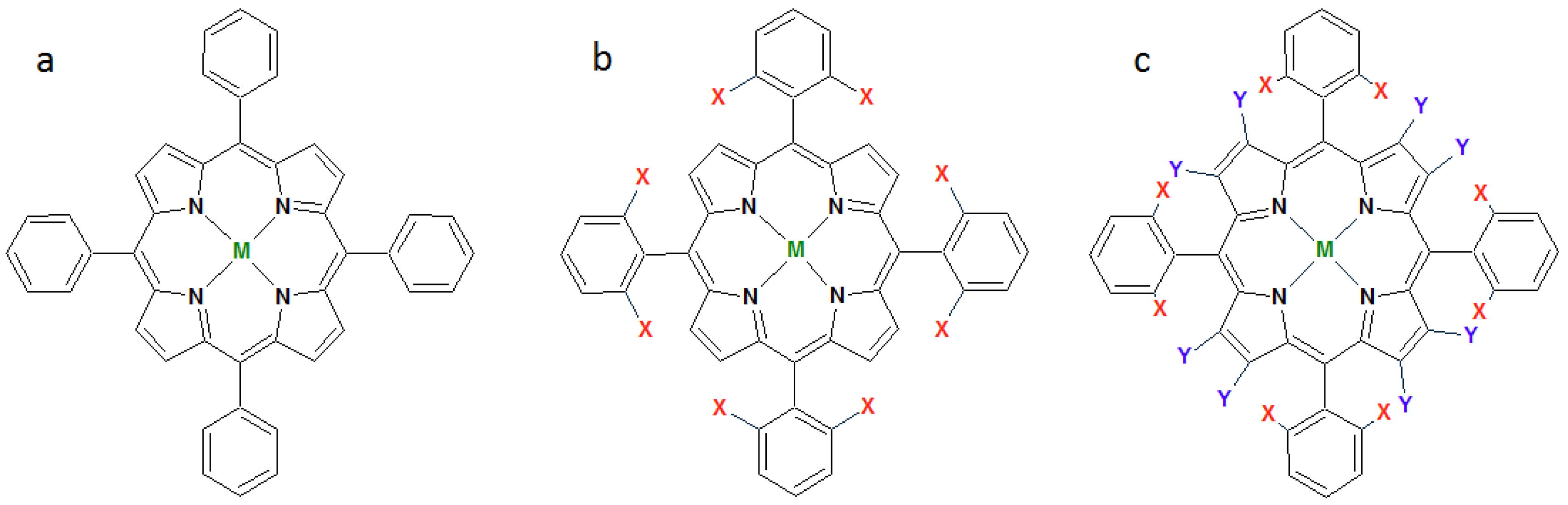
1.2. Metalloporphyrins as Catalysts for Oxidation Reactions in Homogenous Phase
2. Advances in the Field of Coordination Polymers and Metal-Organic Frameworks
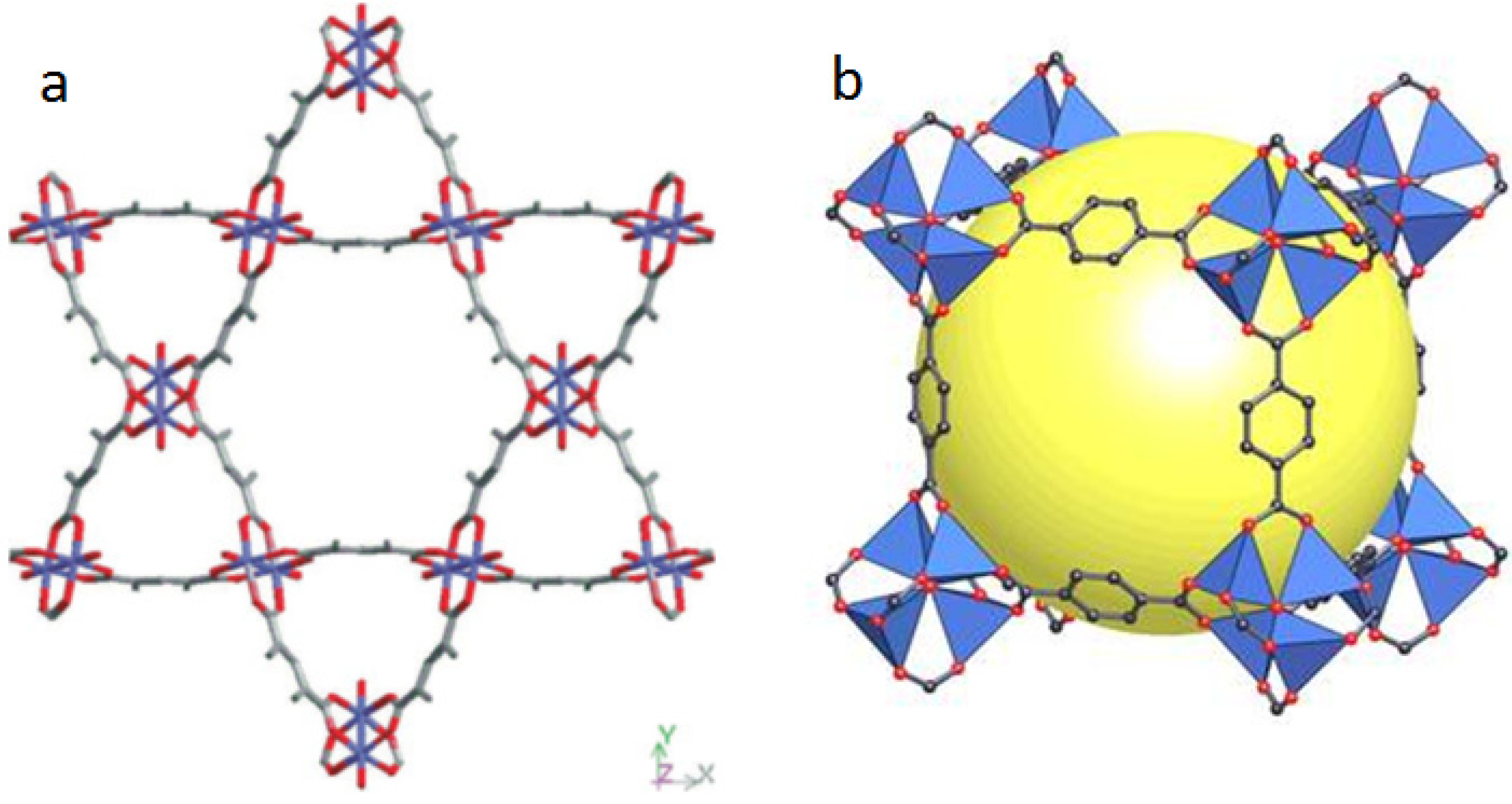
2.1. Strategies to Synthesize MOFs
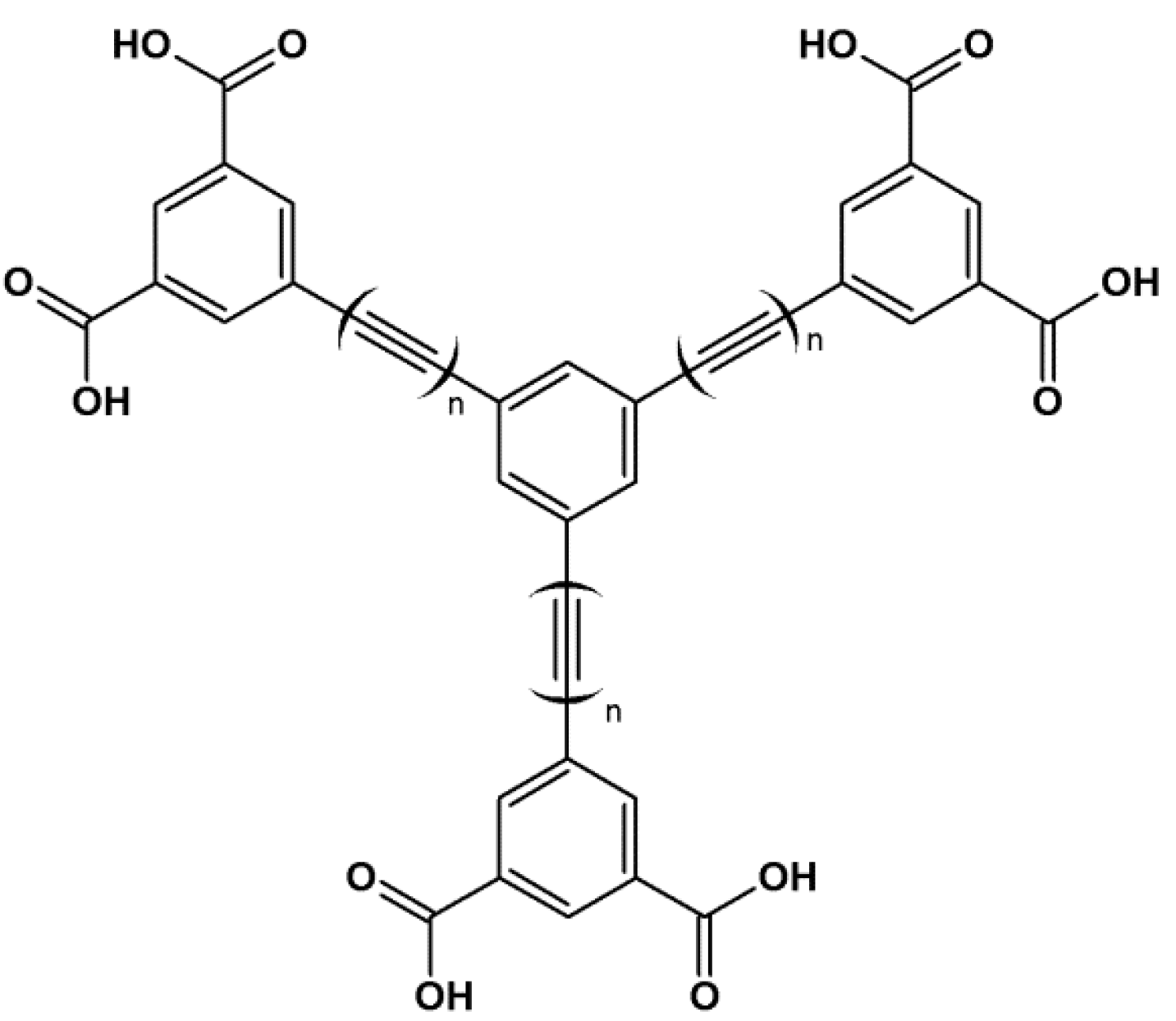
3. MOFs with Porphyrins as Building Blocks
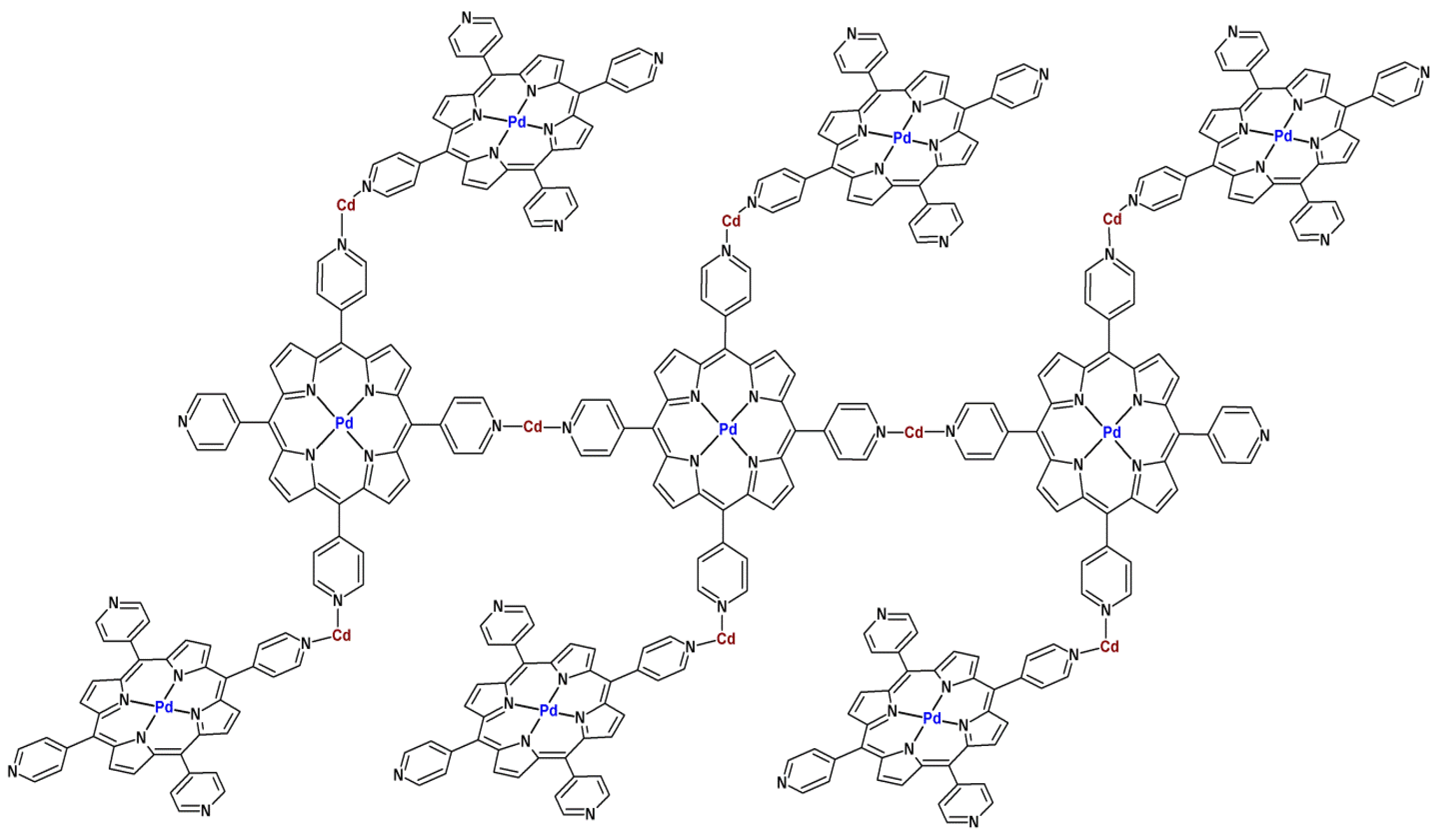
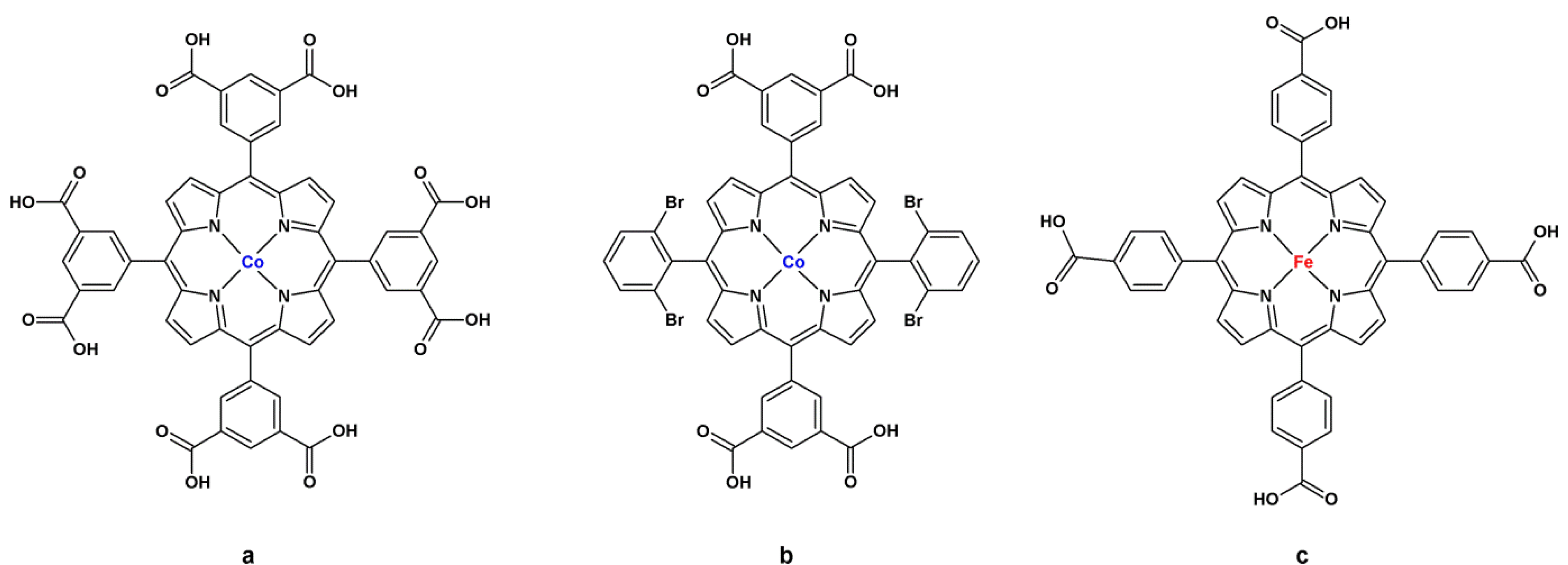
4. Activity of Metalloporphyrin-Based MOFs in Heterogeneous Catalysis
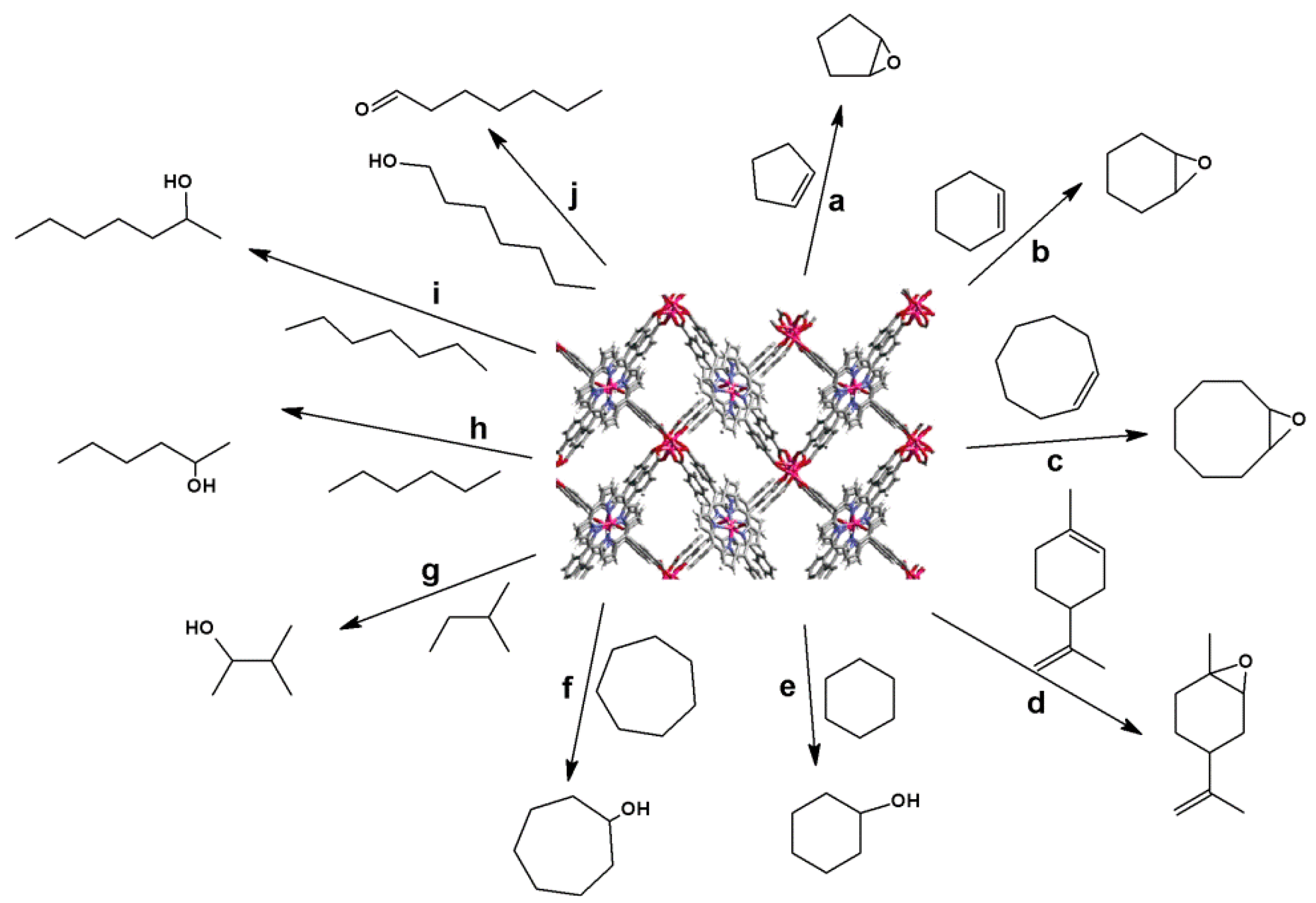
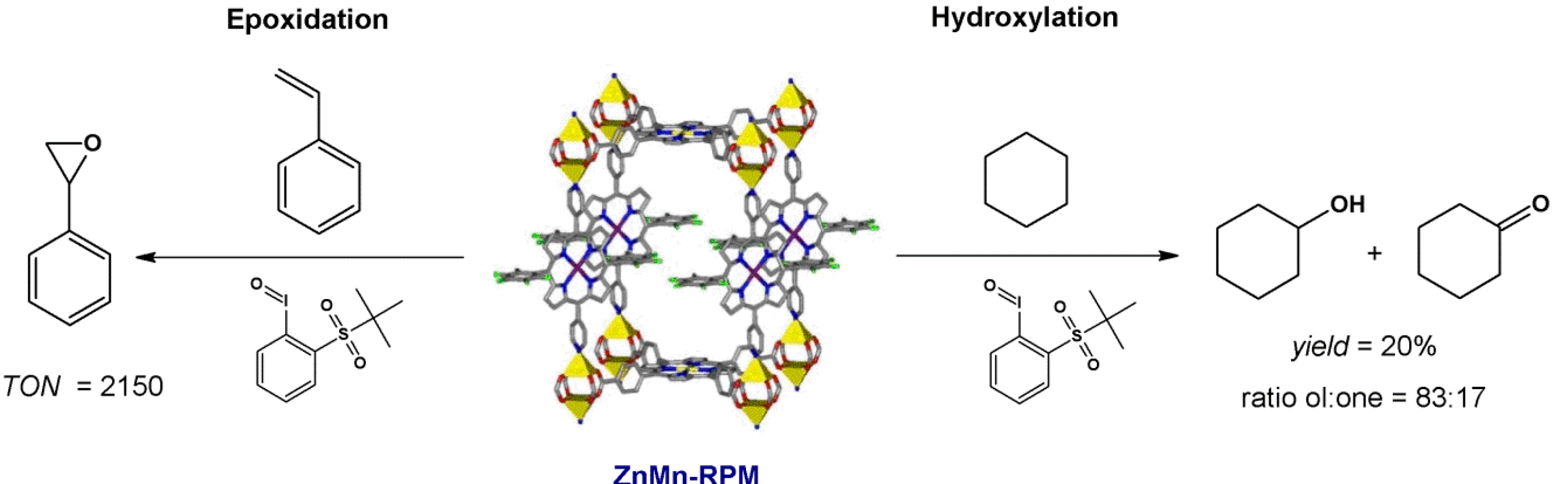
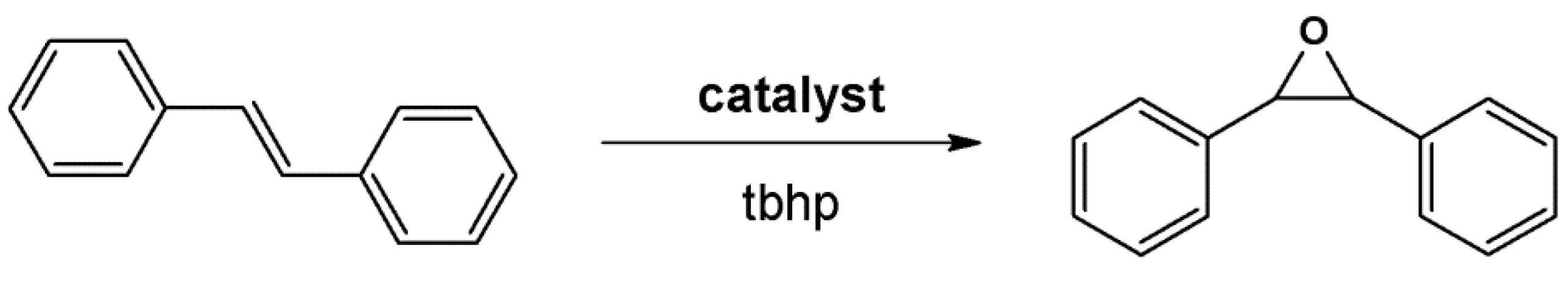
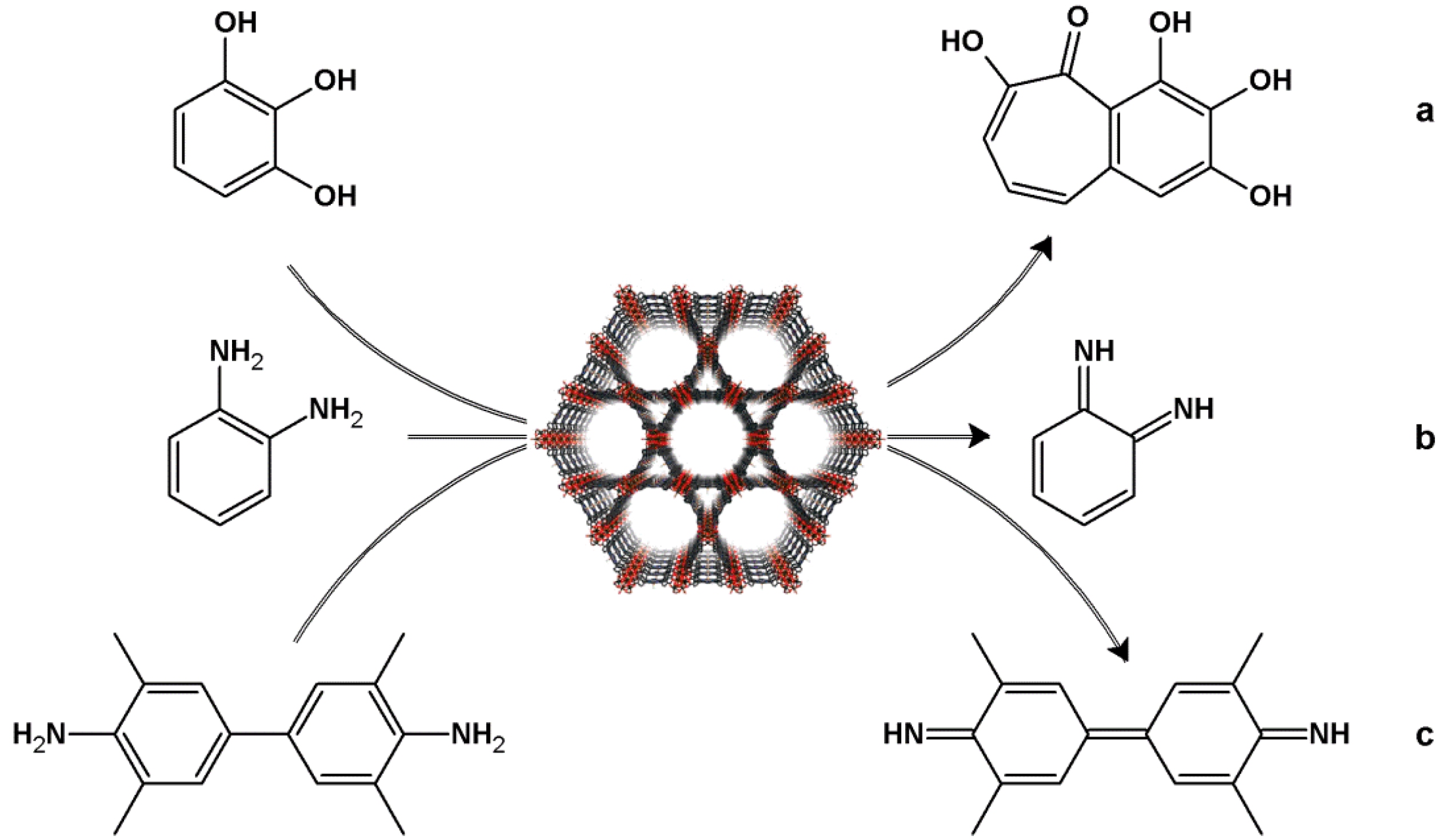
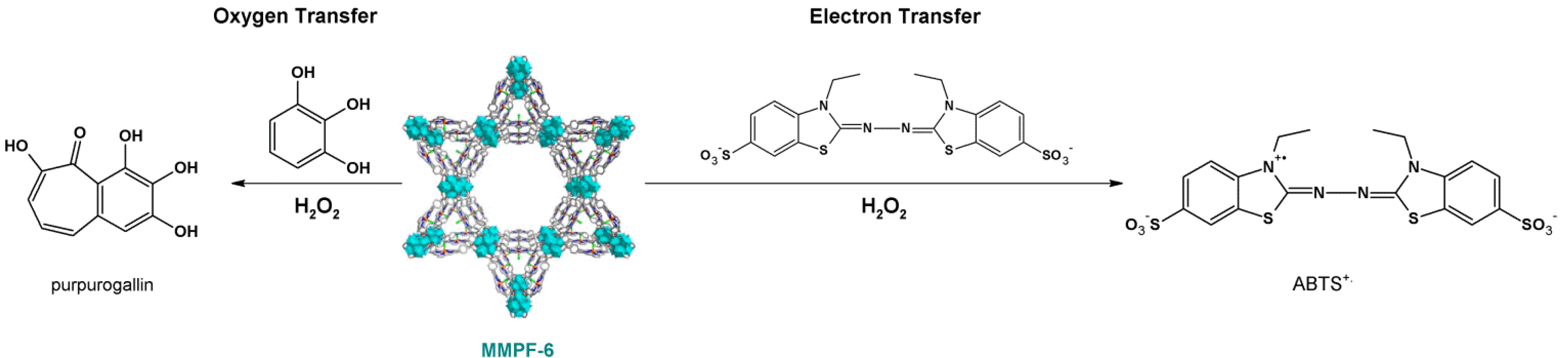
4.1. Oxidation Reaction
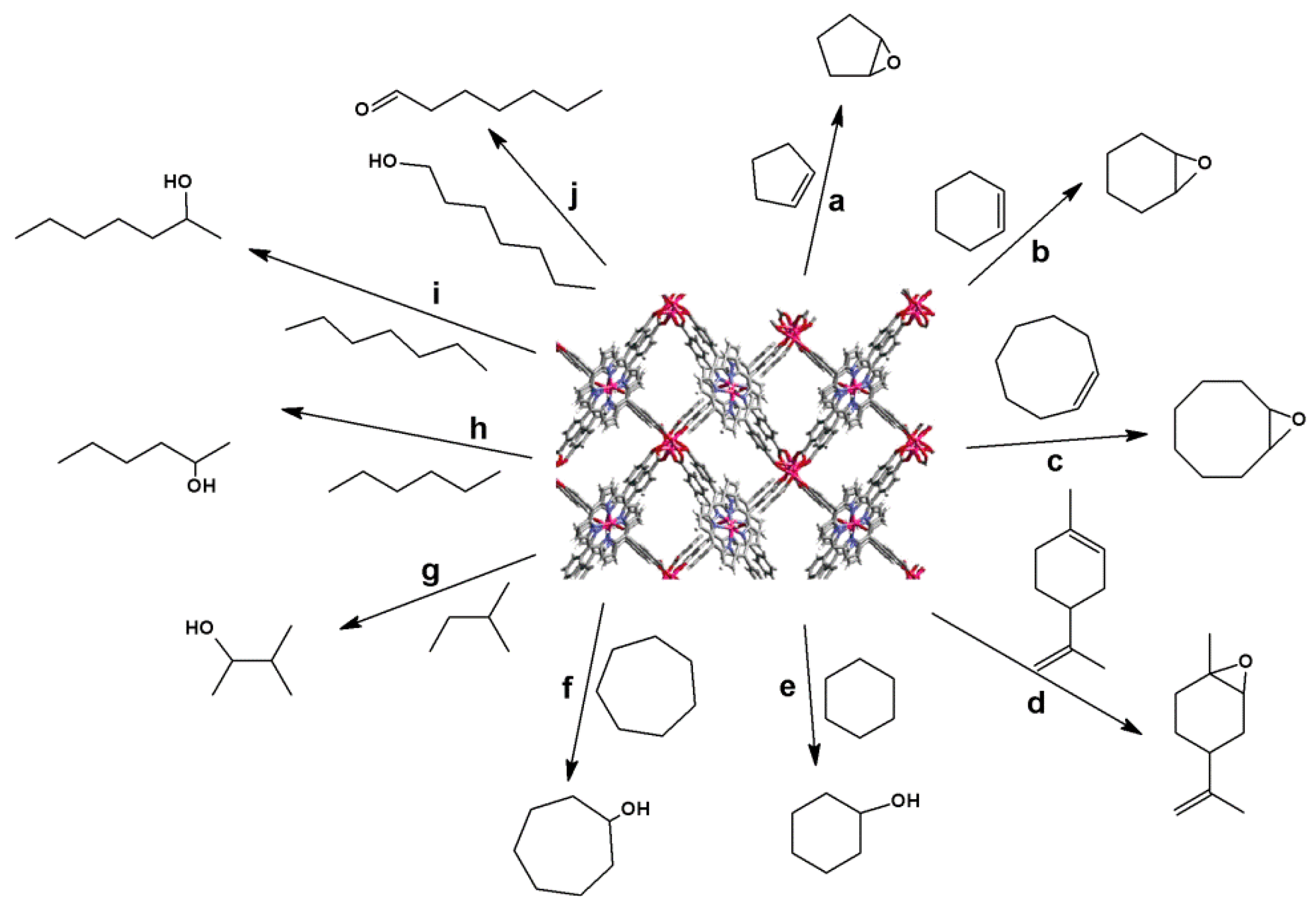
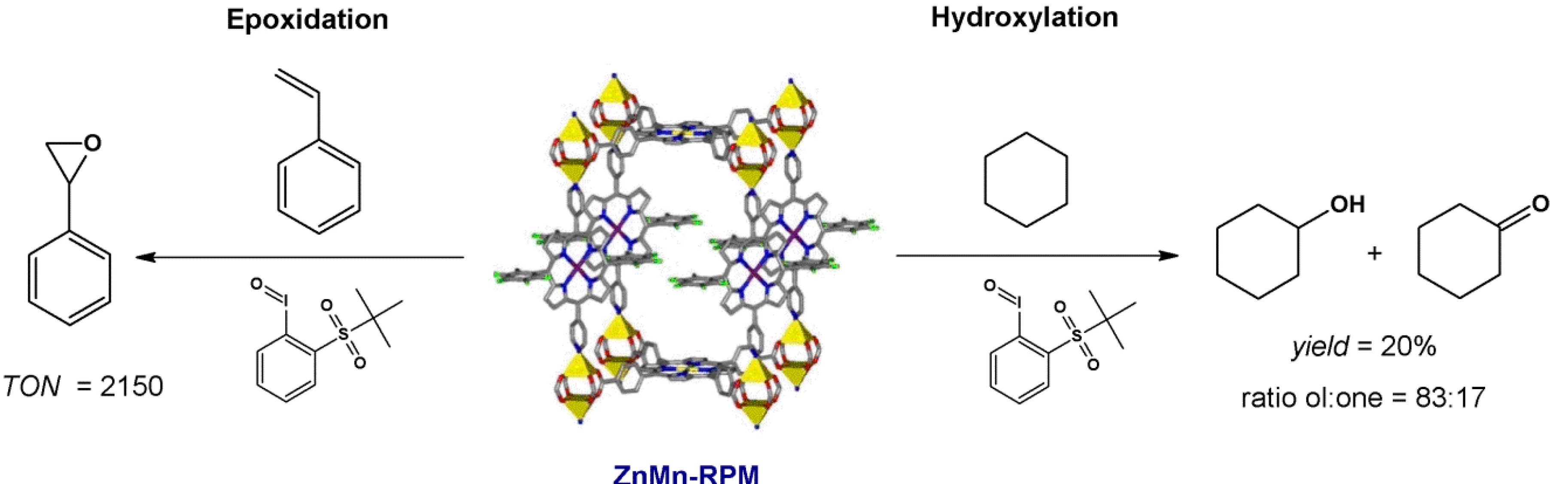
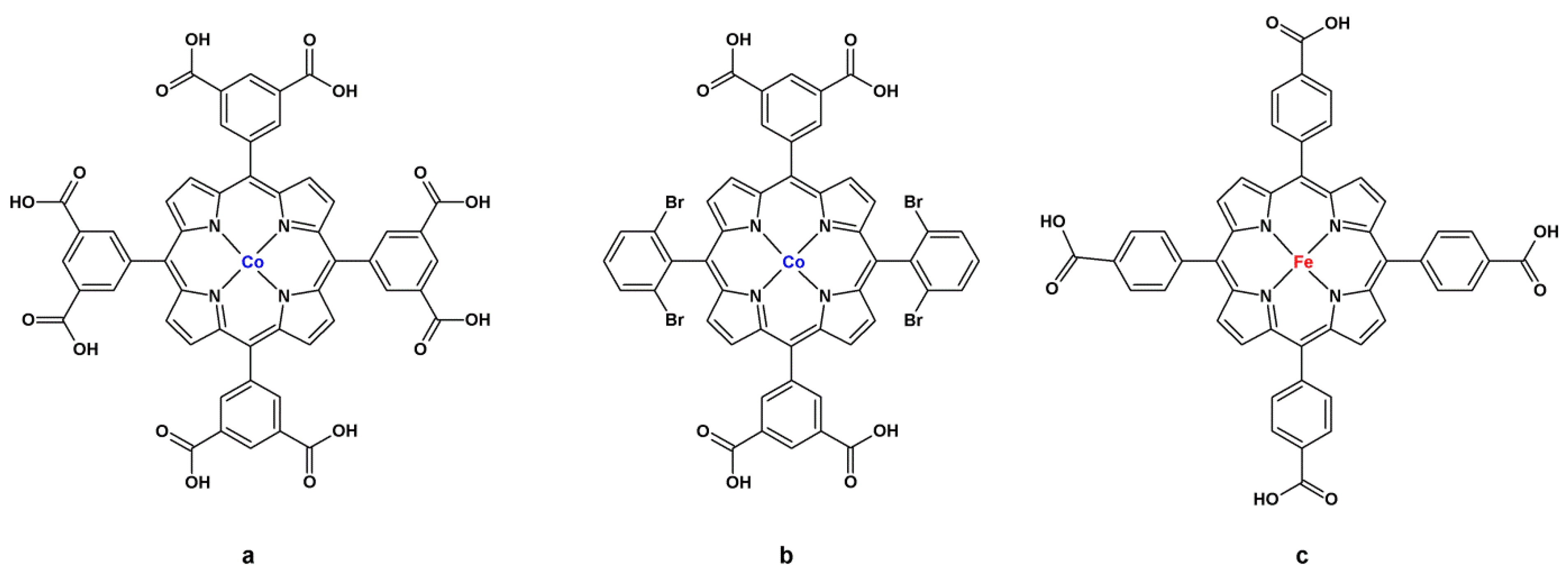
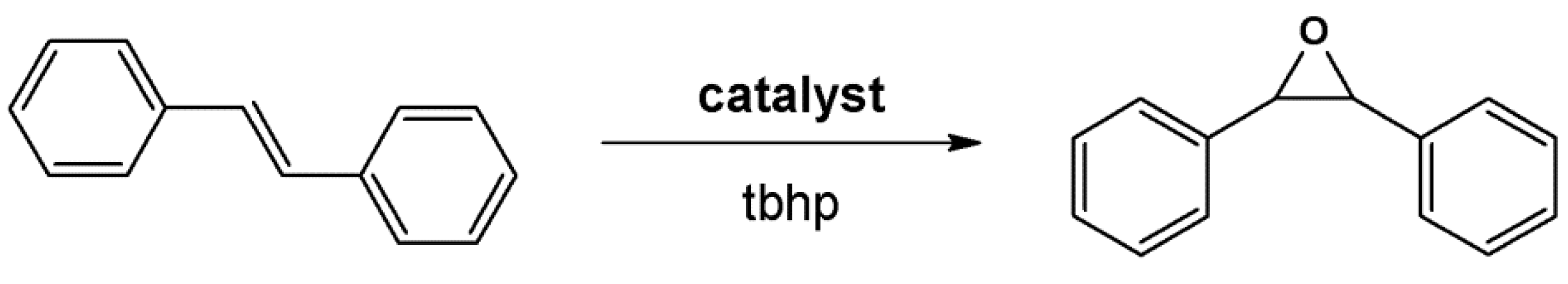
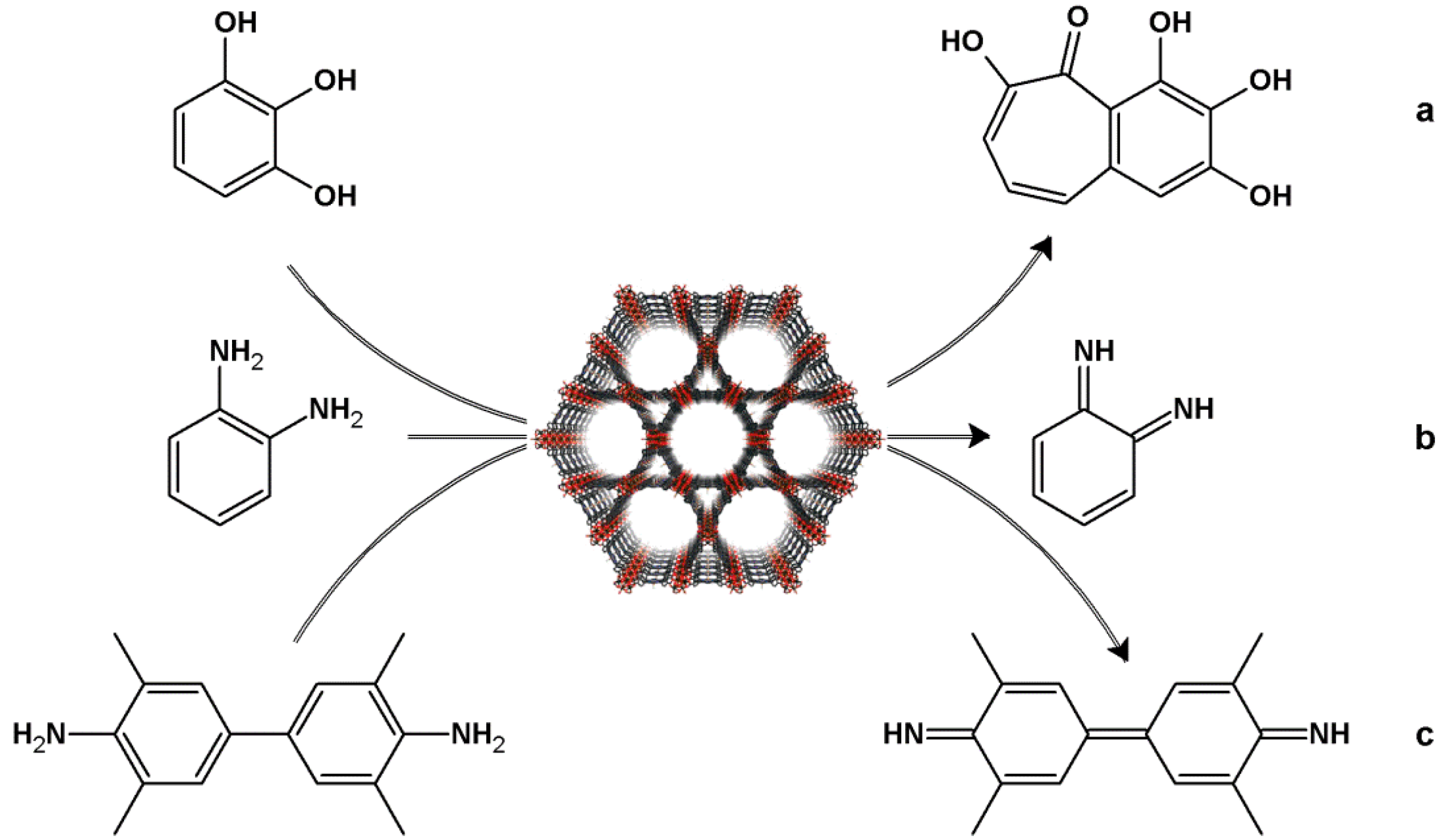
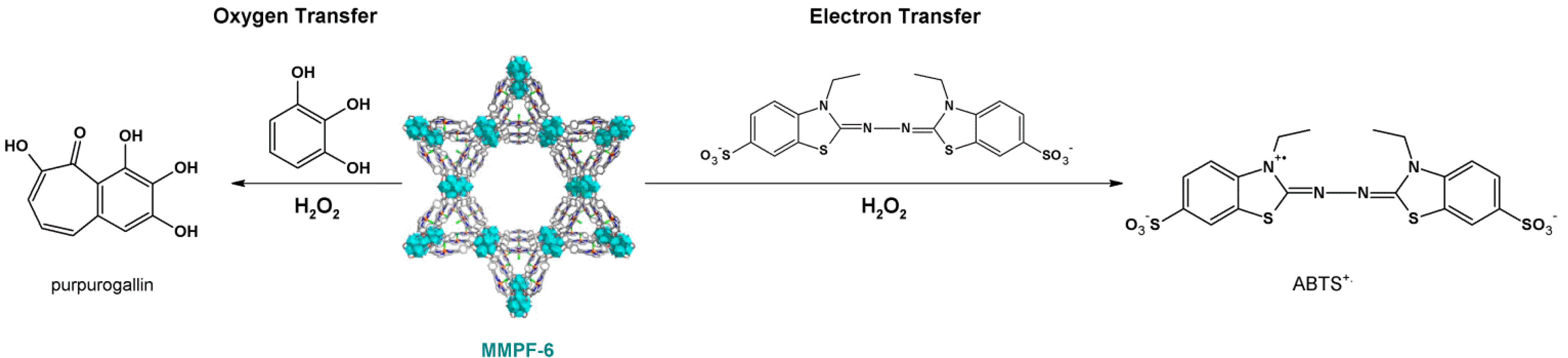
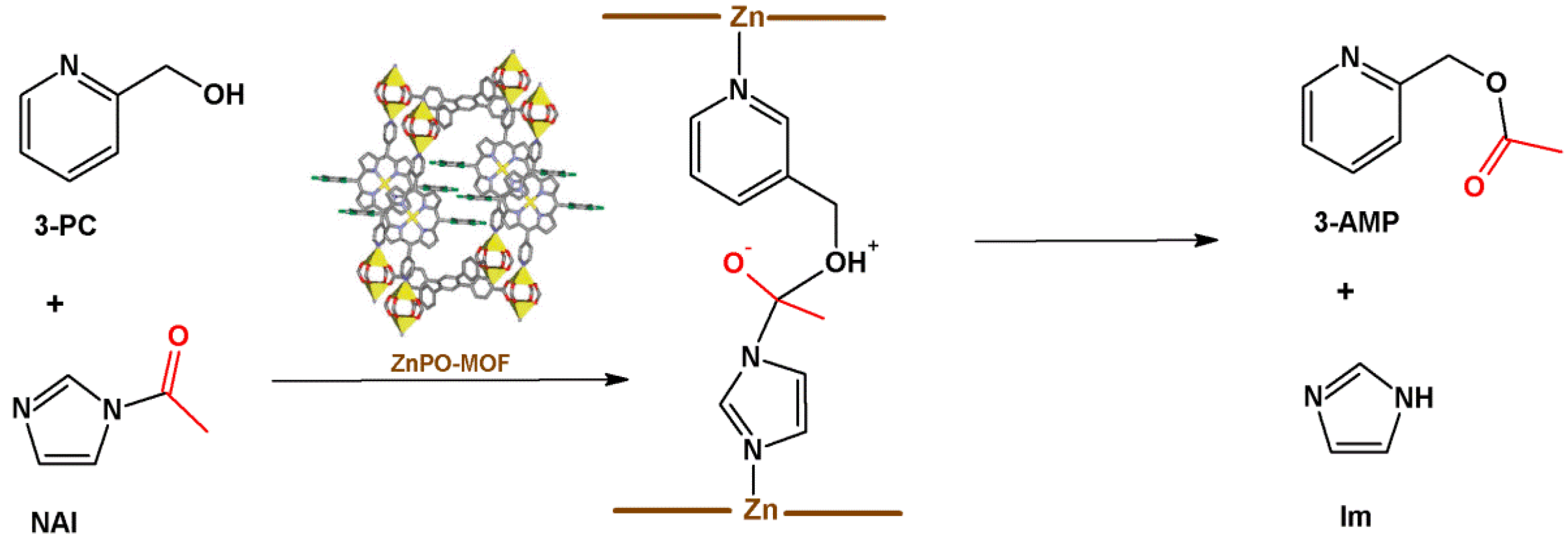
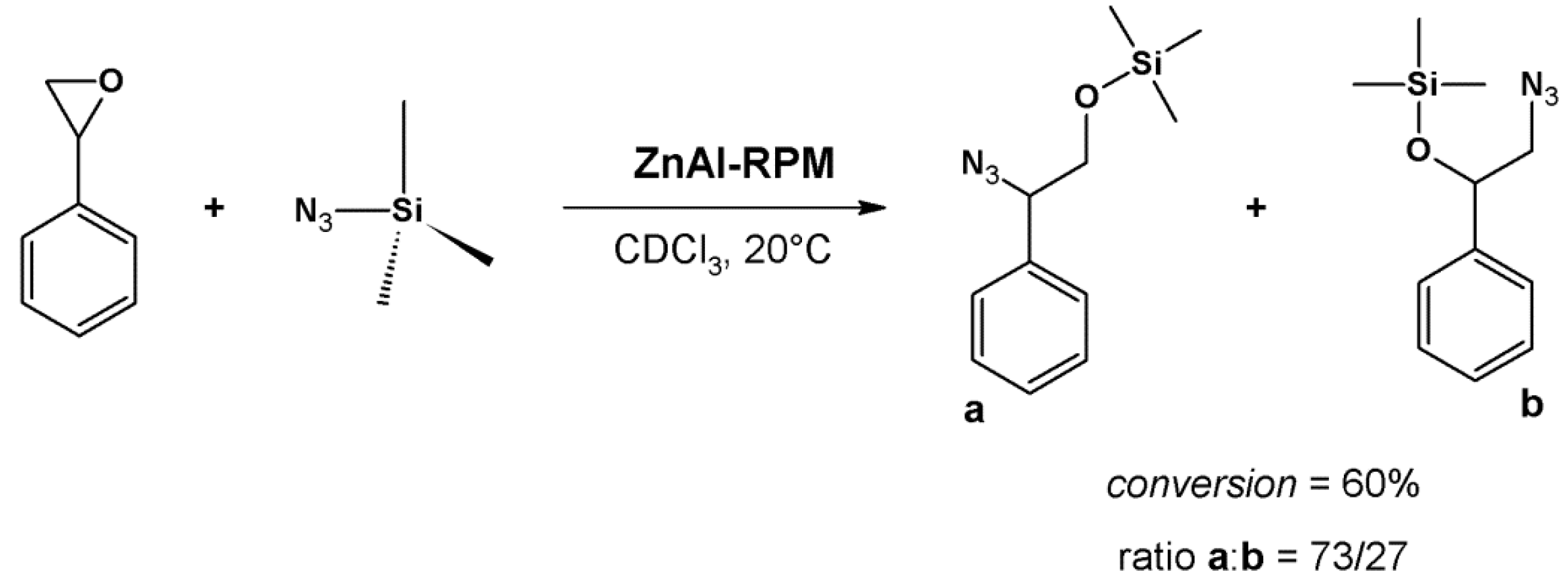
4.2. Lewis Acid Catalysis
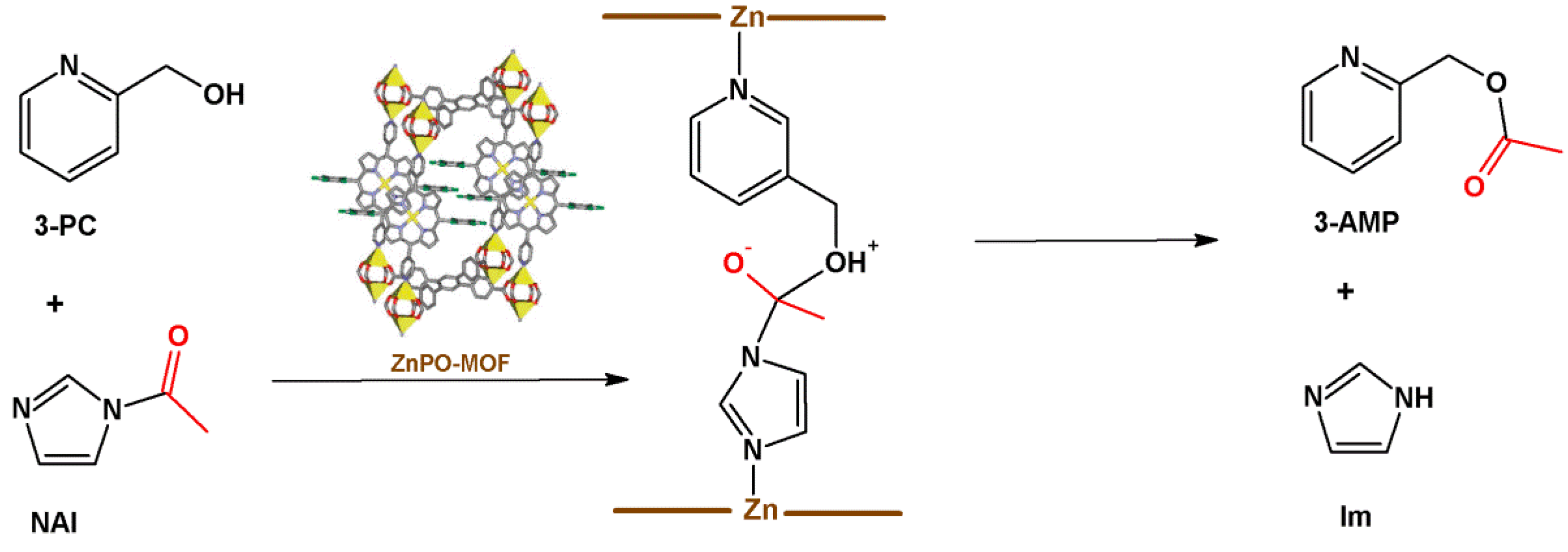
- (i)
- Lewis acid activation: preliminary coordination of NAI to a zinc-porphyrin site withdraws the electron density from the carbonyl group, generating the negative charge (Figure 12, center).
- (ii)
- Concentration of the substrates: ZnPO-MOF cavities can concentrate the substrates and increase the reaction rate without changing the activation energy.
- (iii)
- Preferred orientation of reactants: ZnPO-MOF cavity size (Zn-Zn distance = 11.6 Å) allows a pair of 3-PC and NAI to align, providing the ideal orientation for the reaction to take place.
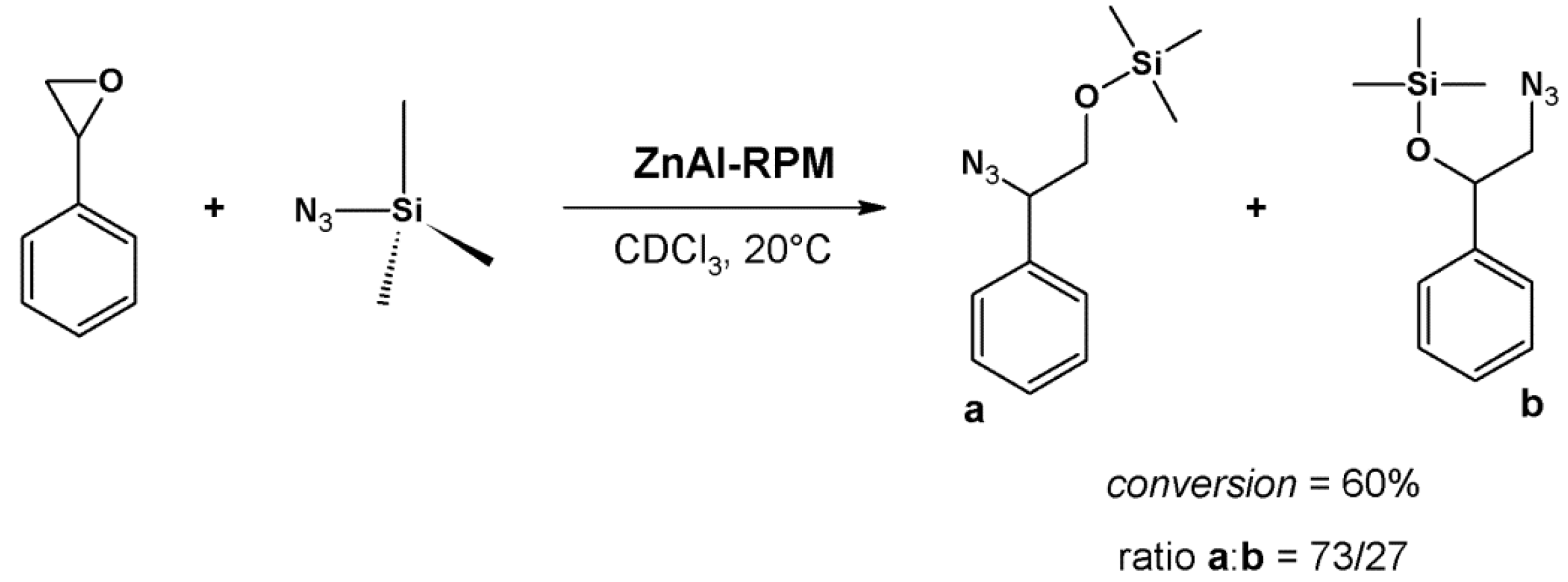
5. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Mansuy, D. A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. C. R. Chim. 2007, 10, 392–413. [Google Scholar] [CrossRef]
- Katagiri, M.; Ganguli, B.N.; Gunsalus, I.C. A Soluble Cytochrome P-450 Functional in Methylene Hydroxylation. J. Biol. Chem. 1968, 243, 3543–3546. [Google Scholar]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Mansuy, D. The great diversity of reactions catalyzed by cytochromes P450. Comp. Biochem. Physiol. C: Toxicol. Pharmacol. 1998, 121, 5–14. [Google Scholar]
- Lindsay Smith, J.R.; Iamamoto, Y.; Vinhado, F.S. Oxidation of alkanes by iodosylbenzene (PhIO) catalysed by supported Mn(III) porphyrins: Activity and mechanism. J. Mol. Catal. A: Chem. 2006, 252, 23–30. [Google Scholar] [CrossRef]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- DiNello, R.K.; Chang, C.K. 7—Isolation and Modification of Natural Porphyrins. In The Porphyrins; David, D., Ed.; Academic Press: New York, NY, USA, 1978; pp. 289–339. [Google Scholar]
- Dolphin, D.; Traylor, T.G.; Xie, L.Y. Polyhaloporphyrins: Unusual Ligands for Metals and Metal-Catalyzed Oxidations. Acc. Chem. Res. 1997, 30, 251–259. [Google Scholar] [CrossRef]
- Senge, M.O.; Richter, J. Synthetic transformations of porphyrins—Advances 2002–2004. J. Porph. Phthal. 2004, 8, 934–953. [Google Scholar] [CrossRef]
- Horn, S.; Dahms, K.; Senge, M.O. Synthetic transformations of porphyrins—Advances 2004–2007. J. Porph. Phthal. 2008, 12, 1053–1077. [Google Scholar] [CrossRef]
- Adler, A.D.; Longo, F.R.; Kampas, F.; Kim, J. On preparation of metalloporphyrins. J. Inorg. Nucl. Chem. 1970, 32, 2443. [Google Scholar] [CrossRef]
- Lindsey, J.S.; Schreiman, I.C.; Hsu, H.C.; Kearney, P.C.; Marguerettaz, A.M. Rothemund and Adler-Longo reactions revisited: Synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987, 52, 827–836. [Google Scholar]
- Kadish, K.; Smith, K.M.; Guilard, R. The Porphyrin Handbook; Academic Press: New York, NY, USA, 1999. [Google Scholar]
- Wickramasinghe, A.; Jaquinod, L.; Nurco, D.J.; Smith, K.M. Investigations on the directive effects of a single meso-substituent via nitration of 5,12,13,17,18-pentasubstituted porphyrins: Syntheses of conjugated β-nitroporphyrins. Tetrahedron 2001, 57, 4261–4269. [Google Scholar] [CrossRef]
- Ali, H.; van Lier, J.E. Phenylselenyl halides: Efficient reagents for the selective halogenation and nitration of porphyrins. Tetrahedron Lett. 1991, 32, 5015–5018. [Google Scholar]
- Turk, H.; Ford, W.T. Epoxidation of styrene with aqueous hypochlorite catalyzed by a manganese(III) porphyrin bound to colloidal anion-exchange particles. J. Org. Chem. 1991, 56, 1253–1260. [Google Scholar] [CrossRef]
- Brantley, S.E.; Gerlach, B.; Olmstead, M.M.; Smith, K.M. Vinyl group protection in porphyrins and chlorins: Organoselenium derivatives. Tetrahedron Lett. 1997, 38, 937–940. [Google Scholar] [CrossRef]
- Kvíčala, J.; Beneš, M.; Paleta, O.; Král, V. Regiospecific nucleophilic substitution in 2,3,4,5,6-pentafluorobiphenyl as model compound for supramolecular systems. Theoretical study of transition states and energy profiles, evidence for tetrahedral SN2 mechanism. J. Fluor. Chem. 2010, 131, 1327–1337. [Google Scholar] [CrossRef]
- Knör, G. Spontaneous nucleophilic addition of hydroxide ions to the meso-position of high-valent antimony-oxo porphyrin complexes. J. Inorg. Biochem. 2001, 84, 297–299. [Google Scholar] [CrossRef]
- Kadish, K.M.; Han, B.C.; Franzen, M.M.; Araullo-McAdams, C. Syntheses and spectroscopic characterization of (T(p-Me2N)F4PP)H2 and (T(p-Me2N)F4PP)M where T(p-Me2N)F4PP = the dianion of meso-tetrakis(o,o,m,m-tetrafluoro-p-(dimethylamino)phenyl)porphyrin and M = cobalt(II), copper(II), or nickel(II). Structures of (T(p-Me2N)F4PP)Co and meso-tetrakis(pentafluorophenyl)porphinatocobalt(II), (TF5PP)Co. J. Am. Chem. Soc. 1990, 112, 8364–8368. [Google Scholar] [CrossRef]
- Jiménez-Osés, G.; García, J.I.; Silva, A.M.G.; Santos, A.R.N.; Tomé, A.C.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Mechanistic insights on the site selectivity in successive 1,3-dipolar cycloadditions to meso-tetraarylporphyrins. Tetrahedron 2008, 64, 7937–7943. [Google Scholar] [CrossRef]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and epoxidation catalyzed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Rocha Gonsalves, A.M.D.A.; Johnstone, R.A.W.; Pereira, M.M.; Shaw, J.; Sobral, A.J.F.D.N. Metal-assisted reactions. Part 22. Synthesis of perhalogenated prophyrins and their use as oxidation catalysts. Tetrahedron Lett. 1991, 32, 1355–1358. [Google Scholar]
- Hoffmann, P.; Labat, G.; Robert, A.; Meunier, B. Highly Selective Bromination of Tetramesitylporphyrin: An easy access to robust metalloporphyrins, M-Br8TMP and M-Br8TMP. Examples of application in catalytic oxygenation and oxidation reactions. Tetrahedron Lett. 1990, 31, 1991–1994. [Google Scholar] [CrossRef]
- Gross, Z.; Simkhovich, L. Hydroxylation of simple alkanes by iodosylbenzene is catalyzed more efficiently by second than by third generation iron(III) porphyrins. Tetrahedron Lett. 1998, 39, 8171–8174. [Google Scholar] [CrossRef]
- Doro, F.G.; Smith, J.R.L.; Ferreira, A.G.; Assis, M.D. Oxidation of alkanes and alkenes by iodosylbenzene and hydrogen peroxide catalysed by halogenated manganese porphyrins in homogeneous solution and covalently bound to silica. J. Mol. Catal. A: Chem. 2000, 164, 97–108. [Google Scholar] [CrossRef]
- CarvalhoDa-Silva, D.; Mac Leod, T.C.O.; de Faria, A.L.; dos Santos, J.S.; de Carvalho, M.E.M.D.; Rebouças, J.S.; Idemori, Y.M.; Assis, M.D.D. Carbamazepine oxidation catalyzed by manganese porphyrins: Effects of the β-bromination of the macrocycle and the choice of oxidant. Appl. Catal. A. 2011, 408, 25–30. [Google Scholar] [CrossRef]
- Guedes, A.A.; Smith, J.R.L.; Nascimento, O.R.; Guedes, D.F.C.; Assis, M.D. Catalytic activity of halogenated Iron Porphyrins in Alkene and Alkane oxidations by iodosylbenzene and hydrogen peroxide. J. Braz. Chem. Soc. 2005, 16, 835–843. [Google Scholar] [CrossRef]
- Bartoli, J.F.; Brigaud, O.; Battioni, P.; Mansuy, D. Hydroxylation of linear alkanes catalysed by iron porphyrins: particular efficacy and regioselectivity of perhalogenated porphyrins. J. Chem. Soc., Chem. Commun. 1991, 440–442. [Google Scholar]
- Silva, D.C.D.; DeFreitas-Silva, G.; Nascimento, E.D.; Rebouças, J.S.; Barbeira, P.J.S.; Carvalho, M.E.M.D.D.; Idemori, Y.M. Spectral, Electrochemical, And catalytic properties of a homologous series of manganese porphyrins as cytochrome P450 model: The effect of the degree of β-bromination. J. Inorg. Biochem. 2008, 102, 1932–1941. [Google Scholar] [CrossRef]
- de Freitas Silva, G.; da Silva, D.C.; Guimarães, A.S.; do Nascimento, E.; Rebouças, J.S.; de Araujo, M.P.; de Carvalho, M.E.M.D.; Idemori, Y.M. Cyclohexane hydroxylation by iodosylbenzene and iodobenzene diacetate catalyzed by a new β-octahalogenated Mn–porphyrin complex: The effect of meso-3-pyridyl substituents. J. Mol. Catal. A: Chem. 2007, 266, 274–283. [Google Scholar] [CrossRef]
- Thomas, J.M.; Raja, R.; Sankar, G.; Bell, R.G. Molecular Sieve Catalysts for the Regioselective and Shape-Selective Oxyfunctionalization of Alkanes in Air. Acc. Chem. Res. 2001, 34, 191–200. [Google Scholar] [CrossRef]
- Cook, B.R.; Reinert, T.J.; Suslick, K.S. Shape-selective alkane hydroxylation by metalloporphyrin catalysts. J. Am. Chem. Soc. 1986, 108, 7281–7286. [Google Scholar] [CrossRef]
- Nakagaki, S.; Machado, G.S.; Halma, M.; dos Santos Marangon, A.A.; de Freitas Castro, K.A.D.; Mattoso, N.; Wypych, F. Immobilization of iron porphyrins in tubular kaolinite obtained by an intercalation/delamination procedure. J. Catal. 2006, 242, 110–117. [Google Scholar]
- Papacídero, A.T.; Rocha, L.A.; Caetano, B.L.; Molina, E.; Sacco, H.C.; Nassar, E.J.; Martinelli, Y.; Mello, C.; Nakagaki, S.; Ciuffi, K.J. Preparation and characterization of spherical silica-porphyrin catalysts obtained by the sol–gel methodology. Colloids Surf. A 2006, 275, 27–35. [Google Scholar] [CrossRef]
- Machado, G.S.; Arízaga, G.G.C.; Wypych, F.; Nakagaki, S. Immobilization of anionic metalloporphyrins on zinc hydroxide nitrate and study of an unusual catalytic activity. J. Catal. 2010, 274, 130–141. [Google Scholar] [CrossRef]
- Halma, M.; Aparecida Dias de Freitas Castro, K.; Taviot-Gueho, C.; Prévot, V.; Forano, C.; Wypych, F.; Nakagaki, S. Synthesis, characterization, and catalytic activity of anionic iron(III) porphyrins intercalated into layered double hydroxides. J. Catal. 2008, 257, 233–243. [Google Scholar]
- Lichtenberger, F.; Nastainczyk, W.; Ullrich, V. Cytochrome P450 as an oxene transferase. Biochem. Biophys. Res. Commun. 1976, 70, 939–946. [Google Scholar] [CrossRef]
- De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Oxidation of styrene and of some derivatives with H2O2 catalyzed by novel imidazolium-containing manganese porphyrins: A mechanistic and thermodynamic interpretation. J. Mol. Catal. A: Chem. 2011, 345, 1–11. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Gonçalves, A.R.; Pereira, M.M.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Epoxidation reactions with hydrogen peroxide activated by a novel heterogeneous metalloporphyrin catalyst. J. Mol. Catal. A: Chem. 2006, 256, 321–323. [Google Scholar] [CrossRef]
- Benedito, F.L.; Nakagaki, S.; Saczk, A.A.; Peralta-Zamora, P.G.; Costa, C.M.M. Study of metalloporphyrin covalently bound to silica as catalyst in the ortho-dianisidine oxidation. Appl. Catal., A. 2003, 250, 1–11. [Google Scholar] [CrossRef]
- Barros, V.P.; Faria, A.L.; MacLeod, T.C.O.; Moraes, L.A.B.; Assis, M.D. Ironporphyrin immobilized onto montmorillonite as a biomimetical model for azo dye oxidation. Int. Biodeter. Biodegr. Int. Biodeter. Biodegr. 2008, 61, 337–344. [Google Scholar]
- Emmert Iii, F.L.; Thomas, J.; Hon, B.; Gengenbach, A.J. Metalloporphyrin catalyzed oxidation of methyl yellow and related azo compounds. Inorg. Chim. Acta 2008, 361, 2243–2251. [Google Scholar] [CrossRef]
- Serra, A.C.; Docal, C.; Rocha Gonsalves, A.M.D.A. Efficient azo dye degradation by hydrogen peroxide oxidation with metalloporphyrins as catalysts. J. Mol. Catal. A: Chem. 2005, 238, 192–198. [Google Scholar] [CrossRef]
- Banfi, S.; Cassani, E.; Caruso, E.; Cazzaro, M. Oxidative cleavage of plasmid bluescript by water-Soluble Mn-Porphyrins and artificial oxidants or molecular oxygen. Bioorg. Med. Chem. 2003, 11, 3595–3605. [Google Scholar] [CrossRef]
- Leal, O.; Anderson, D.L.; Bowman, R.G.; Basolo, F.; Burwell, R.L., Jr. Reversible adsorption of oxygen on silica gel modified by imidazole-attached iron tetraphenylporphyrin. J. Am. Chem. Soc. 1975, 97, 5125–5129. [Google Scholar]
- Gao, B.; Chen, Y.; Lei, Q. Hydroxylation of cyclohexane with molecular oxygen catalyzed by highly efficient heterogeneous Mn(III) porphyrin catalysts prepared by special synthesis and immobilization method. J. Inclusion Phenom. Macrocyclic Chem. 2012, 74, 455–465. [Google Scholar] [CrossRef]
- Ucoski, G.M.; Castro, K.A.D. d. F.; Ciuffi, K.J.; Ricci, G.P.; Marques, J.A.; Nunes, F.S.; Nakagaki, S. Use of iron and manganese porphyrins in solution and immobilized on silica obtained by the sol–gel process as catalyst in the oxidation of organic substrates. Appl. Catal., A. 2011, 404, 120–128. [Google Scholar] [CrossRef]
- Castro, K.A.D.F.; Halma, M.; Machado, G.S.; Ricci, G.P.; Ucoski, G.M.; Ciuffi, K.J.; Nakagaki, S. Preparation of Catalysts based on Iron(III) Porphyrins Heterogenized on Silica obtained by the Sol-Gel Process for Hydroxylation and Epoxidation Reactions. J. Braz. Chem. Soc. 2010, 21, 1329–1340. [Google Scholar]
- Nakagaki, S.; Halma, M.; Bail, A.; Arízaga, G.G.C.; Wypych, F. First insight into catalytic activity of anionic iron porphyrins immobilized on exfoliated layered double hydroxides. J. Colloid Interf. Sci. 2005, 281, 417–423. [Google Scholar] [CrossRef]
- van der Made, A.W.; Smeets, J.W.H.; Nolte, R.J.M.; Drenth, W. Olefin epoxidation by a mono-oxygenase model. Effect of site isolation. J. Chem. Soc., Chem. Commun. 1983, 1204–1206. [Google Scholar]
- Gandini, M.E.F.; Neri, C.R.; Vinhado, F.S.; Minorin, T.S.; Nascimento, O.R.; Serra, O.A.; Iamamoto, Y. Modified Silicas Covalently Bounded to 5,10,15,20-Tetrakis(2-hydroxy-5-nitrophenyl) Porphyrinato Iron(III). Synthesis, Spectroscopic and EPR characterization. Catalytic studies. J. Braz. Chem. Soc. 2008, 19, 344–351. [Google Scholar] [CrossRef]
- Sacco, H.C.; Ciuffi, K.J.; Biazzotto, J.C.; Zuccki, M.R.; Leite, C.A.P.; Nascimento, O.R.; Serra, O.A.; Iamamoto, Y. Synthesis of manganese porphyrinosilica imprinted with templates using the sol-gel process. J. Non-Cryst. Solids. 2000, 273, 150–158. [Google Scholar] [CrossRef]
- Battioni, P.; Cardin, E.; Louloudi, M.; Schöllhorn, B.; Spyroulias, G.A.; Mansuy, D.; Traylor, T.G. Metalloporphyrinosilicas: A new class of hybrid organic-inorganic materials acting as selective biomimetic oxidation catalysts. Chem. Commun. 1996, 2037–2038. [Google Scholar]
- Pires, S.M.G.; Paula, R.D.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Santos, I.C.M.S.; Tomé, A.C.; Cavaleiro, J.A.S. A new silica-supported manganese chlorin as a biomimetic oxidation catalyst. Catal. Commun. 2009, 11, 24–28. [Google Scholar]
- Takagi, S.; Eguchi, M.; Tryk, D.A.; Inoue, H. Porphyrin photochemistry in inorganic/organic hybrid materials: Clays, layered semiconductors, nanotubes, and mesoporous materials. J. Photochem. Photobiol., C 2006, 7, 104–126. [Google Scholar] [CrossRef]
- Smith, J.V. Definition of a zeolite. Zeolites 1984, 4, 309–310. [Google Scholar] [CrossRef]
- Nakagaki, S.; Xavier, C.R.; Wosniak, A.J.; Mangrich, A.S.; Wypych, F.; Cantão, M.P.; Denicoló, I.; Kubota, L.T. Synthesis and characterization of zeolite-encapsulated metalloporphyrins. Colloids Surf., A 2000, 168, 261–276. [Google Scholar] [CrossRef]
- Santos, J.S.D.; Faria, A.L.; Amorin, P.M.D.S.; Luna, F.M.L.; Caiado, K.L.; Silva, D.O.C.E.; Sartoratto, P.P.C.; Assis, M.D. Iron(III) porphyrin covalently supported onto magnetic amino-functionalized nanospheres as catalyst for hydrocarbon and herbicide oxidations. J. Braz. Chem. Soc. 2012, 23, 1411–1420. [Google Scholar] [CrossRef]
- Aparecida Vidoto, E.; Silvia Monsalves Moreira, M.; da Silva Vinhado, F.; Jorge Ciuffi, K.; Rangel Nascimento, O.; Iamamoto, Y. Immobilization of β halogenated ironporphyrin in the silica matrix by the sol–gel process. J. Non-Cryst. Solids. 2002, 304, 151–159. [Google Scholar] [CrossRef]
- Cady, S.S.; Pinnavaia, T.J. Porphyrin intercalation in mica-type silicates. Inorg. Chem. 1978, 17, 1501–1507. [Google Scholar]
- van Damme, H.; Crespin, M.; Obrecht, F.; Cruz, M.I.; Fripiat, J.J. Acid-base and complexation behavior of porphyrins on the intracrystal surface of swelling clays: Meso-tetraphenylporphyrin and meso-tetra(4-pyridyl)porphyrin on montmorillonites. J. Colloid Interf. Sci. 1978, 66, 43–54. [Google Scholar] [CrossRef]
- de Lima, O.J.; de Aguirre, D.P.; de Oliveira, D.C.; da Silva, M.A.; Mello, C.; Leite, C.A.P.; Sacco, H.C.; Ciuffi, K.J. Porphyrins entrapped in an alumina matrix. J. Mater. Chem. 2001, 11, 2476–2481. [Google Scholar] [CrossRef]
- Corma, A.; García, H.; Llabrés i Xamena, F.X. Engineering metal organic frameworks for heterogeneous catalysis. Chem. Rev. 2010, 110, 4606–4655. [Google Scholar] [CrossRef]
- Rowsell, J.L.C.; Yaghi, O.M. Metal-organic frameworks: A new class of porous materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Matsubara, I.; Higuciu, T.; Saito, Y. The Crystal Structure of Bis(adiponitrilo)copper(I) Nitrate. Bull Chem. Soc. Jpn 1959, 32, 1221–1226. [Google Scholar]
- Hoskins, B.F.; Robson, R. Design and construction of a new class of scaffolding-like materials comprising infinite polymeric frameworks of 3D-linked molecular rods. A reappraisal of the zinc cyanide and cadmium cyanide structures and the synthesis and structure of the diamond-related frameworks [N(CH3)4][CuIZnII(CN)4] and CuI[4,4',4'',4'''-tetracyanotetraphenylmethane]BF4.xC6H5NO2. J. Am. Chem. Soc. 1990, 112, 1546–1554. [Google Scholar] [CrossRef]
- Robson, R.; Abrahams Brendan, F.; Batten Stuart, R.; Gable Robert, W.; Hoskins Bernard, F.; Liu, J. Crystal Engineering of Novel Materials Composed of Infinite Two- and Three-Dimensional Frameworks. In Supramolecular Architecture; American Chemical Society: Washington, DC, USA, 1992; Volume 499, pp. 256–273. [Google Scholar]
- Batten, S.R.; Hoskins, B.F.; Robson, R. Two Interpenetrating 3D Networks Which Generate Spacious Sealed-Off Compartments Enclosing of the Order of 20 Solvent Molecules in the Structures of Zn(CN)(NO3)(tpt)2/3.solv (tpt = 2,4,6-tri(4-pyridyl)-1,3,5-triazine, solv = ~3/4C2H2Cl4.3/4CH3OH or .apprx.3/2CHCl3. 1/3CH3OH). J. Am. Chem. Soc. 1995, 117, 5385–5386. [Google Scholar] [CrossRef]
- Yaghi, O.M.; Li, H. Hydrothermal Synthesis of a Metal-Organic Framework Containing Large Rectangular Channels. J. Am. Chem. Soc. 1995, 117, 10401–10402. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O'Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of metal-organic frameworks (MOFs): Routes to various MOF topologies, Morphologies, and Composites. Chem. Rev. 2011, 112, 933–969. [Google Scholar] [CrossRef]
- Jung, D.-W.; Yang, D.-A.; Kim, J.; Kim, J.; Ahn, W.-S. Facile synthesis of MOF-177 by a sonochemical method using 1-methyl-2-pyrrolidinone as a solvent. Dalton Trans. 2010, 39, 2883–2887. [Google Scholar]
- Klinowski, J.; Almeida Paz, F.A.; Silva, P.; Rocha, J. Microwave-Assisted Synthesis of Metal-Organic Frameworks. Dalton Trans. 2011, 40, 321–330. [Google Scholar] [CrossRef]
- Mueller, U.; Puetter, H.; Hesse, M.; Wessel, H.W. Method for electrochemical production of A crystalline porous metal organic skeleton material. WO/2005/049892, 6 February 2005. [Google Scholar]
- Friscic, T. New opportunities for materials synthesis using mechanochemistry. J. Mater. Chem. 2010, 20, 7599–7605. [Google Scholar] [CrossRef]
- Pichon, A.; Lazuen-Garay, A.; James, S.L. Solvent-free synthesis of a microporous metal-organic framework. CrystEngComm 2006, 8, 211–214. [Google Scholar] [CrossRef]
- Qiu, L.-G.; Li, Z.-Q.; Wu, Y.; Wang, W.; Xu, T.; Jiang, X. Facile synthesis of nanocrystals of a microporous metal-organic framework by an ultrasonic method and selective sensing of organoamines. Chem. Commun. 2008, 3642–3644. [Google Scholar]
- Tanabe, K.K.; Cohen, S.M. Postsynthetic modification of metal-organic frameworks—a progress report. Chem. Soc. Rev. 2011, 40, 498–519. [Google Scholar]
- Champness, N.R. The future of metal-organic frameworks. Dalton Trans. 2011, 40, 10311–10315. [Google Scholar] [CrossRef]
- Reticular Chemistry Structure Resource. Available online: http://rcsr.anu.edu.au/ (accessed on 28 March 2013).
- O’Keeffe, M.; Peskov, M.A.; Ramsden, S.J.; Yaghi, O.M. The Reticular chemistry structure resource (RCSR) database of, and symbols for, Crystal nets. Acc. Chem. Res. 2008, 41, 1782–1789. [Google Scholar] [CrossRef]
- Tranchemontagne, D.J.; Mendoza-Cortes, J.L.; O'Keeffe, M.; Yaghi, O.M. Secondary building units, Nets and bonding in the chemistry of metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1257–1283. [Google Scholar]
- Farha, O.K.; Hupp, J.T. Rational design, Synthesis, Purification, and activation of metal−organic Framework materials. Acc. Chem. Res. 2010, 43, 1166–1175. [Google Scholar] [CrossRef]
- Farha, O.K.; Eryazici, I.; Jeong, N.C.; Hauser, B.G.; Wilmer, C.E.; Sarjeant, A.A.; Snurr, R.Q.; Nguyen, S.T.; Yazaydın, A.Ö.; Hupp, J.T. Metal–Organic framework materials with ultrahigh surface areas: Is the sky the limit? J. Am. Chem. Soc. 2012, 134, 15016–15021. [Google Scholar] [CrossRef]
- Murray, L.J.; Dinca, M.; Long, J.R. Hydrogen storage in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1294–1314. [Google Scholar]
- Hu, Y.H.; Zhang, L. Hydrogen storage in Metal–Organic frameworks. Adv. Mater. 2010, 22, E117–E130. [Google Scholar] [CrossRef]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar]
- Li, J.-R.; Sculley, J.; Zhou, H.-C. Metal–Organic frameworks for separations. Chem. Rev. 2011, 112, 869–932. [Google Scholar]
- Min, K.S.; Suh, M.P. Silver(I)−Polynitrile network solids for anion exchange: Anion-Induced transformation of supramolecular structure in the crystalline state. J. Am. Chem. Soc. 2000, 122, 6834–6840. [Google Scholar] [CrossRef]
- Ni, Z.; Jerrell, J.P.; Cadwallader, K.R.; Masel, R.I. Metal−Organic frameworks as adsorbents for trapping and preconcentration of organic phosphonates. Anal. Chem. 2007, 79, 1290–1293. [Google Scholar] [CrossRef]
- Xiong, R.; Fern, J.T.; Keffer, D.J.; Fuentes-Cabrera, M.; Nicholson, D.M. Molecular simulations of adsorption and diffusion of RDX in IRMOF-1. Mol. Simul. 2009, 35, 910–919. [Google Scholar] [CrossRef]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Van Duyne, R.P.; Hupp, J.T. Metal−Organic framework materials as chemical sensors. Chem. Rev. 2011, 112, 1105–1125. [Google Scholar]
- An, J.; Geib, S.J.; Rosi, N.L. Cation-Triggered Drug Release from a Porous Zinc−adeninate Metal−organic framework. J. Am. Chem. Soc. 2009, 131, 8376–8377. [Google Scholar] [CrossRef]
- Horcajada, P.; Serre, C.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Metal−Organic frameworks as efficient materials for drug delivery. Angew. Chem., Int. Ed. 2006, 45, 5974–5978. [Google Scholar]
- Taylor-Pashow, K.M.L.; Rocca, J.D.; Xie, Z.; Tran, S.; Lin, W. Postsynthetic modifications of Iron-Carboxylate nanoscale Metal−Organic frameworks for imaging and drug delivery. J. Am. Chem. Soc. 2009, 131, 14261–14263. [Google Scholar] [CrossRef]
- Lee, C.Y.; Farha, O.K.; Hong, B.J.; Sarjeant, A.A.; Nguyen, S.T.; Hupp, J.T. Light-Harvesting Metal–Organic frameworks (MOFs): Efficient strut-to-strut energy transfer in bodipy and Porphyrin-based MOFs. J. Am. Chem. Soc. 2011, 133, 15858–15861. [Google Scholar]
- Cho, S.-H.; Ma, B.; Nguyen, S.T.; Hupp, J.T.; Albrecht-Schmitt, T.E. A metal-organic framework material that functions as an enantioselective catalyst for olefin epoxidation. Chem. Commun. 2006, 2563–2565. [Google Scholar]
- Kitaura, R.; Onoyama, G.; Sakamoto, H.; Matsuda, R.; Noro, S.-I.; Kitagawa, S. Immobilization of a Metallo Schiff Base into a Microporous Coordination Polymer. Angew. Chem., Int. Ed. 2004, 43, 2684–2687. [Google Scholar]
- Szeto, K.C.; Kongshaug, K.O.; Jakobsen, S.; Tilset, M.; Lillerud, K.P. Design, Synthesis and characterization of a Pt-Gd metal-organic framework containing potentially catalytically active sites. Dalton Trans. 2008, 2054–2060. [Google Scholar]
- Dong, G.; Bing-guang, Z.; Chun-ying, D.; Xin, C.; Qing-jin, M. A novel ferrocene-barium sandwich sheet-shaped coordination polymer and its solid-state electrochemistry. Dalton Trans. 2003, 282–284. [Google Scholar]
- Horikoshi, R.; Mochida, T.; Moriyama, H. Synthesis and characterization of Redox-Active coordination polymers generated from ferrocene-containing bridging ligands. Inorg. Chem. 2002, 41, 3017–3024. [Google Scholar] [CrossRef]
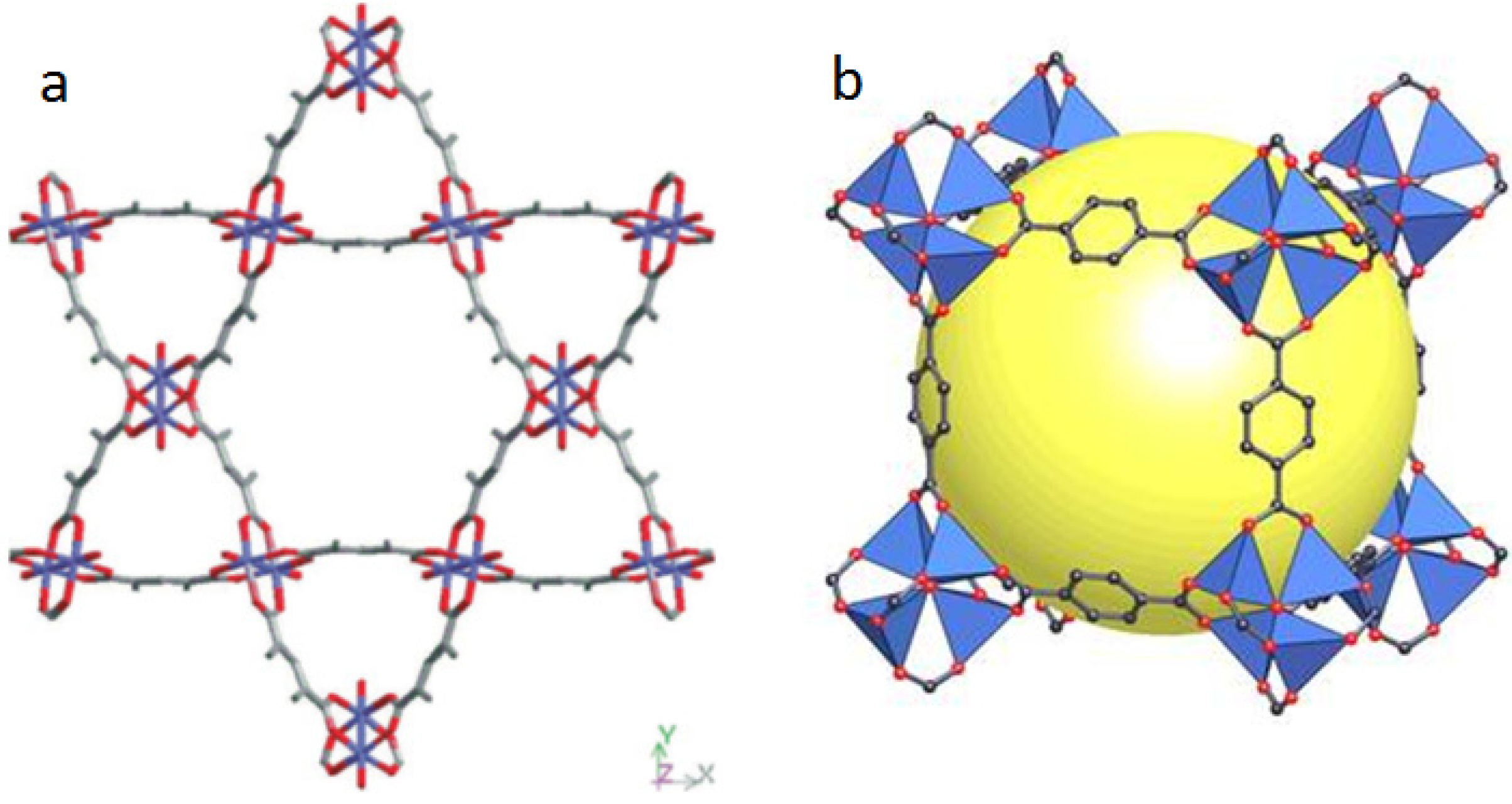
- Abrahams, B.F.; Hoskins, B.F.; Robson, R. A new type of infinite 3D polymeric network containing 4-connected, peripherally-linked metalloporphyrin building blocks. J. Am. Chem. Soc. 1991, 113, 3606–3607. [Google Scholar] [CrossRef]
- Suslick, K.S.; Bhyrappa, P.; Chou, J.H.; Kosal, M.E.; Nakagaki, S.; Smithenry, D.W.; Wilson, S.R. Microporous Porphyrin Solids. Acc. Chem. Res. 2005, 38, 283–291. [Google Scholar] [CrossRef]
- Burnett, B.J.; Barron, P.M.; Choe, W. Recent advances in porphyrinic metal-organic frameworks: Materials design, Synthetic strategies, and emerging applications. CrystEngComm 2012, 14, 3839–3846. [Google Scholar] [CrossRef]
- Choi, E.-Y.; Barron, P.M.; Novotny, R.W.; Son, H.-T.; Hu, C.; Choe, W. Pillared porphyrin homologous series: Intergrowth in metal-organic frameworks. Inorg. Chem. 2008, 48, 426–428. [Google Scholar]
- Farha, O.K.; Shultz, A.M.; Sarjeant, A.A.; Nguyen, S.T.; Hupp, J.T. Active-site-accessible, Porphyrinic metal−organic framework materials. J. Am. Chem. Soc. 2011, 133, 5652–5655. [Google Scholar]
- Takaishi, S.; DeMarco, E.J.; Pellin, M.J.; Farha, O.K.; Hupp, J.T. Solvent-assisted linker exchange (SALE) and post-assembly metallation in porphyrinic metal-organic framework materials. Chem. Sci. 2013, 4, 1509–1513. [Google Scholar]
- Wang, X.-S.; Meng, L.; Cheng, Q.; Kim, C.; Wojtas, L.; Chrzanowski, M.; Chen, Y.-S.; Zhang, X.P.; Ma, S. Three-dimensional porous metal–metalloporphyrin framework consisting of nanoscopic polyhedral cages. J. Am. Chem. Soc. 2011, 133, 16322–16325. [Google Scholar] [CrossRef]
- Wang, X.-S.; Chrzanowski, M.; Kim, C.; Gao, W.-Y.; Wojtas, L.; Chen, Y.-S.; Peter Zhang, X.; Ma, S. Quest for highly porous metal-metalloporphyrin framework based upon a custom-designed octatopic porphyrin ligand. Chem. Commun. 2012, 48, 7173–7175. [Google Scholar]
- Wang, X.-S.; Chrzanowski, M.; Gao, W.-Y.; Wojtas, L.; Chen, Y.-S.; Zaworotko, M.J.; Ma, S. Vertex-directed self-assembly of a high symmetry supermolecular building block using a custom-designed porphyrin. Chem. Sci. 2012, 3, 2823–2827. [Google Scholar] [CrossRef]
- Meng, L.; Cheng, Q.; Kim, C.; Gao, W.-Y.; Wojtas, L.; Chen, Y.-S.; Zaworotko, M.J.; Zhang, X.P.; Ma, S. Crystal engineering of a microporous, Catalytically active fcu topology MOF using a custom-designed metalloporphyrin Linker. Angew. Chem., Int. Ed. 2012, 51, 10082–10085. [Google Scholar] [CrossRef]
- Chen, Y.; Hoang, T.; Ma, S. Biomimetic catalysis of a porous iron-based metal-metalloporphyrin framework. Inorg. Chem. 2012, 51, 12600–12602. [Google Scholar] [CrossRef]
- Wang, X.-S.; Chrzanowski, M.; Wojtas, L.; Chen, Y.-S.; Ma, S. Formation of a metalloporphyrin-based nanoreactor by postsynthetic metal–ion exchange of a polyhedral-cage containing a metal–metalloporphyrin framework. Chem. Eur. J. 2013, 19, 3297–3301. [Google Scholar] [CrossRef]
- Lee, J.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal-organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-Catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef]
- Nam, W.; Kim, I.; Lim, M.H.; Choi, H.J.; Lee, J.S.; Jang, H.G. Isolation of an Oxomanganese(V) porphyrin intermediate in the reaction of a manganese(III) porphyrin complex and H2O2 in aqueous solution. Chem. Eur. J. 2002, 8, 2067–2071. [Google Scholar]
- Groves, J.T.; Kruper, W.J.; Haushalter, R.C. Hydrocarbon oxidations with oxometalloporphinates. Isolation and reactions of a (porphinato)manganese(V) complex. J. Am. Chem. Soc. 1980, 102, 6375–6377. [Google Scholar] [CrossRef]
- Kameyama, H.; Narumi, F.; Hattori, T.; Kameyama, H. Oxidation of cyclohexene with molecular oxygen catalyzed by cobalt porphyrin complexes immobilized on montmorillonite. J. Mol. Catal. A: Chem. 2006, 258, 172–177. [Google Scholar]
- Goifman, A.; Gun, J.; Gitis, V.; Kamyshny, A., Jr.; Lev, O.; Donner, J.; Börnick, H.; Worch, E. Pyrolysed carbon supported cobalt porphyrin: A potent catalyst for oxidation of hydrogen sulfide. Appl. Catal., B 2004, 54, 225–235. [Google Scholar]
- Appleton, A.J.; Evans, S.; Smith, J.R.L. Allylic oxidation and epoxidation of cycloalkenes by iodosylbenzene catalysed by iron(III) and manganese(III) tetra(dichlorophenyl)porphyrin: the marked influence of ring size on the rate of allylic oxidation. J. Chem. Soc., Perkin Trans. 2 1996, 281–285. [Google Scholar] [CrossRef]
- Thomas, J.M.; Hernandez-Garrido, J.C.; Raja, R.; Bell, R.G. Nanoporous oxidic solids: The confluence of heterogeneous and homogeneous catalysis. Phys. Chem. Chem. Phys. 2009, 11, 2799–2825. [Google Scholar] [CrossRef]
- Shultz, A.M.; Farha, O.K.; Hupp, J.T.; Nguyen, S.T. A Catalytically active, permanently microporous MOF with metalloporphyrin struts. J. Am. Chem. Soc. 2009, 131, 4204–4205. [Google Scholar]
- Friedermann, G.R.; Halma, M.; de Freitas Castro, K.A.D.; Benedito, F.L.; Doro, F.G.; Drechsel, S.M.; Mangrich, A.S.; Assis, M.D.D.; Nakagaki, S. Intermediate species generated from halogenated manganese porphyrins electrochemically and in homogeneous catalysis of alkane oxidation. Appl. Catal., A. 2006, 308, 172–181. [Google Scholar] [CrossRef]
- Choi, E.-Y.; Wray, C.A.; Hu, C.; Choe, W. Highly tunable metal-organic frameworks with open metal centers. CrystEngComm 2009, 11, 553–555. [Google Scholar] [CrossRef]
- Feng, D.; Gu, Z.-Y.; Li, J.-R.; Jiang, H.-L.; Wei, Z.; Zhou, H.-C. Zirconium-metalloporphyrin PCN-222: Mesoporous metal–organic frameworks with ultrahigh stability as biomimetic catalysts. Angew. Chem., Int. Ed. 2012, 51, 10307–10310. [Google Scholar]
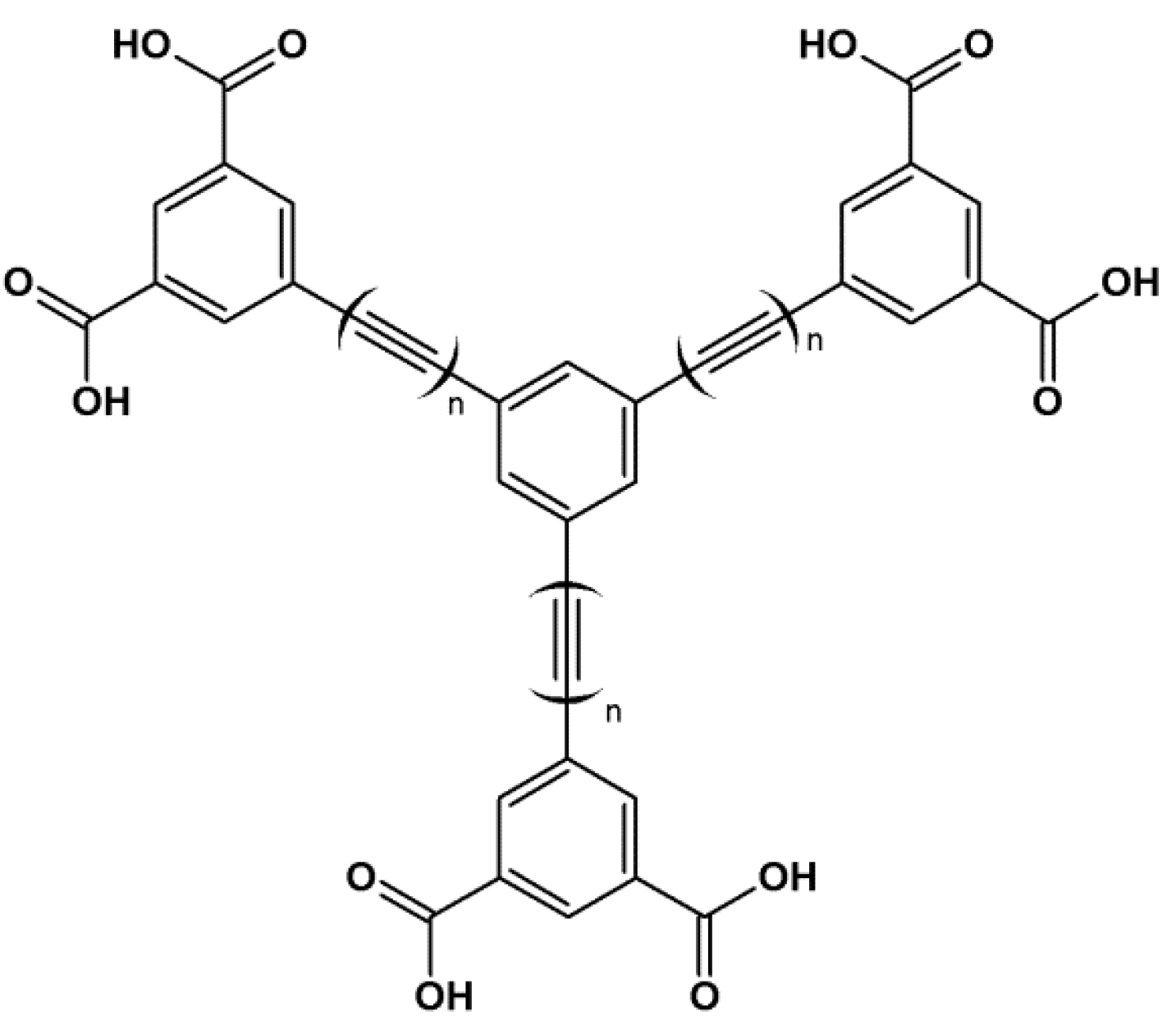
- Yang, X.-L.; Xie, M.-H.; Zou, C.; He, Y.; Chen, B.; O’Keeffe, M.; Wu, C.-D. Porous metalloporphyrinic frameworks constructed from metal 5,10,15,20-Tetrakis(3,5-biscarboxylphenyl)porphyrin for highly efficient and selective catalytic oxidation of alkylbenzenes. J. Am. Chem. Soc. 2012, 134, 10638–10645. [Google Scholar]
- Lee, D.H.; Kim, S.; Hyun, M.Y.; Hong, J.-Y.; Huh, S.; Kim, C.; Lee, S.J. Controlled growth of narrowly dispersed nanosize hexagonal MOF rods from Mn(III)-porphyrin and In(NO3)3 and their application in olefin oxidation. Chem. Commun. 2012, 48, 5512–5514. [Google Scholar]
- Xie, M.-H.; Yang, X.-L.; Wu, C.-D. A metalloporphyrin functionalized metal-organic framework for selective oxidization of styrene. Chem. Commun. 2011, 47, 5521–5523. [Google Scholar] [CrossRef]
- Zou, C.; Zhang, T.; Xie, M.-H.; Yan, L.; Kong, G.-Q.; Yang, X.-L.; Ma, A.; Wu, C.-D. Four metalloporphyrinic frameworks as heterogeneous catalysts for selective oxidation and aldol reaction. Inorg. Chem. 2013, 52, 3620–3626. [Google Scholar] [CrossRef]
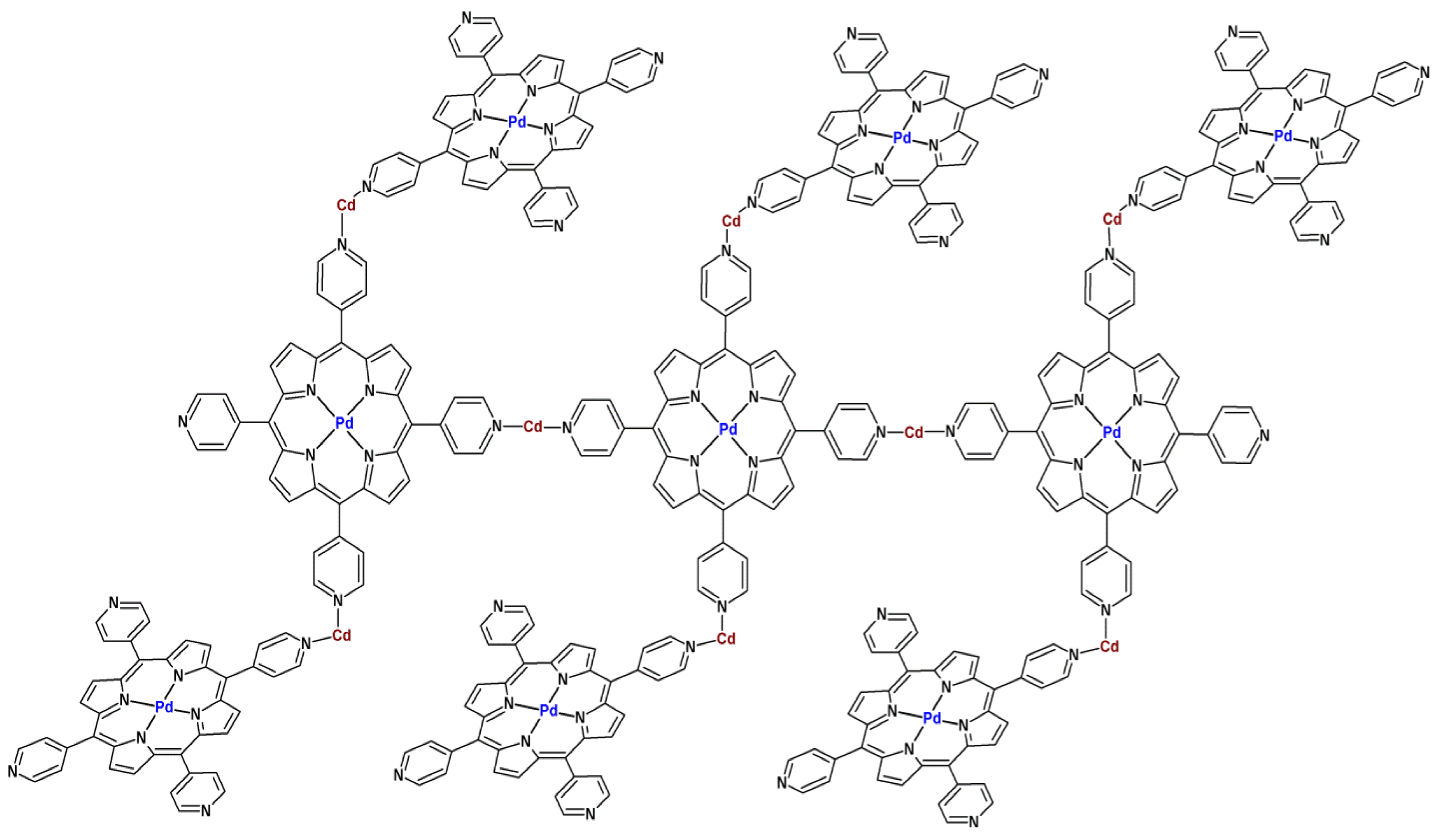
- Oliveri, C.G.; Gianneschi, N.C.; Nguyen, S.T.; Mirkin, C.A.; Stern, C.L.; Wawrzak, Z.; Pink, M. supramolecular allosteric cofacial porphyrin complexes. J. Am. Chem. Soc. 2006, 128, 16286–16296. [Google Scholar]
- Roy, S.; George, C.B.; Ratner, M.A. Catalysis by a Zinc-porphyrin-based metal-organic framework: From theory to computational design. J. Chem. Phys. C 2012, 116, 23494–23502. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakagaki, S.; Ferreira, G.K.B.; Ucoski, G.M.; Dias de Freitas Castro, K.A. Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks. Molecules 2013, 18, 7279-7308. https://doi.org/10.3390/molecules18067279
Nakagaki S, Ferreira GKB, Ucoski GM, Dias de Freitas Castro KA. Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks. Molecules. 2013; 18(6):7279-7308. https://doi.org/10.3390/molecules18067279
Chicago/Turabian StyleNakagaki, Shirley, Gabriel Kaetan Baio Ferreira, Geani Maria Ucoski, and Kelly Aparecida Dias de Freitas Castro. 2013. "Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks" Molecules 18, no. 6: 7279-7308. https://doi.org/10.3390/molecules18067279
APA StyleNakagaki, S., Ferreira, G. K. B., Ucoski, G. M., & Dias de Freitas Castro, K. A. (2013). Chemical Reactions Catalyzed by Metalloporphyrin-Based Metal-Organic Frameworks. Molecules, 18(6), 7279-7308. https://doi.org/10.3390/molecules18067279